
Abstract
Significance:
Epidemiological studies indicate that metabolic disorders are associated with an increased risk for Alzheimer's disease (AD). Metabolic remodeling occurs in the central nervous system (CNS) and periphery, even in the early stages of AD. Mitochondrial dysfunction has been widely accepted as a molecular mechanism underlying metabolic disorders. Therefore, focusing on early metabolic changes, especially from the perspective of mitochondria, could be of interest for early AD diagnosis and intervention.
Recent Advances:
We and others have identified that the levels of several metabolites are fluctuated in the periphery before their accumulation in the CNS, which plays an important role in the pathogenesis of AD. Mitochondrial remodeling is likely one of the earliest signs of AD, linking nutritional imbalance to cognitive deficits. Notably, by improving mitochondrial function, mitochondrial nutrients efficiently rescue cellular metabolic dysfunction in the CNS and periphery in individuals with AD.
Critical Issues:
Peripheral metabolic disorders should be intensively explored and evaluated for the early diagnosis of AD. The circulating metabolites derived from mitochondrial remodeling represent novel potential diagnostic biomarkers for AD that are more readily detected than CNS-oriented biomarkers. Moreover, mitochondrial nutrients provide a promising approach to preventing and delaying AD progression.
Future Directions:
Abnormal mitochondrial metabolism in the CNS and periphery is involved in AD pathogenesis. More clinical studies provide evidence for the suitability and reliability of circulating metabolites and cytokines for the early diagnosis of AD. Targeting mitochondria to rewire cellular metabolism is a promising approach to preventing AD and ameliorating AD-related metabolic disorders.
I. Introduction
Alzheimer's disease (AD), the most common type of dementia, is a progressive and irreversible neurodegenerative disease, characterized by a progressive loss of learning, memory, orientation, language, comprehension, judgment, and intellectual performance (8, 430). This disease was first observed in 1906 by Dr. Alois Alzheimer, who found abnormal plaques and fibrillary pathology in the brain of his patient Auguste Deter, who died due to an atypical mental illness (7). In the United States, ∼5.8 million patients are living with AD, the majority of whom are older than 65 years. Statistics show that in 2010, about 10 million people suffered from AD in China, which is the country with the largest number of patients in the world. The prevalence of AD in China was estimated at 3.21% among people aged ≥65 years (181). AD is the sixth leading cause of death in the United States: Deaths from AD have increased ∼1.5-fold, whereas deaths from others have decreased during the past two decades.
Once clinically diagnosed, the patient has an average life expectancy of 3 to 9 years. In the clinic, only a few Food and Drug Administration drugs have been approved for the treatment of AD, including donepezil, rivastigmine, galantamine, and memantine; however, these drugs have limited abilities to reverse AD progression and only temporarily improve the quality of life for patients (8, 158).
Pathologically, AD is characterized by Aβ and neurofibrillary tangles (NFTs) in the brain. Although there is no conclusion about the molecular mechanism(s) of AD, possibilities include (i) beta amyloid (Aβ) and amyloid plaques, (ii) hyperphosphorylation of the Tau protein and NFTs, (iii) oxidative stress and inflammation, (iv) and mitochondrial dysfunction (Fig. 1).
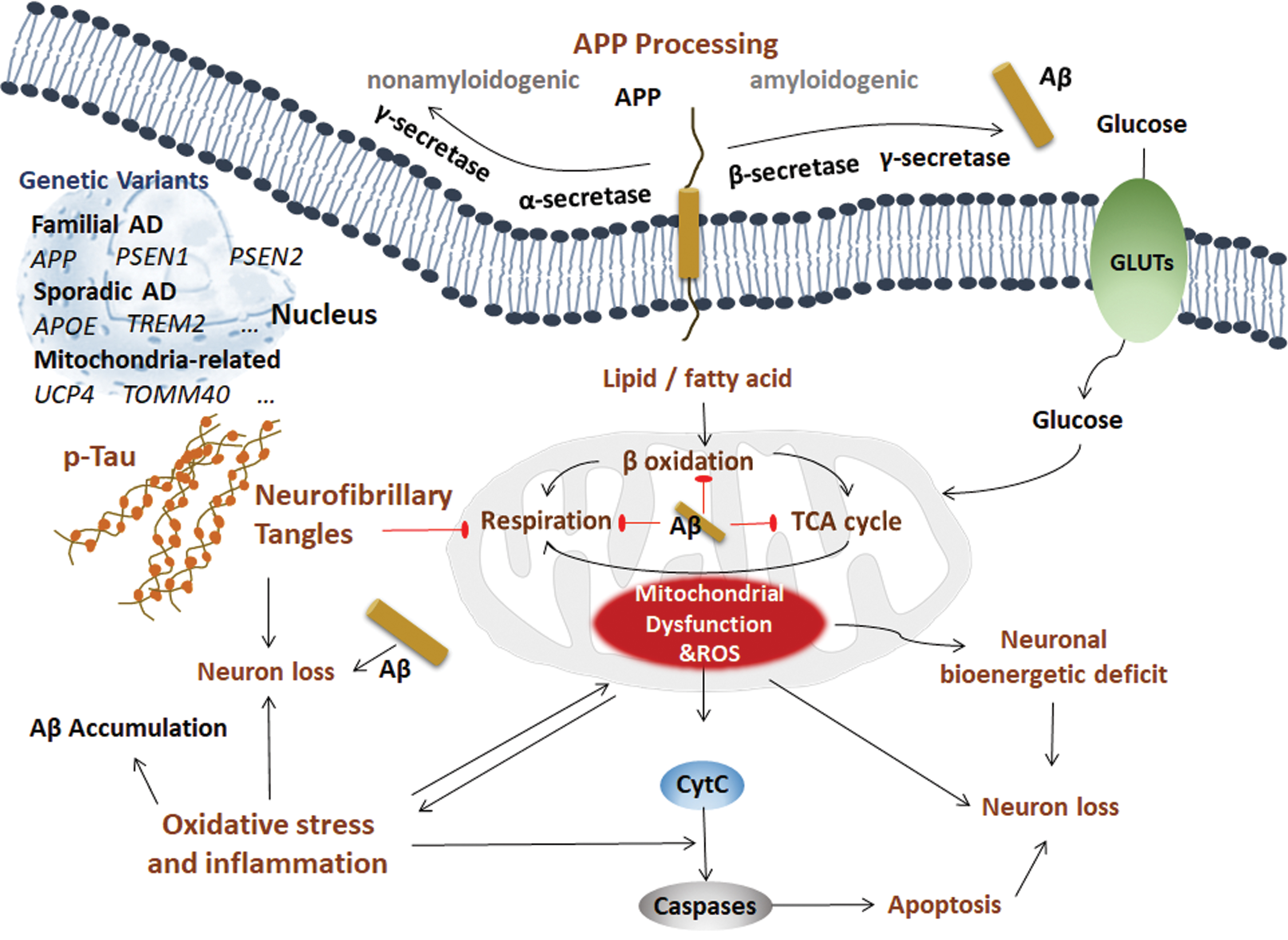
A. Aβ and amyloid plaques
AD can be divided into three main types: early-onset familial AD (FAD), early-onset sporadic AD, and late-onset sporadic AD. The early-onset FAD subtype accounts for ∼1% of all AD cases, with 326 total families worldwide living with this type of AD. The genetic variants in autosomal dominant FAD include genes encoding the transmembrane protein amyloid beta precursor protein (APP), presenilin 1 (PSEN1, also known as PS1), and presenilin 2 (PSEN2) (311, 336, 394), which are all involved in APP processing.
