
Abstract
Aims:
Influenza A virus hemagglutinin (HA) binding to sialic acid on lung epithelial cells triggers membrane fusion and infection. Host thiol isomerases have been shown to play a role in influenza A virus infection, and we hypothesized that this role involved manipulation of disulfide bonds in HA.
Results:
Analysis of HA crystal structures revealed that three of the six HA disulfides occur in high-energy conformations and four of the six bonds can exist in unformed states, suggesting that the disulfide landscape of HA is generally strained and the bonds may be labile. We measured the redox state of influenza A virus HA disulfide bonds and their susceptibility to cleavage by vascular thiol isomerases. Using differential cysteine alkylation and mass spectrometry, we show that all six HA disulfide bonds exist in unformed states in ∼1 in 10 recombinant and viral surface HA molecules. Four of the six H1 and H3 HA bonds are cleaved by the vascular thiol isomerases, thioredoxin and protein disulphide isomerase, in recombinant proteins, which correlated with surface exposure of the disulfides in crystal structures. In contrast, viral surface HA disulfide bonds are impervious to five different vascular thiol isomerases.
Innovation:
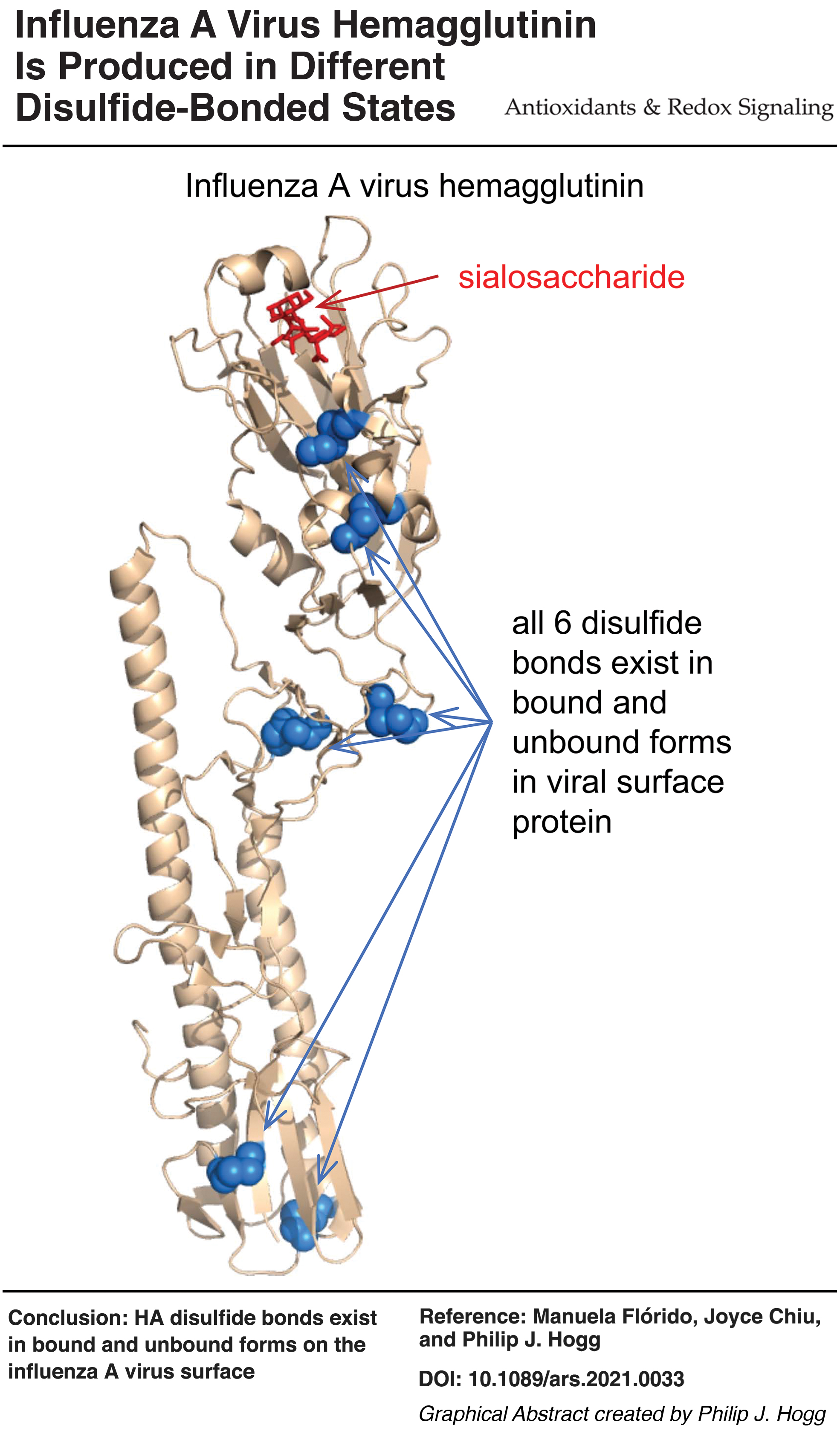
It has been assumed that the disulfide bonds in mature HA protein are intact and inert. We show that all six HA disulfide bonds can exist in unformed states.
Conclusion:
These findings indicate that influenza A virus HA disulfides are naturally labile but not substrates for thiol isomerases when expressed on the viral surface.
Introduction
Influenza A virus infections, mostly caused by circulating H1N1 and H3N2 strains, are estimated to cause 0.29–0.65 million deaths per year (23) and have significant economic impact around the world. These viruses replicate in the respiratory tract, leading to acute self-limiting infection and have health outcomes ranging from mild respiratory disease to fatal pneumonia (13).
Hemagglutinin (HA), the major viral glycoprotein in influenza viruses, is expressed as a non-covalently linked homo-trimer at the surface of the virion and initiates infection by binding to sialic acid on epithelial cells and inducing membrane fusion, enabling the virus entry into the host cell (9). HA is synthesized as a precursor HA0 that to become active requires cleavage by host proteases into HA1 (the receptor-binding domain) and HA2 (the membrane-fusion domain) subunits, which remain linked through an inter-domain disulfide bond (47). On entry of the virus into the host cell, the low pH of the endosome triggers HA conformational change, exposing the HA2 domain that mediates fusion between viral and endosomal membranes. This leads to release of the viral ribonucleoprotein complexes into the cytoplasm and their translocation to the nucleus, where they undergo transcription and replication (14).
(Color images are available online)
Thiol/disulfide exchange has an important role in the entry into host cells of several viruses (24, 34, 38, 51, 54). The oxidoreductases, or thiol isomerases, are factors containing a cysteine pair at the catalytic site that can transfer electrons and reduce or oxidize disulfides or dithiols in other proteins. Although these proteins are found in high amounts in the endoplasmic reticulum where they participate in the folding of nascent proteins, they are also secreted and found at the cell surface where they mediate thiol–disulfide interchange reactions in secreted proteins to control function via conformational changes (17). For example, protein disulfide isomerase (PDI) is secreted by activated platelets and binds to the platelet surface, where it cleaves two allosteric disulfides in the GP1bα receptor that enhances ligand binding (33).
