
Abstract
Significance:
Cancer cells are stabilized in an undifferentiated state similar to stem cells. This leads to profound modifications of their metabolism, which further modifies their genetics and epigenetics as malignancy progresses. Specific metabolites and enzymes may serve as clinical markers of cancer progression.
Recent Advances:
Both 2-hydroxyglutarate (2HG) enantiomers are associated with reprogrammed metabolism, in grade III/IV glioma, glioblastoma, and acute myeloid leukemia cells, and numerous other cancer types, while acting also in the cross talk of tumors with immune cells. 2HG contributes to specific alternations in cancer metabolism and developed oxidative stress, while also inducing decisions on the differentiation of naive T lymphocytes, and serves as a signal messenger in immune cells. Moreover, 2HG inhibits chromatin-modifying enzymes, namely 2-oxoglutarate-dependent dioxygenases, and interferes with hypoxia-inducible factor (HIF) transcriptome reprogramming and mammalian target of rapamycin (mTOR) pathway, thus dysregulating gene expression and further promoting cancerogenesis.
Critical Issues:
Typically, heterozygous mutations within the active sites of isocitrate dehydrogenase isoform 1 (IDH1)R132H and mitochondrial isocitrate dehydrogenase isoform 2 (IDH2)R140Q provide cells with millimolar
Future Directions:
Uncovering further molecular metabolism details specific for given cancer cell types and sequence-specific epigenetic alternations will lead to the design of diagnostic approaches, not only for predicting patients' prognosis or uncovering metastases and tumor remissions but also for early diagnostics.
Preface
Specific cancer cells, notably grade II/III glioma (35, 36), secondary glioblastoma (127), and acute myeloid leukemia (AML) (16, 60, 103, 144) cells, exhibit heterozygous point mutations in the active sites of cytosolic isocitrate dehydrogenase isoform 1 (IDH1) (8, 11) or isoform 2 (IDH2), localized in the mitochondrial matrix (65, 189). The resulting heterodimeric enzymes play a neomorphic role since they form the oncometabolite
Links have been established between the patient's IDH1 or IDH2 mutation pattern, molecular mechanisms of the alternated epigenetic niche, and reprogrammed metabolism for predicting prognoses for various cancers. The neomorphic activity of mutated IDH1 or IDH2 enzymes causes a dramatic elevation of 2HG levels, which themselves are sufficient to promote gliomagenesis (35) or leukemogenesis in hematopoietic cells through the maintenance of dedifferentiation and increased proliferation (99). Also, a key component of the hypoxia-inducible factor (HIF) pathway, the enzyme prolyl hydroxylase domain-2 (PHD2/EglN1), has been found to be activated by
In contrast, at low concentrations, both 2HG enantiomers participate in not yet fully elucidated metabolic pathways, which may be associated with the regulation of cell proliferation and other functions. A big question is whether “intermediate levels” of 2HG also provide neomorphic effects, such as those naturally produced by nonmutated IDH1 and IDH2; and
Surprisingly, 2HG can be found in a very wide concentration range. The concentration of 2HG can reach between 1 and ∼30 mM in grade II/III gliomas (30, 36, 60), whereas “intermediate levels” of both 2HG enantiomers would be in the 10–100 μM range and their effects in these levels should be further studied. There is no doubt that an imbalance in 2HG formation/degradation very frequently accompanies a specific cancer metabolism. In turn, the reprogrammed metabolism may lead to the further accumulation of 2HG, reaching higher concentrations than in physiological states.
In this review, I briefly discuss the known metabolic pathways involving 2HG, the concomitantly reprogrammed metabolism, oxidative shifts in redox homeostasis, and the effects resulting from 2HG accumulation promoting cancerogenesis, as well as the role of 2HG in interactions of tumors with the immune system.
The main metabolic changes connected to increased 2HG enantiomer levels induce alternations in redox homeostasis, such as decreasing NADPH/NADP+ and NADH/NAD+ ratios, increased reactive oxygen species (ROS) formation, or decreased antioxidant defense. Possible roles of these states are discussed. The states are not only passive reflections of the altered metabolism but also represent factors that can further accelerate metabolic and other functional or pathological changes. Last but not least, I will attempt to speculate on a possible utilization of 2HG as a prognostic/diagnostic marker, discussing a specific example of breast cancer.
Metabolism of 2HG
Isocitrate dehydrogenases IDH1 and IDH2 as sources of r -2HG
Canonical reactions of wild-type IDH1 and IDH2