The APP gene is located on chromosome 21, and the mutations include A201V (344), A235V (295), D243N (295), E246K (111), E296K (295), P299L (295), R468H (347), A479S (111), K496Q (344), A500T (347), Y538H (285, 344), V562I (344), E599K (344), T600M (347), P620A (295), P620L (344), T663M (347), E665D (306), V669L (Seoul) (20), KM670/671NL (283), A673T (306), A673V (79), H677R (179), D678H (52), D678N (403), E682K (472), K687N (187), A692G (154), E693G (188), E693K (383), E693Q (221), D694N (128), L705V (297), G708G (21), G709S (347), A713T (47), T714A (304), T714I (210), V715A (70), V715M (11), I716F (139), I716M (32), I716T (391), I716V (94), V717F (284), V717G (51), V717I (125), V717L (285), T719N (168), T719P (121), M722K (416), L723P (211), and K724N (392).
PSEN1 and PSEN2 are located on chromosomes 14 and 1 and contain 13 exons and 12 exons, respectively, and more than 300 mutations have been reported in PSEN, which represents the most common genetic variation in AD (214). On the other hand, PSEN2 is very rarely mutated and is related to AD (44). Mutations in the APP gene trigger the overproduction of neurotoxic and aggregation-prone forms of Aβ peptides by shifting the cleavage of APP toward amyloidogenic processing (24). The PSEN1 and PSEN2 genes aggravate the production ratio of Aβ42 by regulating the γ-secretase-mediated cleavage of APP (24). However, the majority of sporadic AD cases have no such mutations in APP, PSEN, and PSEN2 (186), indicating a different or additional mechanism underlying AD pathogenesis.
APP is a transmembrane protein that can be processed in two distinct ways: nonamyloidogenic processing and amyloidogenic processing. In the nonamyloidogenic processing method, APP is sequentially cleaved by α-secretase and γ-secretase (PS1) into a secreted C-terminal fragment (sAPPα), p3 and amyloid intracellular domain. In amyloidogenic processing, APP is sequentially cleaved by β-secretase (β-site APP cleaving enzyme 1 [BACE1]) and γ-secretase, producing toxic Aβ fragments. This sequential cleavage occurs on the plasma membranes of neurons, adipocytes, and hepatocytes (463, 473). Aβ deposition in the central nervous system (CNS) serves as the most significant pathological hallmark of AD at patient autopsy (8). Under pathological conditions, amyloidogenic processing predominates, and AD is thus initiated by an imbalance between the formation and degradation of Aβ, leading to the deposition of Aβ and subsequent disruption of synapse and neuronal function (423), representing the so-called Aβ hypothesis (151). Moreover, Aβ has expanded roles in other peripheral organs (412).
The Aβ peptide constitutes 37 to 43 amino acids, most of which are Aβ40 and Aβ42. Aβ42 differs from Aβ40 by two extra isoleucine and alanine residues at the C-terminus. Aβ42 is the major component of plaque deposition, whereas Aβ40 is the major component of cerebrovascular Aβ (137). Aβ42 is more amyloidogenic and has a higher cellular toxicity than Aβ40 (319). A higher Aβ40/Aβ42 ratio may promote the construction of cerebral amyloid angiopathy over parenchymal deposition (156, 443). The Aβ peptide can be cleared under normal conditions but accumulates as neurotoxic Aβ-derived diffusible ligands in AD pathogenesis. Aβ peptide deposition by the self-assembly and aggregation of 4 kD Aβ peptides not only shows strong neurotoxicity related to neuronal degeneration but also induces a series of pathological events, such as astrocyte and microglia activation, blood-brain barrier (BBB) destruction, and microcirculation changes, leading to neurodegeneration and death in AD (349). Aβ can be degraded by insulin degrading enzyme (IDE), neprilysin (NEP), and other degrading enzymes (339).
The most common variant in sporadic AD patients is apolipoprotein E (APOE), which not only increases the severity of FAD but also increases the susceptibility to sporadic AD (172). ApoE is a lipid-binding protein that modulates triglyceride and cholesterol transportation in the liver, brain, and other tissues. The APOE type 4 allele (APOE4) is the main genetic risk factor for sporadic AD (64), whereas the APOE2 genotype has been identified as a protective factor for late-onset sporadic AD (63). Mechanically, ApoE has an Aβ-binding motif and serves as a clearance protein to induce toxic Aβ degradation in lysosomes.
According to genome-wide association studies, other rare genetic variants accounting for late-onset AD have been identified, including genes encoding triggering receptor expressed on myeloid cells 2 (TREM2), complement C3b/C4b receptor 1 (CR1), bridging integrator 1 (BIN1), phosphatidylinositol binding clathrin assembly protein (PICALM), clusterin (CLU), adenosine triphosphate (ATP) binding cassette subfamily A member 7 (ABCA7), and CD33 molecule (CD33) (113, 138, 152, 160, 213). These genetic variants are all related to Aβ deposition; for example, TREM2 and CR1 play a key role in the removal of cell debris and Aβ clearance (138).
Although preclinic and clinical studies show that Aβ pathology is the major characteristic of AD, many clinical trials focusing on clearing Aβ and decreasing Aβ production have failed (8, 162, 209, 234, 405), indicating that track-Aβ-only is insufficient for AD treatment and that additional mechanisms are essential for the pathogenesis of AD.
