
Abstract
Significance:
The mitochondrial oxidative phosphorylation (OXPHOS) system, comprising the electron transport chain and ATP synthase, generates membrane potential, drives ATP synthesis, governs energy metabolism, and maintains redox balance. OXPHOS dysfunction is associated with a plethora of diseases ranging from rare inherited disorders to common conditions, including diabetes, cancer, neurodegenerative diseases, as well as aging. There has been great interest in studying regulators of OXPHOS. Among these, ATPase inhibitory factor 1 (IF1) is an endogenous inhibitor of ATP synthase that has long been thought to avoid the consumption of cellular ATP when ATP synthase acts as an ATP hydrolysis enzyme.
Recent Advances:
Recent data indicate that IF1 inhibits ATP synthesis and is involved in a multitude of mitochondrial-related functions, such as mitochondrial quality control, energy metabolism, redox balance, and cell fate. IF1 also inhibits the ATPase activity of cell-surface ATP synthase, and it is used as a cardiovascular disease biomarker.
Critical Issues:
Although recent data have led to a paradigm shift regarding IF1 functions, these have been poorly studied in entire organisms and in different organs. The understanding of the cellular biology of IF1 is, therefore, still limited. The aim of this review was to provide an overview of the current understanding of the role of IF1 in mitochondrial functions, health, and diseases.
Future Directions:
Further investigations of IF1 functions at the cell, organ, and whole-organism levels and in different pathophysiological conditions will help decipher the controversies surrounding its involvement in mitochondrial function and could unveil therapeutic strategies in human pathology. Antioxid. Redox Signal. 37, 370–393.
Introduction
Mitochondria are essential intracellular organelles of eukaryotic cells that are involved in a large number of cellular processes such as energy production, oxidation of fatty acids, regulation of oxidative stress, the balance between cell death and survival, and calcium homeostasis. Given the importance of these processes, essentially any disease, as well as the process of aging itself, could, theoretically, be related to mitochondrial dysfunction.
Mitochondrial dysfunction is the result of defective mitochondrial quality control or impaired cellular respiration, particularly in the oxidative phosphorylation (OXPHOS) machinery, which comprises the electron transport chain (ETC) and ATP synthase. Defects in mitochondrial functions are primarily caused either by genetic factors from either the nuclear or the mitochondrial genome or by multiple environmental factors. For instance, mitochondrial function can be affected by adverse effects of drugs, infections, an unhealthy lifestyle (e.g., a Western diet, smoking, lack of physical activity), or the body's reduced capacity to manage stress.
Mitochondrial dysfunction leads to bioenergetic deficiencies and oxidative stress that are early indicators of declining health, even before diseases become apparent. Hence, understanding the mechanisms regulating mitochondrial functions and their importance in human pathologies is of clear clinical relevance.
Inhibitory factor 1 (IF1) is a nuclear-encoded endogenous inhibitor of the mitochondrial ATP synthase (121). For long, IF1 has been considered to be a unidirectional inhibitor of ATP synthase, acting only by inhibiting its hydrolase activity (i.e., the ATPase activity) (11). However, recently, a number of other roles for IF1 have been proposed, including the possibility that IF1 could also inhibit the synthetic activity of ATP synthase.
This unexpected ability of IF1 to inhibit ATP synthase activity led to studies that showed a potential action of IF1 as regulating mitochondrial energy metabolism and metabolic byproducts in a way that reprograms energy metabolism toward enhanced aerobic glycolysis and that favors mitochondrial dysfunction. In addition, ATP synthase is ectopically expressed on the plasma membrane of various cell types (further referred to as ecto-F1-ATPase), and IF1 has been detected in human serum (97), thus indicating a possible extended function of IF1. These recent observations have led to IF1 being viewed as a potent regulator of the mechanism age-associated diseases and cancer.
The aim of this review is to provide an overview of the current understanding of the role of IF1 in mitochondrial functions, health, and diseases.
Mitochondrial ATP Synthase and IF1: Structure and Regulation
Molecular structure and catalytic functions of mitochondrial ATP synthase
Mitochondrial ATP synthase (EC 7.1.2.2), also referred to as F1-FO ATPase or complex V, is the last complex in the OXPHOS cascade. In the OXPHOS cascade, the electrons derived from biological oxidations are transferred to the respiratory chain complexes to reduce oxygen and to generate the electrochemical proton gradient at the inner membrane that ATP synthase uses to produce ATP from ADP and inorganic phosphate [see Walker (153) and Walker (154) for recent reviews].
Structurally, ATP synthase is a multisubunit complex that represents the smallest rotary motor in nature. Briefly, it is composed of 15 subunits in mammals, amounting to a total molecular mass of ∼600 kDa, and it can be divided into two main domains: a domain located within the inner membrane (FO) that acts as a proton (H+) channel, and a globular catalytic domain situated in the mitochondrial matrix (F1). These two domains are linked together by a central and a peripheral stalk [see Yoshida et al. (168) for a review].
Mechanistically, in normally respiring mitochondria, ATP synthesis is driven by an influx of H+ into the mitochondrial matrix through FO and is catalyzed by three αβ-subunit heterodimers of F1 (153, 154) (Fig. 1a). As ATP synthase is a reversible nanomotor, it can also run “in reverse,” acting as an ATP-consuming proton pump (an ATPase), pumping H+ back into the intermembrane space (IMS). This reverse situation occurs when the proton motive force (PMF) generated by the ETC collapses during hypoxic or ischemic conditions and allows the mitochondrial membrane potential (ΔΨm) to be rescued at the expense of ATP (Fig. 1c).
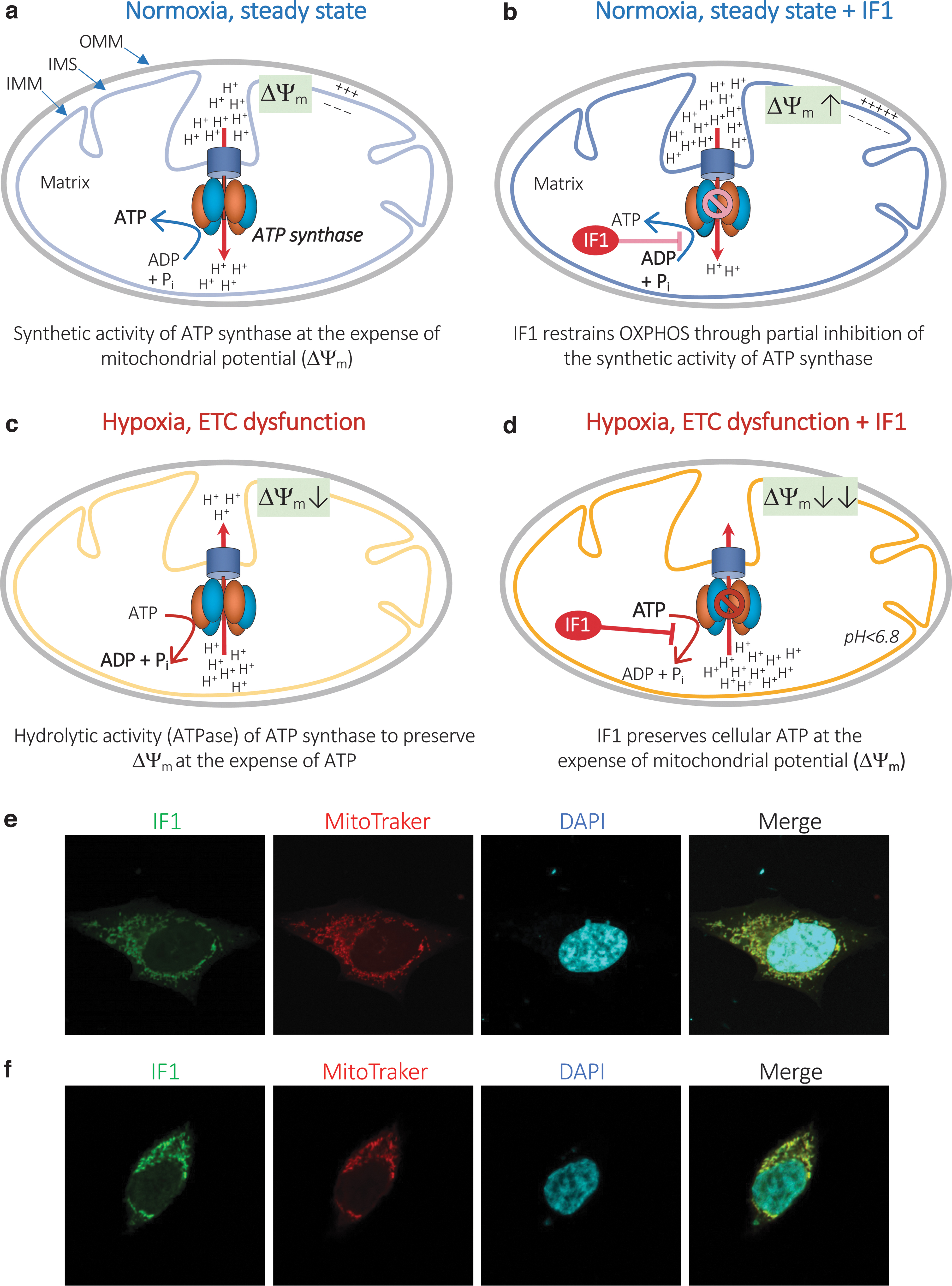
Mitochondrial ATPase IF1: gene, protein, and regulation
Mitochondrial ATP synthase is regulated at the molecular level by an endogenous, nuclear-encoded, small protein called ATPase IF1 (Fig. 1e, f). Since its identification in 1963 (121), the structure and mechanism of action of IF1 on ATP synthase activity have been extensively studied, notably by Sir John E. Walker and collaborators [see Walker (154) for a detailed review]. IF1 is highly conserved across species, and IF1 interspecies inhibition of ATP synthase has been reported (98). However, the present review will focus on IF1 in mammals and humans, in particular.