Cytosolic and peroxisomal IDH1 (94 kDa, EC 1.1.1.41) (111, 190) as well as the mitochondrial isoform IDH2 (94 kDa, EC 1.1.1.42) (22, 89) are homodimeric enzymes, which reversibly catalyze the oxidative decarboxylation of isocitrate (IC) into 2OG and CO2, using the cofactors NADP+ and Mg2+ (133). In contrast, the reverse reaction of reductive carboxylation then consumes NADPH and with CO2 can transform 2OG to IC. This complete reaction of IDH2 is followed by citrate efflux from mitochondria, typically upon reductive carboxylation glutaminolysis (72, 73, 115, 116, 154, 195) (Fig. 1). However, an incomplete reaction, not requiring CO2, leads to a simple NADPH-driven reduction of 2OG to 2HG.
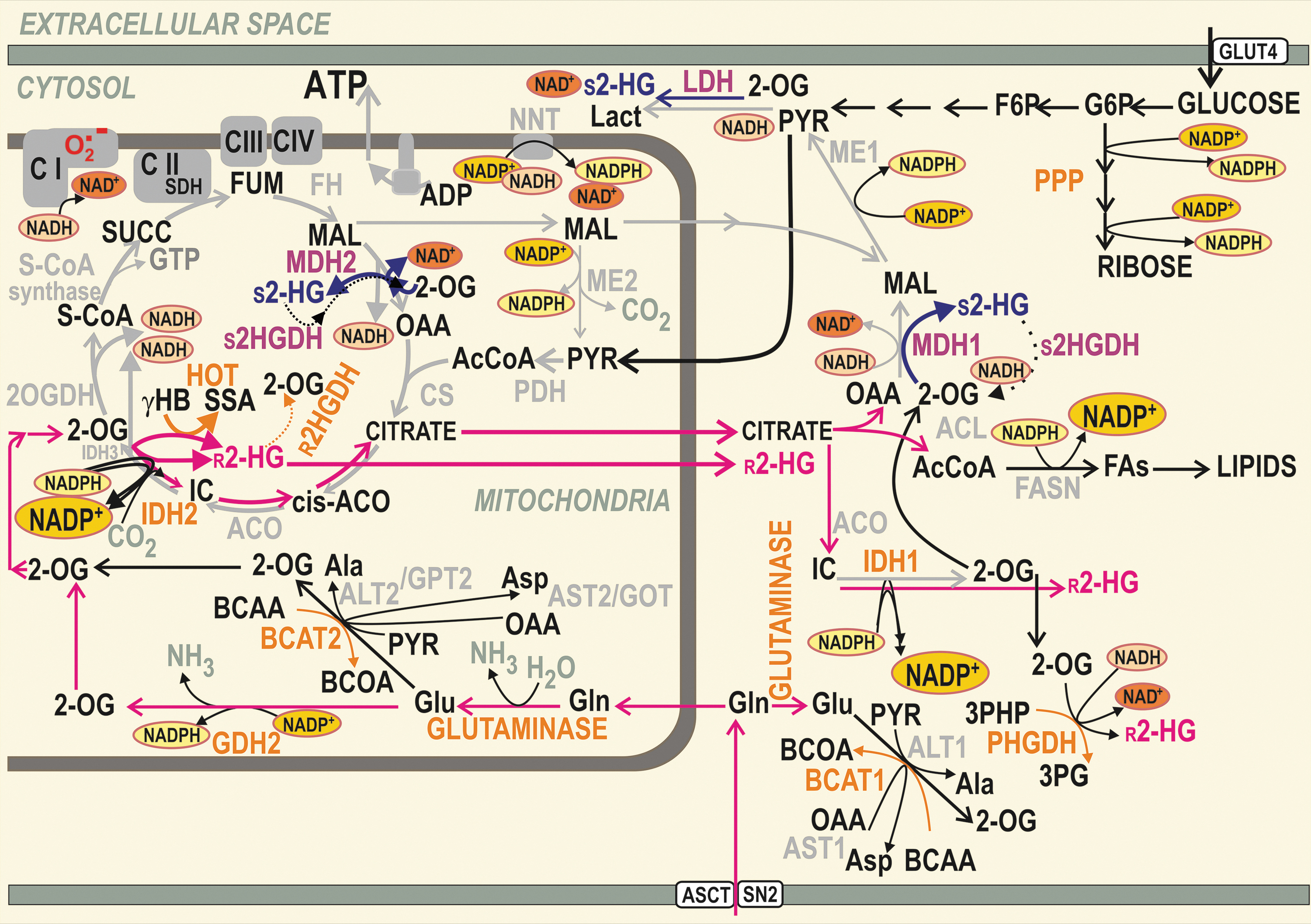
r -2HG formation by wild-type IDH1 and IDH2
There is no more controversy over whether the wild-type (wt) IDH1/2 enzyme is capable of such a reaction. We were among the first in demonstration that wt IDH2 produces 2HG (155). Also, the transfection of cells with wt IDH1 or wt IDH2 selectively increased
Glioblastoma SF188 cells also produce 2HG at hypoxia, again despite lacking the IDH1/2 mutations (183). Also, estrogen receptor-negative (ER−) breast carcinoma HTB-126/Hs 578T cells, and epithelial adenocarcinoma MDA-MB-231 cells, contain 2HG in the absence of IDH2 mutations and its formation substantially dropped upon IDH2 silencing (155). In hypoxia,
It should be investigated whether each wt IDH1/2 molecule forms
Mutant IDH1 and IDH2 as sources of r -2HG
In human grade II/III gliomas (35, 36, 74, 76, 180), secondary glioblastomas (127), AML (16, 60, 103, 144), cholangiocarcinoma, chondrosarcoma (2), and in other cases of different tumor types (20, 54, 135, 192), heterozygous somatic missense mutations were found in IDH1 arginines of the catalytic site, such as (bold for most abundant)
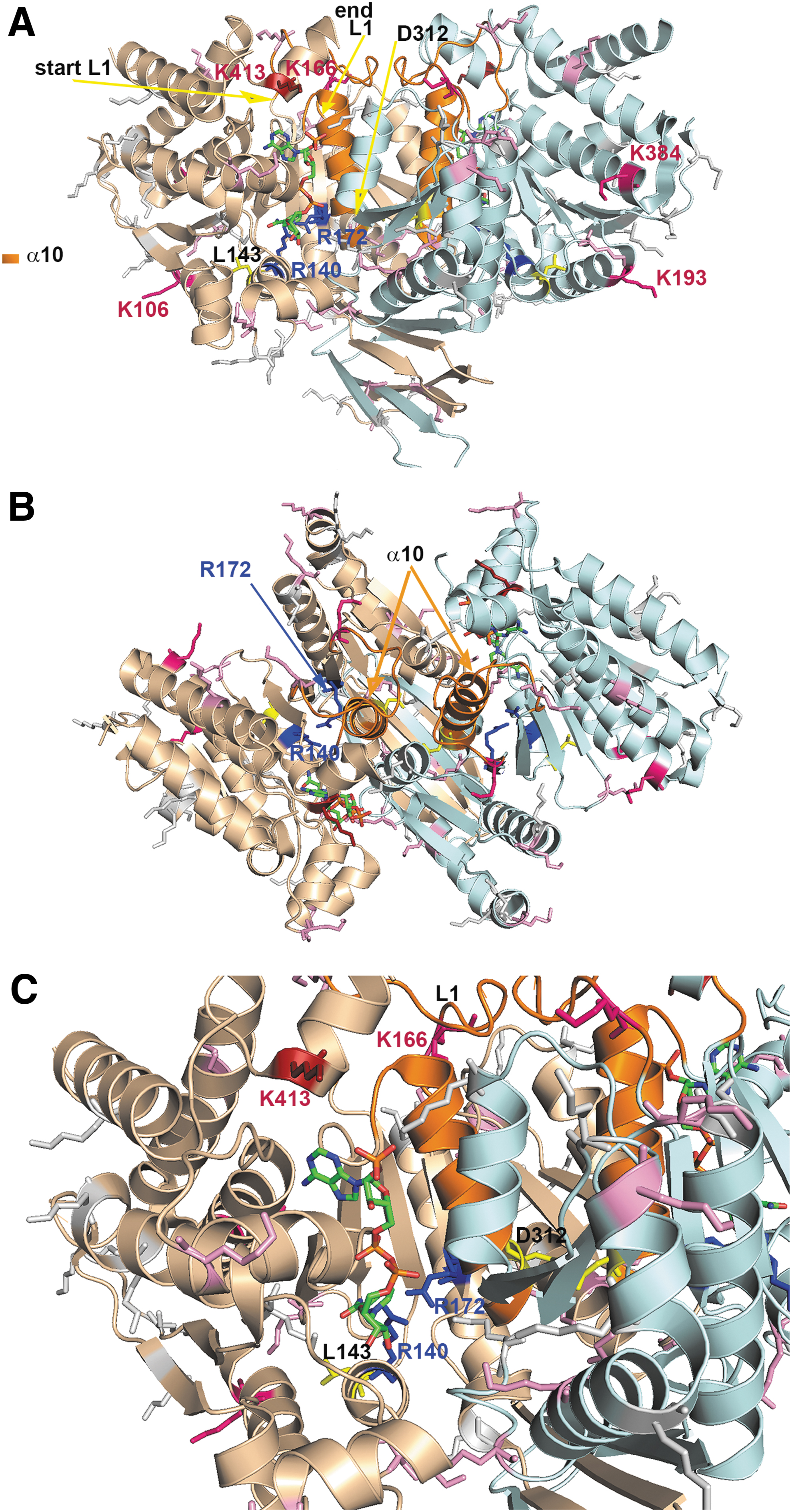
In wt enzymes, arginines form hydrogen bonds with both the α and β carboxyl of IC and thus ensure IC binding (158, 187). Substitutions of arginines decrease affinity for IC binding but increase it for NADPH (35, 36). With IDH1R132H, the resulting mutation prevents conformational changes between the initial IC binding and a pre-transition state (190). The IDH1R132H enzyme is thus set to the so-called closed/active conformation (36), where H132 cannot interact with N271 of a “regulatory segment” (“segment α10”), that is, α-helix 271–286 (187), causing a 300-fold decrease in the catalytic efficiency relative to the wt IDH1 enzyme and a 1000-fold loss of affinity (38) for Mg2+.
Since
Three phenotypes were characterized: The first phenotype involved depleted 2OG but moderate
Specific inhibitors of mutant IDH1/2
The resulting changes induced by 2HG in chromatin and the cell differentiation state are mostly reversible (54, 99). Hence, in principle, they could be reverted by specific inhibitors for mutant IDH1/2. Indeed, specific inhibitors have been developed for mutant IDH1 (15, 38, 92, 122, 130, 131, 173, 184) or mutant IDH2 enzymes (179). Usually, they do not bind the mutated arginines except to an allosteric pocket of each monomer, which is not accessible in wt enzymes (35). Since in mutant IDH1 enzymes a regulatory segment α10 (187) is destabilized, and hence only partially ordered (184), there is an open accessible pocket space for the inhibitor. Moreover, bound Mg2+ protects the inhibitor binding to the wt enzyme. These properties determine which inhibitors are specific for the mutant IDH1 enzyme.
In contrast, IDH2 mutants are targeted by different drugs. This is because the IDH2 mutant is set to the closed/inactive conformation with stabilized α10. A specific IDH2 inhibitor was developed to bind to the IDH2 dimer interface (179). Surprisingly, tumors targeted by the specific IDH1 inhibitors have the ability to switch their mutagenesis toward unmutated IDH2, which is not affected, and vice versa (63, 69).
Nevertheless, AG-881 from Agios Pharmaceuticals was claimed to inhibit both mutant IDH1/2 in a common allosteric pocket (102). The inhibitor of mutant IDH1 ivosidenib (40, 130) and mutant IDH2 enasidenib (159, 160) exhibited positive responses in patients with relapsed or refractory gliomas, intrahepatic cholangiocarcinomas, and chondrosarcomas (48, 130) in phase I/II clinical trials. Ivosidenib was also tested in AML patients, but acquired resistance for these mutants was frequently developed (118). Enasidenib also induced remissions of AML (3, 159).
It seems that specific inhibitors should be designed for each mutation. For example, the mutant IDH1R132Q was 105-less sensitive to mutant inhibitors than IDH1R132H (108). This was explained by the conformation of α-helices more closely resembling the wt enzyme.
