
Abstract
Significance:
Nuclear factor erythroid 2 (NFE2)-related factor 2 (NFE2L2, or NRF2) is a transcription factor predominantly affecting the expression of antioxidant genes. NRF2 plays a significant role in the control of redox balance, which is crucial in cancer cells. NRF2 activation regulates numerous cancer hallmarks, including metabolism, cancer stem cell characteristics, tumor aggressiveness, invasion, and metastasis formation. We review the molecular characteristics of the NRF2 pathway and discuss its interactions with the cancer hallmarks previously listed.
Recent Advances:
The noncanonical activation of NRF2 was recently discovered, and members of this pathway are involved in carcinogenesis. Further, cancer-related changes (e.g., metabolic flexibility) that support cancer progression were found to be redox- and NRF2 dependent.
Critical Issues:
NRF2 undergoes Janus-faced behavior in cancers. The pro- or antineoplastic effects of NRF2 are context dependent and essentially based on the specific molecular characteristics of the cancer in question. Therefore, systematic investigation of NRF2 signaling is necessary to clarify its role in cancer etiology. The biggest challenge in the NRF2 field is to determine which cancers can be targeted for better clinical outcomes. Further, large-scale genomic and transcriptomic studies are missing to correlate the clinical outcome with the activity of the NRF2 system.
Future Directions:
To exploit NRF2 in a clinical setting in the future, the druggable members of the NRF2 pathway should be identified. In addition, it will be important to study how the modulation of the NRF2 system interferes with cytostatic drugs and their combinations.
Introduction
The redox balance in cells is tightly regulated to meet physiological needs by balancing the abundance of pro-oxidants and reductants. In cancer, the redox balance is commonly dysregulated. However, whether the redox imbalance induces or inhibits cancer formation and cell growth is rather controversial, as the pro- or anticancer effects vary between different cancers and may change as a function of the cancer type (48, 49, 73 –75, 80, 95, 180, 183, 218, 225, 227, 295). Nuclear factor erythroid 2 (NFE2)-related factor 2 (NFE2L2, commonly referred to as NRF2) is a transcription factor that plays a crucial role in maintaining the expression of antioxidant genes. Therefore, NRF2 is vital in maintaining the cellular redox balance.
NRF2 is present in model organisms such as Caenorhabditis elegans and Drosophila melanogaster, which have an antioxidant system similar to mammals, suggesting that the NRF2 pathway represents an evolutionary conserved cytoprotective program (71). However, NRF2 signaling spans beyond redox signaling toward metabolic pathways and the induction of collective cellular changes exemplified by cancer metabolism. In this review, we set out to provide a critical focus on NRF2 activity in cancer. We discuss the aberrant activation of NRF2 and its critical cellular targets.
The Molecular Determinants of NRF2 Signaling
The molecular structure of NRF2
NRF2 is a basic leucine zipper domain (bZIP) transcription factor and contains a Cap'N'Collar structure. NRF2 comprises 605 amino acids and has a calculated molecular weight of 68 kDa. However, recent studies claim that NRF2 may have a higher apparent molecular weight (up to ∼130 kDa) (155, 292). In our hands, the actual molecular weight of NRF2 and the proportions between the different molecular weight isoforms vary between experimental models. Thus, the use of validated antibodies is crucial in investigating NRF2.
The NRF2 transcription factor contains seven conserved NRF2-ECH homology (Neh) domains, known as Neh1-Neh7 (Fig. 1A). The Neh1 domain allows the binding of NRF2 to the antioxidant response sequences (electrophilic responsive/antioxidant-response elements [EpRE/ARE]; 5′-RTGABNNNGCR-3′). The Neh2 domain, the major regulatory domain located in the N-terminal region of NRF2, is essential for the Kelch-like ECH-associated protein 1 (KEAP1)-dependent degradation of NRF2 (see later). The Neh2 domain possesses ETGE and DLG motifs, which are responsible for direct interaction with the negative regulator, KEAP1 (191, 192, 287). The Neh3, Neh4, and Neh5 domains are transactivation domains. Neh3 is located in the C-terminal region of NRF2 and binds to the chromo-ATPase/helicase DNA binding protein family member CHD6, which functions as an NRF2 transcriptional coactivator (204). Neh4 and Neh5 are responsible for NRF2-mediated ARE transactivation by binding to a transcriptional coactivator CREB-binding protein (130). The Neh6 domain is a serine-rich region that regulates NRF2 stability. The redox-insensitive Neh6 domain consists of two motifs (DSGIS and DSAPGS) that are responsible for the binding of β-transducin repeat-containing protein (β-TrCP). Phosphorylation of the DSGIS motif by glycogen synthase kinase 3 (GSK3) enhances the ability of β-TrCP to ubiquitinate NRF2 (37, 223). The Neh7 domain was shown to interact with the retinoic acid receptor X receptor α (RAR α), which represses NRF2 target gene expression (297).
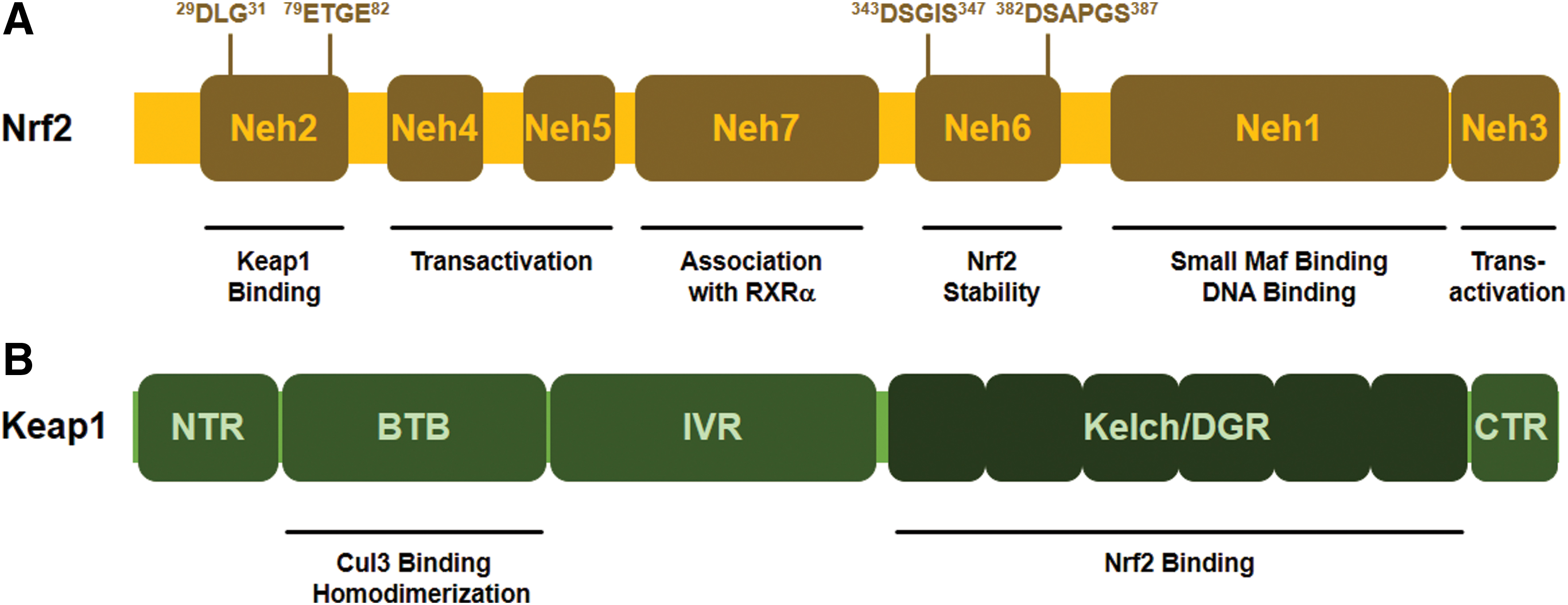
The repressor protein of NRF2, KEAP1; the canonical activation of the KEAP1-NRF2 pathway
The main function of NRF2 is to protect the cells against exogenous and endogenous insults elicited by oxidative stress, which impairs cell function by damaging intracellular lipids, proteins, nucleic acids, and carbohydrates (45). KEAP1 is a repressor of NRF2 located mainly in the cytoplasm, which acts as a substrate adaptor for Cul3-containing E3 ubiquitin ligase (108, 202, 280). E3 ubiquitin ligase is responsible for the ubiquitination of NRF2 followed by its degradation in the proteasomes, resulting in low cellular NRF2 levels under unstressed conditions (41, 145, 332).
KEAP1 contains five domains, including the amino-terminal N-terminal region, the Broad complex/Tramtrack/Bric-a-brac (BTB) domain, the intervening region (IVR), the six Kelch/double glycine repeats (DGR), and the carboxy-terminal (CTR) domain (Fig. 1B). The BTB domain interacts with Cul3 and is required for KEAP1 dimerization (347). The IVR contains several cysteine residues and regulates the activity of KEAP1 (207). The Kelch/DGR domain maintains the interaction between NRF2 and KEAP1 (191). The DGR region recognizes ETGE and DLG motifs in the Neh2 domain of NRF2 protein.
Under normal conditions, NRF2 interacts with KEAP1 through the Neh2 domains (DLG and ETGE motifs), which drive NRF2 to the Cul-3-RBx1-E3 (Cullin-3-ring box-1 E3) ubiquitin ligase complex for degradation (108, 202, 280). KEAP1 is a cysteine-rich protein; in humans, it contains 27 cysteine residues that can be modified by different oxidant and electrophilic compounds (54, 190). The highly reactive cysteine residues, Cys151, Cys171, Cys273, and Cys288, are located in the BTB-IVR domains of KEAP1 protein. Cysteines are susceptible to oxidation and modification (e.g., alkylation or Michael addition) of the cysteine thiols of KEAP1 and NRF2 during oxidative stress is a key step in NRF2 activation (184). Cys151 of KEAP1 was found to facilitate NRF2 activity, whereas Cys288 was shown to diminish NRF2 activity (94, 321). Modification of redox-sensitive cysteines (Cys119, Cys235) of NRF2 prevents KEAP1 recognition and binding (93). Modification of those residues alters the conformation of KEAP1, causing detachment of the DLG motif (low-affinity binding site) from the KEAP1-NRF2 complex. However, NRF2 remains in association with KEAP1 through the ETGE motif (high-affinity binding site), according to the hinge and latch model by Tong et al. (287, 288). This steric change, when the DLG motif of KEAP1 releases NRF2, prevents the degradation of NRF2.
