
Abstract
Significance:
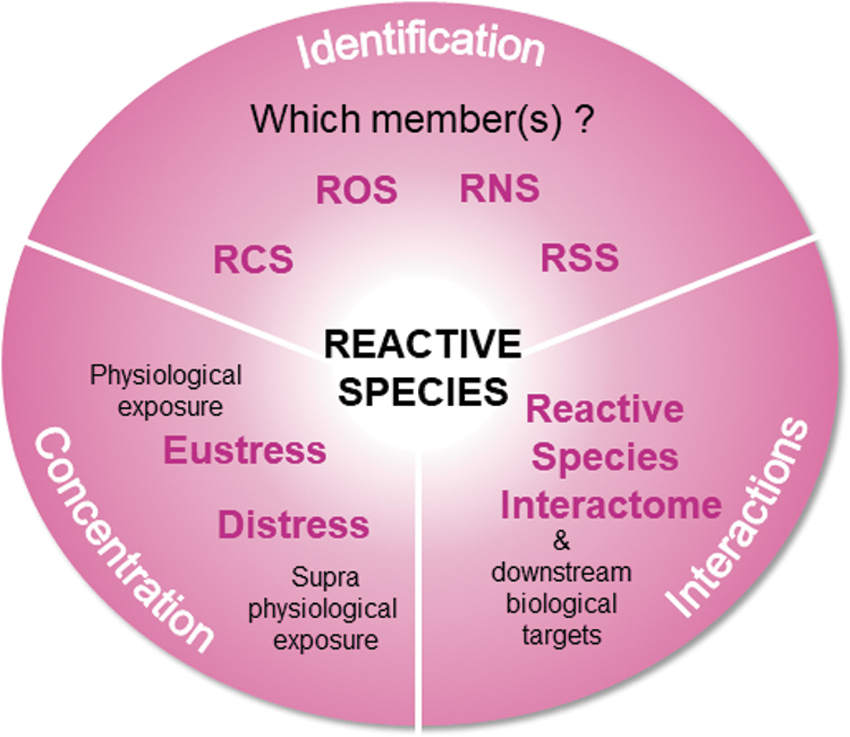
Redox pioneer Helmut Sies attempted to explain reactive species' challenges faced by organelles, cells, tissues, and organs via three complementary definitions: (i) oxidative stress, that is, the disturbance in the prooxidant-antioxidant defense balance in favor of the prooxidants; (ii) oxidative eustress, the low physiological exposure to prooxidants; and (iii) oxidative distress, the supraphysiological exposure to prooxidants.
Recent Advances:
Identification, concentration, and interactions are the most important elements to improve our understanding of reactive species in physiology and pathology. In this context, the reactive species interactome (RSI) is a new multilevel redox regulatory system that identifies reactive species families, reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive sulfur species, and it integrates their interactions with their downstream biological targets.
Critical Issues:
We propose a united view to fully combine reactive species identification, oxidative eustress and distress, and the RSI system. In this view, we also propose including the forgotten reactive carbonyl species, an increasingly rediscovered reactive species family related to the other reactive families, and key enzymes within the RSI. We focus on brain physiology and pathology to demonstrate why this united view should be considered.
Future Directions:
More studies are needed for an improved understanding of the contributions of reactive species through their identification, concentration, and interactions, including in the brain. Appreciating the RSI in its entirety should unveil new molecular players and mechanisms in physiology and pathology in the brain and elsewhere.
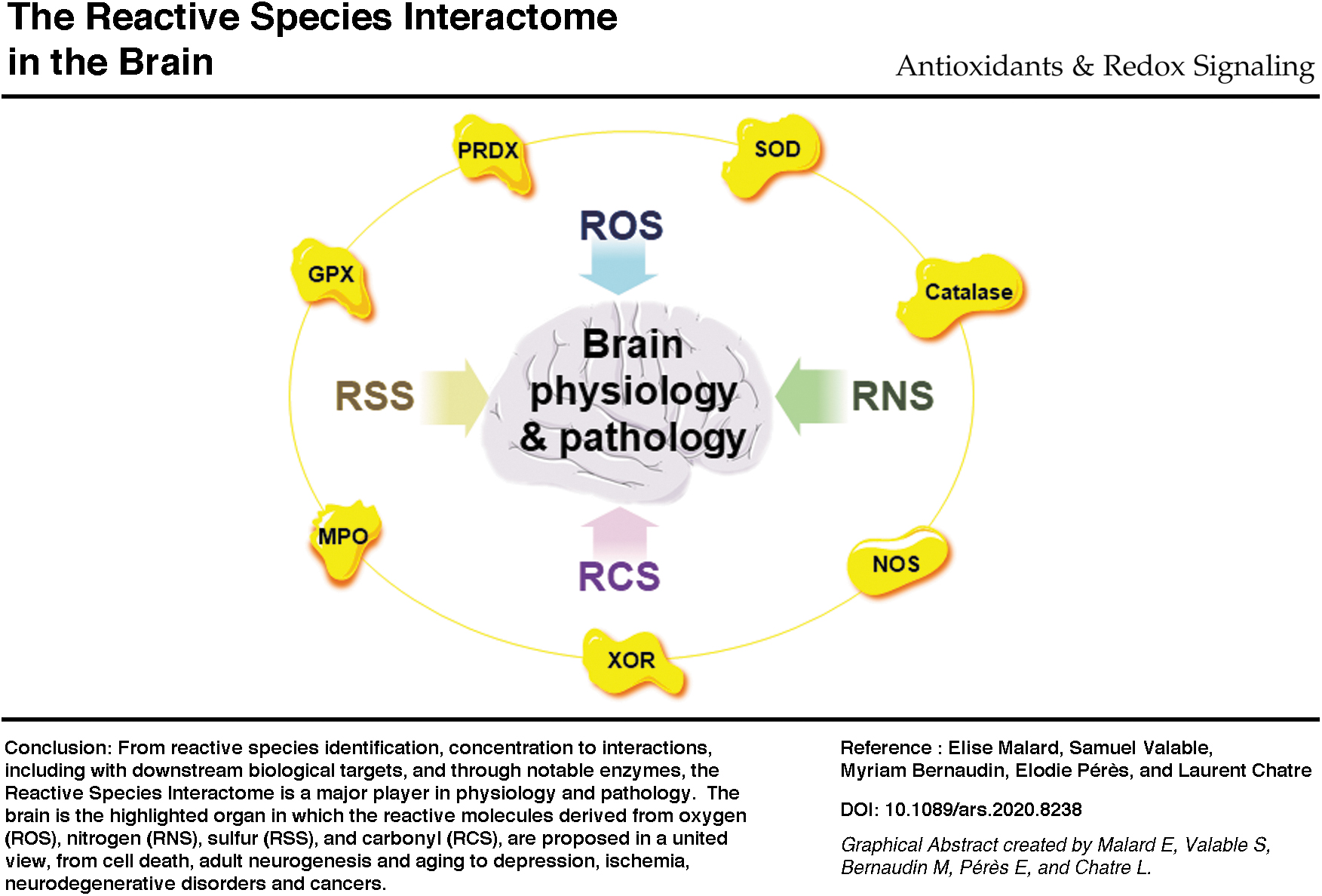
(Color images are available online).
“Remember to look up at the stars and not down at your feet. Try to make sense of what you see and wonder about what makes the universe exist. Be curious. And however difficult life may seem, there is always something you can do and succeed at. It matters that you don't just give up.” Stephen William Hawking
Introduction
“Stress is the spice of life” is a famous quote by Selye (210), who, in 1925, originally, defined stress as a general adaptation syndrome with three stages: alarm reaction, resistance, and exhaustion (208 –210). Subsequently, redox pioneer Helmut Sies helped the concept of stress evolve, especially regarding the prooxidant–antioxidant balance and the concentration-dependent functions of stress molecules. After proving that the prooxidant reactive oxygen species (ROS) hydrogen peroxide (H2O2) is normally produced in intact eukaryotic cells by studying the steady-state level of the antioxidant defense enzyme catalase, Sies defined oxidative stress in 1985 as “a disturbance in the prooxidant-antioxidant balance in favor of the former” (114, 216, 219, 221). This definition was updated in 2007 as “an imbalance between prooxidants and antioxidants in favor of the prooxidants, leading to a disruption of redox signaling and control and/or molecular damage” (222). In 2015, Sies legitimately argued for clearly naming the individual prooxidants instead of applying only the generic labels of oxidative stress and ROS (217). Finally, in 2017, Sies defined oxidative eustress as “low physiological exposure to prooxidants” with specific targets involved in redox signaling, and oxidative distress as “supraphysiological exposure to prooxidants” with unspecific targets, disrupted redox signaling, and damaging molecule modifications (218, 220).
From physiology to pathology, likely related to tissue-specific metabolome, excessive stress is, indeed, a major risk factor in the lungs, liver, kidneys, heart, and brain, the metabolically active organs that are vital for survival (46, 122, 142, 238). In humans, the brain represents ∼2% of the total body weight but it consumes 20% of the total basal body oxygen and 20% of the body's energy. It is one of the most metabolically active of all organs (52, 196). Importantly, the brain is also highly susceptible to oxidative stress, specifically oxidative distress, for several reasons (52). One of these is due to the multifaceted roles of reactives species themselves, especially superoxide anion (O2 .−) and H2O2, in redox signaling (52). Another source of oxidative distress is H2O2 generated by the metabolism of neurotransmitter dopamine via monoamine oxidases (MAO) and by oxidative phosphorylation (OXPHOS) in mitochondria (52). Low endogenous antioxidant defense in the brain relative to other organs, especially low catalase and reduced glutathione (GSH) content, makes the brain susceptible to oxidative distress (52). Mitochondria, including those in neurons and microglia, the resident immune cells in the brain, perform oxygen-dependent OXPHOS for O2 .− and bioenergetic metabolite ATP generation, contributing to oxidative distress (52, 236). They have roles in many processes such as cell death and calcium homeostasis that are involved in synaptic plasticity (52, 263). Neuronal activity and death also occur when the brain is enriched in redox-active transition metals ferrous ion (Fe2+) and cuprous ion (Cu+) relative to other organs (52). Further, enriched unsaturated lipids, including omega-3 docosahexaenoic acid, in the brain cause distress due to their susceptibility to lipid peroxidation related to cell death, including ferroptosis (52). Finally, brain susceptibilty is region- and time-dependent, depending on the condition that induces oxidative distress and the endogenous antioxidant capacity (104, 121, 248). The following four areas of the brain are the most susceptible to oxidative stress: the cerebral cortex, the largest site of neural integration involved in many functions including motor function, language processing, and planning; the hippocampus, the dedicated region for neurogenesis during adulthood with its major role in learning and memory; the striatum, which coordinates cognition including decision making, action planning, and motivation; and the cerebellum, which coordinates motor movements (104, 121, 248).
This review focuses on reactive species identification, concentration, and interactions, including those through the brain's key enzymes. We urge the scientific community to consider together ROS, reactive nitrogen species (RNS), reactive sulfur species (RSS), and likely the forgotten reactive carbonyl species (RCS) to improve our understanding of oxidative eustress and distress in physiology and pathology.
Identification, Concentration, and Interactions of the Reactive Species
We propose here a united view to (i) name the reactive species, that is, identification; (ii) consider the concentration-dependent functions of the different reactive species, that is, concentration; and finally (iii) integrate the different reactive species in a dynamic interactome, that is, interactions. In 2017, this dynamic interactome was named the reactive species interactome (RSI), a multilevel integrative concept underappreciated in physiology and pathology (53). The RSI is defined as the interactions between reactive species and downstream biological targets, prevalently cysteine thiols in proteins, the so-called redox switches, and also many other elements including lipids and DNA (20, 53). The RSI-related structural and functional modifications of antioxidant defense enzymes, transcription factors, molecule transporters, phosphatases, kinases, and other proteins will change adaptations from short-term to long-term and even induce alterations (20, 53). Therefore, sensors and transducers of any changes in the intracellular and extracellular environments, including those related to bioenergetic metabolism, redox signaling, and nutritional status, should be considered reactive species (20, 53). The RSI is still poorly supported by original publications, and there is massive research potential in studying the interactome. We consider the strategy focusing on identification, concentration through oxidative eustress and distress, and interactions through the RSI regarding how stress should be considered in physiology and pathology (Fig. 1).
Endogenous Key Sources and Interactions of ROS, RNS, RSS, and RCS
Considering the sources of the main endogenous reactive species, including intra- and inter-species interactions and those with their downstream targets, will improve our understanding of oxidative stress phenomena. In a nonexhaustive manner, we overview key sources and interactions of ROS, RNS, RSS, and the neglected RCS to strengthen the relationship between themselves and show that the RSI needs to integrate the RCS family and key enzymes for a thorough understanding (Fig. 2).