Other enzymes producing r -2HG
Hydroxyacid-oxoacid transhydrogenase/alcohol dehydrogenase iron-dependent isoform 1
Physiological mitochondrial metabolism involves both 2HG enantiomers (87). In mammalian mitochondria, hydroxyacid-oxoacid transhydrogenase (HOT), also known as alcohol dehydrogenase iron-dependent isoform 1 (ADHFE1; EC 1.1.99.24), forms
Glutathione-dependent glyoxylases
In mammalian cells,
Phosphoglycerate dehydrogenase
Also, human phosphoglycerate dehydrogenase (PHGDH; EC 1.1.1.95) has been reported to form
Enzymes producing s -2HG
Lactate dehydrogenase
A noncanonical or side function of several enzymes also leads to the formation of
At acidic pH, 2OG binds more stably to LDHA, with a concomitantly enhanced
Malate dehydrogenase
Degradation of 2HG and other reactions
Degradation of 2HG
The catabolism of 2HG diminishes its levels (45). The specific degradation of
The recombinant
Other reactions
Also, human glutamine synthetase ensures the amidation of
Regulations and Signaling by 2HG
2HG and redox homeostasis in carcinogenesis
Redox homeostasis related to canonical IDH1 and IDH2 reactions
IDH1 supplies 2OG for cytoplasmic and nuclear dioxygenases that require 2OG as a co-substrate (66) and regenerates extramitochondrial NADPH, which is required for lipid biosynthesis and antioxidant protection. IDH1 also supplies NADPH for the constitutively expressed NADPH oxidase isoform 4 (NOX4), producing hydrogen peroxide (H2O2). Together with malic enzyme (ME) 1 and two enzymes of the pentose phosphate pathway (PPP; glucose-6-phosphate dehydrogenase [G6PDH] and 6-phosphogluconate dehydrogenase), IDH1 contributes to the cytosolic NADPH pool, being the main NADPH source in the brain and several other tissues (10).
Also, the typical IDH2 mode is oxidative, that is, NADP+-dependent oxidative decarboxylation converting IC to NADPH and 2OG. This “forward Krebs cycle” direction is the typical reaction in nonmalignant cells. However, both enzymes are reversible, changing directions depending on the IC to 2OG and NADP+ to NADPH ratios, and presence of Mg2+ and CO2. This reversibility links these enzymes to redox homeostasis (Fig. 3). Thus, IDH2 may act in a “reverse” Krebs cycle mode in the reductive carboxylation reaction (72, 73, 115, 116, 154, 195) (Fig. 1). This reductive reaction of IDH2 as well as IDH1, including a side
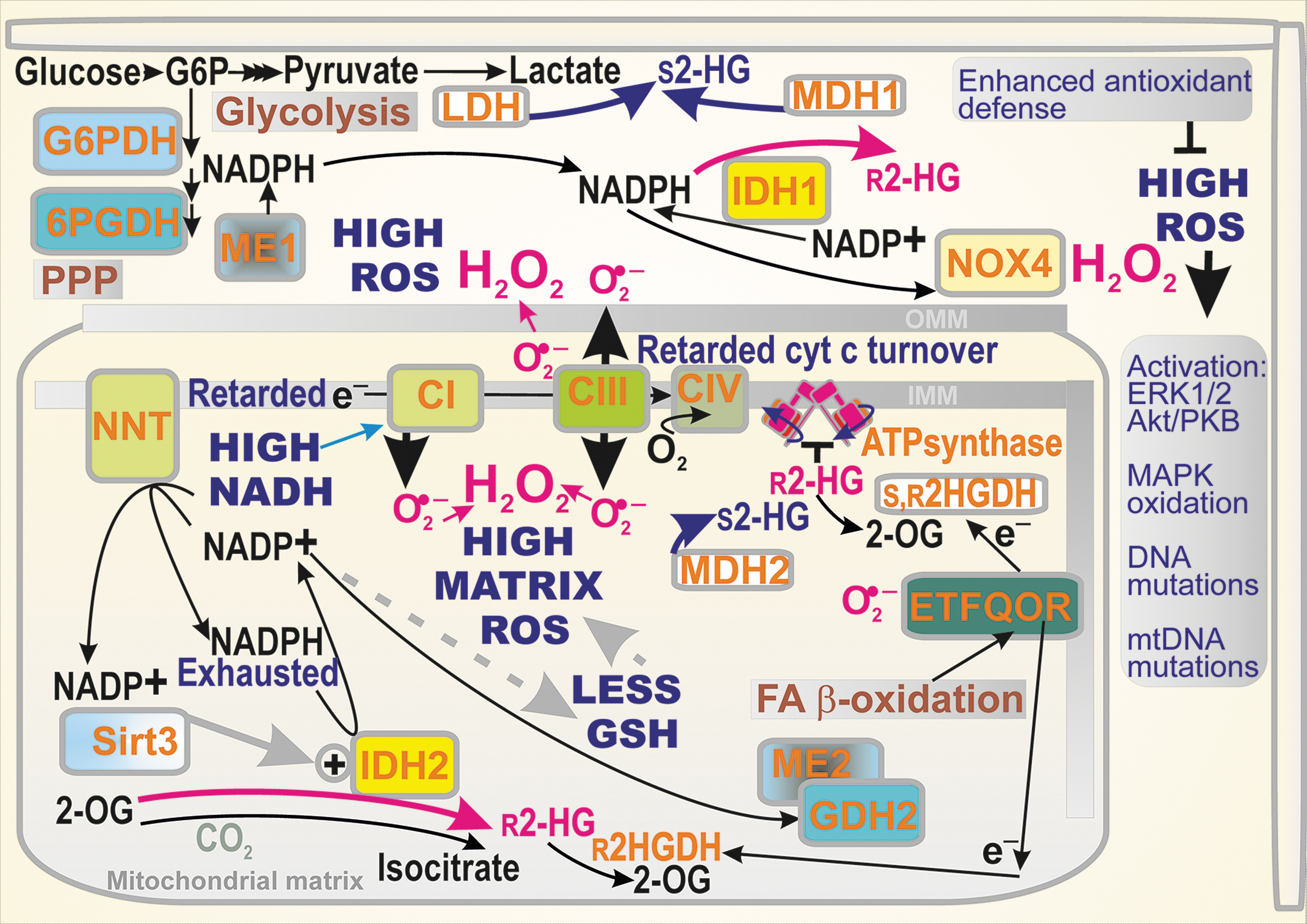
In contrast, since NADPH is produced in the oxidative mode, such a “normal” IDH2 reaction thus substantially contributes to keeping the mitochondrial matrix in a reduced redox state and consequently prevents oxidative damage (75, 81, 82). IDH2 supplies the mitochondrial NADPH pool together with nicotine nucleotide translocase (NNT), ME2 (mitochondrial), and glutamate dehydrogenase (GDH) (Figs. 1 and 3). This pool serves for the regeneration of mitochondrial antioxidant systems, reduced glutathione (GSH) and reduced thioredoxin by glutathione reductase and thioredoxin reductase, respectively (72).
Consequently, IDH2 plays an important role in the ROS homeostasis (72) and in the prevention of apoptosis (61), such as that induced by heat shock (150) or in neuroprotection (87). Interestingly, a self-perpetuating antioxidant effect of IDH2 stems from the fact that deglutathinylation activates IDH2 at the prevailing reduced matrix glutathione level (81). In turn, the glutathionylation of IDH2 inhibits its activity when there is a substantial amount of oxidated glutathione in the mitochondrial matrix (81).
The regular Krebs cycle enzyme IDH3, structurally distinct from IDH2, then converts NAD+ irreversibly to NADH. The IDH3 reaction is allosterically positively regulated by Ca2+, ADP, and citrate, and negatively regulated by ATP, NADH, and NADPH (149). When the OXPHOS glutaminolysis takes place in cancer cells, the aconitase-IDH3 segment is frequently inactive (72, 195). This results in a decrease in the substrate pressure (NADH/NAD+) and mitochondrial superoxide formation.
2HG affecting redox homeostasis
As discussed above, the predicted general effect of modes of
Also, when aerobic glycolysis predominates and LDH or MDH1, 2 are allowed to provide a parasitic formation of
Only a disbalance leads to the so-called oxidative stress when ROS production significantly and permanently exceeds the antioxidant mechanisms (125). A general oxidative stress in a cell arises when the function of redox buffers and antioxidant enzymes is diminished, so that they no longer possess the ability to detoxify the produced ROS. A permanent character distinguishes this stress from repeatable redox signals. The direct pathological consequences are due to the oxidative stress, which reaches a certain threshold when there is an accumulation of oxidative products of biological constituents (oxidative modification of lipids by nonenzymatic lipid peroxidation or oxidative modification of proteins, such as carbonylation). This may initiate programmed cell death, such as apoptosis. Of course within a tumor, apoptosis would retard its growth. The tumor cells prevent this regress by overexpressing antioxidant systems.