During cellular stress, Sekhar and co-workers (251) suggested that the binding between KEAP1 and Cul3 can also be disrupted, leading to NRF2 escape from proteasomal degradation. NRF2 can also be degraded and eliminated by KEAP1-independent mechanisms with the help of β-TrCP, which can recognize and bind to phosphorylated NRF2 and trigger its ubiquitination and degradation (37, 223, 224). A third degradation procedure has been reported in cirrhotic liver, where endoplasmic reticulum stress triggers E3 ubiquitin ligase (namely, HRD1) interaction with NRF2 domains, promoting NRF2 elimination (313). Subsequently, free NRF2 translocates to the nucleus where it dimerizes with transactivation partners [for instance, Small Maf protein family (293) or Jun (289, 328)] and binds to the promoter region of EpRE/ARE-response genes, inducing their transcription (101, 219). After NRF2 exerts its effect in the nucleus, it is phosphorylated by tyrosine kinases that promote NRF2 nuclear export and cytosolic degradation (110).
Role of NRF2 in antioxidant defense
There are more than 200 NRF2-dependent target genes identified in humans (186, 346). A more recent study suggests that there might be more than 1000 genes regulated by NRF2 (186). The majority of NRF2-dependent target genes encode for detoxifying enzymes to overcome oxidative damage of DNA, proteins, and lipids, for example, genes involved in the synthesis of glutathione, antioxidant proteins, drug-metabolizing enzymes, transporters, and numerous other stress response proteins [for review, see Hayes and Dinkova-Kostova (92) and Suzuki et al. (277)] (Fig. 2).
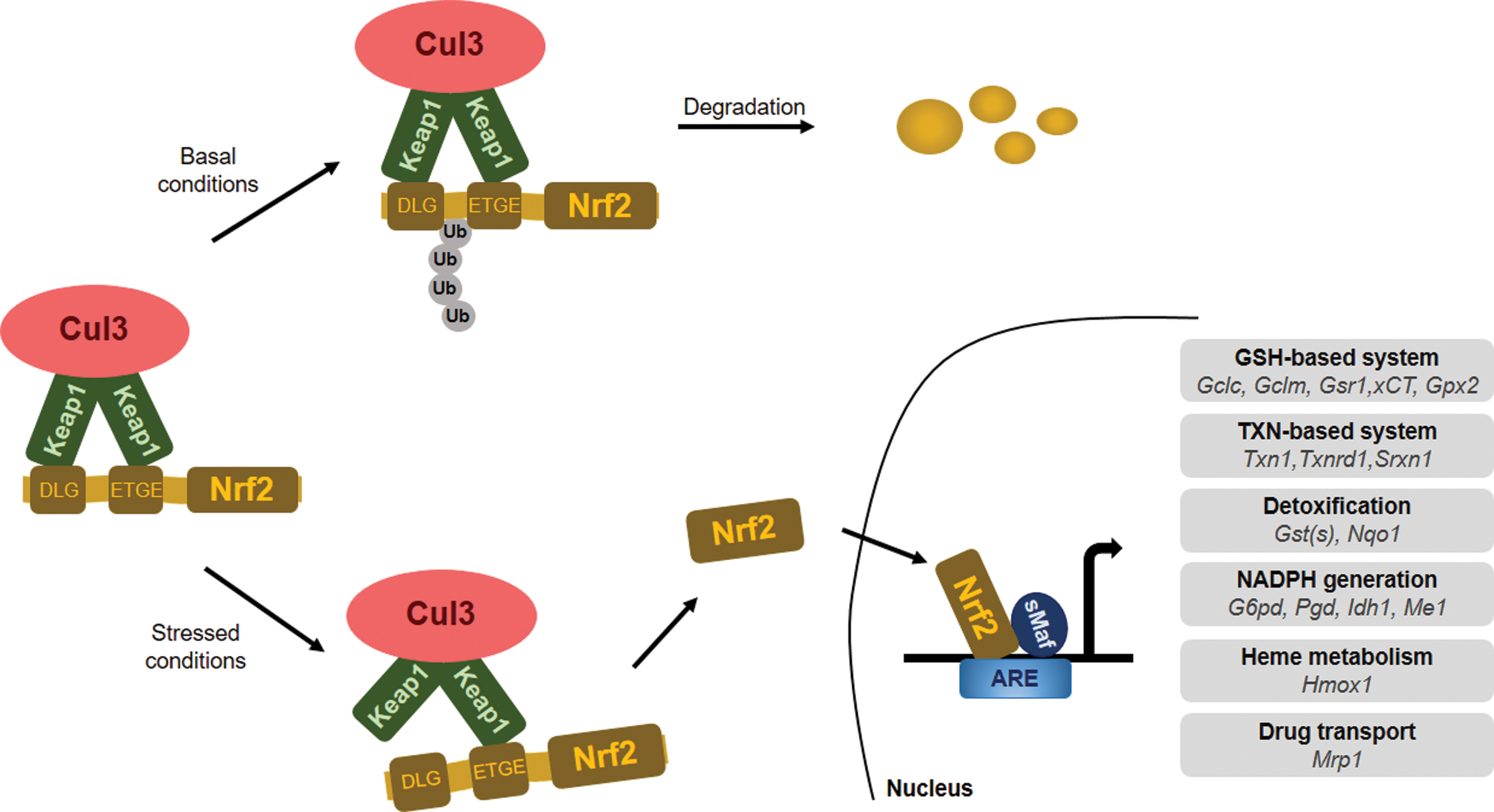
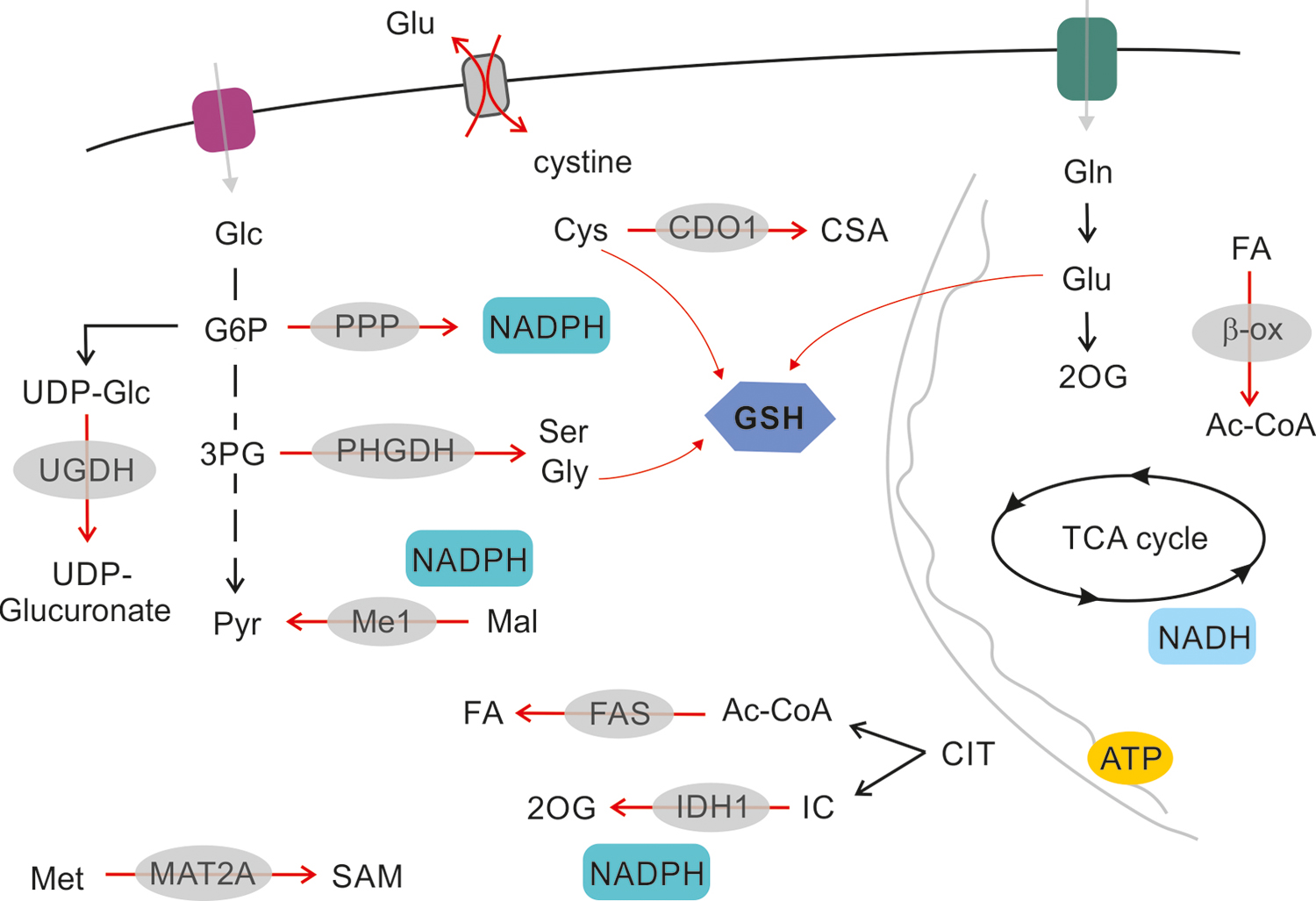
Oxidative, nitrosative, and electrophilic stress promote NRF2 stabilization and activity. NRF2 maintains the appropriate intracellular reduced glutathione (GSH)/oxidized glutathione (GSSG) ratio by maintaining glutathione synthesis and glutathione reduction. This is achieved through controlling the expression of glutamate-cysteine ligase catalytic (GCLC) and modifier (GCLM) subunits of the glutamate-cysteine ligase (GCL) complex, which collectively catalyze the rate-limiting step in GSH synthesis (99). NRF2 also regulates the expression of glutathione reductase 1 (Gsr1) (282). Further, NRF2 regulates the expression of the SLC7A11 (subunit of the cystine/glutamate antiporter system Xc−), which imports cystine into cells in exchange for glutamate, and hence, regulates cysteine and glutamate availability in cells (163). Finally, NRF2 regulates the pentose-phosphate shunt and through that NADPH availability that is needed for reducing GSSG (4, 35, 92, 159, 334).
Besides the regulation of glutathione levels, NRF2 controls the expression of a broad set of enzymes that play a role in the detoxification of H2O2, peroxide radicals, and oxidized thiols, including glutathione peroxidase 2 (Gpx2) (88), thioredoxin 1 (Txn1), thioredoxin reductase 1 (Txnrd1), sulfiredoxin 1 (Srxn1) (2, 90), and glutathione S-transferases (282). Production of NADPH, which is used as a cofactor by many antioxidant systems in redox reactions, is also under the control of NRF2. The expression of glucose-6-phosphate dehydrogenase (G6pd), 6-phosphogluconate dehydrogenase (Pgd), isocitrate dehydrogenase 1 (Idh1), and malic enzyme 1 (Me1) is NRF2 dependent. Another prominent cytoprotective enzyme regulated by NRF2 is heme oxygenase 1 (HMOX1), the enzyme catalyzing the breakdown of heme molecules (7) and catalase, which reduces H2O2 to water and oxygen.
