
Abstract
Significance:
Polysulfide species (i.e., R-S n -R′, n > 2; and R-S n -H, n > 1) exist in many organisms. The highly nucleophilic nature of hydropersulfides and hydropolysulfides contributes to the potent antioxidant activities of polysulfide species that protect organisms against oxidative and electrophilic stresses.
Recent Advances:
Accumulating evidence suggests that organic polysulfides (R-S n -R′) readily undergo alkaline hydrolysis, which results in formation of both nucleophilic hydrosulfide/polysulfide (R-S n−1H) and electrophilic sulfenic acid (R′SOH) species. Polysulfides maintain a steady-state equilibrium that is driven by hydrolysis even in aqueous physiological milieus. This unique property makes polysulfide chemistry and biology more complex than previously believed.
Critical Issues:
The hydrolysis equilibrium of polysulfides shifts to the right when electrophiles are present. Strong electrophilic alkylating agents (e.g., monobromobimane) greatly enhance polysulfide hydrolysis, which leads to increased polysulfide degradation and artifactual formation of bis-S-bimane adducts in the absence of free hydrogen sulfide. The finding that hydroxyl group-containing substances such as tyrosine efficiently protected polysulfides from hydrolysis led to development of the new alkylating agent, N-iodoacetyl
Future Directions:
Using precise methodologies to achieve a better understanding of the occurrence and metabolism of polysulfide species is necessary to gain insights into the undefined biology of polysulfide species. Antioxid. Redox Signal. 36, 327–336.
Introduction
Many investigations have confirmed the presence of hydropersulfides (RSSH) and higher order polysulfide species (i.e., R-S n -R′, n > 2; and R-S n -H, n > 1, with R = cysteine, glutathione, or proteins) in various organisms, including bacteria and mammals (2, 14, 24, 26, 30, 34, 35, 37, 39, 40). Accumulating evidence has suggested that these reactive sulfur species are involved in regulation of diverse physiological and pathological phenomena (14, 39, 40). For instance, cysteine persulfide (CysSSH) can serve as an important intermediate in the biosynthesis of sulfur-containing molecules, such as iron–sulfur clusters, biotin, and lipoic acid, by donating its sulfane sulfur atom (23, 47). CysSSH is also reportedly involved in regulation of tRNA methylthiolation and insulin secretion (44). Reactive sulfur species also exist as glutathione-bound forms (e.g., glutathione persulfide [GSSH]) and protein-bound forms (2, 24). Because of their strong nucleophilicity and reducing ability, reactive sulfur species can act as powerful antioxidants and play important roles in cells by regulating oxidative stress and redox signaling (15, 16, 24). Recent studies also suggested that reactive sulfur species can have potent anti-inflammatory activity in cells and in vivo (45, 48, 49). In bacteria, persulfides and polysulfides offer protection from external bactericidal insults caused by immune cell responses and antibiotics (29, 36). Thus, bacteria-specific persulfide-producing machinery may become an attractive target for development of antibacterial drugs (29, 36).
Cysteinyl-tRNA synthetase (CARS) has been demonstrated to act as an effective CysSSH-producing enzyme (as CPERS) (2). This CysSSH-producing ability is conserved from bacteria to mammalian cells (2). In mammalian cells, two different CARSs exist: cytosolic CARS1 and mitochondrial CARS2 (9, 18). Biochemical analyses with recombinant enzymes showed that both CARS1 and CARS2 possess potent CysSSH-producing activity (2). As an intriguing finding, CysSSH derived from CARS2 may play an important role in maintaining the mitochondrial membrane potential by acting as an electron acceptor in the electron transport chain (2, 14, 33). During this process, CysSSH is suggested to be metabolized to hydrogen sulfide (H2S), sulfite, sulfate, and other oxidized sulfur species, with this process depending on the catalytic actions of sulfide:quinone oxidoreductase and ethylmalonic encephalopathy protein 1 (ETHE1; also known as persulfide dioxygenase) in mitochondria (2, 14). Sulfide metabolism is thereby tightly controlled by the activity of these different enzymes. A recent study demonstrated that impairment of sulfide metabolism and concomitant persulfide accumulation in mitochondria led to mitochondrial dysfunction associated with hypoxia-induced brain injury (32).
Chemical biology studies of hydropersulfides and polysulfides have revealed that hydropersulfides are superior to thiols as nucleophiles and reducing agents (15, 16). Hydropersulfides can be readily deprotonated so that the pK a values are 1.5–4 orders of magnitude lower than pK a values of the corresponding thiols: the typical pK a values of thiol species are 8–9, whereas pK a values of hydropersulfide species are 4–7.5 (11, 42). A deprotonated perthiolate anion is significantly more nucleophilic compared with the corresponding thiolate anion (24, 38, 42). Polysulfide species may also be more susceptible to alkaline hydrolysis of their sulfur–sulfur bonds compared with disulfide species (2, 8, 19, 28). This unique property makes polysulfide chemistry and biology more complex than previously believed. In this review article, we discuss the hydrolysis-driven equilibrium of polysulfide species, with particular emphasis on its modulation by electrophiles. Full understanding of the chemistry and chemical biology of the equilibrium of polysulfide hydrolysis is indispensable not only for development of analytical methods for polysulfide species but also for elucidation of the fundamental impacts and physiological roles of polysulfide species formed endogenously.