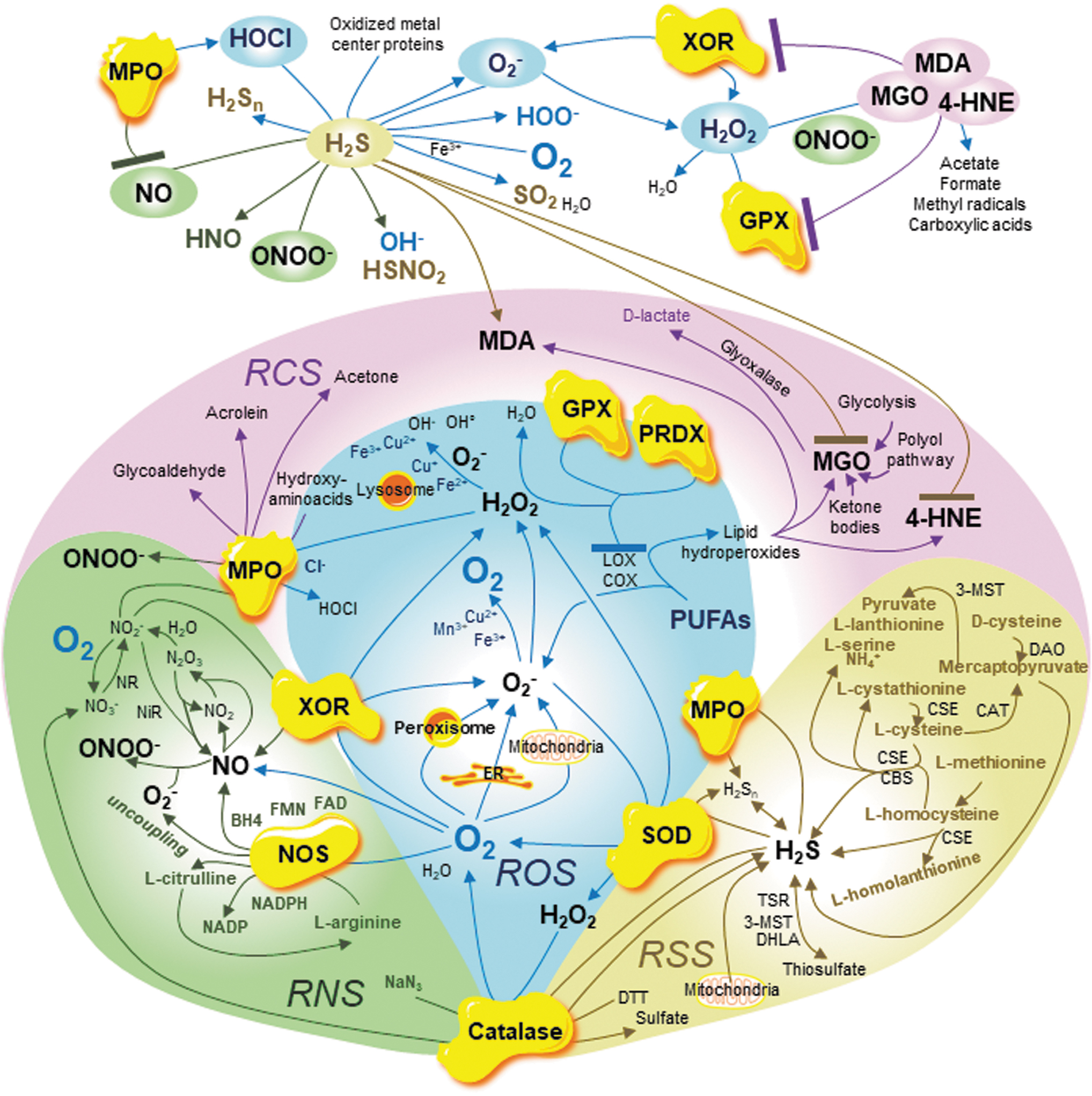
Three reactive species sources include (i) organelles, (ii) enzymes, and (iii) chemical reactions between reactive species and with transition metal ions. To elaborate key connections between the reactive species families, including those with key enzymes, we summarize ROS, RNS, RSS, and RCS metabolism.
ROS Metabolism
ROS initiates the generation of O2-derived reactive species by using the precursor O2 to form O2 −, then H2O2, hydroxyl radicals OH., OH−, and many other derivative species (20, 53, 223, 225) (Fig. 2). As in the metabolism of other reactive species families, ROS metabolism is regulated by enzyme activities and chemical reactions that are tightly linked to organelle functions. Mitochondria, peroxisome, the endoplasmic reticulum (ER), and lysosome are key organelles in this regard (20, 53, 64, 223, 225). From O2, O2 .− is mainly generated by (i) OXPHOS complexes I and III and other proteins such as glycerol 3-phosphate dehydrogenase and 2-oxoglutarate dehydrogenase in mitochondria; (ii) fatty acid β-oxidation, iron-sulfur flavin hydroxylase xanthine oxidoreductase (XOR) and urate oxidase (UO) in the peroxisomal matrix, and electron transport chain in the peroxisomal membrane; and (iii) nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, including NADPH oxidases (NOXs) and dual oxidases, and the microsomal monooxygenase (MMO) system, which contains cytochrome P450 oxidase in the ER (225). In addition, nonheme iron-containing dioxygenases, lipoxygenases, and cyclooxygenases also generate O2 .− from polyunsaturated fatty acids (PUFAs) (225). Then, O2 .− reduces key transition metal ions iron Fe3+, copper Cu2+, and manganese Mn3+, which generate O2 (101, 225, 247). Importantly, O2 .− is also metabolized to generate H2O2, one of the most stable ROS (20, 53, 223, 225). Without developing all H2O2 sources, including spontaneous dismutation of O2 ., polyamine catabolism with copper-containing diamine oxidase and thymidine catabolism, mitochondrial MAO, copper-dependent lysyl oxidases, peroxisomal enzymes such as XOR, UO, and acyl-CoA oxidases, and the ER oxidoreductin-1 related to glutathione metabolism GSH/oxidized glutathione (GSSG) and MMO system, it is important to highlight that one of the major antioxidant enzymes catalyzes the reduction of O2 .− to H2O2, superoxide dismutase (SOD) (53, 78, 100, 223, 225).
H2O2 is then metabolized to generate other ROS, O2, and dihydrogen monoxide (H2O), and regulate transition metal ion reactivities (53, 100, 223, 225). Among several enzymes, glutathione peroxidases (GPXs), through GSH/GSSG metabolism, and peroxiredoxin (PRDX) generate H2O from H2O2 (181, 225). Two vital enzymes also must be highlighted, the important antioxidant defense enzyme catalase and the heme enzyme myeloperoxidase (MPO). Catalase, the most abundant peroxisomal antioxidant enzyme, dismutates H2O2 to O2 and H2O (53, 100, 168, 174, 221). MPO catalyzes the oxidation of Cl− by H2O2 to hypochlorous acid (HOCl) (83, 178). In addition, H2O2 reacts with transition metal ions Fe2+ or Cu+ to generate OH., OH− and corresponding reactive Fe3+ or Cu2+ in the Fenton reaction (118, 225). The Fenton reaction with Fe2+ occurs mainly in the lysosome, an essential acidic organelle for cellular degradation and recycling of proteins, DNA, RNA, lipids, and carbohydrates (64). This lysosomal reaction induces lysosomal stress, leakage, and multiple intracellular dysfunctions, including bioenergetic metabolism and autophagy alterations (64). Through another reaction, the Haber-Weiss reaction, H2O2 also reacts with O2 .− to generate OH., OH−, and O2 (118, 225). Finally, multiple antioxidant defense systems such as glutaredoxin, OXPHOS complex IV cytochrome c oxidase, glutathione (GSH/GSSG), and vitamins contribute to regulating the entire ROS metabolism (53, 223, 225). This partial view of ROS metabolism will be completed later with regard to SODs, catalase, XOR, and MPO that serve as the four key enzymes.
RNS Metabolism
Closely related to iron homeostasis, RNS metabolism is the generation of nitric oxide (NO)-derived reactive species mainly from L-arginine and NO synthase (NOS) activities (20, 53, 74, 76, 120, 140) (Fig. 2). From L-arginine, O2 and cofactors tetrahydrobiopterin (BH4), NADPH, flavin mononucleotide, and flavin adenine dinucleotide, NO is generated by three isoforms of NOS: neuronal NOS (nNOS, NOS I), constitutively expressed in the central nervous system; inducible NOS (iNOS, NOS II), which is expressed in many cell types including macrophages and dendritic cells; and endothelial NOS (eNOS, NOS III), which is mainly expressed in endothelial cells (13, 74, 76, 120, 225). While nNOS and eNOS are calcium-dependent enzymes, iNOS is a calcium-independent enzyme (74, 76).
Using O2, oxidation of L-arginine by coupled NOS with cofactors including NADPH generates NO, NADP, and L-citrulline, which is also an L-arginine precursor through the citrulline-NO cycle (12, 74, 76, 120). NO then reacts with O2 to generate nitrogen dioxide (NO2) and with NO2 to form dinitrogen trioxide, which reacts with water to produce nitrite (NO2 −) or dissociates to form NO and NO2 (74, 76, 120). Nitrite oxidation with O2 leads to the generation of nitrate (NO3 −) (74, 76, 120). NO is also generated by the following process: Nitrate reductase catalyzes the reduction of nitrate into nitrite, and nitrite reductase reduces nitrite into NO (120). The RNS metabolism also includes other reactive species such as nitroxyl, nitrous acid, hydroxylamine, and ammonia that are not discussed here (120). This partial view of RNS metabolism will be completed later regarding NO as the key reactive species and NOS as the key enzyme.
RSS Metabolism
The RSS are derived from hydrogen sulfide (H2S) (20, 29, 53, 128, 171, 272) (Fig. 2). From essential amino acid L-methionine recycling and transsulfuration pathway with L-homocysteine and L-cysteine, the RSS-related enzymes cystathionine b-synthase (CBS), cystathionine-γ-lyase (CSE here, a.k.a. CGL), cysteine aminotransferase (CAT), and 3-mercaptopyruvate sulfurtransferase are the key enzymes involved in H2S production (29, 53, 128, 157, 171, 172, 233, 272). First, L-methionine catabolism leads to L-homocysteine biosynthesis (29, 128, 171, 233, 272). Cysteine-γ-lyase then converts L-homocysteine to L-homolanthionine and H2S (29, 128, 171, 233, 272). The CBS and CSE enzymes convert L-homocysteine and L-cysteine to L-cystathionine and H2S, and L-cystathionine is further converted to L-cysteine by CSE (29, 128, 171, 233, 272). In addition, L-cysteine is metabolized either by CBS to produce H2S, L-serine, and L-lanthionine; by CSE to generate H2S, ammonium, and pyruvate; or by CAT to 3-mercaptopyruvate (29, 128, 157, 171, 233, 272). The enzyme CAT is also both an aspartate aminotransferase (AST) and a glutamate oxaloacetate transaminase (GOT) that generates α-ketoglutarate (α-KG) and aspartate from oxaloacetate, and glutamate in mitochondria (GOT2) catalyzes the reverse reaction in the cytosol (GOT1) (155, 157). Cystine, the dimeric form of L-cysteine, is converted by CSE to L-thiocysteine, producing H2S through thiol (RSH)-dependent reactions (171, 233). The 3-mercaptopyruvate, which is also produced by the conversion of D-cysteine by D-amino acid oxidase and that spontaneously generates H2S, is the substrate of 3-MST to generate pyruvate, and finally, bound sulfane sulfur (171, 233). This sulfur generation results in the release of H2S through the thioredoxin (TRX) enzyme and dihydrolipoic acid (DHLA) (171, 233). This pathway is the standard for H2S generation, although TRX and DHLA are part of the unconventional pathway (171). In this pathway, H2S is generated from thiosulfate (S2O3 2−) through reductant DHLA or thiosulfate reductase coupled to GSH oxidation or 3-MST (171). Associated with GSH/GSSG and NADPH/NADH systems, nonenzymatic reactions through thiocysteine persulfides and polysulfides also generate H2S (29). Moreover, mitochondrial OXPHOS complex I is a source of H2S due to its accessory sulfurtransferase subunit (171). Finally, not discussed here, volatile organic sulfides such as carbon disulfide, carbonyl sulfide, and sulfur dioxide, and acid-labile sulfide in iron centers, including cytochromes and aminothiols such as cysteinylglycine, are unconventional sources of H2S (171).
Once H2S is produced, as its accumulation is toxic, H2S catabolism occurs by oxidation to form thiosulfate (29, 128, 171). This reaction also occurs in mitochondria, where sulfide quinone oxidoreductase oxidizes H2S to produce persulfide (29, 128, 171). Sulfite and sulfate are also derived from H2S and related to thiosulfate (29, 128, 171). Thiosulfate is converted into sulfite through thiosulfate sulfurtransferase or rhodanese and then into sulfate, and sulfite contributes to H2S generation (29, 128, 171). H2S catabolism also occurs by methylation to produce methanethiol, and then dimethylsulfide by thiol S-methyltransferase (29). Finally, H2S is oxidized into hydrogen thioperoxide, sulfinic acid, and finally sulfonic acid (128). Although many RSS are compelling, we will not discuss all the species and pathways. Two species we will discuss are hydrogen polysulfides (H2Sn), including persulfides (H2S2). These species are of increasing research interest, and their production and catabolism conventional and unconventional pathways (171). Finally, polysulfides are unstable and their catabolism tends to generate H2S (171). This partial view of RSS metabolism will be completed later, considering H2S as the notable reactive species.
RCS Metabolism
The sprawling network of the electrophilic, highly reactive, often forgotten RCS family is partially illustrated (Fig. 2). The key RCS members worthy of investigation include glyoxal (GO), acrolein, glucosone, acetaldehyde, glyceraldehyde-3-phosphate (G3P), and the three most abundant malondialdehydes (MDA), methylglyoxal (MGO)s, and 4-hydroxy-trans-2-nonenal (4-HNE) (211). The most abundant RCS are derived from PUFA peroxidation into lipid hydroxyperoxides through nonenzymatic reactions (4, 8, 187, 211). Fortunately, PUFA peroxidation is reversible due to notable enzymes GPX and PRDX (94, 211). The RCS are also produced by other nonenzymatic reactions such as oxidative modifications of amino acids (including threonine) and glycation of proteins through reduction of carbohydrates (including glucose) (158, 211).
Notably, RCS, including MGO, are generated as metabolic intermediates during glycolysis and polyol processing (205, 211). MGO is mainly generated during glycolysis through nonenzymatic catabolism of triose phosphates G3P, dihydroxyacetone-phosphate, and the glycation pathway to ultimately produce advanced glycation end products (AGEs) (205, 211). The AGEs and lipoxidation end products, also induced by RCS, are irreversible toxic products (205, 211). The end-product fructose from the polyol glucose-sorbitol-fructose pathway is also a precursor of MGO through triose phosphate formation (205, 211). Finally, MGO is generated by ketone body catabolism, including oxidation of acetone (205, 211). Fortunately, the glyoxalase defense system counteracts the accumulation of MGO by converting MGO into D-lactate (205, 211).
Long seen as side products from ROS after lipid peroxidation, careful examination of RCS shows that this family strengthens the connections between ROS, RNS, and RSS families in such a way that the RCS family could be the missing piece of the “stress puzzle” that will change perspectives and contribute to revealing new pathways. This partial view of RCS metabolism has presented the notable reactive species MDA, MGO, and 4-HNE and the key enzymes, GPX as well as PRDX.