Natural and synthetic inductors and inhibitors of NRF2 signaling
Other small-molecule inhibitors and activators are vital in fine-tuning NRF2 activity (184). In cancer, certain intermediates of cell metabolism can modulate the NRF2-KEAP1 system by introducing post-translational modifications into NRF2 or KEAP1 that impact NRF2 function or stability. For instance, the mutation of fumarate hydratase in papillary renal cell carcinoma leads to fumarate accumulation, which induces KEAP1 succination and NRF2 stabilization (209). The inhibition of glycolysis and concomitant build-up of glycolytic intermediates in several cell lines promote NRF2 activity through methylglyoxal-induced (elimination product of dihydroxyacetone phosphate and glyceraldehyde-3-phosphate) formation of a covalent bond between arginines and cysteines (so-called MICA modification) that leads to the dimerization of KEAP1 (23). In glucose-free conditions, a decrease in KEAP1 O-glycosylation with β-N-acetylglucosamine leads to NRF2 accumulation in breast cancer cells (30). Finally, glycation destabilizes NRF2 via increased proteasomal degradation in hepatocellular carcinoma (HCC) (245).
Several artificial NRF2 activators function by disrupting KEAP1-NRF2 interaction, including tert-butylhydroquinone (166) or RA839 (310). Removing KEAP1 from the KEAP1-NRF2 complex leads to stabilization and activation of NRF2. The inhibition of the proteasome (e.g., by MG132) can prevent the degradation of NRF2 (149). A food additive and pro-carcinogen, butylated hydroxyanisole, can induce NRF2 (179) and NAD(P)H quinone dehydrogenase 1 (NQO1), which boosts the expression of 8-Oxoguanine glycosylase (OGG1) (268). Increased OGG1 prevents the oxidative catabolism of 4-hydroxyestradiol (4-OHE2) to the catechol estrogen quinone metabolites, which readily reacts with DNA to produce depurinated adducts. Superphysiological concentrations of vitamin C can also decrease the nuclear/cytosolic NRF2 ratio, indicating NRF2 activation (200).
A nucleoside analog antimetabolite, Cordycepin, downregulates NRF2 signaling and, consequently, increases the cytoplasmic levels of oxygen-centered reactive species (ROS). Cordycepin was employed as a radiosensitizer in breast cancer in a preclinical study (56). Although this study did not elucidate a mechanism of action, Cordycepin may induce caveolin-1 (Cav-1) expression (119). Cav-1 can suppress the NRF2-elicited effects by reducing manganese-dependent superoxide dismutase (MnSOD) expression, an important downstream target of the NRF2 pathway (86). In agreement with this theory, epidemiologic nested case
The n-6 and n-3 polyunsaturated fatty acids (PUFAs) serve as substrates for cyclooxygenases that synthesize PUFA-derived hormones. One of the final products of the cyclooxygenase-2 (COX-2) pathway, 15-deoxy-prostaglandin J2 (15d-PGJ2), is switched on by inflammatory stimuli (258). In breast cancer cells, NRF2 translocation to the nucleus is induced by 15d-PGJ2, resulting in enhanced interaction between NRF2 with ARE (161) to drive the expression of a variety of phase 2 detoxifying and antioxidant enzymes (131). Nuclear accumulation and the transactivation of NRF2 appears to be dependent on the presence of the characteristic cyclopentenone moiety in 15d-PGJ2 (161). The electrophilic α,β-unsaturated carbonyl group, constituting the cyclopentenone ring of 15d-PGJ2, is anticipated to bind covalently to specific cysteine residues of the KEAP1 molecule, resulting in the liberation and nuclear translocation of NRF2 (59).
There is a firm link between the etiology of several cancers and bacterial dysbiosis (65, 67, 76, 77, 98, 118, 150, 196). In breast cancer, bacterial metabolites (estrogen metabolites, short chain fatty acids, lithocholic acid [LCA], and cadaverine) were shown to play key roles in regulating the behavior of cancer cells by influencing breast cancer stemness, anticancer immunity, cancer cell migration, metastasis formation, epithelial-to-mesenchymal transitions, and cancer cell metabolism (109, 149, 150, 194 –196). Apparently, the capacity of the microbiome to synthesize these metabolites is reduced in breast cancer as compared with controls (76, 150, 196). The secondary bile acid, LCA was shown to exert cytostatic properties on breast cancer cells. Further, bacterial LCA production is largely reduced in the early stages of breast cancer (196). LCA reduces NRF2 expression, and the consequent reduction in antioxidant defenses exerts cytostatic properties in breast cancer. This mechanism is ineffective in nontransformed cells (149).
Hormones regulating NRF2 signaling
Hormones, such as gonadotropins and sex steroids (follicle-stimulating hormone, luteinizing hormone, estrogen), can promote NRF2 signaling through the induction of ROS [measured by dichlorofluorescein (DCF)—please note that DCF autooxidation may produce reactive species and, hence, provide a false positive result (124, 236)] in ovarian cancer (167). Follicle-stimulating hormone can further activate hypoxia-inducible factor 1 (HIF1) and vascular endothelial growth factor (VEGF) to promote tumor angiogenesis (339). Hormone regulation of NRF2 was found in ovarian cancer and may also play a major role in the development of other hormone-related cancers such as prostate and breast cancer (122).
Noncanonical NRF2 activation
Noncanonical NRF2 activation is based on the presence of proteins that activate NRF2 transcription by inducing the disassembly of the NRF2-KEAP1 complex. Certain ETGE motif-containing proteins can compete with KEAP1 for dimerization with NRF2, to prevent NRF2 degradation. Proteins that activate NRF2 include Wilms tumor gene on the X chromosome (WTX, AMER1) (25), partner and localizer of BRCA2 (PALB2) (181), dipeptidyl peptidase 3 (DPP3) (89), Cyclin-dependent kinase 20 (CDK20) (300), sequestosome 1 (p62) (107), p21 (31), and additional proteins identified in (89). The spectrum of competing proteins might represent a divergence of oxidative stress-related and -unrelated cellular signals, resulting in NRF2-induced cytoprotection.
Although several noncanonical activators of NRF2 were described, only a few have been studied in the context of cancer etiology, including kidney (25), breast (178), and lung (89) cancers, and osteosarcoma (181). DPP3 contains an ETGE motif, which binds to the Kelch domain of KEAP1 and activates NRF2 (89) in breast cancer cells in response to ROS. Overexpression of DPP3 in MCF7 cells causes accumulation of NRF2 and promotes resistance to H2O2 and diquat (178). Hast et al. (89) performed TCGA-based data mining analysis in tumor gene expression datasets to assess DPP3 expression in relation to NFE2L2 and KEAP1. DPP3 expression and copy number were increased in NFE2L2 wild-type and KEAP1 mutant tumors as compared with NFE2L2 mutant or KEAP1 wild-type tumors. This implicates that DPP3 enhances NRF2 activity under conditions when KEAP1 activity is attenuated by hypomorphic mutations. Consequently, the function of NRF2 is amplified even though hypomorphic KEAP1 activity is still able to suppress NRF2. On the other hand, NRF2 mutations often lead to hyperactivation of NRF2 and eliminate the need for additional amplifying factors. Further, this phenomenon might apply to other ETGE-containing proteins, too.
More recent research (32) identified family with sequence similarity 129, member B (FAM129B) as another positive regulator of NRF2 signaling, active in several cancer cell lines. FAM129B contains DLG and ETGE sequences, and FAM129B expression correlates with poor clinical outcome in breast and lung cancers (32). Similarly, PALB2 competes effectively with KEAP1 for NRF2 binding and the overexpression of PALB2 induces NRF2 levels and transcription via interaction with the nuclear NRF2-KEAP1 complex (181). The depletion of PALB2 in vitro impairs NRF2 function.
Another example of noncanonical activation of NRF2 is p21, a cyclin-dependent kinase inhibitor. In the cell model of colorectal cancer, p21 can recognize and directly bind to the DLG/ETGE motif of NRF2 to prevent NRF2 degradation. Hence, the expression of antioxidant genes, such as HMOX1 an NQO1 (31), in response to H2O2 treatment is enhanced. NRF2, in turn, can elevate p21 expression by binding to the promoter region of p21, as demonstrated in a study focused on lung carcinoma with deficient p53 (111).
During the initiation of the autophagy cascade, p62 binds to KEAP1 and initiates NRF2 transactivation of target genes. As an autophagy substrate, p62 can also interfere with NRF2-KEAP1 interaction through binding to the DLG/ETGE motif and activating NRF2 (107). The role of p62 in NRF2 transactivation was confirmed in hepatic adenoma (107), HCC (261), breast and ovarian cancers (233, 315), and in glioma (340). In contrast, in normal hepatocytes, NRF2 is degraded under the same conditions (317). The expression of p62 and oligomerization in the cytosol is also a negative prognostic factor in HCC recurrence (317). In breast cancer, p62-dependent NRF2 activation induces the proportions of CD44-positive cancer stem cells (CSCs) that are associated with an aggressive phenotype with enhanced tumor growth and elevated therapeutic resistance (239).