The active site dithiols/disulfides of PDI exist predominantly in reduced configuration on the activated platelet surface (6). A fraction of the PDI secreted by human fibroblasts also exists in reduced configuration on the cell surface, which correlates with a reduction of disulfide bonds in cell surface proteins (27). These observations indicate that secreted PDI has reducing potential. These factors are frequently upregulated during infection (10, 17) and have been implicated in the conformational changes in proteins of enveloped viruses that are required for cell entry (3, 24, 34, 51).
The role of thiol isomerases in HIV infection is best understood. HIV entry into host lymphocytes is mediated by attachment of the gp120 subunit of the gp160 viral envelope coat protein to cell surface CD4, and after conformational change, binding to co-receptors CXCR4 or CCR5. PDI and/or thioredoxin cleave specific disulfide bonds in HIV gp120 on interaction with CD4 and CXCR4/5, resulting in conformational changes that facilitate entry of the virus into host cells (3, 4, 16, 35, 38).
Innovation
It has been assumed that the disulfide bonds in mature HA protein are intact and inert. We show that all six HA disulfides can exist in unformed states, although the bonds are not substrates for thiol isomerases when HA is expressed on the viral surface.
The role of the thiol isomerases, ERp57, PDI and ERp72, in influenza A virus HA maturation and transit to the cell surface has been established. Treatment of mice with ERp57 inhibitor, LOC14, impaired disulfide formation and oligomerization of HA and viral load in lung epithelial cells, whereas lung-specific ablation of ERp57 was associated with a decrease of viral burden and lung inflammation in infected mice (10). Nitazoxanide, another inhibitor of ERp57 (40), blocked HA terminal glycosylation and transport to the cell surface (43), whereas knockdown of ERp57 inhibited disulfide bond formation and maturation of HA in fibroblasts (46) and replication of influenza A virus in lung epithelial cells (42). PDI inhibitors or knockdown of PDI or ERp72 impair the replication of influenza viruses in Madin-Darby canine kidney (MDCK) and A549 cells (29).
There is also evidence that secreted thiol isomerases affect mature HA function at the viral or host cell surface. Systemic treatment with the membrane-impermeable PDI inhibitor, isoquercetin, reduced influenza A viral replication and lung pathology in infected mice (30). In this study, we have examined the redox state of the disulfide bonds of mature influenza A virus HA and the susceptibility of these bonds to cleavage by vascular thiol isomerases, including PDI. We find that mature HA naturally exists in different disulfide-bonded states, which are impervious to vascular thiol isomerases when HA is expressed on the viral surface. These findings may have implications for the role of different HA covalent forms in influenza A virus infection.
Results
The disulfide landscape of HA is generally strained, and some bonds exist in unformed states in crystal structures
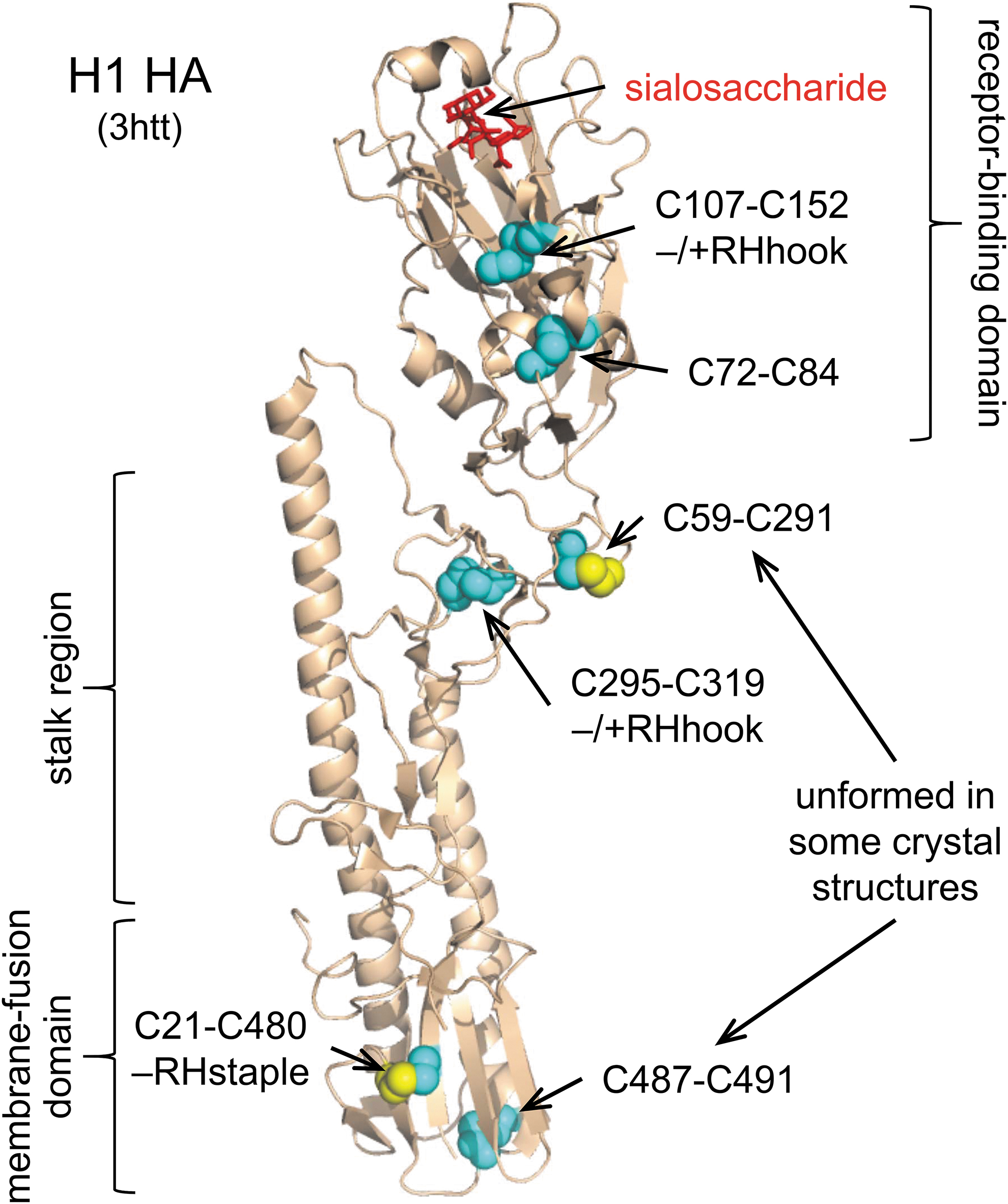
The HA molecules contain six disulfide bonds; two in the receptor-binding domain that interacts with sialic acid, two in the stalk region, and two in the membrane-fusion domain (Fig. 1). Disulfide bonds are classified based on the geometry of the five dihedral angles that describe the cystine residue and can adopt 20 different conformations (45). Two of the 20 disulfide conformations, the –RHstaple and –/+RHhook bonds, are naturally strained and have high potential energy due to stretching of the S-S bond and neighboring α angles (12, 45, 56). It is noteworthy that three of the six HA disulfides have high energy –RHstaple or –/+RHhook conformations (Table 1). These conformations are also associated with a subset of disulfide bonds, the allosteric disulfides, that control mature protein function when cleaved or formed (12). Using the residue numbering for H1 (Uniprot identifier P03452), the C107-C152 disulfide in the receptor-binding domain near the sialic acid binding pocket and the C295-C319 bond in the stalk region has a –/+RHhook conformation, whereas the C21-C480 bond in the membrane-fusion domain has a –RHstaple conformation. The equivalent bonds in the H2 and H3 isotypes also have these conformations (Table 1). This finding suggests that the disulfide landscape of HA is generally strained. We next inspected X-ray structures of the H1, H2, and H3 proteins for disulfide bonds that are present in some molecules of a protein crystal but absent in others. We hypothesized that such disulfides might naturally exist in unformed states (i.e., present as free thiols) in the native protein (41).