The functional importance of IF1 is indicated by its presence in all tissues. Human IF1 is encoded by the ATPIF1 gene (ENSG00000130770), which is located on chromosome 1. Alternative splicing occurs at the ATPIF1 gene locus, and three transcript variants encoding distinct isoforms have been identified (58). IF1 is highly expressed in tissues with high energy demands, such as heart, liver, kidney, and brain (37), and it is overexpressed in carcinomas from various organs, including colon, lung, breast, ovaries, and liver (46, 55, 66, 131, 133). Interestingly, IF1 is expressed in human mesenchymal stem cells, but not in osteogenic differentiated cells, thus suggesting that IF1 is a stemness marker involved in maintaining quiescence (132). Regarding regulation of ATPIF1 gene expression, nuclear factor-κB (NF-κB) has been shown to be a direct transcriptional regulator of IF1 expression (145).
IF1 is translated as a precursor protein that undergoes removal of the N-terminal 25-residue presequence when it is imported into the mitochondria (53, 58, 72, 161) (Fig. 2). IF1 isoform 1 (NCBI Reference Sequence: NP_057395.1, encoded by variant 1) corresponds to a mature protein of 81 amino acids in length (∼9.5 kDa), whereas IF1 isoform 2 (NCBI Reference Sequence: NP_835497.1, encoded by variant 2) and isoform 3 (NCBI Reference Sequence: NP_835498.1, encoded by variant 3) have a shorter and distinct C-terminus compared with isoform 1, resulting in mature proteins of 46 amino acids (∼5.2 kDa) and 35 amino acids (∼3.9 kDa), respectively (Fig. 2).
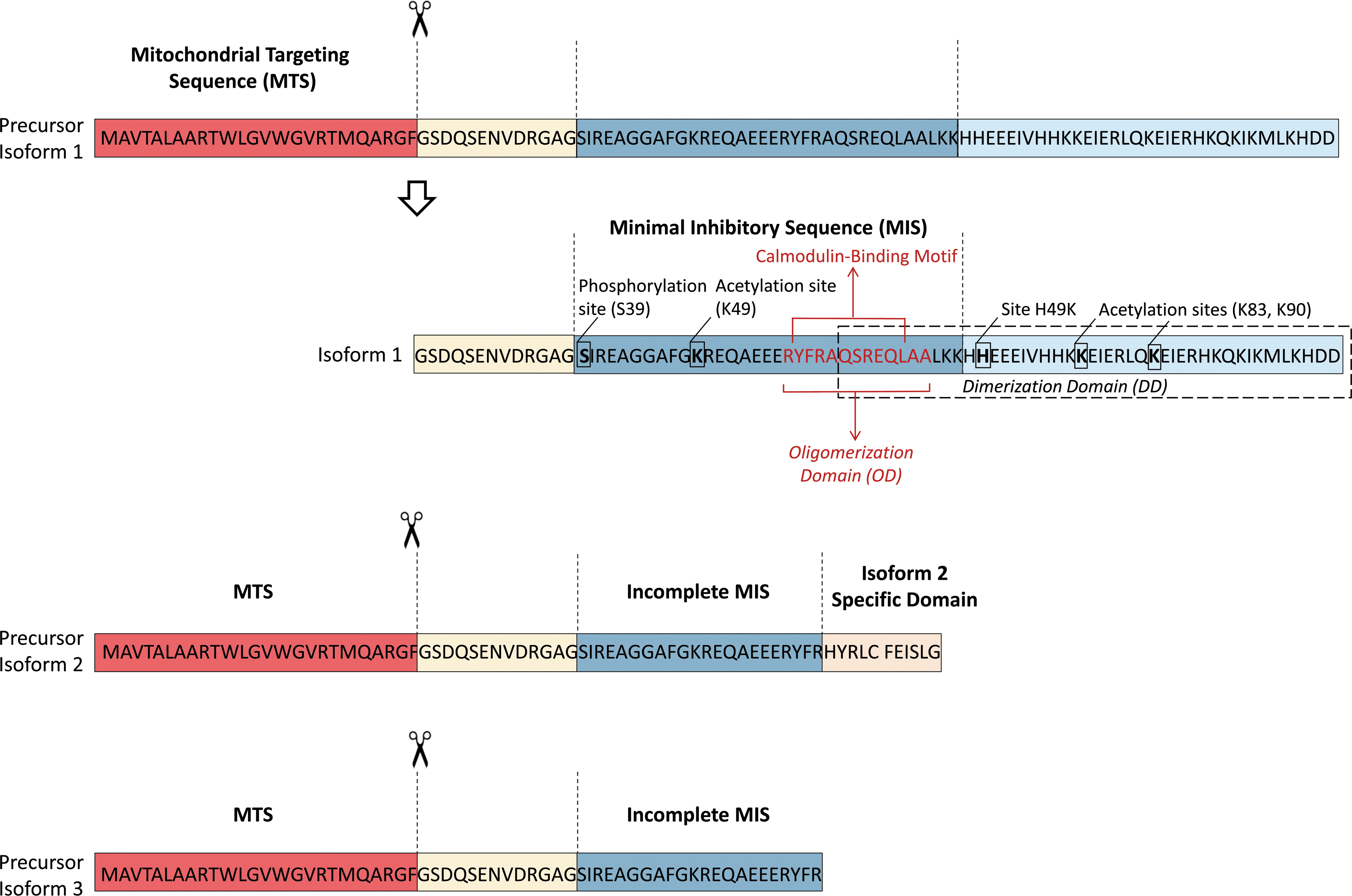
Based on mRNA expression levels, isoform 1 is the major isoform expressed in human organs, including liver, heart, kidney, and brain (mainly neurons) (37, 44). IF1 binds to the catalytic interface between the α and the β subunit of the F1 domain (12, 13). The minimal inhibitory sequence of IF1 has been shown to comprise amino acid 14–47 of isoform 1, with the adjacent amino acid 10–13 and amino acid 48–56 sequences helping to stabilize the ATP synthase and IF1 complex (122). Thus, isoforms 2 and 3, which do not contain the complete minimal inhibitory sequence (Fig. 2), are unlikely to have ATP synthase inhibitory activity, and their functional roles are presently entirely unknown. From here onward, IF1 will refer to isoform 1 only.
IF1 has been shown to have a short half-life (∼100 min) in colon cancer cells, which is indicative of rapid turnover of the protein (131). Whether the overexpression of IF1 in cancer cells is due to its defective degradation is still a matter of debate, but it could be postulated that IF1 has an even shorter half-life in normal cells (<100 min).
Although not yet fully elucidated, there are several studies regarding post-transcriptional regulation of IF1 (52). The immediate early response gene X-1 (IEX-1) interacts with IF1 to target its degradation by an unidentified protease in CHO cells (140). However, the role of its human orthologue, IER3, has not been confirmed (131) and no other proteases involved in IF1 degradation have been identified to date (132). Hypoxia-inducible factor 1-alpha (HIF1-α) and the synthetic steroid pregnenolone-16 α-carbonitrile (PCN) may upregulate the expression of IF1 in rat liver to maintain homeostasis during toxic stress (71, 76). The β1-blocker nebivolol has recently been reported to increase the expression of IF1, independently of ATPIF1-mRNA abundance (109).
Calmodulin (CaM) has been shown to interact with IF1 at amino acid positions 33–42 in a pH- and Ca2+-dependent manner, thus indicating that an IF1-CaM complex formed outside the mitochondria could possibly regulate IF1 trafficking or activity (29, 112, 118). In agreement with this hypothesis, interaction between IF1 and CaM has been reported at the plasma membrane, and this is thought to modulate the ability of IF1 to inhibit the hydrolytic activity of ecto-F1-ATPase (28) (see IF1 and ecto-F1-ATPase section). Also, in vitro studies reported a possible interaction between IF1 and coupling factor 6 (CF6), which is a subunit part of the peripheral stalk that links the F1 and FO complexes together (80). It was proposed that IF1 could counteract CF6-induced apoptosis and senescence, although the underlying mechanisms governing these effects remain poorly characterized (80, 111).
IF1 has been reported to be acetylated on the highly conserved lysine residues K49, K83, and K90 (69, 125) (Fig. 2). Interestingly, the acetylation level of K49 and K90 seems to depend on the mitochondrial NAD+-dependent deacetylase sirtuin-3, whose activity improves OXPHOS, attenuates oxidative stress damage, and counteracts cellular senescence (69, 125). Finally, the most documented post-transcriptional regulatory mechanism for IF1 activity is its phosphorylation by the β-adrenergic/cAMP/PKA signaling pathway (54). In particular, IF1 phosphorylation at Ser39 by PKA (Ser14 in its mature form, Fig. 2) abrogates its binding and inhibitory activity on ATP synthase. This inhibitory effect was lost when the inactive phosphomimetic (S39E) mutant of IF1 was overexpressed in human colorectal carcinoma HCT116 cells and primary human skeletal muscle cells (45, 54). These results indicate that the inhibitory action of IF1 on ATP synthase may require its dephosphorylation. Nevertheless, the short half-life of IF1 could result in rapid reestablishment of the level of active dephosphorylated IF1 (131).
The inhibitory activity of IF1 is controlled by self-association, which is influenced by the Ca2+ concentration and the pH (8). In normally respiring mitochondria, the pH of the mitochondrial matrix is ∼7.9 and IF1 is mainly organized as an inactive tetramer, as the residues that bind to F1 are masked in this state (11). However, during conditions of oxygen deprivation such as ischemia or on mitochondrial depolarization, the mitochondrial matrix pH becomes more acidic (144). At a pH value below 7.0, the formation of IF1 dimer is favored, uncovering the N-terminal IF1-inhibitory domains that can concomitantly interact with two F1 domains, blocking a dimeric F1-FO complex (11).
Therefore, the inhibitory effect of IF1 on ATP synthase is likely to be optimal during hypoxic or ischemic conditions, when the enzyme acts as an ATP hydrolase (see IF1 and Mitochondrial Bioenergetics section). Interestingly, a mutation in the structure of bovine IF1 (histidine 49 to lysine—referred to as IF1-H49K) results in a pH-independent active IF1 dimer (11, 137) (Fig. 2). This shows how the pH and protonation of IF1, in particular the histidine(s) residues, determine its structure and activity. These dynamic properties of pH-dependent structural organization of IF1 led to it being considered for a long time as a unidirectional inhibitor of ATP synthase that only acted by inhibiting its hydrolase activity (i.e., the ATPase activity) (11). However, several recent studies support the possibility that IF1 may also inhibit ATP synthesis in normally respiring mitochondria (see the IF1 and Mitochondrial Bioenergetics section).