Role of NRF2 in Cancer
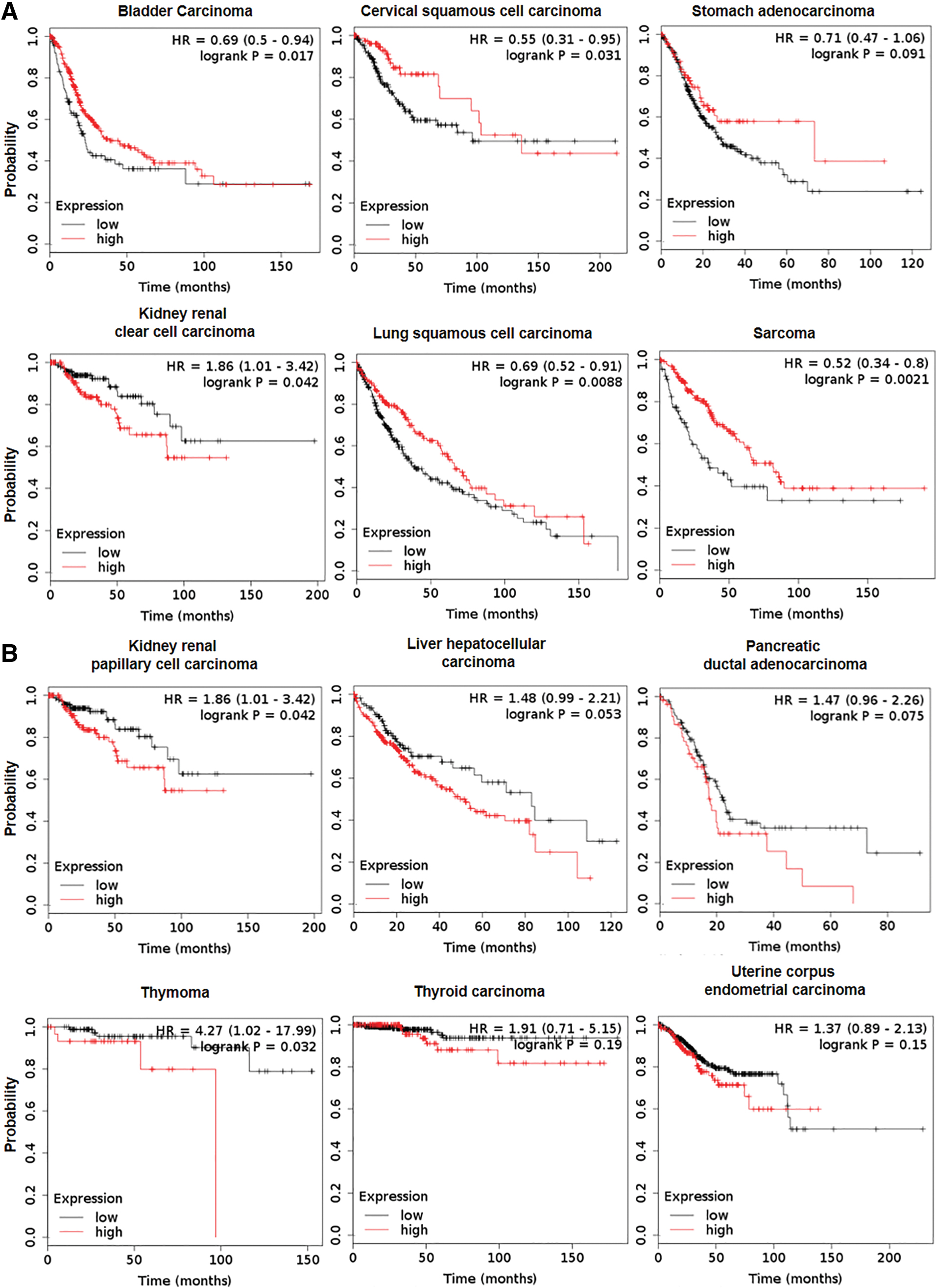
Cancers and cancer cell behavior can be characterized by cancer hallmarks (85), which contribute to the ability to sustain proliferation and to evade cell death. NRF2, as a strategic cytoprotective factor, positively regulates several cancer hallmarks, including the promotion of metastasis, evasion of cell death, and metabolic changes. In normal cells, activation of NRF2 complies with the period of acute detoxification; whereas in cancer cells, a newly described phenomenon called “NRF2 addiction” occurs (144). NRF2 addiction refers to sustained NRF2 signaling due to genetic, epigenetic, or general oncogenic signaling. There is growing experimental evidence that NRF2 activation enhances cancer growth and increases therapy resistance (14, 177) due to the expression of phase 1 and phase 2 drug-metabolizing enzymes (92) and multidrug-resistance-associated transporters (Mrps) (185), which support metastasis formation (100, 246), cancer growth (52, 232), and poor survival (115, 178) (Fig. 3 and Table 1). It is interesting to note that, despite the large number of examples where NRF2 activation is associated with negative outcomes, NRF2 overexpression can support longer survival in certain cancers (58, 75, 80, 95, 128, 156, 172, 180, 277) (Fig. 4). In this article, we will review the molecular prerequisites of aberrant NRF2 signaling in cancer.
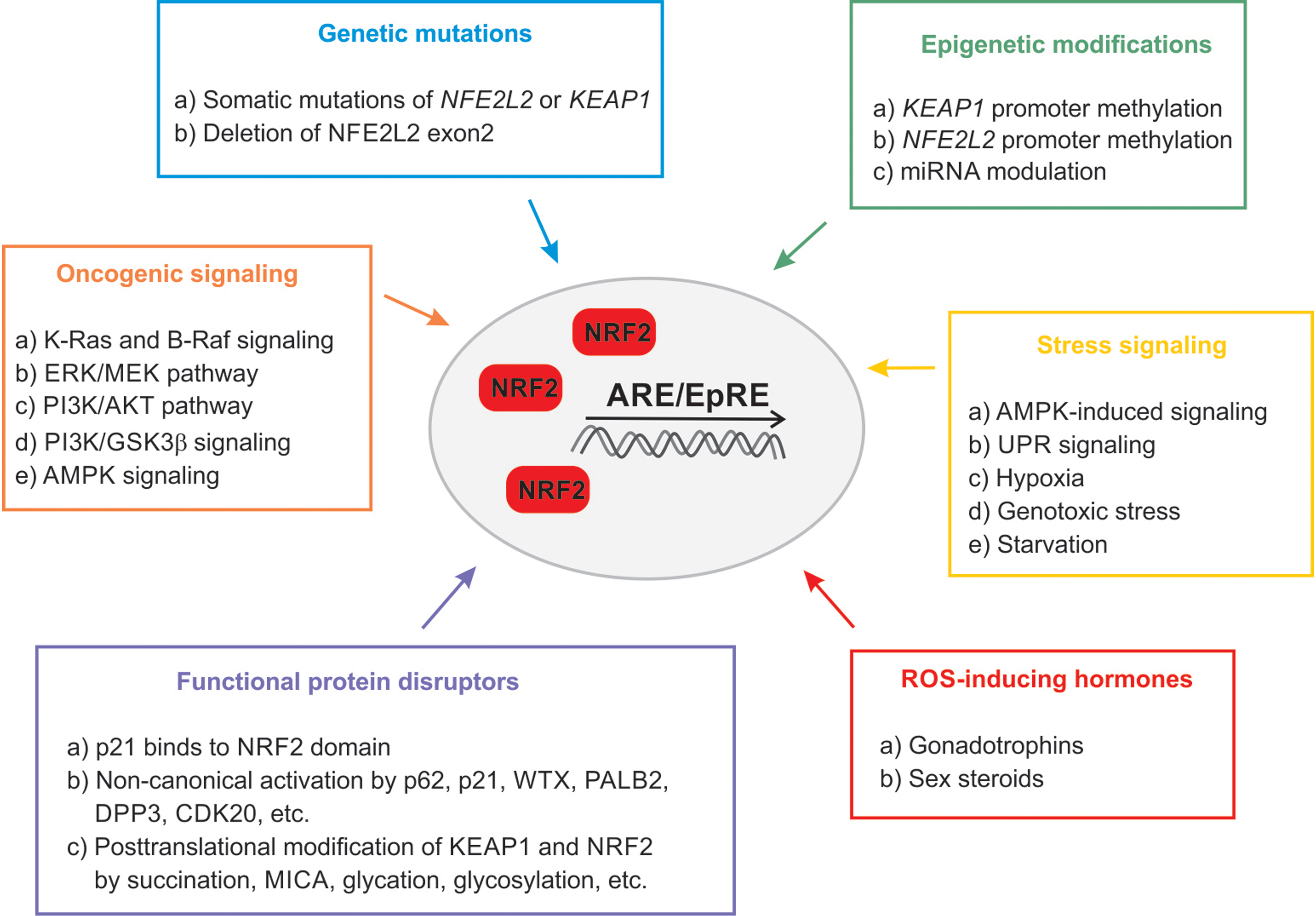
Aberrant Activation of NRF2 in Cancer and NRF2-Addicted Cancer Types
AKT, protein kinase B; AML, acute myeloid leukemia; AMPK, AMP-activated protein kinase; BRCA2, breast cancer type 2 susceptibility protein; CDK20, cyclin-dependent kinase 20; DPP3, dipeptidyl peptidase 3; FH, fumarate hydratase; GSK3β, glycogen synthase kinase 3β; LOF, loss-of-function; MEK, mitogen-activated protein kinase kinase; NSCLC, nonsmall-cell lung carcinoma; p62, sequestosome 1; PALB2, partner and localizer of BRCA2; PI3K, phosphoinositide 3-kinases; ROS, oxygen-centered reactive species; WTX, Wilms tumor gene on the X chromosome.
Genetic mutations in KEAP1 and NRF2
The occurrence of somatic mutations in KEAP1, NRF2, or Cul3 is the most well-known mechanism of sustained NRF2 activation in cancer (193). According to public genomic databases, KEAP1 and NFE2L2 seem to be the most affected genes in lung cancer, followed by esophageal and endometrial cancer, as summarized (134). Studies assessing statistics of genetic alterations in lung cancer (226) demonstrate frequent loss-of-function (LOF) mutations in KEAP1 (213) and loss-of-heterozygosity at the genetic locus of KEAP1 (266). KEAP1 missense or nonsense mutations have been also identified in malignant melanoma (198), thyroid cancer (44), HCC (38), endometrial carcinoma (311), and breast (203, 271) and ovarian cancers (148). In summary, mutations of KEAP1 that confer loss of function or hypomorphic function induce NRF2 activity.
Alleles of NRF2 that lead to its activation have been described as well. The genetic mutation of NFE2L2 affecting the DLG/ETGE motif of the Neh2 domain was shown in a lung cancer cell line (259), renal cell carcinoma (208), HCC (81), skin cancer (142), and esophageal carcinoma (257). A unique genetic mutation, deletion of the NFE2L2 exon 2, was found to stabilize NRF2 and enhance its activity in nonsmall-cell lung carcinoma (NSCLC) and head-neck cancer (78). Moreover, copy number loss of Cul3 or inactivating mutation of RBx1, which controls NRF2 degradation, was shown in esophageal (170) and ovarian cancer (188), as well as in thyroid (187) and renal cell carcinoma (208).