The Metabolism and Interactome of Notable Reactive Species and Key Enzymes
Connections between the notable reactive species, families (including multiple ROS, NO, H2S, and MDA, MGO, and 4-HNE), and key enzymes (SOD, catalase, XOR, MPO, NOS, GPX, and PRDX) reinforce the united concept of RSI highlighted in our overview (Fig. 2).
In a recent demonstration of this connectedness, Kenneth Olson and colleagues showed in a concentration- and oxygen-dependent manner that SODs oxidize H2S into polysulfides, including persulfide H2S2, and to a lesser extent trisulfane (H2S3) and pentasulfane (H2S5), which may be an ancient mechanism to detoxify or manage RSS (173). Thus, both ROS and RSS metabolism are regulated by SODs. Moreover, in a pH-dependent manner, catalase is also involved in RNS metabolism: In the presence of H2O2, catalase oxidizes sodium azide (NaN3), an inhibitor of mitochondrial cytochrome c oxidase in OXPHOS complex IV and catalase, into at least RNS NO2 − (nitrite) and NO3 − (nitrate) (168). In addition, the Olson research group recently proposed that catalase is also a sulfide–sulfur oxido-reductase during normoxia and hypoxia, as a sulfide oxidase by metabolizing H2S under the influence of H2O2, as a sulfide reductase generating H2S from dithiothreitol, and by eliminating polysulfides H2Sn in a concentration- and oxygen-dependent manner (174). Thus, ROS, RNS, and RSS metabolism are altogether regulated by catalase. The enzyme XOR is found in peroxisomes, where, in addition to generating O2 −, it catalyzes the reduction of nitrite and nitrate into NO, thus regulating ROS and RNS metabolism (225). The MPO enzyme is also a major NO scavenger that upregulates iNOS activity by reducing feedback inhibition of NO, and oxidizing nitrite to form NO2 or peroxynitrite (ONOO−), the so-called MPO-H2O2-nitrite system (23, 79). It has also been recently proposed that MPO is a key regulator of RSS metabolism by oxidizing H2S with H2O2 or O2 in the absence of H2O2 (83, 178). Further, the MPO-H2O2-chloride system generates from hydroxy-amino acids several RCS such as aldehyde, acetone, and acrolein (211). Thus, ROS, RNS, RSS, and RCS metabolism is regulated by MPO.
NO reacts with O2 − to form ONOO−, and H2S to form nitroxyl, thus linking ROS, RNS, and RSS metabolism (171, 225, 258). Importantly, NOS activity directly links ROS and RNS, and indirectly RSS through NO (76, 197, 225, 258). Indeed, coupled NOS with cofactors leads to NO generation, whereas uncoupled NOS produces O2 −, thus fueling O2 −-related reactive species including the peroxynitrite-positive feedback loop, and reduces NO bioavailability, thus impacting NO reactive species targets such as H2S (76, 197, 225, 258). Together, NO and NOS regulate at least ROS, RNS, and RSS metabolism.
H2S sits at the crossroads of ROS, RNS, RSS, and RCS metabolism: It reacts with O2 in the presence of transition metal ion Fe3+ to form sulfur dioxide and water, with O2 − to form hydroperoxyl, with oxidized metal center proteins, mostly hemes including iron-heme cytochromes, to form O2 −, with HOCl to form polysulfides, and with ONOO− to form thionitrite and OH− (109, 164, 171, 258). H2S can efficiently scavenge at least the aldehyde 4-HNE at physiological concentrations, meaning oxidative eustress in neuronal cells, protecting cells against MGO-induced damage in keratinocytes, and increasing MDA levels in the rat cerebral cortex (31, 206, 266).
Finally, to complete these examples, the notable species and enzymes in RCS metabolism, that is, MGO, MDA, 4-HNE, GPX, and PRDX, link together ROS, RNS, RSS, and RCS metabolism. H2O2 oxidizes the MGO and aldehydes, including MDA and 4-HNE, to form carboxylic acids (165, 201). Moreover, the RCS are oxidized by H2O2 and ONOO− to form adducts and intermediate products, ultimately forming acetate, formate, and methyl radicals (165, 201). In addition, MDA and 4-HNE inhibit XOR activity through direct interactions with XOR oxidase and dehydrogenase forms, whereas a GSH increase blocks this inhibitory effect (48). The highly reactive dicarbonyl MGO inactivates GPX enzymes by direct interactions with the glutathione binding site (185). Importantly, GPX and PRDX enzymes regulate one of the most important sources of RCS, lipid hydroperoxides (9, 16, 137, 161, 195, 269). Specifically, the selenoenzymes GPX4 and PRDX, including PRDX4, which are sensors and regulators of peroxides, neutralize lipid hydroperoxides (9, 16, 137, 161, 195, 269).
Due to its direct reactions with ROS, RNS, and RSS and key enzymes, the RCS family should be considered as important as ROS, RNS, and RSS families in the RSI.
Notable Reactive Species, Key Enzymes, and the RSI in the Brain
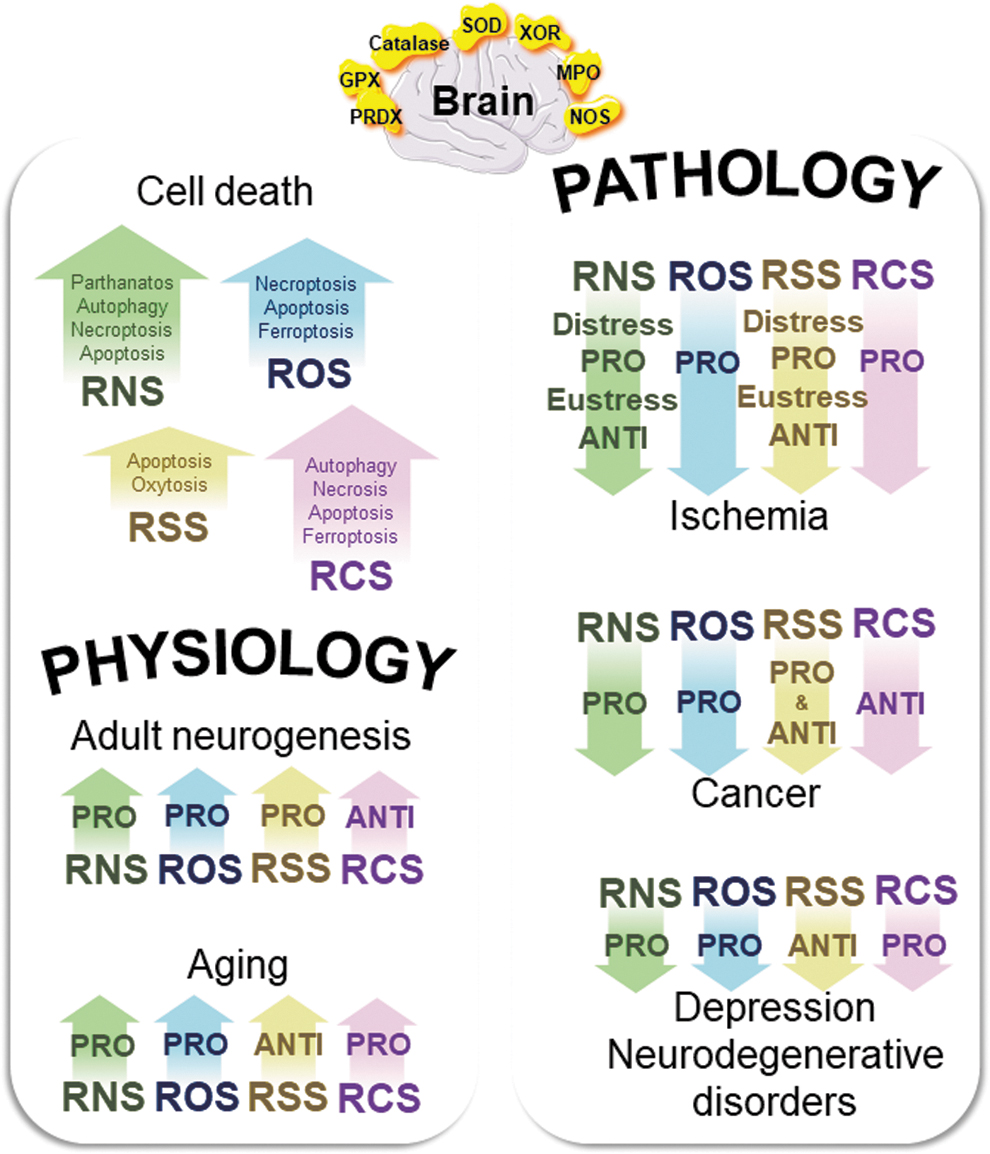
As developed in the introduction, we are focused on the brain, which, of all organs, is one of the most metabolically active and sensitive to oxidative distress. From physiology to pathology, from oxidative eustress to distress, considering (i) our strategy of identification–concentration interactions; (ii) the notable reactive species and key enzymes; and (iii) descriptions of individual and puzzling implications of ROS, RNS, RSS, and RCS, we combined key findings to reveal the RSI (including RCS) potential in the brain. We concentrate the brain physiology discussion on adult neurogenesis, cell death and aging, and regard the brain pathology on depression, ischemia, neurodegenerative disorders, and cancers.
Physiology: The Brain Adult Neurogenesis
In mammals, throughout life, neurogenesis, which occurs in several brain regions such as the hippocampal dentate gyrus and lateral ventricles, is defined as “the formation of new neurons from neural stem cells (NSC) and progenitor cells” (19, 146). The existence of adult neurogenesis, one that occurs in adulthood, has been intensively debated due to discrepancies between different studies using histology and multiple biomarkers (19, 146). Historically, in 1998, adult neurogenesis was observed continuously, at least in the human hippocampus (70). Then, after lengthy debates, in 2019, persistent adult neurogenesis in the hippocampal dentate gyrus was confirmed in healthy adult humans up to 90 years old, (146, 159). As solid evidence is growing in support of human adult neurogenesis (in the hippocampus and elsewhere), the field has moved toward reconciliation of various neurogenesis biomarkers and open questions on the molecular mechanisms regulating neurogenesis, including through oxidative stress (146, 159, 275).
The RSI in Brain Adult Neurogenesis
The neurogenesis field is currently investigating more than 20 different biomarkers (146, 275). An increasing number of studies highlight the involvement of several reactive species in brain adult neurogenesis, although the molecular mechanisms between reactive species and neurogenesis biomarkers require further investigation (Fig. 3). First, differentiation during adult neurogenesis induces oxidized glutathione accumulation and at least mitochondrial O2 − overgeneration (251). Transient oxidative distress due to overgeneration of mitochondrial O2 − promotes NSC and progenitor cell differentiation and, therefore, adult neurogenesis (251). High levels of H2O2 are then required for regular NSC and progenitor cells' self-renewal, a process related to doublecortin (DCX), Nestin, and Forkhead box O (FOXO) proteins (130). For example, the self-renewal process is modulated by the tumor suppressor and metabolic regulator Phosphatase and tensin Homolog (PTEN) protein, which regulates DCX and Nestin expression, and which is a negative regulator of phosphoinositide 3-kinase (PI3K)-AKT-mTORC1 pathway (130). The PTEN protein inactivation through overoxidation due to H2O2 promotes NSC and progenitor cells' self-renewal (130). Moreover, the inhibition of H2O2 overgeneration counteracts NSC proliferation (97). Oxidative distress due to H2O2 activates adult neurogenesis. Interestingly, from the antioxidant defense side, SOD deficiency induces a reduction in adult neurogenesis at all stages, from self-renewal to differentiation related to DCX, neurogenic differentiation (NeuroD), and glial fibrillary acidic protein (GFAP) biomarkers (107). In addition, NSC and progenitor cell proliferation are increased by the accumulation of catalase in mitochondria (138). Finally, MPO inhibition increases adult neurogenesis through at least DCX and sex-determining region Y box 2 (Sox2) stimulation (271). SOD, catalase, and MPO are key enzymes that are used to investigate adult neurogenesis.