IF1 and Mitochondrial Bioenergetics
IF1 and mitochondrial bioenergetics during conditions of hypoxia and OXPHOS dysfunction: role in the preservation of cellular ATP
ATP is life's major energy-carrying molecule, and most of it is produced by mitochondrial ATP synthase through OXPHOS. For instance, the ATP production rate of mitochondrial OXPHOS is 18-fold higher compared with that of aerobic glycolysis. In OXPHOS, ETC complexes, which comprise complex I to IV, catalyze the transfer of electrons from energy-rich molecules (NADH or FADH2) to oxygen, and ATP synthase generates ATP by exploiting the energy released by the ETC complexes.
If oxygen is unavailable to pick up these electrons, as is the case under hypoxic/ischemic conditions, the H+ electrochemical gradient across the mitochondrial inner membrane is lost, leading to acidification of the mitochondrial matrix and reversal of ATP synthase activity to ATP hydrolysis. This hydrolytic response, called F1-ATPase activity, pumps H+ back into the IMS and preserves the ΔΨm, the disruption of which can induce apoptosis (75) (Fig. 1c). However, if the hydrolytic activity is prolonged, the wastage of ATP can lead to cellular energy collapse, and thus have the opposite effect from that expected, precipitating apoptosis or necrosis (34). In this context of oxygen deprivation, IF1, which has a higher activity at low pH (see the Mitochondrial ATP Synthase and IF1: Structure and Regulation section), has been shown to prevent ATP synthase hydrolytic activity at the expense of the ΔΨm (11, 16) (Fig. 1d).
This function of IF1 in preserving ATP levels under conditions whereby the cell experiences oxygen deprivation was thus logically proposed to play a protective role during myocardial ischemia or ischemic stroke (see the IF1 and Cardiovascular Diseases section).
The protective role of IF1 against cell death in other pathological conditions that occur when OXPHOS is deficient has remained controversial. Instead, IF1 deletion in mouse hepatocytes protected from cell death by maintaining the ΔΨm at the expense of ATP when OXPHOS dysfunction was pharmacologically induced by the complex III inhibitor antimycin (25). Moreover, Rho0 cells, which lack mitochondrial DNA and have dysfunctional OXPHOS (21), were unexpectedly found to not undergo apoptosis in response to apoptotic stimuli (70). This protection of Rho0 cells from apoptosis could be attributed to ATP synthase, for which the ATPase activity was shown to maintain the ΔΨm (9) unless IF1 was overexpressed (90). Thus, IF1 is likely to promote apoptosis when OXPHOS is deficient.
IF1 and mitochondrial bioenergetics under normoxic conditions: role in mitochondrial energy reprograming
It was thought until recently that IF1 could selectively inhibit the ATPase activity of ATP synthase under conditions of oxygen deprivation, without affecting the synthesis of ATP during OXPHOS under normoxic conditions. However, several recent studies have found that IF1 could inhibit both the synthetic and the hydrolytic activity of ATP synthase. In particular, Gu et al. recently reported that tetramers of ATP synthase internally linked by IF1 dimers appear to be in an inhibited state (64), thus supporting the possibility that IF1 can also inhibit ATP synthesis in normally respiring mitochondria (Fig. 1b).
Additional cellular studies have corroborated the unexpected ability of IF1 to inhibit the synthetic activity of ATP synthase. In mice, tissue-specific expression of the constitutively active IF1-H49K variant (11) has been shown to induce mitochondrial hyperpolarization (i.e., a higher ΔΨm) as a result of the inhibition of ATP synthase, thus promoting a metabolic shift from mitochondrial OXPHOS to aerobic glycolysis in neurons (44), hepatocytes (134), and hepatoma cells (133). The same was observed when native IF1 was overexpressed in mouse intestinal epithelium (47), primary human skeletal muscle cells from obese individuals (45), and rat insulinoma INS1-E cells (79). Accordingly, the silencing of ATPIF in INS-E cells cultured under normoxic conditions has been reported to be associated with higher OXPHOS activity and cellular ATP levels compared with control cells (78).
Further, the silencing of ATPIF in rat liver epithelial F258 cells prevents the glycolytic shift, known as the Warburg effect (156), induced by the carcinogen benzo[a]pyrene (B[a]P) (66). Altogether, these data suggest that IF1 inhibits the synthetic activity of ATP synthase during physiological OXPHOS (Fig. 1b), thus reprogramming the energy metabolism toward enhanced aerobic glycolysis. Such metabolic reprogramming is likely to promote metabolic dysfunction, chronic inflammation, and cancer development (see in IF1 in Diseases section).
The ability of IF1 to inhibit the synthetic activity of ATP synthase and compromise OXPHOS in normally respiring mitochondria may depend on the cellular energy demand, the metabolic state, and the cellular models. For instance, under normoxic conditions, it has been proposed that IF1 could inhibit the residual ATPase activity of the enzyme (48, 158) or promote ATP synthase dimerization and the modeling of mitochondrial cristae (6, 16) (see the IF1 and Mitochondrial Fitness section), which would result in an increase in the OXPHOS rate.
IF1 and Mitochondrial Fitness
Oxidative stress
Aside from its role in bioenergetics, OXPHOS activity also contributes to the production of reactive oxygen species (ROS). Indeed, in addition to proton influx by ATP synthase, there is a degree of proton leakage that leads to the production of superoxide radical (O2 •−) as a byproduct of complex I and II, although other sites also contribute to its production (106). O2 •− is then typically converted into hydrogen peroxide (H2O2) by manganese superoxide dismutase (SOD2), which can then be reduced to water by glutathione peroxidase or peroxiredoxins in most mammalian cell types, along with catalase in heart and liver cells (49, 128, 148).
The ROS play central roles as second messengers mediating the cellular response to exogenous and endogenous signals (126). However, OXPHOS dysfunction leads to increased production of mitochondrial ROS (mtROS), which cause oxidative damage and contribute to a number of pathologies and to the process of aging itself (35).
Several studies have explored the role of IF1 in mtROS production in relation to its ability to inhibit the synthetic and the hydrolytic activity of ATP synthase under normoxic and hypoxic conditions, respectively (see IF1 and Mitochondrial Bioenergetics section).
Under normoxic conditions, IF1-mediated inhibition of ATP synthase has been shown to promote mitochondrial hyperpolarization and to inhibit OXPHOS activity, thereby leading to enhanced O2 •− production (44, 46, 131). An increase in protein carbonylation has been observed when IF1 was overexpressed, which is consistent with a higher basal production of mtROS (44, 46, 131). Interestingly, IF1 overexpression does not appear to affect H2O2 levels, the ratio of reduced to oxidized glutathione (GSH/GSSG), or the expression of SOD2 and catalase (46, 47), indicating that IF1 promotes a moderate level of ROS-mediated signaling in mitochondria.
The IF1-mediated ROS signaling pathway has been shown to trigger nuclear reprogramming and tumor development (Fig. 3). This has been found to occur in various human cancer cell lines. The IF1 generated a moderate level of ROS signaling in these cell lines, which did not result in the induction of an oxidative stress response, although it did protect the cells from chemotherapy-induced apoptosis (131). This study reported that the apoptosis resistance was due to the activation of NF-κB and increased expression of the anti-apoptotic mitochondrial protein B cell lymphoma-extra large (Bcl-xl) (131) (Fig. 3).
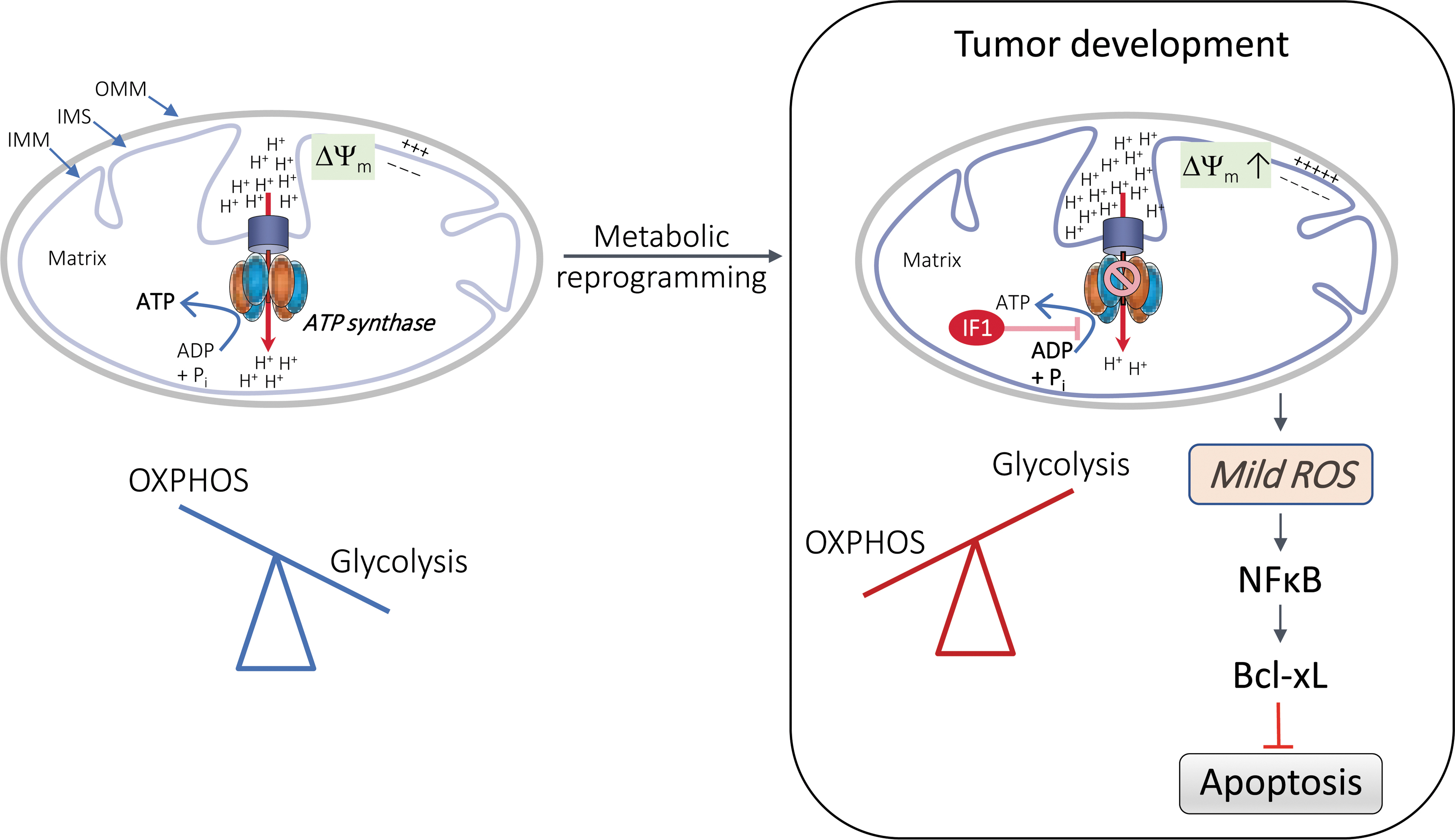
Likewise, in colon cancer cells, ATPIF1 triggers an ROS-mediated retrograde prosurvival and proliferative response by promoting activation of the NF-κB signaling pathway (46). Accordingly, ATPIF1 silencing in human glioma and hepatocellular carcinoma cell lines has been reported to result in inhibition of the expression of NF-κB and to lead to increased E-cadherin expression and reduced vimentin expression, thus limiting cell migration and invasion (83, 145, 160).