Conformations of the Hemagglutinin Disulfide Bonds
The disulfide conformations were determined by using the Disulfide Bond Analysis tool (47) and PDB identifiers 2wrg, 1ruz, 1ru7, 1ruy, 1rv0, 1rvt, 1rvz, 1rvx, 3hto, 3htp, 3htq and 3htt for H1 HA, 4w8n, 2wrf, 2wrd, 2wr7, 2wrb, 2wrc, 2wr5, 2wre, 2wr0, 2wr2, 2wr3, 2wr4, 2wr1, 3ku5 and 3ku3 for H2 HA, and 4we4, 4we5, 4we6, 4we8, 4we9, 6mzk, and 3vun for H3 HA. Cysteine numbering is based on protein sequences from Uniprot identifiers P03452 for H1 HA, Q20W19 for H2 HA, and P03437 for H3 HA.
The C59-C291 bond in H1 HA exists in +/−LHhook conformation in the 3httA, 3htqA, 3htpA, 1ru7CEIK, 3htoA, 2wrgJLH, 1rvzACEGIK, and 1rvxACEIK structures, and in +/−RHstaple conformation in 1ru7AG, 1ruzJHL, 1ruyJHL, 1rv0HJL, and 1rvtJHL structures.
The C19-C477 bond in H2 HA exists in −RHstaple conformation in the 2wrfABCDEFGHI, 2wrdABC, 2wr7BC, 2wrbB, 2wrcBC, 2wr5ABC, 2wreABC, 2wr0ABC, 2wr2ABC, 2wr3BC, 2wr4ABC, and 2wr1ABC structures, and in −LHhook conformation in the 2wr3A structure.
The C57-C288 bond in H2 HA exists in −RHstaple conformation in the 4w8nACE, 2wrfABCDEFGHI, 2wrdABC, 2wr7ABC, 2wrbABC, 2wrcABC, 2wr5ABC, 2wreABC, 2wr0C, 2wr2AC, 2wr3ABC, 2wr4ABC, 2wr1ABC, 3ku5A, and 2KU3A structures and in +/−LHhook conformation in the 2wr0AB structures.
The C484-C488 bond in H2 HA exists in −/+LHhook conformation in the 3ku5B, 3ku3B, 2wrfABCDEFGHI, 2wrdABC, 2wr7AB, 2wrbAB, 2wrcABC, 2wr5CB, 2wreABC, 2wr0ABC, 2wr2ABC, 2wr3B, 2wr4BC, and 2wr1ABC structures, in −LHhook conformation in 2wr7C, 2wr5A, 2wr3AC, and 2wr4A structures, and in +LHhook conformation in the 4w8nBDF structures.
HA, hemagglutinin; PDB, Protein Data Bank.
Two of the six disulfides in H1 (C59-C291 and C487-C491) and H2 (C57-C288, and C105-C149) were found to exist in formed or unformed states in crystal structures of the proteins (Table 2). For example, the C59-C291 H1 bond is unformed in one molecule of a protein crystal, whereas the C487-C491 is unformed in 12 molecules of three different protein crystals. The distance between the sulfur atoms of the unpaired cysteines of the broken bonds was clearly in excess of the equilibrium bond length of 2.038 Å (50), confirming that the disulfide bonds are unformed and not erroneously assigned. The limitations of this analysis are the nonnative conditions used to obtain the protein crystals, although no reducing agent was present during crystallization, potential differences in the qualities of the structures, and the possibility that some unformed bonds are due to cleavage by X-rays during data collection (48). Nevertheless, the information does suggest that one or more of the HA disulfides may be labile in the mature protein. This theory was tested by measuring the redox state of recombinant H1 and H3 proteins by differential cysteine alkylation and mass spectrometry. We chose to study these Has, as the circulating influenza A virus are mostly H1N1 and H3N2 strains.
Hemagglutinin Structures Containing Unformed Disulfide Bond(s)
Cysteine numbering is based on protein sequences from Uniprot identifiers P03452 for H1 HA and Q20W19 for H2 HA.
Although the C487-C491 disulfide was indicated as formed in the structures, the sulfur–sulfur bond lengths (range of 2.11–2.80 Å) were well in excess of the equilibrium bond length of 2.038 Å (50), indicating that they were erroneously assigned.
Recombinant HA H1 and H3 proteins contain unformed disulfide bonds
Briefly, the unpaired cysteine thiols in purified recombinant HA proteins (H1N1 A/Puerto Rico/8/1934 and H3N2 A/Aichi/2/1968) were alkylated with a carbon-12 version of 2-iodo-N-phenylacetamide (12C-IPA) (Fig. 2A), the protein isolated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 2B), the disulfide bonds reduced with dithiothreitol, and the resulting free cysteine thiols alkylated with a carbon-13 isotope of IPA (13C-IPA) (8). The HA was digested with trypsin/chymotrypsin and peptides quantified by HPLC, and identity was established by mass spectrometry (Supplementary Fig. S1). The levels of the different redox forms of the cysteines were determined from the relative abundance of peptides labeled with 12C-IPA and/or 13C-IPA (Supplementary Tables S1 and S2). The results are expressed as % of the HA disulfide that is unformed in the population. We have used the term “unformed” disulfide bond, as opposed to “reduced” disulfide bond, as reduced implies that the bond was formed at some point and later reduced, for which there is no evidence.
