
Abstract
Significance:
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus causing coronavirus disease 2019 (COVID-19), affects every aspect of human life by challenging bodily, socioeconomic, and political systems at unprecedented levels. As vaccines become available, their distribution, safety, and efficacy against emerging variants remain uncertain, and specific treatments are lacking.
Recent Advances:
Initially affecting the lungs, COVID-19 is a complex multisystems disease that disturbs the whole-body redox balance and can be long-lasting (Long-COVID). Numerous risk factors have been identified, but the reasons for variations in susceptibility to infection, disease severity, and outcome are poorly understood. The reactive species interactome (RSI) was recently introduced as a framework to conceptualize how cells and whole organisms sense, integrate, and accommodate stress.
Critical Issues:
We here consider COVID-19 as a redox disease, offering a holistic perspective of its effects on the human body, considering the vulnerability of complex interconnected systems with multiorgan/multilevel interdependencies. Host/viral glycan interactions underpin SARS-CoV-2's extraordinary efficiency in gaining cellular access, crossing the epithelial/endothelial barrier to spread along the vascular/lymphatic endothelium, and evading antiviral/antioxidant defences. An inflammation-driven “oxidative storm” alters the redox landscape, eliciting epithelial, endothelial, mitochondrial, metabolic, and immune dysfunction, and coagulopathy. Concomitantly reduced nitric oxide availability renders the sulfur-based redox circuitry vulnerable to oxidation, with eventual catastrophic failure in redox communication/regulation. Host nutrient limitations are crucial determinants of resilience at the individual and population level.
Future Directions:
While inflicting considerable damage to health and well-being, COVID-19 may provide the ultimate testing ground to improve the diagnosis and treatment of redox-related stress diseases. “Redox phenotyping” of patients to characterize whole-body RSI status as the disease progresses may inform new therapeutic approaches to regain redox balance, reduce mortality in COVID-19 and other redox diseases, and provide opportunities to tackle Long-COVID. Antioxid. Redox Signal. 35, 1226–1268.
Introduction
Stress is not what happens to you, but how you react to it.
Hans Selye
In the midst of every crisis, lies great opportunity.
Albert Einstein
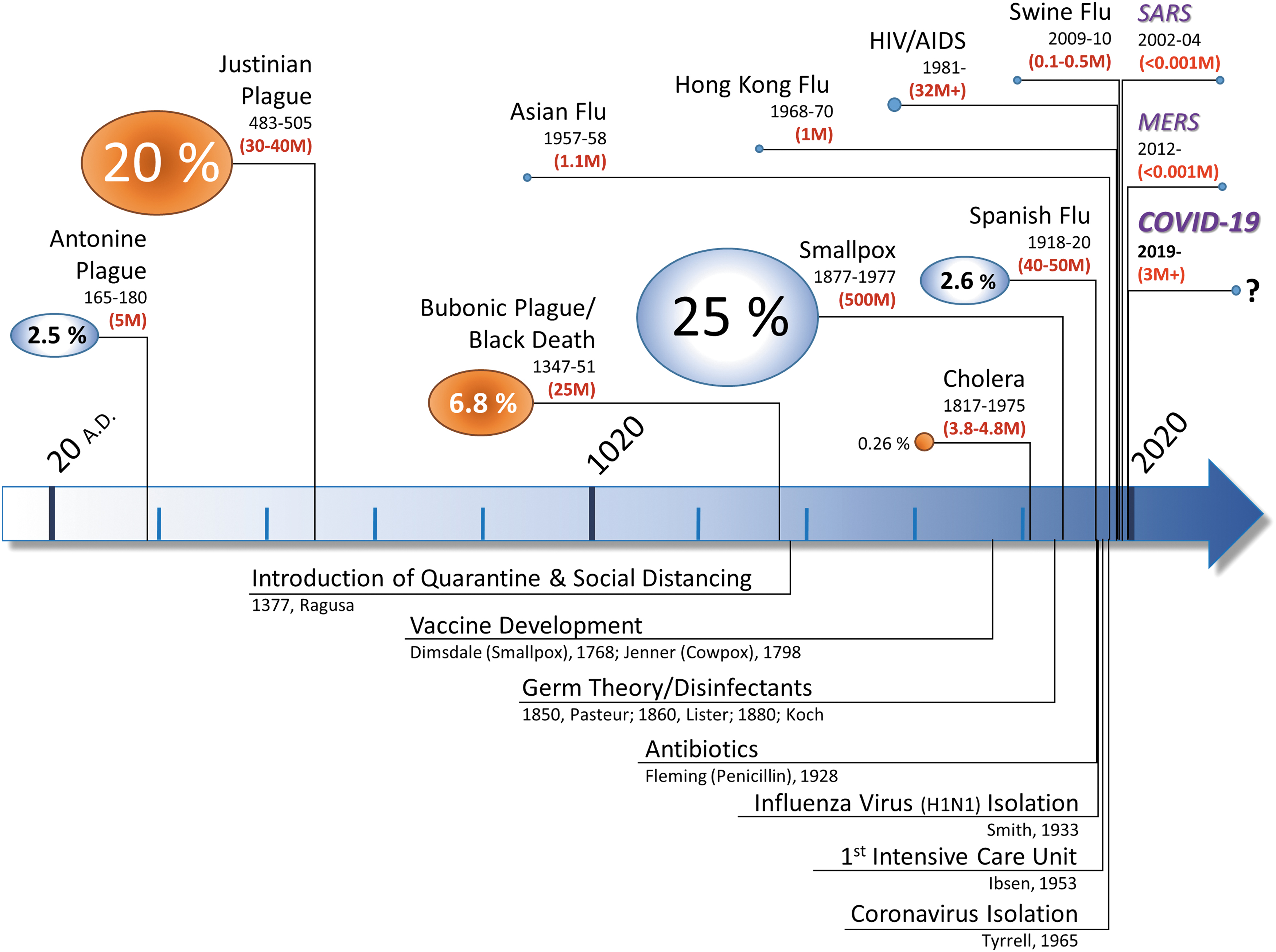
Here, we consider the nature of human infection with “severe acute respiratory syndrome coronavirus 2” (SARS-CoV-2), the virus that causes coronavirus disease 2019 (COVID-19) and holds the world hostage, as an example of a stress response that challenges our ability to maintain redox balance at the whole-body level. With > 230 million confirmed infections and almost 5 million deaths globally (490) and third/fourth waves driven by new variants advancing through South Asia, Europe, and the Americas, the threat of contracting this virus continues to evoke fear and misery, affecting every aspect of human life (Fig. 1 and Supplementary Data for a comparison of COVID-19 to earlier pandemics and additional contextual information). All of the systems within which humans interact, including commercial and financial markets, health care, travel, and recreational activities, are stressed in ways never witnessed before. These changes to our external environment engender stress and elicit reciprocal changes in behavior and the internal physiological milieu, causing fatigue and affecting mental well-being, even in uninfected individuals. The current pandemic has de facto become a worldwide social experiment (372). Coping with the associated changes in metabolic demand requires adaptation at multiple levels of biological organization, including hormonal, neuronal, and metabolic systems, with coordination at the cellular and whole-body level.
Box 1. Coronaviruses: From the Common Cold to Pandemics
The discovery of viruses (as filterable submicroscopic agents causing infectious disease in tobacco plants) dates back more than a century (249). Viruses are not living entities but obligate intracellular parasites containing either DNA or RNA. Their replication depends on exploiting the hosts' cellular machinery, and they can only multiply in dividing cells. As with other viruses, the evolutionary origin of coronaviruses remains obscure (19, 89, 451). Six coronavirus strains were previously known to infect humans—all contain an enveloped single strand of positive-sense RNA inside a lipid membrane decorated with envelope proteins and petal-shaped glycoprotein spikes, yielding an appearance resembling the solar corona on electron microscopy images (403). Four coronavirus strains (229E, OC43, NL63, and HKU1), identified in the 1960s and 1970s (425), cause the “common cold.” These are usually self-limiting infections of the upper respiratory tract producing relatively minor symptoms in immunocompetent hosts (403). Two other strains, severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), which can cause potentially fatal respiratory infections, were only recognized in the past two decades. After the discovery of human coronaviruses, further investigations revealed the importance and enormous variety of coronaviruses causing diseases in animals (191). Both SARS-CoV and MERS-CoV are believed to have originated in bats before transferring to humans [thereby classifying as zoonotic diseases (253)]. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is believed to have transferred to humans via an intermediate animal host in a similar way (224), and its future will depend on whether it can establish a reservoir in an animal population (251). Although sharing many similarities with SARS-CoV and MERS-CoV, SARS-CoV-2's rapid progression around the world (Supplementary Data) demonstrates that it possesses significantly higher infectivity while managing to escape clearance. What triggers viruses to cross species is unclear, but structural inter-species similarities of proteins used by viruses to gain cellular access such as angiotensin converting enzyme 2 (ACE2) for SARS-CoV/SARS-CoV-2 (80) and environmental pressures are important contributors. The increased frequency with which new pathogens and pandemics emerged in the past century (Fig. 1) is testimony to the impact that humans have had on their environment, perturbing ecosystems as well as provoking climate change and biodiversity loss (14, 62, 89, 173, 324). A recent census of the Earth's biosphere composition revealed that the mass of humans is already an order of magnitude higher than that of all wild mammals combined (22); by comparison, the total mass of SARS-CoV-2 virions on Earth is estimated to be <1 kg (377).
The pattern and magnitude of the response to any challenge (or “stressor”) is determined by the specific demand placed on the system. Challenges vary in nature and intensity across a wide spectrum. Our ability to maintain health relies on mounting a response that is similarly varied, proportionate, and “fit for purpose.” Although milder perturbations are accommodated locally, for more severe challenges the response is escalated through higher levels of organization to a fully integrated whole-body response. The interaction of the processes at play during stress escalation is understood, partly, at individual levels of biological organization, but how these horizontal and vertical responses are coordinated in time and space remains to be defined. At the biochemical level, this system uses cascading chemical reactions of short-lived small redox-active molecules for sensing and signaling purposes defined as the “Reactive Species Interactome” (RSI) (84) (Supplementary Fig. S1). This system is fundamental to an organism's ability to achieve bodily homeostasis while adapting to changes in demand. It accomplishes this by using reactive small chemicals (specifically, reactive oxygen, nitrogen, and sulfur species; ROS, RNS, and RSS) and the “cellular stress response” to integrate inputs generated by interaction with its environment (214, 215) while maintaining electron balance according to a set of principles outlined in the “Redox Code” (188). Given the interdependencies of the many interconnected systems within the human body, COVID-19 is a “complex systems” disease that has the potential to overwhelm the body's ability to cope and maintain homeostasis.
Humans and pathogens have been coevolving for millennia. The ability of viruses to modulate redox-sensitive pathways in the host is increasingly recognized as being essential for each stage of their life cycle (202) (see Boxes 1 and 2 for context on coronaviruses and redox-related aspects). The more we learn about COVID-19, the more it appears to represent a redox-based disease. Knowledge about what determines someone's susceptibility to infection; the clinical effects of COVID-19; the risk factors for severe disease and poor outcome; and even the success/failure of potential therapies all point to the critical involvement of electron exchange processes. Early in the pandemic, oxidative stress was proposed as playing a determinant role in the pathogenesis (64, 97, 220), but alone this appears unremarkable given that ROS production is an established feature of all viral (and bacterial) infections (53, 175, 369). Enhanced oxidant production usually protects plants and animals from invasion by bacterial pathogens (by providing a burst of ROS from nicotinamide adenine dinucleotide phosphate [NADPH] oxidase [NOX], specifically NOX2, in phagosomes). Mammalian NOX stimulation, however, impedes viral clearance whereas abrogating ROS production improves influenza-induced lung infection (391, 438), suggesting that oxidative stress promotes viral pathogenicity. NOX2 activation appears to play a central role in infections with single-stranded RNA/DNA viruses (420) and SARS-CoV-2-induced sequelae such as thrombosis (436) and vascular inflammation (283). Marked elevation of multiple cytokines (cytokine storm) and the effectiveness of dexamethasone in improving patient survival suggested that an overreaction of the immune/inflammatory response is important to disease development in more severe cases (although that may only be part of the story). Why, with SARS-CoV-2 infection, immune hyperactivity does not principally affect the young as in the Spanish Flu (Fig. 1) pandemic (207, 412) is unclear. Curiously, molecules at opposite ends of the redox spectrum [e.g., highly reducing molecular hydrogen (H2) (311); and hydrogen sulfide (H2S) (76), and the archetypal oxidants oxygen (O2) and ozone (O3) (134, 157)] are all either being used clinically or have been proposed to confer benefit in the treatment of COVID-19. What determines virulence, selectivity, and involvement of extrapulmonary organ systems largely remains unclear, although the identification of angiotensin converting enzyme 2 (ACE2) as the primary receptor for SARS-CoV-2 quickly focused attention on the renin angiotensin system (RAS). However, distinct from other viruses, SARS-CoV-2 evades many defence pathways that usually guard the human host against harm, striking a finer balance between deadliness and replication efficiency.
Box 2. Intracellular Life Cycle of Severe Acute Respiratory Syndrome Coronavirus 2 and Implications for Redox Signaling
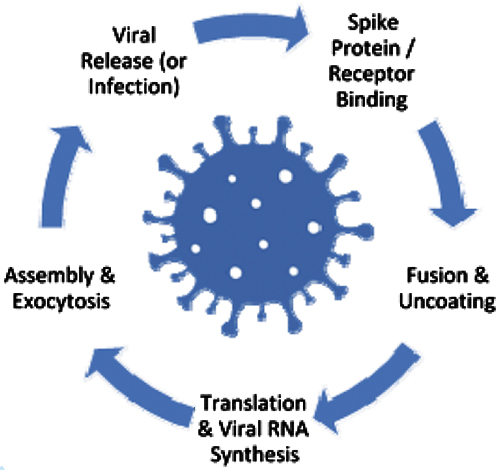 The intracellular life cycle of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is incompletely understood (437). The redox reactions affecting individual stages of this process are summarized next; acute and chronic virus-induced reactive oxygen species (ROS) production has been reviewed (67, 404) and appears to be highly compartmentalized.
The intracellular life cycle of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is incompletely understood (437). The redox reactions affecting individual stages of this process are summarized next; acute and chronic virus-induced reactive oxygen species (ROS) production has been reviewed (67, 404) and appears to be highly compartmentalized.