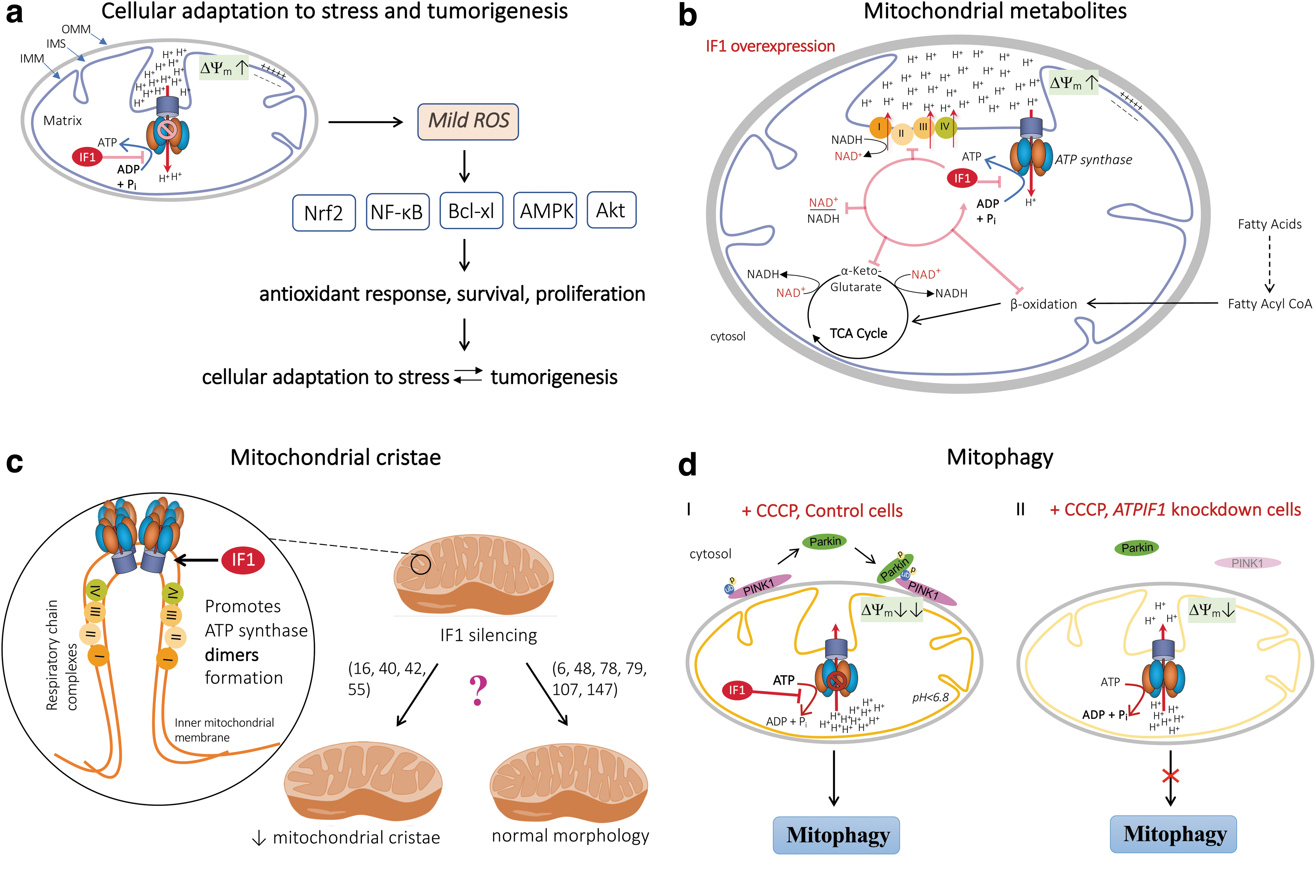
The oncogenic role of IF1 has also been confirmed in a mouse model that overexpresses the constitutively active IF1-H49K variant in hepatocytes (134). This study showed that IF1 promoted the development of hepatocarcinoma by activation of the energy sensor 5′ AMP-activated protein kinase (AMPK) and the NF-E2 p45-related factor 2 (Nrf2) antioxidant response, presumably through ROS-mediated inactivation of Kelch-like ECH-associated protein 1 (Keap1) and translocation of Nrf2 to the nucleus (116, 134). Overall, these studies indicate that the inhibitory effect of IF1 on OXPHOS triggers an ROS-mediated retrograde response in mitochondria, which increases resistance to cell death and contributes to the acquisition of a cancer phenotype (Figs. 3 and 4a, see also IF1 in Diseases section).
Aside from its detrimental role in protecting against cell death in cancer, it has been proposed that IF1-mediated induction of moderate mtROS levels could protect the intestine from inflammation in a mouse model of colitis in which IF1 is overexpressed in the intestinal epithelium (47). In this model, ATP synthase inhibition by IF1 promoted a metabolic shift from OXPHOS to aerobic glycolysis in colonocytes, and this was associated with the recruitment and polarization of intestinal macrophages to an M2 anti-inflammatory phenotype. It was proposed that, mechanistically, this innate immune response is driven by the mtROS-mediated induction of NF-κB signaling. Although the mtROS level was unchanged in the colon of IF1 transgenic mice compared with littermate controls, this hypothesis is supported by enhanced carbonylation of intestinal proteins and inhibition of the M2 anti-inflammatory phenotype when mtROS were quenched (47).
The IF1-induced metabolic reprogramming and mtROS signaling has also been assessed in vivo in cortical neurons (44). It was reported that the production of O2 •− was enhanced in neurons isolated from mice overexpressing the constitutively active IF1-H49K variant compared with wild type. An increase in protein carbonylation in neuron and brain homogenates was also observed, consistent with a higher production of mtROS in IF1 transgenic mice. These mice did not exhibit any symptoms of neurological dysfunction, but they were protected from quinolinic acid-induced excitotoxicity.
However, the involvement of IF1-mediated mtROS production in the protection against neuronal cell death is questionable since quinolinic acid treatment by itself dramatically induced the formation of ROS (32). Instead, the apoptosis-resistance was attributed to activation of the prosurvival protein kinase b (Akt)/p70S6K pathway and Bcl-xl-mediated protection from cell death after IF1-mediated metabolic reprogramming (44).
Overall, it appears that IF1 promotes mtROS production under normoxic conditions. Mechanistically, the data suggest that the IF1-mediated ROS signaling pathway triggers nuclear reprogramming that results in the activation of defense mechanisms that prevent cell death (Fig. 4a). Although this mechanism could allow for a physiological concentration of ROS to be maintained to ensure cellular responses to exogenous and endogenous signals, it could be detrimental in some pathological conditions such as tumorigenesis and age-related diseases for which chronic inflammation and oxidative stress are common features. For instance, an increase in mtROS production due to dysfunctional OXPHOS reduced the lifespan of aged mice (127), whereas another study reported that the generation of mtROS positively affected lifespan extension in worms (135).
The anti-oxidant status is also critical for maintaining proper activity of the mitochondrial respiratory chain complexes. Thus, a decline in GSH level dramatically reduces the threshold below which inhibition of complex I activity compromises respiration and ATP synthesis (33). Disturbances in the antioxidant/oxidant balance affecting respiratory complex activities and energy threshold in mitochondria are likely to contribute to the development of neurodegenerative disorders (15). More generally, this dilemma/debate is consistent with the “Dr. Jekyll and Mr. Hyde” effect of ROS that can be beneficial or detrimental for health, depending on the ROS origin, concentration, and experimental detection method, as well as the pathophysiological conditions and the animal species (110, 141).
A low level of oxygenation in tissues is a common characteristic of various pathological conditions such as ischemic disorders, chronic obstructive pulmonary disease, inflammatory disease, and cancer. In this regard, the relationship between IF1 and cellular ROS levels has been explored under in vitro hypoxic cell culture conditions (139). In this study, hypoxia alone was associated with a pronounced decrease in cellular ROS and mtROS, whereas IF1 silencing partially prevented the decline in ROS levels induced by hypoxia. Also, when mitochondrial respiration was arrested by rotenone, a condition that elicits mtROS generation, IF1 overexpression in HeLa cells hindered mtROS production (40).
Thus, during conditions of hypoxia and OXPHOS dysfunction, IF1 is likely to prevent mtROS production. Whether this process strictly depends on the ability of IF1 to inhibit the hydrolase activity of ATP synthase in those conditions will require further investigation (see also section IF1 and mitochondrial bioenergetics during conditions of hypoxia and OXPHOS dysfunction: role in the preservation of cellular ATP).