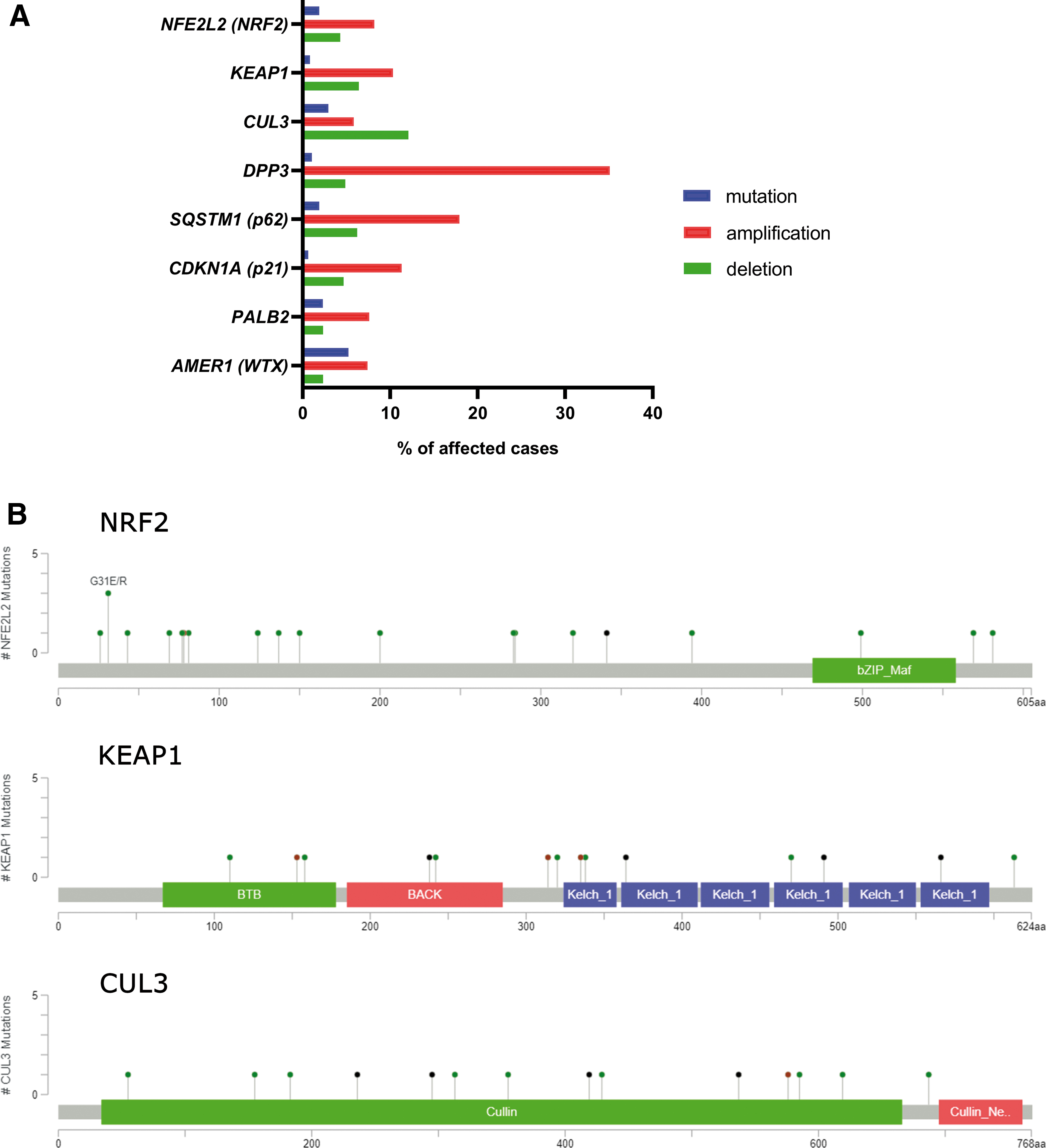
A recent study identified two single nucleotide polymorphisms of KEAP1 associated with shorter relapse-free survival in breast carcinoma (87). A study by Kovács et al. (149) showed that NRF2 overexpression and the downregulation of downstream pro-oxidant genes negatively correlates with patient survival in nontriple negative breast cancer. Our search in the TCGA-BRCA dataset revealed prominent amplification of DPP3 and p62 genes, which suggests amplification of NRF2 activity in breast cancer by noncanonical mechanisms (Fig. 5).
Epigenetic modifications of KEAP1 and NRF2
Epigenetic modification in KEAP1 or NFE2L2 promoter regions can also contribute to NRF2 stabilization in many cancers (122). Both hypo- and hypermethylation of the NFE2L2 promoter have been reported. Hypomethylation, due to the reduced expression of EZH2 methyltransferase, is associated with NRF2 overexpression in colon cancer (125). Hypermethylation of the NFE2L2 promoter is associated with prostate cancer (135) and leads to reduced NRF2 activity, a rare example among malignant diseases. Bromodomain protein 4 (BRD4) was implicated in the transcriptional regulation of KEAP1. Thus, BRD4 might represent a strong modulator of the NRF2/KEAP1 pathway in prostate cancer (105).
Hypermethylation of the KEAP1 promoter (CpG islands) is catalyzed by methyl-CpG-binding domain protein 1 and ubiquitin-like, containing PHD and RING finger domains-1 (UHRF1), leading to reduced KEAP1 protein levels and augmented expression of NRF2-dependent genes (3, 331). KEAP1 hypermethylation is associated with lung (301), colorectal (84), breast (16), prostate (337), thyroid (187), and renal (61) cancers, as well as malignant glioma (201).
The regulatory role of microRNAs in NRF2 signaling
NRF2 and KEAP1 levels can be modulated at the post-transcriptional level through altering mRNA splicing or microRNA (miRNA) levels. In the breast cancer cell line, MCF-7, miR-28 inhibits NRF2 action without the contribution of KEAP1 (324). The miRNAs, miR-507, miR-634, miR450a, miR129–5p miR-340, miR-146b, and miR144, were reported to directly suppress NRF2 activation in different cancer types, including acute myeloid leukemia (AML) (274), breast cancer (104), and esophageal cancer (320). In HCC, miR-144 suppressed NRF2 levels (344); whereas in neuroblastoma, the same miRNA induced ROS-induced apoptosis (341). Recent studies demonstrate that miRNAs promote migration and support malignant transformation in different neoplasias. For example, miR-155 promotes lung cancer (28) and miR93a and miR153 drive the malignant transformation of the breast (269, 296). Last but not least, NRF2 can influence and modulate the expression of numerous miRNAs in tumors, creating a complex feedback-regulatory system in humans (253).
Several miRNAs were shown to modulate KEAP1, NRF2, and target gene translation or transcription. The miRNAs, miR-432-3p and miR-200a, impair KEAP1 translation in esophageal and breast carcinoma (6, 58); whereas miR-7 and miR-148b increase the expression of NRF2-dependent antioxidant genes (HMOX1 and GCLM) in neuroblastoma (123) or elevate ROS production due to the repression of HIF1 and NRF2 expression (220), respectively.
Oncogenic signaling
Although NRF2 activity promotes cancer progression by various mechanisms, as previously discussed, we should emphasize that NRF2 accumulation can only contribute to malignant proliferation in the presence of active oncogenic signaling and, therefore, we refer to its role as the oncogenic driver. Keap1-deficient mice develop cancer only in the presence of oncogenic mutations, such as those in Kras/Hras or Tp53 (113, 234). The contribution of NRF2 to carcinogenesis differs between cancer types. For example, the Kras:Tp53:Keap1 triple mutation is associated with an aggressive phenotype in lung carcinoma in ∼30% of cases, whereas in the pancreas this combination of mutations does not initiate cancer but leads to fibrosis (83).
Recent evidence suggests that cancer-associated signaling pathways can influence the activity of NRF2 by increasing its mRNA level rather than improving its stability. In this respect, K-Ras and B-Raf signaling pathways can enhance NRF2 mRNA levels to promote ROS elimination (52). K-Ras promotes NRF2 transcription through the extracellular signal–regulated kinase (ERK)/mitogen-activated protein kinase kinase (MEK) pathway in NSCLC (279) and renal carcinoma (13). The importance of Ras signaling is highlighted by the findings that H-Ras-activated HO-1 overexpression is abolished by ERK inhibitors or NFE2L2 knockdown (13). In K-Ras-associated malignancies, the elevated expression of antioxidant genes (HMOX1 and NQO1) is associated with an aggressive phenotype, whereas the elimination of NRF2 disrupts malignant progression and restores sensitivity to cancer therapy (254). NRF2 signaling also actively improves cancer survival by maintaining independence of growth factors in K-Ras-associated malignancies; in pancreatic ductal adenocarcinoma (PDAC), NRF2 signaling is upregulated, resulting in improved cancer survival via maintaining phosphorylated levels of EGFR and ERBB2, and by maintaining intact mRNA translation efficiency (33). Interestingly, NFE2L2 knockdown in normal organoids was tolerated, whereas NRF2 knockdown was detrimental in tumor-derived organoids.
The nuclear accumulation of NRF2 increases when different proliferative pathways are active (263, 278). Phosphoinositide 3-kinases (PI3K)/protein kinase B (AKT) activation is associated with elevated NRF2 mRNA levels and nuclear accumulation of NRF2, leading to increased cell proliferation, survival, and metabolic reprogramming (197). In breast cancer, PI3K/AKT activation, in response to estradiol, is accompanied by elevated expression of NRF2 and, consequently, increased expression of a large set of antioxidant genes (79). As mentioned earlier, GSK3 can phosphorylate NRF2 and induce its degradation by the Cult1-βTrCP complex. When the PI3K-AKT pathway is active, AKT phosphorylates and inactivates GSK3, leading to inactivation of Cult1-βTrCP-driven NRF2 degradation in cancer cells (37). Further, in Keap1-deficient mice, PI3K-AKT activation leads to NRF2 nuclear accumulation and NRF2-driven cell proliferation of hepatocytes (263).
NRF2 in unfolded protein response
Accumulation of unfolded proteins in the lumen of endoplasmic reticulum, termed endoplasmic reticulum stress, activates the unfolded protein response (UPR) (235). Endoplasmic reticulum stress and UPR are frequently found in tumor cells due to nutrient deficiency, hypoxia, and exposure to therapeutic agents. The association of UPR with the KEAP1/NRF2 pathway has been investigated in both cancer cells and normal cells. Starvation activates NRF2 through AMP-activated protein kinase (AMPK) and through protein kinase R-like endoplasmic reticulum kinase (PERK), which is located in the endoplasmic reticulum membrane and induced during UPR (40 –42). NRF2 is phosphorylated by PERK, which leads to the dissociation of the NRF2/KEAP1 complex and increased expression of ARE genes to promote survival via reduced ROS levels (42).
In cancer, the PERK-NRF2 pathway drives endoplasmic reticulum stress-resistance and multidrug resistance as demonstrated in colorectal cancer (243), multiple myeloma (276), and AML cells (8). Further, in oral (tongue) squamous carcinoma cells, heat shock 70 kDa protein 5 (GRP78) and PERK-dependent activation of NRF2 induced Warburg-type metabolism and maintained cancer-initiating cells independently of ROS production (27). Moreover, fibroblast growth factor 19, which is overexpressed in HCC, can promote the cytoprotective FGFR4/GSK3β/NRF2 axis to protect against endoplasmic reticulum stress (281).