Binding
For enveloped viruses such as SARS-CoV-2, genome entry into the host cell involves two key steps: (i) virion binding to a cell surface receptor and (ii) fusion with the cell membrane; the redox considerations for SARS-CoV and the involvement of thioredoxin and protein disulfide isomerase have been previously reviewed (114). Specific domains in the S1 subunit of the viral spike protein (S-protein) bind peptidase domains of the host cellular receptor, angiotensin converting enzyme 2 (ACE2) (468). Four disulfide bonds between multiple cysteine residues in the receptor binding motif of the S-protein govern the affinity for this reaction (221). The binding affinity is significantly impaired when all four of these bonds are reduced to sulfhydryl groups (150); a hinge-like conformational change in the protein structure makes binding impossible unless the protein is in the less stable “up” configuration that allows the binding site to be accessible (462). The absence of a particular redox-active disulfide in the ACE2 protein structure of animals not susceptible to infection by SARS-CoV-2 suggests that it is important for receptor recognition and potentially for the virus' ability to jump species; it may also be a site that is susceptible to modulation by host redox status (390).
Fusion and Uncoating
After the binding to specific receptor proteins, many viruses use redox-induced structural changes to initiate fusion of viral and host membranes to gain cellular entry (404), commonly involving changes in thiol/disulfide balance as described earlier. Unusually though, SARS-CoV has demonstrated the ability to fuse and enter cells even in the presence of sulfhydryl and oxidoreductase inhibitors (227). Since SARS-CoV-2 shares many structural and functional similarities with SARS-CoV, this marked redox insensitivity to cellular fusion may be a key aspect of this new coronavirus that allows it to enter cells more readily even in inflamed host environments (for specific changes to the redox landscape in inflammation see Box 3 and “Endothelial Dysfunction” section). SARS-CoV-2 binds to human ACE2 with a higher affinity than SARS-CoV (379) and enters cells by endocytosis, a process during which ACE2 translocates from the cell surface to the endosomal lumen. The redox-active endosomes (redoxosomes) involved are important for homeostasis and stress signaling, and their function is modulated via cytokine-mediated signal transduction pathways (394). Influenza A virus has been shown to activate endosomal NOX2, resulting in ROS generation, which suppresses host cell antiviral signaling and Toll-like receptor 7 (TLR7)-driven immunity via modification of a high-conserved cysteine residue on the ectodomain of TLR7 (420); the latter is essential for B cell activation and antibody production (and thus viral clearance). Since this mechanism operates with all viral strains entering cells via endocytosis, it is likely to be used by SARS-CoV-2 as well.
Viral RNA and Protein Synthesis
Coronavirus RNA is one of the largest known viral RNAs, coding for two polyproteins, and viral gene expression is a highly regulated process; it includes viral and host proteins to perform RNA polymerization, proofreading and final capping, with critical cofactors that enhance the efficiency of RNA replication (354). Immediately on entry, 16 nonstructural proteins are co- and post-translationally released through proteolytic cleavage of the polyproteins by cysteine proteases. Fifteen of these proteins go on to hijack the endoplasmic reticulum (ER) membrane and form the viral replication and transcriptome complex, and other interactions between these proteins and host cell factors initiate the biogenesis of the viral replication organelles (437). A number of RNA viruses, including SARS-CoV-2, target the mammalian selenoprotein, thioredoxin reductase, using an antisense knockdown mechanism that interferes with host ribonucleotide reductase activity required for DNA synthesis, a viral strategy to maximize RNA synthesis (414). This may further increase virulence when combined with selenium deficiency (see Nutrition section).
Assembly, Exocytosis, and Release
After replication, essential viral components are assembled for transmission first in the host cell's ER and Golgi apparatus (382). The most error-prone process in gene expression is protein folding, and accumulation of misfolded proteins elicits “ER stress,” which affects a variety of cellular signaling processes, including redox homeostasis, energy production, inflammation, and apoptosis (56). Protein folding in the ER is a highly coordinated, chaperone (e.g., protein disulfide isomerase)-controlled process regulated by NOX activity (226). The ER plays an important role in the quality control of secretory protein production by providing a favorable environment for oxidative protein folding. Both folding and subsequent post-translational modification, including palmitoylation and glycosylation of the envelope and S-proteins [which is presumed to occur partly in attempts to evade host immune attack but is also important for folding/receptor binding; (60)], are under similarly tight redox control (29, 126). Since the ER is also a key site for lipid biosynthesis (184), lipid composition is sensitive to redox alterations. Oxidative protein folding in the ER is a key ROS producer [that the S-protein induces ER stress and chemokine upregulation was recognized with SARS-CoV already in 2007; (434)], the intensity of which is sensed by redoxosomes (472). Although SARS-CoV-2 infection triggers the “unfolded protein response,” how the virus avoids early induction of apoptosis allowing it to maximize replication (127) is not fully understood. Once ready to be packaged, virions enter the cellular secretory pathway before being released into the extracellular space through exocytosis—itself a process that is tightly regulated by nicotinamide adenine dinucleotide phosphate (NADPH) concentration, thioredoxin and glutaredoxin activities (176). After release, the cycle starts over again as the infection propagates throughout the host.
Apart from exploring each of these aspects in turn, we also consider the importance of nutrition in dealing with stressful challenges and integrating intra- and extracellular redox status. Finally, we discuss how “redox phenotyping” and the determination of redox/nutritional status along the illness trajectory might be used to characterize host responses and identify the determinants of individual resilience.
Dealing with Stress: General Considerations, Significance of the Reactive Species/Redox Interactome, and the Vulnerability of Complex Self-Organizing Systems
In earlier perspectives (84, 360), we explored how the response to stressors could have emerged as an integral feature of the evolutionary process and how the complex capabilities in humans reflect the layers that were selected throughout evolution as “fit for purpose,” enabling survival over extended periods of time. We and others have argued that since life emerged in a sulfur-rich environment, the ability to effectively handle sulfur must have played a fundamental role in the development of these survival attributes (84, 308). Earlier, we proposed that nitric oxide (NO) must have played a crucial role in evolution by acting as an antioxidant before its widespread use as a biological signaling agent (113) [NO is an important mediator of homeostatic processes and host defence mechanisms (284, 285); its role in COVID-19 has been reviewed (111), and pertinent aspects of its discovery/action profile are provided in the Supplementary Data]. We have used these ideas to develop the concepts of the “Reactive Species Interactome” (RSI) (84, 87), the “Redox Interactome,” and the notion of the “Redox Landscape” (360) (Supplementary Fig. S1 and Box 3).
Box 3. Sensing Stress: The “Reactive Species Interactome” Versus “Redox Systems Interactions”
The way Redox Biology unfolds in living entities can be envisioned along two dimensions for which we coined two distinct concepts.
The Horizontal Dimension: The Redox Species Interactome
Redox agents are not “standard” biomolecules: They are “Reactive Species.” As such, they permanently react with each other, extending a web of interactions throughout the cellular space. They are unstable, changing sequentially in chemical nature, and continuously convert into other redox-active species.
Redox interactions are the essence of biological chemistry, hence all biomolecules are prone to react with redox agents, fashioning an extended “Redox Landscape.”
Redox molecules are involved in many physiological processes, often assuming pleiotropic roles. This is the case also for nitric oxide (NO), a physiological “swiss army knife”-like all-purpose signaling molecule, which is also involved in antioxidant defence and associated with oxidative stress-related immune responses (Supplementary Data).
Together, this constitutes a Redox Landscape that integrates many biomolecules and processes, which are continuously synchronized in “real time.” The singularity of the reactive species interactome (RSI) implies that each redox stress will always impact the entire network, modifying the system as a whole, affecting many physiological processes, and potentially overturning the originally “assigned” function of particular molecules within the system.
The RSI defines what has been called “Systems Chemical Biology” (309): different processes taking place in parallel, using a pool of shared redox entities that diffuse and connect all of these biological modules with each other. By maintaining a redox milieu that is suitable for many different physiological processes within the same tissue environment, the RSI affords the synchronization and stabilization of various redox modules in different cellular loci, allowing specific and otherwise independent processes to take place. The enormous diversity of the RSI accommodates many different redox biologies, with sufficient plasticity to adapt to environmental change (87).
In this context, oxidative stress has to be contained locally, using antioxidant defence systems, to maintain an appropriate redox status at each local level. NO, for example, cannot serve as a signaling molecule under “oxidative stress” conditions, because it is rapidly converted into nitrogen dioxide (NO2) and peroxynitrite (ONOO−). NO also stabilizes redox regulation by preventing other redox agents from flipping into another role and ensuring the integrity of redox processes. Thus, the entire regulation of the “horizontal space” essentially relies on the driving force of chemical reactions and basic redox biochemical processes.
The Vertical Dimension: Redox Systems Interactions (the “Redox Interactome”)
These many different processes, unrelated in purpose, are connected through the use of the same redox language, relying on shared redox agents (words) and chemistry (grammar). It is the same language that is utilized horizontally within the RSI, which connects all redox agents and processes. However, processes and molecular structures relying on redox biochemistry are taking place at different levels of biological organization, ranging from the basic chemical reaction between biomolecules in the cytosol, via the electron transfer chains embedded within organelle membranes, to the complex redox signaling machinery that governs communication between cells and tissues. This relies on a series of increasingly complex structures that are integrated within what we defined as the “Redox Interactome” (360). This Redox Interactome provides an architecture that allows different levels of redox biology to be tuned and synchronized via (i) horizontal interactions providing information about the status of each redox module and allowing specific regulation of each redox-based physiological function (metabolism, bioenergetics, immune system, vascular homeostasis, cell cycle, neural communication, etc.); and (ii) vertical interactions supported by redox agents acting as signaling mediators through the “Redox Spine” (360).
This complex Redox Architecture (360) provides the organism with an integrative Redox Regulation System that allows the accommodation of different stresses both within and across levels of biological organization (transcription, translation, post-translational modification, metabolism, signaling, bioenergetic regulation, and organ function).
Although the significance of reactive species and free radical biology for health and disease is widely appreciated, an understanding of electron exchange processes at the systems level (Redox Systems Biology) is still at its infancy (46, 154, 385, 444). Insight into the organizing principles of seemingly robust biological systems such as the human body may also help understand complex diseases (205) such as COVID-19. Paradoxically, the same features that confer biological robustness can also give rise to vulnerability from unusual threats (205). Although it has been appreciated that redox regulation is a multisystems capability engaging lung, liver, and kidneys (380), how redox balance is achieved at the whole-body level remains largely unknown. Our bodily redox regulatory system comprises an interconnected system of individually robust redox modules, including HIF-1, Keap-1/nuclear factor erythroid 2-related factor 2 (Nrf2), nuclear factor kappa light chain enhancer of activated B cells (NF-κB)/IkB, Nrf2/antioxidant response elements (ARE), and thioredoxin (Trx)/thioredoxin reductase (TrxR), each of which exerts optimal function at distinct, different redox potentials. Flavin catalysts (FMN/FAD) and the NAD(P)H/NAD(P)+ couple to support cellular energy production/metabolism, antioxidative defence, and redox balance (188, 432, 442, 465). Physiologically, ROS act as pleiotropic local signaling agents (386), with specific roles in metabolic and inflammatory signal transduction (119) and redox homeostasis (385). In some cases, this is linked to oxygen availability: for example, ROS activate the HIF-1α promoter via a functional NF-κB site (39) whereas the HIF system switches cells from oxidative to glycolytic metabolism, reducing mitochondrial ROS formation and increasing production of NADPH and glutathione under hypoxic conditions (358). The nature of any response is determined by a limited number of fundamental building blocks, from which an almost limitless pattern of responses is possible, with the character of the response determined by the particular building blocks recruited—this is the essence of complex systems (162, 281). The dynamic interaction between those redox modules will have evolved in a self-organizing manner, poised around a “critical state” far from equilibrium. In such systems, even minor perturbations can lead to avalanche phenomena, resulting in either a switch in the mode of operation or destabilization and catastrophic failure. This so-called “self-organising criticality” is the only mechanism known to generate complexity (18). Such “cascading failure scenarios” are well known in complex biological networks and man-made systems such as electrical power grids and telecommunication systems (281); they even extend to the chemistry of redox flow batteries (459), demonstrating that coupled chemical reactions themselves can be a source for vulnerability (Box 4). These considerations may help explain the individual tragedy of a failing human body as well as the related societal, financial, and health care systems being driven to the brink of collapse during the current pandemic (162, 281). Here, we use these considerations to explore how the RSI may help in providing an understanding of SARS-CoV-2's mode of operation and to review the redox-related changes this environmental stressor evokes while spreading through the human host. Although extensively covered in the medical literature, we start with a brief overview about the disease presentation to provide context (see Supplementary Data for information about its emergence).
Box 4. Vulnerability Patterns Specific to Redox Systems
Life may have evolved to rely on redox chemistry as a result of its highly efficient support of biological processes, allowing biological structures to act and interact, with nitric oxide (NO) playing a central role (Supplementary Data). The inherent complexity of the redox “network of networks” creates vulnerability to oxidative stress in both the horizontal and vertical dimensions.
The Horizontal Dimension: Janus-Faced Agents
The specificity of redox agents is linked to their ability to simultaneously play distinct biological roles.
- Oxidative stressors, with stress arising directly and/or indirectly from the outer or inner milieu (electron transfer chain inhibitors, ultraviolet/X-Ray radiation, redox-cyclers)
- Redox defence agents: Redox-active molecules and enzymes are the major biological tools used for antioxidant defence (thiol-based molecules, NO, metalloproteins)
- Signaling and communication mediators: Most of the adaptive (against stresses) signaling processes rely on redox molecules (NO/hydrogen peroxide [H2O2]) and redox reactions (oxidation, nitrosation, nitrosylation).
These redox agents can easily switch activity. NO is an antioxidant per se, but can both mediate signaling alerts and, in an oxidative milieu, also rapidly be flipped into a stressor (such as nitrogen dioxide [NO2] or ONOO−). Their physiological activity is determined by the properties of their milieu. A change in milieu will change both their nature and function, and what had earlier acted as a signaling agent or an antioxidant guardian suddenly turns into an oxidative stressor, a traitor. This is also true for the entire redox system, including enzymes, organelles, and processes. For example, NO-synthases turn into superoxide producers in an oxidative (tetrahydrobiopterin-oxidizing) milieu; once released, NO will turn into peroxynitrite in a superoxide-rich environment; NO signaling causes harmful excitotoxicity phenomena on long-term nitric oxide synthase (NOS) activation; and even nitrosylation chemistry, which underpins much of the physiological signaling of NO, can trigger harmful processes. The chemical biology of NO may shift from “physiological functioning” to “harmful action.”