Metabolic byproducts
NAD+ is a crucial mediator of energy metabolism and redox state, acting as a cofactor for NAD+-dependent enzymes, including oxoglutarate dehydrogenase complex, lactate dehydrogenase (LDH), deacetylases, sirtuins (SIRTs), and poly (ADP-ribose) polymerases (PARPs). It is also involved in mitochondrial homeostasis, mitophagy, and the response to DNA damage, and it has also a critical role in redox signaling (31, 43).
In mitochondria, NAD+ is reduced to NADH by pyruvate dehydrogenase and the tricarboxylic acid cycle enzymes malate dehydrogenase and α-ketoglutarate dehydrogenase (αKGDH). NADH is a substrate of mitochondrial ETC, acting as the principal electron donor to complex I (NADH dehydrogenase), thereby supporting mitochondrial OXPHOS to generate ATP. OXPHOS dysfunction results in a decline in the NAD+/NADH redox state and it may force cells to rely on glycolysis via LDH to regenerate NAD+, thus favoring a Warburg phenotype (156).
Interestingly, IF1 overexpression in primary human skeletal muscle cells from obese individuals has been reported to be associated with a significant reduction in the NAD+/NADH ratio (45). In these cells, it was also observed that there was a decrease in the cellular level of α-ketoglutarate (αKG), which is a Krebs cycle metabolite and an intermediate in carnitine biosynthesis that is involved in various fundamental processes—central metabolism, collagen synthesis, epigenetic regulation, and stem cell proliferation (169).
Fatty acid oxidation was also impaired in these IF1-overexpressing cells (45). Overall, this study suggests that inhibition of ATP synthase activity by IF1 induces the accumulation of ETC substrates such as NADH at the expense of a number of Krebs cycle metabolites (Fig. 4b). One could expect that such perturbations in the production of mitochondrial metabolites could result in a decline in health in patients with metabolic syndrome (7).
Mitochondrial cristae remodeling and apoptosis
Mitochondria are dynamic organelles that can modulate their morphology and OXPHOS function depending on the energy demand of the cell (27). This modulation can be achieved via the cristae, which are deep invaginations of the inner mitochondrial membrane that contain the OXPHOS complex (27, 120). This particular structural organization not only limits the diffusion of molecules that are important for the OXPHOS system, but it also prevents activation of the mitochondrial apoptosis pathway by sequestering cytochrome c (Cyt c) (27). A dynamic rearrangement of the cristae shape, called “cristae remodeling,” occurs during apoptosis to allow the release of Cyt c (27).
One of the essential factors for cristae formation is the dimerization of ATP synthase (27, 146). Indeed, the dimerization of ATP synthase contributes to the formation and preservation of the normal cristae architecture, together with an increase of OXPHOS performance (65, 146). Young normal cells are characterized by cristae that protrude deeply into the matrix (85), whereas during aging there are significant modifications of the cristae shape and a decrease in ATP synthase dimers (27, 142).
The proposed role of IF1 in mitochondrial cristae formation is based on the observation that silencing of this protein decreases the density of mitochondrial cristae (Fig. 4c), whereas IF1 overexpression leads to an increase in the density (16, 42). Consistent with the rise of cristae density when IF1 is overexpressed, the progression of cellular apoptosis was limited by a delay in the release of Cyt c (42). The proposed mechanism for IF1's contribution to mitochondrial cristae formation is that IF1 promotes dimerization of ATP synthase (16, 51) and that it inhibits OMA1-dependent proteolytic cleavage of the dynamin GTPase optic atrophy 1 (OPA1), thereby impeding apoptotic cristae remodeling (40).
Nevertheless, several studies are not in keeping with a role of IF1 in mitochondrial remodeling and apoptotic cell death as they showed that the mitochondria had a normal morphology in cells in which the IF1 protein was either knocked down (6, 48, 78, 107, 147) (Fig. 4c) or overexpressed (79). A recent study reported that overexpression of the constitutively active IF1 H49K variant led to alteration of the spatiotemporal organization of ATP synthase without any change in its dimerization state and was associated with aberrant mitochondrial cristae (158).
Overall, the effect of IF1-mediated ATP synthase dimerization on cristae ultrastructure preservation and enhancement of mitochondrial OXPHOS activity is controversial (Fig. 4c). Instead, there is much more evidence that IF1 tends to decrease mitochondrial OXPHOS activity by inhibition of the ATP synthetic activity (Fig. 1b), whereas it favors apoptosis by promoting loss of the ΔΨm when OXPHOS is impaired (Fig. 1d, see also IF1 and Mitochondrial Bioenergetics section).
Mitophagy
Dysfunctional or damaged mitochondria are cytotoxic by releasing cell death factors and producing ROS. Mitophagy is a selective autophagic program that degrades entire mitochondria when they become dysfunctional (e.g., when the ΔΨm is disrupted) or in response to different stress stimuli such as oxidative stress and nutrient starvation (91, 113). Mitophagy is, thus, a fundamental mitochondrial quality control mechanism, essential for maintaining proper cellular function (108). There is also increasing evidence pointing to a role of mitophagy in age-related conditions, as this process diminishes during aging and age-related diseases (4, 24).
Among the different mitophagy regulatory pathways, the best studied involves the phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1, encoded by PINK1) and the E3 ubiquitin-protein ligase Parkin (encoded by PRKN, previously PARK2) (113). On mitochondrial dysfunction, as a result of ΔΨm depolarization, PINK1 is targeted to the outer membrane of the mitochondrion where it is activated and sequentially recruits and phosphorylates Parkin (5). Parkin then ubiquitinates mitochondrial surface proteins to recruit p62 or other molecules that interact with light chain 3 (LC3) for autophagosome engulfment of the dysfunctional organelle (5). This mitophagy-dependent pathway could be important because mutations of Parkin impair mitochondrial quality control, leading to Parkinson's disease (82). After several steps, the autophagosome engulfs the dysfunctional organelle (5).
A genomic study in HeLa cells has revealed that IF1 is required for the PINK1-parkin mitophagy pathway (73). Under conditions of mitochondrial depolarization induced by carbonyl cyanide m-chlorophenyl hydrazone (CCCP, a widely used inducer of mitophagy), the IF1-mediated inhibition of the hydrolytic activity of ATP synthase promoted loss of the ΔΨm and subsequent outer mitochondrial membrane PINK1 accumulation and Parkin recruitment. In the absence of IF1, the ATPase activity of the ATP synthase was sufficient to restore the ΔΨm at the expense of the cellular ATP, thus abolishing the required signal for Parkin recruitment to mitochondria and inhibiting mitophagy (Fig. 4d).
Moreover, the levels of IF1 in cells that rely heavily on OXPHOS (e.g., neurons) are upregulated in ischemia/re-oxygenation models (101). This increase in the cellular level of IF1 was found to be essential to promote PINK1/Parkin-mediated mitophagy, thus pointing to a role of IF1 in the coordination of pro-survival mitophagy to prevent hypoxia injury after restoration of oxygenation (101).
IF1 and ecto-F1-ATPase
A cell surface protein complex related to mitochondrial ATP synthase
The subunits of ATP synthase are expressed ectopically on the plasma membrane of a range of human and animal cell types (20, 99, 150). The majority of studies to date indicate the presence of α and β subunits of the F1 catalytic domain, although some have reported the presence of the entire monomeric F1-FO ATP synthase complex containing both the nuclear and the mitochondrial DNA-encoded subunits (3, 96, 124). These findings make the hypothesis that the ATP synthase subunits are assembled at the plasma membrane very unlikely and instead suggest that the enzyme is first assembled in the mitochondria and then routed as a whole to the cell surface by a yet-to-be elucidated trafficking pathway (150) (Fig. 5). Due to its cell surface expression, the enzyme has been called cell surface or ecto-F1-ATPase/ATP synthase, although the exact structure of this ectopically expressed complex has not been determined definitively.
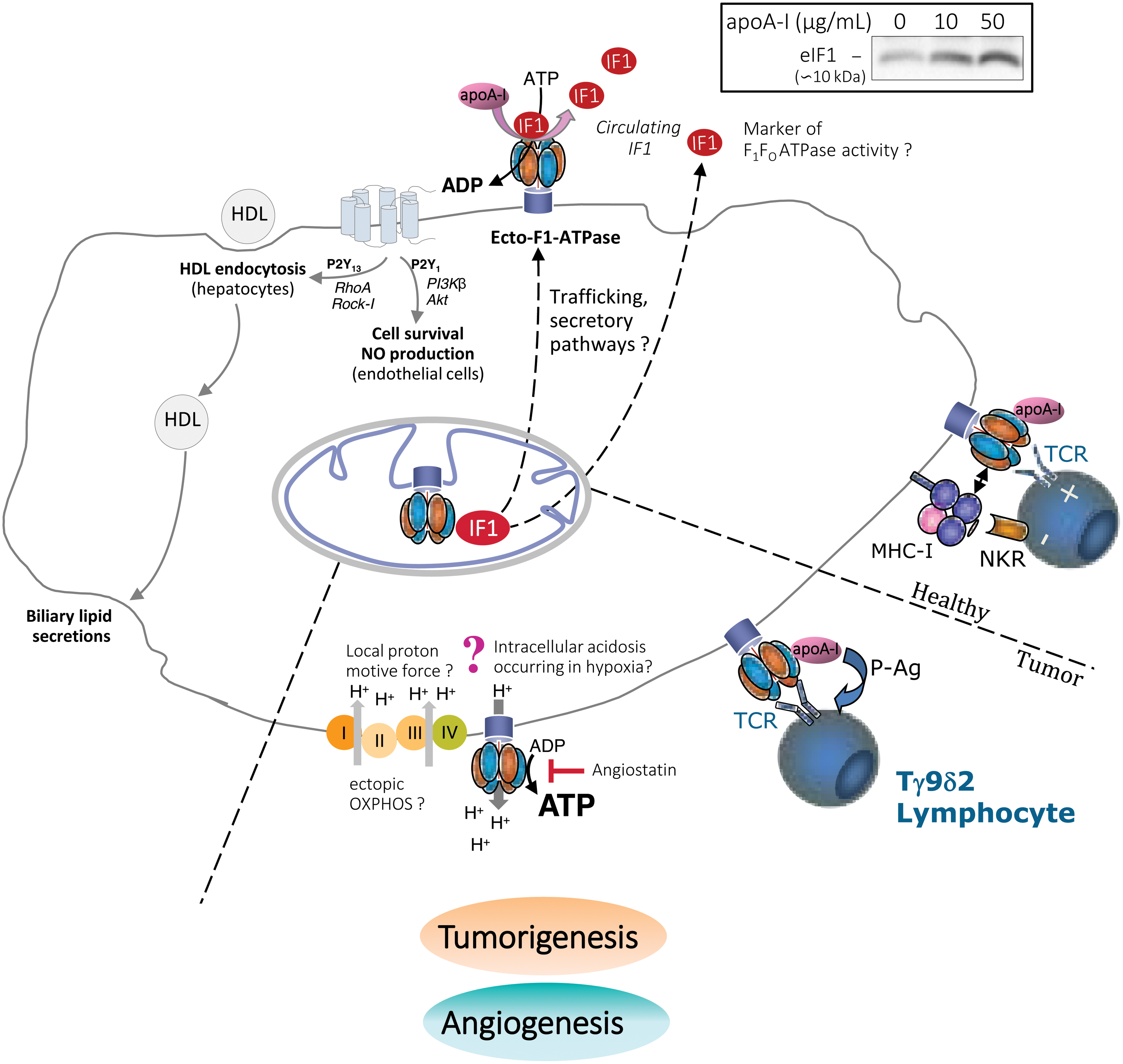
Localization and activity of ecto-F1-ATPase
The F1 catalytic domain faces the extracellular space (Fig. 5) at the cell surface and is mainly localized in cholesterol-rich raft domains/caveolae (3, 81, 88). In keeping with this, its expression has been positively associated with the membrane cholesterol level (81, 124, 155).