Notably, some genetic variants of AD are involved in not only classic Aβ pathology but also cellular metabolism (e.g., ABCA7, APOE4, CLU) and inflammation (e.g., CLU, CD33). Although there is no consistent evidence of mitochondrial DNA (mtDNA) variants in AD (171, 312), mitochondria-related variants were identified in AD. For example, the genetic variant in mitochondrial uncoupling protein 4 (UCP4), which encodes the mitochondrial inner membrane transporter uncoupling protein and is responsible for mitochondrial energy metabolism, markedly affects the susceptibility to AD (274). Variants in intron 6 of translocase of outer mitochondrial membrane 40 (TOMM40), which encodes the central pore of the translocase of the mitochondrial outer membrane and controls protein entry into the mitochondria, are associated with the risk of AD (456). With the advanced technologies for genetics and omics studies, an increasing number of genetic variants can be identified to uncover the mechanisms and new treatments for AD.
1. Aβ and mitochondria
Several studies have suggested that Aβ can directly affect mitochondrial function (6, 10, 36, 84, 248, 303). The amino terminus of APP enters mitochondria depending on the mitochondrial outer membrane translocase TOM40 and inner membrane translocases TIM23 and TIM44, whereas the carboxy terminus of APP cannot, resulting in blockage of the mitochondrial protein import channel (10). This blockage hinders other proteins from entering the mitochondria, causing decreased cytochrome oxidase activity, increased production of reactive oxygen species (ROS), and mitochondrial dysfunction (10). Aβ itself can bind to alcohol dehydrogenase (ADH) in mitochondria to form Aβ-binding alcohol dehydrogenase (ABAD), promote ROS production, and damage mitochondrial function (248). Aβ also regulates Ca2+ homeostasis, leading to opening of the mitochondrial permeability transition pore (mPTP) to decrease the mitochondrial membrane potential, matrix swelling, and respiratory damage, which can be reversed by several compounds, such as the mitochondrial osmotic transfer channel inhibitor cyclosporine A (CsA), adenosine diphosphate (ADP), and oligomycin. Notably, ADP together with oligomycin works better in brain mitochondria than CsA (6, 36, 303). Mitochondria are the core producers of ROS in the cell, and Aβ is partially targeted to mitochondria to promote ROS and mitochondrial dysfunction (84, 248).
B. Hyperphosphorylation of the Tau protein and NFTs
Another significant pathological hallmark of AD is NFTs, which indicates the severity of AD (134, 177). Although NFTs exist in many diseases, such as AD, ischemia, and stroke, their pathology is distinct among each disease. Tau is the primary component of NFTs in AD and contains microtubule-binding domains, projection domains, and proline-rich regions with multiple phosphorylation sites (29, 217).
The Tau protein is an unfolded and soluble protein that assembles and stabilizes microtubules to maintain neuronal function. In the brains of AD patients, Tau undergoes multiple post-translational modifications, such as hyperphosphorylation and glycosylation (134, 413). The most prominent post-translational modification of Tau in AD is hyperphosphorylation. Thus far, numerous phosphorylation sites have been identified, including Ser199, Ser202, Ser262, Ser422, Ser396, Thr205, and Thr231 (18, 351). Several kinases, including cyclin-dependent kinase 5 (CDK5), mitogen-activated protein kinase (MAPK), and GSK-3β, have been demonstrated to regulate Tau phosphorylation (62, 277). Hyperphosphorylated Tau forms Tau dimers, which then form Tau oligomers. The aggregated and misfolded Tau oligomers form paired helical filaments, which eventually form abundant NFTs. The hyperphosphorylated Tau is toxic to synapses and neurons and impairs cognitive function (62). The N-glycosylation of Tau inhibits its binding with microtubules (413). Another type of glycosylation, O-GlcNAcylation, is negatively correlated with Tau phosphorylation, indicating an effect opposite that of N-glycosylation (230).
Aβ also promotes Tau accumulation. Crossing transgenic APP mice with Tau mice induces neurotoxicity, indicating that tauopathy is also required for Aβ toxicity (334). In addition, the Aβ42 oligomer induces progressive tauopathy in neurons (369). The prolyl isomerase Pin1 is downregulated by oxidation in AD. The deficiency of Pin1 promotes glycogen synthesis kinase 3 (GSK3) to hyperphosphorylate Tau, leading to tangle formation and neuronal cell death (305). On the other hand, Pin1 also binds to the p-Thr668 motif in APP to increase Aβ production (305). Apart from Aβ, tauopathy may lead to cognitive dysfunction through gray matter loss in AD (27). Although the mechanistic relationship between mitochondria and tauopathy is elusive, the hyperphosphorylation of Tau is known to be associated with altered mitochondrial distribution, increased abnormal mitochondrial trafficking, and impaired mitochondrial dynamics and functions (190, 204, 260).
C. Oxidative stress and inflammation
Systemic inflammatory responses and oxidative stress have been recognized as two other moderators and biomarkers of AD. Oxidative stress and inflammation are largely attributed to mitochondrial dysfunction in the brain and periphery, leading to energy metabolism failure and neurological disorders (321). Oxidative stress and inflammation induced by mitochondrial damage occur before AD pathological features, such as Aβ deposition, and accelerate AD progression (451). Although inflammation, oxidative stress, and related mitochondrial dysfunction are not specific to AD, they overlap extensively with AD pathogenesis and contribute to AD progression. On the one hand, mitochondria-associated oxidative damage and the inflammatory response are early key factors in the development of AD. On the other hand, Aβ, as the most typical pathological feature of AD, directly dampens mitochondrial structure and function, which promotes oxidative stress and inflammation in neurons, and further facilitates the pathogenesis of AD.
D. Mitochondrial dysfunction
The “Mitochondrial cascade hypothesis” and “mitochondrial bottleneck hypothesis” suggest that mitochondrial dysfunction is an essential early and primary event in AD pathogenesis (73, 378, 380). Swerdlow and Khan proposed the mitochondrial cascade hypothesis to show that mitochondrial dysfunction is a primary event that causes all AD pathogenic changes, including Aβ deposition, NFT formation, neuron death, and synaptic loss (379 –381). In early research, several groups observed different types of mitochondrial abnormalities in the brains or peripheral tissues of patients with AD and animal models (22, 157, 248, 270, 354, 419). Using genetic AD animal models, many studies have shown that mitochondrial bioenergetic deficits occur in these models at very early ages, and mitochondrial dysfunction precedes Alzheimer's pathology (85, 448). Studies by us and others have shown that targeting mitochondria to improve mitochondrial biogenesis, remodeling mitochondrial fusion/fission, or promoting mitochondrial respiration function with pharmaceutics and nutrient approaches efficiently slow down or reverse the pathogenesis of AD (233, 234, 356). Taken together, these studies indicate that mitochondrial dysfunction plays a key role in the pathogenesis of AD.