Such a dramatic “redox swing” already happened at least once during the course of evolution. During the Great Oxidation Event (Fig. 4A), poisonous oxygen became the central driver of bioenergetics for the majority of living systems on the Earth, reflecting the shift from a reducing environment to an oxidative one. Consequently, prolonged and massive redox stress (e.g., an “oxidative storm”) will lead to three parallel but synergistic effects by:
- exhausting the buffer and antioxidant defences, thereby increasing vulnerability of the redox network;
- amplifying redox stress by subverting redox defences into oxidative aggressors;
- disrupting redox signaling and disabling major physiological processes (related to metabolism, bioenergetics, and cellular homeostasis).
The latter is probably the most impactful. A dissipation of the “redox chaos” leads to failure of all redox signaling systems that control many physiological processes. As a result, a functional “NO shutdown” will lead to a failure not only of vascular function and tissue perfusion but also of neurotransmission, immune function, gastrointestinal motility, cellular bioenergetics, and many other processes.
The Vertical Dimension: Losing Cellular and Organismal Communication
Massive and prolonged oxidative stress, as elicited by an acute infectious insult on top of a chronic inflammatory process, poses a significant threat beyond oxidative damage and local system failure. It might jeopardize the whole organism by disrupting the backbone of its central communication/signaling system, its language. Redox biology is not simply a question of Yin/Yang, a balance between oxidants/reductants that has to be maintained around a specific setpoint to preserve the correct functioning of redox-based physiological processes. Redox chemistry is the “lingua franca” that is used to support and integrate many different functions (360) (Box 3). All of these functions are using the same shared signaling system based on redox chemistry. Preserving the integrity of the internal redox communication system is vital for survival. Maintaining an appropriate redox balance might be an important “first-line defence” at the scale of a specific redox module (cell/organelle, or process/activity) but is insufficient to preserve the whole redox communication system from sustained and/or large redox stress. As the stress increases in amplitude and/or changes in nature, the organism must proportionately adapt by mounting countermeasures to cope with these challenges. A robust redox regulation system is required to maintain the integrity, internal coherence, and constancy of the language in the face of such a threat.
Such regulation systems cannot solely be based on self-equilibrating redox chemistry and the mobilization of antioxidant defence. We postulated (360) that increasingly sophisticated environmental challenges have been a major driving force for the complexification of form and function, leading to multiple layers of redox regulatory modules that we defined as the “Redox Interactome” (Box 3). These different devices are synchronized/tuned through a “Redox Spine” that integrates within a unique Redox Architecture all redox processes and multiscale structures [molecules, proteins, membranes, vacuoles, organelles, cells, tissues, organs, organism(s), populations] by providing a sophisticated Redox Regulation System. All regulatory devices are inserted (Russian doll-like) into one another, allowing the delivery of proportionate and targeted answers to increasingly challenging stresses (affecting gene/cell repair machinery, antioxidant enzymes, transcription, metabolic reconfiguration, sulfur and nitrogen cycle shifts, apoptotic pathways, etc.).
Such complexity can become a liability and translate into a vulnerability in the face of a new challenge such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). If, despite the multiplicity and redundancy of regulatory systems, the redox chemistry changes such that the organism loses the ability to communicate across systems, the survival of the organism is threatened due to loss of the cohesiveness that defines its identity.
Clinical Features of COVID-19 and Risk Factors
COVID-19 is highly transmissible [particularly via airborne droplets and aerosols indoors; (329)], with a mortality rate considerably greater than that of the “common cold” and marked demographic, socioeconomic, and geospatial variation (56). Clinical presentations vary widely; the most frequent symptoms are dry cough, high fever, and shortness of breath (similar to previous severe acute respiratory syndrome [SARS] and Middle East respiratory syndrome [MERS] outbreaks), and loss of smell and taste. Headaches, myalgia, nausea, and diarrhea are rarer symptoms (142, 164). However, up to 75% of infected individuals are completely asymptomatic—“superspreaders” unknowingly mass-distributing the virus (130, 322). At least one-third of all SARS-CoV-2 infections originate from asymptomatic people (310).
Disguising as respiratory illness, COVID-19 is a complex multisystems disorder (352). Pneumonia is the most common lung injury inflicted (133); severe cases cause acute respiratory distress syndrome (ARDS) with profound but paradoxically silent hypoxemia (unhelpfully called “happy hypoxia”) that is poorly understood but distinctive for COVID-19 (88, 133, 421). Acute kidney injury, neurological complications, and cardiovascular involvement are common (78, 155, 172, 254, 301, 402, 429); venous and arterial thromboembolic complications are central to the disease. Pulmonary embolism, deep vein thrombosis, or ischemic stroke is frequent, especially in severe disease and often despite anticoagulant prophylaxis (258, 263). Similarly, cerebral microhemorrhage is an increasingly recognized complication (103), associated with worse prognosis (258) and—being more inflammatory-mediated—distinct from other coagulation disorders (145). Increased plasma D-dimer levels, a sensitive fibrin degradation marker indicative of increased coagulation and oxidative stress (257), correlate with morbidity and mortality (471).
Although all ages are susceptible to infection, the median age of hospitalization is mid-50s to early 60s. Although increasing numbers of young people have become infected in recent months (37), older age (accompanied by increased inflammation and enhanced ROS production) remains the biggest risk factor for poorer outcome (72, 142, 164, 304). Other risk factors include male gender and preexisting comorbidities (52, 185, 319) associated with oxidative stress and/or metabolic dysfunction such as hypertension, asthma, and obesity (72, 142, 164). The same risk factors may limit the capacity to mount a humoral immune response to vaccination (321). Both COVID-19 and diabetes can exacerbate each other's severity (416). A possible link with menopause suggests that estrogen may explain the lower mortality risk in women (292). Poverty and ethnicity are additional risk factors (454); Black, Asian, and other ethnically diverse populations, who display proportionately higher oxidative stress (287), exhibit greater risk for severe disease that cannot be explained by metabolic, socioeconomic, or behavioral factors alone (338, 407).
The incidence of respiratory viral infections tends to vary seasonally, affecting exacerbations of other chronic respiratory conditions (456); it remains to be seen whether SARS-CoV-2 infections follow the same pattern (204, 211, 241). This is of interest considering that serum 25-hydroxyvitamin D [25-(OH)D] levels, largely determined by exposure to sunlight and diet, at time of hospital admission are associated with COVID-19 disease state and mortality (96, 140). However, 25-(OH)D is also a negative acute-phase reactant (441); thus, circulating levels may not adequately reflect vitamin D status in the presence of inflammation. Of note, sunlight also stimulates the release of NO from storage forms in the skin to increase systemic NO availability and reduce blood pressure, possibly via modulation of whole-body redox status (74, 240, 450). Inherited glucose-6-phosphate dehydrogenase (G6PD) deficiency, oxidative stress, and (suspected) vitamin D deficiency may explain racial inequalities in mortality (185), and globally COVID-19 has followed the 4°C–12°C isotherm (362). Seasonal peaks in chest exacerbations also correlate with air pollution exposure (e.g., nitrogen dioxide [NO2], carbon monoxide [CO] and O3); in Milan, air pollution and lower temperatures were associated with excess mortality from the 2016–2017 influenza epidemic (293) and COVID-19 in 2020 (487). SARS-CoV-2 infection correlated positively with ambient O3 and negatively with NO2 levels, possibly due to effects on viral aerosol particle diffusion; SARS-Cov-2's spike protein (S-protein) (Box 2) may facilitate attachment to ambient fine particulates, aiding transmission (487). Exposure to other environmental stressors, such as industrial chemicals, may further increase COVID-19 severity through mitochondrial and immune toxicity (143, 463). Traffic-related air pollution perturbs metabolic, inflammatory, and redox pathways, with the arginine metabolome particularly affected (235). Since arginine is the main substrate for nitric oxide synthases (NOS), this observation may have important implications for endogenous NO formation and its role as antioxidant, protecting sulfur-based redox signaling (see section “Redox Regulation at Local and Whole-Body Level”).
Peculiar Persistence of Symptoms: Long-COVID/Post-Acute Sequelae of COVID-19/Post-COVID Syndrome
The acute symptoms of many viral infections may be short-lived, although the virus remains latent within the body and can be reactivated (HIV, Eppstein-Barr, Varicella). For patients with COVID-19, including younger ones with mild presentations and asymptomatic cases, there are reports of persistent or late-onset effects known as Long-COVID, post-acute sequelae of COVID-19, or post-COVID syndrome (15, 252, 295, 424). Descriptions of reoccurrences also suggest the possibility of reinfection, relapse, or inflammatory rebound (141). A special case is the “Multisystem Inflammatory Syndrome in Children” (MIS-C), also called “Pediatric Multisystem Inflammatory Syndrome” (PMIS or PIMS), a rare but potentially serious illness resulting from delayed hyperinflammatory immune responses in previously asymptomatic children/adolescents (25, 356).
Consistent with earlier reports from Italy (57), an analysis of >1700 hospitalized patients with COVID-19 from China revealed that three quarters had at least one symptom 6 months after acute infection, with fatigue or muscle weakness and sleep difficulties being the most common, followed by anxiety/depression (163); also, lung and kidney function remained impaired in patients with more severe infections. In addition, cardiac involvement with ongoing cardiac inflammation, independent of preexisting conditions and severity of the acute illness, has been reported (332). Symptoms in U.S. outpatients with milder disease include cognitive impairment (“brain fog”) (242), preventing many from returning to work. The largest observational study to date, including almost 48,000 patients with COVID-19 in England (15), confirmed that individuals discharged from hospital have elevated rates of mortality, readmission, and multi-organ dysfunction; this greater risk was neither confined to the elderly nor uniform across ethnicities. Why this occurs so frequently with SARS-CoV-2 is unclear. With millions of infections worldwide, Long-COVID may become a substantial long-term burden, adding to the management of acute infections and losses of life/livelihoods, by impairing health-related quality of life of huge numbers of people. Similarities between Long-COVID and symptoms of individuals previously infected by Ebola or Chikungunya virus (295, 370) suggest the possible involvement of oxidative stress. We hypothesize that these conditions result from a disturbance of redox-dependent metabolic, bioenergetics, and inflammatory signaling networks governing the innate immune response and affecting RSI operation. It may be caused by cofactor loss within a central redox hub, shifts in redox equilibria to different setpoints, or by a re-routing of electron flow through alternative redox nodes (291) to maintain whole-body redox balance. A return to previous functionality may not occur spontaneously, even once inflammation resolves, and may necessitate nudging metabolic regulation in a direction that reestablishes the dynamic balance.
Viral Acquisition, ACE2, and the Renin–Angiotensin Redox Signaling Axis
The specificity of viruses for particular cell types/tissues (viral tropism) is an important determinant for infection outcome. COVID-19 interferes with redox processes throughout the disease course, starting from the moment the virus enters the cell. SARS-CoV-2 gains access to host cells via endocytosis after an interaction between its S-protein and cellular ACE2 after priming by tissue-specific transmembrane serine proteases, including TMPRSS2 (161); unlike with SARS-CoV, cell entry of SARS-CoV-2 is preactivated by the endosomal proprotein convertase FURIN, reducing its dependence on cellular proteases (379). These processes are well recognized and have been reviewed extensively (40, 393, 401). However, how SARS-CoV-2 perturbs redox networks, affects organelle/cell/tissue function and NO availability and how this may affect the RSI merits further discussion. A significant portion of the perturbations experienced is related to oxidative stress caused by the viral replication process itself and the way the virus uses the human redox system for its own purposes (see Box 2 and below). Each of these steps has the potential to affect operation of the local RSI and whole-body redox balance by altering the chemistry of the reaction cascade that connects the reactive species with the thiol targets involved in stress sensing and metabolic adaptation (Supplementary Fig. S1). The ROS production (e.g., due to ACE2 internalization, endoplasmic reticulum [ER] stress, or inflammation) leads to reduced NO availability with a switch from antioxidant to prooxidant chemistry and shifts in redox setpoints (affecting redox-sensitive transcription factor, enzyme and transporter expression/activity), albumin depletion after acute-phase reactions translating into impaired extracellular redox buffer/thiol transport capacity, and malnutrition-induced cofactor/nutrient limitations (affecting electron flow through the redox network). All these changes can occur simultaneously at multiple levels with distinct consequences in different cells/tissues/organs, dependent on their enzyme/transporter activity and receptor density required to fulfill their specific physiological role.
A pathogen that affects lung function is often assumed to be contracted mainly via the upper and lower airways, but this is not necessarily the case with SARS-CoV-2 (481). It may infect human hosts via the oral and/or intestinal mucosal epithelium after the ingestion of contaminated food (as known for influenza) or the conjunctival and nasal epithelium (168). At all sites, epithelial cells play a critical role as mediators and regulators of innate and adaptive immune responses (366). Recent investigations highlighted the significance of the oral cavity and salivary glands for transmission and disease propagation (166). Although interactions between the viral S-protein and ACE2 are important for infection of many cell types with SARS-CoV-2, several alternative receptors and enhancing mechanisms have been identified (16, 77, 259, 330, 337) but little is known of their connection to the redox system. In the human lung, the tyrosine-protein kinase receptor AXL rather than ACE2 may be the key cellular receptor for SARS-CoV-2 infection (445); its activation elicits ROS accumulation (165).
Both viral S-protein and host cellular ACE2 are densely glycosylated (11), highlighting the importance of glycan-mediated interactions. Glycobiology plays fundamental roles in health and disease, specifically immunity, protein folding, and infection with enveloped viruses (346, 388, 447). Glycan-mediated interactions between the S-protein and the epithelial/endothelial glycocalyx may facilitate viral adhesion and contribute to infection with SARS-CoV-2 (77, 330, 440). Besides its role as a receptor for SARS-CoV-2, ACE2 is important for cellular redox control. Here, oxidative stress and redox regulation meets glycobiology, an intersection defined as “glyco-redox” (201, 411). ACE2 is one of three proteases of the RAS signaling pathway, which has been highlighted as a novel therapeutic target for COVID-19 (401). Several metabolic effects of RAS and the expression/activity of ACE/ACE2 show sexual dimorphism (208, 453). Indeed, many of the distinguishing features of COVID-19 appear to be related to how SARS-CoV-2 interacts with this system and its downstream redox signaling cascades. At the same juncture, oxygen transport, hypoxic signaling, and ROS production all intersect, allowing the virus to establish itself in the host while evading multiple elements of the innate immune response that usually keep viral infections at bay.