Ecto-F1-ATPase activity occurs in the extracellular space and potentially affects the extracellular ADP/ATP levels, which is consistent with the well-defined role of the ectopic enzyme in mediating downstream cell signaling via purinergic receptors (see next paragraph and Fig. 5). Under physiological conditions, ecto-F1-ATPase only has a hydrolytic function and thus contributes to the generation of extracellular ADP (96, 98). This can be explained by the potential of the plasma membrane, which ranges from −40 to −80 mV, rendering the ecto-F1-ATPase unable to synthesize extracellular ATP under physiological conditions (99). When the extracellular pH was artificially increased or the intracellular space was acidified, ecto-F1-ATPase was able to drive cellular proton extrusion accompanied by extracellular ATP synthesis (96).
Along with ATP synthase, subunits of the respiratory chain complexes were found to be present in lipid-rafts (81, 167). Although the respiratory chain activity at the plasma membrane was shown to produce extracellular ROS (88), it is not certain whether it generates a PMF sufficient to drive the synthesis of ATP by ecto-F1-ATPase. Instead, many groups have reported that extracellular ATP synthesis is due to adenylate kinase, which uses two molecules of ADP to produce ATP and AMP (39, 150, 165).
Overall, the data suggest that ecto-F1-ATPase is mostly active by producing extracellular ADP from ATP hydrolysis. It may only synthesize ATP in particular metabolic settings such as the intracellular acidosis that occurs in hypoxia (1, 143).
Signaling pathways mediated by ecto-F1-ATPase and regulation by IF1
The best-characterized signaling pathways mediated by ecto-F1-ATPase are those that take place in hepatocytes, endothelial cells, and cancer cells.
In hepatocytes, our group has demonstrated that apolipoprotein A-I (apoA-I), which is the main apolipoprotein in high density lipoprotein (HDL) particles, binds with high affinity to the β subunit of ecto-F1-ATPase, thereby resulting in increased hydrolysis of extracellular ATP into ADP (98). The extracellular ADP that is generated selectively activates the P2Y13 purinergic receptor, resulting in RhoA/ROCK1 signaling that controls HDL endocytosis by an unknown low-affinity receptor (74, 95) (Fig. 5).
This process is regulated in a precise manner by extracellular nucleotide metabolism. Cell surface activity of adenylate kinase consumes the extracellular ADP generated on activation of ecto-F1-ATPase and it downregulates HDL endocytosis (39). Similarly, adenine nucleotide translocase activity transports ADP into the cell, thereby resulting in a decrease in the level of extracellular ADP at the expense of its production by ecto-F1-ATPase (17). Importantly, the ecto-F1-ATPase/P2Y13-mediated HDL endocytosis pathway in hepatocytes contributes to excess cholesterol removal by the liver and hence protects against atherogenesis in mice (93). Interestingly, p2y13 genetic variant (rs3732757) was associated with higher circulating IF1 levels (151), supporting the notion that ecto-F1-ATPase and P2Y13 act in close coordination in humans to promote hepatic HDL endocytosis.
In endothelial cells, the hydrolytic activity of ecto-F1-ATPase is also increased by apoA-I (123) but the downstream signaling pathway differs profoundly from the earlier mentioned pathway in hepatocytes. The extracellular ADP that is generated stimulates the P2Y1 receptor followed by PI3Kβ/Akt pathway signaling, thereby promoting cell survival and proliferation (18) (Fig. 5). A similar process had been observed in human endothelial progenitor cells (EPCs), which led to enhancement of their angiogenic capacity (62).
The sequence apoA-I/ecto-F1-ATPase/P2Y1 also promotes nitric oxide (NO) production by endothelial cells, thereby controlling vascular tone in vivo in mice (14). Finally, coupling of ecto-F1-ATPase to the P2Y12 receptor promotes HDL transcytosis by endothelial cells, which is an important step in HDL reaching the macrophage foam cells underneath the endothelium (19). Overall, these functions of ecto-F1-ATPase in endothelial cells are likely to protect from endothelial cell damage occurring during the initial phases of atherogenesis. Pharmacological activation of ecto-F1-ATPase could represent a promising approach for enhancing vascular endothelial protection.
Aside from these two important roles in hepatocytes and endothelial cells, ecto-F1-ATPase has also been implicated in tumor cell recognition and proliferation. Vγ9/Vδ2 T lymphocytes recognize ecto-F1-ATPase on the surface of tumor cells through their T cell receptors, and apoA-I enhances the cellular response (138). Mechanistically, ecto-F1-ATPase binds and converts phosphoantigens produced by tumor cells to nucleotide derivatives to present them to Vγ9/Vδ2 T lymphocytes (104) (Fig. 5). Importantly, ecto-F
Remarkably, most of the aforementioned cellular events mediated by ecto-F1-ATPase could be specifically inhibited by treatment with exogenous IF1, either as its constitutively active IF1-H49K variant or as its inhibitory domain IF110–40 (14, 17 –19, 84, 98, 123). For instance, in hepatocytes, treatment with IF110–40 decreases HDL endocytosis-mediated by ecto-F1-ATPase (17). Similarly, in endothelial cells, treatment with IF1-H49K inhibits the effect of apoA-I on NO production and consequent vasorelaxation (14). Native IF1 has been reported to be present at the plasma membrane of hepatocytes (28) and endothelial cells (30). It has been proposed that ecto-IF1 can constitutively bind ecto-F1-ATPase and partially inhibit its hydrolase activity (60).
One could hypothesize that some other ligands, such as apoA-I, could activate ecto-F1-ATPase by dissociating IF1 from its inhibitory binding site on ecto-F1-ATPase. This hypothesis is supported by our observation that incubation of HepG2 cells with apoA-I, at 4°C to block cellular secretory pathways, promotes IF1 release into the extracellular medium (Fig. 5, upper left inset).
Overall, ecto-F1-ATPase activity is involved in numerous physiological and pathophysiological functions, thus making it an attractive therapeutic target (see IF1 in Diseases section). Depending on the cell type, distinct signaling pathways could be activated, thereby inducing distinct cellular events. However, in many instances, the signaling pathways mediated by ecto-F1-ATPase appear to rely on its ATP hydrolase activity, which generates extracellular ADP and that can be inhibited by IF1.
IF1 in Diseases
IF1 in cancer and angiogenesis
Cancer is primarily a disease of energy metabolism, linked to impaired mitochondrial function and cellular respiration. Tumor cells undergo metabolic reprogramming to support their high metabolic rate. The most well-known metabolic alteration in cancer, in normoxic and hypoxic conditions, is the energy shift from OXPHOS to glycolysis, with increased glucose uptake [the Warburg effect (156)]. Cancer cells exhibit metabolic heterogeneity and flexibility, however, depending on the tumor type, microenvironment, nutrients, and oxygen availability (77). Thus, the Warburg effect is not observed across all tumor types and it exists more as glycolysis–OXPHOS cooperation during disease progression (77).
Several in vitro and in vivo studies have reported that mitochondrial IF1 favors tumorigenesis. Under aerobic conditions, the ability of IF1 to inhibit the synthetic activity of ATP synthase promotes tumor progression by favoring a Warburg phenotype (55, 66), activation of an ROS-mediated retrograde prosurvival and proliferative response (46, 131, 134), or by preservation of cristae structures to limit apoptotic cell death signaling (40, 42) [see previous sections on mitochondrial bioenergetics and fitness, as well as Esparza-Moltó and Cuezva (36), Faccenda et al. (41), and Sánchez-Aragó et al. (130) for reviews].
Alternatively, under hypoxic conditions, IF1-mediated inhibition of ATP synthase hydrolase activity can protect cancer cells from cytosolic ATP depletion. Mitochondrial IF1 has been reported to be overexpressed in various types of carcinomas (colon, lung, breast, ovarian, and liver) (46, 55, 66, 131, 133) and it is considered to be a biomarker of poor prognosis in patients with glioblastoma (160); hepatocarcinoma (145); or carcinoma of the bladder (157), lung (50), and stomach (166). By contrast, IF1 overexpression in colon and breast carcinomas is indicative of a favorable prognosis, thus suggesting tissue-specific functions of IF1 (61, 131). Although the underlining mechanisms of IF1 overexpression in carcinomas have not yet been elucidated, they could be related to a defect in its degradation (132) or exposure to environmental carcinogens (66).
