
Abstract
Significance:
While atherosclerosis is an almost inevitable consequence of aging, food preferences, lack of exercise, and other aspects of the lifestyle in many countries, the identification of new risk factors is of increasing importance to tackle a disease, which has become a major health burden for billions of people. Iron has long been suspected to promote the development of atherosclerosis, but data have been conflicting, and the contribution of iron is still debated controversially.
Recent Advances:
Several experimental and clinical studies have been recently published about this longstanding controversial problem, highlighting the critical need to unravel the complexity behind this topic.
Critical Issues:
The aim of the current review is to provide an overview of the current knowledge about the proatherosclerotic impact of iron, and discuss the emerging role of non-transferrin-bound iron (NTBI) as driver of vasculotoxicity and atherosclerosis. Finally, I will provide detailed mechanistic insights on the cellular processes and molecular pathways underlying iron-exacerbated atherosclerosis. Overall, this review highlights a complex framework where NTBI acts at multiple levels in atherosclerosis by altering the serum and vascular microenvironment in a proatherogenic and proinflammatory manner, affecting the functionality and survival of vascular cells, promoting foam cell formation and inducing angiogenesis, calcification, and plaque destabilization.
Future Directions:
The use of additional iron markers (e.g., NTBI) may help adequately predict predisposition to cardiovascular disease. Clinical studies are needed in the aging population to address the atherogenic role of iron fluctuations within physiological limits and the therapeutic value of iron restriction approaches. Antioxid. Redox Signal. 35, 387–414.
Introduction
Atherosclerosis, one of the leading causes of morbidity and mortality in Western countries, is a complex pathological process affecting arterial walls. Oxidative stress plays a crucial role in atherosclerosis, since free radicals promote lipoprotein (low-density lipoprotein [LDL]) oxidation and therefore accelerate its progression. In this context, iron can be detrimental through the generation of free radicals and reactive oxygen species (ROS) (231). In 1981, Sullivan proposed the “iron hypothesis,” according to which iron overload promotes cardiovascular disease (CVD) and, on the contrary, iron deficiency exerts a protective action (209 –213). Multiple experimental and clinical observations provided evidence of a key role of iron in atherogenesis, while others provided conflicting data in this regard. Especially, the absence of increased incidence of atherosclerosis in patients with hereditary hemochromatosis (HH), afflicted by serious life-long iron overload, was perceived as a “paradox” and considered the most persuasive evidence against the iron hypothesis (214, 231). In this review, I provide an overview of the most recent findings and studies, which addressed this longstanding controversial aspect. Importantly, I will show how novel experimental approaches and animal models helped in the last decade to uncover mechanistically the contribution of iron overload to atherosclerosis. While here I will describe predominantly recent findings, a more comprehensive overview of older studies is summarized in a previously published review on this topic (231).
Regulatory Mechanisms of Iron Homeostasis
Systemic iron homeostasis
Due to its potential toxicity, iron levels are tightly regulated in the body. Iron is absorbed by duodenal enterocytes from the diet and recycled by reticuloendothelial macrophages from senescent red blood cells (RBCs) through the membrane iron exporter ferroportin (FPN). Systemic iron fluxes are controlled by a major regulatory system, the hepcidin–FPN axis. The peptide hormone hepcidin, by binding and mediating FPN occlusion, internalization and degradation, controls FPN-mediated iron export (96, 147). Hepcidin levels are regulated by several stimuli: while iron levels and inflammation induce hepcidin synthesis, erythropoiesis and hypoxia exert a negative regulation of the hormone. Therefore, in condition of iron deficiency and stimulated erythropoiesis, hepatic hepcidin expression is decreased to promote dietary iron influx into the circulation through FPN. When body iron levels are elevated or an infection occurs, hepcidin expression is increased to reduce FPN-mediated systemic iron influx and thus overall body iron as well as iron availability for microorganism growth. Once released systemically, iron is bound to the iron carrier transferrin (Tf) and delivered through transferrin receptor 1 (TfR1)-mediated endocytosis to cells throughout the body where it is utilized as cofactor in heme and nonheme enzymes. Erythropoiesis is the process that uses most iron, whereby erythroblasts acquire up to 30 mg iron per day to synthesize hemoglobin (Hb) and provide de novo RBCs (96, 147).
Non-transferrin-bound iron and non-hemopexin-bound heme
Upon iron overload conditions, either inherited (e.g., HH) or acquired (e.g., transfusion dependence), serum iron levels and Tf saturation (>50%) are elevated, which lead to the formation of “free” nontransferrin-bound iron (NTBI). Within the NTBI pool, the labile plasma iron (LPI) represents the toxic reactive fraction, which is loosely bound to plasma proteins and highly pro-oxidant. NTBI and LPI can accumulate intracellularly through the metal transporter ZIP14 (147). Therefore, tissues expressing high levels of ZIP14 and/or low levels of FPN are prone to develop iron overload in presence of NTBI. Similar to NTBI, Hb and heme released upon hemolysis become toxic when their circulating levels override the binding capacity of the Hb scavenger haptoglobin (Hp) and heme scavenger hemopexin (Hx), leading to the formation of non-Hp bound Hb (NHBHB) and non-Hx bound heme (NHBH). While the Hb–Hp and heme–Hx complexes are selectively taken up by macrophages and hepatocytes, respectively, NHBHB and NHBH freely enter cells in a nonspecific manner, causing heme overload and tissue damage (46, 193, 219, 229, 230). In iron overload or hemolytic conditions, the vasculature might be exposed to elevated NTBI, NHBHB, and NHBH. Accumulation of unshielded iron, Hb, and heme in vascular cells elicits an adaptive response, whereby the cytoprotective enzyme heme oxygenase 1 (HO-1) is induced. Pro-oxidant stimuli as well as the substrate heme are able to trigger HO-1 induction. HO-1 exerts a cytoprotective and antioxidant action by promoting carbon monoxide and biliverdin release as a result of heme catabolism (19, 20). Heme-derived iron is oxidized from its toxic ferrous (Fe2+) to nontoxic ferric (Fe3+) form by the ferroxidase of the ferritin heavy chain (H-Ft) and then safely stored into ferritin light chain (18, 19).
Cell iron homeostasis
Cellular iron homeostasis is controlled post-transcriptionally by the iron regulatory protein (IRP)/iron regulatory element (IRE) system. The RNA-binding IRPs 1 and 2 (IRP1, IRP2) interact with conserved cis-regulatory hairpin structures termed IREs, which are located in the 5′ or 3′ untranslated regions (UTRs) of target mRNA transcripts (96, 147). IRP binding to the 5′ UTR IREs of mRNAs encoding for iron storage and export molecules such as ferritin and FPN inhibits their translation, whereas their binding to the 3′ UTR IREs of TFR1 mRNA prevents TfR1 degradation. Therefore, in iron-depleted cells, the IRE–IRP system reduces the expression of molecules involved in iron export and storage, and increases the stability of the main receptor responsible for cell iron uptake, TfR1 (96, 147).
Overall, a fine tuning of both systemic and cellular iron levels orchestrated by the hepcidin/FPN axis and the IRE/IRP system is key to the maintenance of iron homeostasis. The pathological formation of free forms of heme and iron, NHBH and NTBI, eventually leads to systemic and cellular iron overload, causing oxidative stress, cell damage, and organ dysfunction.
Novel Evidence on the Vasculotoxic and Atherogenic Impact of Iron
What we have learned from human studies
Association between iron status and atherosclerotic CVD
While a clear association exists between systemic iron deficiency and CVD, including coronary artery disease (CAD) and myocardial infarction (MI), more complex and less clear is the association between iron overload and CVD emerging from trials and clinical studies. Some studies support a role for iron in atherosclerosis and found a link between body iron status and CVD, whereas others failed to observe any association (231). The Brunick study indicates serum ferritin as one of the strongest predictors of carotid artery disease (119). Likewise, a positive association between iron status measured as serum ferritin, iron levels or Tf saturation, and risk of cardiovascular events has been described in several studies (99, 125, 140, 145, 170, 172, 186, 217, 221, 246). Specifically, a 10 mg/L increase in serum ferritin levels raises by 3% the probability of having at least two atherosclerotic plaques (4). Indeed, patients with severe CAD show higher serum iron levels compared with those with normal, mild, and moderate CAD (13).
Electron paramagnetic resonance spectroscopy and inductively coupled plasma mass spectroscopy analyses revealed that iron accumulates in human arteries and carotid lesions, and its levels positively correlate with cholesterol deposition (206). Iron accumulation in arterial tissue is elevated in subjects with high body iron stores (132). Moreover, plaque iron loading positively correlates with plaque vulnerability and propensity to rupture: advanced plaques in patients with carotid atherosclerosis show in fact higher iron content together with signs of cap rupture and increased number of inflammatory macrophages (91, 248). The association between serum ferritin and atherosclerosis is potentiated by hyperlipidemia, smoking, inflammation, and gender, suggesting a synergistic effect of iron with other classical factors predisposing to CVD (145, 186).
Iron may have cardiovascular implications for pathological conditions that show an altered iron status and a propensity to CVD as a result of disease complication, ranging from diabetes and metabolic syndrome to transfusion-dependent anemia and chronic kidney disease (155). Diseases associated with both heme and iron overload, such as β-thalassemia and sickle cell disease, show underlying cardiovascular alterations. These individuals develop systemic Hb and heme overload consequent to hemolysis, and iron overload consequent to ineffective erythropoiesis, hepcidin downregulation and chronic transfusion requirement (181, 233). Together with NTBI, unshielded Hb and heme might exert vasculotoxic, proatherogenic, and prothrombotic effects due to their pro-oxidant and proinflammatory action (193, 231, 233). This is currently believed to be a major mechanism promoting vascular alteration and vasculopathy in sickle cell disease and thalassemia and underlying complications, including vaso-occlusive crisis, acute chest syndrome, increased blood pressure, hypertension, altered cardiac function, and stroke (113 –115). A major cause of death in β-thal patients is congestive heart failure due to transfusional iron overload. While patients with hemoglobinopathies develop to some extent vascular complications, such as arterial and venous thromboembolic events, stroke (2, 43, 45, 90, 93, 94, 198), their atherosclerotic mortality is somehow limited. This is likely explained by the constant exposure of these patients to iron chelation therapy and their favorable lipid profile, with low cholesterol levels (8, 178, 179, 196). Premature atherosclerosis is more frequent in children who often show dyslipidemia associated with high triglycerides (29, 118, 198).