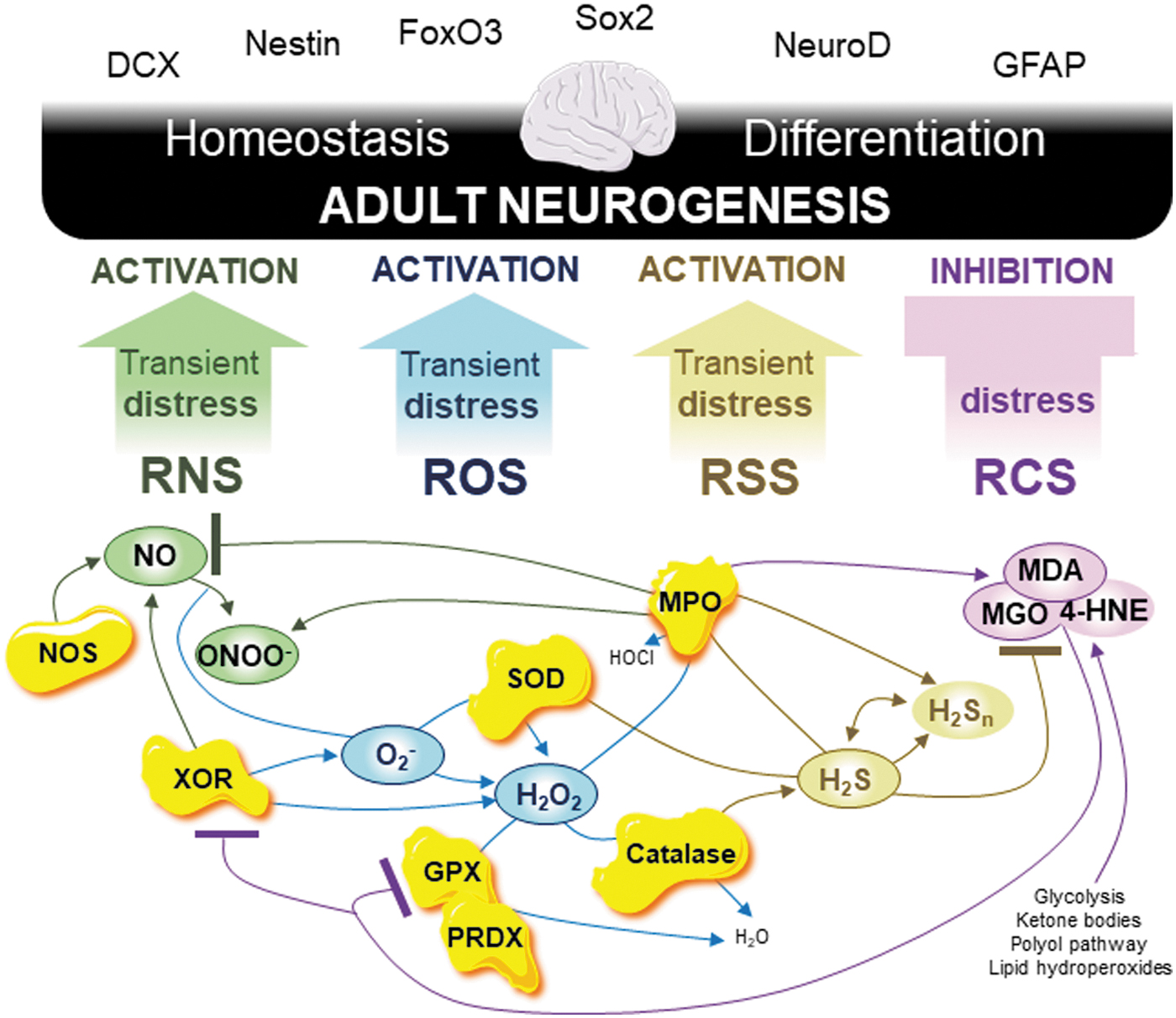
Increasing the complexity, NO generated by neuronal NOS is a negative regulator of NSC and progenitor cell proliferation in the healthy brain whereas its overgeneration after brain damage promotes proliferation and then adult neurogenesis (72, 160, 177). The PTEN and AKT proteins are inactivated by nitrosylation mediated by NO (188). Although NO has a dual role in adult neurogenesis, transient ONOO− accumulation promotes NSC and progenitor cell self-renewal, proliferation, and even differentiation through activating the hypoxia-inducible factor 1-alpha (HIF-1A) signaling pathway (43). Transient oxidative distress due to ONOO− activates adult neurogenesis. Finally, the H2S promotes NSC and progenitor cell proliferation and differentiation in association with increased phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) to activate the kinase activity and downstream targets (141, 245). All this evidence indicates that transient oxidative distress mediated by ROS, RNS, and RSS metabolism activates adult neurogenesis.
However, this is not the case for RCS metabolism. The accumulation of MDA due to lipid peroxidation inhibits NSC and progenitor cell proliferation, and MGO impairs neurogenesis by inducing cell death of NSC (47, 183). Therefore, oxidative distress mediated by RCS inhibits adult neurogenesis. Moreover, regarding key enzymes GPX and PRDX, the situation is even more complicated. Although GPX1 does not regulate adult neurogenesis, other GPXs such as GPX4 increase adult neurogenesis (191, 244, 269). The same occurs for PRDXs as PRDX1, PRDX2, and PRDX4 stimulate adult neurogenesis whereas PRDX6 inhibits adult neurogenesis (268).
In adult neurogenesis, considering interactions between the four reactive species' metabolism, including through key enzymes, the RSI is fully committed and reveals all its potential when integrated with RCS metabolism (Fig. 2).
The RSI in Brain Cell Death
Brain vulnerability increases due to neuroinflammation and decreases in neuroplasticity, dysregulation of neuronal activities, and alteration of bioenergetic metabolism (153). Brain vulnerability is also exacerbated by increased intracellular oxidation, damage, and cell death in neurons, astrocytes, oligodendrocytes, and microglial cells, at the intersection of brain physiology and disorders (153, 175). Cell death is an inevitable consequence of cell life, and there are at least 12 types of cell death (80) (Fig. 4). In the brain, apoptosis, necrosis including necroptosis, oxytosis, and ferroptosis (two names of the same pathway or two highly similar pathways), autophagy-dependent cell death, and parthanatos poly ADP-ribose polymerase 1-dependent cell death are the main cell death types that are all initiated by bioenergetic metabolism dysregulation and oxidative distress (34, 135, 170).
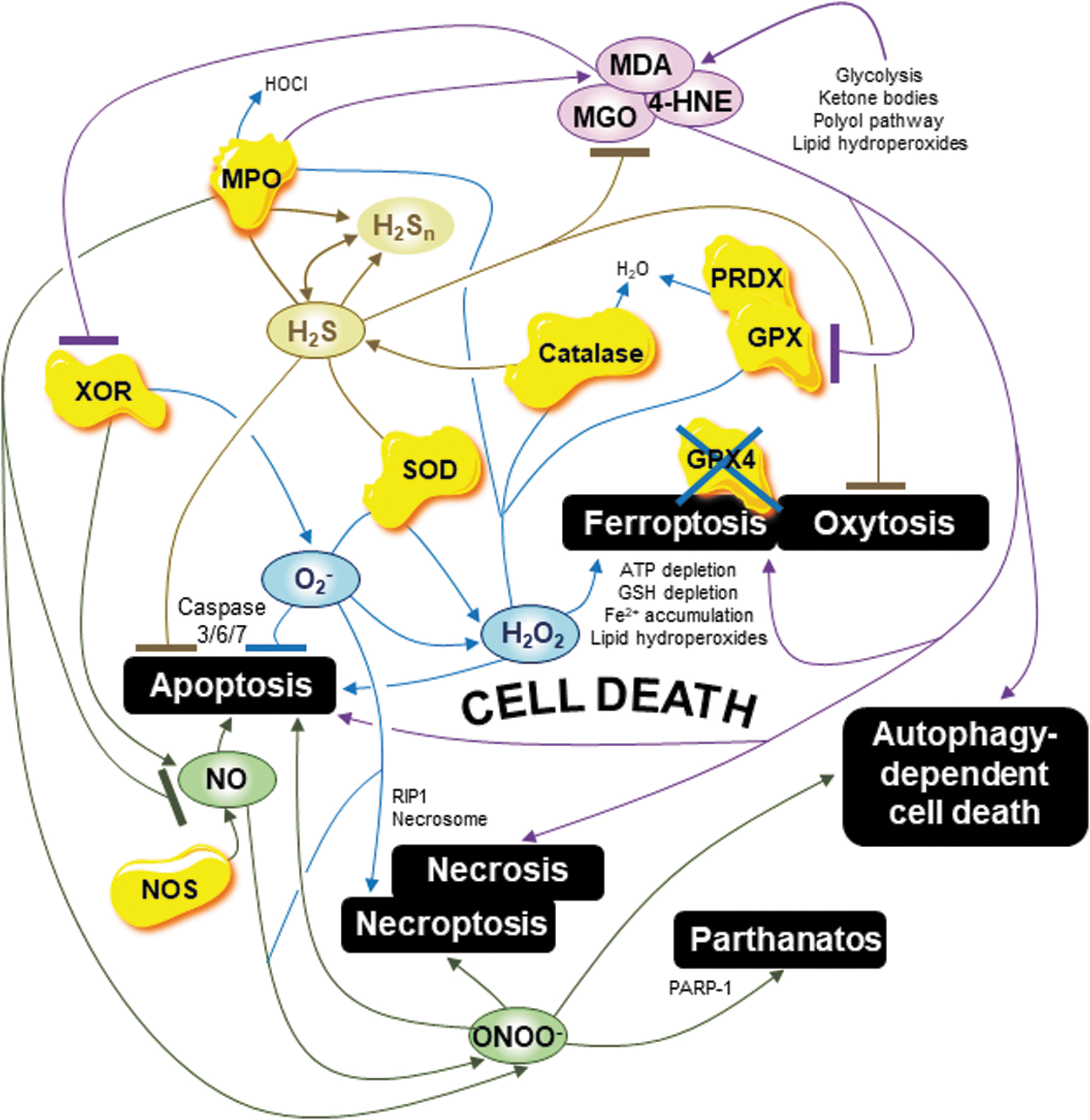
First, in the brain, mitochondrial O2 − is a major regulator of necroptosis by oxidizing the key necroptosis factor receptor-interacting protein kinase 1 (RIP1) and activating the necrosome (59). This regulation is exacerbated by hyperglycemia that induces a shift from apoptosis to necroptosis through mitochondrial O2 −, which both oxidizes necroptosis RIP1 and inactivates apoptosis factors caspases-3/6/7 (59). In other words, oxidative distress mediated by ROS O2 − triggers necroptosis and inhibits apoptosis. In addition, in the brain, H2O2 accumulation initiates the main cell death types, including ferroptosis, the newly discovered nonapoptotic cell death type characterized by accumulation of H2O2 as well as cytosolic adenosine triphosphate (ATP) depletion, active iron Fe2+ accumulation, antioxidant GSH depletion, GPX4 inactivation, and ultimately, by the deadly increase in lipid peroxidation, the most important source of RCS (34, 63, 135, 170). The excitatory neurotransmitter glutamate induces ferroptosis at high concentrations in the brain, positioning ferroptosis as an emerging major cell death nexus linking iron metabolism, lipid metabolism, bioenergetic metabolism, antioxidant metabolism, and reactive species metabolism associated with glutamate toxicity (150, 229). Moreover, the key enzyme SOD regulates apoptosis as an increase in SOD1 activity delays or even prevents apoptosis in neurons (92). Interestingly, the role of SOD1 in apoptosis is tightly linked to NO as NO mediates apoptosis, and NO–SOD balance has been shown as a key determinant in apoptosis initiation (49). An increase in the key enzyme catalase also prevents apoptosis by metabolizing H2O2 and ultimately regulating apoptosis factors, including Bax and B cell lymphoma 2 (Bcl-2) in neurons (69, 224). The key enzyme XOR also regulates both apoptosis and necroptosis through O2 − overgeneration (18). Finally, the enzyme MPO regulates apoptosis and ferroptosis through H2O2 and HOCl modulation, and MPO inhibition reduces apoptosis and ferroptosis in the brain (37, 213). The key enzymes SOD, catalase, XOR, and MPO merit further investigation in studying brain cell death.
Oxidative distress mediated by the RNS family also stimulates the main cell death types in the brain. Accumulation of NO induces apoptosis through the nitrosylation of key proteins involved in apoptosis signaling, including caspases 3/8/9 and Bcl-2, and accumulation of ONOO− activates apoptosis, necroptosis, parthanatos, and autophagy-dependent cell death in neurons, including through tyrosine nitration of heat shock protein 90 (Hsp90), α-synuclein, cytochrome c, and through DNA damage (188, 192). In contrast to RNS, oxidative distress mediated by H2S and sulfite is neuroprotective by inhibiting apoptosis and oxytosis. Anti-inflammatory and anti-apoptotic, H2S physiologically and pharmacologically reduces cell-death type activation, including after a brain injury, even though very high levels of H2S can disrupt mitochondrial function and trigger at least apoptosis in neurons (99, 274). Sulfite also protects neurons against oxytosis, a cell death type due to H2O2 accumulation, GSH depletion, lipoxygenase activation, and calcium influx (124, 135).
Finally, oxidative distress mediated by RCS is also a major cell death-type regulator to consider, at least in the brain. The MGO triggers apoptosis in neurons (55, 61, 86). Further, high levels of 4-HNE and MDA due to increased lipid peroxidation lead to apoptosis, necrosis, autophagy-dependent cell death, and, most importantly, ferroptosis (55, 61, 86). Interestingly, the key enzyme GPX1 protects immature neurons against H2O2-mediated apoptosis, and GPX4 is crucial in controlling ferroptosis (207). The key enzyme PRDX6 protects astrocytes and microglia against ferroptosis (154, 180).
Altogether, oxidative distress mediated by ROS, RNS, and RCS activates several brain cell death types, that is, apoptosis, necrosis including necroptosis, autophagy-dependent cell death, and ferroptosis; whereas oxidative distress mediated by RSS inactivates at least apoptosis and oxytosis. Therefore, considering both the RSI in its entirety, including the RCS family, and key enzymes at the intersection of the reactive species families will reveal all RSI potential in brain cell death. This information is important in understanding roles and switching between different cell death types in physiology, including aging and pathology (Fig. 4).
Is RSI the 11th Hallmark of Brain Aging?
Brain aging has a signature of 10 hallmarks: neuroinflammation, oxidative damage, adaptive stress response signaling alteration, bioenergetic metabolism dysregulation, mitochondrial dysfunction, aberrant neural network activity, neuronal calcium homeostasis dysregulation, stem cell exhaustion, molecular waste disposal impairment, and DNA repair impairment (153, 264) (Fig. 5). Multiple pathways are involved in brain aging through many different biomarkers not listed here to focus our overview on the reactive species themselves. Impaired adult neurogenesis, overactivation of cell death including apoptosis, and cellular replicative senescence are involved in brain aging (153, 264).