Equally important, activation of cell proliferation by ecto-F1-ATPase has been reported in various carcinomas. In cancerous colonic epithelial cells, the peptide hormone product of the glycine gene (glycine-extended gastrin [G-gly]) activates the hydrolytic activity of ecto-F1-ATPase and promotes the growth and proliferation of carcinoma cells (84). Of note, blocking ecto-F1-ATPase with an exogeneous constitutively active IF1 variant decreases G-gly-induced cell growth (84). Similar to apoA-I, G-gly may activate ecto-F1-ATPase by dissociating the inhibitory IF1 from ecto-F1-ATPase, although the presence of IF1 at the plasma membrane of tumor cells has not been well documented.
A similar activation of proliferation by ecto-F1-ATPase has been observed in lung and breast carcinoma and myeloid leukemia cell lines (22, 23, 114, 159). Some studies have attributed the effect of ecto-F1-ATPase on tumorigenesis to the production of extracellular ATP, which could accommodate the increased energy requirement (Fig. 5).
The bioenergetic law governing the synthetic activity of ecto-F1-ATPase casts doubt, however, on this activity in the tumor microenvironment. As previously mentioned, the synthesis of ATP by ecto-F1-ATPase is strictly driven by a proton flux from the cytosol to the plasma membrane that provides the required chemical energy (96). In extracellular acidosis, the H+ gradient would not be conducive to ecto-F1-ATPase-mediated ATP synthesis. Alternatively, despite the acidic tumor microenvironment, ectopic respiratory chain activity at the plasma membrane could generate a local inverse H+ gradient that promotes ATP synthesis (81, 88, 167).
Ecto-F1-ATPase has been identified as a player in tumor angiogenesis due to its presence on endothelial cells and its interaction with angiostatin, which is a protein with anti-angiogenic properties (105). By inhibiting the ectopic enzyme, angiostatin can reduce the amount of ATP, lower the intracellular pH, and reduce cell survival and proliferation (Fig. 5) (26).
The IF1 has been proposed to act in a similar manner, although evidence that IF1 and angiostatin are competitive inhibitors is still lacking (10). Although treatment with exogeneous IF1 can limit endothelial cell proliferation, it does not inhibit endothelial cell tube formation. Instead, IF1 has been shown to antagonize the angiogenesis-promoting effect of apoA-I in human EPCs (62), which could be rather detrimental to the promotion of neovascularization by EPCs after ischemia and the repair of injured or damaged endothelium (62). Finally, although constitutive ecto-IF1 has been shown to be associated with ecto-F1-ATPase at the cell surface of endothelial cells (30), whether it plays a role in limiting endothelial cell proliferation remains to be determined.
IF1 and neurodegenerative diseases
Life expectancy has increased considerably over the past century, thus leading to a rise in the incidence of neurodegenerative diseases. The main neurodegenerative disorders (Alzheimer's disease [AD], Parkinson's disease, and amyotrophic lateral sclerosis) are age-related, and mitochondrial dysfunction is a recurring element in these pathologies (92). This is not surprising, as neuronal activity requires a very substantial input of energy, which renders neurons highly dependent on mitochondrial OXPHOS.
Neurons are characterized by a high level of IF1 expression (44), with the levels of this protein surpassing those of ATP synthase (37). It has been shown that IF1 plays a pivotal role in the neuronal and visual development of zebrafish and mice by limiting apoptosis and excessive microglial activity (100). Further, Esparza-Moltó et al. have shown that a lack of IF1 in mouse neurons leads to impaired cognitive functions, whereas mice exhibit increased learning abilities when IF1 is overexpressed (38). The authors explain this effect through the influence of IF1 on ATP synthase dimer formation, ATP synthase activity, mitochondrial proton leakage, ΔΨm, and mtROS-induced nuclear responses (38).
The extracellular ATP level in the brain has been proposed to be a factor in AD progression (136). Molecules involved in the production of β-amyloid plaques can block ecto-F1-ATPase, which is present at the plasma membrane of neurons and astrocytes, by interacting with the α subunit (136). The proposed mechanism includes reduction of extracellular ATP production, changes in the intracellular/extracellular pH, and Ca2+ signaling (136). Interestingly, the molecules involved (amyloid precursor protein and amyloid β-peptide) share a high degree of homology with IF1 (136).
The IF1 levels in neurons are upregulated in ischemia/re-oxygenation models, thus pointing toward a role of IF1 in cell self-protection and preservation (101). The neuroprotective effects of IF1 could be attributed to inhibition of the hydrolytic activity of F1-ATPase, along with its role in mitophagy (101). Interestingly, the human IF1-H49K mutant form has been shown to rewire neuronal metabolism in mice, thereby resulting in increased aerobic glycolysis (44). This model led to a decrease in ATP in the brain, but the preconditioning state partially protected the animals' brains against an excitotoxin by activation of the Akt/p70S6K and PARP pathways, along with Bcl-xL (44). However, it remains to be determined how this research can be translated to patients and what the exact role of IF1 is in age-related neurodegenerative diseases.
IF1 and cardiovascular diseases
ATP synthase is a major consumer of ATP under ischemic conditions (63, 67). With regard to its function under hypoxic conditions (see IF1 and Mitochondrial Bioenergetics section), IF1 was originally assumed to be involved in myocardial ischemia–reperfusion injury, which is a common pathophysiological process and therapeutic approach in patients with coronary heart disease, by preventing (at least to a certain extent) ATP hydrolysis by mitochondria.
This hypothesis is supported by in vitro models of ischemic injury that alter the expression of IF1, showing that overexpression of the protein has a protective effect against cell death, whereas a significant increase in cell death was observed after suppression of IF1 (16). Accordingly, the expression level of IF1 was found to be inversely associated with the hydrolytic activity of ATP synthase in goat preconditioned myocardium (115, 119).
However, the protective role of IF1 in the acute and late phases of ischemia–reperfusion remains to be demonstrated (68, 152). Indeed, ATPIF1 deletion in mice appears to provide protection from cardiac hypertrophy by a mechanism that prevents mitochondrial depolarization and that activates autophagy (163). Conversely, overexpression of IF1 in primary cardiomyocytes promotes hypertrophy and has been associated with a shift from respiration to glycolysis, along with an increase in mitochondrial oxidative stress (117). This effect of IF1 on cardiomyocyte hypertrophy development has been ascribed to a reduced mitochondrial capacity to store Ca2+, thereby leading to activation of Ca2+/calmodulin protein kinase II (CaMKII), independently of IF1 binding to ATP synthase (117).
Overall, under normal respiratory conditions, it appears that IF1 promotes mitochondrial dysfunction and cardiac hypertrophy. The upregulation of IF1 observed in mouse and human dysfunctional hearts may be a maladaptive response (117, 163).
The hydrolytic activity of plasma membrane ecto-F1-ATPase contributes, through purinergic signaling, to several atheroprotective functions mediated by HDL, such as hepatic cholesterol removal and endothelium repair (see IF1 and ecto-F1-ATPase section and Fig. 5).
Until now, the inhibitory action of IF1 on ecto-F1-ATPase activity has been assessed by using an exogenous constitutively active IF1-H49K isoform, which has been shown to impair HDL atheroprotective activities (14, 18, 19, 98). However, it could be hypothesized that endogenous ecto-IF1, the presence of which has been reported in hepatocytes and endothelial cells (28, 30), would favor atherogenesis by constitutive inhibition of ecto-F1-ATPase activity, thereby counteracting the HDL atheroprotective effects. In keeping with this, in acute cholestasis, which is a condition associated with impaired hepatic HDL endocytosis, ecto-F1-ATPase activity has been found to be reduced in hepatocytes due to an increase in ecto-IF1 bound to the enzyme (60).
IF1 and metabolic diseases
Excess nutrient consumption directly impacts mitochondria by increasing the concentration of free fatty acids, hyperglycemia, and increased ROS production (86). Downregulation of mitochondrial biogenesis has been reported in obese animal models and humans (102). Proteomic analysis of human skeletal muscle mitochondria has revealed obesity-induced downregulation of the α and β subunits of ATP synthase (45). The same study found that IF1 was highly upregulated, thereby resulting in an increase in the IF1/β-F1-ATPase ratio and indicating that the inhibition of OXPHOS is an attribute of obesity (45). Interestingly, this ratio had a moderate positive correlation with the body mass index, but not other factors, such as age, fasting plasma glucose, and insulin (45).
Diabetes is a chronic condition characterized by the disruption of glucose metabolism either by an autoimmune response against the β-pancreatic cells (type 1) or by impaired insulin signaling (insulin resistance) due mostly to lifestyle risk factors (type 2 diabetes [T2D]) (94). Mitochondrial dysfunction has been identified as an important factor in T2D as it is associated with insulin resistance (103, 129), especially in skeletal muscle (2).
It has been reported that obese patients with diabetes have a higher IF1/β-F1 ATPase ratio and a lower level of inactive phosphorylated IF1 compared with healthy individuals, which results in a lower synthetic activity of ATP synthase (45). It has been proposed that IF1 can negatively impact the progression of T2D as its overexpression in skeletal muscle cells stimulates tumor necrosis factor alpha secretion (45), which is a cytokine that promotes insulin resistance (164), and it affects mitochondrial β-oxidation (altering α-ketoglutarate and L-carnitine metabolism), thereby increasing the cellular content of nonesterified fatty acids (45).
In a model of β-pancreatic cells (INS-1E cells), silencing of IF1 led to increased insulin secretion and glucose sensitivity (78), whereas overexpression downregulated glucose-stimulated insulin secretion (79). Reducing the inhibitory activity of IF1 should be possible in vivo by the activation of PKA-induced IF1 phosphorylation (54). The cAMP/PKA signaling pathway has already been proposed as a target for T2D and glycemic control (162).
Unlike the earlier mentioned studies highlighting the potential deleterious effects of mitochondrial IF1 on glucose homeostasis and metabolic health, a recent study has reported that IF1 treatment stimulated glucose uptake in skeletal muscle cells through different mechanisms, involving, among others, ROS production and a rise in extracellular ATP, with the latter possibly related to IF1-mediated inhibition of ecto-F1-ATPase (89). Chronic treatment with recombinant IF1, which solely targets ecto-F1-ATPase, improved glucose tolerance in db/db mice with type 2 diabetes (89). In addition, recombinant IF1 restrained high fat diet-induced obesity in C57BL/6 mice by regulating food intake through hypothalamic signaling in a sex-specific manner, since this impacted only male and ovariectomized female mice (87).