Among the studies that do not support the iron hypothesis, the iron and atherosclerosis study (FeAST) trial failed to demonstrate a beneficial effect of reduced body iron stores on peripheral arterial disease-related mortality and MI/stroke (249, 250). However, in a subset of patients who died during the FeAST study a correlation between levels of circulating ferritin, inflammatory biomarkers, and mortality has been observed (59). A recent study suggested that increased iron status is protective against some forms of atherosclerotic disease and cardiovascular mortality, whereas it promotes the risk of venous thromboembolism (83).
The hemochromatosis paradox and potential explanations
A major challenge to the iron paradigm was the observation that individuals affected by HH, a life-long iron overload disease, do not apparently show increased risk of cardiovascular events. In HH, the hepcidin/FPN regulatory axis is disrupted due to mutations in hepcidin regulators (high Fe2+ protein [HFE], transferrin receptor 2 [TfR2], hemojuvelin), in hepcidin itself or FPN. This results in inadequate hepcidin levels and constant FPN-mediated systemic iron influx, leading to iron overload (147). Whereas some clinical studies showed an increased risk of MI, and cerebrovascular or cardiovascular mortality in HFE-mutated patients (175, 183, 220), most of them showed no association between parameters of iron status and atherosclerotic CVD (65, 67, 143, 148, 161, 215, 224). A matter of debate is the cholesterol level in HH patients, which was found increased (232) and decreased in different studies (156). While decreased cholesterol might explain the lack of increased CVD predisposition, these patients present clear iron-aggravated markers of vascular dysfunction and oxidative stress, which are rescued by therapeutic phlebotomy (232). HH patients bear in fact elevated oxidized lipids and proteins, increased soluble adhesion molecules and cytokine levels, structural alterations in arterial muscle media layer, reduced nitric oxide (NO), and impaired endothelium-dependent vasodilation (70, 77, 111, 232). These alterations might accelerate CVD only in combination with an unfavorable lipid profile in HH subjects, suggesting that iron is a “modifier” rather than per se a trigger of atherosclerosis.
Another explanation of incoherent findings in HH comes from the altered tissue iron distribution in these patients. Low hepcidin in HH induces macrophage iron depletion by enhancing cell iron efflux. Iron-depleted macrophages likely show antiatherogenic and anti-inflammatory properties. Therefore, reduced macrophage iron content has been proposed as mechanism counteracting the atherosclerotic action of elevated systemic iron and the key for the lack of CVD predisposition in HH (214, 215). Mechanistic insights on the atheroprotective role of iron-depleted macrophages are described below in the section “Iron and Macrophages”. This “adjusted hypothesis” therefore implies that tissue and cell iron distribution has a role in iron-aggravated atherosclerosis, whereby macrophage iron retention due to elevated hepcidin represents a risk factor for CVD (215). Indeed, some studies found a positive correlation between serum hepcidin levels and all-cause and cardiovascular mortality in patients with CAD (130) and plaque instability in patients with MI (78, 79, 190). However, to exclusively rely on hepcidin as marker for the association between iron and CVD is susceptible of misleading conclusion, as hepcidin could reflect the iron status (high hepcidin = iron deficiency; low hepcidin = iron overload) as well as an adaptation to the current iron status (high hepcidin = iron overload triggers hepcidin induction; low hepcidin = iron deficiency blocks hepcidin synthesis). This level of complexity helps explain why other studies found no or opposite association between hepcidin and CVD (87, 159, 180). Finally, the observation of a continuous and inverse correlation between hepcidin levels and CAD mortality suggests a dominant role of elevated systemic iron over macrophage iron in promoting CVD (87).
Spectrum and parameters of iron status in CVD
While most of the clinical studies to assess the validity of the iron hypothesis have been performed in patients with CVD or altered iron status, a nuanced understanding of how small fluctuations of iron parameters within the physiological limits predispose to atherosclerosis in the healthy population is still lacking. A recent human population study reported for the first time a correlation between circulating ferritin levels and carotid-intima media thickness in a large healthy cohort of 692 children (171). Importantly, a J-shaped association of systemic iron status and cardiovascular mortality has been recently described in CAD patients (87, 163). The observation that CAD patients at increased risk fall into the two opposite extremes of the iron spectrum points toward the notion that iron-associated cardiovascular risk results from a misbalance in iron homeostasis—either iron overload or deficiency (Table 1). This helps explain the conflicting results obtained from clinical studies, where the definition of a “protected window” of iron levels across studies is difficult to determine.
Range of Parameters of Iron Status and Risk of Cardiovascular Disease Mortality
The range of iron parameters, including serum iron, Tf saturation, serum ferritin, NTBI, and LPI, is reported for individuals with a normal iron status, iron deficiency, and iron overload. The range of iron parameters for individuals with no risk and increased risk of CVD mortality is indicated according to the J-shaped association by Grammer et al. (87). Values of NTBI and LPI are extrapolated from the preclinical study by Vinchi et al. (232) as no clinical studies are available. Being NTBI and LPI detectable almost exclusively in iron overload conditions, their association with CVD has to be considered upon iron overload only.
CVD, cardiovascular disease; HH, hereditary hemochromatosis; LPI, labile plasma iron; NTBI, nontransferrin-bound iron; Tf, transferrin.
The discrepancy of epidemiological studies may also derive from the scarce reliability of parameters of iron status, their variability and fluctuations due to multiple modifiers—for example, age, gender, diet—and the limited consideration of treatment regimen. The use of markers of iron status which poorly reflect the systemic and tissue iron burden may have biased the conclusions of several studies. While serum ferritin is considered a marker of tissue iron content, its levels are highly affected by inflammation, and do not reflect the iron saturation of Tf or the amount of NTBI or catalytically active LPI (108, 135). Additional markers may be required, such as NTBI or vascular iron levels, to adequately predict predisposition to CVD (Table 1). The treatment regimen of iron-loaded patients (e.g., anti-inflammatory drugs, iron depletion strategies) should be critically considered as it may mask the cardiovascular risk imposed by iron. As an example, most of HH patients are on iron depletion therapies, the underestimation of which might have biased the outcome of several studies.
In particular, NTBI together with its reactive fraction LPI likely represent more reliable measures of the circulating iron fraction capable of proatherogenic action. Importantly, NTBI is increased not only in iron overload conditions but also in other diseases such as diabetes and metabolic syndromes. Therefore, the use of these parameters may help identify those patients and link iron dyshomeostasis to accelerated CVD also in nonprimarily iron overload conditions (6). Along this line, an association between the frequency of hemochromatosis mutations in HFE and an increased risk of CAD was observed in diabetic patients, suggesting that elevated systemic iron exacerbates the CVD risk imposed by diabetes (189). Overall, clinical studies specifically monitoring NTBI and LPI modulation and their connection with CVD are urgently needed to assess the validity of these parameters of iron status as better predictor of cardiovascular events (6, 184). The lack of coherence among human studies mirrors the complex relationship existing between the iron status and cardiovascular functionality, where the two extremes of the iron spectrum imply an impairment of the cardiovascular system and translate into propensity to CVD. Further investigations are required to specifically address the association of alternative parameters of iron status—NTBI and LPI—with cardiovascular events in iron-loaded individuals and patients with CVD as well as in the healthy populations. Considering the implications of NTBI and LPI in CVD emerging from animal studies, the assessment of these iron markers in clinical studies will provide new indications on the more relevant iron forms with vasculotoxic and atherosclerotic properties.
What We Have Learned from Animal Models
Experimental evidence on the role of iron in atherosclerosis mainly derives from the analysis of “artificial models” of iron overload, whereby animals have been experimentally overloaded with iron through infusion or supplemented diet. By contrast, for decades, atherosclerosis has remained understudied in pathophysiological models of iron overload, including animal models of genetic iron overload (e.g., HH mouse models). Recent works addressed the development of atherosclerosis in mouse models of hemochromatosis, providing new mechanistic insight into the proatherogenic cellular and molecular mode of action of iron. In this section, I will review the major findings obtained from the analysis of mouse models of iron overload, achieved either by genetic manipulation or by iron treatment.
Lesson from animal models of genetic iron overload
Mouse models with disrupted hepcidin/FPN regulatory axis have been recently analyzed to dissect the relationship between iron overload and atherosclerosis. These models include the following: (i) FPNwt/C326S knock-in mice—a mouse model of type IV HH; (ii) hepcidin knockout mice—a mouse model of type II HH; and (iii) flatiron mice—a mouse model of classical FPN disease (Table 2).
Lessons from Animal Models
Atherosclerosis phenotype, hallmarks, similarities and differences, and corresponding human disease of animal models used to study the impact of iron overload in CVD.
ApoE, apolipoprotein E; FPN, ferroportin; Ft, ferritin; Hb, hemoglobin; HO, heme oxygenase; Hp, haptoglobin; Hx, hemopexin; ip, intraperitoneal; SCD, sickle cell disease; VSMC, vascular smooth muscle cell.