Alkaline Hydrolysis of Disulfides
Hydrolysis of disulfide bonds (evident primarily under strong alkaline conditions) was studied with cystine, oxidized glutathione (GSSG), and proteins, and findings showed that hydrolysis of disulfides (RSSR) produces reduced thiols (RSH) and sulfenic acid (RSOH) species, as shown in Reaction (1) (5, 10, 12):
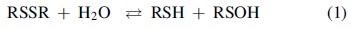
At physiological or neutral pH, equilibration in Reaction (1) shifted far to the left. Hydrolysis could be accelerated and driven to completion by including thiol-reactive reagents such as HgCl2 and p-hydroxymercuribenzoate in the solution (5). For example, hydrolysis of the disulfide bond of GSSG occurs only when the pH is higher than 9 and the solution contains thiol-reactive reagents such as p-hydroxymercuribenzoate (5).
Hydrolysis-Driven Equilibrium of Sulfur–Sulfur Bonds in Polysulfides
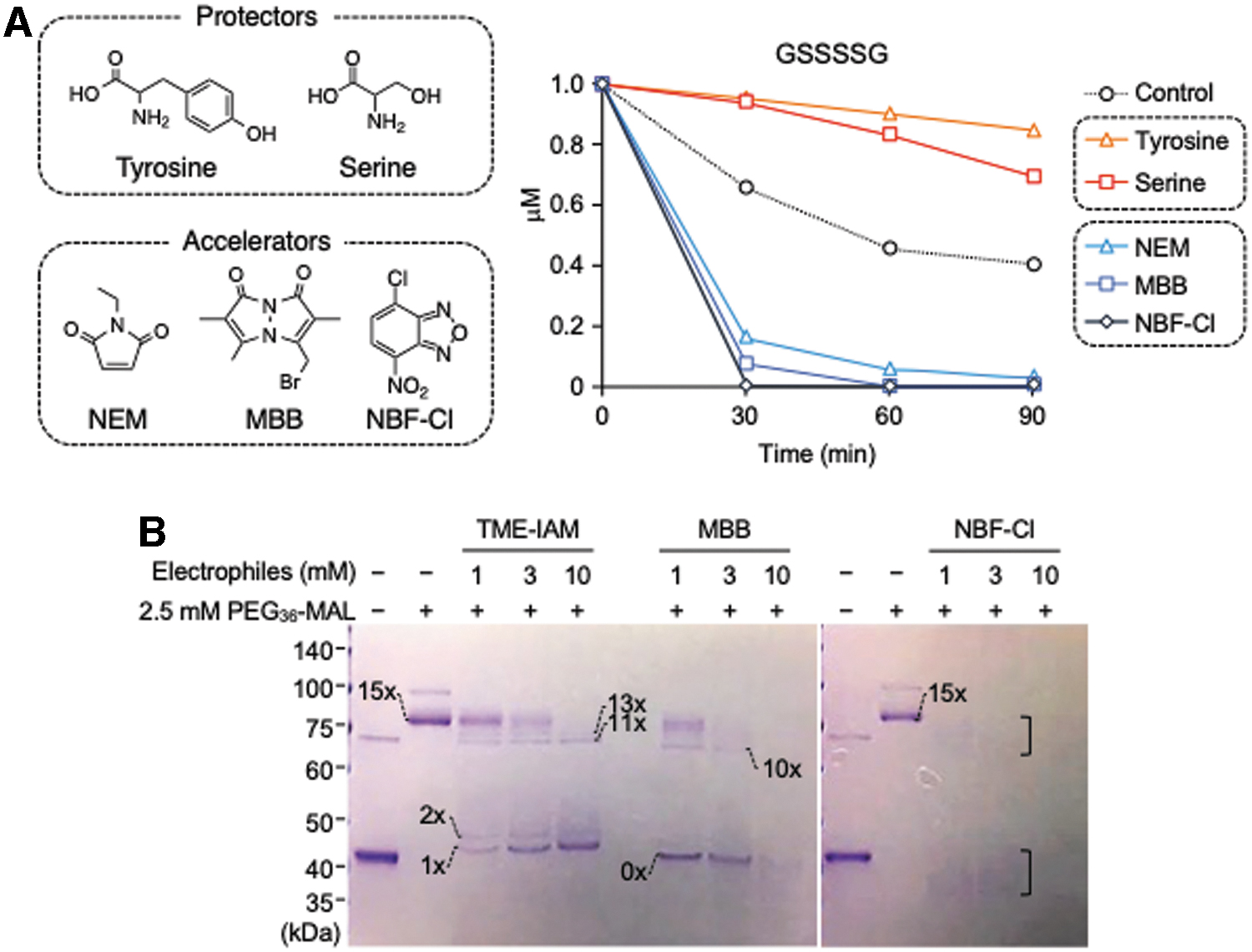
Consistent with this disulfide reactivity, sulfur–sulfur bonds in symmetrical disulfides are reportedly stable in aqueous media with neutral pH even in the presence of strong electrophilic thiol-reactive reagents such as iodoacetamide (IAM), monobromobimane (MBB), and N-ethylmaleimide (NEM) (2, 8). According to careful and detailed studies with glutathione polysulfides (GS n G, n > 2) (2) and alkylated polysulfides (RS n R, n > 2) (8), however, sulfur–sulfur bonds in polysulfides become susceptible to hydrolysis even under weak alkaline and physiological conditions, compared with such bonds in disulfides. The hydrolysis of polysulfides, which already occurs at physiological pH, results in formation of reduced thiols or hydropolysulfides (RS n−1H) and RSOH or polysulfenic acid (RS n−1OH) [Reaction (2); Figs. 1 and 2] (19).