Two major biological functions that occur in mitochondria are the tricarboxylic acid cycle (TCA cycle, also known as the Krebs cycle) and oxidative phosphorylation (OXPHOS) (266). Although the three major nutrients, glucose, lipids, and proteins, have different metabolic pathways in vivo, they all share the common intermediate metabolite acetyl-CoA, which is further utilized by the TCA cycle to produce NADH (313). NADH is further used as a substrate for OXPHOS to produce ATP. The high-order assembly of respiratory chain complexes in the inner mitochondrial membrane allows mitochondria to respond to energy conversion to promote efficient electron transfer (141). The process of electron transfer in the respiratory chain is coupled to ADP phosphorylation to generate ATP. The ATP-synthase activity is also significantly lower in AD patients in the stage that is characterized by entorhinal and transentorhinal tauopathy with no clinical symptoms than in age-matched controls. Targeted for oxidative stress, the mitochondrial ATP-synthase α subunit is lipoxidized distinctly in the entorhinal cortex in the AD cases. This study specifies that the energy production system in mitochondria may be one pathway that is impaired in the very early stages of AD (390). A reduced cytochrome c oxidase (COX) subunit 4 in mitochondrial electron transport chain links amyloid deposition and mitochondria dysfunction in the dystrophic neurites of senile plaques. However, a preserved COX subunit 4 in neurons is associated with NFTs (310).
Epidemiological studies indicate that hyperglycemia and hyperlipidemia are major risk factors for neurodegenerative diseases, especially AD (69, 201, 226). The 2019 World Health Organization (WHO) guidelines for the reduction of cognitive decline and dementia risks recommended the management of diabetes in adults and the management of dyslipidemia at midlife to reduce the risk of cognitive decline and/or dementia (425). Indeed, we and others have recently shown that deficits in glucose and lipid metabolism occur early and before significant cognitive decline (412).
Apart from their key role in bioenergy metabolism, mitochondria represent the “integration” center of signal transduction and regulate epigenetics, stem cells, and the differentiation, initiation, and execution of apoptosis. Mitochondria are capable of sensing cellular stress and helping cells adapt to challenging microenvironment conditions, giving cells a high degree of plasticity to promote their growth and survival. These abilities suggest that mitochondria are uniquely significant in age-related diseases, especially in AD progression (378).
Each suggested hypothesis functions in the AD pathogenesis feedback loop, and they coparticipate with each other at multiple levels during AD progression. Further, mitochondria serve as the bridge among glucose metabolism, lipid metabolism, oxidative stress, inflammation, and AD pathogenesis. Mitochondrial dysfunction and mitochondrial remodeling may provoke AD pathogenesis and lead to AD progression. A potential role of mitochondria at the core of metabolism during AD progression deserves more attention (Fig. 2).
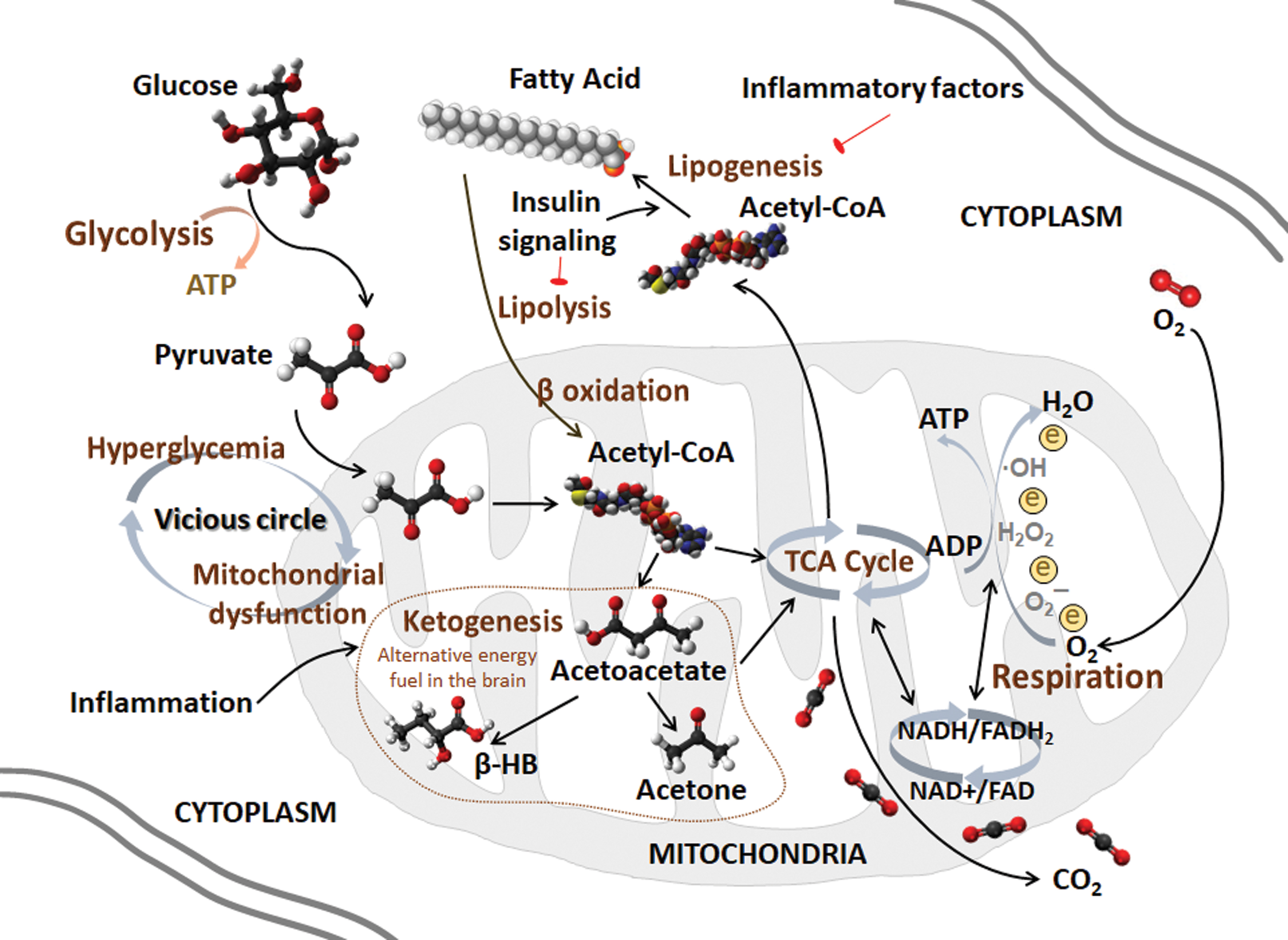
II. Glucose Metabolism in AD
Glucose is the only energy source that can cross the BBB, and it provides energy for normal neuronal functions. Numerous studies have identified impairments in glucose utilization and metabolism in the brain and peripheral tissues of AD patients and animal models. Alterations in glucose metabolism occur with the accumulation of oxidative damage before AD pathology in the brains of AD patients and animal models (89, 227, 272, 279).