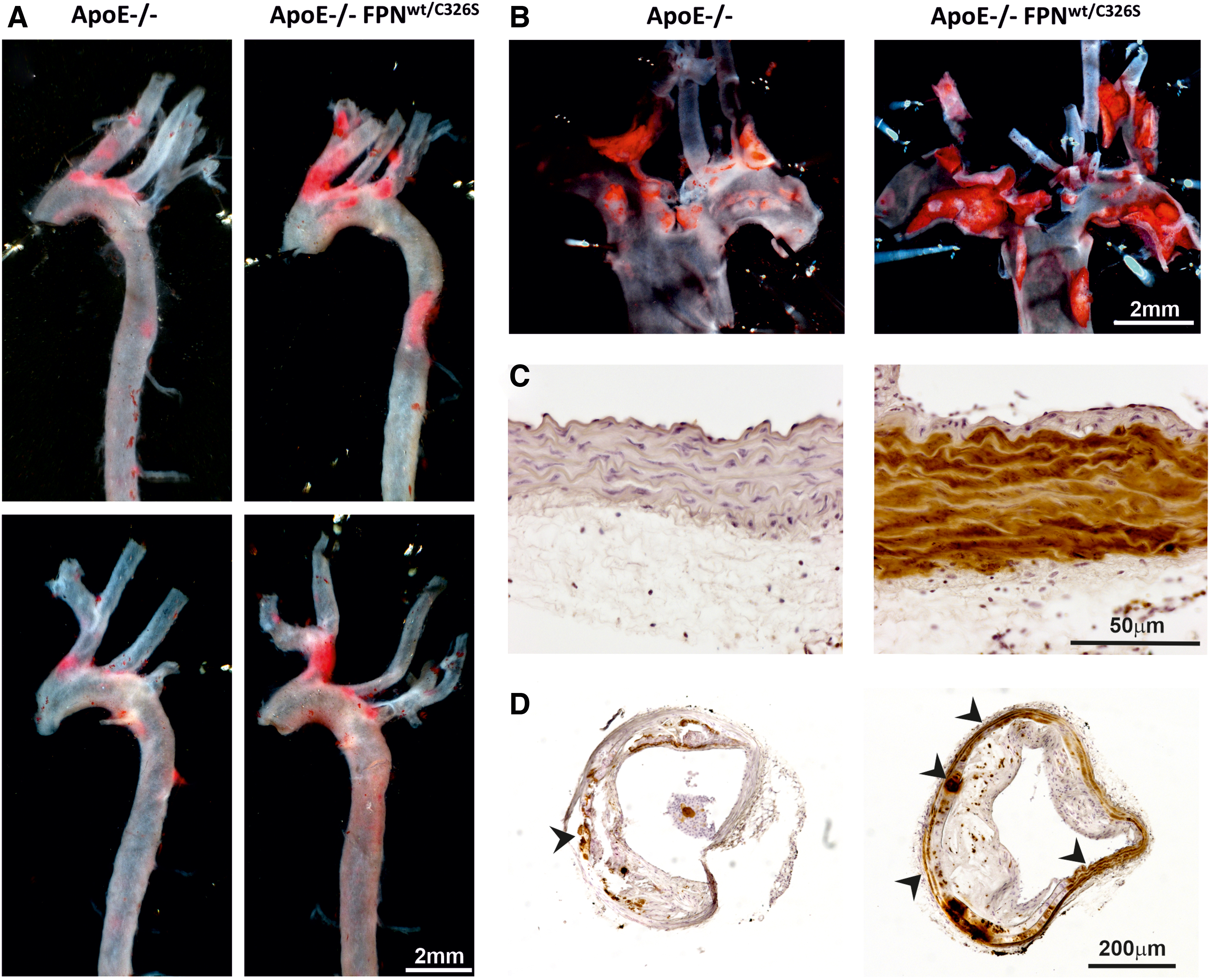
Vinchi et al. have recently assessed the role of iron overload in atherosclerosis by analyzing a mouse model carrying a heterozygous gain-of-function mutation of FPN (FPNC326S) (232). This dominant mutation confers resistance to hepcidin binding and leads to the disruption of the hepcidin–FPN axis by preventing hepcidin-mediated FPN internalization and degradation. The disruption of this regulatory mechanisms results in constitutive iron efflux from macrophages and increased iron uptake by duodenal enterocytes. Indeed, this model is hallmarked by elevated Tf saturation, NTBI formation, and tissue iron overload (7). The presence of the FPNC326S mutation in the apolipoprotein E (ApoE)-null genetic background (ApoE-null FPNC326S mice) results in exacerbated atherosclerosis, whereby atherosclerotic lesion area and numbers are significantly higher compared with ApoE-null control animals (Fig. 1A, B). A hallmark of this model is the massive iron deposition in major arteries (e.g., aorta), consequent to NTBI uptake (Fig. 1C, D). While this work does not speak about the role of iron within physiological limits, it documents the relevance of NTBI in atherogenesis, and its contribution to pro-oxidant and proinflammatory mechanisms with atherogenic effect (232).
By contrast, Malhotra et al. recently showed that a different model of hemochromatosis, the hepcidin-null (Hamp-null) mouse on low-density lipoprotein receptor (Ldlr)-null genetic background, presents with less severe atherosclerosis than the Ldlr-null control mouse (89, 134). The antiatherogenic effect of hepcidin deletion was mostly attributed to the reduced proinflammatory activation of intraplaque iron-depleted macrophages.
Interestingly, FPNC326S and Hamp-null mice would have been expected to phenocopy in regard to atherosclerosis development, based on the considerations that both models (i) have elevated NTBI, (ii) show tissue iron accumulation, and (iii) bear macrophages with reduced intracellular iron content (88, 232). However, the opposite phenotypes observed should be considered in light of major differences between these models: (i) while FPNC326S mice have been analyzed on the ApoE-null genetic background, which spontaneously promotes plaque development, Hamp-null mice required a high-fat diet to develop atherosclerotic plaques in the Ldlr-null background. In this second model, the diet might contribute to atherosclerosis in a synergistic way together with iron; (ii) LDL levels are different in the two models, being reduced in Ldlr-null Hamp-null mice and slightly increased in ApoE-null FPNC326S mice at 6 months of age. Lower LDL levels might be due to increased cell cholesterol efflux from iron-depleted macrophages but also to different intake from the high-fat diet in Ldlr-null Hamp-null mice (which was not explored in the study). Understanding why LDL levels are different in these models might uncover novel mechanisms whereby iron controls cholesterol levels; (iii) NTBI levels and arterial iron deposition have not been directly compared in these mouse models. Hypothetically, different levels of NTBI and arterial iron content between ApoE-null FPNC326S and Ldlr-null Hamp-null mice would explain the opposite atherosclerotic phenotypes (e.g., if we assume a higher NTBI in ApoE-null FPNC326S mice, the antiatherogenic benefit of macrophage iron depletion might be masked by NTBI detrimental effects); (iv) these models strongly differ in the circulating levels of hepcidin. While the Ldlr-null Hamp-null mouse is hepcidin deficient, the ApoE-null FPNC326S mouse has high levels of hepcidin, which, despite inactive on FPN, is still induced by iron overload. The different levels of hepcidin in these models likely call for extra functions of hepcidin, independent from its FPN-related action and potentially proatherosclerotic; (v) while the Ldlr-null Hamp-null model is completely deficient for hepcidin, therefore having the lowest intracellular iron content possible in macrophages, the ApoE-null FPNC326S mouse may still have residual macrophage iron due to the presence of the FPNC326S mutation in heterozygosity. The presence of residual intracellular iron in macrophages may limit their protective effect in atherosclerosis; (6) finally, the effect of low macrophage iron may be “time dependent,” providing an antiatherogenic action in the early stages of atherosclerosis through anti-inflammatory macrophage phenotypic switching, and a detrimental action in later stages of the disease, whereby hypoxia-triggered vascular endothelial growth factor (VEGF) production by iron-depleted macrophages promotes angiogenesis, vasopermeabilization, and intraplaque inflammatory cell recruitment (88, 169). Despite constantly elevated systemic iron levels, ApoE-null FPNC326S mice show aggravated atherosclerosis at 6 months of age with no obvious differences at earlier age (232). We speculate that the presence of iron-depleted macrophages might provide a certain degree of protection in ApoE-null FPNC326S mice at earlier stages of the disease, until a more advanced stage whereby NTBI toxicity becomes dominant. At the same time, these mice show intraplaque angiogenesis and hemorrhages at older ages (12–18 months), compatible with a detrimental effect of iron-depleted macrophages in advanced atherosclerosis. Whether this duality holds true in Ldlr-null Hamp-null mice requires further exploration as atherosclerosis was not analyzed beyond 5 months of age in this model.
Overall, these models highlight the complexity of the interplay existing between circulating NTBI and intracellular macrophage iron in regard to atherosclerosis progression. Along this line, the two hemochromatosis models are hallmarked by high NTBI and macrophage depletion, which renders difficult the dissection of the contribution of the single components to atherosclerosis. To overcome this issue, Kautz et al. (116) have analyzed atherosclerosis in the flatiron (ffe) mouse, a model of classical FPN disease, carrying the heterozygous H32R missense mutation of FPN (FPNH32R), which causes a dominant negative mistrafficking of the iron exporter and results in macrophage iron overload (260). This model on ApoE-null background allowed one to assess the specific role of elevated macrophage iron content in atherosclerosis. Contrarily to other observations, this study failed to show aggravated atherosclerosis in ApoE-null ffe mice (116). These findings argue against a significant detrimental action of iron-loaded macrophages in atherosclerosis and are seemingly in line with other studies, which rather support a critical role of systemic iron in disease progression (232). Nevertheless, the possibility that the detrimental effects triggered by macrophage iron overload are overall less relevant compared to the beneficial effects induced by macrophage iron depletion cannot be ruled out.
Lesson from animal models of exogenous iron overload
Animal models of experimentally induced iron overload have been used to study the role of iron in atherosclerosis (Table 2). The administration of exogenous iron or iron-enriched diet in experimental animals generates iron overload in a nonspecific manner, likely affecting both the vasculature and macrophages, and thus generally addressing the role of NTBI in atherosclerosis. Whereas an earlier study reported diminished atherosclerosis in ApoE-null mice challenged with dietary iron overload (121), Hu et al. recently showed that an iron-enriched diet aggravates atherosclerosis in ApoE-null mice (101). Likewise, iron infusion (either iron dextran or iron sucrose) promotes the development of atherosclerosis in rabbits and ApoE-null mice (10, 54, 124, 137). A single study showed opposite results with iron dextran in rabbits (55). Although the majority of these studies support a proatherosclerotic role of iron, conflicting data may result from variable parameters and different experimental approaches, including the extent and length of iron treatment, the amount of iron and fat present in animal diet, the age of the animal at the moment of treatment and plaque analysis.