Elements of the RAS signaling pathway appeared more than 400 million years ago (121), long before the emergence of mammals; its importance for homeostasis is evidenced by its evolutionary conservation across species. It is intimately connected to the autonomic and neuroendocrine stress response mediated via the hypothalamic-pituitary-adrenal (HPA) axis (7). The RAS comprises circulating and local tissue components of two counter-regulatory pathways that both start with the cleavage of hepatically produced angiotensinogen into angiotensin 1 (Ang-I) by renin, a protease released by renal cells in response to neural stress signaling (171, 317) (Fig. 2). From here on, the system diverges into two antagonistic arms that both intersect with the function of the RSI inasmuch as Ang-I acts as a critical redox-sensing node. We propose that the RAS and the RSI act in concert as a highly sensitive—although tissue dependent—oxygen-sensing switch, with variable sensitivity determined by prior stress exposure and a SARS-CoV-2-induced shift toward a pro-inflammatory state. Inflammation is accompanied by a shift in NO availability, oxygen consumption, and redox chemistry. Since the major oxygen-consuming cell organelles, the mitochondria, also express angiotensin receptors (110) and their oxygen consumption is under control of NO (Supplementary Data), it is not difficult to see how RAS alterations due to aging (428), inflammation/glutathione depletion (104), and/or viral infection may cause mitochondrial dysfunction.
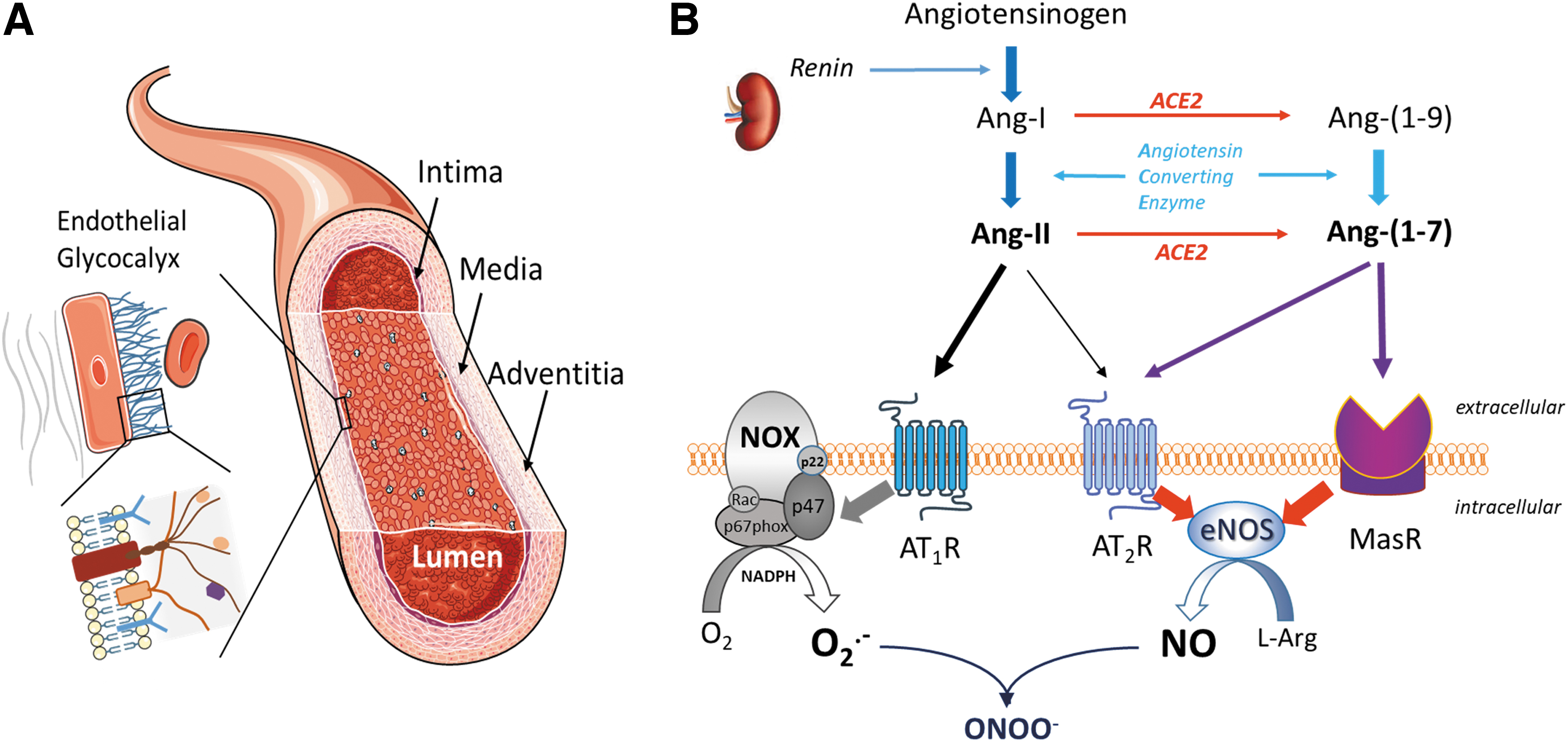
ACE (a protease homologue of ACE2) cleaves Ang-I into angiotensin 2 (Ang-II); Ang-II acts mainly through angiotensin type-1 receptor activation (118), a pro-inflammatory signaling pathway associated with ROS production, collagen deposition, vascular smooth muscle cell growth/migration, cytokine production, and inflammation (73, 92), resulting in endothelial dysfunction (244, 419). Alternatively, ACE2 converts Ang-I into angiotensin 1–9 [Ang-(1 –9)] to act on the G-protein coupled receptor Mas [interestingly, Mas was discovered as part of a system that controls the import of proteins into mitochondria (17), reinforcing the notion that infection with SARS-CoV-2 can affect mitochondrial function]; its activation triggers NO release (359) and is associated with antioxidant, anti-inflammatory, and antiproliferative effects. ACE2 also converts Ang-II into angiotensin 1–7 [Ang-(1 –7)]—closing the negative feedback loop in this signaling cascade (Fig. 2). In health, this translates into a finely tuned equilibrium of ACE/ACE2-dependent pro- and antioxidant mediators that allow swift adjustment of local redox status as needed. This equilibrium is dramatically distorted after SARS-CoV-2 infection by the virus “hijacking” ACE2 as its main binding protein to gain cell access (433), which results in receptor internalization and leaves the pro-oxidant actions of Ang-II unopposed. Unlike ACE deficiency, acquired or inherited deficiency in ACE2 activity significantly increases tissue and circulating Ang-II concentrations, and reduces Ang-(1 –7) levels; this suggests that ACE2 rather than ACE is the dominant enzyme that determines RAS balance (419). In skeletal muscle, ACE2 deficiency is associated with increased lipid accumulation secondary to ER stress and mitochondrial dysfunction (55) and Ang-II mediated shifts in energy metabolism from fatty acid oxidation toward glucose utilization (31). ACE2 deficiency is also associated with increased cardiovascular morbidity (418).
The lungs are a major source of ACE activity and Ang-II synthesis. Ang-II is initially protective in early pulmonary insults, inducing pulmonary vasoconstriction to prevent shunting, preserve gas exchange, and minimize hypoxemia (203). However, high Ang-II concentrations increase pulmonary vascular permeability, resulting in pulmonary edema, potentially worsening lung injury (171). Given that pulmonary cells also express ACE2, all the necessary components for a pulmonary oxygen-sensitive redox switch are colocated within the main organ that allows oxygen (and other gases) to enter and leave the body. ACE2, the viral entry point in both SARS and COVID-19, also seems to be the most important nodal regulator for systemic Ang-(1 –7) concentrations and thus extracellular redox status via the Ang-(1 –7)/Mas receptors (MasR) signaling axis. Extracellular redox status can regulate intracellular ROS generation (174). Blood cells use the RSI to adjust their function according to environmental cues (including shear stress and redox status). ROS are, in fact, also important intraplatelet signaling entities, with NOX2 playing a key role (436, 466); in conjunction with NO and H2S, they regulate platelet aggregation, adhesion, and platelet/leukocyte interactions (106, 335). The RAS-mediated alterations in RSI signaling may be linked to COVID-associated coagulopathy, inflammation, and immune hyperactivity by affecting the clotting process, endothelial leukocyte adhesion/rolling, macrophage polarization, and T cell and erythrocyte function.
Global loss of ACE2 precipitates severe ARDS in mouse models (171) and enhances Ang-II-induced renal oxidative stress, inflammation, and fibrosis, resulting in greater renal injury (483); the latter may explain the renal abnormalities observed in patients with COVID-19 (320). Interestingly, a common insertion polymorphism in the ACE genome has been associated with decreased mortality from ARDS—often described as the archetypal hypoxemic critical illness (256). The same polymorphism is also associated with improved performance at high altitude, where the resulting systemic hypoxemia is an inescapable physiological challenge (461). The higher population frequency of this ACE insertion polymorphism also correlates strongly with both lower SARS-CoV-2 infection and COVID-19 mortality rate (467).
ACE2 is widely expressed across human cells [including macrophages and dendritic cells; data for platelets are inconsistent; (2, 54)], but particularly prevalent on epithelial cells of the small intestine and the lungs (148); specifically, alveolar epithelial type II cells, which have important immunomodulatory functions and produce surfactant (409). Pulmonary surfactant lines the alveolar surface, reduces surface tension, increases pulmonary compliance, and modulates innate immune function through its antioxidant, anti-inflammatory, and signaling effects (115). Even though the original SARS virus and SARS-CoV-2 gain access to cells through the same receptor (213), a key difference in the severity of the resultant disease (apart from variations in binding affinity to the S-protein) may be related to the fact that only SARS-CoV-2 is able to significantly suppress endogenous surfactant production after entering alveolar epithelial cells, type 2 (AE2) cells (409). This would seem to provide a strong rationale for surfactant replacement therapy in patients with COVID-19 (105). The ACE2/MAS axis also modifies several aspects of acute and chronic inflammation (112), and NOX-mediated formation of superoxide (O2 −) and hydrogen peroxide (H2O2) as well as oxygen itself can increase ACE and RAS activity (422, 477). Increased ACE activity and angiotensin type-1 receptor activation, as in ARDS, lead to enhanced ROS production (477). Loss of protective surfactant or ACE2 activity after SARS-CoV-2 mediated inactivation may have similar effects in COVID-19.
The situation may be even more complex as in alveolar cells ACE2 is coexpressed with elements of the kinin-kallikrein and coagulation system (383). In addition to cell-bound ACE2, a soluble isoform circulates in blood that originates from (ROS-induced) shedding of the enzymatically active ectodomain of ACE2 by the metalloproteinase ADAM-17 (485), which also contributes to the shedding of inflammatory cytokines (333). ADAM17 and TMPRSS2 compete for ACE2 processing, but only cleavage by the latter promotes virus-driven cell entry (158). Collectrin, a distant member of the ACE family, is another transmembrane glycoprotein expressed in endothelial cells (ECs); despite lacking a catalytic domain (and therefore not modulating angiotensin concentrations), it plays a critical role in blood pressure homeostasis (469). It shares with ACE2 the ability to act as a chaperone for cellular
Epithelial–Endothelial Barrier Disruption and Endothelial Inflammation
The respiratory barrier is an active safeguard and regulator of defence against airborne particles, microbial pathogens, and viruses (229). Facing the airspace, alveolar epithelial cells have a glycocalyx layer rich in heparan sulfate and chondroitin sulfate that promotes alveolocapillary barrier integrity (147); site-specific sulfation of the disachharide repeats confers a negative charge imparting specific binding to positively charged protein residues and regulates their function through electrostatic interactions. Since many bacterial and viral pathogens [including SARS-CoV-2; (77)] use heparan sulfate as receptor, epithelial shedding of this layer may have been evolutionarily beneficial by disrupting the propagation of infection (147). However, it comes at a price: Glycocalyx shedding aggravates pulmonary barrier dysfunction by disrupting epithelial tight junctions in ARDS (233); because its composition/function has been characterized only recently, nothing is known about its interaction with surfactant or its role in COVID-19. Nrf2, the master regulator of cellular antioxidant gene expression, protects alveolar epithelial cells against influenza A-induced injury (209), and its activation may also reduce SARS-CoV-2 infection severity (90, 270). Indeed, Nrf2 gene expression was suppressed in biopsies of patients with COVID-19, and Nrf2 agonists can induce an interferon (IFN)-independent antiviral program that limits viral replication and suppresses pro-inflammatory responses (306). The alveolar epithelium is in close contact, via its fused basal lamina, with the pulmonary capillary endothelium to ensure efficient gas exchange. Although ECs also express ACE2 (430), levels in human cells may be insufficient to allow direct infection with SARS-CoV-2 (262). However, cultured ECs may not be an ideal model to study these processes because of in vivo/in vitro differences in glycocalyx expression (69).
The pulmonary endothelium has recently been recognized as a key orchestrator of ARDS (274), and the roles of ROS and RNS in pulmonary endothelial barrier function have been reviewed (289). Postmortem examination of lung tissue from patients with ARDS due to either influenza A (H1N1) or SARS-CoV-2 infection revealed diffuse alveolar damage and perivascular T cell infiltration in both cases. However, histological examination of pulmonary vessels demonstrated widespread microangiopathy and thrombosis (considerably higher in patients with COVID-19) frequently accompanied by signs of angiogenesis (4).
Intracellular viral defences are largely dependent on IFN activation; IFN-β is usually one of the first cytokines produced by the lung in response to viral invasion (449). Its release is stimulated by eNOS-mediated NO production (144). Viral antagonism of cellular IFN production via blockade of nuclear import has already been observed with SARS-CoV (389) but appears to be more efficient with SARS-CoV-2 (475). Low IFN1 and IFN3 in combination with exuberant inflammatory cytokine production (“cytokine storm”) form part of the immune-evasion strategy of SARS-CoV-2 and is observed in COVID-19 (35, 408). Endothelial dysfunction is characterized by compromised eNOS activity, resulting in impaired vascular integrity and coagulopathy (236, 415) (besides being a potent vasodilator, NO also has antioxidant, anti-inflammatory, and antiviral activity; see Supplementary Data). The mechanisms involved in SARS-CoV-2 induced thromboembolic complications involve interactions between inflammation and hypoxia, with crosstalk between the RAS, kinin-kallikrein, coagulation, and complement systems, in which the endothelium plays a central role (219). More recently, H2S bioavailability has been implicated in endothelial dysfunction (443). Lower serum H2S concentrations have been measured in non-survivors compared with survivors of COVID-19 (348), and H2S donors have been proposed as potential treatment in COVID-19 (76). H2S also has anti-inflammatory effects that seem to be mediated, in part, via an upregulation of endothelial ACE2 expression (238).