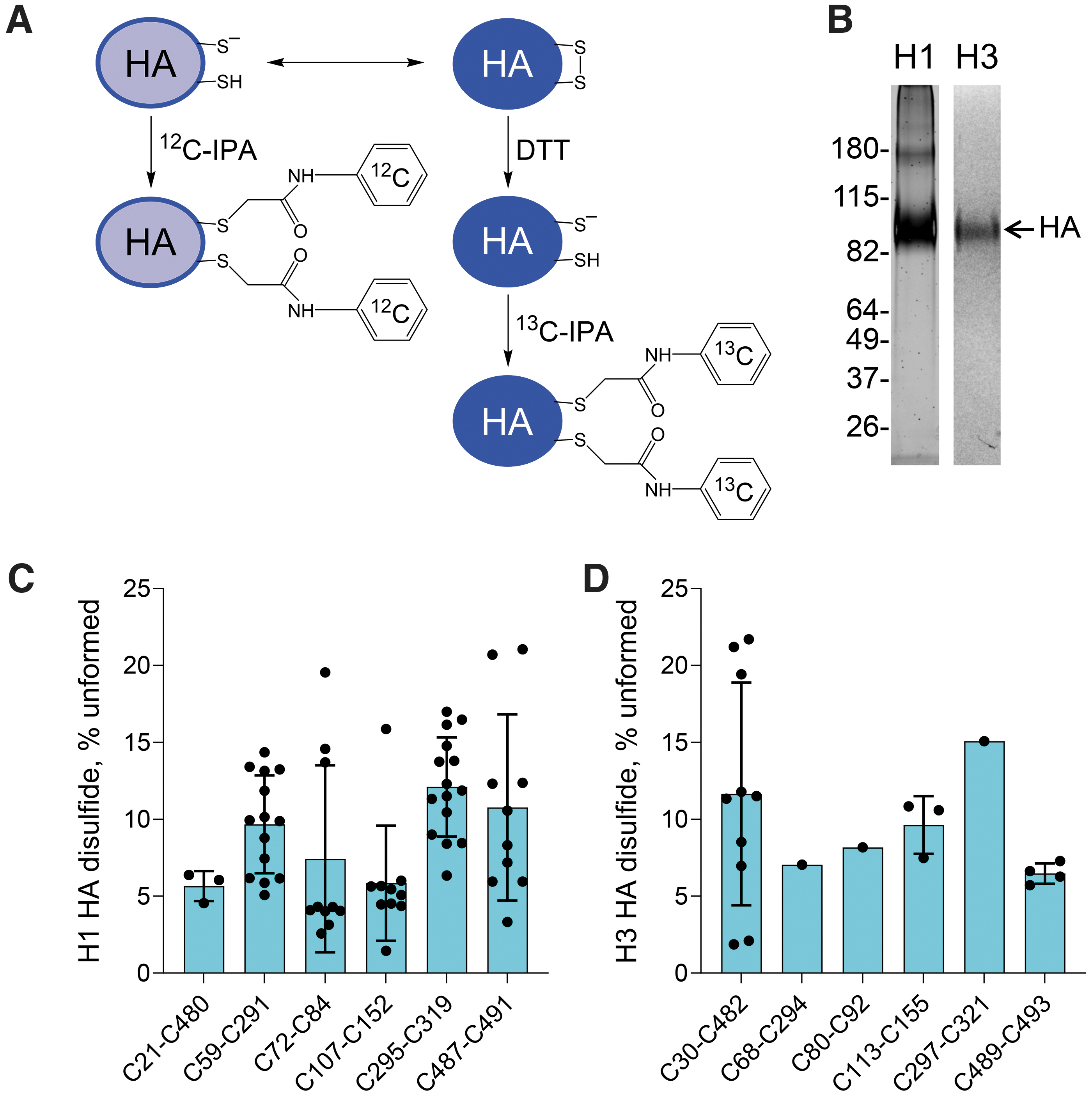
We were able to quantify the redox state of all six HA disulfides in both isotypes. For the H1 isotype, peptides containing both cysteines of four of the disulfides were resolved and analyzed (C72-C84, C107-C152, C295-C319, and C487-C491), whereas a single cysteine was analyzed for the C21-C480 and C59-C291 disulfide bonds (Supplementary Table S1). For the H3 isotype, peptides containing both cysteines of two of the disulfides were resolved and analyzed (C30-C482 and C489-C493), whereas a single cysteine was analyzed for the C68-C293, C80-C92, C113-C155, and C297-C321 disulfide bonds (Supplementary Table S1). All six HA disulfide bonds exist in formed and unformed states in the recombinant proteins (Fig. 2C, D). The mean redox state of the six H1 disulfides ranged from 5.8% ± 3.7% unformed for the C107-C152 disulfide to 12.1% ± 3.2% unformed for the C295-C319 bond, whereas the mean redox state of the six H3 disulfides ranged from 6.5% ± 0.7% unformed for the C489-C493 disulfide to 15.1% unformed for the C295-C319 bond. This result indicates that all six disulfides exist in unformed states in about 1 in 10 HA molecules. We previously analyzed the disulfide status of the platelet integrin receptor, αIIbβ3, using the same methodology employed herein for HA. Twenty of the 24 disulfide bonds in the β3 subunit were >95% formed, whereas the other four bonds were 5%–15% unformed (39). This result supports our finding of variability of HA disulfides being a result of its biology and not a technical artefact of the experiments.
This result was for purified recombinant HA proteins produced in human embryonic kidney cells, and it was important to test whether the same phenomenon applies for viral surface HA. We employed the H1N1 influenza A virus strain A/Puerto Rico/8/1934 (PR8) for this experiment.
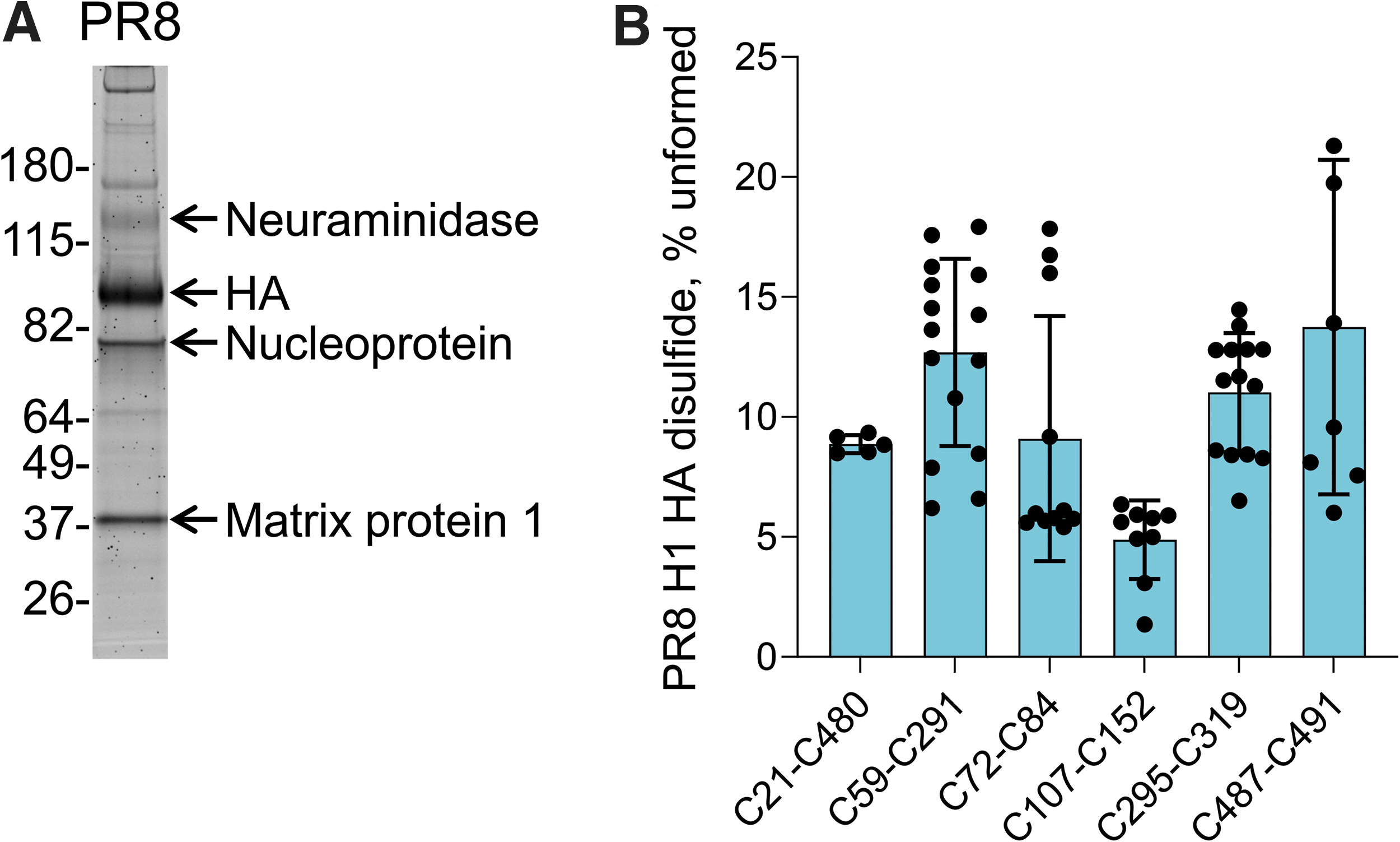
Viral surface HA H1 contains unformed disulfide bonds
The PR8 virus was propagated in MDCK cells, purified by sucrose gradient centrifugation, and labeled with 12C-IPA and the HA protein was resolved by SDS-PAGE (Fig. 3A and Supplementary Fig. S2). The redox state of the PR8 H1 disulfides was indistinguishable from the redox state of the same purified recombinant H1 protein. All six PR8 HA disulfide bonds exist in formed and unformed states: Mean redox state ranged from 4.9% ± 1.6% unformed for the C107-C152 disulfide to 13.7% ± 6.8% unformed for the C487-C419 bond (Fig. 3B).
