
Abstract
Significance:
Under homeostatic conditions, the endothelium dynamically regulates vascular barrier function, coagulation pathways, leukocyte adhesion, and vasomotor tone. During sepsis and acute inflammation, endothelial cells (ECs) undergo multiple phenotypic and functional modifications that are initially adaptive but eventually become harmful, leading to microvascular dysfunction and multiorgan failure.
Critical Issues and Recent Advances:
Sepsis unbalances the redox homeostasis toward a pro-oxidant state, characterized by an excess production of reactive oxygen species and reactive nitrogen species, mitochondrial dysfunction, and a breakdown of antioxidant systems. In return, oxidative stress (OS) alters multiple EC functions and promotes a proinflammatory, procoagulant, and proadhesive phenotype. The OS also induces glycocalyx deterioration, cell death, increased permeability, and impaired vasoreactivity. Thus, during sepsis, the ECs are both a significant source and one of the main targets of OS.
Future Directions:
This review aims at covering the current understanding of the role of OS in the endothelial adaptive or maladaptive multifaceted response to sepsis and to outline the therapeutic potential and issues of targeting OS and endothelial dysfunction during sepsis and septic shock. One of the many challenges in the management of sepsis is now based on the detection and correction of these anomalies of endothelial function.
Introduction
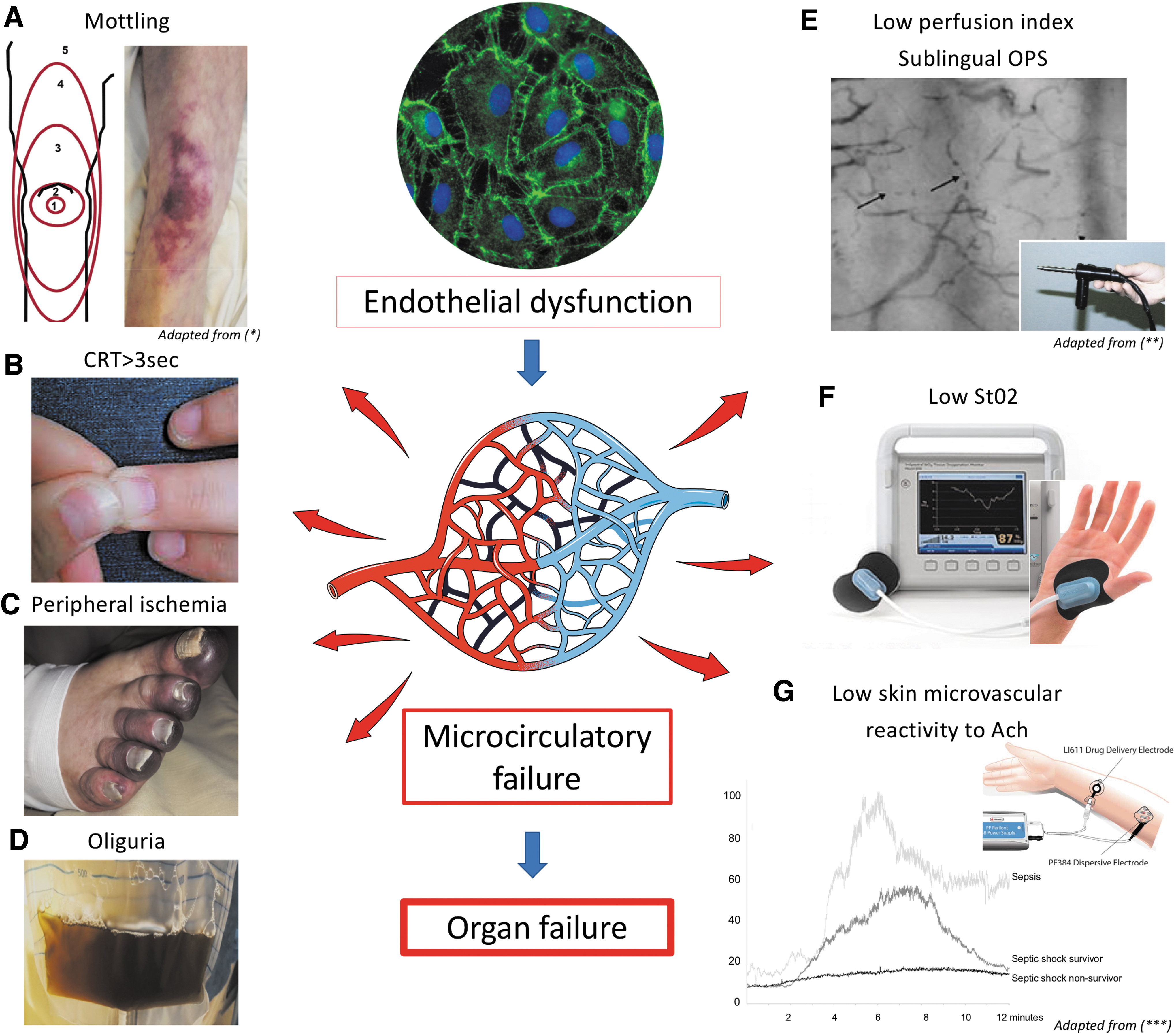
Septic shock is defined as a condition whereby the circulation cannot deliver adequate blood flow to meet the tissue's metabolic demand and/or cellular metabolism is impaired, ultimately leading to organ dysfunction (138). Microcirculatory anomalies are involved throughout the course of septic shock, and signs such as skin mottling on the knee area, prolonged capillary refill time (CRT), central-to-peripheral temperature gradient, low tissue oxygen saturation (StO2), or abnormal sublingual perfusion index correlate with organ failure severity and are predictive of intensive care unit (ICU) mortality (7, 40, 47, 68). The substratum of microcirculatory failure is characterized by endothelial dysfunction, which is often referred to as sepsis-induced endotheliopathy. Indeed, beyond a simple malfunction, the endothelium during sepsis presents with multiple phenotypic and functional modifications that are initially adaptive but eventually become harmful.