A. Glucose uptake deficit
Glucose cannot be synthesized or stored in the brain. Dr. Magistretti discovered that glycogen in the brain is enriched in astrocytes, indicating that glucose from the blood first enters and is stored in astrocytes (41, 253). This finding has led to the initial discovery of glucose transport, storage, and transformation pathways in the brain. Thus, subsequent series of studies showed that when neurons are activated, they release glutamate signals, stimulate astrocytes to break down glycogen, and convert glucose into pyruvate and, finally, lactic acid, which is released extracellularly by a monocarboxylic acid transporter and taken up by neighboring neurons to produce ATP (254).
Glucose uptake is markedly reduced in early-stage AD patients, suggesting that decreased glucose metabolism and increased steady-state concentrations of glucose are early signs of AD (69, 178, 368, 370). The age and sex-adjusted hazard ratios of AD in diabetic individuals were reported to be 1.94 times higher than those in the control group, indicating that diabetes itself is an independent risk factor for AD (298).
Using fluoro-2-deoxy-
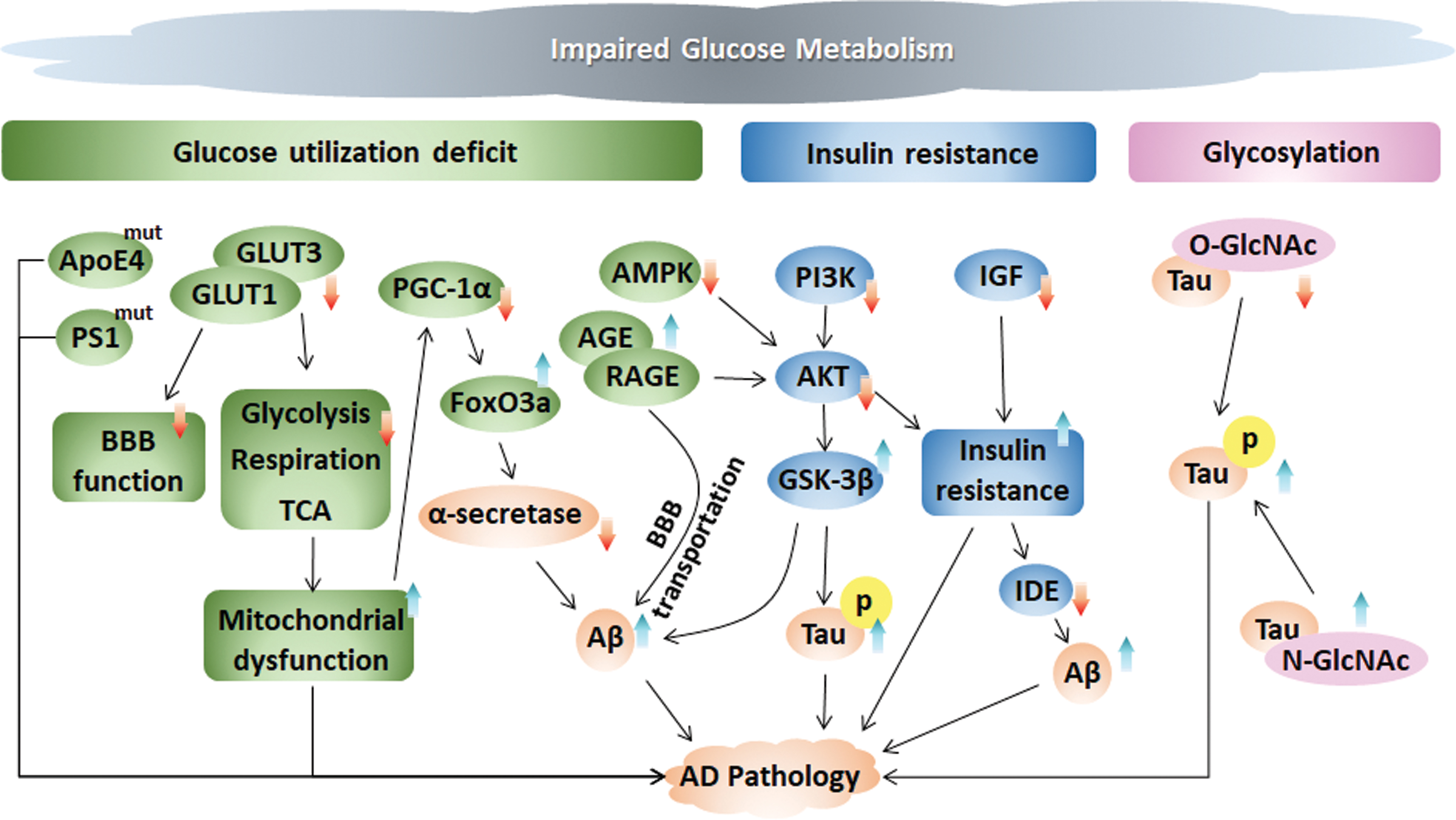
The polar, hydrophilic glucose molecules require specific transporters to cross the cellular membrane. Glucose uptake functions via glucose transporters (GLUTs) at the BBB to transport glucose between the brain and periphery. The BBB is mainly formed by endothelial cells, astrocytes, and the basement membrane (195). Glucose transporters GLUT1 and GLUT3 are major GLUTs required for neuronal and glial glucose transport via the BBB (401, 402). GLUT1 is encoded by the Slc2a1 gene, and GLUT1 at the BBB mediates glucose transport to the brain. Decreased GLUT1 levels were found in the AD brain, and overexpression of GLUT1 was shown to ameliorate neuronal activity and metabolism, alleviate cognitive dysfunction, and increase lifespans in an AD mouse model and a Drosophila model (294, 427). Crossing transgenic AD mice with GLUT1-deficient mice worsens BBB function and cognitive function (427). Two isoforms of GLUT1 exist: a 55 kD isoform in brain endothelial cells that make up the BBB and a 45 kD isoform in astrocytic endfeet. The capacity of GLUT1 in microvascular membranes of the BBB is dramatically higher than that in astrocytes (364). Astrocytes are the key cellular support for BBB integrity, and GLUT1 deficiency in endothelia, but not in astrocytes, promotes BBB breakdown (427). GLUT3, mainly expressed in axons and dendrites, is expressed at lower levels in AD patients than in nonaffected individuals and is associated with AD pathology (365). Activation of the cysteine protease Calpain I induces GLUT3 proteolysis and inhibits glucose uptake and O-GlcNAcylation in the AD brain (135).