Protein disulfide bonds can be cleaved by thiol isomerases, and humans constitute more than 20 of these factors. Host thiol isomerases have been implicated in influenza A virus infection as well as in infection by HIV and Newcastle disease virus. We examined whether the vascular thiol isomerases cleave the HA disulfide bonds. This experiment tested the accessibility of the HA bonds to thiol isomerases and their possible involvement in influenza A virus infection.
Disulfide bonds in recombinant HA proteins are reduced by vascular thiol isomerases
Thioredoxin and PDI are the archetypal thiol isomerases, and PDI has been implicated in influenza A virus infection (30). Recombinant monomeric HA proteins (H1N1 A/Puerto Rico/8/1934 and H3N2 A/Aichi/2/1968) were incubated with 2- or 10-fold molar excess of thioredoxin or PDI for 30 min, and the redox sate of the HA bonds was determined. There was a dose-dependent reduction of four of the six HA H1 (C21-C480, C59-C291, C295-C319, C487-C491 bonds) (Fig. 4A) and H2 (C30-C482, C68-C294, C80-C92, C297-C321) (Fig. 4B) disulfides. For example, the state of the C295-C319 HA H1 bond increased from 12.1% ± 3.2% to 37.4% ± 2.4% unformed on incubation with a 10-fold molar excess of thioredoxin, whereas the C30-C482 HA H3 bond increased from 11.6% ± 7.2% to 23.6% ± 5.3% unformed. The extent of reduction was higher for thioredoxin than PDI, which equates with the redox potentials of these factors. The active site dithiol/disulfide of thioredoxin has a redox potential of −270 mV (32), whereas the redox potentials of the two catalytic dithiols/disulfides of PDI are −191 mV (a domain) and −190 mV (a′ domain) (5). Thioredoxin, therefore, is a better reductant than PDI.
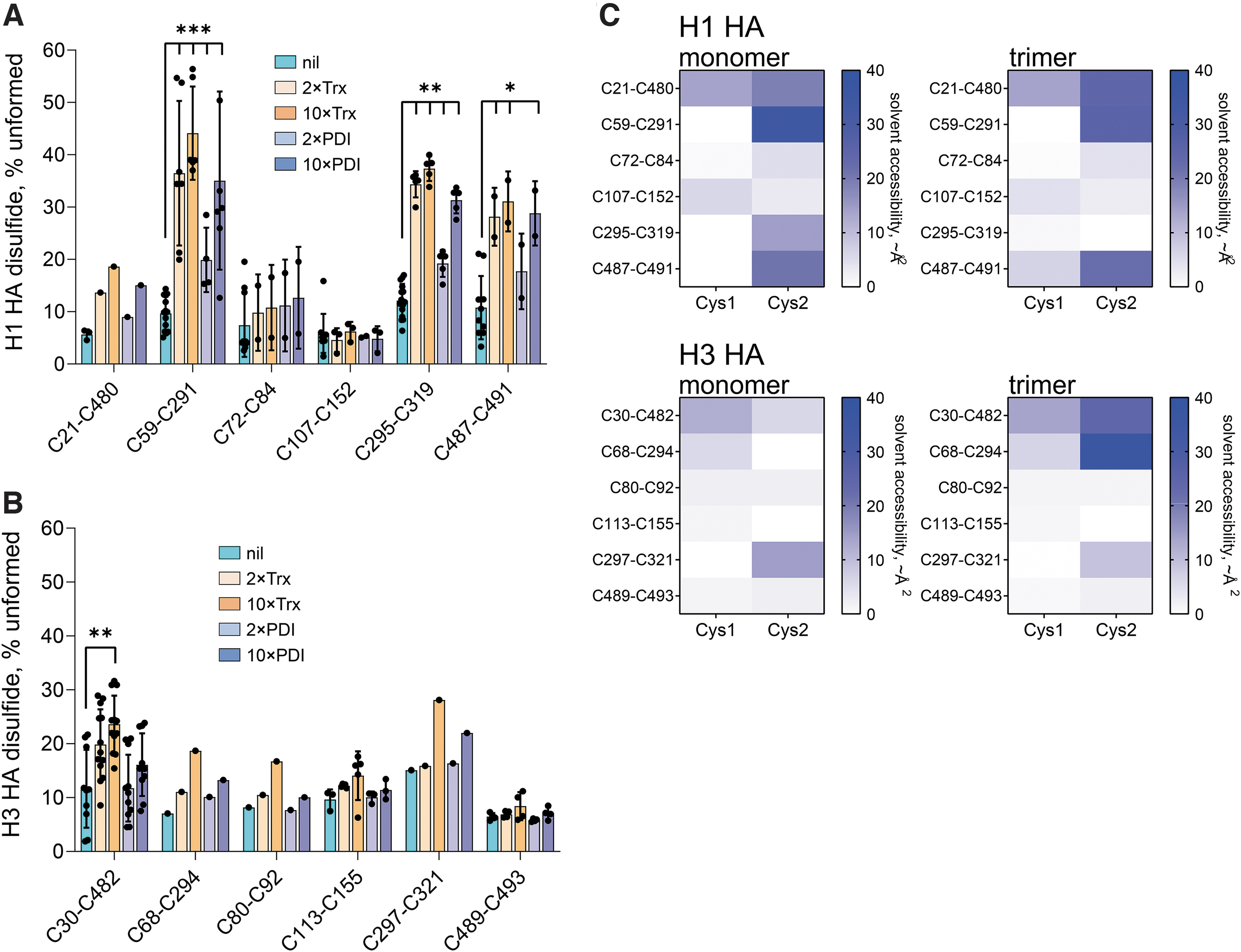
We examined whether the differential reduction of the HA disulfides correlated with their surface exposure in crystal structures. Thiol isomerases require access to at least one of the substrate disulfide bond cysteines to affect redox state, and considering their size, the target disulfides in substrate proteins are predicted to be on or near the protein's surface. Heat maps of the solvent accessibility of the 12 H1 and H3 HA disulfide bond cysteines in monomeric and trimeric crystal structures are shown in Figure 4C. Solvent accessibility of the disulfides in the crystal structures generally reflected whether the bonds are cleaved by thioredoxin or PDI. The two disulfides not appreciably cleaved in thioredoxin or PDI in monomeric HA H1, C72-C84 and C107-C152, have the lowest mean solvent accessibility of the six bonds: 4.0 and 4.5 Å2, respectively. Similarly, the two disulfides not appreciably cleaved in thioredoxin or PDI in monomeric HA H3, C113-C155 and C489-C493, have low mean solvent accessibility values of 0.5 and 3.0 Å2, respectively.