During sepsis, the engagement of pattern-recognition receptors (PRRs), including Toll-like receptors (TLRs), drive endothelial cell (EC) reprogramming toward a proinflammatory, proapoptotic, proadhesive, and procoagulant phenotype. In addition, sepsis causes glycocalyx (GCX) damage, dysregulated microcirculatory vasoreactivity, capillary leak, and impaired tissue perfusion (83).
During sepsis, oxidative stress (OS) plays a role in promoting adative responses to hypoxia, bacterial clearance, and postinjury endothelial repair processes. However, sepsis can cause an imbalance between reactive oxygen species (ROS) and reactive nitrogen species (RNS), and the antioxidant system, which, in addition to sepsis-induced mitochondrial dysfunction, can, in a vicious cycle, enhance the multiple aspects of endothelial dysfunction. Indeed, OS contributes substantially to sepsis-induced endotheliopathy by playing a role in impaired vasomotricity, augmenting leukocyte and platelet adhesion to ECs and capillary permeability, and promoting cell death and a procoagulant state of the endothelium. This review aims at covering the current understanding of the role of OS in the adaptive or maladaptive multifaceted response to acute inflammation, and therapeutic avenues targeting OS and endothelial alterations during septic shock.
Endothelial Dysfunction Is a Major Driver of Organ Failure in Sepsis
Sepsis is one of the main reasons for ICU admission and is responsible for more than 10 million deaths each year worldwide (131). Over the past two decades, clinical studies have shown that endothelial and microvascular dysfunction are primary causes of tissue hypoperfusion and organ failure. Further, the presence and intensity of microcirculatory abnormalities during septic shock have been linked, by multiple teams, to organ failure and poor outcome regardless of blood pressure or cardiac output. In 2002, De Backer et al. used an orthogonal polarization spectral (OPS) imaging technique to investigate the sublingual microcirculation in 10 healthy volunteers, 10 acutely ill patients without sepsis, and 50 patients with severe sepsis. They demonstrated, for the first time in humans, that sepsis causes altered microcirculatory flow, with reductions in vessel density and in the proportion of perfused vessels (47). These factors compromise tissue perfusion, and the intensity of these alterations correlates with organ failure and death. Since these pioneering observations, a large number of studies have shown that the identification of microcirculatory abnormalities using simple skin signs [mottling (7), prolonged CRT (6, 72)] or precision morphological tools [OPS (81), laser Doppler (24)] is predictive of the severity of organ failure and survival. Therefore, current resuscitation strategies target the restoration of endothelial function and preservation of the microcirculation and thereby promote adequate tissue perfusion and oxygen delivery, with the ultimate goal of reducing organ failure and improving sepsis outcomes. Figure 1 summarizes the cutaneous clinical signs and some bedside clinical tools used to assess the microcirculatory function during septic shock.
The Endothelium Is Both a Source and a Target for OS: A Vicious Circle
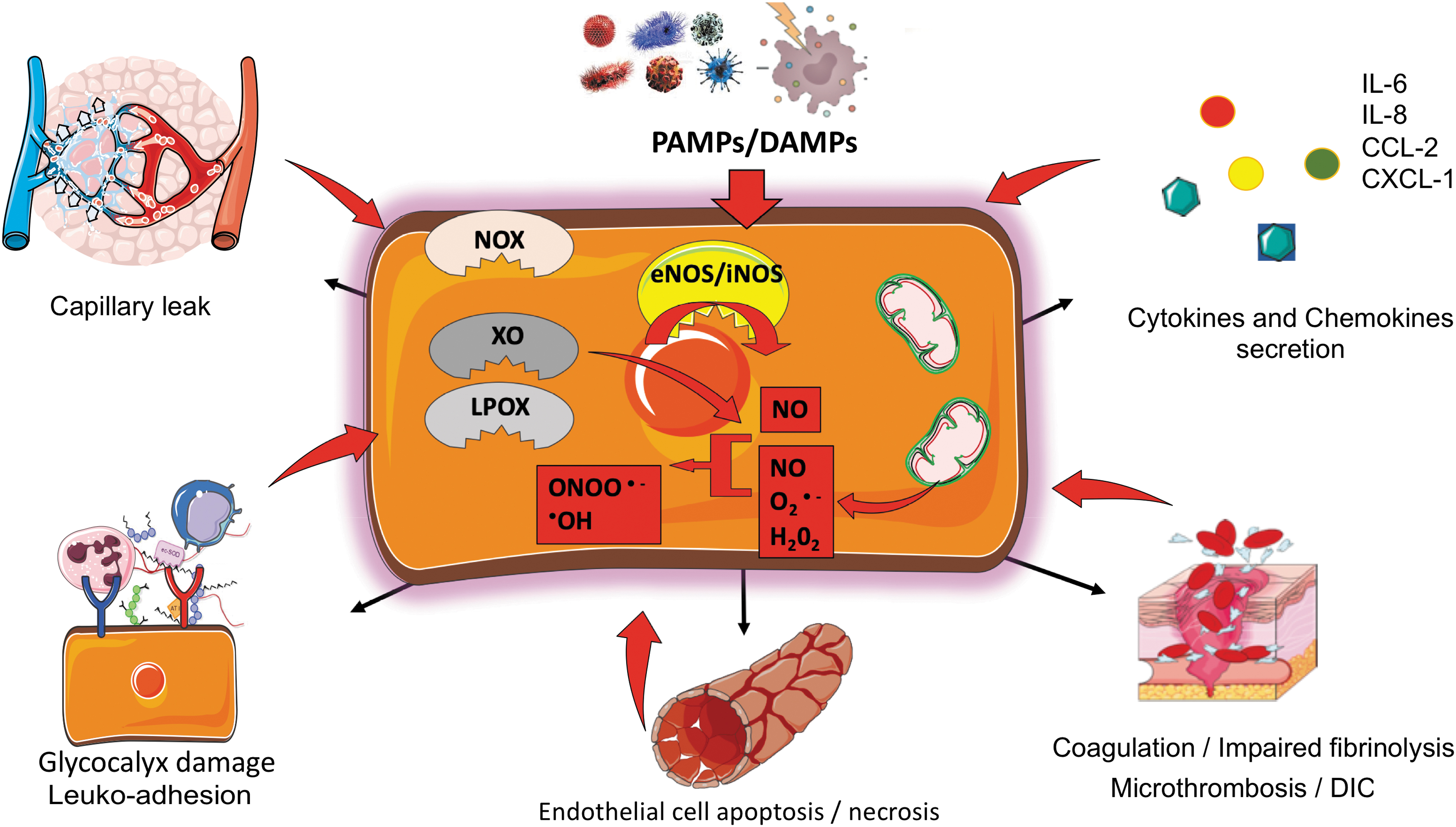
The paradigm governing the relationship between OS and sepsis-induced endotheliopathy is that of pathogenic reciprocity. Under homeostatic conditions, there is a balance between the formation of reactive oxidizing/oxygen species and their removal by endogenous antioxidant scavenging of toxic compounds of the endothelium (64). The ROS are continuously produced in EC metabolism: Superoxide (O2 •−) is immediately transformed by superoxide dismutase (SOD) into hydrogen peroxide (H2O2) and then transformed by catalase and peroxidase into water (H2O). In addition to SOD, mammals are equipped with various enzymatic systems (glutathione/glutathione reductase and thioredoxin/thioredoxin reductase) or nonenzymatic antioxidants (vitamins A/C/E) to counterbalance the effect of oxidants. This balance is called the “redox homeostasis.” Under inflammatory conditions, the endothelial cytotoxicity of OS is the combination of excessive production of ROS (O2 •−, H2O2, and hydroxyl radicals: •OH) and RNS (e.g., peroxynitrite [ONOO−]), and inadequate antioxidative systems including decreased SOD and catalase activity, reduced glutathione (GSH) accumulation, and potential deficiencies of vitamins (63).