A specific line of effects of oxidative stress is concerned with oxidative modifications of DNA and of more vulnerable mitochondrial DNA (mtDNA). Physiological mechanisms exist for DNA repair. However, in cancer (stem) cells, excessive DNA oxidation in synergy with insufficient DNA repair leads to the occurrence of somatic mutations, which are prerequisites for the origin of the primordial cancer cells. When the impairment of normal autophagy and notably autophagic mechanisms dealing with mitochondria also lead to the accumulation of products that were supposed to be cleared, this must have serious consequences for the cell. Again, this acts against carcinogenesis.
2HG affecting redox signaling
ROS manifest dual functions as cancer promoters and cancer suppressors (124). The regulation of redox reactions impacts RAS-RAF-MEK1/2-ERK1/2 signaling related to carcinogenesis (154). Also, NADPH oxidases are ROS sources that promote or modulate this pathway. In contrast, redox signaling is involved in the p38 mitogen-activated protein kinase (MAPK) pathway that suppresses cancer by oncogene-induced senescence, inflammation-induced senescence, replicative senescence, contact inhibition, and DNA-damage responses (154). Nevertheless, MAPK also plays a procarcinogenic role (55). Another branch of redox signaling initiated with electrophiles is provided by the KEAP1-nuclear factor erythroid 2-related factor (NRF2) transcriptome upregulation of antioxidant and other genes (138).
Elevated ROS are able to control the transition from proliferating to quiescent phenotypes and to signal the end of proliferation. Suppression of these higher ROS levels in tumor cells should allow sustained proliferation. The upstream elements responsible for H2O2-induced extracellular-related kinase (ERK) 1/2 and protein kinase B (Akt) activation remain poorly characterized, but a potential role has been postulated for receptor and nonreceptor protein tyrosine kinases as triggers that initiate such events (124). The pathway involving PI3K and Akt is also redox-regulated through the oxidation of cysteine residues in phosphatases (e.g., phosphatase and tensin homolog [PTEN] and protein phosphatases 1 and 2). Akt then regulates an array of downstream targets including pro- and antiapoptotic members of the BCL2 family, caspase-9, forkhead box protein O (FOXO) family members, GSK-3β, and mammalian target of rapamycin (mTOR) (32).
It was also suggested that the cancer cell phenotype persists because of selective MAPK oxidation in mitochondria (55). Thus, H2O2 reportedly induces MAPK transfer to mitochondria, where it co-localizes with upstream kinases (MAPKKs). Subsequent oxidation of conserved cysteines in MAPK results in MAPK-MAPKK translocation to nuclei with consequent ERK1/2 and p38-JNK1/2 activation and a concomitant increase in ERK1/2-mediated cell proliferation and p38-JNK1/2-mediated cell cycle arrest (55). It has been hypothesized that because “dysfunctional” mitochondria in cancer cells may not generate excess ROS, the above-mentioned MAPK oxidation is disrupted and cells remain in proliferation mode.
Lower mitochondrial respiration is triggered by metabolic constraints and, along with the accumulation of mutations in mtDNA in some tumors, is associated with high-level ROS generation in mitochondria (71). This promotes genetic instability in tumors and favors growth, chemotherapeutic escape, and tumor stage progression.
As mentioned above, a larger extent of NADPH depletion results in a disbalance of redox equilibria toward oxidative stress. Of course, its actual occurrence depends on simultaneous changes in antioxidant defense, which can even be improved, for example, by the activation of NRF2-mediated expression of the antioxidant proteins. An increased flux via PPP was also found to support
Specific redox homeostasis in hypoxia
In hypoxic cells, the ratio of NADH/NAD+ (substrate pressure) increases (57). This leads to enhanced superoxide formation at the flavin IF site of Complex I and perhaps also by mitochondrial dehydrogenases (14). Moreover, as a direct consequence of HIF transcriptome reprogramming promoting aerobic glycolysis (the Warburg phenotype), the suppressed OXPHOS is usually linked to a slow Krebs cycle turnover, but an increased accumulation of 2OG (183). Since the acidification also increases due to the enhanced lactate formation and carbonic anhydrase reaction in hypoxia, conditions are set for the described parasitic reactions of LDH and MDH, forming
The role of mitochondrial sirtuins in regulation of IDH2
Lysine acylation is a common reversible post-translational modification associated with regulatory mechanisms of enzymes and proteins in general. Most frequently, acetylation, malonylation, succinylation, glutarylation, and so on, leads to the inhibition of protein function since they eliminate the positive charge of lysine (152). Mitochondrial lysine deacetylation is controlled by the NAD+-dependent deacetylase sirtuin 3 (SIRT3) (39, 142, 174). SIRT3 activity promotes OXPHOS and catabolic metabolic pathways and, due to its NAD+ dependence, is controlled by the redox state.
A higher substrate pressure deactivates SIRT3, whereas SIRT3 should be activated during the operation of redox shuttles and/or OXPHOS glutaminolysis, when the substrate pressure is lower (NAD+ higher). The ablation of SIRT3 causes pleiotropic effects in cancer but typically SIRT3 acts as a tumor suppressor protein (82, 193). Loss of SIRT3 leads to increase in proliferation and tumor growth, resulting from the concomitantly increased mitochondrial superoxide formation (164).
Acetylated IDH2 exhibits a reduced activity and, in turn, SIRT3-mediated deacetylation elevates the forward NADP+-dependent IDH2 reaction (194). The deacetylation of IDH2 prevents the oxidated state of the mitochondrial matrix milieu and helps to maintain the mitochondrial glutathione levels. Moreover, IDH2 acetylation was associated with a disturbance of the homodimeric IDH2 structure (200). Thus, the IDH2 K413Q mutant, simulating acetylation in the sense of the positive charge vanishing, also exhibited a reduced dimerization (200). There is also disagreement over the acetylation itself (156). Suggestions include the acetylation resulting from a nonenzymatic (uncatalyzed) reaction of acetyl-CoA at alkaline pH, which typically occurs in the matrix of respiring mitochondria (177).
SIRT3 was suggested as a target in breast cancer since higher SIRT3 expression was correlated with a poorer prognosis for patients with grade III breast carcinoma (171). Besides the reported interference with redox homeostasis (169) and mitochondrial biogenesis (170), this phenomenon may also stem from SIRT3-mediated activation of 2HG production by IDH2 (156). Also, another mitochondrial sirtuin, SIRT5, was found to ensure the desuccinylation of IDH2 to activate the enzyme (199). Consistent with the antioxidant role of IDH2, the ablation of SIRT5 led to increasing cellular ROS.
The role of sirtuin 1 in regulation related to 2HG
Among all sirtuins 1–7 (isoforms 3,4,5 being mitochondrial), the NAD+-dependent deacetylase sirtuin 1 (SIRT1) deacetylates numerous proteins mainly in the nucleus and also in the cytosol. Consequently, SIRT1 is involved in numerous cellular regulations (e.g., transcription factors, p53, FoxO proteins, PPARγ, PGC1α, and nuclear factor kappa-light-chain-enhancer of activated B cells [NF-κB]), including histones. SIRT1 generates nicotinamide, while the acetyl group of the protein substrate is transferred to cleaved NAD, generating O-acetyl-ADP ribose (137). In several types of cancer, SIRT1 is elevated and may serve as a tumor promoter. In contrast, in certain situations, SIRT1 may act as tumor suppressor (137).
Possible signaling mediated by r -2HG
The development of malignancy is inevitably related to metabolic reprogramming. Studies of cancer-specific metabolism have demonstrated that besides the shifts in metabolic pathways, certain metabolites play an information signaling role. Notably, Krebs cycle substrates and derived metabolites such as succinate, fumarate, itaconate, acetyl-CoA, and both enantiomers of 2HG exhibit such a nonmetabolic signaling function (138). The latter refers to the inducer-mediated event resulting in an altered expression of specific sets of genes or changes in the epigenome. This aspect will be described in the next sections for 2HG. The finding (21) that 2HG activated the mTOR pathway is one such signaling role. However, there are sure to be other information signaling pathways affected by 2HG enantiomers.