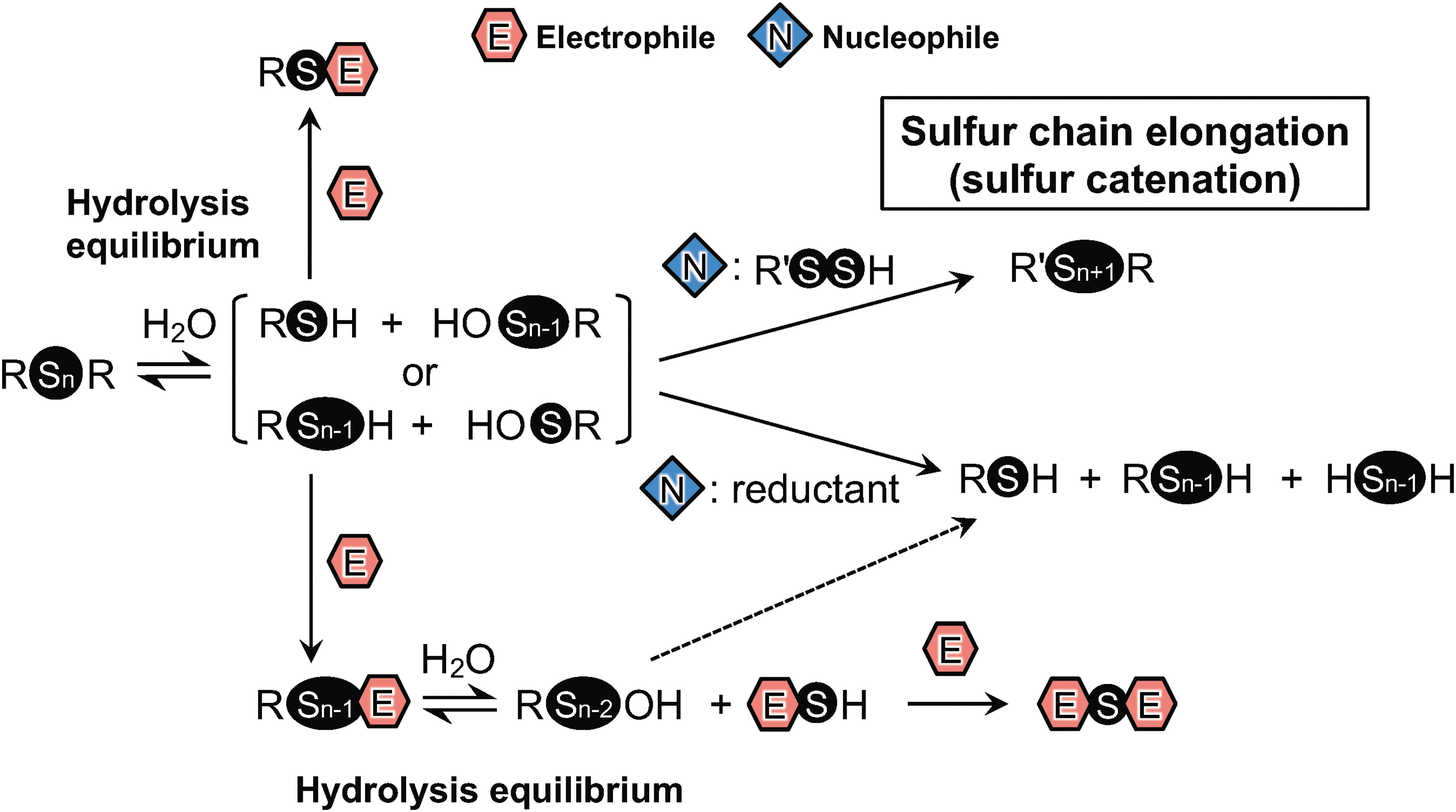
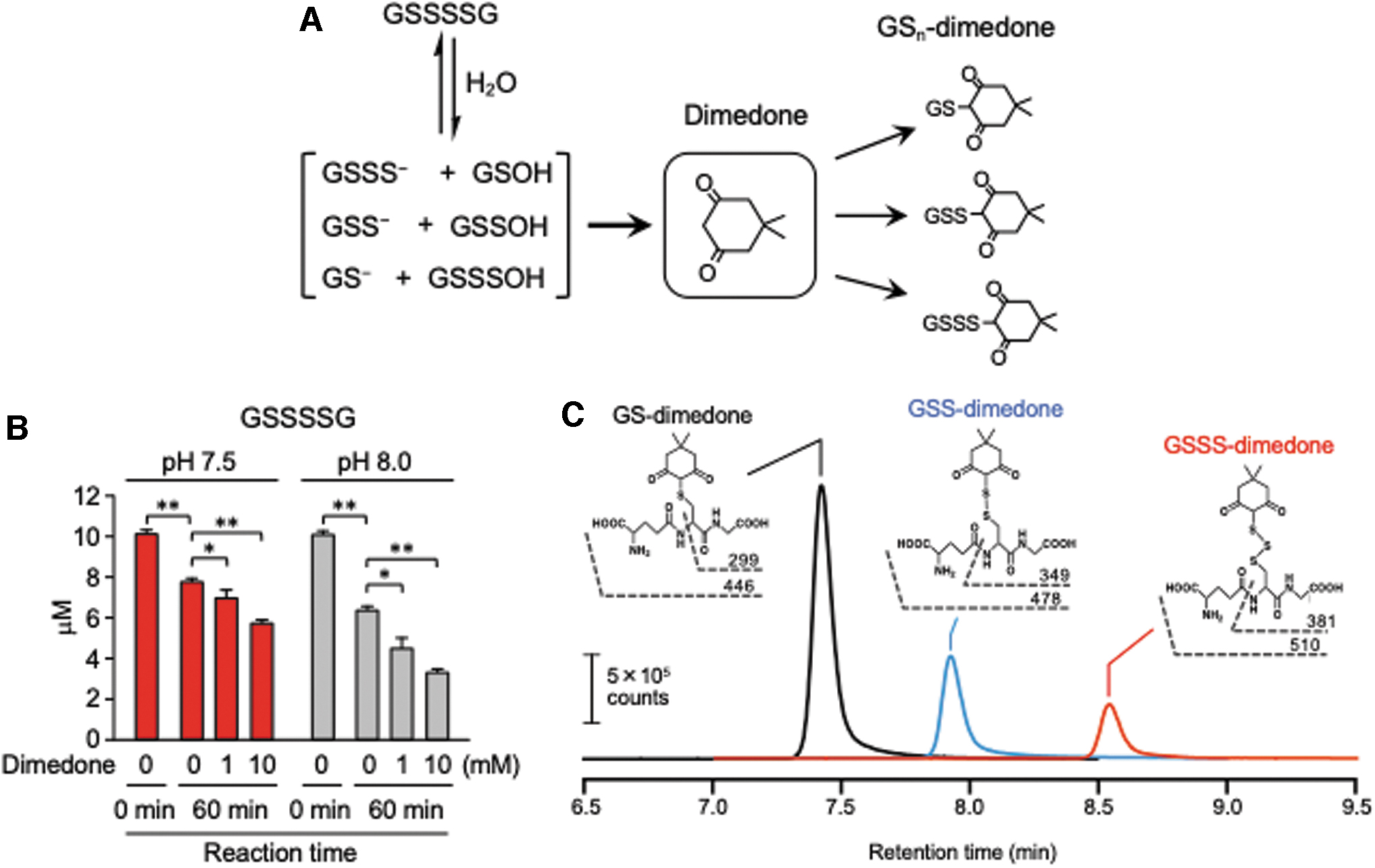
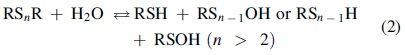
Electrophilic thiol-reactive reagents react with and capture thiols and hydropolysulfides, which drive hydrolysis (hydrolysis equilibrium shifts to the right). In fact, we observed that the hydrolysis of glutathione polysulfides was greatly enhanced by addition of strong electrophiles such as NEM, MBB, and 4-chloro-7-nitrobenzofurazan (NBF-Cl), with concomitant formation of glutathione–electrophile adducts (Figs. 1 and 3) (2). As with symmetrical polysulfides, hydrolysis of asymmetrical polysulfides (RS n R′, n > 2) was also noted (2, 8). Ab initio calculations suggested that the RSOH-reactive reagent, dimedone, does not react directly with polysulfide species, but only with RSOH/RS n−1OH (22). Consistent with this notion, formation of RSOH/RS n−1OH was supported by the finding that addition of dimedone could also shift the hydrolysis equilibrium equation to the right (Fig. 2) (2, 8, 19).
RSOH can react with thiols to form disulfides (3, 4). In eukaryotic and prokaryotic cells, appreciable amounts of GSSH have been found. For instance, high micromolar levels of GSSH were detected in mouse tissues, including the brain, liver, and lung (2, 24). The reaction between RS n−1OH and such hydropersulfides (R′SSH) will result in formation of longer chain polysulfides (R′S n+1R) (Fig. 1). Thus, polysulfide hydrolysis may play an important role in initiating sulfur chain elongation (sulfur catenation), as Figure 1 illustrates.