Also, during sepsis, similar to conventional innate immune cells, such as neutrophils and macrophages (51), EC produce large amounts of ROS and RNS. PAMPs (pathogen-associated molecular patterns) and DAMPs (damage-associated molecular patterns) increase endothelial OS production via engagement of PPR, including TLRs, in addition to tissue hypoxia and reperfusion via several distinct mechanisms (36). Mitochondrial dysfunction (4, 35), xanthine oxidase (XO), uncoupled NO synthases (NOS), cytochrome P-450 enzymes, lipoxygenases, and NADPH oxidases all contribute to superoxide production by EC.
The mitochondrial respiratory chain is considered the primary source of O2 •− during critical illness (112). Mitochondria generate energy by producing adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) and typically, OXPHOS results in up to 1%–4% of O2 being incompletely reduced, leading to O2 •− formation (77). Moreover, proinflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-1α, and lipopolysaccharide (LPS) may directly induce mitochondrial O2 •− production in human umbilical vein endothelial cell (HUVEC) via a ceramide-dependent pathway, and in lung arterial ECs via upregulation of NADPH oxidase (76, 111). Corda et al. also reported that plasma from septic patients decreases endothelial GSH in HUVECs, increasing ROS endothelial production and ultimately endothelial injury (39).
Sepsis causes alterations in mitochondrial signaling pathways controlling function and population. Inflammatory conditions, and metabolic disorders (hypoxia, hyperglycemia, reperfusion) and metabolic reprogramming toward aerobic glycolysis can reduce the activity and/or the maximum capacity of the mitochondrial respiratory chain complex I, III, and IV (55, 59), although inflammation is sometimes associated with increased mitochondrial OXPHOS, particularly in the short term. These derangements in mitochondrial function lead to elevated production of O2 •−.
Finally, sepsis causes over-expression of an inducible endothelial form of NOS that significantly increases NO production. During septic shock, eNOS (endothelial NO synthase) function is altered. In addition to NO, during sepsis, eNOS also produces large amounts of superoxide anion. This phenomenon is named NOS uncoupling, as superoxide generation mainly occurs when NOS is not coupled with its cofactor or substrate (52, 103). NO itself has a specific effect by inhibiting complexes I and IV of the respiratory chain (26, 32). Nitrosylation of cellular proteins and peroxynitrite production may indirectly lead to harm. These products induce inhibition of the respiratory chain and, therefore, direct mitochondrial toxicity, which fuels a vicious circle. Indeed, the simultaneous production of O2 •− and NO can be 1000-fold upregulated. At these high concentrations, NO can compete with SOD and react with O2 •− to generate ONOO− (27, 137). Moreover, NO and ONOO− can also be directly produced by the mitochondria via mtNOS (an isoform of nNOS that is expressed in the mitochondria) (95, 127). ONOO− causes rapid oxidation of sulfhydryl and thioether groups, as well as nitration and hydroxylation of aromatic compounds such as tyrosine, tryptophan, and guanine. These modifications are reported to induce numerous cellular function alterations, such as decreased protein activity, alteration of the mitochondrial respiratory chain, peroxidation of membrane lipids, and dysfunction of Na+/K+ membrane pumps ATPase, and even direct damage to deoxyribonucleic acid (DNA) (144).
Collectively these mechanisms result in the disbalance of the endothelial redox homeostasis. The resultant OS disrupts multiple facets of microvascular endothelial function such as vasomotor tone, coagulation, GCX integrity, barrier function, leukocytes, and platelet adhesion, and it ultimately promotes cell death. In return, some of these alterations increase OS, which further amplifies endothelial dysfunction and injury and ultimately causes organ dysfunction and failure (Fig. 2). Consistent with this paradigm, some clinical studies have reported a positive correlation between OS, mitochondrial dysfunction, and severity in septic shock (25, 43, 76, 78).