The Endothelium as a Nexus of Whole-Body Redox Regulation
The vascular endothelium is the body's largest organ (by surface area and weight), providing an important interface between blood and tissues. It is crucial to the function of every organ, metabolically highly active (417), and important for homeostasis by regulating barrier function, blood fluidity, and tissue/organ supply with essential nutrients, including fatty acids (268). Together with virus-induced oxidative stress (Box 2), COVID-19-induced endothelial dysfunction may affect lipid synthesis/handling (368), the ability of the immune system to distinguish self from nonself, and contribute to the formation of prothrombotic anti-phospholipid autoantibodies (488) and blood clots reminiscent of the “antiphospholipid syndrome” (99). Moreover, the endothelium controls angiogenesis and blood cell trafficking through the vascular wall (9). Despite adequate oxygen availability, its bioenergetic demands are met mostly through glycolysis (334). Endothelial release of short-lived mediators, including NO, H2S, and prostacyclin, regulates vascular function and platelet activation while orchestrating host defence mechanisms via immune cell modulation. Regulation of barrier function after inflammatory cell recruitment by the lymphatic endothelium not only affects fluid drainage but also changes immune responses by removal of inflammatory mediators from tissues (5). In the pulmonary microcirculation, angiopoietins have been recognized as important regulators of vascular barrier integrity. Activation of their receptor, Tie2 by angiopoietin-1 reduces whereas Tie2 blockade by angiopoietin-2 increases inflammation and permeability in sepsis, with prognostic and pathogenic roles in pneumonia (146). Tie2 is coupled to eNOS to regulate vascular reactivity and protect ECs from damage (43), and abrogation of Tie2 signaling results in disseminated intravascular coagulation (387).
The endothelium is the “gatekeeper of vascular health” (50). To fulfil this role, mechanosensors translate blood flow-mediated shear stress into variations in cellular NO production and Nrf2 activity (70, 265). The NO/Nrf2 relationship is bidirectional: NO stimulates Nrf2 translocation in the endothelium (44), possibly via S-nitrosation of Keap-1 (232); Nrf2 upregulation also prevents eNOS uncoupling (153), but laminar shear stress also upregulates inducible nitric oxide synthase (iNOS) in the endothelium via NF-κB signaling (312). Together, shear stress and the RAS play important roles in endothelial homeostasis. Shear stress counteracts the inflammatory EC phenotype also via suppression of the TAK1 pathway while restoring endothelial redox signaling via ERK5 activation (228). Pulsatile shear stress reduces the expression and activity of ACE but it upregulates ACE2 expression to maintain normal EC function (392). These effects are achieved by a cooperation between extracellular matrix components on either side of the EC (48).
The luminally facing endothelial glycocalyx (Fig. 2A) is an important modulator of EC function (93, 282). This negatively charged hair/gel-like surface layer comprises glycoproteins (syndecan, glycipan, CD44) with glucosaminoglycan side-chains (heparan sulfate, chrondroitin sulfate), long-chain hyaluronic acid, and glycolipids; it regulates fluid homeostasis, albumin transcytosis (218), and permeability (455), and it minimizes adhesion and emigration of blood cells. Glycocalyx sialic acid residues regulate EC redox state via modulation of shear stress-induced Nrf2-mediated signaling (331). This is of relevance considering that sialic acids are emerging as important players in the pathophysiology of SARS-CoV-2 (330), their incorporation into cell membranes is influenced by the diet (see “The Importance of Nutrition for Stress Resilience” section) and linked to complex immune responses (318). The function of this fragile structure is compromised in inflammation or hyperglycemia-induced oxidative stress in patients with diabetes and sepsis, largely via ROS-mediated activation of matrix metalloproteinases (10) and consecutive cleavage (shedding) of membrane-anchoring proteins, including syndecans. It is also impaired in patients who are critically ill with COVID-19 (395). It may take days to rebuild after damage; however, glycocalyx protection/restoration can be modulated by the administration of heparin [stimulating synthesis and sulfation of heparan sulfate proteoglycans in an eNOS/NO-dependent manner; (267)], N-acetylcysteine (300), and glucocorticoids (68, 131), providing alternative explanations for the effectiveness of these drugs in COVID-19 (see “Current Challenges and Treatment of COVID-19” section). On the abluminal side of the vasculature, ECs are attached to a differentially glycosylated extracellular matrix that separates them from the vascular smooth muscle. The adventitia surrounding the blood vessel acts as another important layer for retrieval, integration, storage, and release of key regulators of vascular function (396). Thus, it appears that the endothelium is a critical nexus for bidirectional control of redox regulation, from “inside-out” to “outside-in.”
It has been argued that the later stages of COVID-19 are a vascular and essentially an endothelial disease (236, 384), suggesting that ECs represent the cornerstone of organ dysfunction, vascular leakage, and initiation of coagulation in SARS-CoV-2 infection (136, 325, 361, 415). The EC metabolism is modulated by circulating cytokines to a pro-inflammatory defence phenotype, which triggers a switch in cell signaling that is characterized by enhanced ROS production with consecutive changes in NO metabolism (Box 3), endothelial dysfunction, enhanced expression of cell adhesion molecules, stimulation of coagulation, and altered barrier function. In addition, inflammation promotes a reallocation of arginine availability within cells, lowering NO output [(288); see Nutrition section]. Although compensatory increases in NO formation under these conditions are typically attributed to iNOS induction, a prolonged high-output production from constitutive eNOS has been demonstrated in endothelial inflammation (245). Endotheliitis is not unique to COVID-19, but also a hallmark of cytomegalovirus-associated atherosclerosis (231), acute allograft rejection (290, 353), and chronic viral hepatitis (471); it can even be induced by certain drugs. Persistent endothelial inflammation may push cell signaling beyond a stage that is not easily rescued. Fittingly, a recent review explores the role of EC resilience in cardiovascular health and disease (132).
Both vascular and endothelial barrier function are under tight redox control (129, 427), and ECs can rapidly adjust their local redox environment by adjusting NO fluxes. However, little is known about the endothelium's role in regulating whole-body redox status. What is known, however, is that endothelial redox imbalance affects not only endothelial junctional integrity (482) but also trans-plasma membrane electron transport in ECs (272). We propose that regulation of extracellular redox status is another key feature of the endothelium that is achieved by integrating neuronal and endocrine signals, highlighting the importance of the extracellular compartment as a conveyor belt-like conduit for interorgan communication. Any change in endothelial redox control might thereby have an immediate effect on all circulating immune-competent cells (as their activity is dependent on the redox status of lymph/plasma) and platelets. If substantiated, this would confirm and extend the critical importance of ECs in redox-related disease processes and might be of critical relevance for SARS-CoV-2 infectivity, which is suspected to be modulated by host redox status [(390); see also Box 2].
The ECs of different vascular beds show phenotypic heterogeneity that has given rise to the description of systems-level features of endothelial regulation (344). Given its significance for gas exchange in lung alveoli, this heterogeneity has recently attracted attention in connection with tissue regeneration after influenza-induced acute lung injury (299). Intriguingly, the endothelium appears to display “swarm intelligence” (the organized behavior of a large community without the need for a coordinating global organizer) in that it can simultaneously detect multiple noisy, mixed low- and high-intensity signals and elicit coordinated actions in response (261). Such integrative ability in response to dynamically changing inputs is essential to the successful operation of complex network activities (281). Disruption of this function by changes in the “Redox Landscape” (Box 3) may trigger the transition from mild to severe phenotype in COVID-19. By using the endothelium as its main dissemination conduit, evolution appears to have found a way for SARS-CoV-2 to spread very efficiently—after an initial insult in the upper/lower airways, a rapid breach of epithelial/endothelial barrier function offers rapid dissemination along the entire vascular tree with concomitant redox activation, facilitating viral propagation accompanied by systemic spread of oxidative damage and inflammation. Viral spread may be complemented by alveolar macrophages (usually a frontline defence against respiratory pathogens and important to the initiation and resolution of inflammation) acting as “Trojan horses” on migration to other tissues (2). Since endothelial dysfunction has also been hypothesized to play a role in physical frailty (12), perpetuating elements of virus-induced endotheliitis and systemic inflammatory disturbance, this may also explain why some patients go on to develop muscle weakness (see “Peculiar Persistence of Symptoms” section).
The Advanced Glycation Endproduct/Receptor for Advanced Glycation Endproduct Axis and Other “Sweet” Connections Between Redox and Stress Signaling
Another area where glycan structure and function are modulated by changes in redox status has been linked to SARS-CoV-2 infection (397) and is related to “advanced glycation endproducts” (AGEs), the formation of which is enhanced in inflammation. The AGEs are products of a nonenzymatic reaction of reducing sugars with protein amine residues (specifically, lysine and arginine). In diabetes, AGE formation is increased due to higher glucose levels, triggering accelerated atherosclerosis with enhanced expression of leukocyte adhesion molecules, procoagulant activity, and vascular permeability (23). However, AGEs can also form in the absence of hyperglycemia (439), particularly with advanced age. The major protein adduct excreted by macrophages, AGE-albumin (49), circulates in blood, can accumulate in the extracellular matrix of tissues, and contributes to chronic disease (196). Their interaction with the cell membrane receptor for AGEs (RAGE) may be another pathway through which oxidative stress is amplified in COVID-19. RAGE is a pattern recognition receptor of the immunoglobulin superfamily, which regulates energy expenditure in response to environmental and metabolic stress (169). Its activation also results in mitochondrial dysfunction (446). RAGE is highly expressed in the lung (45), where it mediates pulmonary inflammatory responses (303). In preclinical models of viral pneumonia, RAGE activation is detrimental (486), causing cell apoptosis and further aggravation of bronchial epithelial and vascular endothelial inflammation via NF-κB signaling.
Glycation reactions are modulated by the local redox environment, enhanced by oxidation (glycoxidation), and can promote further oxidative stress by triggering the formation of stress-induced transcription factors and pro-inflammatory mediators, including acute-phase proteins and cytokines (426). In addition to their endogenous formation, AGEs are also derived from foods. The AGEs together with other toxins such as “advanced lipid oxidation end products” (ALEs) perpetuate the inflammatory response. The ALEs are adducts of reactive aldehydes (derived from the peroxidation of unsaturated fatty acids) with biological substrates and are considered a significant risk factor to ill health (195). These products also affect redox regulation by modulating Nrf2, peroxisome proliferator-activated receptor gamma (PPARγ), activator protein 1 (AP1), and NF-κB activities and-by uncoupling oxidative phosphorylation-result in mitochondrial dysfunction with increased ROS production (100). The ratio of AGE to the soluble form of RAGE is a predictor of endothelial function (192).
Hyperglycemia is common in critical illness and often linked to poor outcome. Assuming causality, this has led to widespread adoption of tight glucose control, despite evidence that this can also worsen outcome (297). In fact, both hypo- and hyperglycemia at admission are associated with higher mortality in patients with COVID-19 (206), independent of diabetic status (59). The finding that insulin treatment can increase mortality in patients with COVID-19 and type 2 diabetes mellitus (T2DM) (473) suggests that in severe stress hyperglycemia can be protective. These seemingly counterintuitive findings may be reconciled by considering stress hyperglycemia to be an essential survival response (255). In acute illness, stress-induced hyperglycemia may be an evolutionarily preserved adaptive response that increases the host's chances of survival by serving as fuel (for e.g., brain and macrophages) and/or by providing extra reducing power in the form of NADPH (via the pentose phosphate pathway), and interference with these functions causes harm (of note, the activities of both NOS and NOX enzymes, and thus regulated NO and superoxide output, are dependent on NADPH availability). However, hyperglycemia also triggers mitochondrial ROS production, promoting glycolysis through HIF-1α stabilization and amplifying monocytic cytokine production—all changes facilitating SARS-CoV-2 replication (79, 315). COVID-19 can increase the risk of developing ketosis and ketoacidosis, further worsening oxidative stress (234). However, ketone bodies such as β-hydrobutyrate also have antioxidant, anti-inflammatory, and immunomodulatory effects mediated via Nrf2 stimulation and NF-κB/inflammasome inhibition, and ketogenic diets may ameliorate COVID-19 via these mechanisms (315). Several ketone bodies are readily transported in and out of cells, contributing to redox-mediated metabolic regulation (82) beyond the AGE/RAGE axis.
Endothelial Dysfunction: From Oxidative Stress to a Full-Blown Redox Tsunami
Endothelial NO production and function decline with age (225). Endothelial dysfunction is a hallmark of COVID-19 and is associated with impaired NO availability secondary to enhanced oxidative stress, an established risk factor for mortality in chronic, noncommunicable diseases (117). Many different processes contribute to promoting oxidative stress in COVID-19, but—importantly—the changes in oxidative poise provoked by infection with SARS-CoV-2 happen rapidly and can be of considerable magnitude; this leaves complex homeostatic systems (with interdependency for the same substrates) little opportunity to readjust. Greater viral loads elicit more pronounced oxidative stress more quickly, translating into greater vulnerability to perturbation of the RSI with a higher risk of failure.
The effects of oxidative stress on complex redox systems are not as simple as a yin-yang balance. Oxidative stress triggers a change in Redox Landscape (Box 3) rather than a linear shift of associations. The redox network itself as well as the shape and precision of redox communication change altogether. Thus, complex signaling interrelationships become challenged such that the shared redox language becomes “non-sensical,” with messages from remote organ systems becoming “difficult to interpret.” This would lead to the initiation of a positive feedback cycle, resulting in an “Oxidative Storm” rather than a “Cytokine Storm,” with eventual failure in redox communication—an oxidative stress-triggered “Redox Tsunami” at which stage the system would become unstable and collapse (Fig. 3).