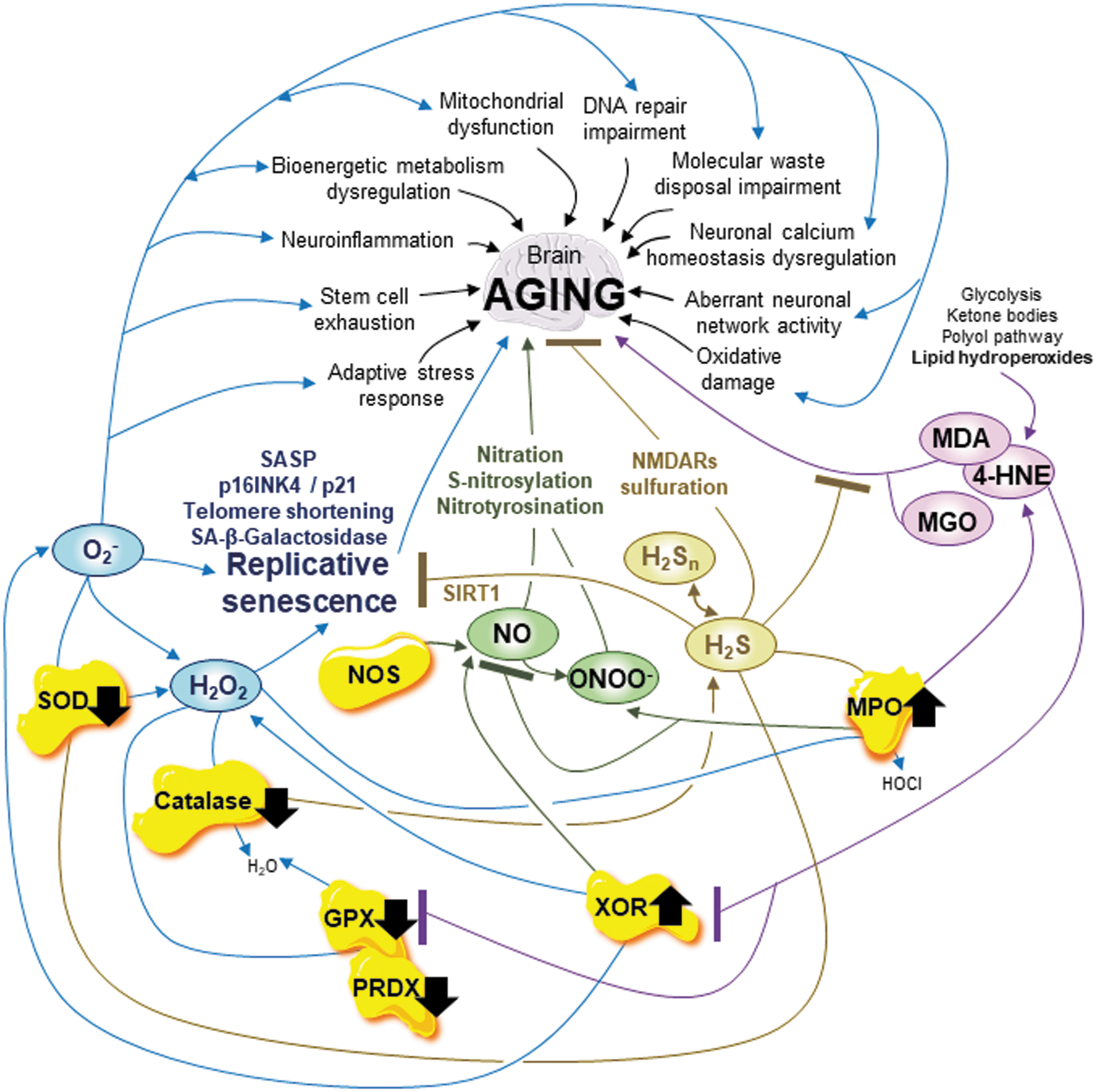
It is important to remember that replicative senescence is probably a key driver of aging (14, 153, 273). Replicative senescence, a powerful anticancer mechanism, is a stable cell cycle arrest associated with telomere shortening, senescence-associated beta-galactosidase (SA-β-gal) activity, kinase inhibitor proteins p16INK4 and p21 activation, and a senescence-associated secretory phenotype (SASP) including proteases, growth factors, and pro-inflammatory cytokines (133, 264). Senescence is both a physiological and pathological process related to several reactive species and is implicated in aging, including that in postmitotic neurons and oligodendrocytes, as recent evidence argues that senescence occurs in these cells (14, 133, 153).
We propose that dysregulated RSI is the 11th hallmark of brain aging that links cellular replicative senescence, cell death types, adult neurogenesis, and the 10 established hallmarks (Fig. 5).
First, at a minimum, mitochondrial O2 − accumulation and antioxidant defense decline are the major events that trigger the 10 hallmarks of brain aging (153). Oxidative distress mediated by mitochondrial O2 − in the brain, including the frontal cortex, hippocampus, and amygdala, initiates brain aging (228). Interestingly, in mitotic astrocytes, highly glycolytic compared with other glial cells, mitochondrial O2 − does not strongly contribute to cellular senescence whereas its contribution is significant in other glial cells (228). Overgeneration of H2O2 also leads to senescence of mitotic astrocytes with SA-β-gal activity and probable SASP, and mitotic microglial cells with telomere shortening, SA-β-gal activity, and probable SASP (153, 256). Compared with astrocytes, postmitotic neurons, which are much less glycolytic, and thus more dependent on mitochondrial function, generate substantial mitochondrial O2 − (27, 228). Moreover, overgeneration of H2O2 seems to lead to senescence of human neural progenitor cells and postmitotic neurons with SA-β-gal activity and probable SASP characteristics (153, 256, 273). All this evidence indicates that oxidative distress mediated by H2O2 initiates brain aging. In addition, key enzymes SOD, catalase, XOR, and MPO play key roles in aging, including in the brain as brain aging is associated with progressive decline in SOD and catalase activities. The use of SOD/catalase mimetics may contribute to reversing age-related declines including in cognitive function, and as XOR and MPO activities dangerously increase with aging (50, 87, 243, 250). Antioxidant defense decline and oxidative distress mediated by ROS are clearly pro-aging, including in the brain.
The RNS metabolism is also actively involved in brain aging, although heterogeneously in nitrosative vulnerability depending on brain cell types (26, 27, 228). Mitotic glycolytic astrocytes are not vulnerable to NO and ONOO− whereas postmitotic neurons, which are more dependent on mitochondrial respiration, are vulnerable to both NO and ONOO− (26, 27, 228). NO, both an anterograde and a retrograde neurotransmitter in synapses, and ONOO− contribute to protein nitration, nitrosylation, and, consequently, brain aging (188). Aging, including in the brain, is associated with nitrative damage, and nitrosylation of amyloid β-peptides, and proteins involved in regulation of immune response, including S100, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (188). Oxidative distress mediated by RNS is pro-aging, including in the brain.
Reactive sulfur species metabolism is also actively involved in brain aging through H2S and H2Sn (polysulfide), which are suggested to act as gliotransmitters (147, 149). Oxidative distress mediated by H2S and H2Sn supports brain function, including memory formation during aging by modulating N-methyl-D-aspartate subtype glutamate receptors through sulfuration (147, 149). The NMDARs are excitatory glutamate receptors involved in neuronal plasticity, and learning and memory. They are dysregulated during brain aging and numerous brain pathologies (147, 149). In addition, H2S also exerts anti-aging properties by inhibiting ROS and RNS generation, and by activating sirtuin 1 (SIRT1), a major anti-aging and anti-senescence nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylase found in the brain (277). Oxidative distress mediated by RSS is anti-aging, including in the brain.
Finally, oxidative distress mediated by 4-HNE and MDA is pro-aging. The 4-HNE and MDA are overgenerated by increased lipid peroxidation and accumulate in tissues and organs with age, including in the brain (35, 115, 153). However, with no clear molecular explanation, a progressive decrease in MGO levels associated with an increase in aldolase reductase expression, which metabolizes MGO, occurs during brain aging, thus suggesting an unsolved key role of MGO in brain aging (127). Moreover, during brain aging, reduced GSH decreases whereas oxidized GSSG increases, and key enzymes GPXs (including GPX4 activities) as well as PRDXs (including PRDX6) decline (15, 45, 71, 145, 176, 281). Interestingly, GPX4 activity plays a key role at the intersection of apoptosis and ferroptosis, an important topic in aging research, including that exploring the brain (15, 96, 270). An increase in GPXs and PRDXs expression and activity that improve detoxification of lipid hydroxyperoxides is a promising route of anti-aging therapy exploration to limit detrimental oxidative distress due to 4-HNE and MDA in brain aging.
Considering that antioxidant defense decline and oxidative distress mediated by ROS, RNS, and 4-HNE and MDA are pro-aging whereas oxidative distress mediated by RSS is anti-aging, including in the brain, the entire RSI, including RCS, has to be further investigated. A dysregulated RSI could be the 11th hallmark of brain aging (Fig. 5).
Conclusion on the RSI in Brain Physiology
Through adult neurogenesis, cell death, and aging in the brain, we showed that the entire RSI should be considered instead of only one reactive species family. Knowing (i) exactly which reactive species is active in, that is, identification, and (ii) the physiological or supraphysiological concentration, that is, concentration integrating oxidative eustress and distress, will reveal all the RSI potential in physiology, including in the brain.
Reactive species family roles vary in the physiological process, and a family is not solely detrimental or beneficial. Oxidative distress mediated by ROS, RNS, and RSS promotes adult neurogenesis, whereas oxidative distress mediated by RCS inhibits adult neurogenesis. Oxidative distress mediated by ROS, RNS, and RCS triggers apoptosis, necrosis, autophagy-dependent cell death, and ferroptosis, whereas that mediated by RSS inhibits at least apoptosis and oxytosis. Oxidative distress mediated by ROS, RNS, and RCS is pro-aging, whereas that mediated by RSS is anti-aging. Together, the RCS family is coordinated with the other reactive species families, and therefore only the RSI, including RCS, is fully committed in brain physiology.
Finally, key enzymes SOD, catalase, XOR, MPO, GPXs (including GPX4), and PRDXs require further examination in light of the entire RSI, including RCS in brain physiology. In a physiological process, the global redox effect is created by the combination between (i) effects from different reactives species families in different concentrations, (ii) interactions between the different families, and (iii) activities of the key enzymes that contribute to the RSI dynamics. This axiom is true in physiology and pathologies.
The RSI in Mental Disorder Depression
Oxidative stress, or better, oxidative distress, plays a significant role in mental disorders from addiction to anxiety, depression, bipolar disorder, post-traumatic stress disorder, dementia, and schizophrenia affecting almost 1 billion people worldwide (38, 189, 240). Depression affects 300 million people worldwide, thus being the leading mental disorder, and by 2030, depression is predicted to be the second leading cause of illness worldwide, after human immunodeficiency virus infection/acquired immunodeficiency syndrome and followed by ischemic heart disease (102, 152).
Different complementary approaches such as transcriptomic, proteomic, metabolomic, and epigenetic depression, including major depression, have been progressively better characterized to improve treatments, although complex molecular mechanisms are involved (230). More than 15 biomarkers are predictive of this mental disorder, including a reduction in gray matter volume, overgeneration of the stress hormone cortisol, dopamine and noradrenaline reduction, glutamate accumulation, neuroinflammation, tryptophan metabolism alterations, brain-derived neurotrophic factor (BDNF), and vascular endothelial growth factor (VEGF) reduction, and possibly genetic polymorphisms (230) (Fig. 6). The antioxidant defense and reactive species and related targets are also increasingly considered biomarkers for depression, although the molecular mechanisms are still missing (230).
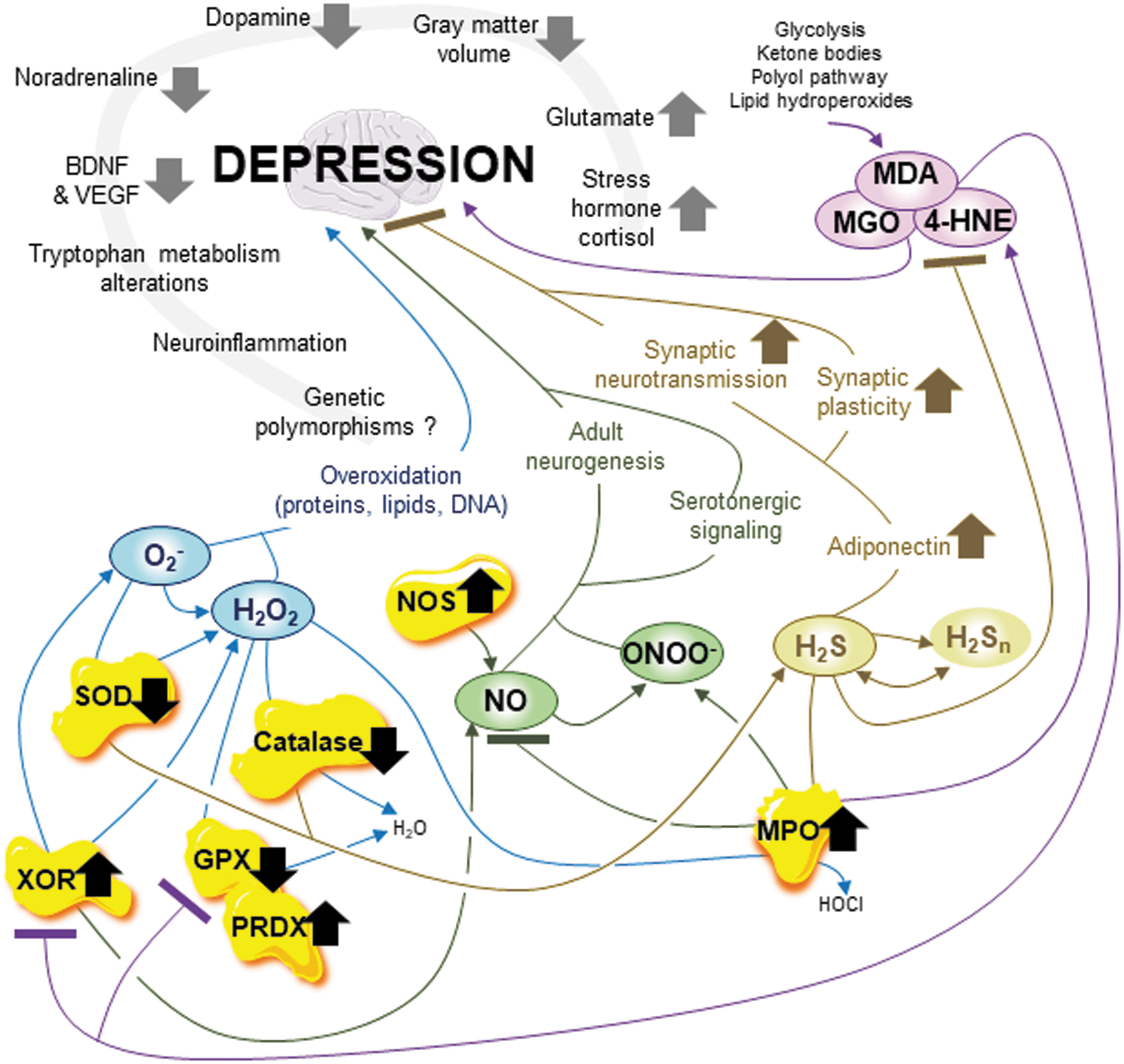
The entire RSI, including RCS, should be considered in the study of mental disorders, including depressive disorder, to improve the understanding of the targets and the molecular mechanisms (Fig. 6). First, an imbalance between O2 − and H2O2 due to specific SOD2 genetic polymorphisms increases the risk of depression (54). Overgeneration of ROS, antioxidant defense alterations including SOD dysfunction, and decrease in GSH levels are involved in major depressive disorder through harmful overoxidation of proteins, lipids, and DNA (143, 199, 249). Specific genetic polymorphisms of catalase are also associated with an increased risk of depression (199). A decrease in SOD and catalase activities and increased XOR levels are often found in depressed patients compared with healthy individuals (249). The XOR activity also increases in recurrent depressive episodes in many brain regions, including the hippocampus and thalamus (156). In addition, the accumulation of MPO enzyme in serum is a biomarker of immune dysregulation associated with major depressive disorder (246). Altogether, an imbalance between ROS, oxidative distress mediated by ROS, and altered SOD, catalase, XOR, and MPO enzymes exert a pro-depressant effect.