Electrophilic thiol-reactive reagents (E) can react with thiols to form an electrophile–thiol adduct (RSE) (Fig. 1). The RSE thus formed is stable, and no additional reaction with electrophiles and nucleophiles will occur. Electrophile–polysulfide adducts (RS n−1E), however, particularly those with more than two sulfur atoms, are susceptible to alkaline hydrolysis, which leads to formation of secondary metabolites, as shown in Figure 1. Electrophile–sulfide adducts then react with electrophiles to form bis-sulfur–electrophile adducts (ESEs). ESEs also form in the reaction of electrophilic thiol-reactive reagents with H2S. For instance, the strong electrophile, MBB, was used to detect H2S by means of measuring the formation of the ESE of MBB (41, 46). However, as just mentioned, ESEs may form in the absence of free H2S in reactions of polysulfides with electrophiles. Thus, analysis using strong electrophiles, such as MBB, NEM, and NBF-Cl, to speciate a complex mixture of reactive sulfur species is quite difficult (Figs. 1 and 2) (2, 8, 19, 28). While the harsh electrophiles are not suited to assess free sulfide levels, a value for total sulfide is reportedly obtained using a strong electrophilic probe such as NEM whenever persulfides and polysulfides are present, as pointed out by Sutton et al. (43). Thiol-reactive reagents that are suitable for the precise analysis of reactive sulfur species are discussed in detail below.