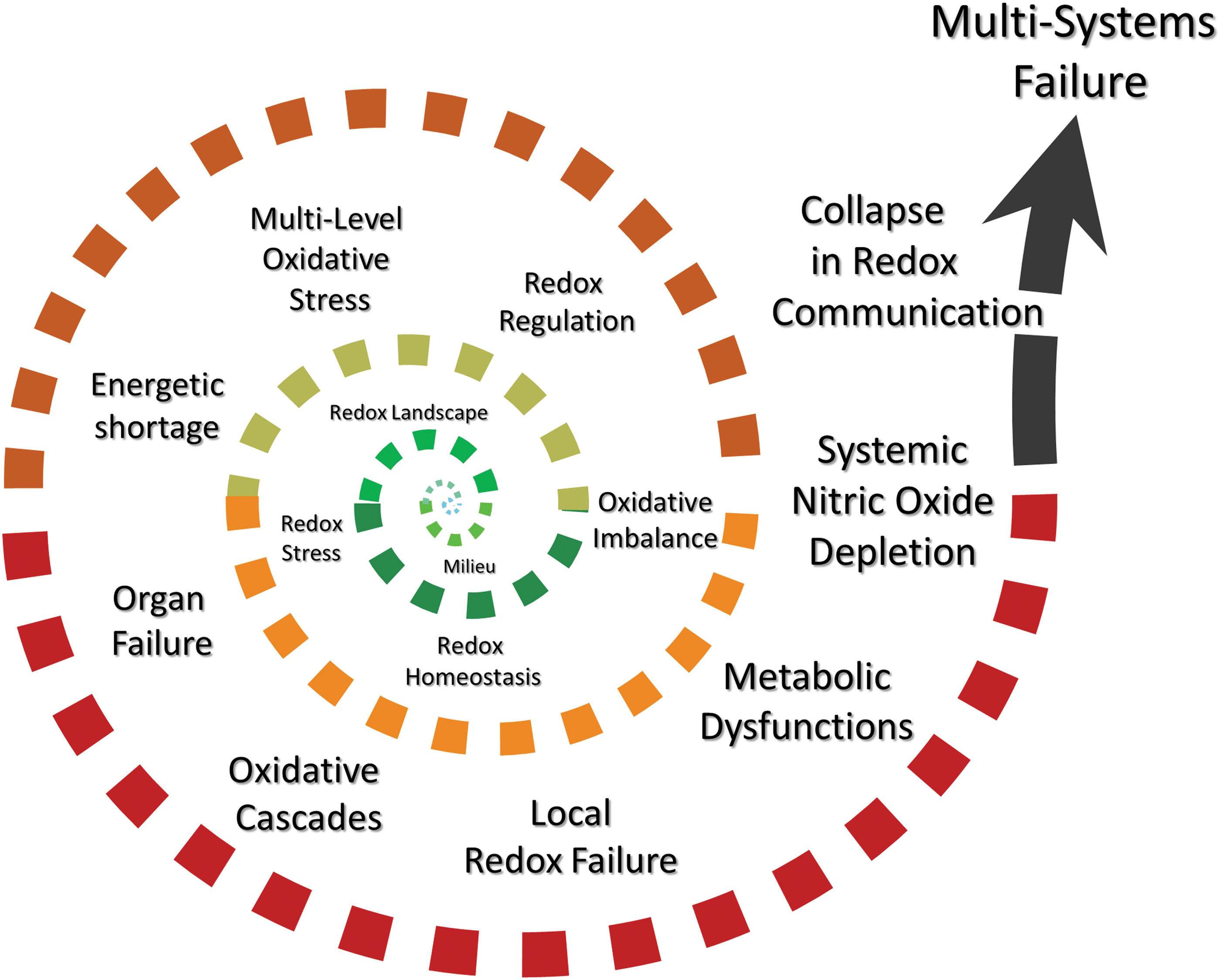
Complex redox systems comprise chains of coupled nonlinear reactions. As little as three components, that is, NO, O2, and either a thiol (glutathione) or an amine (tyrosine), can form networks of surprising complexity that give rise to a dazzling array of reaction products (222); reactions of H2S/polysulfides with biologically relevant oxidants are of similar complexity (26). Even the seemingly simple reaction between NO and superoxide to form peroxynitrite (ONOO−) (314) (Fig. 2) levels of which are controlled by mitochondrial thioredoxin reductase-2 (194) shows a peculiar bell-shaped characteristic for oxidation and nitration (189). Similarly, NO and H2S—in the presence of O2—react to form a cascade of reaction products, including nitroxyl, S/N-hybrid species such as nitrosopersulfide (SSNO−), polysulfides and sulfoxides such as sulfite, thiosulfate, and sulfate (83, 85), explaining the observed cooperative interactions between NO and H2S in multiple organ systems (198). One product of these interactions, SSNO−, shows remarkable resistance to reduction (38) and targets Keap-1/Nrf2 (86); others such as polysulfides have powerful antioxidant/anti-inflammatory properties that interfere with Toll-like receptor (TLR) signaling (479, 480). In such complex systems, product composition is highly dependent on temperature, pH, and flux rate of each reaction partner. The same compounds are integral constituents of the RSI, important for stress signaling and adaptation of cellular metabolism and mitochondrial function (Supplementary Fig. S1). The principles of self-organized criticality not only explain the overall robustness of redox networks, but they also offer insight into their vulnerability to failure (281). Networks of networks, with distinct interdependencies, are inherently vulnerable to abrupt collapse due to the “global rule of activation” (280). For redox biology, this translates into an understanding whereby damage to a single critical cofactor [e.g., tetrahydrobiopterin for NO production by NO-synthases; (27)] may lead to the collapse of an entire system, as following an acute-on-chronic burst of oxidative stress protection against over-oxidation depends on the integrity of functions such as the sulfur-based relays of the RSI.
Redox Regulation at Local and Whole-Body Level: Possible Role of NO in Protecting Sulfur-Based Stress Signaling
For electron exchange reactions to take place redox couples do not necessarily need to be in close proximity, but the organism as a whole must maintain electron/redox balance. The human body is an elaborate multiplexed “network of networks” (347), a “superorganism” not only comprising an astonishing variety of highly differentiated cells/organs with different metabolic needs (and specific nutritional requirements for optimal function) but also harboring a highly diverse microbial flora in the intestine, on oral/gut mucosal, lung epithelial, and skin surfaces, all of which contribute to homeostasis by interacting with metabolism and redox regulation. Reductionist science has unraveled much of the diversity in redox-regulatory systems, provided a detailed inventory of key contributors to cellular redox status, and identified several important redox hubs (e.g., glutathione, pentose phosphate pathway, HIF-1, NF-κB, Nrf2, thioredoxin). However, little is known about how these redox modules integrate at the whole-body level. Besides the vulnerability of complex systems in general, redox-regulatory systems have their own unique vulnerabilities (Box 4). NO plays a central role as a local signaling agent and cytotoxic entity (for an overview of its many functions, see Supplementary Data); however, its significance may be far more fundamental than currently appreciated, considering its early role in evolution (113). In addition to small-molecule antioxidants (such as water-soluble ferrous ions, ascorbate, urate and glutathione, as well as lipophilic ubiquinone and α-tocopherol), classical antioxidant enzymes (subject to regulation by Nrf2) are believed to provide the bulk of protection of sulfur-based redox signaling governing intermediary metabolism, mitochondrial function, and sensing/adaptation to stress against ROS-mediated oxidative damage. Before the advent of aerobic respiration, however, ROS-processing enzymes such as catalase and SOD may have fulfilled different roles and oxidized H2S into polysulfides (307), which are powerful antioxidants in their own right (125). At that time, NO may not have fulfilled any of its contemporary physiological roles either but rather acted as an antioxidant (113). This primordial function of NO may still operate in contemporary organisms, but it does not become apparent unless Nrf2 is abrogated (109) or the “more sophisticated” antioxidant systems fail. Organ failure secondary to a cytokine-driven “oxidative storm” (Fig. 3) may be protected by maintaining the integrity of endogenous NO formation. Tonic NO release may provide a protective shield against over-oxidation of thiols, the nanotransducers of cellular redox signaling (81), by acting as a guardian of sulfur-based stress-signaling circuits (Fig. 4B). In viral infection, reduced NO availability may be an early indicator of greater vulnerability to redox dysfunction and pharmacologically targetable (e.g., by stimulation of endothelial production, exogenous NO-donors, or NO inhalation).
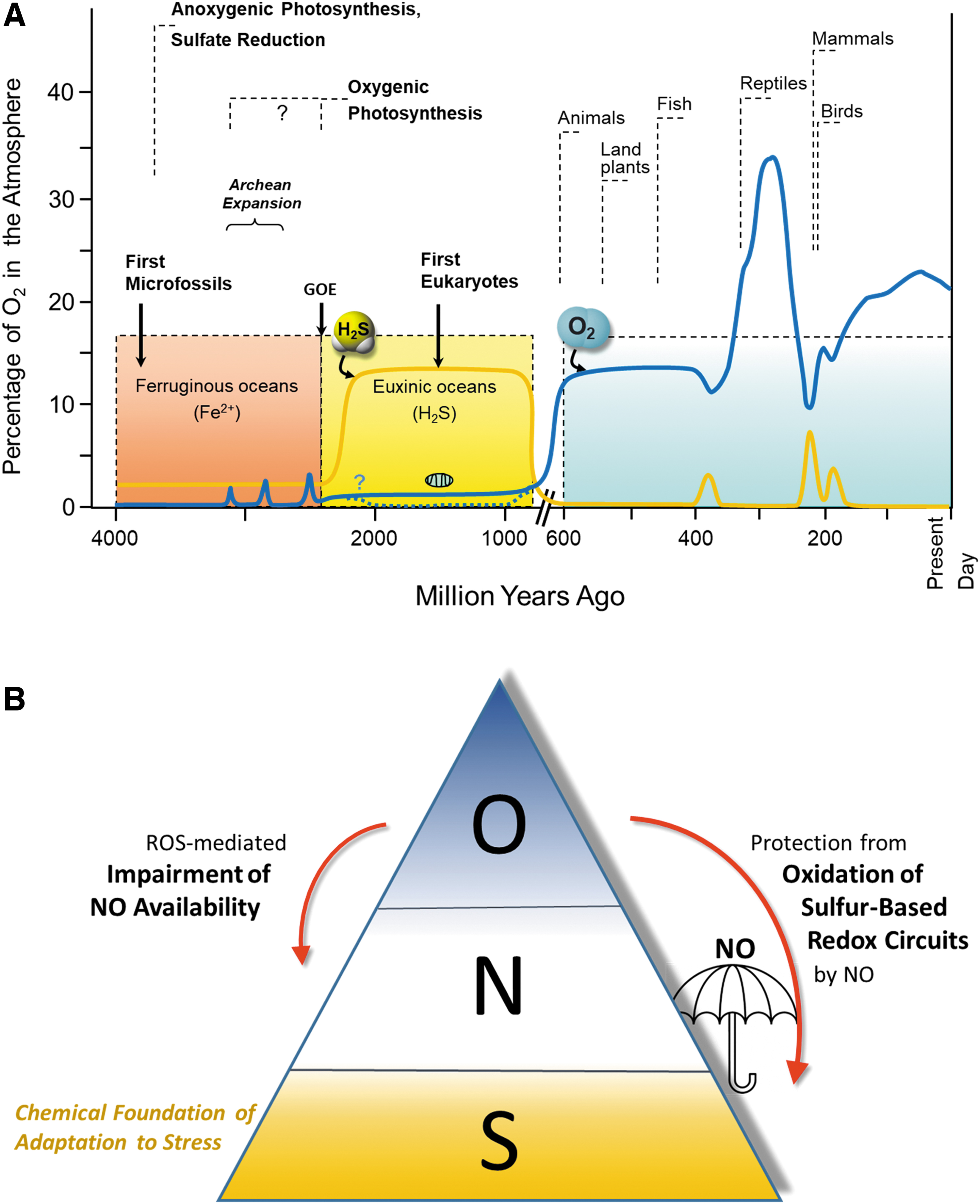
Current Challenges and Treatment of COVID-19
Despite how much has been learned about COVID-19 since its emergence and the tremendous success with developing vaccines in record time, no specific drug treatments exist; availability and risk profiles of the different vaccines are uncertain; and logistics for a global vaccination effort remain challenging (357). Besides extremely rare complications, vaccine efficacy is an additional concern considering the pace at which “escape variants” carrying mutations at multiple sites are emerging in different parts of the world. Those mutations also include SARS-CoV-2's S-protein, leading to variants with an enhanced binding affinity for ACE2 that can translate into greater transmissibility, viral load, and deadliness (66, 71, 122, 340). Inevitably, variants of this kind quickly come to dominate infection dynamics and magnitude. A higher affinity of the viral receptor binding domain for ACE2 may translate into a greater perturbation of local and whole-body redox regulation through the sort of mechanisms discussed earlier. Whether RNA-based vaccines aimed at boosting antibodies against the viral S-protein also produce oxidative stress, although only transiently, by triggering ER stress (Box 2) and ACE2 internalization is unknown. Encouragingly, mounting evidence suggests that immunization against SARS-CoV-2 not only protects vaccinated individuals from developing severe disease but also reduces the risk of onward virus transmission. Nevertheless, vaccination can only be part of the exit strategy. New pharmacological approaches are required, and the search for new drug targets and drug repurposing efforts are ongoing (47, 95, 102, 139, 381).
Numerous investigational drugs have been tested in small/medium-sized trials, but interpretation of study results is often limited due to study size or design. Several large randomized, controlled Phase-II clinical trial platforms launched to fast-track research into potential COVID-19 treatments, including the UK-based ACCORD, RECOVERY, and the adaptive REMAP/CAP, have been particularly effective in quickly delivering meaningful results (51, 458), whereas the international nature of SOLIDARITY and DISCOVERY proved more challenging to swift patient enrolment. Despite these successes, no therapeutic approach has yet emerged that successfully targets SAR-CoV-2 itself; so far, only drugs that modulate the immune or inflammatory host response have demonstrated convincing benefit. Preliminary results also suggest that patients who are critically ill with COVID-19 benefit from tocilizumab (IL-6 receptor antagonist and immunosuppressant) therapy (457), whereas those with milder disease and less inflammation do not (400). Dexamethasone successfully reduced 28-day mortality, but only in patients requiring supplemental oxygen therapy (342); hydrocortisone and inhaled budesonide also appear to provide protection (8, 13, 339). Administration of high concentrations of supplemental oxygen (± ventilatory support) has become the mainstay of treatment in patients requiring hospital admission. Such therapies may fuel oxidative stress-mediated tissue damage and modulate immune responses (149, 250); in these cases, the benefit of steroids may be due to downregulation of oxidative stress and lesser RSI perturbation. The mechanism(s) through which protection is provided remain unclear. Glucocorticoids can inhibit cytokine-induced iNOS expression (336, 406), which can enhance rather than ameliorate stress hormone-induced oxidation/nitrosation and DNA damage (116). Dexamethasone induces oxidative stress by suppressing constitutive eNOS expression (363). At the same time, it contributes to redox homeostasis by promoting antioxidative defenses via Nrf2, upregulating G6PD and thus glutathione and NADPH synthesis; this effect is mediated by promoting a shift from the reduced to the oxidized form of KEAP1 (33, 34). It also alters gene expression of mitochondrially encoded respiratory chain complexes and antioxidant enzymes (294) and facilitates cellular ascorbate uptake (124). Importantly, dexamethasone suppresses T cell function by enhancing NOX2-mediated ROS production by anti-inflammatory macrophages (210). Clearly, dexamethasone's profile of action is complex and involves multiple redox elements.