Metabolic regulations
The most important effect of 2HG lies in the ability to inhibit the chromatin-modifying enzymes (see the Antagonism of 2HG in epigenetic changes section). This effect dysregulates the cell's gene expression, which otherwise supports differentiation in normal nonmalignant cells. As a result, 2HG promotes carcinogenesis by stabilizing malignant cells in an undifferentiated state similar to stem cells (51, 100, 144, 173, 186). Numerous other effects of 2HG stem from the dysregulated metabolism, which we will discuss first.
General effects of 2HG
There is a very wide range of cancer cells. One can expect (140) that 2HG might also activate the NRF2, downregulate p53 (73), inactivate pyruvate dehydrogenase (PDH) enzymes (143), and decrease the demethylation of DNA and histones, causing the so-called hypermethylation (20, 29, 34, 35, 37, 51, 54, 88, 98, 99, 100, 135, 144, 164, 173, 186). The resulting metabolic reshuffling typically involves increased glutaminolysis (46) and may also produce an increase in glycolysis (HIF activation even under aerobic conditions) and an increase in fatty acid β-oxidation (140). As a rule, this is accompanied by a typically increased ROS production and dysregulation of redox homeostases and redox signaling. Moreover, in Caenorhabditis elegans, 2HG was found to inhibit ATP-synthase, a phenomenon that can also occur in glioma cells with mutant IDH1/2 (52).
Synergy of 2HG-producing enzymes with other enzymes
A synergy exists for other enzymes with processes producing 2HG. A typical example is mitochondrial glutaminase, a key enzyme of glutaminolysis. For example, AML cells are dependent on glutamine and exhibit an increased expression of glutaminase, specifically its isoform GLS1 (glutaminase 1) (106). When glutaminase was inhibited, glutamate levels decreased and the growth of AML cells was inhibited. Also, 2HG concentrations declined specifically in AML cells with IDH1/2 mutants (106). Importantly, the inhibition of glutaminase mostly suppresses tumorigenesis, at least partially.
Branched-chain amino acids (BCAA) are metabolized by BCAA aminotransferases (cytosolic BCAT1 and mitochondrial BCAT2), transferring their α-amino group to 2OG. A key role of BCAT1 in AML was determined by experiments with BCAT1 ablation, which increased 2OG and this in turn increased PHD2-mediated HIF-1α degradation (134). As a result, leukemic cells lost the ability to survive and stopped their growth. In contrast, BCAT1 overexpression caused DNA hypermethylation via ten-eleven translocation methylcytosine dioxygenase (TET) due to the decreased 2OG (see the Antagonism of 2HG in epigenetic changes section).
Lipoyl transferase 1 (LIPT1) is another enzyme whose deficiency elevates 2HG levels. LIPT1 is essential for the lipoylation of PDH subunit E1, which forms acetyl CoA from pyruvate and thiaminepyrophosphate. Therefore, LIPT1 tunes the balance between the oxidative and reductive glutaminolysis (115, 116), promoting the oxidative mode at a higher activity of LIPT1 (119). The ablation of LIPT1, such as in patients with lactic acidosis, causes a blockage of pyruvate oxidation by PDH, consequently increasing the pyruvate conversion to lactate and transamination of alanine with pyruvate by the aminotransferase reaction. Since 2OG dehydrogenases should also be lipoylated for a proper function, 2OG-dehydrogenase (2OGDH) is also blocked. This results in increases in glutamate and proline.
Altogether, the ablation of LIPT1 leads to the elevation of both 2HG enantiomers, due to the ongoing shift toward reductive carboxylation glutaminolysis. In this reaction, its first step can synthesize
When 2OGDH and/or lipoic acid synthase (LIAS) were ablated, both
Antagonism of 2HG in epigenetic changes
Inhibition of 2OG-dependent dioxygenases
A small molecule, such as 2HG, influences enzymes requiring 2OG as a substrate or co-factor. Since a class of such enzymes regulates epigenetics, the 2HG dysregulation of epigenetics is the ultimate mechanism of accelerated cancerogenesis (20, 29, 34, 35, 37, 54, 98, 164), besides activation of mTOR pathway (Figs. 4 and 5). There are >70 2OG-dependent dioxygenases that promote histone demethylation when functional. Theoretically, all of them may be targeted by both inhibitory 2HG enantiomers, hence 2HG should increase histone methylation up to a hypermethylated state (88, 99, 186).
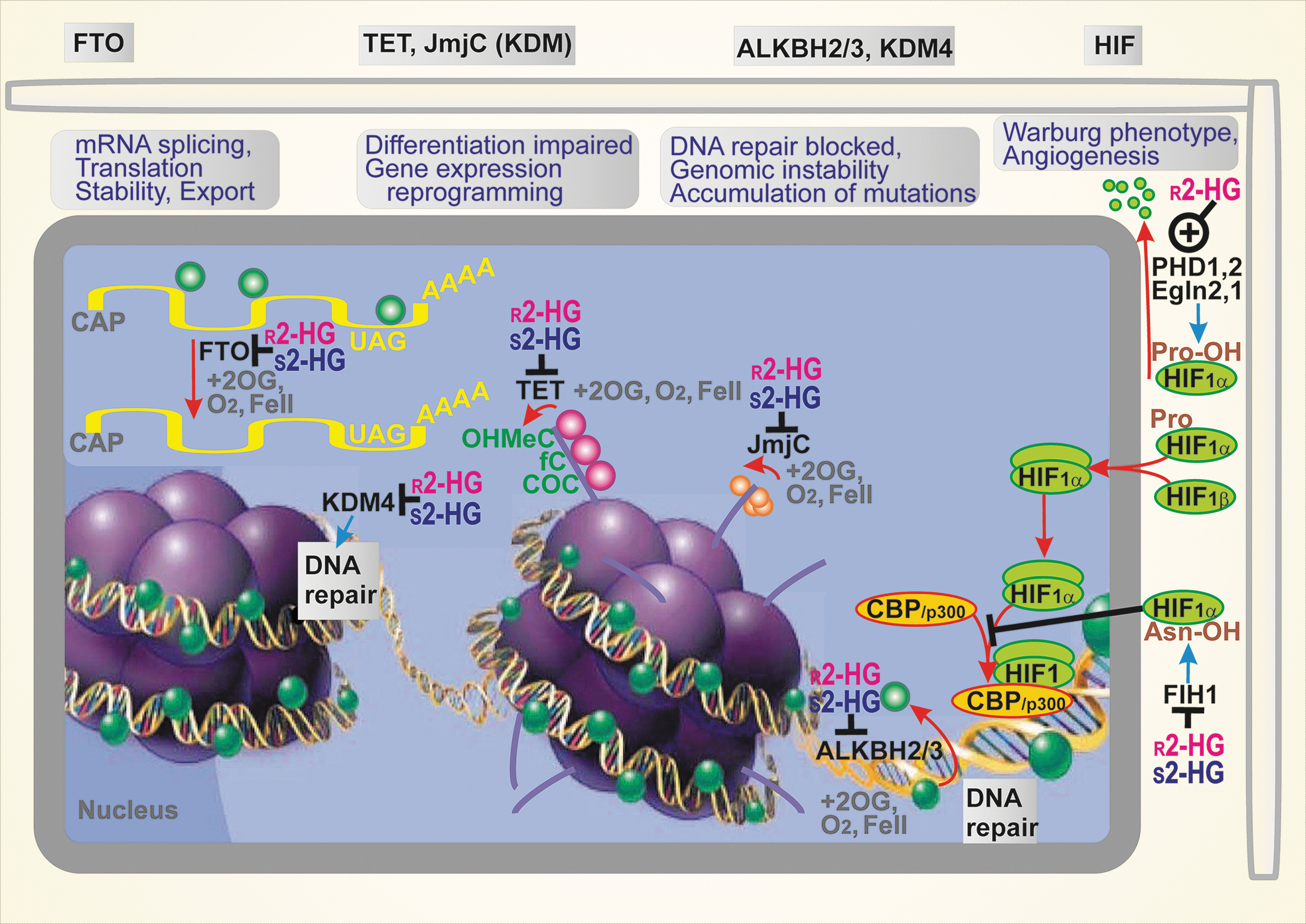
In particular, the myeloid tumor suppressor TET DNA demethylases, such as TET1, TET2, and TET3, provide 5-methyl-cytosine hydroxylation followed by the formation of 5-formyl-cytosine and 5-carbonylcytosine (186). The resulting base pair mismatches with guanine are subjected to base excision repair, leading to the demethylation of DNA. For example, TET2 is potently inhibited by
The other enzyme family targeted by
Elevations in DNA and histone methylation induced by 2HG have other important consequences, such as defects in DNA repair by homologous recombination (162). Both 2HG enantiomers inhibit DNA repair enzymes of the AlkB family, ALKBH2 and ALKBH3 (25). These defects sensitize the inhibition of poly(ADP-ribose) polymerase (PARP). Hence, the use of PARP inhibitors was suggested as another therapeutic strategy for cancers with mutant IDH1/2 (114, 162).