Vascular Tone and OS
The OS directly alters vascular tone by modulating NO bioavailability or signaling. Animal models of sepsis revealed significantly increased levels of NO in the first hours of sepsis secondary to increased tissue iNOS (inductible NO synthase) expression (149). However, eNOS uncoupling limits the bioavailability of NO because of immediate transformation in ONOO−, which is believed to participate in microcirculatory failure by limiting the capillary vasodilation that is necessary to maintain organ perfusion. Consistent with this concept, experimental studies on rat coronary arteries revealed that application of ONOO− prevented vasodilatation induced by acetylcholine or isoprotenol (156). Concomitantly, the production of NO by iNOS increases levels of two potent vasoconstrictive factors, endothelin-1 and thromboxane A2 (50, 70). Several clinical studies reported increased NO levels in early sepsis and altered bioavailability (45, 46, 162). However, difficulties with measuring NO coupled with the dynamic regulation in its production and bioavailability during the different phases of sepsis add complexity to the understanding of the role of NO in sepsis. These factors may partly explain the negative results of clinical studies attempting to inhibit or increase its production during sepsis (described below).
OS and Coagulation
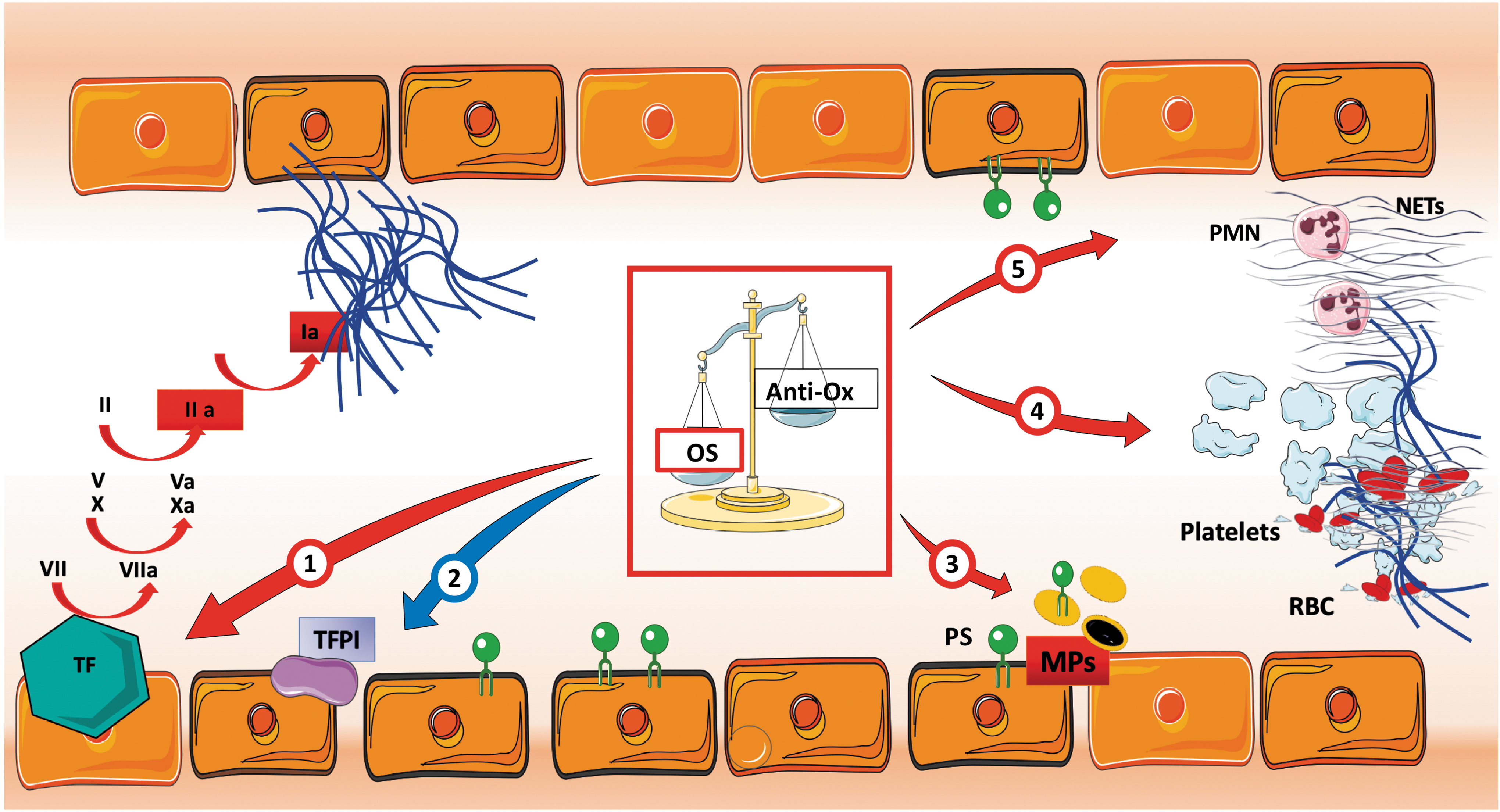
Under homeostatic conditions, the GCX layer, as well as anticoagulant molecules produced by EC, such as tissue factor pathway inhibitor (TFPI), EC protein C receptor, and thrombomodulin, prevent microvascular thrombosis and maintain optimal blood fluidity (20). The EC-anticoagulant functions are impaired during acute inflammation and sepsis, which leads to platelet adhesion and imbalances in the formation and breakdown of thrombin, and ultimately to the development of micro thrombosis (34). Experimental studies provide insight into the crucial role of redox regulation of the endothelium in triggering or promoting dysregulation of the coagulation pathways. The OS has been reported to directly inactivate TFPI on EC (37). Treatment of EC with ROS-generating systems (xanthine/XO or H2O2) for 24 h resulted in reduced activity of TFPI, without significant changes at the mRNA level, suggesting a direct action on TFPI structure (28, 105). Tissue factor (TF) is a primary initiator of the extrinsic coagulation cascade. During homeostasis, the active form of TF is absent or poorly expressed on the surface of EC, and it is maintained in an inactive “crypted” form. During acute inflammation, post-transcriptional modifications and membrane phosphatidylserine (PS) exposure convert crypted TF to fully active TF (5, 97). Active TF has a high affinity to FVII/FVIIa and FX/FXa, which creates a procoagulant environment resulting in the generation of thrombin, fibrin formation, and platelet activation (33, 152). In 2018, Ebert et al. demonstrated that deficiency of antioxidative protein paraoxonase-2 (PON2) causes vascular inflammation and a procoagulant endothelial phenotype via deregulated redox homeostasis. They observed that PON2 −/− mice display superoxide accumulation in EC and a proinflammatory phenotype (higher IL-6 and CCL-2 production) at baseline. They hypothesized that the antioxidant role played by PON2 controlled the decryption of TF (48). Also, both PON2 deficiency and OS decrypt TF procoagulant activity and trigger coagulation (9, 65). Finally, EC and platelet-derived ROS enhance platelet activation and adhesion and promote coagulation during inflammation (38, 93, 150).