In addition to downregulating inflammation, immune hyperactivity, and oxidative stress, the beneficial effects of tocilizumab and hydrocortisone in COVID-19 may also involve preservation of barrier function by protecting the endothelial glycocalyx (68, 170). Curiously, also H2 inhalation attenuates endothelial glycocalyx damage in preclinical models (410). In COVID-19, heparin is often administered for its antithrombotic and anti-inflammatory effects (239); in vitro, it has also been shown to reduce glycocalyx shedding by interacting with heparan sulfate (328). Thanks to the possibility of noninvasively monitoring the sublingual endothelial glycocalyx and to quantify products of glycocalyx shedding (such as syndecan-1 and hyaluronic acid) in blood treatment strategies to preserve the endothelial glycocalyx may become an important therapeutic target and protect endothelial function/extracellular redox balance in COVID-19 (305, 405, 460).
However, not all anti-inflammatory approaches have been successful. As a pulmonary vasodilator that improves arterial oxygenation in high-altitude pulmonary edema (365) while also exerting anti-inflammatory effects (223), inhaled NO (iNO) has been repeatedly trialed in classical ARDS, but with limited success—despite transiently improving oxygenation, no reduction in overall mortality has been demonstrated (6, 355). However, because NO also displays anti-viral activity against SARS-CoV and influenza viruses in vitro (199, 345), iNO emerged as a potential therapy in COVID-19, particularly for critically ill patients with lung injury and refractory hypoxemia, with mixed success (152, 243, 413). Short bursts of high concentrations are believed to be particularly efficacious (151). However, NO affects many different targets (Supplementary Data), is rapidly inactivated by superoxide, and converted to peroxynitrite under oxidative stress conditions (Fig. 2B), particularly in the lungs of older people where SOD3 is strongly downregulated (3). NO may, therefore, need to be administered together with SOD or an SOD-mimetic. Whether much lower doses of NO early in the infection could preserve endothelial function and prevent catastrophic collapse of redox signaling (by preventing chronic oxidative stress turning into an “oxidative storm”) has not been tested. It seems likely that any success of redox-based therapies would depend on the right drug being delivered to the correct target site, at the right time in the most appropriate patient group (271). There may be merit in shifting whole-body redox status to a more reduced state in attempts to inhibit viral replication efficiency and prevent ROS-mediated tissue damage, considering that viral infection promotes oxidative stress and SARS-CoV-2 thrives in oxidative conditions. However, as the complexity of signaling systems affected by COVID-19 increases, mere administration of high doses of a single antioxidant (e.g., ascorbate, N-acetylcysteine, epigallocatechin gallate) (41, 58, 75, 94, 156) to an already stressed system may only further perturb redox signaling rather than protect from insult. Similarly, certain drug combinations may work in some patients, but not in others. In this context, patient stratification according to their stress phenotype would be an important consideration.
Redox Phenotyping for Risk Stratification and Treatment of COVID-19
A prerequisite for the development of an effective therapeutic strategy for any redox disease is an understanding of whole-body redox regulation and its implications for the individual in relation to the disease. This requires tools with which the whole-body system can be interrogated. Biochemical readouts of global RSI status would be needed to guide therapeutic efforts. In this context, plasma/serum concentrations of total free thiols, a simple oxidative stress readout that reflects the concentration of the single free cysteine thiol of albumin (20, 193), low-molecular-weight thiols (137), or the ratio of reduced over oxidized cysteine/glutathione (316), all seem pertinent points of departure as integrative markers of extracellular redox status. In severe COVID-19, low albumin levels (237) are often associated with enhanced mortality (435); low plasma/serum free thiol concentrations may reflect stress on the “redox communication highway” that enables synchronization throughout the body (84) (Supplementary Fig. S1). Redox status shows sexual dimorphism and becomes more oxidized with age (138, 323), at the expense of reduced antioxidant capacity. Moreover, each patient's composite redox status is different, depending on genetic phenotype, gut and skin microbiota composition, and exposome history. Therefore, an individualized approach (personalized redox phenotyping/metabolomics) (84, 91) is required to select the most appropriate intervention. Using multiple redox-related readouts, together with deep metabolomic/lipidomic phenotyping, in a small (COVID-unrelated) cohort of critically ill patients revealed discrete redox profiles between survivors and non-survivors (264). The pattern of changes in erythrocytes and plasma of the same patients revealed considerable heterogeneity, with opposing alterations in redox status in different compartments for some readouts, indicative of the complexity of regulation to achieve whole-body redox balance. At its simplest, we consider that a “traffic light” system with three levels of “redox stresses” would be useful for clinical triaging in the first instance (Box 5).
Box 5. Redox Status Triage System
 Future approaches may use a simple traffic light system such as the one suggested below to select an intervention that is most appropriate to the whole-body redox status of the patient.
Future approaches may use a simple traffic light system such as the one suggested below to select an intervention that is most appropriate to the whole-body redox status of the patient.
Green
Small-amplitude oxidative stress, not challenging physiological redox regulation. The Redox Landscape remains stable and synchronized, redox-based functions are preserved, and defence systems are operational. It might be worth considering use of redox-active interventions such as antioxidants and/or cofactors such as tetrahydrobiopterin.
Amber
Imbalanced redox state due to marked oxidative stress; vulnerable system no longer operating within normal homeostatic ranges. Any kind of intervention may provoke further derangement. It might be advisable to decrease the intensity of redox fluxes by, for example, lowering administered oxygen concentrations or using nitric oxide synthase (NOS) inhibitors to limit peroxynitrite formation while concomitantly providing supplementary nitric oxide (NO) by either inhalation of NO or infusion of an NO-donor. In this case, NO would not be applied to improve ventilation/perfusion matching but as a disease-modifying intervention.
Red
Major failure of the bodily redox system, with local redox processes deranged, dysfunctional communication between organ systems, and little opportunity to regain control. Aggressive pharmacological treatment must be used with caution, and the best option may be to support organ systems under immediate challenge in the least perturbing way to allow the endogenous redox network time to regain an appropriate equilibrium.
To disentangle the complexity of the many parallel redox reactions and the challenges of maintaining whole-body balance in the face of new stressors, as in COVID-19, there would be value in gaining further insight into the sequence of events by mapping redox changes in time, together with nutritional status and markers of inflammatory and organ/tissue damage throughout the disease process. A powerful approach (253), developed by Schneider and coworkers (367, 423), traces individual “health curves” to describe “disease space” and “disease tolerance” by characterizing individual host immune responses from the start of infection to remission (Fig. 5). Much of the related clinical information is routinely gathered and could readily be enriched by measurements of biochemical readouts of RSI/Redox Interactome status. The feasibility of such personalized “Redox Phenotyping” has recently been demonstrated in healthy individuals subjected to graded physical exercise in the absence or presence of hypoxic stress (91). Such analyses could be readily enhanced by also mapping cytokine profiles and other immune-related readouts in patients with COVID-19.
What Determines Disease Resilience?
Typically, children have milder disease than older individuals, but why do some people die from COVID-19 whereas others remain asymptomatic? We tell the old and vulnerable to shield themselves yet hear stories of the successful recovery of centenarians and the tragic deaths of young and previously healthy people—the question of what determines individual resilience against viral infection (and other stressors) has never been more relevant. The way redox chemistry changes with aging may provide important clues and inform future treatment regimens. Together with a greater endothelial NO production, providing a larger capacity to deal with oxidative perturbations, and greater efficiency in achieving redox balance may protect the young from severe COVID-19 complications and perhaps even explain the global disparity in health outcomes across ethnicities (144, 197). In the context of age/comorbidity-associated chronic inflammatory processes, the body's stress defence systems might reach their critical limits earlier and fail when a certain viral load is reached or excessive oxidative stress (e.g., upon hyper-oxygenation) is experienced. Metabolic and mitochondrial regulation, RAS, endothelial mediators, and the microbiome all contribute to regulation, with the RSI and the “Redox Spine” synchronizing electron exchange processes across all organs. At this juncture, adequate nutrition becomes central to resilience by providing buffering capability and an adequate reserve of the necessary building blocks and cofactors to secure metabolic flexibility and maintain the RSI/Redox Interactome functional at cellular and whole-body levels. A holistic systems approach (applying integrated omics tools in combination with advanced data processing) may be required to understand the interplay between nutrition and oxidative stress and to fully disentangle the operational principles of physiological redox networks (98).
The Importance of Nutrition for Stress Resilience
Food and nutrition matter (186, 327), yet much of the chemical composition of our diet remains unmapped (21). Malnutrition may account for a considerable portion of the socioeconomic and geographical inequality observed during this pandemic (491). A diet rich in fruit and vegetables has long been known to be beneficial for pulmonary health (32), but the multisystems nature of COVID-19 requires wider consideration. Here, we provide a brief overview of the fundamental principles that enable the metabolic flexibility required to cope with stress.
Effects of stress on nutrient interchange
A fundamental feature of life is that of structured and organized processes in which every cell has a persistent demand for energy to ensure its structural integrity and enable a wide range of functions (360). This, together with a mix of nutrients, supports a pattern of dynamic biochemical interchange that secures viability in an ever-changing environment. In humans, energy is derived from food predominantly as a mix of macronutrients, carbohydrate, lipid, and protein; these are oxidized in a similar proportion to that consumed to ensure maintenance of whole-body balance, body composition, and functional capability (179). Alteration of metabolic priorities during infection (and other stresses) changes patterns of demand for energy and nutrients. The interdependencies of macronutrients are complex because, in addition to serving as energy sources, dietary protein acts as a source of amino acids and other nitrogen-containing compounds, carbohydrates as a resource for complex alterations in glycobiology, and lipids for membrane structure and function, together with other molecular roles. Each class has wide metabolic engagement as precursors for messengers, signals, and refined structural entities. Each of the associated metabolic processes has its own energy-requiring dynamic to maintain homeostasis and thereby achieve a stable “milieu interieur” as components of a complex interacting system (179). The reciprocal relationship between immune function and nutrition is captured by the emergent concepts of nutritional immunology and immunometabolism (260, 326). Immunometabolism is under redox control (291) and modulated by the extracellular redox environment. The latter affects the redox status of cell-surface proteins, including transporters, receptors, and enzymes, and consequently nutrient absorption and secretion (286), which explains why SARS-CoV-2-induced oxidative stress (Box 2) has major effects not only on immune reactivity but also on nutrient processing (Fig. 6).
The pattern of energy and macronutrients consumed seldom matches the pattern needed to meet the demand closely, and sophisticated metabolic interconversion pathways operate to achieve the balanced mix required for function on an hour-to-hour and day-to-day basis. There is also a critical contribution from the dynamic interchange of nutrients/metabolites with the host's microbiome, especially in the lower bowel (182, 187). These rich interchanges are regulated to protect homeostasis and support a reserve functional capability that can be called upon when needed (431). The dietary provision of micronutrients also meets the need to maintain a range of fundamental functions such as substrate, cofactors, and signaling molecules. Each has a role and an importance related to its particular chemical character and the context.
An adequate reserve capability and capacity to support metabolism, and the flexibility to adjust the pattern of nutrient interchange and response to any internal/external perturbation is the hallmark of health. Ill health supervenes when either the capacity to maintain the reserve is reduced below a critical level, or the reserve is overwhelmed by external challenges (179, 448). Resilience is characterized by the robustness of multilayered nutritional interrelationships; the flexibility conferred by the retention of functional capacity in excess of immediate needs; and protected capacity for critical functions together with seemingly redundant pathways and processes that can be called on under exceptional circumstances. There is a cost for supporting resilience, and the manifestations when the system is challenged may be characterized as “stress of the stress response” (179).
The metabolic response to any challenge is calibrated to the type of stressor, its location within the body, intensity, and duration; hence, the nutritional demands and the appropriate response are variable (Fig. 7A). The nutritional integrity of subcellular compartments coming together as an intracellular response may extend to embrace the local microenvironment but can be escalated to a whole-body response with recruitment and engagement of higher levels as the buffering capacity required to cope with increases (360). Systemically, it may be necessary to draw on reserves mobilized from muscle, bone, or adipose tissue to access sources of energy, macronutrients, vitamins, minerals, or trace elements to support the demands of progressively greater stressors. The whole-body response represents a major reorganization of metabolic priorities as a direct consequence of the integrated hormonal, cytokine, and chemokine communication system, themselves responsive to and modulated by the availability of nutrients. Loss of appetite is a feature of a more substantial systemic response and enables major shifts in the availability and utilization of endogenous reserves to meet the new metabolic priorities. The liver plays a coordinating role [perhaps explaining why patients with chronic liver disease have higher COVID-19-related in-hospital mortality; (128)], and as the demands of the acute-phase response supervene, the limitation on production and availability of nutrient transport proteins further limits the delivery of nutrients to the tissues, including the immune system, where the cellular demands are reordered. These changed cellular and metabolic priorities, together with increased but unbalanced losses of nutrients from the body in urine and stool, place a drain on reserves, leading to depleted pool sizes. This incurs a whole-body debt that has to be replenished during convalescence, when the regular diet may no longer be adequate to support usual function and correct deficits; the latter may contribute to “Long-COVID.”