NRF2 regulating specific aspects of cancer metabolism
Cancer metabolism is usually perceived in the context of maintaining high energy standards for growing cells and includes enhanced catabolic pathways, enhanced biosynthetic pathways, and maintenance of redox homeostasis associated with high substrate turnover. The primary function of NRF2 is still the regulation of oxidative damage. However, since a number of NRF2 targets are rate-limiting enzymes in the respective pathways, the function of NRF2 logically encompasses metabolic rewiring also. Indeed, several metabolic pathways in NRF2-overexpressing cancer cells have been described as elevated, including the pentose-phosphate pathway (PPP), and nucleotide-, glutathione- (234), and serine synthesis in a lung cancer (51) model. In this section, we draw attention to certain metabolic pathways that tend to be affected by NRF2 signaling as a primary or secondary function, and we evaluate the role of NRF2 in cell survival based on metabolic rewiring (Fig. 6).
Strategic role of p62 in NRF2 signaling: autophagy and metabolic rewiring
A considerable body of evidence points toward a link between NRF2 and autophagy, which represents a conserved strategy to overcome starvation and nutrient deprivation (162, 329) by recycling cellular macromolecules and organelles for essential metabolic substrates (222). During autophagy, p62 functions as a marker of proteins and organelles that are targeted for degradation by the autophagosome. The NRF2 signaling pathway is activated by p62 through the noncanonical mechanism connecting NRF2 to a spectrum of conditions related to starvation.
In HCC, Parkin-induced autophagy promotes p62 phosphorylation on Ser349 by a mechanistic (or mammalian) target of rapamycin complex 1 (mTORC1), leading to a concomitant rise in NRF2 protein expression (106). In fact, p62 binds to the STGE motif of KEAP1 with low affinity, which increases when p62 is phosphorylated. Hence, phosphorylation of p62 effectively induces NRF2 expression during autophagy (146). The p62-dependent NRF2 activation also occurs in a carfilzomib- (proteasome inhibitor) resistant model of multiple myeloma (228).
Other reports contradict the induction of NRF2 in autophagy by p62. A Crispr-based screen focused on finding novel negative regulators of NRF2 signaling (133) also identified a number of autophagy-related genes in human kidney HK2 cells. However, subsequent functional analyses demonstrated that changes in the phosphorylation of p-p62 do not necessarily induce NRF2 in HK2 cells, indicating that the autophagy-NRF2 loop can proceed in a p62-independent manner. In addition, the p62-NRF2 axis is not activated in a TCS2-deficient kidney cancer model, where upregulated levels of p62 do not activate NRF2 (153). Thus, NRF2 function in autophagy requires tissue- and context-specific evaluation. On the other hand, in Atg7-deficient mice, NRF2 activation remains p62 dependent and enhances cellular growth of HCC in 3D cultures (107).
The p62-NRF2 axis also interacts with signaling pathways through other pathways besides mTORC1. In HCC, H2O2 and hypoxia-induced ROS accumulation and NRF2 activation are p62 dependent (317). In this case, p62 is activated by phosphorylation at S28 by ketohexokinase-A (KHK-A), a splice isoform of phosphofructokinase that possesses protein kinase activity and KHK-A activation depends on AMPK activation (317). These changes collectively lead to NRF2-mediated gene expression and contribute to the development of HCC in xenograft animals (317). AMPK, a well-known energy sensor, can also influence NRF2 activity in breast cancer directly, as AMPK phosphorylates NRF2 and facilitates its nuclear accumulation (120). In MCF-7 and T47D breast cancer cells, AMPK activity, induced by glucose deprivation during nutrient starvation, is necessary for the expression of NRF2-driven genes (294). In addition, in breast cancer, a positive feedback loop between p62 and NRF2 has been demonstrated. The S349 residue of p62 can be phosphorylated by class III PtdIns3 lipid kinase (VPS34), whereas VPS34 induces NRF2-dependent expression of p62 and induces the noncanonical activation of NRF2 (117). Moreover, VPS34 promotes PKCδ-dependent phosphorylation of p62, also on S349, that not only strengthens the interaction between NRF2 and p62 in response to caspase inhibitor treatment but also activates downstream signaling pathways leading to tumor formation, including the MEK/ERK pathway (117).
In terms of generic carbon rewiring for energy metabolism, HCC expressing pS349–p62, reprogram both glucose and glutamine metabolism, marked by the induction of the glucuronate pathway and glutamine-derived glutathione synthesis in an NRF2-dependent manner (242). The resulting increase in glutathione levels is vital to support cancer growth and chemoresistance. Activation of NRF2 is also critical for the survival of breast cancer cells in glucose-deprived conditions, as NRF2 induction protected cells from autophagy and oxidative stress (294). Under glucose-free conditions, NRF2 enhances cellular survival in a p62-independent fashion (248). The authors suggest that the mechanism is initiated by ATP deficiency followed by increases in the levels of NQO1 (248). In accordance with this suggestion, reduced ATP induces NRF2 activity, causing an alteration in redox balance, as recently described in lung carcinoma cells (248).
To summarize, a substantial body of evidence supports the notion that NRF2 can be upregulated by p62, which connects NRF2 signaling to various means of signal transduction, such as AMPK and mTORC1. Consequently, NRF2 induction supports growth in a spectrum of cancer types.
NRF2 regulated ferroptosis and metabolic rewiring through SLC7A11 expression
Ferroptosis is a form of programmed cell death that is dependent on iron and independent of caspases, which is induced specifically by oxidative stress (55). In principle, inhibition of SLC7A11, depletion of intracellular cysteine and glutathione, and lipid peroxidation suppress GPX4 activity and initiate ferroptosis. NRF2 regulates the expression of genes involved in the prevention of ferroptosis, including iron metabolism, NADPH synthesis, glutathione reduction, and nonessential amino acid interconversion. The SLC7A11 gene codes for xCT protein, a subunit of a membrane-localized cystine-glutamate antiporter (Xc−), which imports a cysteine dimer (cystine) in exchange for glutamate. SLC7A11 is a direct target of NRF2, activating transcription factor 4 (ATF4) (286), and ATF3 (298) and is also a prominent target of NRF2 signaling and ferroptosis in a number of malignancies.
Consistent evidence demonstrates that the NRF2 transcriptional response, involving the expression of SLC7A11 and xCT, is protective against ferroptosis. This implies that NRF2-dependent transcription programs lead to escape from ferroptosis and, in turn, support cell growth. xCT is induced by H2O2 and NRF2 activation in MCF7 breast cancer cells, with a concomitant increase in GSH synthesis (82). Also, in KrasG12D, TP53−/− lung adenocarcinoma cells, the expression of the KEAP1 LOF mutant induces SLC7A11, which boosts GSH and reduces ROS levels (247). Moreover, the repression of xCT impairs colony formation in Ras-transformed cell models (169). As a result, xCT induction due to NRF2 signaling confers a refractory phenotype in response to the ferroptosis-inducing agents, erastin or RSL3, in glioma cell models (62). Further, NRF2 expression coincides with worse clinical outcome in glioma patients. Recent evidence also indicates that depletion of ovarian tumor family member deubiquitinase (OTUB1) (174) or the inactivation of breast cancer type 1 susceptibility protein (BRCA1) associated protein 1 (BAP1) tumor suppressor in breast cancer (338) leads to induction of xCT in an NRF2/hypoxia-inducible factor 1α (HIF1α)-dependent fashion, which protects cancer cells from ferroptosis.
It should be noted that a high level of NRF2 accumulation, which is achieved by the functional impairment of KEAP1 or NRF2 hyperactivation, combined with sustained oncogenic signaling, allows NRF2 to modulate metabolism under pathological conditions. NRF2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming (197). In addition, xCT alters cellular glucose and glutamine utilization to promote cell survival (Fig. 7). The tendency of cells to prefer glucose and glycolysis in response to xCT expression level was demonstrated in several cell models (114). The knockdown of SLC7A11 rescues viability in glucose-free conditions. Inhibition of xCT in nonsmall lung cancer cells improves survival in glucose-free medium, when bioenergetics rely primarily on mitochondrial OXPHOS through glutamine oxidation (114). Increased xCT expression sensitizes breast cancer cells to glucose, but not glutamine withdrawal (262). Thus, the export glutamate via the Xc− system due to the expression of SLC7A11 maintains glucose addiction for survival.
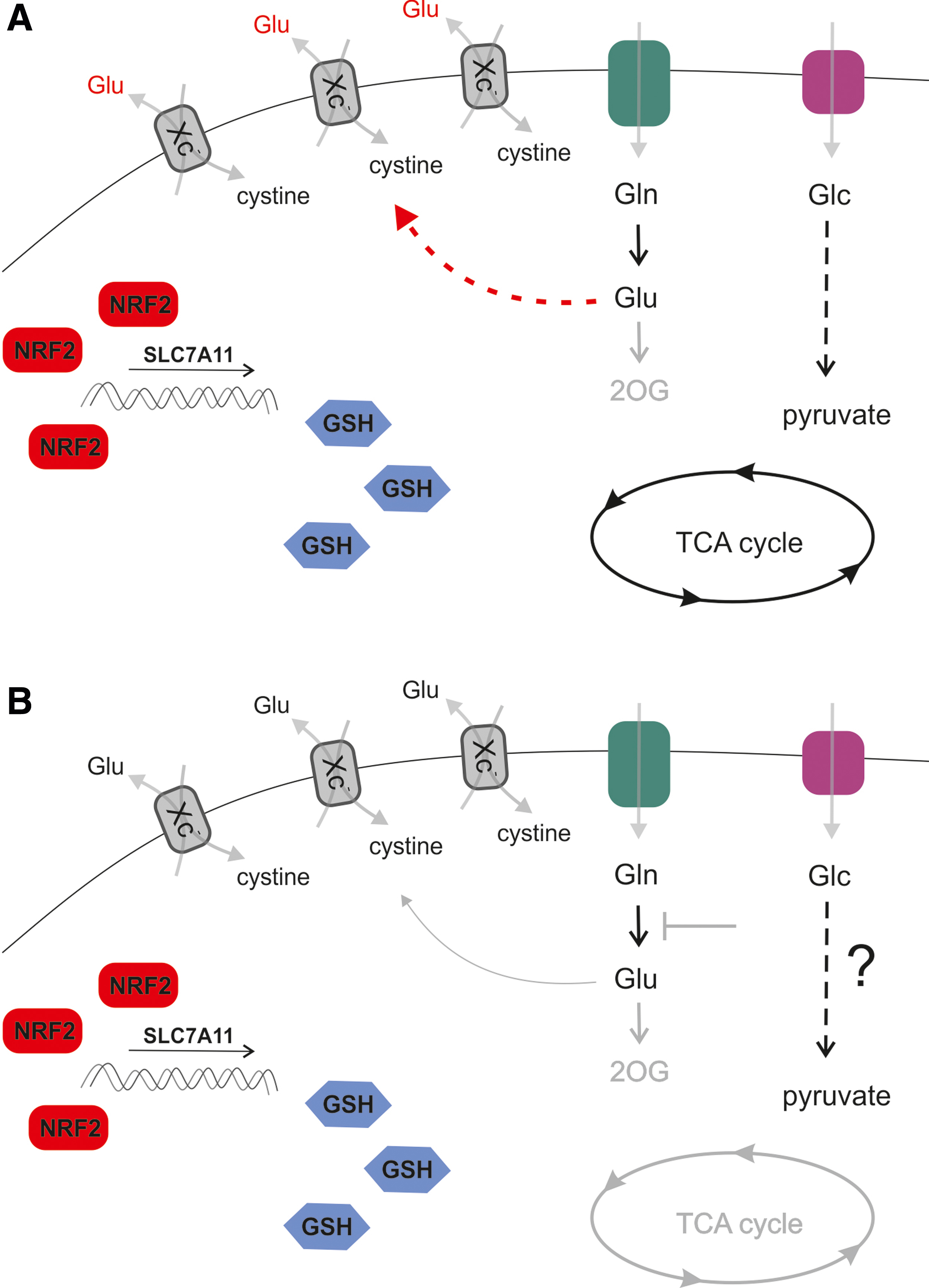
Comparing low- and high-expressing SLC7A11 cell lines revealed that OXPHOS-related gene expression negatively correlates with the expression level of SLC7A11 in breast cancer. In turn, xCT expression improved GSH levels and ROS homeostasis, while diminishing the ability of breast cancer cells to utilize glutamine instead of glucose (262).