Polysulfide Bond Stabilization by Hydroxyl Group-Containing Compounds
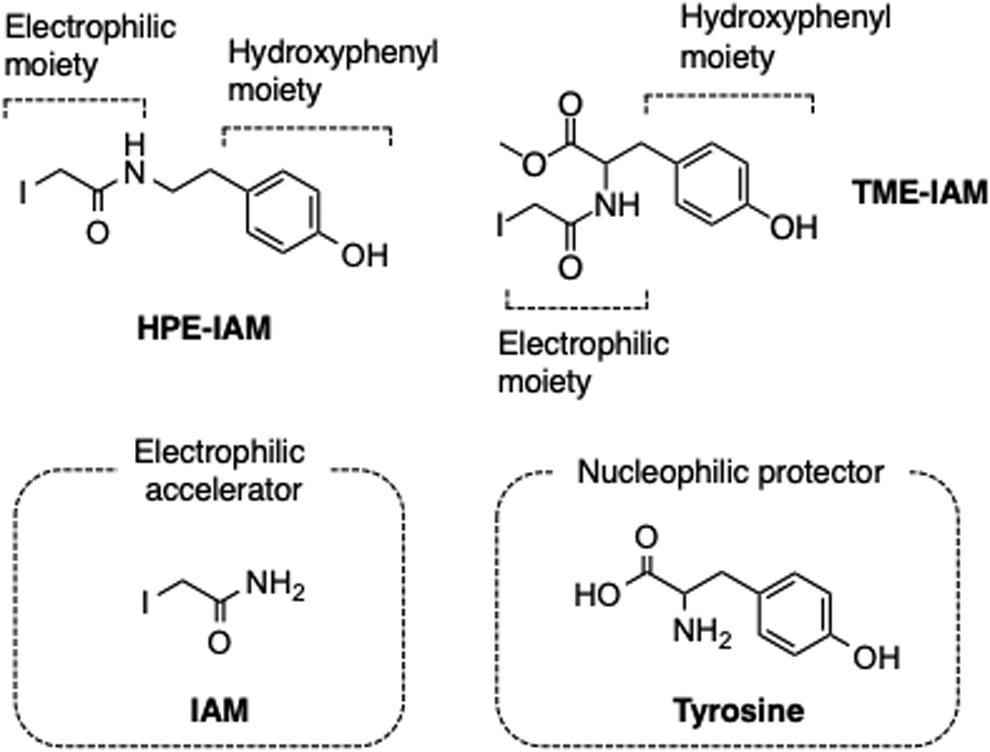
As described above, the hydrolysis equilibrium of polysulfides can be shifted to the right by the presence of electrophiles (Fig. 1). Hydrolysis-driving potentials varied among the electrophiles studied; NBF-Cl, MBB, and NEM were the strongest (hydrolysis accelerators; Fig. 3), whereas IAM derivatives that contain tyrosine-like residues, such as β-(4-hydroxyphenyl)ethyl (HPE)-IAM and N-iodoacetyl
Hydropolysulfide Alkylation in the Analysis of Reactive Sulfur Species
As discussed earlier, hydropolysulfides are reportedly more acidic than the corresponding thiols. For example, the pK a value of GSSH has been determined to be 5.45, which is significantly lower than the pK a value of glutathione—8.94 (7). Because of this acidity, hydropolysulfides exist in their deprotonated state (polythiolate anions, RS n −) at the physiological pH range (7, 15, 17). In addition, RS n − thus formed can be more nucleophilic compared with the corresponding thiolate anion because of an alpha effect (6). Such strong nucleophilicity and reducing ability of hydropolysulfides may contribute to their potent antioxidant and protective activities against cellular insults caused by oxidative and electrophilic stresses (1, 3, 21, 24, 25).
The nucleophilicity of hydropolysulfides (and also RS n −) is also an important issue to consider when one aims to quantitate, speciate, and visualize the presence of hydropolysulfides in biological samples. In many previous studies, thiol-reactive agents, including NBF-Cl, MBB, NEM, and IAM, have been used to alkylate terminal hydropersulfide and hydropolysulfide moieties to form alkylated polysulfides (RS n E) to prevent oxidation of reactive thiols or labile RS n − during extraction or analysis (13, 24, 50). However, as alluded to earlier, those electrophilic alkylating agents (E) have promoted the alkaline hydrolysis of polysulfur chains, which resulted in degradation of both RS n E polysulfide species (Figs. 1 and 3). In addition, those reagents can degrade RS n R in biological samples (Fig. 3B) (2, 19, 28). We should also note that treatment with alkylating agents can result in formation of ESEs from degradation of polysulfides even in the absence of free H2S (Fig. 1). Although the formation of ESEs has been used as an indicator of the presence of H2S (41, 46), more careful attention should be paid to formation of ESEs via hydrolysis-coupled degradation of polysulfides with alkylating agents (2, 8, 19, 28).