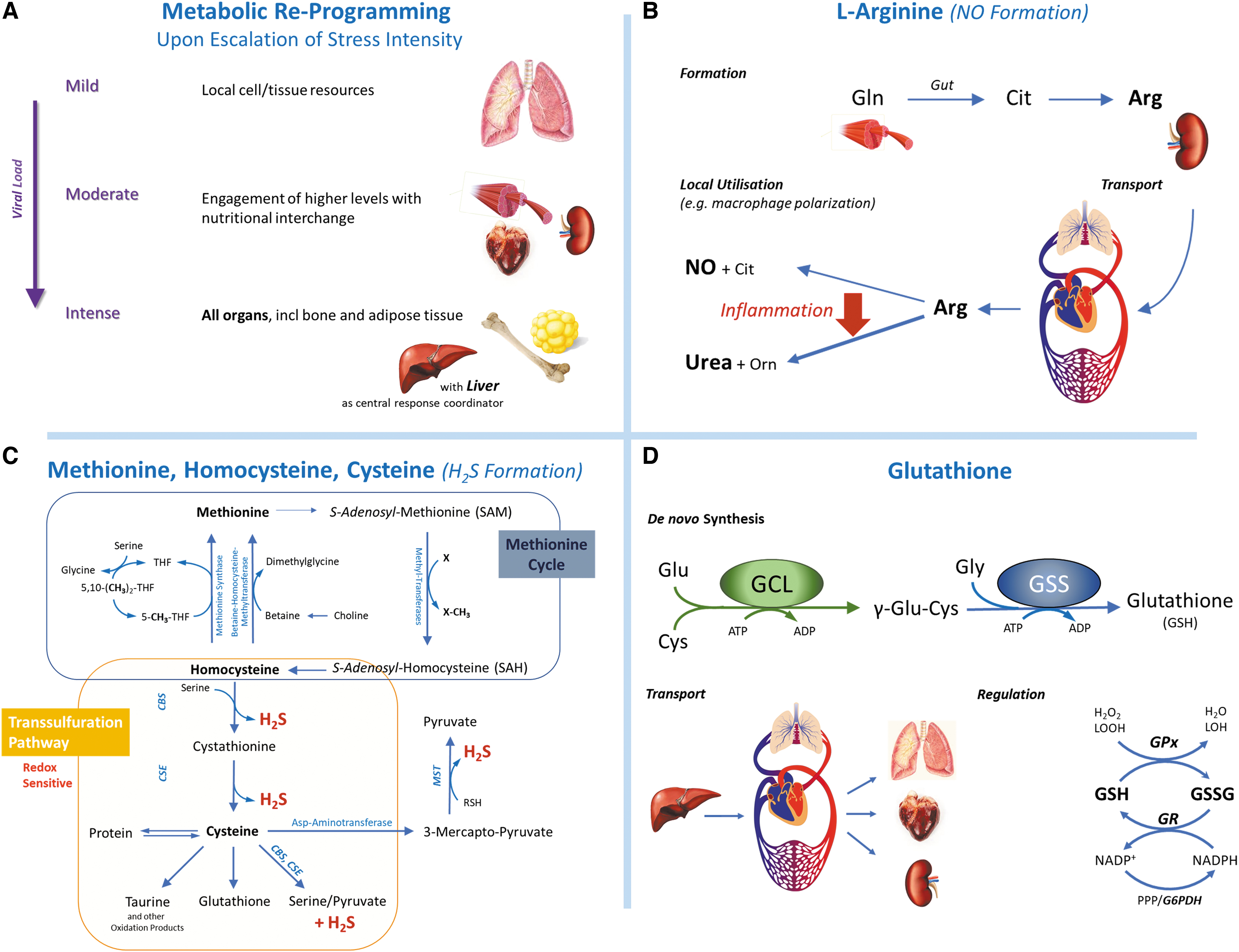
A cardinal feature of the acute-phase response is the ability to maintain appropriate redox responses within the framework of the central coordinating response (215, 216, 375). Most aspects of metabolism and metabolic control are affected, but most important is the ability to protect those pathways that are central to maintaining homeostasis in the face of shifting metabolic demands (375, 470). As a layered defence process, the challenge to the RSI is context-specific but based on the underlying principles of processes enabled by RSS, RNS, and ROS as well as based on other redox-based responses (87, 360). In addition to the availability of specific precursors to produce regulated fluxes of reactive species, oxidative stress also places a particular demand on sufficient reducing power to maintain redox balance. The critical nutritional and metabolic capabilities supported by integration of the glutathione, peroxiredoxin, and thioredoxin systems represent the fundamental determinant capability to provide protection and support/maintain life (28, 30, 376). The pattern of nutrients required for the cellular stress response mechanisms has been clearly characterized at cellular and subcellular levels (215, 216). It has been proposed that there is evidence for a central hub that plays a major role in controlling and determining cell fate (375). In this context, both GAPDH and G6PD (298, 470) appear to play a critical role, and their moonlighting functions may serve to coordinate metabolic rerouting (375), thereby enabling a precision redox response (273) that is “fit for purpose.” The provision of adequate energy and the balanced availability of all nutrients carry the potential to modulate all of these responses.
Specific nutritional considerations and cofactor dependencies for stress signaling and adaptation
Among the wealth of possible processes of importance, perhaps the one that has received least attention is the stoichiometry and quantitative availability of amino acid precursors for the generation of H2S (methionine, choline, glycine, serine, cysteine) (190, 275, 398, 399), NO (arginine, glutamine) (247, 276, 277, 288), and glutathione (glutamine, cysteine, glycine) (183, 248, 266). Except methionine, all other amino acids involved are considered to be dispensable from the diet (180, 266). They have been considered to be “non-essential” from a dietary perspective, as under usual situations they can be formed in adequate amounts by the body (and exchanged between organs) to meet its demands, although this fails to take account of the needs when responding to stress or growth. From a metabolic perspective, it is clear that there is a limit to the endogenous formation of each amino acid, and when this fails to match the needs of unusual/excessive demands the system is challenged at all levels of organization (177). In this situation, the disposal of a specific amino acid to one or another pathway must be constrained, causing additional metabolic stress.
There is an absolute demand for arginine as a precursor for NO formation (247, 276, 277, 288). Although this could be met in part by the diet, most of that needed is derived from endogenous formation requiring metabolic cooperativity among tissues (Fig. 7B), with glutamine derived from muscle being converted to citrulline in the gut, which is the precursor for arginine formation in the kidney (247). Total body demand for arginine is high, and acting as a precursor for NO production is one of many competing pathways (247, 288). During infection, both availability and metabolism of arginine play a determinant role in the direction of macrophage polarization (276, 277). Inflammation is associated with enhanced expression of arginase, which shunts arginine away from NOS to generate urea and ornithine (341), leading to reduced NO production; since this has been demonstrated to occur in macrophages infected with another enveloped RNA virus (476), the same might happen with SARS-CoV-2. When demand for arginine outstrips dietary supply and endogenous formation combined, limited arginine availability is marked by excretion of orotic acid, an intermediate in the pyrimidine pathway (279). In humans, even modestly increased demands for arginine during pregnancy lead to orotic aciduria (178). With serious infection, the substantial increase in demand to enable enhanced NO formation greatly outstrips endogenous generation and thereby limits the flow of arginine to one or more pathway(s), with compromised function (247). A recent metabolomic profiling study demonstrated that plasma arginine levels in patients critically ill with COVID-19 were lower than in healthy controls, and the creatinine/arginine ratio predicted COVID-19-associated death (123).
The formation of H2S through the trans-sulfuration pathway is under control of the HPA signaling axis, increasingly recognized as being critical to metabolic health, fitness, and stress resistance, and credited with the particular benefits afforded by dietary restriction (159, 160). There is close metabolic interdependence (Fig. 7B) of the specific metabolic flow through the re-methylation and trans-sulfuration pathways (190, 275, 398). The re-methylation cycle is sensitive to the availability of cofactors such as folate, B12, riboflavin, vitamin A, copper, zinc and the methyl donors choline and serine. Trans-sulfuration, and H2S generation, requires adequate pyridoxal with methionine as -SH donor and serine as the 3-carbon source for cysteine formation, and endogenous H2S formation is necessary and sufficient to confer stress resistance in experimental models in vivo (160, 275, 399). The vulnerability of older people to poor B-vitamin status and the associated increased mortality is of particular concern for pyridoxal, vitamin B6 (167); because pyridoxal is implicated in immune competence and believed to ameliorate inflammation and oxidative stress, it was hypothesized to also reduce COVID-19 severity (217).
The maintenance of cellular reduced glutathione (GSH) and its redox cycling to GSSG (dependent on reducing equivalents from NADPH, via G6PD and the thiamine-dependent pentose phosphate shunt) is fundamental to cell survival (248, 343). In COVID-19, low GSH is associated with high mortality (200). Glutathione is exclusively produced in the cytosol, from where it is distributed to organelles, including the nucleus, ER, and mitochondria. Aging and a range of pathologies are associated with reduced mitochondrial glutathione content and mitochondrial dysfunction (350). At the whole-body level (Fig. 7D), the majority of glutathione is produced in the liver from where it is transported, via blood, to other tissues (246, 269). Dietary provision of methionine has been shown to sustain redox homeostasis in the liver by providing the cytosolic reducing power via the trans-sulfuration pathway, independent of NADPH availability (108). Production and oxidation as well as reduction and metabolism of glutathione are controlled by different enzyme systems, allowing independent catalytic control of the forward and backward reactions, consistent with the notion that GSH is an important checkpoint in intermediary metabolism (296). The antioxidant selenoprotein, glutathione peroxidase (GPx), functionally links glutathione with selenium, both of which have been implicated as antiviral factors in COVID-19 (414). Selenium exerts its effects largely via the highly reactive selenocysteine, which is incorporated into selenoproteins, and thereby has close links also to alanine/glycine metabolism (selenocysteine lyase) and the Nrf2 system (thioredoxin reductase-1) (42, 63, 371). Glutathione is utilized in a range of pathways, and maintaining its cellular concentration depends on the availability of its precursor amino acids (glutamine, cysteine, and glycine). The formation of glutamyl-cysteine is considered to be rate-limiting for its formation and ensures that available cysteine is held in a bound form within the cell (248). However, any constraint on glycine availability limits GSH formation when demand is high, as indicated by increased urinary excretion of 5-L-oxoproline (181, 183, 266). The availability of precursors for GSH formation is dependent on the overall metabolic state, and there is evidence that glycine can be limiting during states of increased demand (stress, infection) due to reduced endogenous capacity for formation (aging, obesity, hypertension, T2DM) and/or increased consumption (detoxification of xenobiotics; flow to metabolic end pathways such as creatine or heme formation) (181). There is increasing evidence that supplemental glycine improves age-related oxidative stress and glucose homeostasis (101, 373, 374); its success in treating T2DM may, in part, be related to its effects on decreasing endogenous AGE formation (302). Glycine's health protective effects also include anti-inflammatory, immunomodulatory, and antiplatelet activity (364, 484).
Beyond the provision of energy and nutrients: risks for excess metabolic stress/harm and strategies for reclaiming metabolic control
In addition to providing energy and nutrients, diets contain other components, some of which are potentially harmful. Food processing is associated with increased formation of AGEs. Excessive endogenously formed (see section “The Advanced Glycation Endproduct/Receptor for Advanced Glycation Endproduct Axis and other “Sweet” Connections between Redox and Stress Signaling”) and/or dietary AGEs have adverse metabolic effects, and their accumulation can predispose to chronic inflammatory diseases (120, 426). The AGEs are pro-oxidants, and it has been suggested that an aspect of the aging-related increase in redox stress may be directly linked to progressively accumulating AGEs. The AGEs are increased in older patients, those with obesity and/or T2DM and in people of African ancestry living in the United States (349).
Malnutrition is frequent in the elderly (278), and a risk factor for COVID-19 infection and recovery (1, 61, 474). There are many specific dietary interventions that might be considered to address the complex metabolic derangements associated with COVID-19, including zinc, glycine, or arginine in relation to AGEs (302, 378, 452), folate or B12 for remethylation pathways, pyridoxine to enhance the trans-sulfuration pathways, supplements of mixtures of specific amino acids or individual lipids, vitamin D, and others. However, for any particular individual it is currently impossible to identify their specific needs, and inappropriate delivery of nutrients in itself can enhance the degree of metabolic stress. The preferred approach to treatment, care, or prevention follows similar underlying principles as have been enunciated by national/international bodies for severe malnutrition (489, 492), or in the presence of serious viral infection such as Ebola or HIV (493, 494): to provide nutritional support to enable the person to regain metabolic control and regulate their own “milieu interieur.” This can be identified as a three-step process:
Gain metabolic control through the provision of sufficient energy to meet metabolic demand, but neither more nor less (greater than the demand places a stress on the system, especially where there are other nutrient limitations). Correct specific nutrient deficiencies with a balanced diet and, where necessary, supplement to meet requirements (excess of any one nutrient will place stress on other aspects of the system for handling and excretion). Manage, remove, and minimize all other metabolic stressors.
Diet and nutrients can act as powerful metabolic stressors, especially in the acute situation when given in excess of the ability of an already stressed metabolism to cope. In the longer term, greater than usual amounts of specific nutrients may be required to enable and support replenishing reserves that may have been depleted during illness, particularly in the muscle and bone.
What has not been adequately appreciated is the importance of ensuring the nutritional health and well-being of the whole population by taking special care to maximize the nutritional health of the most vulnerable. In poorly nourished individuals with limited antioxidant protection, there is an increased risk of infection with a retrovirus, even when that virus may not be particularly virulent (135). In poorly nourished individuals with a compromised RSI/Redox Interactome, viral replication may give rise to more virulent forms of the virus that can then be passed on and adversely affect even those who enjoy good nutritional health (24, 230). Evidence from China suggests that this (in the form of selenium deficiency) was a factor in the initial development of the COVID-19 pandemic and invites speculation about the emergence of other more contagious forms. To this end, society can only enjoy the level of health permitted by the most vulnerable in the chain of infection (478). Thus, taking good nutritional care of the most vulnerable is in everyone's (self-)interest.
Conclusions
We here explored the opportunities that the current pandemic offers to understand how the human body copes with stress. By focusing on redox-related processes of COVID-19, we outline a path to better understand how SARS-CoV-2 works while at the same time providing new insight into redox regulation and organization of the RSI at the systems level, demonstrating how each consideration adds value to the other. The emergent understanding would be difficult to gain from the study of either one on its own, and the insight gained not only may be of relevance for the health care management of acute infections but also may pave new avenues to tackle Long-COVID and other stress-related diseases. Our holistic perspective should be interpreted as a “call to action,” encourage investigators to focus on disentangling whole-body redox regulation, and determine factors that are critical for resilience—let us not waste the opportunities of the current crisis, as the next pandemic is imminent.
Footnotes
Acknowledgments
The authors are indebted to their families for their support and stimulating discussions with friends and colleagues during one of the most challenging times of their lifetime. M.F. is grateful for previous research support, which helped shape his thinking, from the U.S. National Institutes of Health, the UK Medical Research Council, and the Wellcome Trust and current funding from The Bill & Melinda Gates Foundation (INV-016631).
Author Disclosure Statement
M.F. is a member of the Scientific Advisory Board of AOBiome and a consultant for ASEA Global, Fourth State Medicine, Smith & Nephew, Therabel Pharma, and Vasomune Therapeutics. None of the other authors have any potential conflict of interests to declare.
Funding Information
A.D.C. has received funding through the NIHR Academic Foundation Programme. A.F.C. is funded by the National Institute for Health Research Southampton Biomedical Research Centre. No specific funding was received for preparation of this article.
Supplementary Material
Supplementary Data
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.