HA is expressed as a non-covalently linked homo-trimer on the surface of the virion, a quaternary structure that could impede access by thiol isomerases to the HA disulfide bonds.
Viral surface HA H1 disulfide bonds are not cleaved by vascular thiol isomerases
PR8 was incubated with a 2- or 10-fold molar excess of thioredoxin or PDI for 30 min, and the redox state of the HA bonds was quantified. There was no significant change in the redox state of the HA bonds on incubation with either factor (Fig. 5A). The peptides containing C487 and C491 were not resolved in this experiment. To confirm this result, we tested reduction of the PR8 HA bonds by three other vascular thiol isomerases: ERp5, ERp57, and ERp72.
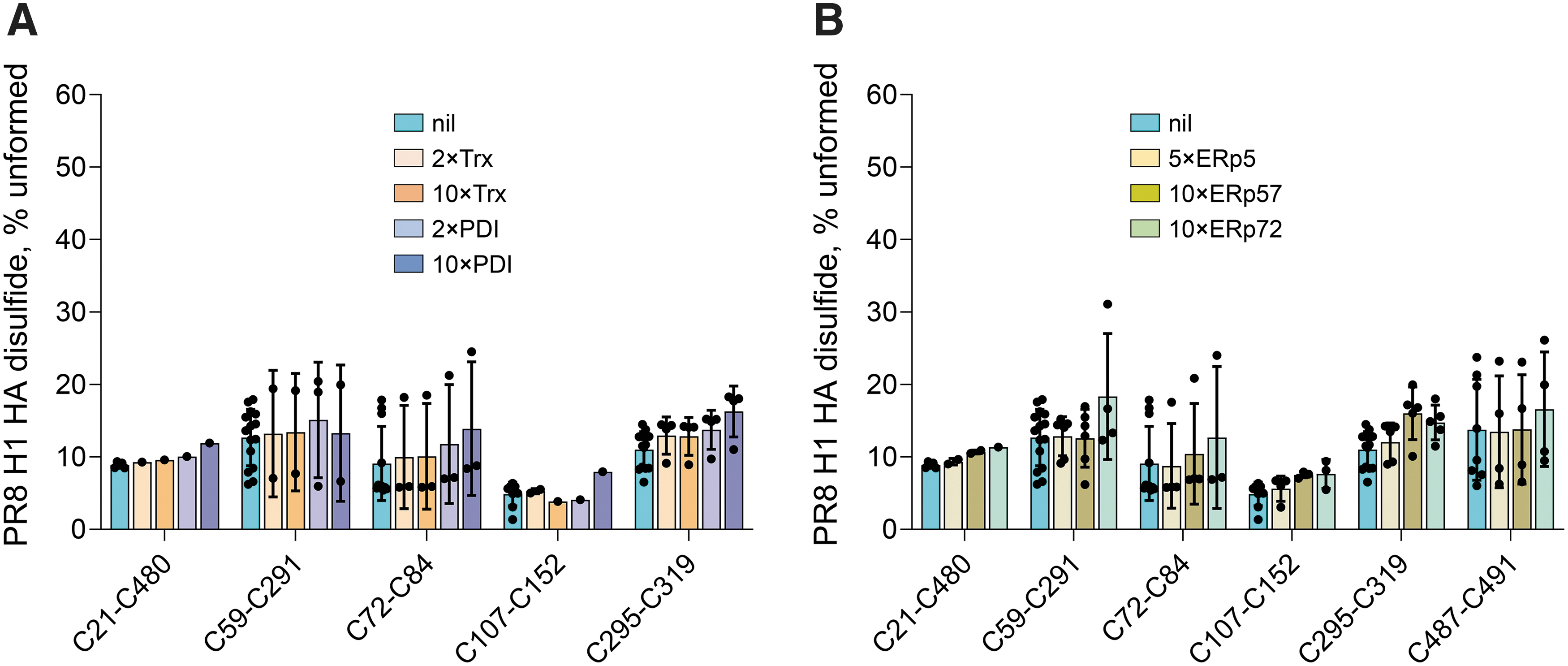
Both ERp5 and ERp72 are more powerful reductants than PDI, whereas ERp57 is a weaker reductant. The redox potentials of the two catalytic dithiols/disulfides of ERp5 are −206 mV (a domain) and −211 mV (a′ domain) (39), whereas the redox potentials of the three catalytic dithiols/disulfides of ERp72 are −217 mV (a domain), −215 mV (a′ domain), and −220 mV (a″ domain) (12). The redox potentials of the two catalytic dithiols/disulfides of ERp57 are −167 mV (a domain) and −156 mV (a′ domain) (18). There was also no appreciable cleavage of the PR8 HA bonds by ERp5, ERp57, or ERp72 (Fig. 5B).
Discussion
Inspection of the crystal structures of influenza A HA protein's revealed that three of the six HA disulfides occur in strained conformations. In addition, two of the six disulfides in H1 and four of the six disulfides in H2 were found to exist in formed and unformed states in some crystal structures of the proteins. These findings suggested that one or more of the HA disulfide bonds may be labile or unformed in the mature protein. Indeed, all six HA disulfide bonds were found to exist in unformed states in approximately 1 in 10 recombinant and viral surface HA molecules. There is precedence for this biochemistry in the blood proteins, fibrinogen and α2-macroglobulin.
Thirteen fibrinogen disulfide bonds and 12 α2-macroglobulin disulfides are 10%–50% and 10%–70% unformed in the blood of healthy human donors, respectively (7). The functional relevance of the disulfide lability for fibrinogen conversion to fibrin polymer has been studied. Disulfides were found to form on fibrin polymerization, and this is important for a stable fibrin matrix. In addition, the disulfide-bonded states of fibrinogen in plasma are influenced by fluid shear forces, indicating that the different covalent states can change in response to an external stimulus (7). Future studies will explore whether physiologically relevant fluid shear forces can also influence the redox state of the influenza A HA bonds.