Interestingly, it is now known that anti-depressant drugs modulate ROS/RNS generation and antioxidant defense (199, 249). Reactive nitrogen metabolism is involved in depression, including major depressive disorder. In depression, the neuronal NOS enzyme accumulates in several brain regions, including the hippocampus, cortex, and hypothalamus (279). Researchers have proposed that the interaction between nNOS and serotonergic signaling, and the link between nNOS and adult neurogenesis are the key deregulated mechanisms in depression (279). Inhibition of nNOS with NG-nitro-L-arginine methyl ester exhibits antidepressant properties, and several weeks of antidepressant treatment reduce NO levels in depressed patients (199, 249, 279). Thus, related to nNOS accumulation, oxidative distress mediated by RNS exerts a pro-depressant effect.
Importantly, by increasing synaptic neurotransmission and synaptic plasticity, the H2S has an antidepressant-like effect (41). This effect is through the induction of adiponectin expression, a hormone involved in glucose and lipid metabolism, that ultimately reduces autophagy and favors synapse formation in the brain, including in the hippocampus (241). Oxidative distress mediated by RSS exerts an anti-depressant effect.
Finally, an increase in lipid peroxidation, the main source of RCS, is associated with depression. Treatment with n-3PUFAs, including docosahexaenoic acid, appears as effective as antidepressant therapy by inhibiting neuronal apoptosis and reducing neuroinflammation (249). The MGO, MDA, and 4-HNE that accumulate in depressed patients are considered biomarkers of depressive disorder, and anti-depressant therapy effectively reduces their levels (62, 143, 198, 249). Oxidative distress mediated by RCS exerts a pro-depressant effect. In several studies, a decrease in key enzyme GPX activity was found in depressed patients compared with healthy individuals, whereas in other studies, no difference was detected (143, 249). Finally, associated with decreased BDNF growth factor in serum, the key enzyme PRDX1 accumulates in depressed patients (276).
Oxidative distress mediated by ROS, RNS, and RCS exerts a pro-depressant effect, whereas oxidative distress mediated by RSS exerts an anti-depressant effect. The entire RSI, including RCS, plays a clear role in depression (Fig. 6).
The RSI in Brain Ischemia
When a blood clot blocks or plugs an artery or vein in the brain, an arterial or venous occlusion, an ischemic stroke occurs, creating a medical emergency for which brain aging is the highest nonmodifiable risk factor (28, 65, 113, 153, 202). Every year, stroke, which includes hemorrhagic stroke (20% of the cases) and ischemic stroke (80% of the cases), accounts for ∼80.1 million cases worldwide and 5.5 million deaths (28, 65, 113, 202). After a stroke, per hour, 120 million neurons die, 830 billion synapses and 714 km/447 miles of myelinated fibers are lost, so the brain is losing nearly 3.6 years of normal aging (202). To treat arterial or venous occlusion, rapid reperfusion with thrombolysis and endovascular thrombectomy is the main therapy, and though it provides benefits, it also presents adverse effects regarding oxidative distress (28). Ischemic stroke provokes rapid oxygen and glucose deprivation, inducing excitotoxicity due to glutamate release, mitochondrial dysfunction, oxidative distress, lipid peroxidation, DNA damage, neuroinflammation, and neuronal cell death through at least apoptosis and necrosis, including necroptosis (57, 153) (Fig. 7). Oxygen deprivation followed by reoxygenation induces an increase in the blood–brain barrier (BBB) permeability, partially through activation of the hypoxia-inducible transcriptional factor HIF-1 and an increase in expression of its target genes such as matrix metalloproteinase 2 and VEGF (171, 208). Oxygen deprivation, resulting in tissue hypoxia, induces oxidative distress due to, a minimum, mitochondrial respiration deterioration and loss (91, 153). Then, rapid oxygen supply, that is, reoxygenation due to reperfusion, exacerbates oxidative distress due to, minimally, mitochondrial respiration (57, 91, 153).
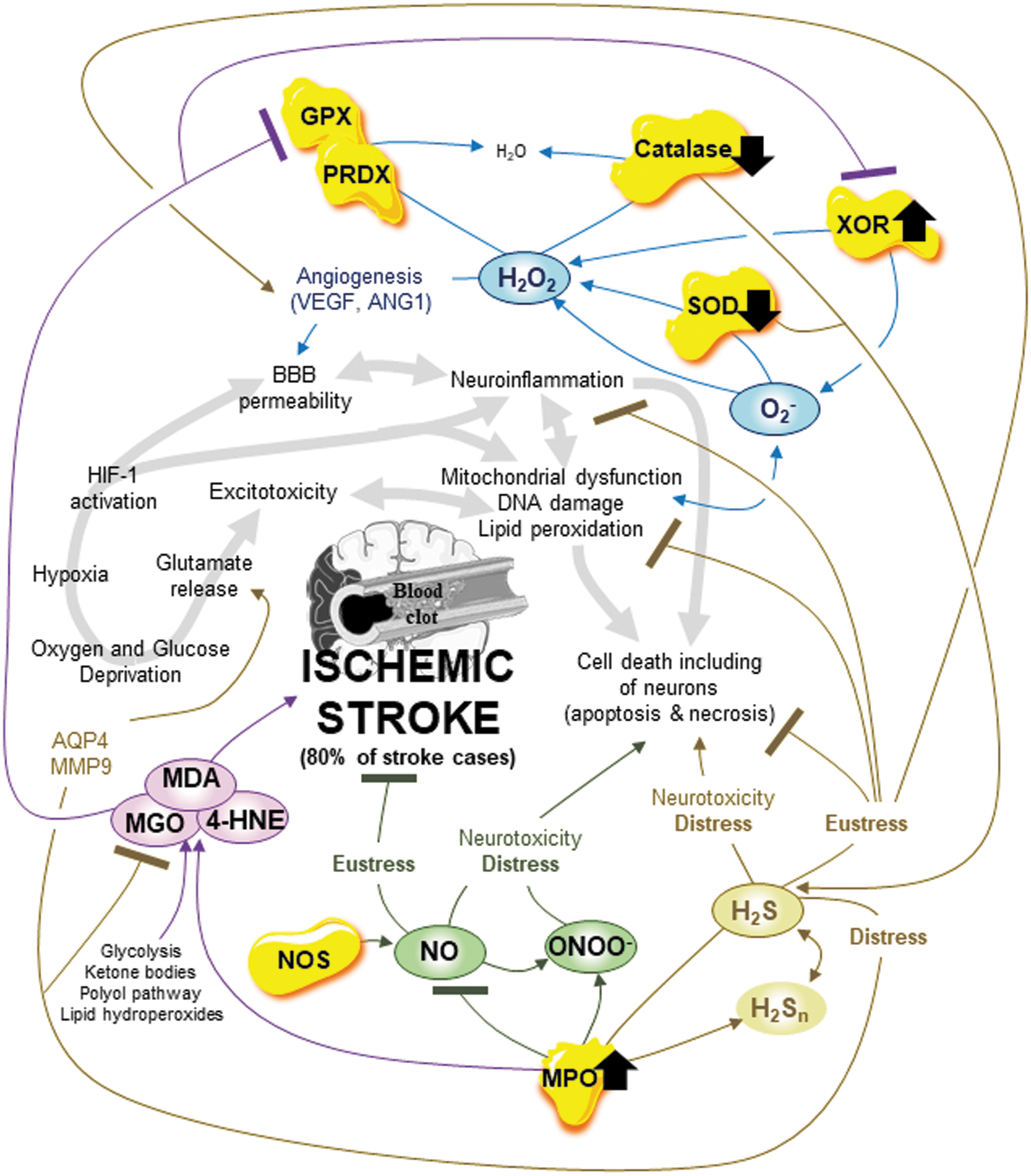
First, during ischemia/reperfusion (I/R), mitochondrial O2 − overgeneration due to oxygen deprivation and then to reoxygenation is a major contributor to the mitochondrial OXPHOS electron leak at complexes I/II and to complex V ATP synthase dysfunction. These two alterations play a critical role in neuronal cell death, including apoptosis (112). In addition, mitochondrial NOXs are important O2 − sources during I/R (112, 150, 151). Notably, although oxidative distress mediated by mitochondrial O2 − is detrimental during I/R, oxidative eustress mediated by mitochondrial O2 − plays a signaling role in activating antioxidant defense and cell survival in postischemic tissues (32, 112, 243, 258, 259, 267). In BBB endothelial cells, changes in H2O2 levels strongly regulate angiogenesis, permeability, and cell death (6). Oxidative eustress mediated by H2O2 induces endothelial tube formation and increases endothelial cell tube length, whereas oxidative distress mediated by H2O2 increases endothelial cell permeability and induces cytoskeleton disorganization and cell death apoptosis, although the cytoskeleton alteration starts during oxidative eustress (6). Moreover, mitochondrial calcium overload during I/R blocks H2O2 catabolism in the brain (112). Regarding the key enzymes, during I/R, SOD and catalase activities decrease in the brain, mainly in the hippocampus and striatum, whereas XOR and MPO activities harmfully increase in the patient (40, 103, 104). Altogether, during I/R, oxidative eustress mediated by ROS is beneficial whereas oxidative distress mediated by ROS and altered SOD, catalase, XOR, and MPO enzymes is detrimental.
The RNS metabolism plays a critical role during I/R through both its reactive species and key NOS enzymes. Depending on cell types, levels, and sources, the NO can be either detrimental or beneficial in ischemic stroke (44). After reperfusion, overgenerated NO by nNOS plays a rapid neurotoxic role (44, 85, 239). Hours after reperfusion, in microglia, astrocytes, and endothelial cells, estimated at 1000 times more than nNOS-related NO, NO overgenerated by activated iNOS also plays a neurotoxic role (44, 85, 239). In contrast, eNOS-related NO plays a neuroprotective role by interacting with ROS metabolism and inducing collateral flow (44, 85, 239). Although the role played by NO is known to vary in ischemic stroke, the debate continues regarding the contribution of nNOS, iNOS, and eNOS, the time course of NO generation from the three NOS, and other factors (42, 44, 85). Moreover, oxidative distress mediated by ONOO− is neurotoxic after ischemic stroke, and blocking it using triterpenoid saponin is suggested as a promising treatment to attenuate the deleterious effects (39). In ischemia, during I/R, the key NOS enzymes are essential regulators of both the detrimental and beneficial roles of NO, and oxidative distress mediated by ONOO− is detrimental.
H2S also contributes to maintaining cerebrovascular homeostasis, including in ischemia but still with controversial effects (66, 90, 112, 166, 254). Indeed, in ischemia, oxidative eustress mediated by H2S is beneficial whereas oxidative distress is detrimental, that is, neurotoxic (66, 90, 112, 166). The benefits of H2S effects are observed through anti-oxidation property via increasing GSH levels, anti-apoptosis by regulating calcium homeostasis and caspase status, anti-necrosis, and attenuation of the deleterious effects of I/R related to mitochondrial function (66, 90, 112). Benefits are also derived through the anti-inflammatory property via inhibiting the release of NO and pro-inflammatory cytokines and proangiogenic property by induction of the expression of the angiogenic factors angiopoietin-1 and VEGF (66, 90, 112). In contrast, the detrimental effect of H2S is through glutamate excitotoxicity and brain edema by negatively modulating aquaporin 4 and matrix metalloproteinase 9 status (66, 90, 112).
Finally, the RCS family is negatively involved in ischemic stroke. In different organs, plasma and serum MGO, which triggers autophagy activation, accumulates along with 4-HNE, considered a plasma biomarker of ischemic stroke, and MDA (33, 68, 73, 134, 169). In addition, activities of GPX, including GPX1 and PRDX, appear critical during and after ischemia, including related to neuroinflammation (57, 84, 108). Thus, in ischemia, oxidative distress mediated by RCS and key enzymes GPX and PRDX are detrimental.
During I/R, oxidative eustress mediated by ROS and RSS, and oxidative distress mediated by eNOS-related NO are beneficial to counteract deleterious effects of I/R. Meanwhile, oxidative distress mediated by ROS, nNOs- and iNOS-related RNS, RSS, and RCS is detrimental. Notably, in other pathologies, the key enzymes SOD, catalase, XOR, and MPO play essential redox roles during I/R (Fig. 7).
Why Did Antioxidant Therapies Fail to Treat Ischemia?