As described above, our recent study clearly showed that incorporation of a phenol (hydroxyphenyl) moiety into an alkylating agent (e.g., HPE-IAM, Fig. 2) markedly reduced the hydrolysis-dependent degradation of polysulfides compared with the corresponding alkylating agent without the phenol moiety (e.g., IAM) (Figs. 4 and 5) (2, 8, 19). On the basis of this observation, recently we successfully developed a new IAM-based alkylating agent (Figs. 4 and 5) (28). We synthesized TME-IAM by using carbodiimide-mediated condensation of iodoacetic acid and TME (Fig. 4) (28).
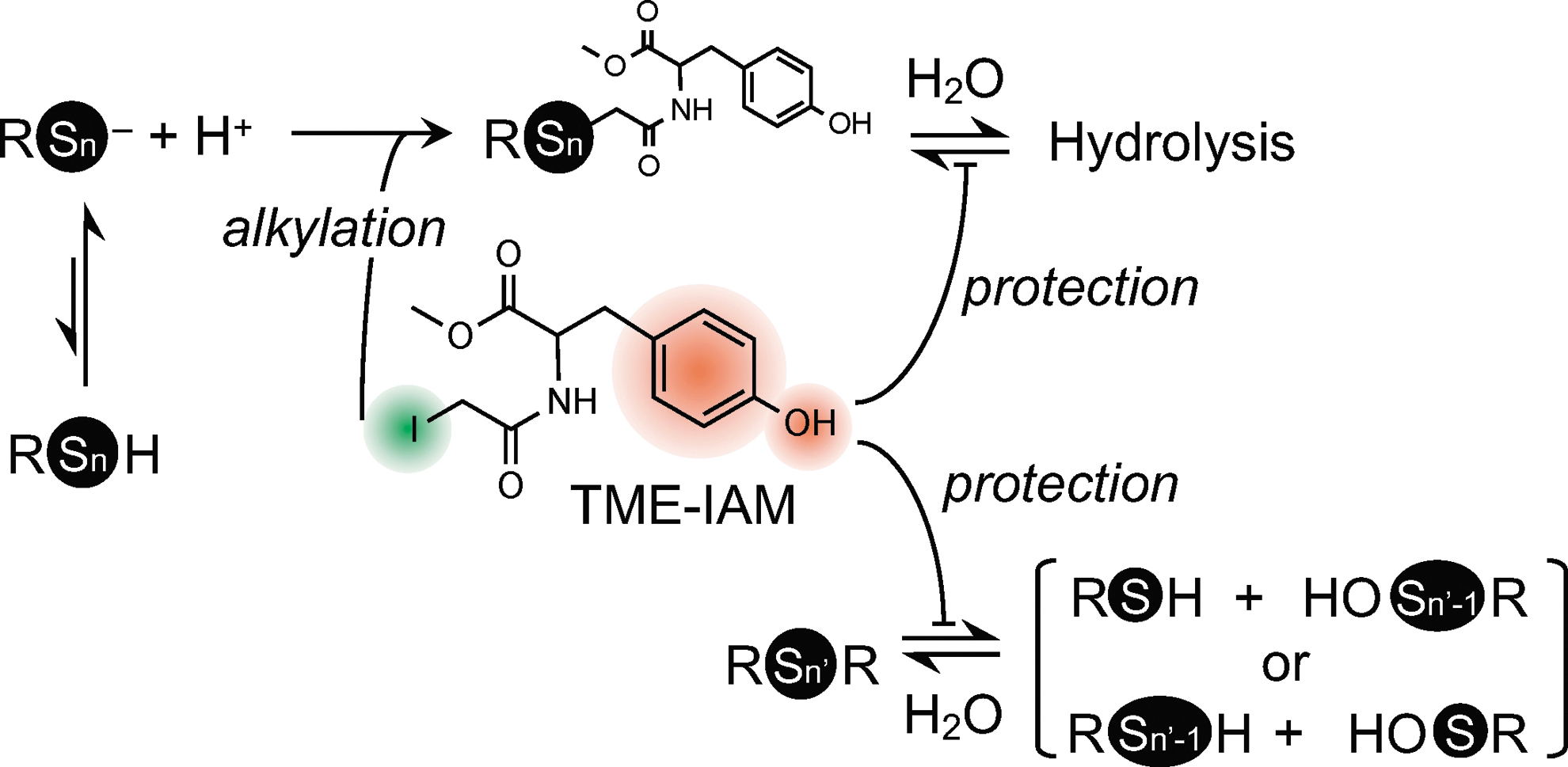
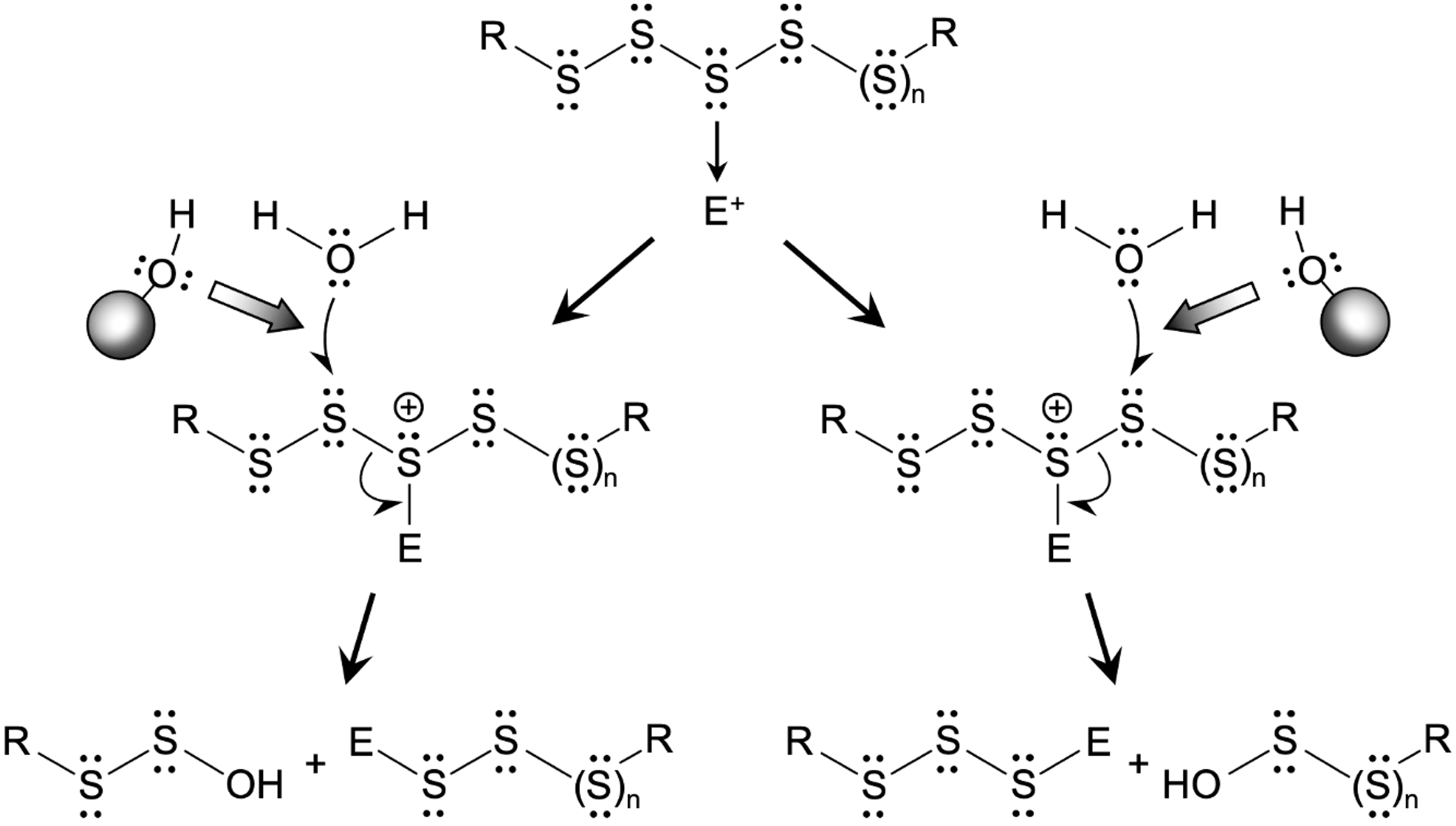
During polysulfide hydrolysis promoted by an electrophile, once an electrophile reacts with one of the sulfur residues in a polysulfide chain, then that sulfur becomes a good leaving group (since it is positively charged) (Fig. 6). At this point, any nucleophile, such as water, could react with a sulfur atom adjacent to the sulfur bound to an electrophile (Fig. 6). This would cause cleavage of the S-S bond and generate sulfenic acid and an electrophile–sulfur adduct (8). The S-S bond cleavage by water occurs in a polysulfide chain, although to a lesser extent in the absence of an electrophile. Therefore, the compounds that possess hydroxyl moieties may compete with the nucleophilic attack by water to reduce hydrolysis-dependent degradation of polysulfides regardless of the apparent involvement of exogenous electrophiles (Figs. 5 and 6).