Probabilities for disulfide bond formation can be employed to estimate numbers of covalent states of HA (20). The mean oxidized (or formed) fraction of the individual HA disulfides represents the probability that the bond is formed in the population of molecules. The probability of an HA state containing any number of bonds is the product of the probabilities for each of the bonds, assuming no special dependence in formation of bonds. For the six values of the mean oxidized state of the viral surface HA bonds (Fig. 3B), this equates to a probability of 0.53. In other words, 53% or just half of the viral surface HA is predicted to be fully disulfide-bonded. However, if disulfide bond formation in the protein has special conditions, such as one of the HA bonds will only form if another HA disulfide has already formed, the incidence of fully oxidized HA will increase (20). On the other hand, this analysis assumes that a fully oxidized HA protein exists, which has not been demonstrated for viral surface HA. The existence of different disulfide-bonded or covalent forms of HA may impact viral infectivity, as some forms may allow for better ligand binding or membrane fusion than other forms. It is also possible that efficiency of host proteolysis of HA0 to HA1 and HA2 (47) is different for the different disulfide-bonded states that could influence infectivity. Future studies will test this hypothesis by comparing the HA disulfide patterns of influenza A virions that readily infect cells with those that failed to do so after brief incubation with MDCK cells.
The redox state of some viral coat protein disulfide bonds plays a role in viral infections, including HIV (16) and Newcastle disease virus (24 –26), and host thiol isomerases have been implicated in redox control of the viral disulfides. Humans make more than 20 thiol isomerases and several of them are exported by activated cells. Those that circulate in blood are referred to as vascular thiol isomerases (17). Seven members of the PDI family (31), two members of the thioredoxin family, and glutaredoxin-1 (22) are secreted into the blood (15) and plasma membrane. TMX1, TMX3, ERp44, and ERp29 are found on the blood platelet surface (21, 49, 55). Thioredoxin is secreted by cells in pathological conditions associated with inflammation, including viral infections (37). Thioredoxin and PDI cleaved four of the six disulfide bonds in recombinant HA H1 and H3 but not viral surface HA. Presumably, the disulfide bonds are not accessible to the thiol isomerases when HA is presented on the viral surface.
It is possible that virion HA has a different glycosylation pattern than the recombinant proteins, which is blocking access by thiol isomerases. Although the recombinant HAs were expressed in human HEK293 cells to allow for more complex glycosylation profiles, it is known that protein glycosylation is dependent on the host-cell type (19). The glycans attached to a protein can be differently modified in the Golgi depending on how they are exposed to the Golgi glycosylation-modifying enzymes. Also, the viral HA will mostly be the cleaved form consisting of the HA1 and HA2 polypeptides linked by a disulfide bridge, whereas the recombinant HA molecules are uncleaved. Another possibility for the lack of effect of thiol isomerases is the quaternary structure of viral surface HA. The protein is expressed as a non-covalently linked homo-trimer on the virion surface that could protect the HA disulfides from external factors. However, our analysis of monomeric versus trimeric HA crystal structures indicates no appreciable differences in the solvent accessibility of the disulfide cysteine residues (Fig. 4C). Most likely, it is the crowding of the glycoproteins on the surface of the influenza A virus that is impairing access of the thiol isomerases to the HA disulfides. The HA molecules have a very low affinity for their ligand (mM dissociation constant), which is overcome by the high viral surface density of about one HA molecule per 100 nm2 of membrane surface. The HA trimers are ∼70 Å wide and separated by a mean distance of ∼100 Å with a range of 0 to ∼160 Å (53). PDI and thioredoxin have diameters of ∼90 Å (52) and ∼40 Å (2), respectively, so their access to viral surface HA disulfide bonds will be severely restricted. Neuraminidase molecules tend to spatially segregate to distinct regions of the virus surface and, thus, they are not likely to greatly impact thiol isomerases' access to HA (50).
Our findings do not support a role for vascular thiol isomerases in viral surface HA function, in contrast to what has been observed for HIV (16) and Newcastle disease virus (24 –26). The absence of cleavage of mature viral HA disulfides by the thiol isomerases tested suggests that the reported role of these factors in influenza A virus infection is restricted to their function as facilitators of thiol/disulfide exchange during maturation of nascent HA proteins in the ER and Golgi systems of infected cells. This conclusion is consistent with the observation that the largely membrane-impermeable PDI inhibitors, bacitracin A and 5,5′-dithiobis(2-nitrobenzoic acid), do not affect influenza A virus replication in MDCK cells (29), but they inhibit the entry of HIV (44) and Newcastle disease virus (24) into host cells. However, the membrane-impermeable PDI inhibitor, isoquercetin, reduced influenza A viral replication in infected mice (30), suggesting that host PDI has a role in some aspect of viral infection.
The mode of viral entry may dictate whether thiol isomerases play a role during the infection process. After the binding of influenza to its receptor on host cells, the virus is internalized by macropinocytosis or by clathrin-mediated and clathrin-independent endocytosis. The viral particles reach the endosomal compartments that then mature into late endosomes and undergo acidification, which is a prerequisite to membrane fusion. The acidification triggers a conformational change in HA, exposing the fusion peptide that coordinates viral–host membrane fusion and release of viral ribonucleoprotein complexes into the cytoplasm (14). Other enveloped viruses do not rely on acidification for activation of their fusogenic mechanisms. Interaction of these viruses with host cell surface receptors triggers conformational changes in the envelope glycoproteins that lead to fusion of the viral and host cell membranes and the entry of viral particles directly into the cytoplasm (36). It is possible that viruses that do not rely on an acidic trigger in endosomes, such as HIV (4, 16, 35, 38), Newcastle disease virus (24, 25), and Sindbis virus (1), may require alternative forms of activation at the cell surface involving thiol isomerases.
In addition, the route of infection may also be a factor. Considering the widespread vascular location of thiol isomerases, their effect on mature viral envelope proteins may be more relevant for viruses that infect by the systemic route, such as HIV, than for viruses that cause a localized pulmonary infection, such as influenza A virus. Since the pandemic SARS-CoV-2 virus causes a mixed pulmonary/systemic infection and the high-affinity ACE2-binding Spike protein is distributed on the virus surface at a low density of one molecule per 1000 nm2 of membrane surface (28), it will be important to analyze the effect of thiol isomerases on SARS-CoV-2 spike protein disulfide bonds and their possible role in viral infectivity.