Lesson from animal models of iron restriction
Iron restriction by low-iron diet and iron chelators
Several studies examined the effect of iron restriction achieved by low-iron diet or iron chelation on atherogenesis (Table 3). In this regard, mice fed a low-iron diet developed smaller atherosclerotic lesions than control littermates (127). Dietary iron restriction likely increases plaque stability via elevated collagen and reduces matrix metalloproteinase-9 expressions in the lesion (126). Consistently, iron chelation by deferoxamine and desferricoprogen limits atherosclerotic lesion development in cholesterol-fed rabbit (144) as well as ApoE-null mice (256). Recently, low-iron diet and iron chelation by deferasirox were found to significantly prevent atherosclerosis progression in iron-loaded ApoE-null FPNC326 mice (232), by lowering arterial iron content, endothelial activation, and inflammation.
Experimental Therapeutic Approach Targeting Iron in Cardiovascular Disease
Approach, atherosclerosis phenotype, treatment or genetic manipulation, similarities and differences of animal models used to study the therapeutic benefit of iron targeting in CVD.
BMP, bone morphogenetic protein; MGP, matrix Gla protein.
Cellular iron depletion by hepcidin modulation
Hepcidin induction is critically regulated by the activation of the bone morphogenetic protein (BMP)/small mother against decapentaplegic (SMAD) signaling pathway. Therefore, inhibitors of hepcidin production such as the small molecule BMP type I receptor inhibitor LDN-193189 have been applied in animal models to reduce macrophage iron content (Table 3). Treatment of ApoE-null mice with LDN-193189 results in significant reduction of macrophage iron and decreased atherosclerotic lesion formation (185). LDN-193189 as well as a soluble receptor–antibody fusion protein acting as BMP ligand trap, ALK3-Fc, reduces vascular calcification, inflammation, and atheroma formation in Ldlr-null mice (60). The overexpression of another BMP signaling antagonist, matrix Gla protein (MGP), similarly decreases atherosclerosis severity and vascular calcification (243). Besides the key role in hepcidin induction, the BMP pathway is a critical mediator of vascular media calcification through vascular smooth muscle cells (VSMCs) osteogenic differentiation (133). Moreover, BMP signaling plays a role in cholesterol homeostasis. LDN-193189 in fact has been found to affect lipoprotein biosynthesis, thus reducing total cholesterol and LDL serum levels (60). Therefore, BMP modulators likely target multiple mechanisms in atherosclerosis, which are not restricted to hepcidin suppression and the modulation of iron levels. In addition, BMP signaling is crucial for the maintenance of bone and cartilage homeostasis, and its inhibition for antiatherosclerotic purposes might trigger a broad range of adverse events (21, 112, 235). Future studies testing drugs that reduce hepcidin expression without interfering with the BMP signaling or that directly counteract hepcidin activity are needed to examine the antiatherosclerotic benefit of hepcidin suppression.
Lesson from animal models of heme overload
Animal models with altered heme homeostasis have been used to better understand the proatherogenic role of heme (Table 2). A recent work showed that Hx deficiency in ApoE-null mice increases systemic free heme, ROS, and proinflammatory high-density lipoprotein, and aggravates atherosclerosis and macrophage infiltration (139). The proatherogenic action of Hb and heme is also demonstrated by studies on Hp polymorphisms, which give rise to the Hp 1-1, Hp 2-2, and Hp 2-1 genotypes. Mice bearing the Hp 2-2 allele show increased intraplaque iron deposition and lipid peroxidation as well as macrophage accumulation, typical features of advanced atherosclerosis (128). These effects are explained by the decreased ability of the Hp 2 allele to inhibit the pro-oxidant and proinflammatory action of Hb (12, 109). Indeed, in humans the Hp 2-2 genotype is associated with an increased risk of CVD and its sequelae such as acute MI (128, 231). Deficiency of the transcriptional repressor Bach1 in mice reduces atherosclerosis burden by upregulating HO-1, further demonstrating heme proatherosclerotic action and the importance of its catabolism (237) (Table 3). Consistent with these observations, HO-1 deficiency promotes while its induction prevents atherosclerosis progression in atherosclerosis-prone ApoE-null or LDLR-null mice (44, 105, 244). HO-1 expression in macrophages seems to be crucial to provide an atheroprotective effect (153) (Tables 2 and 3). Interestingly, the antiatherogenic action of HO-1 is also suggested by the observation that statins, the mainstay treatment for lowering cholesterol, are capable of triggering HO-1 expression both in endothelial cells (ECs) and macrophages through increased promoter activity and HO-1 transcription (5, 42, 146). Statin treatment has been also associated with reduced ferritin levels and hepcidin modulation (9, 40). Overall, these observations suggest that the beneficial role of statins in CVD can be in part mediated by their effects on heme and iron homeostasis.
Current Molecular Mechanisms of Iron-Mediated Vasculotoxicity
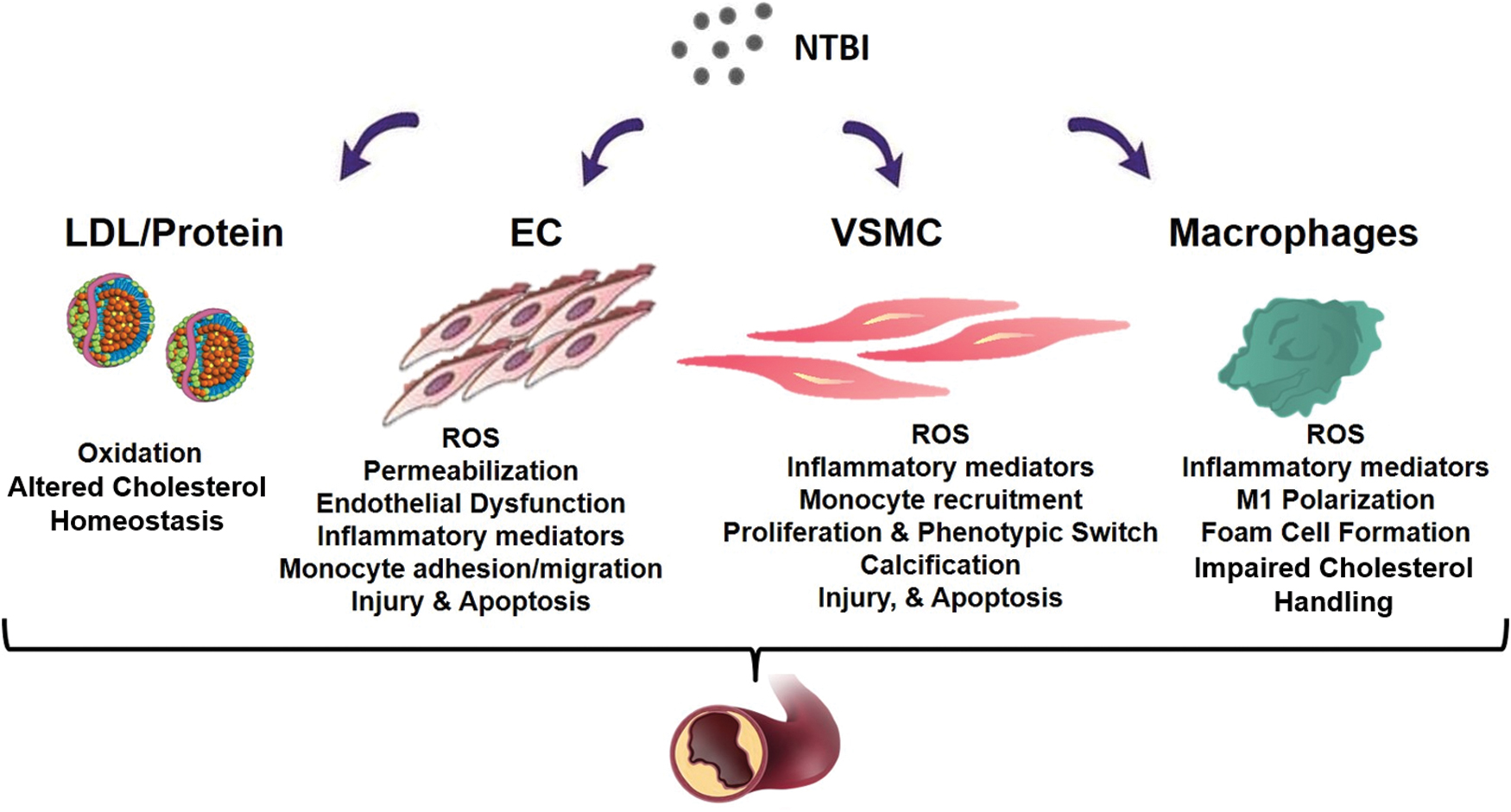
In the following section, I will illustrate how NTBI contributes to atherosclerosis at multiple levels by (i) altering the composition of the serum in a proinflammatory manner; (ii) affecting functionality and survival of endothelial and VSMCs; (iii) promoting foam cell formation; and (iv) inducing angiogenesis, calcification, and plaque destabilization (Fig. 2).
Iron, cholesterol, and oxidative potential
Alterations in iron homeostasis could impact lipid metabolism. However, no consensus exists on whether iron overload increases or lowers cholesterol levels. Recent research, using a plethora of model organisms and data from clinical studies, has revealed novel links between iron homeostasis and lipid-related pathways (Fig. 2). Iron overload increases cholesterol levels by triggering the upregulation of key enzymes involved in the cholesterol biosynthetic pathway (86). The expression of cholesterol 7 α-hydroxylase (Cyp7a1), a major player of cholesterol homeostasis, is modulated by iron through a functional noncanonical IRE in the 3′-UTR of the transcript. Increased hepatic iron content results in cholesterol accumulation through IRE-mediated Cyp7a1 downregulation. Iron modulation of Cyp7a1 unravels a clear implication of iron metabolism in the regulation of cholesterol homeostasis through the IRE/IRP regulatory system (131). In addition, elevated iron levels increase the expression but reduce the secretion of a key lipid carrier, ApoE, potentially resulting in impaired cholesterol transport (33, 57, 182, 242).