This highlights OS's direct role in triggering thrombotic complications and potentially disseminated intravascular coagulation (99). Figure 3 summarizes the direct effect of ROS in sepsis-associated coagulopathy and microvascular thrombosis.
Sepsis- and OS-Induced GCX Damage
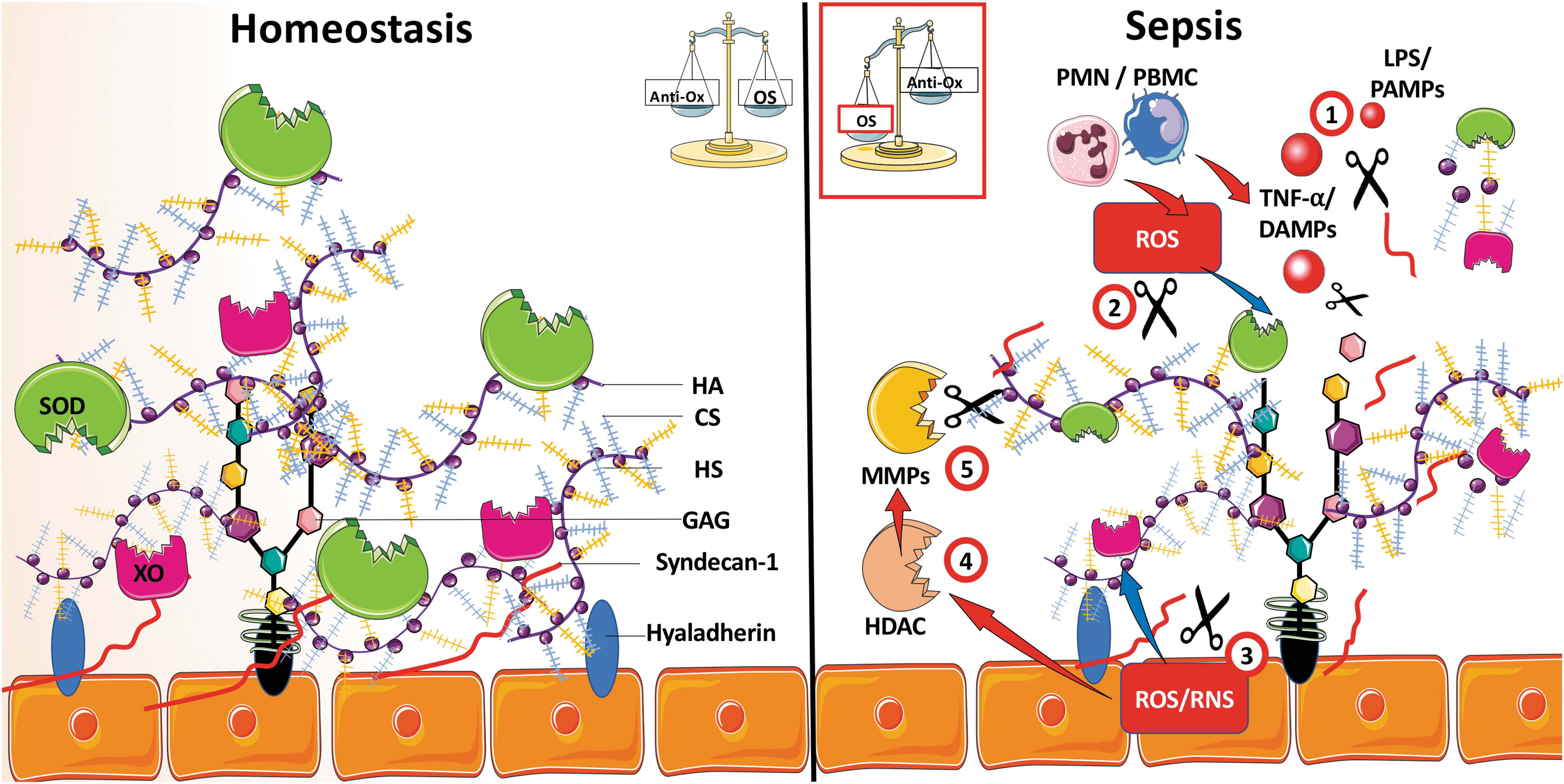
The GCX is a heparan sulfate-rich layer of glycosaminoglycans and proteoglycans that coats the healthy vascular endothelium. The GCX is critically involved in microvascular barrier function maintenance, leukocyte trafficking, local anticoagulation, and capillary rheology (60, 100, 129). Inflammatory conditions cause an early and durable disruption of GCX structure and function (92). Wiesinger et al. observed reduced GCX thickness after challenges with LPS or TNF-α (161) on mice aorta freshly isolated and human cultivated lung microvascular EC. Electron microscopy has recently enabled 3D ultrastructure visualization of capillary GCX at baseline and after LPS challenge in the heart, kidney, liver (114), and lung microvasculature (80) in mice. Kataoka et al. showed that GCX destruction increased macromolecule permeability and leukocyte adhesion in microcirculation (86).
Human studies have reported evidence that shedding of the GCX component occurs in many critical illnesses where OS play a significant role, including in patients with septic shock or postcardiac-arrest syndrome (62) or ischemia–reperfusion syndrome (153). For example, shed GCX components, such as Syndecan-1 and Hyaluronan, are higher in septic patients than healthy control (116, 141) and positively correlate with severity of illness, capillary leak (125, 140), and mortality (10, 74, 79, 113).