Materials and Methods
Quantification of the redox states of HA disulfide bonds
The recombinant influenza A HAs H1N1 [A/Puerto Rico/8/1934 HA(H1N1)] or H3N2 [A/Aichi/2/1968 HA(H3N2)] used in this study are monomers consisting of the extracellular domains (HA1+HA2 uncleaved) fused with a C-terminal polyhistidine tag, expressed in HEK293 cells and purified by Ni-affinity chromatography (SinoBiological, Beijing, China). The HA proteins were incubated with 5 mM 12C-IPA (Cambridge Isotope Laboratories, Tewksbury, Massachusetts) in phosphate-buffered saline (PBS) containing 10% DMSO for 1 h at 22°C in the dark to alkylate unpaired cysteine thiols in the proteins. The proteins were incubated with NuPAGE LDS sample buffer (Life Technologies, Carlsbad, California) for 10 min at 80°C and resolved on 4%–12% Bis Tris NuPAGE SDS-PAGE gel (Life Technologies). The gels were stained with Brilliant Blue (Sigma, St. Louis, Missouri) and destained with 10% methanol, 7% acetic acid solution. The HA bands were excised, sliced, destained with 25 mM NH4CO2/50% acetonitrile solution, dried, and incubated with 40 mM dithiothreitol for 30 min at 56°C. The gel slices were washed with 25 mM ammonium bicarbonate/50% acetonitrile solution and incubated with 5 mM 13C-IPA (Cambridge Isotope Laboratories) for 1 h at room temperature in the dark to alkylate the cysteines involved in disulfide bonds. The slices were washed and dried before deglycosylation with 5 U/mL of PNGase F (Sigma, St. Louis, Missouri) overnight at 37°C. Gel slices were washed with water and dried before sequential digestion with 12.5 ng/μL of chymotrypsin (Promega, Madison, Wisconsin) in 25 mM NH4CO2 and 10 mM CaCl2 for 4 h at 37°C, and with 12.5 ng/μL trypsin (Promega) for further 18 h at 25°C. Peptides were eluted from the slices with 5% formic acid/50% acetonitrile solution. Peptides were cleaned by using C18 Ziptip column (Millipore, Burlington, Massachusetts), dried, and reconstituted in 0.1% formic acid. Liquid chromatography, mass spectrometry, and data analysis were performed as described (7, 11). Briefly, peptides were analyzed on a Thermo Fisher Scientific Ultimate 3000. Two hundred nano gram of peptides was injected and resolved on a 35 cm × 75 μm C18 reverse-phase analytical column with an integrated emitter by using a 2%–35% acetonitrile gradient over 60 min and a flow rate of 250 nL/min. The peptides were ionized by electrospray ionization at +2.0 kV. Tandem mass spectrometry analysis was carried out on a Q-Exactive Plus mass spectrometer by using HCD fragmentation. The data-dependent acquisition method acquired MS/MS spectra of the top 20 most abundant ions with charged state ≥2 at any one point during the gradient.
The MS/MS spectra were searched against the Swissprot reference proteome by using the external search engine Mascot (Version 2.7, Matrix Science) or HA protein sequences (HEMAI24A1 and HEMAI68A0) using Byonic™ (Version 3.0, Protein Metrics). Precursor mass tolerance and fragment tolerance were set at 10 ppm, and the precursor ion charge state was set to 2+, 3+, and 4+. Variable modifications were defined as oxidized Met, deamidated Asn/Gln, N-terminal pyro-Glu or Asp, iodoacetanilide derivative Cys, and 13C-iodoacetanilide derivative Cys with full trypsin and chymotrypsin cleavage of up to three missed cleavages. Only peptides with peptide score >20 (p < 0.01) and error < ± 6 ppm were selected for quantification of relative abundance (Supplementary Tables S1 and S2). The cysteine labeled with 12C-IPA or 13C-IPA has a mass of 133.05276 or 139.07289, respectively, and their monoisotopic mass/charge ratio (m/z) of +2, +3, and +4 was calculated by using MS-Product (Version v 6.2.2, Protein Prospector Tools). The different redox forms of the cysteine residues were quantified from the relative ion abundance of peptides labeled with 12C-IPA and/or 13C-IPA in extracted ion chromatograms generated by using XCalibur Qual Browser software (Thermo Fisher Scientific, Waltham, Massachusetts, “v2.1.0”). The percentage of unformed disulfide bonds was calculated by using the formula:
Reduction of HA protein disulfides by thiol isomerases
To analyze the reduction of HA by thiol isomerases, the HA recombinant proteins or influenza A virus were incubated in PBS for 30 min at 22°C with 1:2 to 1:10 molar ratios of the recombinant thiol isomerases PDI, ERp5, ERp57, or ERp72 immediately before alkylation with 12C-IPA. His-tagged recombinant human thiol isomerases were produced in E. coli and purified to homogeneity as described (39). Just before incubation with HA, the active-site disulfides of the thiol isomerases were reduced by incubation with 10 mM dithiothreitol for 30 min at 22°C and the unreacted DTT were removed on Zeba™ Spin Desalting Columns (7K MWCO; Thermo Scientific).
Propagation and isolation of influenza A viruses
The MDCK cells were cultured in RPMI 1640 (Thermo Fisher Scientific) supplemented with 2 mL L-glutamine, 1 mM MEM sodium pyruvate, 100 U/mL penicillin/streptomycin, 24 mg/L gentamycin, and 10% heat-inactivated FCS until 80% confluency and they were infected with PR8 at an MOI of 0.05. The cells were infected with influenza A virus suspension in minimum volume to cover the cell monolayer for 37°C with gentle shaking for 45 min before the addition of seven- to eight-fold that volume of fresh medium containing 4 μg/mL of trypsin (Worthington Biochemical, Lakewood, New Jersey) and further incubation at 37°C for 72 h to allow propagation of the virus. Supernatants were collected and clarified by centrifugation at 2300g for 15 min at 4°C followed by centrifugation at 51,000g for 90 min (HITACHI CP100NX, rotor P21A2-0009). The pellets were resuspended in PBS and transferred into a 60%–20% sucrose gradient for centrifugation at 107,000g for 2.5 h at 4°C (HITACHI CP100NX, rotor P28S2-1259). The ring containing the virus was collected, diluted in PBS in ratio 1:10, and centrifuged at 107,000g for 1.5 h at 4°C (HITACHI CP100NX, rotor P28S2-1259). The pellets were resuspended in sterile PBS, aliquoted, and stored at −80°C until use. The virus preparation was run on 4%–12% Bis Tris NuPAGE SDS-PAGE gel (Life Technologies); the visible bands were excised, alkylated with 55 mM of iodoacetamide in 25 mM NH4CO2, deglycosylated overnight at 37°C with 5 U of PNGase F, digested overnight with 12.5 ng/μL trypsin at 30°C, and analyzed by LC-MS. Neuraminidase, HA, nuclear protein, and matrix protein 1 were identified by using Mascot (MatrixScience, Boston, Massachusetts).
Statistical analyses
Data were analyzed in GraphPad Prism 8.2.0 software by using Student's t test when comparing two experimental groups or Mixed-Effects Model, with fewer assumptions and correcting for multiple comparisons by controlling the False Discovery Rate by using the two-stage step-up method of Benjamini, Krieger, and Yekuliety, Q = 0.05 when comparing more than 2 experimental groups. Discoveries are noted on graphs with *p < 0 .05, **p < 0.01, and ***p < 0.001. An electronic laboratory notebook was not used.
Footnotes
Authors' Contributions
P.J.H. conceived the study and cowrote the article. M.F. designed and performed the experiments and cowrote the article. J.C. designed and performed the experiments and analyzed the data.
Acknowledgments
The authors thank John Stambas, Deakin University, for PR8, and Carl Feng, University of Sydney, for MDCK cells.
Data Availability
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by the National Health and Medical Research Council of Australia (grant nos. 1110219, 1143400, and 1143398) and a Senior Researcher Grant from the NSW Cardiovascular Research Capacity Program.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.