Interestingly, the effects of
Also, hematopoietic stem cells maintain their stem cell character by maintaining a glycolytic (Warburg) phenotype, but their differentiation requires OXPHOS, as proven by the lack of differentiation upon ablation of the Rieske iron–sulfur protein of mitochondrial Complex III of the respiratory chain (5). Such ablation led to an
Inhibition of necroptosis
Necroptosis is a type of cell death that may be programmed to exhibit a necrotic phenotype (27). The typical mechanism involves induction by tumor necrosis factor-α (TNFα) upon its binding to the TNFα receptor complex. Such a death signal activates the receptor-interacting protein 1 (RIP1) and recruits RIP3, alongside the formation of the so-called necrosome. Subsequently, RIP3 is autophosphorylated and binds the mixed lineage kinase domain-like (MLKL) protein. Phosphorylated MLKL diffuses to the plasma membrane and initiates necroptosis (27). It was demonstrated that 2HG stimulates hypermethylation of the RIP3 promoter (191). It is amplified due to the ability of 2HG to bind to DNA methyltransferase 1 (DNMT1). Consequently, 2HG can inhibit the necroptosis since the levels of RIP3 protein are reduced. As a result, this mechanism belongs to those promoting tumorigenesis.
As for apoptosis, glioma cells containing mutant IDH1 exhibit more apoptosis upon the inhibition of Bcl-xL, thus being more vulnerable to this inhibition (79). Both wt IDH1/2 in the oxidative mode have a profound antioxidant and hence antiapoptotic role. A higher extent of the reductive mode of their reaction then leads to a lower apoptosis protection. This was simulated, for example, by silencing IDH2 in HeLa cells (150). Also, the sensitivity of HeLa cells toward apoptosis induced by ionic radiation (90) as well as TNFα and anticancer drugs was markedly elevated upon silencing IDH2 (80).
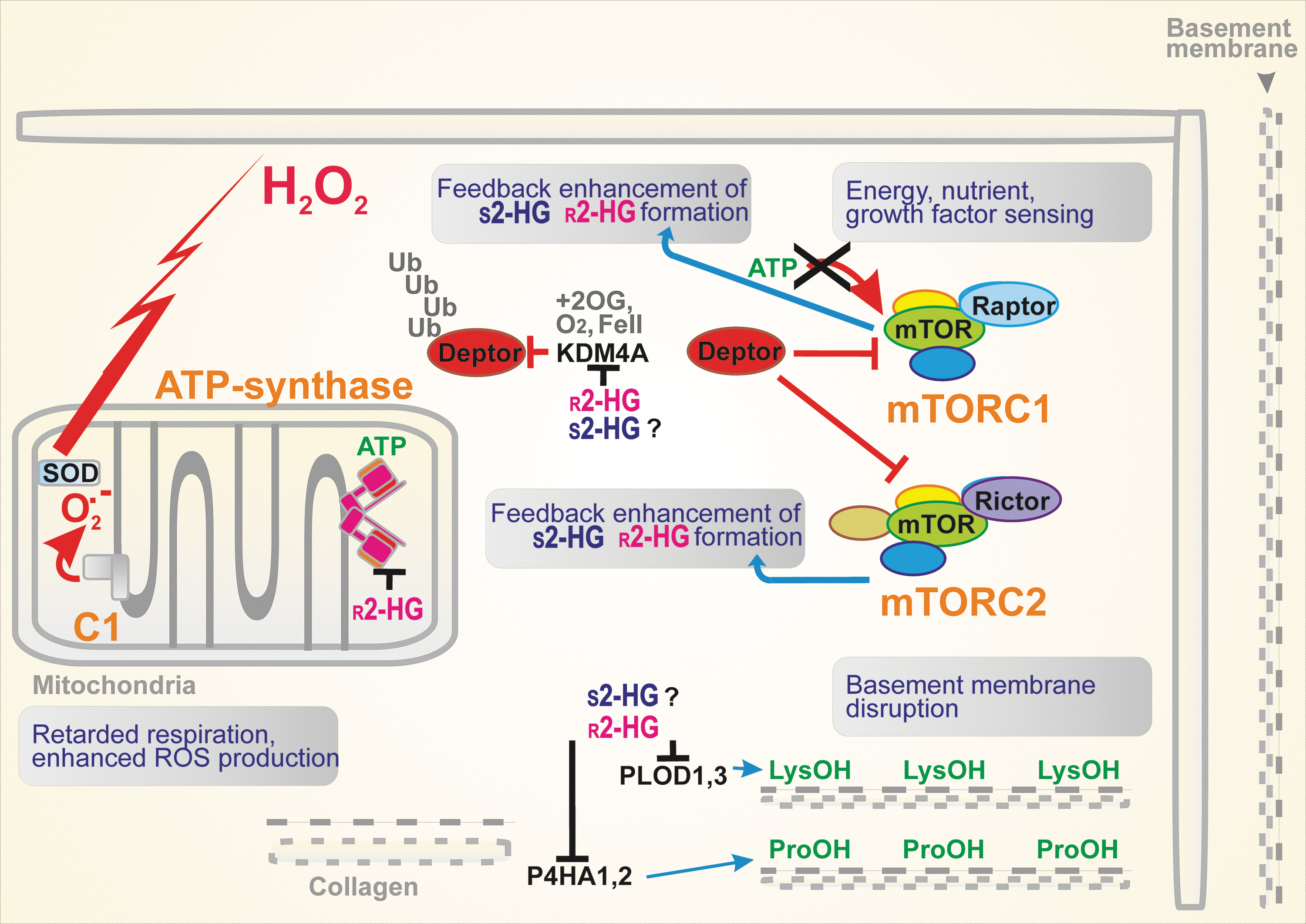
Interference with the mTOR pathway
The mTOR is a serine/threonine kinase, forming complexes with Raptor or Rictor, that is, mTORC1 and mTORC2, respectively. These complexes are regulated by amino acid and energy (ATP) levels. This enables mTORC1 to regulate cell growth and/or autophagy and mTORC2 to determine cell survival (21) (Fig. 5). In cancer cells, both mTORC1/2 are frequently activated by upstream negative modulators disabled by mutations. The modulators of the mTORC1/2 complex, tuberous sclerosis complex TSC1–TSC2 heterodimers, are inhibited by the PI3K/AKT signaling pathway. The TSC2 contains the GTPase-activating protein domain, whereas TSC1 stabilizes the heterodimer. As a result, the TSC1–TSC2 complex downregulates a small G-protein Rheb. Since Rheb is an activator of mTORC1, the mTORC1 activity is inhibited (21).
Since mTORC1/2 activation promotes cancerogenesis, so does the identified 2HG inhibition of KDM4A, a 2OG-dependent dioxygenase of the Jumonji family of lysine demethylases (21). Since KDM4A associates with one of the negative modulators, the DEP domain-containing mTOR-interacting protein (DEPTOR), the 2HG-mediated inhibition of KDM4A releases DEPTOR and activates mTORX1/2. Interference with the mTOR pathway was also reported for C. elegans (52).
Disruption of the cytoskeleton architecture
Procollagen-lysine 2-oxoglutarate 5-dioxygenase PLOD1 and PLOD3 and prolyl 4-hydroxylase P4HA1 and PHA3 stabilize the triple helix of collagen via the respective hydroxylations. Since
Interference with HIF signaling
The initiation of HIF reprogramming of the transcriptome involves the inhibition of prolyl hydroxylase domain enzymes (PHD1/EglN2, PHD2/EglN1, and PHD3/EglN3) by the decreasing oxygen in hypoxia. There is disagreement over whether HIF is stabilized or degraded by 2HG enantiomers (6). Originally,
In vitro
2HG as an Oncometabolite
Promotion of carcinogenesis by 2HG
Gliomas
IDH1/2 mutations should arise during embryonic development due to the somatic mosaic of mutant IDH1/2-expressing cells, such as IDH1 R132H/C/L/S or R100Q and IDH2 R140Q/G/W/L or R172K/G/M/Q/T/S, which are common mutations in gliomas (bold are the most frequent) (20, 180). This is accompanied by loss-of-function mutations of the p53 protein (110). A specific human isoform of glutamate dehydrogenase 2 (GDH2) was also reported to promote glioma. Since unlike GDH1, GDH2 is not inhibited by GTP, this enables the otherwise deficient 2OG input into the Krebs cycle to be replaced by converting glutamate to 2OG (178). Glutamate can be made from 5-oxoproline, resulting from a cleavage of dipeptide metabolites such as γ-glutamyl amino acids. An increased uptake of the latter was found in IDH1R232H p53−/− cells after the overexpression of GDH2, but not GDH1 (178).