Under homeostatic conditions, the intact GCX is a sanctuary of two major antioxidant enzymes. The GCX heparan sulfate has a high affinity for the SOD (16, 84, 85), and sulfated proteoglycans dock active XO (2). Therefore, a healthy GCX can quench and neutralize a high amount of systemic and self-generated free radicals and maintain NO bioavailability, thereby preserving redox homeostasis (91). Conversely, when GCX is damaged, the redox homeostasis is broken. In addition, ROS can directly alter the endothelial GCX density by decreasing the EC surface content of glycosaminoglycans and heparan sulfate proteoglycans, and which amplifies endothelial dysfunction (130). For example, H2O2 of human glomerular EC has been reported to directly increase the cleavage of sulfated glycosaminoglycan, to reduce Trans-endothelial Electrical Resistance, and to alter the selective barrier function to proteins (139). Moreover, oxidative mechanisms, including OS-induced proteoloysis, can also lead to shedding of syndecan-1 (90). Another study demonstrated that OS induced by exposure to H2O2 upregulates the activity of endothelial histone deacetylase and metalloproteinases and, ultimately, amplifies syndecan-1 shedding (8).
Finally, sepsis-induced injury to the GCX, reduced antioxidant defense of the endothelium, and increasing ROS production by the activated EC and leukocytes amplify the phenomenon in a vicious circle and aggravate multiple facets of the endothelial dysfunction (Fig. 4).
Endothelial Inflammation and OS
During sepsis, microbial products and endogenous proinflammatory mediators can directly stimulate EC by binding to PRRs (87, 134), which leads to the activation of intracellular inflammatory pathways mediated by nuclear factor-κB (NF-κB) and the mitogen-activated protein kinases (134, 159). Activation of these signaling pathways drives EC toward a proinflammatory phenotype characterized by secretion of a tremendous amount of IL-6, IL-8, CCl-2, and CXCL-1 (87). The OS has been reported to modulate NF-κB and AP-1, which are two critical transcription factors involved in the proinflammatory switch of ECs (136, 164). In general, reducing agents decreases NF-κB activities, but it conversely enhances AP-1 activity (104). Oxidants, on the other hand, broadly activate NF-κB activity (109, 136), whereas they can also activate AP-1 by activating the JNK pathway via oxidative thiol modification of the apoptosis signal-regulating kinase 1 (124, 133). In microvascular EC, numerous genes that play critical roles in endothelial dysfunction have NF-κB regulation sites in their promoters, including IL-6, IL-8, TNF-α, vascular endothelial growth factor, RANTES, CCL-2, intercellular cell adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), TF, and the nitric oxide synthases. Moreover, ROS and RNS fuel the endothelial inflammation indirectly by promoting the release of DAMPs, which perpetuates the inflammatory stimuli.
OS and Endothelium-Leukocyte Adhesion
The EC regulates leukocyte trafficking through their production of chemokines and expression of adhesion molecules, primarily in the venous part of the microcirculatory network. Integrin family glycoproteins, including CD 62E/P, VCAM-1, PECAM (platelet endothelial cell adhesion molecule-1), and ICAM-1/2, orchestrate leukocyte adhesion, rolling, and crawling, the necessary steps before trans- or paracellular diapedesis (108, 154). In addition, the activation of circulating polymorphonuclear leukocytes facilitates host defense against circulating microorganisms. Luminal expression of these receptors by EC is rapidly upregulated during sepsis or acute inflammation, which enhances the recruitment of phagocytic cells to infectious sites (31). Under pathologic conditions, such as septic shock, tissue infiltration by activated leukocytes contributes to tissue injury and downstream dysfunction and failure of multiple organs. Many experimental data indicate that ROS play an essential role in regulating the production of leukocyte adhesion molecules. For example, overexpression of SOD expression in EC has been reported to limit TNF-α, and to abrogate the upregulation of VCAM-1, E-selectin, and ICAM-1-induced OS (34). Takano et al., based on HUVECs experiments, also reported that EC P-selectin upregulation induced by thrombin or TNF-α is dependent on ROS generated by the EC mainly via the NADPH oxidase (145). These phenotypic modifications of the EC luminal surface, triggered by the endothelial OS and the systemic inflammatory mediators, accelerate the recruitment and the activation of polymorphonuclear leukocytes and macrophages, which are, themselves, powerful generators of ROS. The tissue accumulation of OS from multiple sources can ultimately precipitate organ failure.
OS-Induced EC Death
A large body of evidence supports a key role for ROS and RNS in triggering cell death programs (128, 132). H2O2, •OH, and ONOO− cause cell injuries by oxidizing biological molecules (e.g., lipids) membranes, proteins, DNA, and organelles such as the mitochondria or the endoplasmic reticulum (ER) (23, 66, 127). Therefore, OS can trigger the apoptosis program via the mitochondrial and extrinsic (death receptor and ER) pathways (49, 106, 122). For example, ONOO− can directly permeabilize the mitochondrial outer membrane, triggering the mitochondrial permeability transition, an irreversible process that leads to a cessation of electron transfer and ATP production (23), ultimately provoking cell death (77). ONOO− and H2O2 also cause rapid activation (1–3 h) of the Fas death receptor pathway and caspases-2/7/9 (118). Notably, low doses of ROS have been found to activate cell survival signaling pathways, whereas moderate ROS/RNS concentrations result in apoptosis, and higher concentrations or long exposure have been associated with necrosis. Therefore, the intensity of the redox disbalance will engage EC toward apoptosis or necroptosis, both of which can dramatically enhance endothelial dysfunction. First, apoptotic (and a fortiori necrotic) EC become highly procoagulant. In staurosporine-induced apoptosis experiments, Bombeli et al. reported a rapid exposure of PS and active TF. EC anticoagulant features are impaired in a few hours, and they include reduced production of thrombomodulin, TFPI, and heparan sulfate (19, 21). Necrotic cells release a large amount of DAMPs and PS-positive microparticles that amplify local and disseminated coagulation and inflammation (Fig. 5) (15, 101). Finally, EC death involves loss of structural and membrane properties, which aggravates the sepsis- and ROS-induced permeability, the capillary protein and fluids leakage, and therefore organ injury (61).