Elevated iron levels exacerbate hyperlipidemia and hyperglycemia in animal models, including zebrafish, rats, mice as well as humans (35, 120, 137, 164, 232). Likewise, iron-loaded patients with HH show elevated cholesterol and hypertriglyceridemia (141, 204, 232), which can be improved by therapeutic phlebotomy (37, 232). Conversely, iron deficiency is associated with significantly lower hepatic cholesterol content and reduced serum total cholesterol, LDL, and triglyceride levels in rodents and men (47, 51, 138, 207, 227). The synergistic association of serum ferritin and LDL cholesterol with incident CVD supports the concept of a critical interaction between iron and lipids in CVD progression (119). By contrast, other studies showed an opposite modulation of cholesterol and lipid levels by iron in rodents and men as well as hemochromatotic patients (1, 82, 134, 156, 201). Conflicting findings in hemochromatosis may be explained by altered levels of the liver-specific microRNA 122 (miR-122), which regulate both iron and cholesterol metabolism (38). Decreased miR-122 expression in HFE-null mice and HH patients triggers hepcidin upregulation, and acts as a compensatory response to limit iron uptake and counteract iron overload. In addition, low miR-122 is implicated in the reduction of systemic cholesterol levels by targeting yet unidentified genes (66, 68). Therefore, the degree of iron overload in HH patients might determine the levels of microRNA miR-122, resulting in variable cholesterol levels and explaining the conflicting evidence from studies on hemochromatosis cohorts. The regulatory role of miR-122 provides an additional indication of the close interconnection between the homeostatic pathways of iron and cholesterol. This link is supported by a recent genome-wide association study, showing a significant overlap between genes or loci affecting iron biomarkers and known loci affecting plasma lipids or lipoproteins (28).
As serum lipid profile is affected by many factors, the variations of lipid concentration in the presence of elevated or reduced iron levels may not be related to iron overload/deficiency by itself. Patients with β-thalassemia, who are anemic but show transfusional iron overload, have reduced cholesterol levels, suggesting a further level of complexity in the interaction between anemia, iron overload, and cholesterol biosynthesis/utilization (8, 178, 179). Indeed, anemia is associated with low cholesterol level. Hypocholesterolemia is eventually induced by high erythropoietic activity due to the increased cholesterol demand by proliferating erythroid cells (196).
As additional consideration, iron excess exacerbates hypercholesterolemia in ApoE-null mice but only mildly in wild-type mice (137). Thus, aggravated hypercholesterolemia might result from the synergistic action of an already altered lipid state with iron overload (137, 232). Finally, how either dietary or body iron excess interferes with lipid absorption, and vice versa, is understudied. The observation of reduced LDL levels in Ldlr-null Hamp-null mice receiving a high-fat diet points in this direction, whereby fat intake might be modulated by iron levels (134, 154).
In addition to lipid profile alteration, an extent of NTBI and NHBH proatherogenic action lies in their ability to induce lipid and protein oxidation in cells and serum (15, 34, 76, 137, 232, 247) (Fig. 2). Free labile ferrous iron (Fe2+) gets easily engaged and participates in the Fenton and Haber-Weiss reactions, which ultimately lead to the formation of ROS, including free radical species, superoxide, hydroxyl radicals, and hydrogen peroxide (157). Directly through these reactions or indirectly through ROS, NTBI can induce the peroxidation of polyunsaturated fatty acid, and the resulting by-products, such as reactive aldehydes and gamma-keto aldehydes, likely contribute to the vasculotoxic and proinflammatory effects of NTBI (157). Through similar mechanisms, heme, once liberated from Hb after its oxidation to methemoglobin (17), catalyzes free radical reactions in cell lipid domains (16) and lipoproteins (15), thus triggering a pro-oxidant and cytotoxic environment in the vasculature (104) and atherosclerotic plaques (149). Vascular cells respond to such an insult by upregulating the antioxidant HOs and ferritins (3, 14, 17).
Intriguingly, the appearance of oxidized LDLs (oxLDLs) in ApoE−/− FPNwt/C326S mice precedes the elevation of LDL levels, suggesting that LDL oxidation is an early, if not the first, iron-triggered proatherogenic mechanism (232). The use of the iron chelator desferricoprogen inhibits plaque formation in ApoE-null mice by lowering oxLDL levels and limiting plaque lipid oxidation and deposition oxLDL-induced foam cell formation in the vessel wall (168). Moreover, desferricoprogen preserves endothelial integrity and prevents the upregulation of adhesion molecules, thus blunting monocyte–EC adhesion and inflammation.
To date, two mechanisms of iron-mediated alteration of cholesterol homeostasis have been suggested as proatherosclerotic: the alteration of the lipid profile and the systemic and cellular oxidation of lipids. The multifactorial interaction between iron and cholesterol homeostasis (e.g., iron-mediated regulation of cholesterol enzymes, mir122, ineffective erythropoiesis), and variables that influence cholesterol levels (e.g., diet, exercise, malabsorption, antioxidant systems, gender, ethnicity) add an inevitable complexity to the interpretation of experimental and clinical studies.
Iron and ECs
Iron contributes to the aggravation of atherosclerosis by triggering the proinflammatory activation of the vascular endothelium. This early step in atherogenosis facilitates intraplaque accumulation of LDL and recruitment of circulating monocytes through increased vascular permeability and monocyte–EC adhesion (Fig. 2).
Recently, mechanistic studies showed the involvement of iron-triggered endothelial dysfunction in cardiovascular complications. Endothelial activation and damage are features of iron-loaded ApoE-null FPNC326S mice (232). Exposure of a monolayer of ECs to NTBI-like iron sources (e.g., ferric ammonium citrate, iron nitrilotriacetate) triggers increased expression of adhesion molecules (e.g., vascular adhesion molecule 1 [VCAM1], intercellular adhesion molecule 1 [ICAM-1], E-selectin, P-selectin), secretion of chemoattractants (monocyte chemoattractant protein 1 [MCP-1]), intracellular ROS formation, and cell death (Fig. 3). In vivo this results in vasopermeabilization and increased monocyte adhesion, which likely promote LDL infiltration into the subendothelial space and intraplaque monocyte recruitment followed by foam cell formation (231, 232) (Fig. 2). In addition, iron impairs endothelium-dependent vasorelaxation, and triggers arterial stiffness by inducing ROS formation and decreasing NO bioavailability through reduced endothelial NO synthase expression/activation and/or enhanced NO oxidative consumption (61, 66, 95, 110, 137, 258). NTBI has also the ability to increase VEGF expression in ECs and VSMCs (13). Elevated VEGF has been implicated in atherosclerosis due to its proinflammatory and permeabilizing action on the vascular endothelium (39, 100).
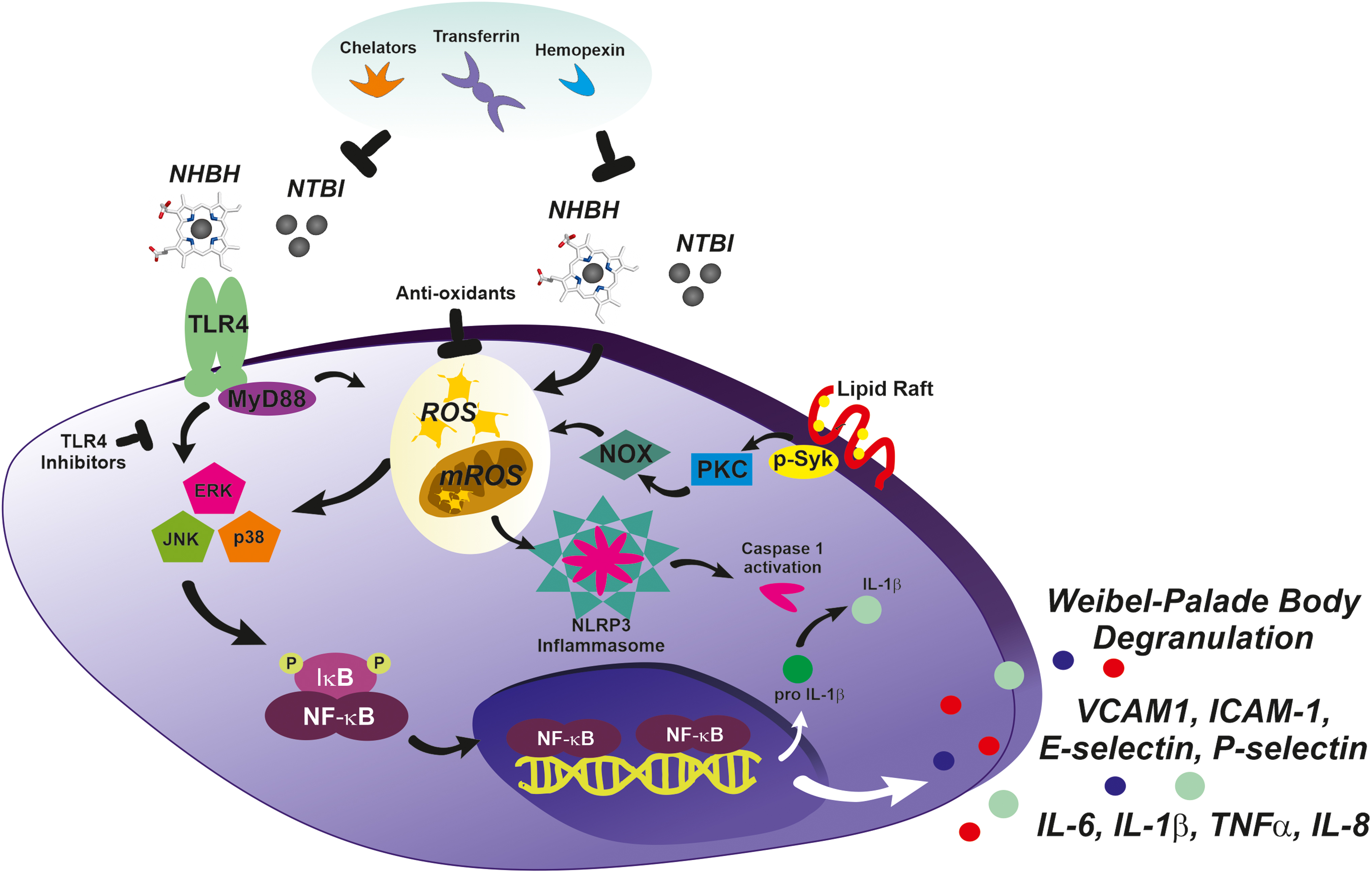
Exposure to free Hb and NHBH similarly impairs the vascular endothelium by inducing toll-like receptor 4 (TLR4)-mediated endothelial dysfunction, NO consumption, and inflammasome activation (24 –26, 46, 71, 160, 167, 193, 200, 219, 229, 230, 233) (Fig. 3). Therefore, NHBH likely contributes to atherosclerosis, especially in advanced plaques where hemorrhages occur (103, 149, 231). NHBH- and NTBI-induced EC apoptosis and ROS production are prevented by heme and iron scavenging through Tf or deferoxamine and Hx, respectively (95, 110, 229). ApoE-null FPNC326S mice administered a low-iron diet or receiving a chelation treatment show normalized markers of endothelial activation (e.g., soluble adhesion molecules, nitrotyrosine, VEGF) (232). By reducing cell iron content and oxidative stress, iron chelation as well as a combined iron chelation–antioxidant therapy significantly limits endothelial damage and vascular dysfunction in animal models (102, 205, 231). Interestingly, chelation also prevents EC activation and adhesion molecule expression induced by inflammatory mediators such as tumor necrosis factor α and lipopolysaccharide (255).