Several therapies introduced to reduce the deleterious effects of I/R are promising. Specific upregulation of mitochondrial antioxidant enzymes Mn-SOD (SOD2) and PRDXs, inhibition of mitochondrial NOX, reduction of oxidative distress with specific mitochondrial antioxidants such as mitoquinone, modulation of mitochondrial redox status using specific microRNAs, and even ROS-scavenging nanoparticles are among the potential therapeutic pathways (112, 185). However, antioxidant therapies targeting ROS have been disappointing as results could not be successfully translated from animal models of stroke, including ischemic stroke, to clinical use (111, 117, 215, 255). For example, uric acid, a natural antioxidant that targets ROS and RNS peroxynitrite, ROS scavengers edavarone, tetramethylpyrazine, FR210575, and even nitrone NXY-059, were neuroprotective and succeeded in treating the deleterious effects of stroke in animal models but failed to prove efficacious at the clinical level (215, 255). After two large randomized and double-blinded clinical trials with more than 5000 patients totally, the promising nitrone NXY-059 did not show any bedside efficacy (215, 255). In addition, although the promising natural plant extracts Egb-761® or Tanakan® from Ginkgo biloba with antioxidant terpenes and flavonoids had neuroprotective effects in animal stroke models, clinical trials did not confirm the results, and larger and robust trials are needed (111, 255). The rationale for using antioxidant therapy, in addition to other therapy against stroke, including ischemic stroke, is to potentially prevent long-term neurodegeneration poststroke by decreasing the risk of Alzheimer's disease (AD) through the reduction of oxidative stress (117).
So, why did antioxidant therapies fail to treat ischemia? First, animal model success does not always translate to clinical trial and bedside success due to differences in complexity, physiology, and pathology, including symptoms and alterations (21). Second, importantly, antioxidant therapies fail due to disappointing targets. Targeting only ROS or RNS is insufficient when the entire RSI is involved with its many reactive species and key enzymes. Targeting ROS, RNS, RSS, and RCS together, considering oxidative eustress and distress effects, and modulating SOD, catalase, XOR, MPO, GPXs, and PRDXs enzymes will be the most powerful therapy to treat ischemia. Regulating the entire RSI will induce key enzyme remodeling, and the best therapy will be one molecule or a combination of molecules directly targeting the heart of the RSI. Therefore, we strongly encourage increased research to better understand the RSI related to the pathophysiology of stroke as the first step to discover the best therapy.
The RSI in Neurodegenerative Disorders
Oxidative distress, bioenergetic metabolism, senescence, and cell death are intriguing and complex key players involved in the so-called neurodegenerative disorders, the leading cause of disability worldwide and a major cause of global deaths. These disorders include Parkinson's disease (PD), AD, vascular dementia, frontotemporal dementia, amyotrophic lateral sclerosis (ALS), Huntington's disease, and spinocerebellar ataxias (14, 32, 75, 81, 153, 232). Adult neurogenetic alteration and neuroinflammation are also involved in neurodegenerative disorders (14, 32, 75, 81, 153, 232) (Fig. 8).
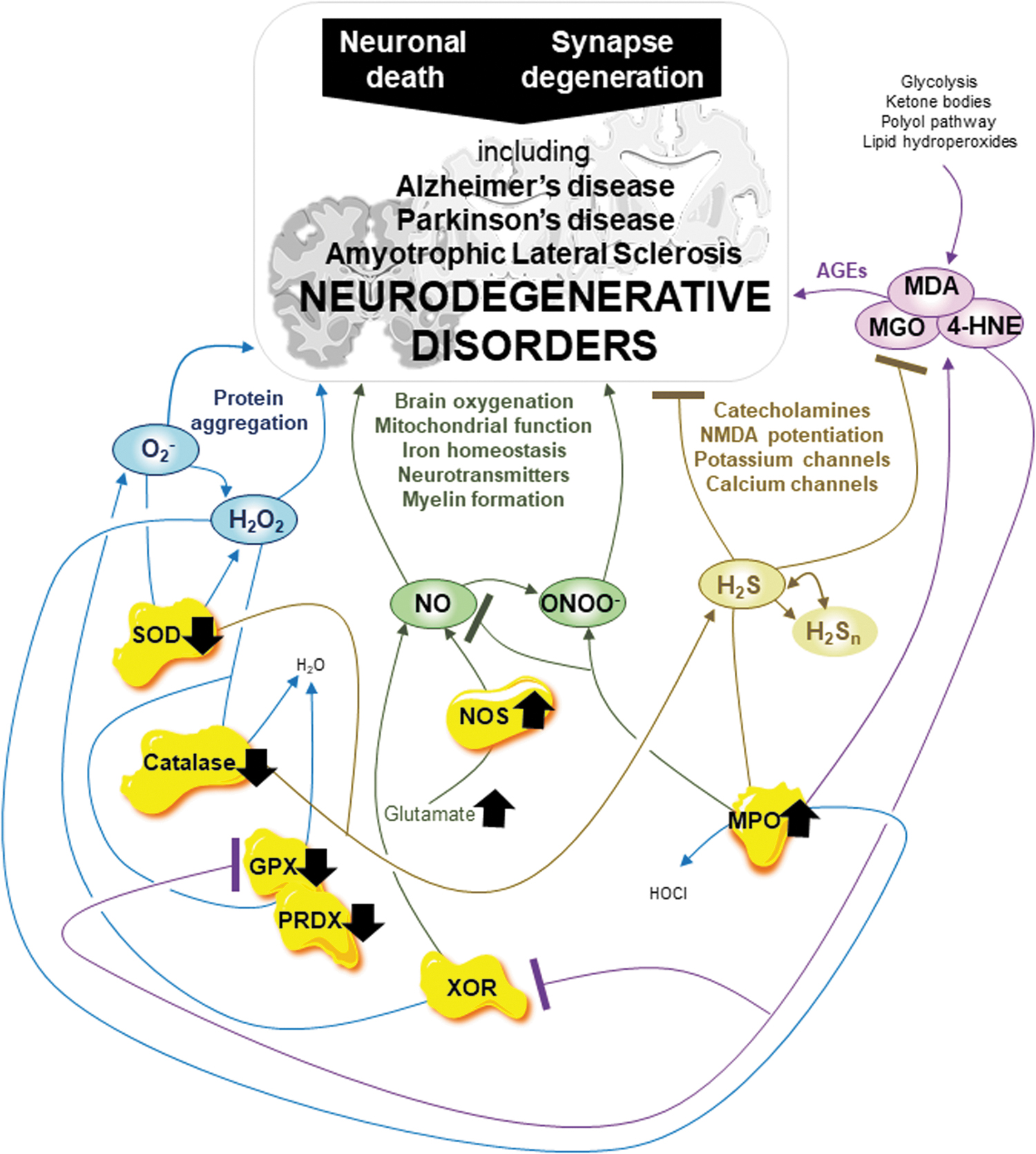
At the molecular level, most neurodegenerative disorders share pathways focusing on mitochondrial function, oxidative distress, the autophagy-lysosome pathway, protein quality control machinery, and finally, neuronal death, therefore implying the involvement of multiple complex biomarkers (81, 204). For example, neuronal death is the PD hallmark mediated by intracellular accumulation and aggregation of the presynaptic protein α-synuclein in Lewy bodies (153). Further, both neuronal death and synapse degeneration are AD hallmarks mediated by amyloid-β (Aβ) clearance processing impairment and phosphorylated tau neurofibrillary tangle aggregation (153). In addition, mitochondrial function, calcium homeostasis, lysosome, and, importantly, redox balance play crucial roles in both neurodegenerative disorders (153). Mitochondrial dysfunction and oxidative distress are promising targets alone or combined to treat several neurodegenerative disorders, including AD and PD (36, 153, 204, 252, 253).
One significant unknown is whether oxidative distress is a cause or consequence of neurodegeneration (7). A better understanding of the RSI, including RCS and the key enzymes, will help answer this question and discover new therapies to treat neurodegenerative disorders, including ALS, PD, and AD (Fig. 8).
First, related to mitochondrial uncoupling and mitochondrial calcium concentration, oxidative distress mediated by O2 − is associated with AD (262). Oxidative distress mediated by H2O2 occurs during the early stages of protein aggregation in AD and dementia (237). In addition, reduced GSH levels are found in ALS, AD, and PD (167). Importantly, the key enzymes SOD, catalase, XOR, and MPO also play complex and crucial roles in neurodegenerative disorders, including ALS, AD, and PD (167). Reduction of both SOD and catalase activities is found in ALS, whereas SOD levels increase in PD (167). In AD, SOD levels increase in the hippocampus and amygdala whereas SOD and catalase levels decrease in the frontal and temporal cortex (167). Inhibition of XOR by oxypurinol (alloxanthine) in a mouse model of AD and by allopurinol in the rat model of PD reduces oxidative distress mediated by ROS, thus suggesting a key role for XOR at least in those neurodegenerative disorders (82). Moreover, in neurodegenerative disorders, including ALS, AD, and PD, increased MPO activity is detrimental (194). Oxidative distress mediated by ROS, in addition to alterations in key enzymes SOD, catalase, XOR, and MPO, is detrimental to neurodegenerative disorders.
The RNS metabolism is also involved in neurodegenerative disorders. Overgeneration of NO and related ONOO− is associated with neurodegeneration through a close link with iron homeostasis, which regulates ferroptosis, brain oxygenation, mitochondrial function, neurotransmitter metabolism, and compact myelin formation (140, 227). NO also regulates iron homeostasis at a minimum through ferritin and ferroportin, and iron levels are strongly increased in Aβ plaques and tau tangles of AD patients and the SN of PD patients (139, 140, 162). In AD, iNOS, eNOSs, and nNOS increases are associated with Aβ deposits in the brain and reduced NO bioavailability in plasma (167). In addition, glutamate, the most abundant neurotransmitter in excess in neurodegenerative disorders, initiates NO overgeneration by nNOS, which strongly induces demyelination, that is, the loss of myelin (139, 140, 162). Thus, in neurodegenerative disorders, oxidative distress mediated by RNS is detrimental.
In addition to ROS and RNS, RSS metabolism must be considered when studying neurodegenerative disorders. H2S is attracting much attention in neurodegenerative disorder research through its effects on neuroinflammation, neurodegeneration-related MAO linked to catecholamines, N-Methyl-D-Aspartate acid (NMDA) potentiation linked to glutamate, potassium channels, calcium channel activity, and finally, oxidative distress (25, 182, 200). In the brain, H2S levels are reduced in AD and PD (182). Interestingly, H2S also inhibits the NO-related angiogenic effect, just as NO inhibits the enzymatic effect of H2S by modulating the activity of the H2S-producing CBS enzyme (25, 182, 200). Moreover, NO, H2S suppresses Aβ plaque formation and deposits in microvessels, including in the brain (25, 182, 200). Although very high levels of RSS are neurotoxic, oxidative distress mediated by RSS is beneficial in fighting neurodegenerative disorders.
Finally, oxidative distress mediated by RCS and key GPX and PRDX enzyme breakdown is detrimental to neurodegenerative disorders. Overgenerated by an excess of lipid peroxidation, MGO, 4-HNE, and MDA accumulate in the brain of ALS, PD, and AD patients, thus resulting in the formation of AGEs (60, 77, 167, 231). In addition, GPXs and PRDXs appear to play important roles in neurodegenerative disorders as neurodegeneration is associated with GPXs and PRDXs breakdown. Although it is unclear whether GPXs levels either increase, decrease, or do not change in ALS, both GPX1 and GPX4, new players in neurodegeneration, are promising targets to activate in PD and AD (30, 167, 214). Activation of PRDXs enzymes is also a promising therapy for ALS, PD, and AD (234).
Present evidence suggests that in neurodegenerative disorders including ALS, PD, and AD, oxidative distress mediated by ROS, RNS, and RCS and altered SOD, catalase, XOR, MPO, GPXs, and PRDXs are detrimental. Interestingly, oxidative distress mediated by RSS is beneficial in the fight against neurodegenerative disorders, although high levels of H2S should be addressed carefully to limit neurotoxic side effects. Once again, the entire RSI, including RCS and the key enzymes, are major players in neurodegeneration and should be considered together in future studies (Fig. 8).
The RSI Is a Promising Target for the Treatment of Neurodegenerative Disorders
Researchers in the neurodegenerative disorder and antioxidant fields need to face the difficult truth that current antioxidant therapies available to treat neurodegenerative disorders have been less than successful. Indeed, all the tested antioxidant drugs that tend to reduce oxidative distress mediated by ROS, increase GSH levels, and increase SOD and catalase activities from in vitro to animal models ultimately failed at clinical trial levels (167). These include edavarone, SOD/catalase mimetics EUK-8 and EUK-134 in ALS, valproic acid, melatonin, selegiline, deferoxamine in PD, vitamin C, resveratrol and curcumin in AD, coenzyme Q10 in PD and AD, and vitamin E (tocopherol) in ALS, PD and AD, and PD (167).
Complexity is one likely reason why a loss in translation occurs from animal models to human physiology (21). Targeting only one reactive species family is not sufficient. The most powerful therapy for neurodegenerative disorders will target ROS, RNS, RSS, and RCS together and the key enzymes that act on them. Therefore, we strongly encourage the research community to improve their understanding of the RSI regarding the pathophysiology of neurodegenerative disorders, including ALS, PD, and AD, as the first step to discovering the best therapy.
The RSI in Brain Cancers
Aging is the biggest risk factor for cancer, which is a leading cause of death globally (56). Cancer is a group of more than 100 malignant diseases throughout the body in which cells grow uncontrollably (56, 125). Glioma accounts for 80% of malignant primary brain cancers (129). They are categorized into three types: oligodendroglioma, ependymoma, and astrocytoma, including the most frequent and the most lethal primary brain cancers, the glioblastoma (129). In addition, it is estimated that malignant secondary brain cancers, called brain metastases, occur in 20% of all the patients with primary cancer with other origins, mainly lung, breast, colorectal, and melanoma cancers (1). In primary brain cancers and metastases, coordination between bioenergetic metabolism and hypoxia plays a critical and harmful role in carcinogenesis, cancer progression, and cancer cell dissemination (22, 98, 179) (Fig. 9).