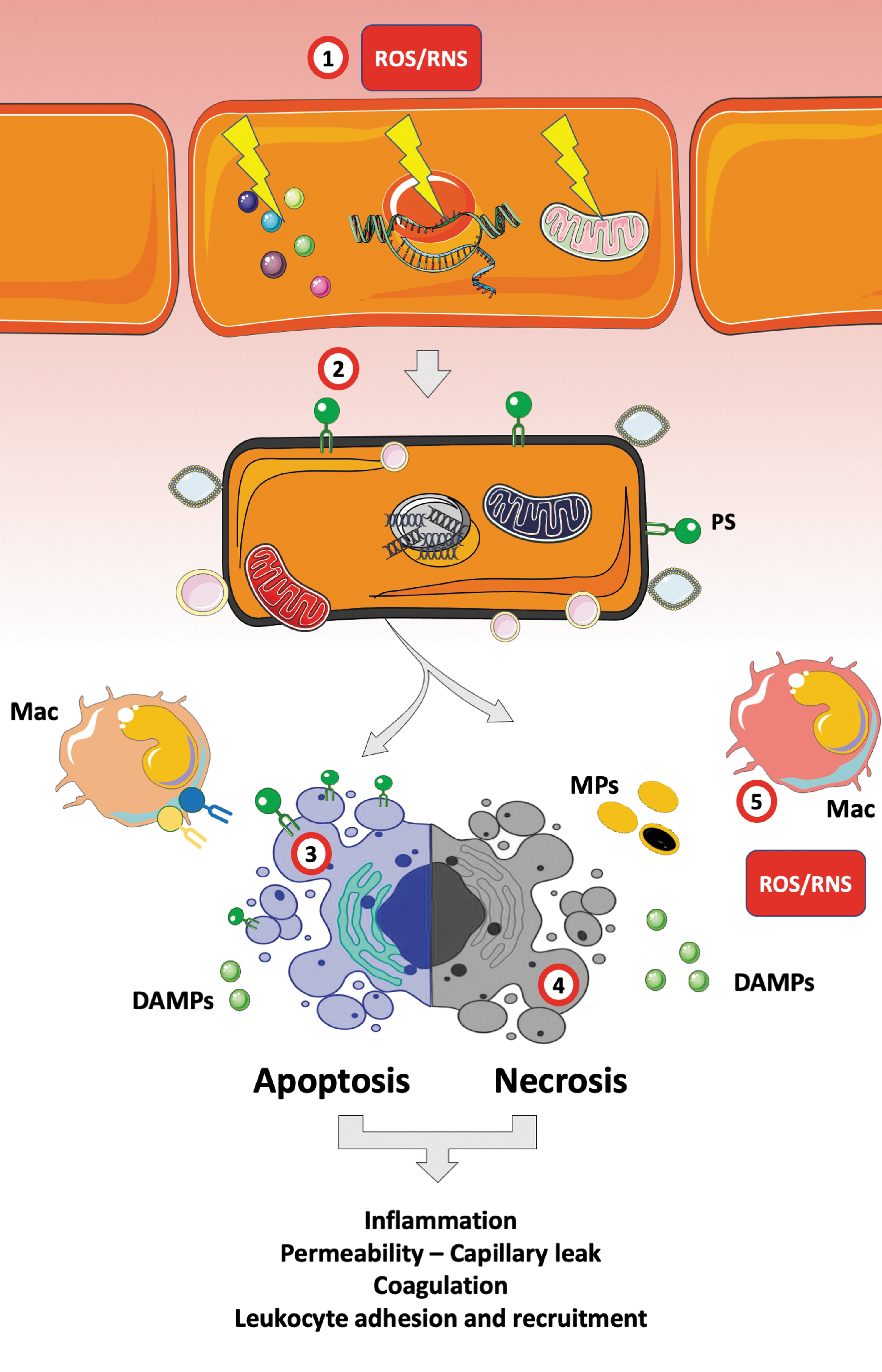
OS as a Goal for an Endothelial-Targeted Therapy in Sepsis
There is currently a focus in septic shock on preserving endothelial function and optimizing the microcirculation (98). Several approaches targeting OS have been explored based on the important role of OS in EC dysregulation during sepsis and acute inflammation (Table 1) (44, 123).
Clinical Trials Targeting Systemic Oxidative Stress or Endothelial-Related Oxidative Stress in Sepsis and Septic Shock
BAL, low-chloride balanced crystalloid solutions; GSH, reduced glutathione; ICU, intensive care unit; IL, interleukin; MDA, malondialdehyde; NAC, N-acetylcysteine; NF-κB, nuclear factor-κB; NOS, NO synthase.
Avoiding hyperoxia
Based on the assumption of improving the delivery of oxygen to the tissue and promoting bacterial defenses via oxidative burst (117, 160), some authors have proposed hyperoxia as a therapy for sepsis (71). Although small studies reported encouraging results in reducing wound infection postsurgery (30, 75), in the hyperoxia and hypertonic saline in patients with septic shock (HYPERS2S) study, the groups receiving mechanical ventilation with FiO2 = 1 had a higher risk of mortality, which resulted in discontinuation of the study before completing full enrollment (13). This deleterious effect of hyperoxia may be partly due to the increase in ROS generation, which has been observed in several critical illnesses such as resuscitated cardiac arrest (89) or acute myocardial infarction (143).
Limiting NO production
Several clinical trials investigated the NOS isoform inhibitors in the years after the recognition of NO as a critical mediator in septic shock-associated vasoplegia (96). Although phase II trials showed improved systemic vascular resistance and better macro hemodynamics, in the phase III trial there was significant excess mortality in the treatment group and the trial was stopped early (14, 102). The complexity of the role of NO between macro/microvessels, the dynamic regulation of iNOS and eNOS, and issues of NO bioavailability during sepsis and septic shock may explain the failure of a basic approach aiming at inhibiting NO production (96). Currently, two clinical trials are underway evaluating methylene blue, as the third line of vasopressor in septic shock (NCT04089072) or in cirrhotic patients with refractory vasoplegia (NCT03120637).