Accordingly, TME-IAM can markedly attenuate the hydrolysis-driven degradation of polysulfides (28). For example, a loss of ∼40% of glutathione tetrasulfide (GSSSSG) was determined after incubation in 100 mM phosphate buffer (pH 7.4) at 37°C for 1 h, and addition of MBB increased degradation, with the total loss being ∼70% (28). In contrast, in the presence of TME-IAM, GSSSSG degradation was almost completely suppressed (28). As an important result, TME-IAM retained its efficient alkylation of hydropersulfide and hydropolysulfide moieties through the IAM functional group. Sulfhydryl alkylation by TME-IAM led to increased hydrophobicity of analytes and thereby sufficient retention during reverse-phase liquid chromatography (LC) separation. In fact, the use of TME-IAM in mass spectrometry-based sulfur metabolomics allowed clear detection of hydropolysulfides such as GSSSH and CysSSSH in mouse tissues (e.g., liver and brain). When compared with data obtained using HPE-IAM, data obtained with the use of TME-IAM showed significantly smaller amounts of bis-S adducts (partly indicative of H2S), higher amounts of various persulfides and polysulfides as well as GSSSG, and higher amounts of polysulfides such as GSSSSG (28). These data suggest that speciation of reactive sulfur species is largely influenced by the particular thiol-reactive alkylating agents used (2, 8, 19, 28). Extensive and careful sulfur metabolomic investigations with the use of alkylating agents such as TME-IAM therefore allow us to achieve a more precise understanding of the complex metabolome of reactive sulfur species in biological systems.