Additional processes in which free Hb and heme might play a proatherosclerotic role are neovascularization and atheroma formation. Because of inappropriate angiogenesis, neovessels that form within the lesion are leaky and prone to rupture, leading to intraplaque extravasation of RBCs (103). Intraplaque hemorrhages are considered features of plaque progression and vulnerability and a critical event in atherosclerosis-associated acute clinical symptoms (142). The advanced atheromatous lesion is hallmarked by a pro-oxidant environment whereby hemolytic RBCs release Hb, which is in turn oxidized to ferri- and ferryl-Hb. These Hb forms and liberated heme and iron promote intraplaque lipid oxidation and endothelial activation and cytotoxicity, further contributing to lesion development (149). Essential to counteract heme/iron-oxLDL cytotoxic action on ECs is the ferroxidase activity of intracellular H-Ft and the scavenging function of extracellular Hp, Hx, and Tf (51, 106).
Iron and VSMCs
Major alterations in VSMCs have been recently implicated in iron-aggravated atherosclerosis (Fig. 2). VSMCs accumulate high amount of NTBI without option to export it, due to the lack of FPN expression. This is in line with the observations that (i) iron accumulation occurs in human arterial tissue and is significantly higher in patients with high plasma ferritin (132); (ii) iron heavily deposits in VSMCs of the aortic media layer of iron-loaded ApoE-null FPNC326S mice (232) (Fig. 1C, D). The association between elevated systemic iron and CVD might at least in part ensue from the detrimental effect of NTBI accumulation in VSMCs of the arterial wall, which contributes to vascular dysfunction and plaque formation. When exposed to NTBI, VSMCs develop iron overload, produce ROS, and undergo apoptosis associated with the release of MCP-1 (Figs. 2–4C). Indeed, apoptotic iron-loaded VSMCs stimulate the recruitment of monocytes to the growing plaque, promoting plaque progression (Fig. 4D) (232). Collagen reduction in atherosclerotic lesions of ApoE-null FPNC326S mice suggests decreased production of extracellular matrix by iron-loaded VSMCs and/or increased matrix degradation due to metalloprotease release by VSMCs or macrophages (Fig. 4A) (232). Since iron overload was found to trigger vascular collagen deposition (61), we speculate that while plaque collagen accumulates at early stages of atherosclerosis, reduced collagen production and/or increased matrix degradation likely occur at more advanced stages, leading to a decreased intraplaque collagen amount as observed in ApoE-null FPNC326S mice. One of the mechanisms potentially implicated in reduced matrix deposition at more advanced stage of atherosclerosis is endoplasmic reticulum (ER) stress. Elevated ER stress markers have been observed in VSMCs of complicated lesions with hemorrhage compared with either atheromas or healthy arteries (80). Interestingly, heme triggers ER stress in human VSMCs, which is inhibited by heme scavenging and antioxidants. By suppressing transforming growth factor beta (TGFβ) expression and collagen production, heme-induced ER stress likely contributes to plaque instability and progression (80). Heme regulation of TGFβ levels in VSMCs is hallmarked by an early induction followed by suppression. This biphasic behavior could explain the differential effect of heme/iron on matrix deposition at different stages of atherosclerosis. Overall, the progressive reduction of the extracellular matrix likely results in fibrous plaque thinning and plaque instability.
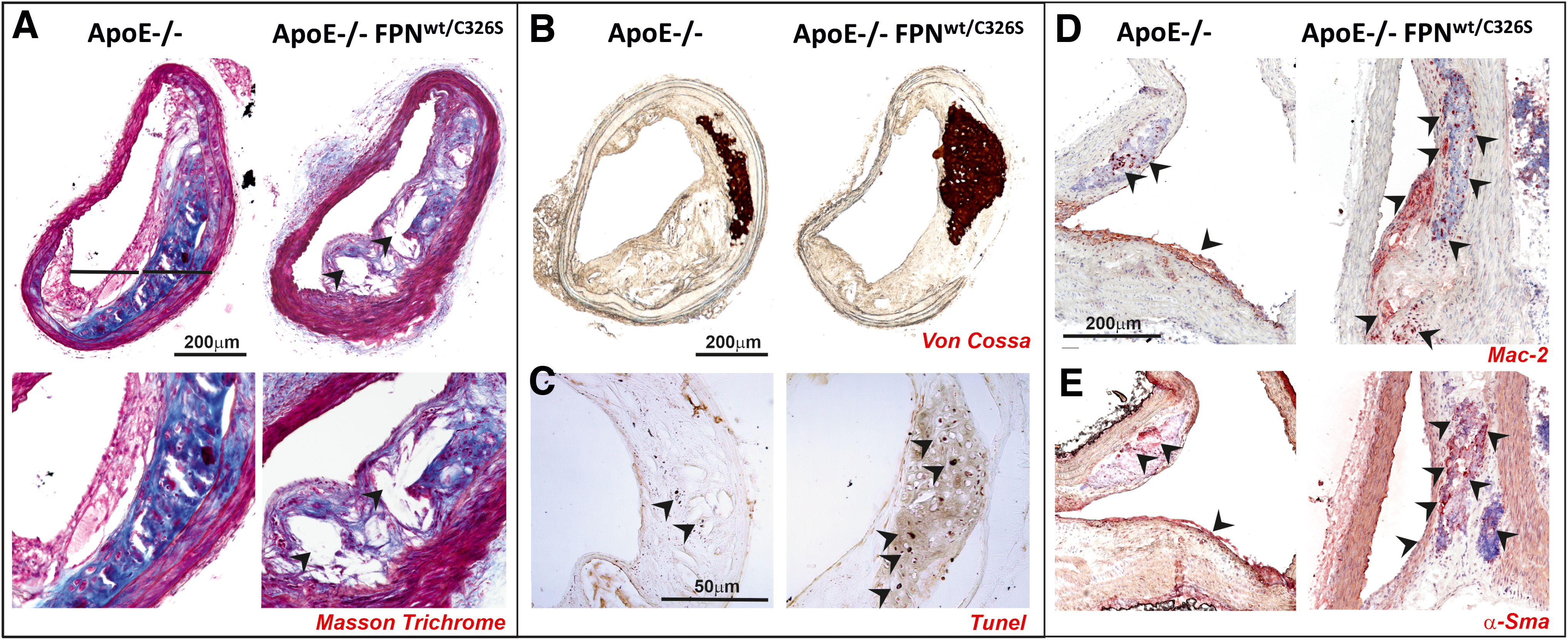
After migrating intraplaque, VSMCs might acquire a macrophage-like phenotype, as indicated by the coexpression of both VSMC and macrophage markers (e.g., α smooth muscle actin [α-SMA] and macrophage 2 protein or Galectin 3 [Mac2]) (see atherosclerotic plaques of ApoE-null FPNC326S mice, Fig. 4D, E) (232). Iron-triggered ROS are potentially implicated in VSMC migration. ROS serve in fact as mediators of promigratory signaling pathways, which control VSMC motility through lamellipodia formation, actin cytoskeleton remodeling, focal adhesion turnover, cell body contraction, and matrix remodeling (187). VSMC-derived macrophage-like cells, by behaving as less-differentiated/specialized cells, may directly promote atherosclerosis via inflammation, poor lipid handling, and impaired VSMC-related functions (including matrix deposition) (22, 27).
VSMCs can also undergo transdifferentiation into osteoblasts and chondrocytes. Whereas oxidative stress has been associated with osteogenic differentiation of VSMCs and calcification, iron has been shown to prevent phosphate-induced vascular calcification, through the inhibition of VSMC osteochondrogenic shift (48 –50, 195). Ferritin was identified as the major protective molecule behind iron-mediated inhibition of mineralization (23, 252, 253). Moreover, iron might negatively regulate calcification by binding and decreasing phosphate levels in VSMCs (252), and by reducing its systemic levels through the induction of the phosphaturic hormone fibroblast growth factor 23 (FGF23) (194). Therefore, excess iron enhances ROS levels, which in turn trigger molecular mechanisms leading to vascular calcification, whereas iron deposition inhibits this process by inducing protective molecules such as ferritin and HO-1. In vivo, the formation of vascular calcification is likely determined by the balance between these mechanisms, whereby the exhaustion of antioxidant system capacity due to excessive iron-driven ROS production acts as calcification trigger.