A study that grouped breast cancer cell lines based on the ability to proliferate in glutamine-free conditions revealed that cells overexpressing SLC7A11 are sensitive to glutamine withdrawal, as glutamate is excessively exported (284). This offers the possibility to target both Xc− and glutamine import in glutamine auxotrophs (284). Another subset of malignancies is unable to upregulate glycolysis to compensate for loss of glutamine as a carbon source (Fig. 7). In accordance with these data, studies using KRASG12V, TP53−/− cell models of lung adenocarcinoma expressing an LOF form of KEAP1, demonstrate the vulnerability of xCT overexpressing cells to glutamine withdrawal. These cell models also have an increased sensitivity to glutaminase 1 inhibitor CB-839 (247), which can be overcome by pretreatment with erastin or knockdown of SLC7A11. In this case, active export of glutamate limits mitochondrial anaplerosis, which, in turn, sensitizes the cells to CB-839 treatment. Thus, a combination of KI696, a compound inhibiting KEAP1-NRF2 interaction, with CB-839 is another plausible strategy to target NRF2 overexpressing cancers. In support of this therapeutic approach, another line of evidence also documents enhanced glutamine dependence due to KEAP1-NRF2 in the KRAS/LKB1/KEAP1 transformed lung cancer (72). Further, a recent study (157) also demonstrated that the NRF2-mediated induction of the Xc− system limits intracellular glutamate content that, consequently, leads to an external dependency on nonessential amino acids (serine, glycine, and asparagine) in cancer cells, rendering cells metabolically inflexible. The antioxidant capacity of cancer cells, indeed, shows an inverse correlation with nonessential amino acids synthesizing capacity. As cells are reliant on nonessential amino acids, a diet that avoids these amino acids can limit cancer cell proliferation.
A recent publication (126) suggested that the NRF2-mediated accumulation of cysteine in nonsmall cell lung cancer may induce metabolic liability. The mechanism of metabolic liability is that cystein in superphysiological concentrations becomes a substrate for cysteine dioxygenase 1 (CDO1), creating cysteine sulfinic acid that apparently hampers cellular proliferation. From a broader perspective, this metabolic inflexibility can be exploited in combating nonsmall cell lung cancer.
NRF2 interference with hypoxia signaling and neovascularization
Hypoxia, a common feature of the tumor microenvironment, leads to the stabilization of HIF1α, which modulates cellular metabolism as an adaptive response (252). HIF1α expression initiates transcriptional programs that lead to survival and overcome hypoxia by inducing angiogenesis, glycolysis, and a decrease in pyruvate oxidation in mitochondria. NRF2 activation supports glycolysis by promoting the stabilization of transcription factor BTB and CNC homology 1 (Bach1) that supports the activity of hexokinase (168, 309). This effect of NRF2 is mediated by induction of HMOX1, which catabolizes heme, the main inhibitor of Bach1. Both NRF2 and HIF1α are critical factors for sensing O2 and are dependent on each other when responding to oxygen tension changes or ROS production. Of note, HIF1α is probably a target of NRF2; an ARE was identified in the HIF1α promoter (152). Strikingly, NRF2 silencing suppresses HIF1α accumulation in breast cancer cells under hypoxic conditions. Thus, HIF1α-mediated metabolic adaptations, including the activation of glycolysis and PPP, are inhibited. Inhibition of metabolic adaptations eventually impairs the viability of NRF2-silenced cancer cells in a hypoxic environment (159). Further evidence suggests that NRF2 is induced in hypoxia due to elevated ROS (250) and the subsequent induction of GCLC and GSH synthesis. Hence, chemoresistance is promoted (11). A recent in vitro study showed that both NRF2 and HIF1α are upregulated in MCF-7 and MDA-MB-231 breast cancer cells, compared with MCF-10A cells (a benign breast epithelial cell line). These results suggest that NRF2 and HIF1α simultaneously contribute to cell proliferation and tumor formation in breast cancer (333). Further, NRF2 can promote glycolysis in breast cancer cells in cooperation with HIF1α (333).
In addition to the well-known angiogenic role of HIF1α to overcome hypoxia, the NRF2 pathway also contributes to neovascularization in cancer models. The induction of NRF2 in endothelial cells promotes cell motility and angiogenesis (102). In addition, the expression of platelet derived growth factor and VEGF, strategic angiogenesis inductors, are regulated by HIF1α and NRF2, as demonstrated in a prostate cancer model (323). Moreover, loss of NRF2 inhibited HIF1α accumulation and VEGF induction, resulting in reduced angiogenesis in colorectal carcinoma cells (140); this effect was mediated by the induction of HIF1α hydroxylation in hypoxia.
NRF2 in the regulation of PPP flux
PPP is upregulated in many types of tumors, including breast cancer where PPP promotes proliferation, differentiation, and survival. Breast cancer cells either overexpress NRF2 or cells, in which KEAP1 is silenced, or upregulate G6pd (318, 334). This observation appears to be generalizable; the activities of G6pd and transketolase (TKT), key PPP enzymes, are commonly increased in cancer cells (318). Oxidative PPP converts G6P, a glycolytic intermediate, into ribulose-5-phosphate and generates NADPH, which is used for GSH regeneration, detoxification, and biosynthesis of lipids. NRF2 directly activates five genes involved in PPP and NADPH production pathways, including G6pd, PGD, TKT, transaldolase 1, and ME1, through binding to AREs (306). The G6pd target for NRF2 is a critical enzyme in the PPP, catalyzing the rate-limiting step that produces NADPH (24).
A recent report showed that NRF2 contributes to the metastatic ability of basal type breast cancer cells through the G6pd/HIF-1α/Notch1 signaling axis. NRF2-dependent G6pd/HIF-1α activation elicits Notch1 signaling by upregulating Jagged1 and Hes1, and, thereby, promoting epithelial-mesenchymal transition (EMT) in breast cancer cells (334). In addition, a clinical study showed that PPP-related proteins are differentially expressed according to the molecular subtype of breast cancer; in particular, G6pd and 6-phosphogluconolactone are highly expressed in HER2-type breast cancer (35). Thus, understanding the role of this pathway in breast cancers, including the effects of targeting this pathway, is needed to clarify the clinical implications of PPP in breast cancers. Benito et al. (19) demonstrated critical rewiring of glucose flux due to the NRF2-induced G6pd and TKT expression; the changes in glucose flux included increased ribose and NADPH synthesis, which are associated with poor outcomes in breast cancer patients. However, in contradiction to previous studies, the NRF2 level was downregulated in all types of breast cancers in the TCGA dataset, suggesting a possible role of NRF2 as a tumor suppressor in breast cancer (35). It is also possible that the result of the RNA sequencing may not reflect the actual activity of NRF2 in cells due to post-translational regulation by ubiquitination and degradation (35).
Methionine adenosyltransferase 2A
Polyamines, such as putrescine, spermine, and spermidine, have unknown biological function and accumulate in cancer cells, resulting in increased proliferation. Upregulated enzymes of polyamine pathways have been implicated in several cancer types (26). Methionine adenosyltransferase (MAT) is an essential cellular enzyme that is ubiquitously expressed in mammalian cells; MAT catalyzes the formation of S-adenosylmethionine, the principal biological methyl donor and the ultimate source of the propylamine moiety used in polyamine biosynthesis (189). Altered expression of methionine adenosyltransferase 2A (MAT2A), an isoform of MAT, in tamoxifen-resistant (TAMR) breast cancer and the role of MAT2A in tumor growth and chemoresistance have been described in the context of NRF2 regulation. MAT2A expression is upregulated in TAMR-MCF-7 cells compared with control MCF-7 cells. Moreover, MAT2A expression, associated with altered NRF2 expression levels, is more frequent in TAM-resistant human breast cancer tissues than in TAM-responsive cases (216). This genetic correlation between MAT and NRF2 has been partially clarified; the promoter of MAT2A contains several potential binding sites for transcription factors, including c-Myb, NRF2, NF-κB, and AP-1 (322), suggesting that NRF2 could be vital in regulating the expression of MAT2A. MAT2A immunoreactivity is significantly higher in TAMR cases of human breast cancer compared with TAM-sensitive cases, and it is associated with the overexpression of NRF2 (215). However, whether this association is direct or acts via another intermediate pathway that links MAT2A and NRF2 is unclear. NF-κB also regulates MAT2A gene transcription, suggesting an underlying mechanism of enhanced NF-κB activity related to miR-146b downregulation, which is associated with NRF2 expression, in TAMR breast cancer (104). Therefore, decreased miR-146b and MAT2A induction could be a new phenotype contributing to the growth of breast cancer cells and tamoxifen resistance in breast cancer cells.