For rigorous and quantitative measurement of metabolites by means of liquid chromatography–tandem mass spectrometry (LC-MS/MS), isotope-labeled standards are spiked into the samples and are necessary to standardize the ionization efficiency of various endogenous metabolites (as in isotope dilution mass spectrometry) (20, 31). One advantage of utilizing TME-IAM as the alkylating agent is that we can readily synthesize isotope-labeled TME-IAM by using commercially available isotope-labeled tyrosine as the starting material (28).
Hydrolysis Equilibrium in Protein Polysulfides and Its Application to the Sulfur Proteome
The hydrolysis-driven polysulfide equilibrium discussed here can be applied for the sulfur proteome to probe into the exact presence and profile of polysulfide chains formed in many proteins. Hydropersulfide and hydropolysulfide residues in proteins (Prot-S n H) are labeled with polyethylene glycol (PEG)-conjugated maleimide to increase the apparent molecular size of proteins (2, 8, 27). On electrophoresis, a mobility shift occurs that depends on the number of PEG labels introduced. Molecular size alterations that are induced by the PEG-maleimide reaction and electrophile treatment can be determined by gel electrophoresis; this protocol is called the PEG-conjugated maleimide-labeling gel shift assay (PMSA) (Fig. 3B) (2, 8, 27). Strong electrophiles such as NBF-Cl, MBB, NEM, and p-chloromercuribenzoic acid readily react with Prot-S n−1H that forms on hydrolysis and thus cause a reduction in the apparent molecular sizes of proteins (Fig. 3B) (2, 8, 27). In fact, extensive hydrolysis and decomposition of protein polysulfidation by harsh electrophiles such as NBF-Cl were clearly observed, as evidenced by the PMSA for alcohol dehydrogenase 5 (ADH5) protein-bound polysulfides (Fig. 3B).
Our seminal work performed earlier established that protein persulfidation and polysulfidation occur not only via post-translational modifications but also via direct incorporation of precursor CysSS n H produced by CARS into tRNA during translation as a part of the translation process (2). Thus, as expected, many types of proteins are endogenously persulfidated and polysulfidated (24). This was unequivocally supported by the finding that all proteins that were investigated, including glyceraldehyde-3-phosphate dehydrogenase, ETHE1, ADH5, and aldehyde dehydrogenase 1 family, member A1, were identified as endogenously polysulfidated by means of the PMSA (2, 8, 27).
Concluding Remarks
Polysulfide species (i.e., R-S n -R′, n > 2; and R-S n -H, n > 1) occur in a wide variety of organisms as low-molecular-weight forms and as proteinaceous forms. The highly nucleophilic nature of hydropersulfide and hydropolysulfide species contributes to the potent antioxidant activities of polysulfide species that protect organisms against oxidative and electrophilic stresses. Recent studies discovered that polysulfide species R-S n -R′ readily undergo alkaline hydrolysis, which results in formation of both nucleophilic hydrosulfide and polysulfide species (R-S n−1H) and electrophilic R′SOH species. As a more important feature, polysulfides can maintain a steady-state equilibrium that is driven by hydrolysis even under physiological conditions at neutral and weak alkaline pH values in vivo. This unique property makes polysulfide chemistry and biology more complex than previously believed. This hydrolysis equilibrium of polysulfides shifts to the right in the presence of electrophiles. In fact, electrophilic alkylating agents that are frequently used for sulfide analysis, such as MBB, greatly enhance the hydrolysis of polysulfides, which leads to increased polysulfide degradation as well as artifactual formation of bis-S-bimane adducts in the absence of free H2S. The finding that hydroxyl group-containing substances such as tyrosine quite efficiently protected polysulfides from hydrolytic degradation led to the development of the new alkylating agent, TME-IAM. TME-IAM efficiently and specifically traps and stabilizes hydropersulfides and hydropolysulfides and protects polysulfide chains from hydrolysis, and, when used with mass spectrometry, TME-IAM allows successful speciation of the reactive sulfur metabolome. PMSA, the assay that relies on the unique hydrolysis equilibrium of polysulfide species, will be a reliable technique for the proteomics of polysulfide-containing proteins. Better understanding of the occurrence as well as metabolism of polysulfide species by means of precise methodologies is necessary to obtain insights into the unclarified biology of polysulfide species.
Footnotes
Acknowledgments
The authors thank J.B. Gandy for her excellent editing of the manuscript. They are also deeply grateful to Professor Jon M. Fukuto (Sonoma State University) for fruitful and critical discussion on the chemistry of polysulfide hydrolysis.
Authors' Contributions
T.S., H.M., and T.A. conceived and wrote the review. T.T., T.M., and H.I. helped in the literature search and drawing of figures. All the authors read and approved the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported, in part, by Grants-in-Aid [for (S), (B), (C), Early-Career Scientists, Challenging Exploratory Research] from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, to Tetsuro Matsunaga (19K07554), Tsuyoshi Takata (20K15983), Tomohiro Sawa (21H02071), Hideshi Ihara (20K21256 and 21H02082), Hozumi Motohashi (20H04832), and Takaaki Akaike (18H05277 and 20K21496); the Japan Science and Technology Agency (JST), CREST, Japan, to Takaaki Akaike (JPMJCR2024); and the Japan Agency for Medical Research and Development (AMED), Japan, to Hozumi Motohashi (JP21gm5010002).