Iron contributes to calcification also in an indirect manner, through (i) the oxidation of lipids and proteins (245), (ii) the increased production of BMP-2 (56, 117), and (iii) the induction of cell apoptosis (188, 192). oxLDL and proteins and BMP-2 promote the differentiation of vascular cells to a calcifying osteoblast-like cell type (56, 245). Apoptotic cells induce arterial matrix calcification by releasing calcifying membrane-bound matrix vesicle (62). Recent findings show indeed that iron boosts rather than suppressing VSMC osteogenic differentiation (117, 151). In support of an active role of iron in triggering vascular calcification, iron overload is associated with more frequent and bigger calcifications in ApoE-null FPNC326S mice (232) (Fig. 3B). In this context, iron-driven apoptosis of ECs and VSMCs is likely a major driver of calcification. The contribution of VSMC osteochondrogenic shift to atherosclerosis remains to be elucidated in this and other models of iron overload.
While most NTBI accumulates in the arterial media layer, calcifications are mainly located intraplaque (232) (Fig. 3B). Likewise, a spatial inverse correlation of iron and calcium was shown within the atherosclerotic lesion (173). However, a link between iron and calcium dyshomeostasis is supported by the association between increased ferritin levels and presence of coronary artery calcium in men (216). Not only VSMCs but also recruited macrophages might be implicated in this process, as indicated by the direct association between calcification and macrophage burden in ApoE-null mice (97).
Another relevant atherosclerotic mechanism consists in vascular remodeling. The impact of iron overload on this process still remains understudied. Evidence from iron-overloaded animals shows that iron increases aortic stiffness and induces vascular remodeling associated with collagen deposition (72, 177). Indeed, the reduced distensibility of the aorta from iron-loaded rats is accompanied by collagen deposition, suggesting that changes in the composition of the wall are responsible for iron-induced stiffness. Overall, iron overload increases the vasoconstrictor response of arteries, associated with altered vascular reactivity and the loss of endothelial modulation of the vascular tone (72, 137). Interestingly, inhibition of the renin–angiotensin system limits vascular remodeling in iron-loaded animals, suggesting a critical role of this pathway in iron-triggered aortic stiffness (72). Nonpathological iron accumulation has been shown to drive VSMC proliferation rather than apoptosis, contributing to vascular remodeling and pulmonary hypertension (174). Iron chelation as well as deficiency of the TfR1 inhibits pulmonary artery SMC proliferation and prevents chronic hypoxia-induced pulmonary hypertension (150, 165, 238).
Iron and macrophages
Macrophages are key cells contributing to plaque development through the evolution into lipid-laden foam cells. Whether and how iron content in intraplaque macrophages impacts atherosclerosis is still a matter of debate. Although available evidence is conflicting, the majority suggests that iron accumulation in macrophages has a detrimental role in atherosclerosis (Figs. 2–5). Iron-loaded macrophages show in fact inflammatory properties and impaired cholesterol handling abilities. Intracellular iron content regulates macrophage polarization by triggering macrophage inflammatory activation (98, 123, 176, 202). The exposure of macrophages to NTBI-induces an M1-like proinflammatory phenotypic switching hallmarked by the expression of inflammatory markers and cytokines (53, 98, 101, 123, 202, 218, 228, 251, 254, 259) (Fig. 5A). Likewise, iron accumulation consequent to FPN deficiency increases the expression of inflammatory cytokines in macrophages (257).
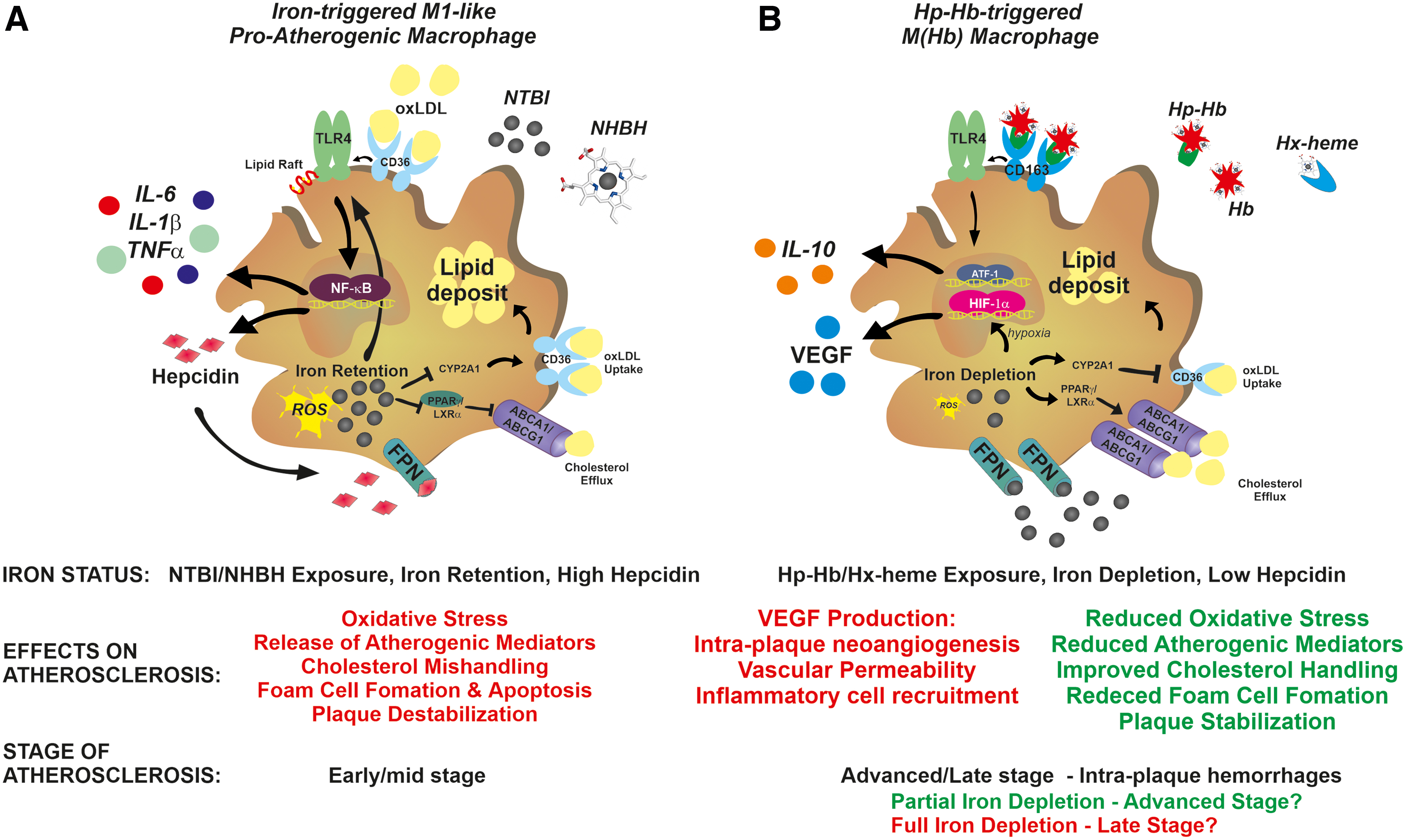
A similar effect is exerted by NHBH and free Hb, which trigger cytokine production, inflammasome activation, and cell death in macrophages (53, 63, 64, 73, 75, 152, 218, 228). Preliminary studies suggest that iron induces macrophage inflammatory switch by triggering a metabolic shift toward glycolysis (101). The inflammatory storm triggered by iron-activated macrophages likely contributes to intraplaque inflammation and lesion progression.
oxLDL exposure induces a specific macrophage phenotype, the Mox macrophage, hallmarked by increased expression of nuclear factor erythroid 2-related factor 2 (Nrf2)-induced redox-regulatory genes and oxidized phospholipid-induced genes (107). In response to oxLDL, macrophages upregulate the oxidative stress response enzyme HO-1 and hepcidin, resulting in cell iron accumulation (129, 136). oxLDL-induced macrophage iron retention is mediated by TLR4/nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) pathway activation, which stimulates the autocrine production of hepcidin (241). Hepcidin-mediated iron accumulation further synergizes with oxLDLs to activate the TLR4 pathway, thus establishing a positive feedback loop. Moreover, macrophage ER stress, which is common in advanced atherosclerosis, plays a role in further increasing hepcidin autocrine production (199, 225). Hepcidin-mediated intracellular iron trapping results in impaired macrophage cholesterol handling, due to enhanced CD36-mediated cholesterol uptake and decreased ABCA1- and ABCG1-mediated reverse cholesterol efflux (Fig. 5). These effects are amplified by exogenous iron sources, such as NTBI-like iron exposure and erythrophagocytosis (122, 234, 241, 256). Iron increases TLR4-dependent macrophage activation by regulating TLR4 trafficking to cell lipid rafts (92). ROS and TLR4 signaling pathway play a critical role in iron-driven inflammatory activation and foam cell formation. Chelators, antioxidants, and TLR4 inhibitors prevent macrophage M1-like phenotypic switching and cholesterol mishandling (129, 218, 228, 241, 257) (Fig. 3).
In vivo, hepcidin overexpression in murine carotid arteries affects plaque composition, increasing intraplaque macrophages and decreasing VSMCs and collagen content (129). This is associated with iron trapping, ROS and cytokine production, and oxLDL accumulation in intraplaque macrophages. Likewise, hepcidin levels and macrophage iron positively correlate with interleukin (IL)-6 and MCP-1 levels and vascular damage in individuals at high risk of vascular disease (223). By contrast, monocytes from HH patients show reduced iron content, and reduced MCP-1 and IL-6 levels (223). Hepcidin deficiency or its pharmacological suppression decreases macrophage iron content and increases cell cholesterol efflux, thus resulting in reduced foam cell formation (134, 185). Reduced macrophage iron content limits ROS formation and increases cholesterol transporter expression, leading to improved cell lipid efflux and reduced foam cell formation.
Overall, these findings indicate that (i) hepcidin is a positive regulator of atherosclerotic plaque destabilization, through the control of macrophage iron content. Elevated hepcidin increases cell iron content, and promotes plaque progression and destabilization by exacerbating macrophage inflammatory cytokine release, intracellular lipid loading, oxidative stress, and cell apoptosis (129, 136, 241). This eventually implies a role not only for systemic but also for local hepcidin production in atherosclerosis, similarly to the local effect that has been recently shown for cardiac hepcidin (226); (ii) iron and lipids act synergistically, and promote foam cell formation by enhancing LDL uptake and impairing iron efflux and cholesterol handling (234, 241).