Typically, millimolar concentrations of 2HG are found in gliomas bearing IDH1/2 mutants (Table 1). In vivo magnetic resonance (MR) imaging using echo-planar spectroscopic imaging dual-readout alternative gradients (DRAG-EPSI) detected 5 mM 2HG before surgery and 3–6 mM after surgery (4). Similar concentrations were found using long echo time MR spectroscopy with semi-localization by adiabatic selective refocusing. It was recognized that gliomas bearing IDH2 mutants accumulated more 2HG than those with IDH1 mutants (9, 148).
Estimated Concentrations of 2-Hydroxyglutarate in Tissues or Cells and Body Fluids
Reported amounts of 2HG were converted to concentrations on the assumption of 1 g being 1 mL and based on 200 μm3 volume of lymphocyte (AML cells).
2HG, 2-hydroxyglutarate; AML, acute myeloid leukemia; CSF, cerebrospinal fluid; DRAG-EPSI, echo-planar spectroscopic imaging dual-readout alternative gradients; IDH, isocitrate dehydrogenase; MALDI-TOF, matrix assisted laser desorption/ionization - time-of-flight;
Matrix assisted laser desorption/ionization - time-of-flight analyses detected
Changes in the expression of other genes affect patient's prognoses and survival since they may induce positive or negative effects. Typically, tumor suppressor genes exhibit an increased expression in gliomas with mutant IDH1, whereas the expression of oncogenes declines (67). For example, gene expression of insulin-like growth factor-binding protein 2 (IGFBP) is downregulated by DNA methylation promoted by 2HG formed by mutant IDH1 (67). Moreover, prognoses are worse for patients with gliomas with a low expression of insulin-like growth factor binding protein 2 (67).
Acute myeloid leukemia
Ivosidenib (commercially Ibsovo) was reported to have a 40% response in AML patients. Nevertheless, since IDH2R140Q is the most frequent mutation found in AML and IDH2 mutations were also found in angioimmunoblastic T cell lymphoma (20, 197), enasidenib was developed as inhibitor of mutant IDH2, inducing molecular remissions (159). It was suggested for AML patients that levels of 2HG in serum exceeding 1 μg/mL might indicate the presence of IDH1/2 mutations. Among a cohort of 200 such patients, about 25% indeed exhibited these IDH1/2 mutations, while a threshold of 0.5 μg/mL was identified for 2HG (16). Elevated 2HG levels were also found in urine, bone marrow aspirates, and aspirate cell pellets. Moreover, the progression of standard chemotherapy was associated with decreasing serum levels of 2HG, supporting a prognostic potential of 2HG (16). The ability of mutant-IDH1 inhibitors to provide effects in AML having mutant-IDH2 stems from the ability to switch their mutagenesis toward unmutated IDH1, which turns to be the right target (63, 69).
Also, the progression of asymptomatic precursor plasma cell malignancies to symptomatic multiple myeloma was associated with elevated 2HG (59). Another lymphoma stems mostly from R172 mutations of IDH2, angioimmunoblastic T cell lymphoma, a subtype of nodal peripheral T cell lymphomas (91).
Breast cancer
Like other types of cancer, breast cancer also undergoes metabolic reprogramming (17, 37, 164, 166, 168) and possesses a modified chromatin and tumor microenvironment in which the antitumor immunity can be suppressed. PHGDH has also been identified as a breast cancer oncogene (96, 132). PHGDH and possibly other sources, such as ADHFE1, besides the nonmutant IDH2 (155) and IDH1 are responsible for elevated 2HG levels in breast carcinoma in the absence of the IDH1/2 mutant enzymes (166, 168). The enhanced 2HG levels correlated with Myc signaling (168). Also, the ablation of ADHFE1 decreased 2HG (168).
It was recently demonstrated that Myc-induced ADHFE1 forming
Other cancer types
IDH mutations were identified in ∼20% of cholangiocarcinomas (12). In a few cases, other types of cancers (78) carried IDH1/2 mutations, such as paraganglioma (53), colon cancer (153), prostate cancer, and lung cancer (146). Chondrosarcomas contain abundant 2HG (2). The kinetics of 2HG formation could be assessed by a hyperpolarized MR imaging technique (141).
In colorectal cancer cells, the epithelial–mesenchymal transition is induced by
IDH mutations may also contribute to prostate cancer since a benign prostatic epithelial is transformed into a malignant one by certain miRNAs, which in turn are promoted by IDH1R132H mutations (196). Also, up to 5% of patient samples of melanoma contained IDH1R132C or IDH1R132S, which co-existed in 3% of samples with NRAS mutations (95).
Elevated IDH1 expression, including the common R132H mutations, was found in non-small-cell lung cancer (NSCLC) cells (188). These mutations induced an elevated migration and proliferation of NSCLC cells, in which the promoter for the glycoprotein fibulin-5 was found to be hypermethylated. Since fibulin-5 is a protein participating in the aggregation and stabilization of complexes in the extracellular matrix, one may speculate that 2HG can also promote cancerogenesis by optimizing the extracellular milieu for tumor growth.
Renal cell carcinomas were recently found to have a decreased expression of
2HG in prevention of immunosurveillance
Immune system within the tumor microenvironment
Recently, a great deal of attention has been paid to immune system cross talk with tumors and metastases (29, 176). Indeed, cancerogenesis progresses not only due to genetic and epigenetic somatic alternations (34) but also due to failed immunosurveillance, at least to some extent (56) (Fig. 6). The following defects can be identified: (i) Impairment of immune cells, leading to the inability of the immune system to recognize cancer cells or cells in premalignant states. (ii) Active secretion by cancer cells of factors causing the above effects as in (i). These factors can act either systematically or locally within the tumor microenvironment. Interestingly, both 2HG enantiomers have been recently found to fulfill such roles. (iii) The transformation of cancer cells or cells in premalignant states so that they expose their cell surface in a manner reducing antigenicity or to be shielded against adjuvancy.