We and others have shown that fluctuations in glucose disturb metabolic sensors, such as adenosine monophosphate-activated protein kinase (AMPK)-mediated energy metabolism and protein kinase B (Akt, also known as PKB)-mediated insulin signaling in both neurons and peripheral organs, which has been identified to trigger GLUTs to increase glucose metabolism and reduce mTOR to increase autophagy, leading to energy metabolic impairment, cognitive decline, and AD pathology (9, 60, 203, 308). Glucose metabolic dysfunction leads to advanced glycation end-product (AGE) production. AGEs represent glycated proteins or lipids that play key roles in aging, diabetes, and AD. In patients with AD, AGEs are mostly present in intracellular NFTs. AGEs also affect neurons via the receptor for advanced glycation end products (RAGE), which is an important mediator of Aβ translocation and Tau hyperphosphorylation via Akt inhibition and subsequent GSK-3β activation (224). Glucose homeostasis can be regulated by the peroxisome proliferator–activated receptor γ (PPAR-γ) coactivator 1α (PGC-1α), a key molecule for mitochondrial biogenesis, activating gluconeogenic metabolic pathways to attenuate high glucose-induced amyloidogenic processing and promote nonamyloidogenic processing by α-secretase through the transcription factor forkhead box O3a (FoxO3a) to influence Aβ pathology in AD (318). The intracerebroventricular injection of Aβ oligomers was shown to trigger peripheral glucose intolerance in transgenic AD mice, whereas the systemic injection of Aβ oligomers failed, indicating that Aβ oligomers in the brain control peripheral glucose homeostasis (61).
The apolipoprotein E4 (ApoE4) genotype is associated with defective glucose utilization in the brains of AD patients (329). A reduced cerebral metabolic rate of glucose utilization is observed in carriers of ApoE4 with AD. Accordingly, high glucose levels were found to be a risk factor for dementia in individuals with the ApoE4 genotype, even among individuals without diabetes (69). In the AD pathological condition, ApoE4 protein fragments escape the normal secretory pathway and enter the mitochondria to bind to the F1-ATPase subunit, thereby reducing energy production. Further, carriers of PS1 gene mutations with FAD have a significantly reduced cerebral glucose metabolic rate, which is associated with a high risk of AD (281).
This evidence detailed earlier indicates that a decrease in glucose metabolism is an early event in the pathological process of AD. Thus, therapy to improve glucose utilization and metabolism may be a promising approach for the treatment of AD.
B. Insulin resistance
Insulin is the systematic hormone regulator that participates in metabolic regulation in many tissues. Rising glucose levels throughout the body trigger insulin release to regulate glucose levels in normal homeostasis. The pathological accumulation of glucose in the blood disturbs insulin actions. Insulin in the brain predominantly crosses the BBB from the periphery, and it functions through the tyrosine kinase pathway in neurons and the periphery. Under normal conditions, insulin binds to insulin receptor (IR), followed by the phosphatidylinositol 3-kinase (PI3K)-mediated phosphorylation and activation of Akt, leading to the phosphorylation and inactivation of GSK3 (43).
Insulin in the brain plays a vital role in learning, memory, neurite growth, and development; facilitates glucose metabolism; and affects tau protein and Aβ processing (58, 67, 328). An in vivo magnetic resonance imaging study showed that insulin could improve neuronal activity and that increased peripheral insulin levels were associated with brain atrophy and cognition in AD (38). For example, impaired insulin signaling leads to a decrease in PI3K activity, thereby reducing Akt activity, which is required for neuronal survival, plasticity, and metabolism, and further increasing GSK3α/β activity promotes Tau phosphorylation and Aβ accumulation (372). In patients with AD, the brain levels of insulin, insulin-like growth factor I (IGF-I), IGF-II, and IGF-I receptor are reduced by nearly 80% compared with those in age-matched controls (372). In the APP/PS transgenic AD mouse model, the disturbed vessel homeostasis may be due to an imbalance in IGF-I and AMPK angiogenic crosstalk, which is reflected by the high IGF-I receptor and p-AMPK levels, impinging cognitive performance in turn (243). Some evidence exists demonstrating that IGF-I modulates the development of AD in two steps. The first step is the reduction of clearance through IGF-I-modulated carrier proteins, leading to Aβ accumulation. The second step is that IGF-I dysfunction by amyloidosis disturbs the MAPK and PI3K/Akt signaling pathways, leading to reduced Aβ trafficking, reduced glucose uptake, increased cell death, and increased Tau hyperphosphorylation. The evidence was not strong enough to support the therapeutic significance of IGF-I for AD (45). Brain resistance to insulin or IGF-I accounts for neuronal tangle formation and amyloidosis in AD. Insulin and IGF-I are potent neuroprotective factors. AD patients show resistance to IGF-I at the BBB. Insulin/IGF-I, on the one hand, stimulates neuronal Aβ release. On the other hand, insulin competes with IDE to contribute to extraneuronal Aβ accumulation (46). Abnormal insulin levels in AD patients repress protein phospholipase 2A (PP2A), which is the phosphatase responsible for Tau dephosphorylation. The intraneuronal accumulation of insulin depends on the hyperphosphorylation of Tau, which induces insulin resistance to progress to tauopathy in AD (338). Intranasal insulin therapy improves memory and cognition and is associated with Aβ42 and Tau/Aβ42 levels in the cerebrospinal fluid (CSF) of AD, especially in early AD patients, supporting the pharmacologic enrichment of insulin signaling for AD therapy (67).