A different macrophage phenotype has been described within areas of intraplaque hemorrhage and neoangiogenesis, characterized by reduced iron content, resistance to foam cell formation, decreased intracellular ROS formation and inflammatory cytokine expression, and increased levels of FPN and the Hp–Hb scavenger receptor CD163 (30, 31, 74) (Fig. 5B). The so called Mheme or M(Hb) macrophages are triggered by exposure to the Hp–Hb complex, and are considered atheroprotective due to their increased cell iron efflux and cholesterol handling abilities and anti-inflammatory properties. Nevertheless, a proatherogenic function of these macrophages has been recently described, adding complexity to the role of macrophage iron content in atherosclerosis (90). Iron depletion in M(Hb) macrophages leads to the stabilization of the hypoxia-inducible factor 1α and the upregulation of hypoxia-regulated VEGF. VEGF release by macrophages is associated with intraplaque angiogenesis, increased vascular permeability, and inflammatory cell recruitment, leading to severe atherosclerosis (88) (Fig. 5B). These findings are in line with the observation of a proatherosclerotic role of VEGF (39, 100) and the severe atherosclerosis phenotype in iron-loaded ApoE-null FPNC326S mice, which show elevated VEGF levels (232). Genetic ablation of CD163 in ApoE-null mice rescues M(Hb)-associated phenotype. Therefore, despite beneficial features such as improved cholesterol handling, macrophage iron depletion might activate other detrimental mechanisms such as hypoxia-dependent ones. In addition, macrophage iron efflux likely increases iron levels in the plaque microenvironments with potential proatherogenic effects on other cell types (e.g., ECs, VSMCs). Remarkably, VEGF appears as a key mediator of iron-aggravated atherosclerosis, being produced by multiple cell types within the atherosclerotic lesion—ECs, VSMCs, and M(Hb) macrophages (88, 125). Indeed, hemorrhagic areas are likely enriched in M(Hb) macrophages actively pumping iron in the plaque microenvironment and resulting in VEGF production by macrophages, VSMCs, and ECs (66, 88). This points toward VEGF as potential target for antiatherosclerotic treatments in iron overload conditions.
Collectively, these findings indicate that the interactions between hepcidin, retained iron, and accumulated lipids are critical for the atherosclerotic role of macrophages. The dual action of macrophage iron depletion in atherosclerosis suggests a potential beneficial effect at early stages of atherosclerosis—associated with improved lipid handling—and detrimental effect in more advanced stages, when intraplaque neovascularization, hemorrhages, and hypoxia occur (52, 89, 240).
Finally, iron might act on other immune cells within the atherosclerotic plaque. Iron and heme have been proven to affect lymphocyte and neutrophil functions (11, 36, 41, 64, 158, 162, 166, 197, 236), but a direct implication of these mechanisms in atherosclerosis still awaits investigation.
Cardiovascular Health As a Result of Iron Balance
In conclusion, the maintenance of iron balance and Tf saturation within proper limits has a critical role in cardiovascular health. The difficult definition of a risk threshold of iron level within the iron spectrum, whereby both iron deficiency and iron overload promote CVD, likely explains the lack of coherence among studies in this area of investigation. The level of complexity is further increased by the multifactorial contribution of iron to atherosclerosis. Emerging evidence suggests that circulating NTBI, by accumulating in vascular cells and macrophages and altering their functions, play crucial roles in the exacerbation of atherosclerotic disease (Fig. 6).
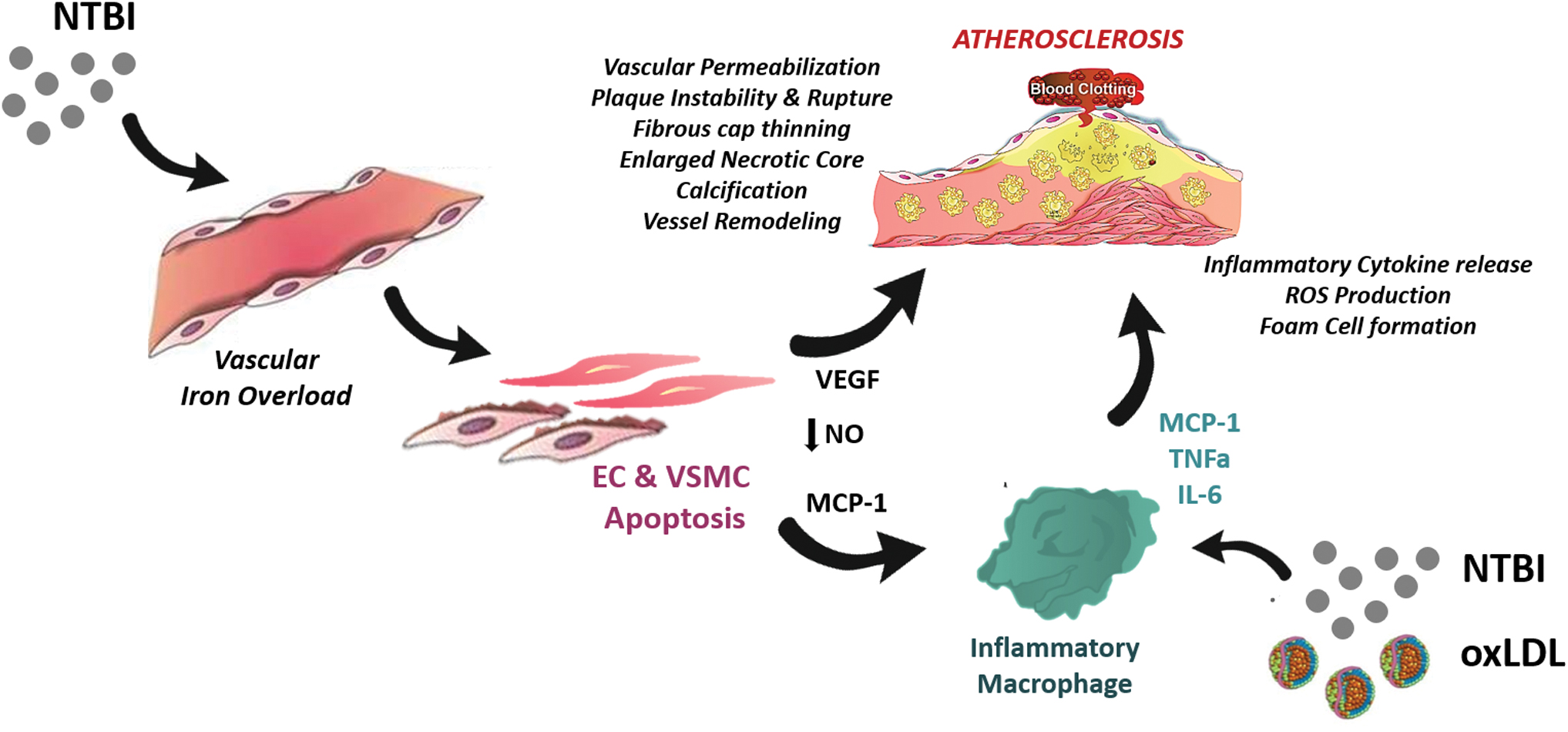
Additional investigations are needed to elucidate what cells within the atherosclerotic plaques and the vessel wall are more sensitive to fluctuations in the catalytic labile iron pool, and better understand at what stage(s) of the disease iron is detrimental. Moreover, the identification of NTBI as a risk factor for atherosclerosis and its use as marker of iron status may help clarify some previously observed controversies, where the distinction between body iron stores and NTBI was not evaluated. Studies are also needed to evaluate the range of LPI which, within the NTBI, is responsible for vasculotoxicity (32, 69).
Despite the potential clinical relevance and the effort made in the last few years to improve available techniques, numerous challenges remain with laboratory standardization and harmonization for the measurement of NTBI and LPI. Nowadays, still few but increasing laboratories have established reproducible and reliable methodology for NTBI and LPI quantification (69, 81, 84, 203). Ten worldwide leading assays—six for NTBI and four for LPI—have been recently compared in the second international round robin for the quantification of serum NTI and LPI (58). All the assays consistently detected elevated NTBI and LPI in patients with untreated HH and β-thalassemia intermedia as well as transfusion-dependent myelodysplastic syndromes and β-thalassemia major. Absolute NTBI and LPI levels differed considerably between assays, with differences lower for LPI than for NTBI measurement (58). Assays showed reproducibility with high between-sample and low within-sample variation. Increased Tf saturation, but not ferritin, correlated and was a good indicator of the presence of circulating NTBI (58). We predict that with methodology improvement, the measurement of these parameters will be more widely adopted in clinical laboratory setting as marker and indicator of iron toxicity, including cardiovascular toxicity.
The use of antioxidants, ranging from vitamin E (alpha-tocopherol), vitamin C, beta-carotene to iron chelation or restriction, has been always considered a potential approach to reduce LDL oxidation and delay atherosclerosis progression. However, this hypothesis has been challenged recently by the failure of most antioxidants to reduce disease progression and clinical events in patients at risk of or with established atherosclerosis (85, 208, 222). An exception to the failure of antioxidants is probucol, a cholesterol lowering drug, which has the benefit to inhibit macrophage accumulation, stimulate re-endothelialization, and prevent VSMC proliferation by inducing HO-1 (191, 239). While HO-1 targeting might represent a valuable approach to elicit antioxidant activity with antiatherosclerotic function, our knowledge about the effect of iron restriction as intervention to limit iron-driven proatherogenic consequences in humans is still limited. Yet, what are the most effective iron restriction approaches and how they could further interact with other antiatherogenic strategies (e.g., statins, anti-inflammatory therapy) remain to be determined. Finally, more studies are required to better address the role of iron fluctuations within physiological limits in atherosclerosis in the healthy population as well as animal models.
Footnotes
Acknowledgments
The author thanks all the coauthors of the article “Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur Heart J 2020,” which is extensively reviewed in this work, and especially Martina U. Muckenthaler and Matthias W. Hentze together with whom this research was shared with passion and enthusiasm for several years.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.