Enhancing antioxidant defenses
N-acetylcysteine
N-acetylcysteine (NAC) has been safely used to treat acetaminophen overdose for decades (151). Its direct antioxidant properties, which increase glutathione synthesis, and its lack of severe side effects make it an excellent candidate to alleviate systemic and endothelial OS in sepsis (12). In vitro, NAC can reduce by about 20% the EC apoptosis induced by plasma from patients with septic shock (78). In mice with endotoxemia or with sepsis induced by cecal ligation and puncture (CLP), NAC administration reduced systemic inflammation (TNF-α, NF-κB) (42) and organ injury (54, 88), and increased survival (17, 157). The administration of high doses of NAC added to glutathione (GSH) has been reported to significantly decrease the peroxidative stress of patients with septic shock as measured by expired ethane, plasma malondialdehyde, erythrocyte deformability, and complement activation (115). Despite a small sample size, the mortality at day 10 in the NAC-treated patients was half that of the untreated group (115). In a randomized controlled pilot trial, NAC infusion reduced NF-κB activation in peripheral blood mononuclear cell (PBMC) and reduced plasma IL-8 (121). Another study reported reported reduced IL-8 levels and better lung oxygenation and compliance in early septic shock in NAC-treated patients. These data suggest a beneficial role of NAC in blunting the inflammatory response to sepsis (142). However, none of these trials focused on endothelial dysfunction, nor did they show a substantial reduction in sepsis mortality with NAC treatment.
Albumin
Albumin is the most abundant endogenous plasma protein and the central extracellular molecule responsible for maintaining the plasma redox balance. The composition of albumin explains its tremendous antioxidant properties, including a free cysteine located in position 34, which has a thiol group that represents 80% of the antioxidant capacity of the plasma, the “binding site 1” attach homocysteine and bilirubin, and its N-terminal portion, which contains DAHK amino acids and is a binding site for pro-oxidant metals (Cu, Fe, Co, and Ni) (146). Moreover, albumin is a carrier of sphingosine-1 phosphate that protects GCX by inhibiting syndecan-1 shedding (3, 163). Several observational studies and meta-analyses have revealed that hypoalbuminemia is an independent risk factor for septic shock mortality (158). Indeed, each decrease of albumin by 10 g/L is associated with significant worse morbidity and a higher risk of death (OR = 2.37). In endotoxemic mice, resuscitation with albumin decreased ROS and RNS production, and it improved vascular reactivity in mesenteric arteries (110).
A recent pilot proof-of-concept study in stabilized septic shock patients showed that infusion of 20% human serum albumin (HSA) improved skin microvascular endothelial-mediated vasoreactivity as compared with saline (67). HSA infusion has been assessed in large clinical trials in septic patients (120). Although it showed the capability to restore a sustained increase in plasma thiols (126), so far it has not improved 28 and 90 day survival rates (29, 82).
Vitamin C
Vitamin C (
In humans, circulating vitamin C concentrations are markedly depleted in patients with sepsis, and they are potentially associated with worse outcomes (22, 57, 58, 135). Therefore, in 2013 the REDOXS study evaluated glutamine and antioxidant vitamins (β-carotene, vitamins C and E) on mortality in a general ICU population. The combination of glutamine or antioxidants did not improve clinical outcomes, and glutamine alone was associated with an increase in mortality among critically ill patients with multiorgan failure (73). Another study investigated the supplemental administration of vitamins C and E in critically ill patients requiring 10 or more days of enteral nutrition. They observed a decreased mortality (45.7% in the antioxidant group and 67.5% in the regular-feeding group; p < 0.05) (41). However, the control group received lower doses of vitamins C and E than usually recommended (50 mg and 10 IU, respectively), and this study highlights the potential harm of vitamin deficiencies. A small retrospective before/after study in patients with severe sepsis and septic shock found that early use of the combination of intravenous vitamin C, corticosteroids, and thiamine reduced progressive organ dysfunction (acute kidney injury) and mortality (107). However, recent large randomized prospective clinical trials failed to confirm these results (53, 56). The results of several clinical trials investigating the potential benefit of vitamin C in sepsis and septic shock are still expected. Table 1 summarizes previous and ongoing clinical trials that investigate the effect of reducing systemic OS or endothelial-related OS in sepsis and septic shock.
Current Challenges and Futures Directions
The EC dysfunction is the primary substratum of microcirculatory failure during sepsis, leading to multiorgan failure and death. Sepsis-induced endotheliopathy is a multifaceted syndrome combining impaired coagulation and fibrinolysis, increased vascular permeability and leukocyte recruitment, and dysregulated vascular tone. Moreover, sepsis is a major inducer of OS in EC. The rupture of the redox balance toward OS in EC enhances all aspects of the sepsis-induced dysfunction, and it can fuel a vicious circle of pro-oxidant condition.
The COVID-19 pandemic has recently increased interest in endothelial dysfunction in severe infections. Although to date there are minimal direct experimental data published that directly show microvascular involvement specific to the betacoronavirus SARS Cov-2 infection, emerging evidence from patients with COVID-19 suggests that EC may critically contribute to the initiation of acute lung injury and propagation of organ failure in severe COVID-19. Pulmonary EC are a potential sanctuary for viral replication, and EC infection could lead to a sepsis-like endotheliopathy characterized by vascular inflammation, capillary leakage, OS, and a procoagulant, proadhesive, and proangiogenesis state (1, 69, 147). The hypothesis that pulmonary EC play an important role in driving COVID-19-induced acute respiratory distress syndrome requires further investigation.
The re-establishment of tissue perfusion is a cornerstone of the early management of septic shock. Therefore, targeting one or more of these pro-oxidant mechanisms might improve microcirculatory function and reduce the development of sepsis-induced organ dysfunction. Although results of many preclinical studies and pilot clinical trials of antioxidant treatment for sepsis have been encouraging, large controlled randomized trials have not yet convincingly shown that antioxidant strategies improve sepsis outcomes. Better identification of septic patients suffering from latent EC dysfunction may help to define those individuals who are more likely to benefit from antioxidant therapies. Finally, a deeper understanding of OS's complex role in sepsis-induced EC dysfunction and downstream shock and organ failure may reveal additional therapeutic targets and strategies.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this work.