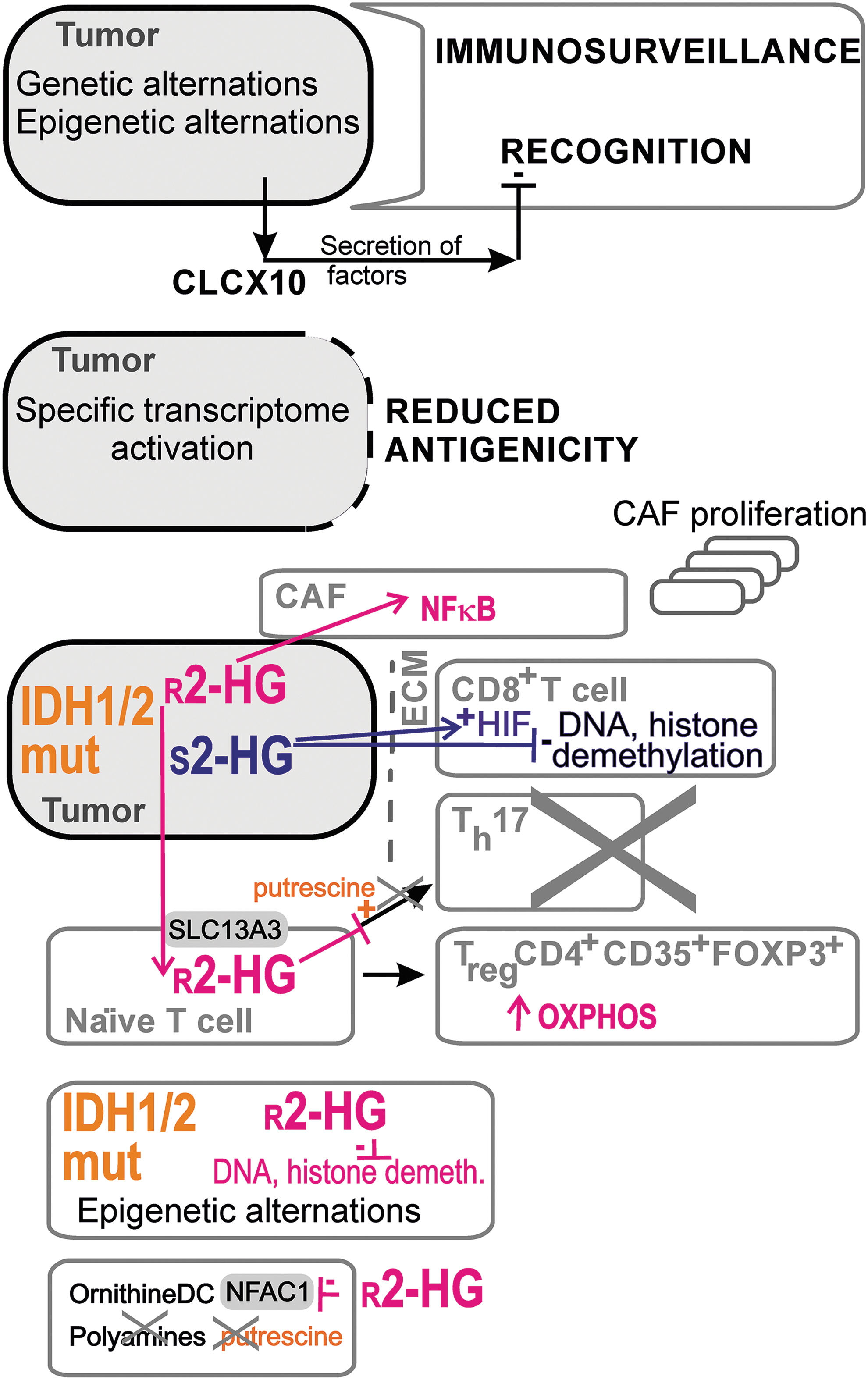
r -2HG effects
Both 2HG enantiomers prevent the immunosuppression of tumors. Thus, several detailed effects of
Finally, nonmalignant cells are affected by
s -2HG effects
Activated mouse CD8+ T cells are affected by
2HG as a possible metabolic marker of cancer
Normal versus pathological levels of r -2HG and s -2HG
In the above sections, it was recognized that the range of
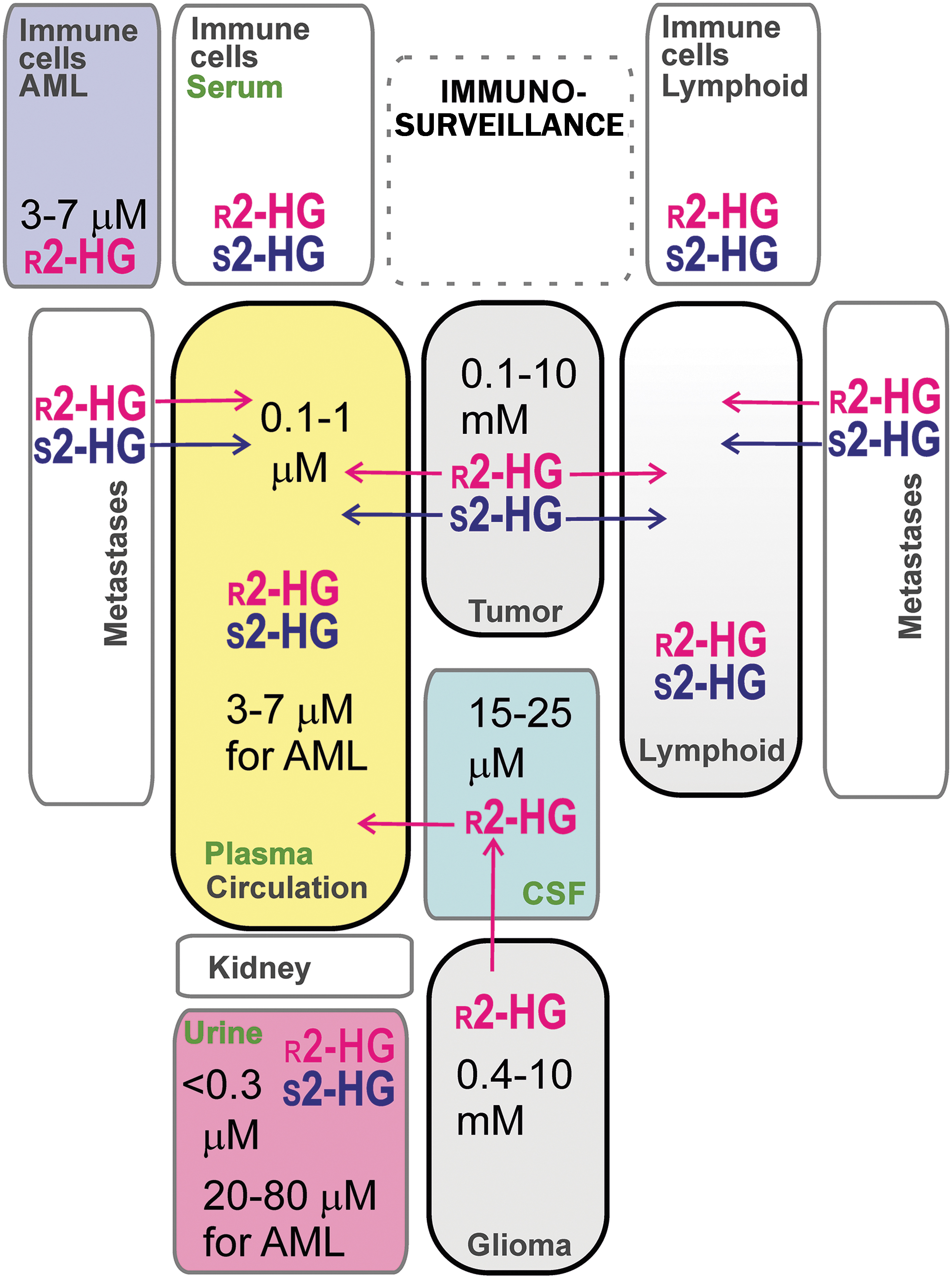
Intermediate concentrations, that is, 50–100 times lower than those found in grade II/III gliomas, were found in ER− breast carcinoma cells, HTB-126/Hs 578T, and epithelial adenocarcinoma MDA-MB-231,cells (155). The leakage of
Focusing on breast cancer, c-Myc-retransformed breast cancer tissues contained substantial levels of 2HG (0.5–20 nmol/mg), despite the absence of IDH1/2 mutations (168). These tumor tissues exhibited global epigenome changes associated with poor prognosis (168). A patient with hormone-receptor (HR+) breast carcinoma exhibited mutant IDH1 R132L within the tumor tissue and cells in lymph nodes, which corresponded to an elevated 2HG concentration in urine (22 ng/mL vs. 1.7 ng/mL in healthy controls) and serum (1979 ng/mL vs. 105 ng/mL in healthy controls) (50). This is comparable to urine 2HG concentrations for gliomas with IDH1/2 mutations (1–14.6 ng/mL), which were found to be much higher than for gliomas with wt IDH1/2 (1–4 ng/mL) (49). Also, serum 2HG concentrations were in a similar range for gliomas with IDH1/2 mutations (33–283 ng/mL), and these were no different from those for patients with gliomas with wt IDH1/2 (35–277 ng/mL) (49).
Also, elevated
Immune cells as sources of r -2HG
Tumor development proceeds in a complex host–tissue microenvironment, in which immune cells play significant pleiotropic roles alongside fibroblasts, the extracellular matrix, and lymphatic vascular networks (157). A distinct type of CD4+ T cells producing interleukin (IL)-17, designated as T helper (Th) 17 cells (105), was found to switch from OXPHOS to aerobic glycolysis with concomitantly elevated levels of
Immune cells as sources of s -2HG
Specifically, due to the activation of the immune system,
Future Perspectives
The normal 2HG metabolism and metabolism of related compounds, such as
2HG's effects on the as yet unidentified particular promoters of genes should be further determined. Promoters of tumor-suppressive miRNA should be investigated with a specific emphasis. They are often large and contain CpG islands to be sensitive to hypermethylation. Currently, unidentified information signaling pathways affected by 2HG enantiomers should be discovered. Uncovering details of 2HG metabolism and signaling in various immune cell types will help to predict cancer recurrence even after tumor excision. All such future knowledge may lead to the establishment of precise diagnostics and/or individual prognoses based on
Conclusions
Both 2HG enantiomers,