NRF2 in the regulation of stemness, EMT, and metastasis
EMT is a trans-differentiation process that enables cell migration. EMT constitutes the initial step of metastatic progression. During EMT, typical markers of the mesenchymal phenotype, such as Vimentin, N-cadherin, Snail, Slug, Twist, and ZEB1, are increased, whereas epithelial markers, such as cell adhesion markers (E-cadherin, MUC-1, and laminin) are lost. Mesenchymal cells also merge phenotypically with circulating tumor cells (CTCs), which are considered a negative prognostic marker of cancer (326). Isolated CTCs usually form clusters, which is a feature of the mesenchymal phenotype (327). In addition, CTCs express the stem cell marker, CD133 (9). On the other hand, mesenchymal
Although research in EMT advanced in recent years, there is persistent controversy regarding the existence of EMT/MET phenotype in CSCs, which are also implicated in tumor maintenance and metastasis. EMT mediators can lead to increments of the CSC phenotype (249). CSC are defined by specific expression of cell surface markers (CD24low, CD44high), the expression of aldehyde-dehydrogenase 1 (ALDH1), and low proliferation rate (50). The typical experimental in vitro models, leading to CSCs enrichment, are “spheroids” cultivated from cell cultures. These spheroids are also used for xenograft studies, due to the improved engraftment compared with conventional 2D cell cultures (129). The overlap of CSC issues with the EMT phenomenon is further underlined by the existence of defined epithelial-like and mesenchymal-like CSCs classes (273). Moreover, a hybrid epithelial/mesenchymal (E/M) state of cells has been described recently as the mixed phenotype preceding full mesenchymal state (151). Nevertheless, studying EMT and CSCs in vivo is rather challenging, given that the phenomenon is typically difficult to capture in real-time, due to the rapid exchange of the markers associated with EMT/MET once the transition is completed.
Information regarding the involvement of NRF2 in EMT, MET, and CSC phenotypes is rather ambiguous, due to the variety of experimental models. NRF2 induction most likely promotes spheroid formation and the CSC phenotype. A study by Luo et al. (180) demonstrated that transition from CD24low CD44high CSCs (mesenchymal-like stem cells) into ALDH+ breast cancer CSCs (epithelial-like stem cells) is accompanied by the NRF2-dependent gene expression signature in response to metabolic perturbation such as 2-deoxyglucose treatment (180). Inhibition of NRF2 and the antioxidant system (Txns and GSH) inhibits sphere formation and cancer growth in vivo (180). In breast cancer cells, the expression of CD44 coincides with higher NRF2 expression and markers of CSC-like cancer cells (239, 240). The induction of NRF2 in CD44+ cells is dependent on the noncanonical activation of NRF2 through p62, and NRF2 induction facilitates spheroid formation and induces chemoresistance to 5-fluorouracil and doxorubicin (239, 240).
Formation of spheroids and xenografts is suppressed after NRF2 silencing in CD44high cells in the same studies (239, 240). Moreover, Kipp et al. (143) used a remarkable fluorescence-based in vitro model of monitoring NRF2 expression to demonstrate that NRF2 is clearly induced during spheroid formation. NRF2 induction by the Txnrd1 inhibitor, auranofin, promotes spheroid growth, evidenced by proliferation and differentiation markers, rather than the quiescent (CSC-like) phenotype. In line with this, Kovács et al. (149) showed that the 4-hydroxynonenal content in breast cancer tissue negatively correlated with the proliferation of cancer cells. On the contrary, another point of view was provided in a study by Bocci (21) showing that the knockdown of NRF2 induces mesenchymal markers and the transition from hybrid E/M to the mesenchymal phenotype. NRF2 expression in TCGA datasets also corresponds with the expression of E/M markers (high ZEB1, high miR-200) and elevated NRF2 expression correlates with a worse prognosis of breast cancer (21).
Another aspect of CSCs is cell migration and invasion, which can also be supported by NRF2 signaling. In cell models of breast cancer, NRF2 overexpression or KEAP1 knockdown promotes cell migration (assessed by wound healing assay) and invasion (transwell invasion assay) through G6pd and possibly Notch1 induction (334). Moreover, the CD24−CD44+ mesenchymal subpopulation of several human breast cancer cell lines is characterized by upregulated TrkB-AKT signaling that suppresses KEAP1 expression (138). In agreement with that, NRF2 accumulation in the nucleus can be prevented by the presence of E-cadherin, which is present in epithelial cells, but not in mesenchymal cells (141). The mechanism of this process requires sequestration of the NRF2/KEAP1 complex in the proximity of the plasma membrane and involves the interaction of NRF2 with β-catenin (141).
Of note, NRF2 is involved in the crosstalk between associated cell species and tumor bulk cells in HCC and PDAC (63). Lactate produced and secreted by cancer cells into the environment and elevated ROS levels induce NRF2 in tumor-associated macrophages, converting them to the M2 (CD163+) polarized phenotype. NRF2 in M2 macrophages was essential for the induction of migration and expression of EMT markers in tumor bulk cells via VEGF signaling, which also induces NRF2 levels.
The role of NRF2 in DNA repair
As previously mentioned, the induction of NQO1 by NRF2 prevents DNA damage (267, 270). NRF2 is responsible for DNA protection at multiple levels and for different kinds of DNA damage inducers, such as irradiation or chemical exposure. Reduction of NRF2 levels sensitizes cells to irradiation, a process involving the decreased ability to repair DNA via homologous recombination, by inhibiting RAD51 levels (112). The study by Jayakumar (112) also indicates additional targets containing ARE sequences that are active in DNA repair, including possible targets of NRF2. NRF2 also regulates expression of ATM and ATR after cysplatin exposure in an ovarian cancer model via stress-induced kinases specific to the DNA damage response (267). In addition, synthetic tripertenoids activate NRF2 and protect cells from radiation damage (60, 139). In this case, the role of NRF is controversial (285) in terms of cancer progression, since NRF2 might prevent DNA damage and cancer progression.
The role of NRF2 in aging and lifespan regulation
In Drosophila, NRF2 expression extends the lifespan and induces tolerance to paraquat treatment (285). A straightforward study (164) investigated NRF2 signaling on naked mole rats and stratified NRF2 expression according to the maximum species lifespan potential (MSLP). The study revealed that NRF2 activity and downstream targets exhibit positive correlation with MSLP, due to the diminished activity or expression of canonical and noncanonical negative regulators. In addition, a spectrum of NRF2 target genes and proteins were recently identified in a high-throughput study; these NRF targets were differentially regulated in the long-lived naked mole rat and the short-lived guinea pig, its close relative (96). Nevertheless, there are contradicting reports concerning the role of NRF2 in longevity. For example, in Nqo1-deficient mice, the lifespan is improved under caloric restriction, but not under ad libitum conditions (53). Taken together, the lifespan-prolonging effect of NRF2 has not been conclusively demonstrated.
Future Prospects
NRF2 is a vital component of the cellular machinery that conserves cellular redox balance through the expression of antioxidant proteins. Reactive oxygen and nitrogen species have a dual nature in carcinogenesis. On one hand, enhanced reactive species production increases the risk for neoplastic diseases by increasing the mutation rate and, hence, increasing the chance of transformation (158). On the other hand, in cancer cells, reactive species negatively regulate cancer cell survival (149, 158). It is also of note that although low NRF2 expression is associated with senescence, the induction of NRF2 expression is apparently associated with cancer cell survival (5, 46, 68, 149, 158, 290, 343). This dual nature of free radicals confers an interesting, ambiguous nature to NRF2 in cancer biology. The pro- or antineoplastic effects of NRF2 are context dependent; thus, the effects of NRF2 depend on the nature and molecular characteristics of the cancer in question. Large-scale studies to carefully evaluate cancer subtypes, where NRF2 signaling can be targeted, should be a focus for the future.
NRF2 promotes several enabling cancer hallmarks, such as metabolism, CSC characteristics, tumor aggressiveness, invasion, and metastasis formation. The role of NRF2 in regulating cancer cell stemness and EMT seems to be a central feature in defining the role of NRF2 in regulating metastasis and progression among the neoplastic processes. The level of NRF2 activity is apparently a switch in metabolism and ferroptosis that enables cancer cells to mobilize nutrients to support growth, enable metabolic flexibility, and avoid cell death. The bulk of cancer cells are sensitive to chemotherapy and are rather reliant on Warburg metabolism, whereas stem cells are metabolically flexible and resistant to chemotherapy (66, 214). Therefore, from a translational point of view, fine-tuning NRF2 activity may help in reprogramming CSCs to become more sensitive to chemotherapy (126). Understanding the complex metabolic rearrangements behind NRF2 activation will be crucial in understanding the potential interference of ROS signaling and metabolic rewiring in cancer and other pathologies.
Immune destruction is a major pathway to eliminate cancer cells, involving the production of reactive species. It remains an open question of whether NRF2 is involved in protecting against immune destruction. Logically, the NRF2-driven antioxidant response is probably protective against immune cell-derived free radicals and, therefore, may be pro-neoplastic. Further, NRF2 activity modulates the function of antigen-presenting dendritic cells and T cells (165, 330), implying a modality to improve anticancer immune response.
There are only a few papers on the interactions between the NRF2 pathway and chemotherapy regimens (12) (Table 2); understanding these interactions can help identify the best therapeutic window for NRF2 activators or inhibitors. Most likely, the picture will be very complex, as some chemotherapeutic agents produce superoxide [e.g., doxorubicin (10, 211, 212)], which interferes with the NRF2 system. Further, the scavenging of free radicals produced by chemotherapeutic drugs is controversial (176). The modulation of NRF2 tone will likely impact the sensitivity of tumors to chemotherapy.
Interactions Between the NRF2-KEAP1 System and Cytostatic Drugs
N/A, not applicable; VEGF, vascular endothelial growth factor.
Values for the Survival Curve on Figure 4
HR, hazard ratio.
The exploitation of the NRF2 pathway clearly holds promise in the clinical setting. Nevertheless, substantial studies are needed to sharply pinpoint the diseases, patient subgroups, and therapeutic schemes where NR2-directed therapies will be beneficial.
Footnotes
Acknowledgment
The authors are grateful to Dr. Karen Uray (University of Debrecen, Department of Medical Chemistry) for the critical revision of the text.
Funding Information
This work was supported by the NKFIH (K123975, PD124110, FK128387, GINOP-2.3.2-15-2016-00006) to P.B., the Hungarian Academy of Sciences (NKM-26/2019) to P.B., the Czech Health Research Council project number NV19-01-00101 to K.S., and institutional funding of IPHYS CAS number RVO67985823. E.M. was supported by the Bolyai fellowship from the Hungarian Academy of Sciences. The research was financed by the Higher Education Institutional Excellence Programme (NKFIH-1150-6/2019) of the Ministry of Innovation and Technology in Hungary, within the framework of the Biotechnology thematic programme of the University of Debrecen. E.M. is supported by the ÚNKP-19-4-DE-79 New National Excellence Program of the Ministry of Human Capacities.