However, as detailed in the previous section “IF1 and ecto-F1-ATPase,” the inhibition of the ecto-F1-ATPase through IF1 administration in mice was clearly detrimental to certain HDL-related metabolic and vascular beneficial functions, such as excess hepatic cholesterol removal and endothelial protection (14, 98, 99).
Altogether, current data indicate that IF1 might have distinct effects on metabolic diseases, depending on the target organ and whether it inhibits mitochondrial ATP synthase or cell surface ecto-F1-ATPase.
Circulating IF1: a biomarker of cardiovascular and metabolic diseases
Using a combination of proteomic approaches, our group was the first to identify IF1 in human serum (58). A specific competitive ELISA immunoassay was first devised to quantify circulating IF1 (59). In a population of normolipemic individuals, the IF1 serum levels had a normal distribution, centered on a median of 490 ng/mL. This competitive measurement has been used in several subsequent studies. To fulfil the growing need for a standardized method measuring circulating IF1, we recently developed a liquid chromatography—tandem mass spectrometry analytical method enabling the accurate quantification of IF1 in human plasma and that could be proposed as a reference method, amenable for clinical practice (56).
In our various studies, serum IF1 was always positively correlated with HDL-cholesterol or apoA-I, but negatively with triglycerides, thus providing further support to the notion of a role of the ecto-F1-ATPase pathway in lipoprotein metabolism (57, 59). In a cross-sectional study, IF1 levels were found to be 20% lower in patients with coronary artery disease (CAD) than in matched control subjects. Serum IF1 was independently associated with CAD following multivariate adjustments on cardiovascular risk factors or markers (59). In a prospective study of 580 patients with established CAD followed up for 11 years, serum IF1 was inversely related to all-cause and cardiovascular mortality (57). This relationship remained significant after adjustments on a large panel of clinical or biological variables associated with the prognosis of CAD.
More recently, Lee et al. identified IF1 as a myokine, with circulating levels being positively correlated with greater muscle mass and negatively associated with age and body fat (89). Exercise stimulation increased circulating IF1 in mice and in healthy humans, suggesting that IF1 is secreted as part of the muscle secretome during contraction (89).
All of these observations suggest that an elevated serum IF1 level should protect against cardiovascular and metabolic disease, which raises the question of its origin. The IF1 is present at the plasma membrane of different cell types, including hepatocytes and endothelial cells, where it binds to and inhibits ecto-F1-ATPase. apoA-I can compete for binding to the enzymatic complex, which is hence activated while IF1 is released into the bloodstream (Fig. 5). Thus, elevated serum IF1 may be associated with high ecto-F1-ATPase activity, which participates in HDL-mediated reverse cholesterol transport and endothelium repair, with these mechanisms thus protecting against atherosclerosis.
However, a mitochondrial origin for serum IF1 can also be considered. Indeed, in muscle cells subjected to ischemic conditions, IF1 is recruited to mitochondrial ATP-synthase/hydrolase to prevent depletion of cellular ATP. Under normoxic conditions, IF1, when no longer recruited, could be released from the mitochondria and into the extracellular space. Hence, elevated serum IF1 could reflect optimal myocardium function, somewhat identical to the impact of muscle exercise on IF1 levels (89). Interestingly, in our observations in patients with CAD, serum IF1 was found to be highly correlated with the left ventricular ejection fraction.
An overview of the IF1 involvement in mitochondrial fitness and diseases is presented in Table 1 and Figure 6.
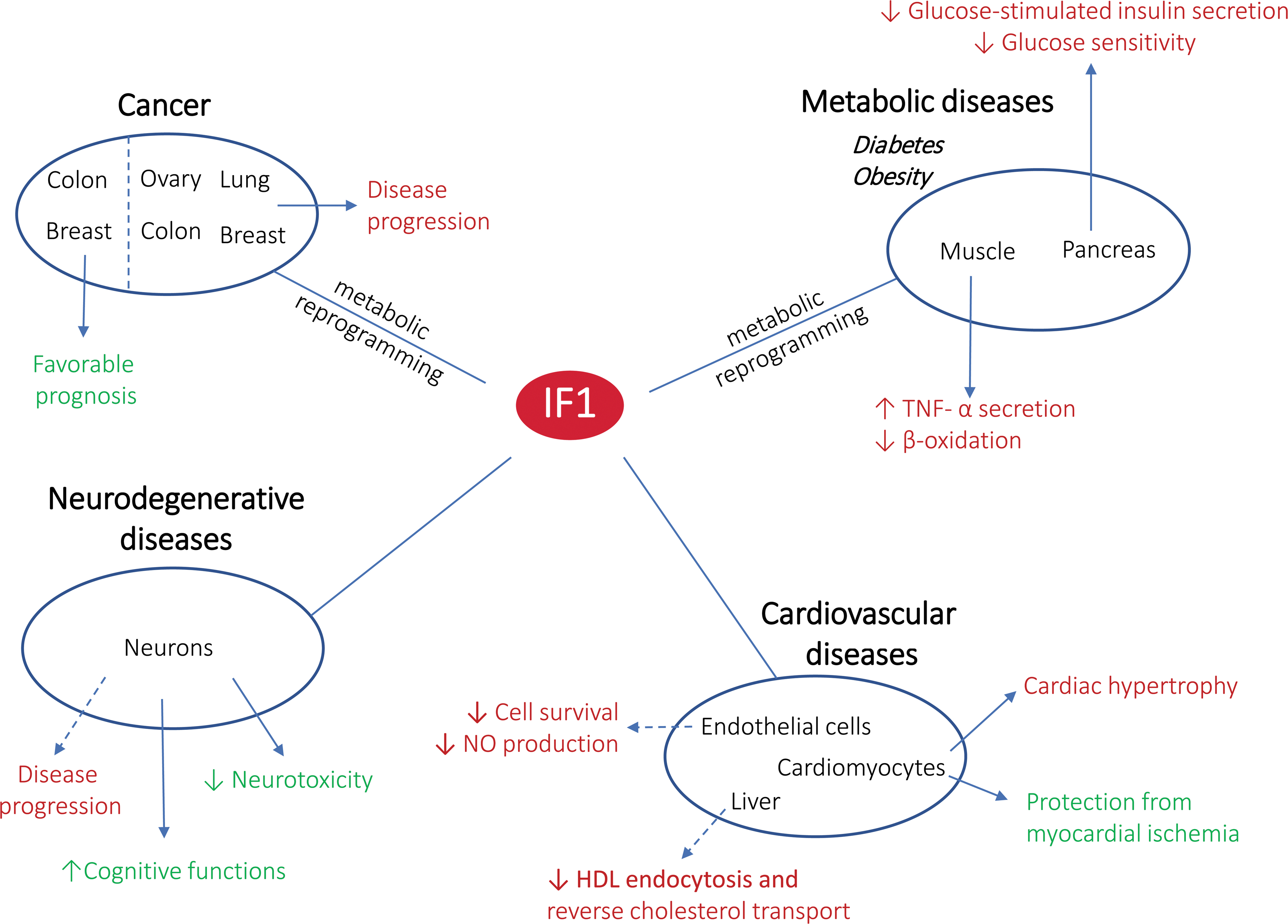
Main Functions of IF1 in Mitochondrial Fitness and Diseases
For each function, the targeted ATP synthase (mitochondrial or cell surface, i.e., ecto-F1-ATPase) is mentioned.
apoA-I, apolipoprotein A-I; BMI, body mass index; Drp1, dynamin-related protein 1; eNOS, endothelial nitric oxide synthase; G-gly, glycine-extended gastrin; GSH, reduced glutathione; HDL, high-density lipoprotein; IF1, inhibitory factor 1; mtROS, mitochondrial ROS; n.d., not determined; NO, nitric oxide; OMA1, OMA1 zinc metallopeptidase; OPA1, optic atrophy 1; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; TNFα, tumor necrosis factor alpha.
Conclusion
Mitochondria are a key organelle, acting as a nexus of bioenergetics, biosynthesis, and signaling pathways. The nuclear-encoded inhibitor of the ATP synthase, IF1, has recently emerged as a multifaceted player and potential therapeutic target. If IF1 was initially considered a unidirectional inhibitor of the ATP synthase's hydrolytic activity, it is currently seen as a versatile protein, involved in mitochondrial homeostasis, energy metabolism, cell fate, and regulation of various cellular pathways through its interaction with ecto-F1-ATPase.
Among other functions, the ecto-F1-ATPase contributes to reverse cholesterol transport and vascular endothelial protection. The IF1-mediated ecto-F1-ATPase regulation could have implications in conditions such as atherosclerosis and cardiovascular diseases. In addition, IF1 appears to be implicated in diabetes and obesity. All these observations indicate that IF1 and/or ecto-F1-ATPase could represent potential targets for the therapy of pathologies associated to the metabolic syndrome. Modulation of mitochondrial and cellular functions by IF1 is also an emerging approach in cancer and neurodegenerative diseases.
The missing pieces in the IF1 biology puzzle are of paramount importance to distinguish between beneficial and detrimental effects of IF1, which is critically important to identify therapeutic strategies and clinical translation.
Footnotes
Authors' Contributions
E.G., T.D., and L.O.M. conceptualized the content, reviewed the literature, wrote the article, and generated figures. B.P. and A.G contributed to the writing of the “Circulating IF1: A Biomarker of Cardiovascular and Metabolic Diseases” section. S.N. contributed to the writing of the “IF1 and Mitochondrial Fitness” section and generated ![]() . L.O.M. obtained the funding.
. L.O.M. obtained the funding.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Inspire Program grants RESPIMITAGING (Reference number: 1901175), THERANOVASC (Reference number: ESR_R&S_DF-000094/2018-003303), and HEPATOCARE (Reference number: R20018BB no. 19014226) from the Region Occitanie Pyrénées-Méditerranée, the European Regional Development Fund (ERDF) (Project number: Inspire MP0022856, THERANOVASC DF-000094/2018-003303), and the Fondation de France (Project number: 00086504—AO Recherche Clinique et fondamentale sur les maladies cardiovasculaires 2018).