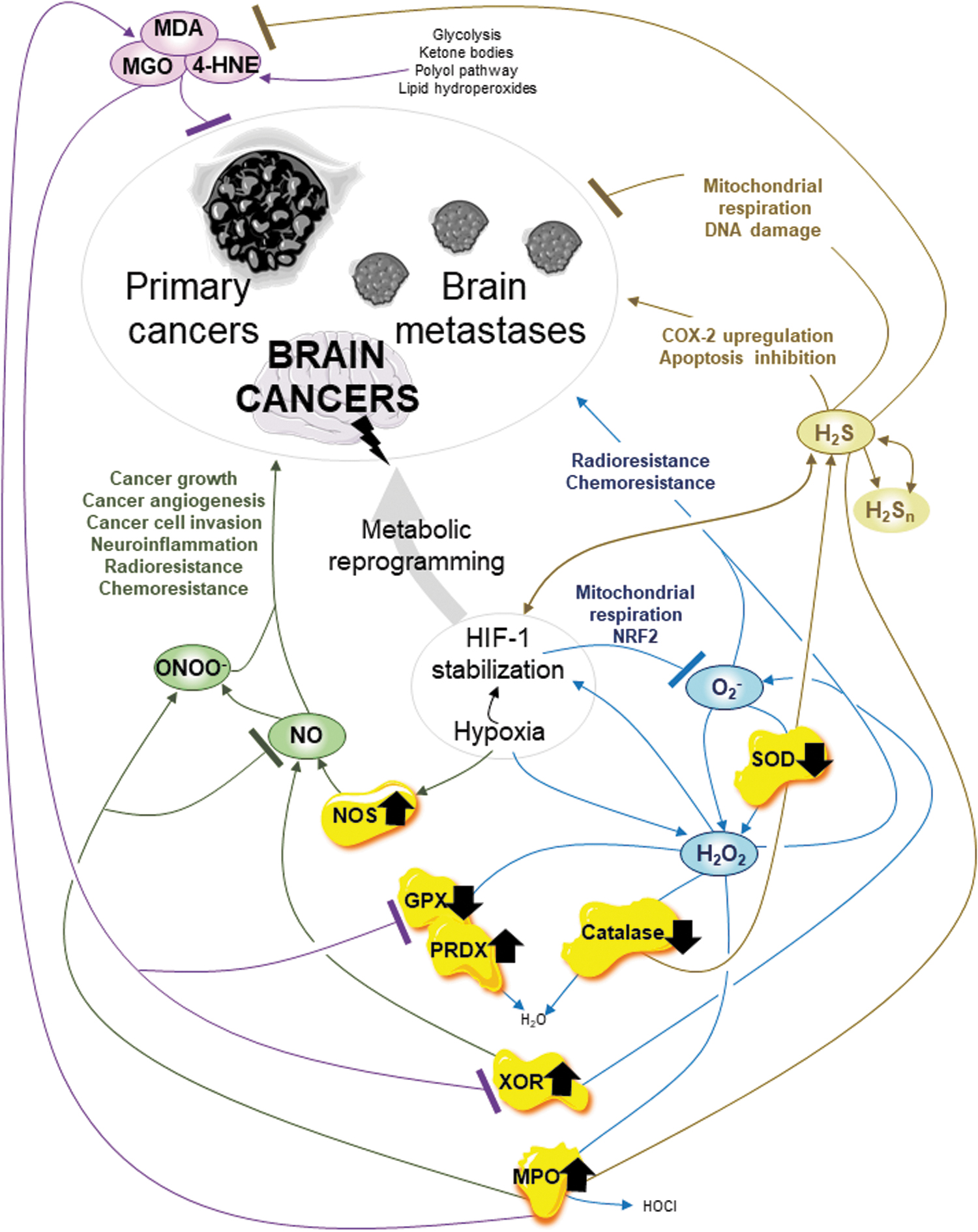
Interestingly, hypoxia, which results from the disequilibrium between oxygen delivery and consumption, is a pervasive stimulus at physiological and pathophysiological levels, including in cancer (3, 67, 163). Hypoxia induces metabolic changes in the cancer cell, in cancer stem cell quiescence, a reversible state of nondividing cells that retain their ability to re-enter cell division, and ultimately, in cancer growth, including in glioblastoma and anticancer therapy resistance such as radiotherapy (3, 10, 67, 88, 163). Associated with hypoxia, the redox homeostasis dysregulation, one of the hallmarks of cancer, increases in antioxidant defense, increases ROS levels, inactivates apoptosis, and provides a metabolic switch from mitochondrial OXPHOS to glycolysis, presenting a complicated picture for anticancer therapies (179). Hypoxia is of particular interest and a still complicated feature in cancer, including brain cancers. Obviously, regarding reactive species, the first concern is always to minimize ROS (Fig. 9).
First, one major concern in cancer, including in the brain, is whether hypoxia modulates ROS generation and whether ROS induce HIF stabilization. Stabilized HIF-1α subunit is translocated to mitochondria to reduce O2 − generation and attenuate mitochondrial dysfunction by improving the OXPHOS function at complex IV by regulating mitochondrial-selective autophagy, and likely by regulating mitochondrial DNA transcription (136). In addition, stabilized hypoxia-inducible factor 2 (HIF-2) stimulates antioxidant gene expression, including the nuclear transcription factor erythroid-2-related factor 2 to reduce O2 − generation in macrophages, whereas mitochondrial O2 − is required to stabilize HIF-2 (58, 212). Moreover, hypoxia induces H2O2 overgeneration (including in cancer), and overgenerated H2O2 induces stabilization of hypoxia factor and related pathways (3, 11, 67, 116). Notably, it has been recently suggested that H2O2 determines the glioma's fate, including in anticancer therapy such as chemotherapy and radiotherapy (280). Finally, in brain glioma, in the hypoxic condition, activities of key enzymes SOD and catalase are reduced whereas XOR and MPO activities are increased to the detriment of the patient (93, 126, 193). Oxidative distress is pro-cancer when associated with transcriptional factor HIFs and related pathways mediated by ROS and altered SOD, catalase, XOR, and MPO.
In cancer, including that in the brain, RNS metabolism also contributes to regulating the hypoxic status and effects (89). Immune system evasion and anticancer therapy resistance occur, in part, due to NO overgeneration induced by hypoxia (17, 24). In glioma, an increase in iNOS expression is a hallmark of neuroinflammation and of chemoresistance, and iNOS expression and activity inhibitors are promising anti-glioma agents (148). Numerous studies showed that NO regulates glioma growth, cancer angiogenesis, cancer cell invasion, and radio- and chemoresistance (5, 226, 235, 242). In addition, ONOO− accumulation in glioblastoma inactivates wild-type p53 function and is associated with an altered inflammatory response in the glioma microenvironment (51). In brain cancer, oxidative distress mediated by RNS is pro-cancer.
The RSS metabolism is as important as ROS and RNS in cancer. In brain cancer, associated with hypoxia, oxidative distress mediated by RSS is pro-cancer, although RSS may be promising radiosensitizers depending on their concentration. H2S has been suggested as a promising radiosensitizer, at least for glioblastoma cells through selective OXPHOS complex III impairment and mitochondrial respiration reduction, an increase in H2O2 levels, and an increase in DNA damage (265). However, H2S also favors C6 glioma cell proliferation and survival through increased cyclooxygenase-2 expression and apoptosis reduction (278). The debate surrounding the anti-cancer and pro-cancer roles of H2S in brain cancers is made more complicated by the puzzling increase in expression of the H2S-producing enzymes CBS and CSE in astrocytomas and brain metastases (132). Hypoxia increases this complexity, as hypoxia induces H2S generation, and H2S regulates HIF-1α subunit expression and stabilization (257).
Finally, RCS metabolism also plays a key role in cancer, including in the brain. MGO induces cell cycle arrest, inhibits cell proliferation, and initiates apoptosis in glioblastoma cells (186). Interestingly, in glioblastoma, 4-HNE and MDA levels decrease, related to high aldehyde dehydrogenase 1A3 levels, which detoxify cancer cells from aldehydes (190, 261). In addition, in glioma, key enzyme GPX activity decreases, whereas a high level of GPX2 is a poor prognosis biomarker in glioblastoma (95, 193). Finally, an increase in PRDX4 activity in glioblastoma promotes cancer proliferation and anticancer therapy resistance (184). Thus, targeting GPX and PRDX as well as inducing oxidative distress mediated by RCS could be promising anti-cancer therapies, including against glioblastoma.
Once again, in brain cancer, the entire RSI is the ideal target through the coordination of the different reactive species and key enzymes (Fig. 9). In the brain, associated with hypoxia, oxidative distress mediated by ROS, RNS, and RSS is pro-cancer, whereas inducing oxidative distress mediated by RCS is a promising anti-cancer therapy. In addition, altered SOD, catalase, XOR, MPO, GPX, and PRDX enzymes are promising targets in anti-cancer therapy, including brain cancer.
Antioxidants and Cancer: Bad and Good News
Anti-ROS antioxidants such as N-acetylcysteine (NAC) and vitamin E, at high concentrations, accelerate lung cancer by disrupting the ROS-p53 axis, as p53 is a tumor suppressor gene involved in stress response after oxidative stress, hypoxia, and ionizing radiation (131, 203). At high levels, NAC and vitamin E also favor malignant melanoma progression and metastasis by deregulating the equilibrium between GSH and GSSG (131, 203). Once again, associated with hypoxia, the delicate equilibrium between reactive species and the antioxidant defense in cancer, including in the brain, is not linked to only ROS. This delicate equilibrium is linked to ROS, RNS, RSS, RCS, and the key enzymes amid the RSI.
A final example of antioxidant activity in many types of cancer is the frequent gain-of-function mutations of the so-called isocitrate dehydrogenase-1 (IDH1) and -2 (IDH2). These enzymes participate in the Krebs cycle, are implicated in multiple metabolic processes, and produce NADPH (NAD phosphate) and α-KG (110, 267). In cancer, mutated IDH1 and IDH2 reduce NADPH and α-KG generation by losing their normal catalytic activity and overproduce 2-hydroxyglutarate (2-HG) by gaining a new function (119). The oncometabolite 2-HG, an antagonist of α-KG, reduces histone and DNA demethylation, promotes cancer microenvironment formation through VEGF upregulation, increases HIF-1α subunit stability, increases ROS generation, and dysregulates H2S generation (105, 106, 110, 123, 144, 260, 267). Thus, gain-of-function mutated IDH1 and IDH2 in cancers (including primary brain cancers) that account for more than 70%–80% of lower-grade glioma and the majority of secondary glioblastoma induce epigenetic reprogramming, bioenergetic metabolism dysregulation (at least both ROS and RSS dysregulation), and finally cancer invasion (105, 106, 110, 123, 144, 260, 267). The RSI should be considered to ultimately design more efficient and powerful anti-cancer therapies in all cancers. Combining RSI modulation therapy with radiotherapy, chemotherapy, nanoparticle therapy, and/or immunotherapy may provide especially promising pathways.
Conclusion on the RSI in Brain Pathology
Through depression, ischemia, neurodegenerative disorders, and cancers in the brain, we demonstrated that the entire RSI, including RCS and key enzymes, should be considered together instead of only one reactive species family. In depression, oxidative distress mediated by ROS, RNS, and RCS exerts a pro-depressant effect, whereas oxidative distress mediated by RSS exerts an anti-depressant effect. In ischemia, oxidative eustress mediated by ROS and RSS and oxidative distress mediated by eNOS-related NO are beneficial to counteract deleterious effects, whereas oxidative distress mediated by ROS, nNOs- and iNOS-related RNS, RSS, and RCS are detrimental. In neurodegenerative disorders, oxidative distress mediated by ROS, RNS, and RCS is detrimental, whereas oxidative distress mediated by RSS is beneficial to fight neurodegenerative disorders, although high levels of H2S should be cautiously addressed to limit neurotoxic side effects. Finally, in brain cancer, associated with hypoxia, oxidative distress mediated by ROS, RNS, and RSS is detrimental, whereas inducing oxidative distress mediated by RCS is a promising anti-cancer therapy. Importantly, in addition to the reactive species, the key enzymes SOD, catalase, XOR, MPO, GPX, and PRDX should be considered in all these brain pathologies.
In a pathological process, what creates the global redox effect is the combination between (i) effects from different reactive species families in different concentrations, (ii) interactions between the different families, and (iii) activities of key enzymes that contribute to the RSI dynamics. The RSI, including RCS and key enzymes, is fully committed in brain pathologies.
Conclusions
Oxidative eustress and distress must be seriously considered in relation to reactive species concentrations. From the most studied ROS, it appears that the RSI in its entirety has to be seriously considered related to reactive species identification and interactions. We highlighted that the RSI, meaning ROS, RNS, RSS, RCS, and key associated enzymes, is fully committed in brain physiology and pathology (Fig. 10). Depending on organ oxygenation, physiology, pathology, and key enzyme dynamics, the complex coordination of the four reactive species families related to their concentrations creates the global redox effect. The ROS, RNS, RSS, and RCS form a link between physiology and pathologies, between the antioxidant defense and the bioenergetic metabolism, and between cell death and cell survival. From adult neurogenesis to cell death, aging, depression, ischemia, neurodegeneration, and finally, cancers, the brain is a vital organ in which the RSI combined in eutress or distress is playing all its potential for mechanistic and therapeutic clues.
Authors' Contributions
E.M. and L.C. wrote the article with support and input from S.V., M.B. and E.P., L.C. supervised the project. All authors discussed the forum review article and contributed to the final article.
Footnotes
Authorship Confirmation Statement
All authors confirm that this article is original, has not been published earlier, and is not currently being considered for publication elsewhere. All authors confirm that the article has been read and approved by all authors and that there are no other people who satisfied the criteria for authorship but are not listed. All authors confirm that the order of authors listed in the article has been approved by all authors. All authors understand that the Corresponding Author is the sole contact for the Editorial process regarding communication, progress, submission(s) of revision(s), and final approval of proofs.
Acknowledgments
The authors thank Omar Touzani, ISTCT, CERVOxy, and Caen for advice and comments on the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The forum review article was supported by the Region Normandie, the Inkermann fund hosted at the Fondation de France, the CNRS, and Université de Caen-Normandie (UNICAEN) the grant reference, which included the different partners, is: RIN-50-2020-META-X.