Insulin resistance is characterized by glucose intolerance and disturbed insulin signaling. Insulin resistance has been confirmed in not only the peripheral tissues of AD patients but also in their brains (385), suggesting that intracerebral insulin signaling impairment may lead to cognitive impairment and that intracerebral insulin resistance may also serve as a biomarker for the risk of neurodegenerative diseases. Insulin resistance appears to be the shared feature of type 2 diabetes and AD, and AD is also known as type 3 diabetes (2). Peripheral insulin resistance is associated with decreased glucose metabolism in the brain, which, in turn, impairs cognitive function (426). The IDE degrades not only insulin but also Aβ. Intracerebral insulin resistance affects the balance of Aβ production and degradation by decreasing IDE (104). Genetic studies in Goto-Kakizaki (GK) rats have shown that IDE mutations impair the degradation of Aβ and induce diabetes (105). IDE expression is much lower in the hippocampi of AD patients carrying APOE4 than in noncarriers (95). ApoE4 is implicated in the pathways of insulin signaling by their shared mechanism. However, how ApoE4 elicits insulin resistance has not been determined. Possible mechanisms by which ApoE4 elicits insulin resistance include mediating cerebral blood volume and postprandial responses (185), trapping IR in the endosomes (468), and altering DNA hydroxymethylation and metabolic pathways of purine metabolism, glutamate metabolism, and the pentose phosphate pathway (184). Taken together, these studies suggest that IDE and ApoE4 may be the possible link between type 2 diabetes and AD.
As the insulin signaling pathway plays an important regulatory role in both diabetes and AD, insulin resistance may be the key bridge to maintaining crosstalk between peripheral tissues and the brain and may be the first step in the association between AD and diabetes (112). Notably, the antidiabetic drugs rosiglitazone and pioglitazone have been shown to reverse cognitive decline in patients with early AD by exerting their protective effects on insulin signaling (333, 424). Another antidiabetic drug, exendin-4, which is a stimulator of glucagon-like peptide 1 (GLP-1) receptors, reverses the insulin resistance induced by Aβ oligomers in the hippocampi of AD patients and improves behavioral cognition (33). Therefore, understanding the connection and underlying mechanism linking diabetes and AD may implicate novel and effective therapeutic targets for AD.
C. Glycosylation
Impaired glucose metabolism between neurons leads to a disturbance in glycosylation. Glucose metabolism is also involved in the post-translational modification of proteins in the hexosamine synthesis pathway to produce O-N-acetylglucosamine (O-GlcNAc). Two highly conserved enzymes mediate glycosylation, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). OGT mediates the addition of O-GlcNAc to serine and/or threonine residues, whereas OGA antagonizes this process and mediates the cleavage of O-GlcNAc. Our studies suggest that enhanced O-GlcNAcylation protects against oxidative stress and promotes mitochondrial homeostasis in aging mice, which provides a possible link between mitochondrial metabolism and AD (466, 467).
Both APP and Tau could be modified by O-GlcNAc modification. Genetic loss of OGT in the mouse forebrain leads to progressive neurodegeneration (408). In AD, deficiencies in GLUT1, GLUT3, and other possible molecules impair glucose metabolism, decrease hexosamine biosynthesis flux, and, subsequently, reduce the production of UDP-GlcNAc. The declined UDP-GlcNAc production decreases the O-GlcNAcylation of Tau. The O-GlcNAcylation of Tau inhibits the binding between Tau and microtubules to promote Tau misfolding, which is most likely mediated by CDK5 and GSK-3β (228). The inhibition of OGA increases the O-GlcNAcylation of Tau and decreases NFT formation in the brains and CSF of AD mice, consequently reducing neuron loss (153, 454). O-GlcNAcylation and phosphorylation antagonize each other to modify Tau, and reduced levels of O-GlcNAcylation on Tau lead to further hyperphosphorylation, promoting NFTs formation (229, 230). Moreover, reduced post-translational modification of O-GlcNAc regulates mitochondrial function, motility, and distribution, suggesting a link between mitochondrial dysfunction and AD (314). Another type of Tau glycosylation, N-glycosylation, in the AD brain stabilizes the modified Tau structure by facilitating the hyperphosphorylation of Tau by kinases and promoting the suppression of Tau dephosphorylation via phosphatases (413).
Protein kinase A (PKA), a cyclic AMP-dependent kinase, plays a pivotal role in cell metabolism, learning, and memory by activating CREB through phosphorylation in AD. In addition to phosphorylation, the PKA catalytic subunits could also be post-translationally modified by O-GlcNAc. The downregulation of O-GlcNAc thereby regulates PKA-CREB signaling, which, in turn, leads to learning and memory deficits in AD (441).
The studies described earlier, including those reporting glucose uptake deficiency, insulin resistance, and glycosylation, indicate that impaired glucose metabolism might be the cause rather than the consequence of AD (Fig. 3).
III. Lipid Metabolism in AD
The adult brain is highly enriched in lipids, which account for ∼20%–25% of the dry weight. These lipids not only constitute the structure of membranes but also provide the infrastructure and function for the membrane system (428). Basically, lipids participate in cell membrane formation, cellular transportation, energy storage, and signal transduction (428). In the liver, insulin signaling controls lipid metabolic events, including stimulating lipogenesis and reducing lipolysis.
Alois Alzheimer first recorded remarkable lipid granule accumulation in the glia of his patient Auguste Deter (108). Altered lipid composition in the cell membranes leads to redistribution of proteins linked to the β-amyloidogenic pathway of lipid rafts in AD (80). The cleavage of APP by the β-secretase protein BACE1 and γ-secretase proteins, including PSEN1, PSEN2, aph-1, pen-2, and nicastrin, occurs in lipid rafts and affects lipid metabolic enzymes, thereby altering subcellular trafficking. Frontal cortex lipid rafts are greatly altered in AD brains from the initial neuropathologic stages of AD, but the underlying mechanism is unknown. However, the changes in lipid rafts were shown to affect both lipid classes and fatty acids, increase the membrane order and viscosity, providing a favorable lipid raft microenvironment for the APP/BACE1 interaction and the amyloidogenic pathway and strengthening AD as a result (100). An age-dependent modification was found in the lipid raft microdomains, thereby affecting signal transduction and protein
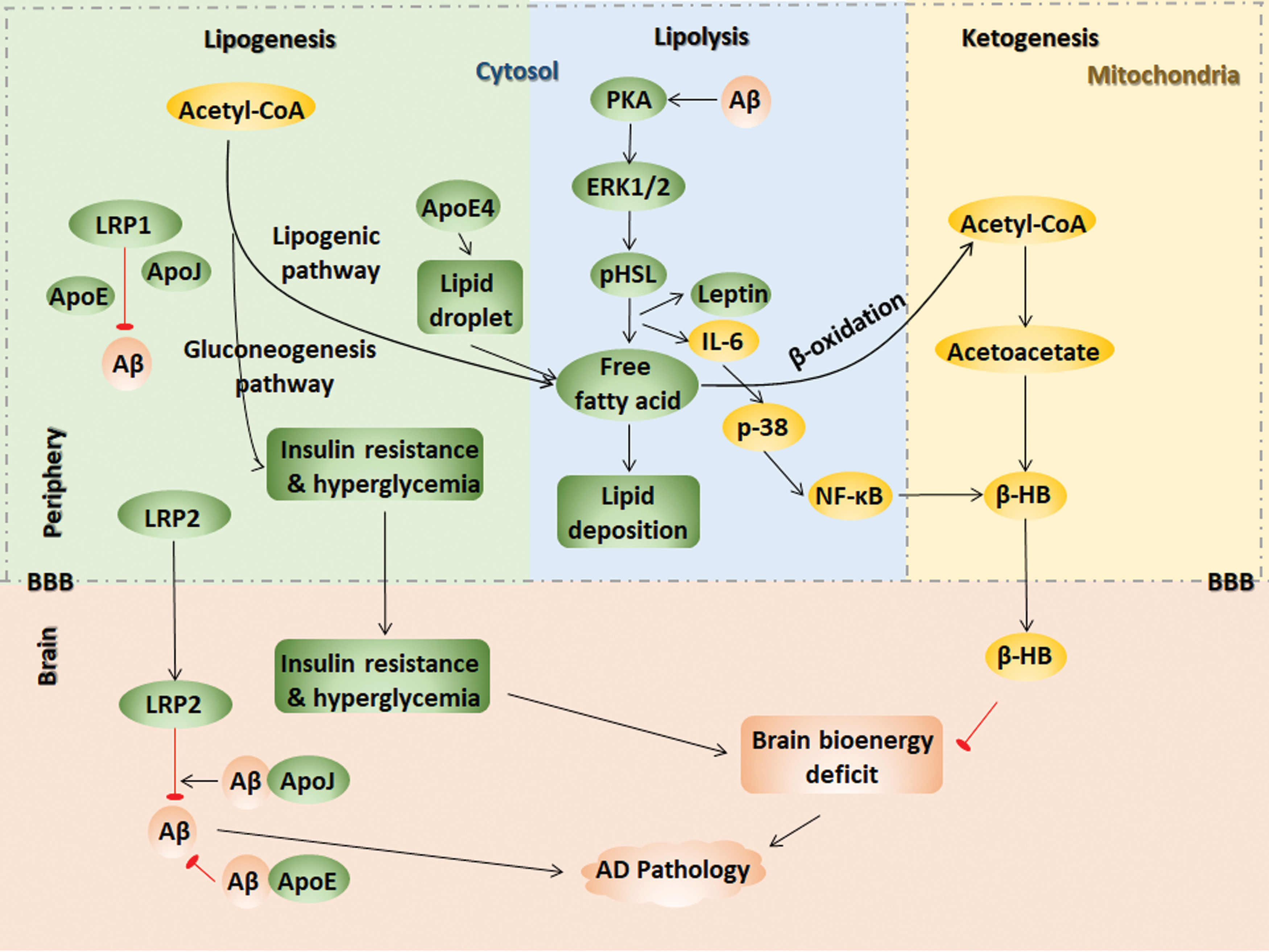
A. Lipid dysregulation as an early signal in AD
Lipid peroxidation occurs as an early event in the progression of AD (265), suggesting that the early prevention of lipid peroxidation products in the presymptomatic phase of AD or before mild cognitive impairment (MCI) could be an effective way to prevent AD. Indicators of lipid peroxidation include thiobarbituric acid reactive substances, Notably, 4-hydroxynonenal and acrolein are elevated in the AD brain (244). Ten lipid metabolites derived from lipidomic approaches have been validated as biomarkers of preclinical AD in blood that could distinguish cognitively normal individuals from cognitively impaired individuals with an accuracy above 90% (264). The biomarkers for AD will be summarized in part V, titled “Mitochondria-centered Metabolic Markers in the Early Diagnosis of AD.”
Lipid-based supplementation has the potential to reduce the risk of AD. Docosahexaenoic acid (DHA), which is a prominent polyunsaturated ω-3 fatty acid efficiently transported across the BBB for the formation of a neuronal membrane structure, could increase the α-secretase cleavage of nonamyloidogenic processes and decrease the amyloidogenic processing activities of β-secretase and γ-secretase. An increased DHA level was found in the early stage of AD by the neurolipidomic study in human AD cases and the DHA levels are progressively manifested as the trilogy of adaptation-overload-failure, leading cells to neurodegeneration (289). DHA serves as an endogenous ligand for retinoic acid receptor and retinoid x receptor, which are responsible for memory function and neurogenesis. DHA activates Ca2+/calmodulin-dependent protein kinase II (CaMKII), which maintains long-term potentiation in the hippocampus, thereby mediating learning and memory functions. DHA increases brain-derived neurotrophic factor (BDNF) in the hippocampus by activating PI3K/Akt signaling to protect synaptic plasticity and nerve cell survival (5). Inflammatory markers contributing to AD can also be reduced by DHA supplementation. A deficiency of DHA in neurons leads to neurodegeneration and cognitive dysfunction. Dietary supplementation with DHA in early stage reverses Aβ oligomerization in the animal model (389); however, this strategy has failed in several clinical trials (109, 322).
DHA intervention has beneficial effects only in the early stage of AD. DHA was shown to enhance the binding between Aβ42 and lipid rafts to promote Aβ degradation. IDE secretion and activity can be increased by DHA, thereby ameliorating AD pathology by increasing Aβ turnover (132).
ApoE4 is a major risk factor for AD and is associated with a high cholesterol level, as ApoE4 delivers cholesterol from astrocytes to neurons, indicating disrupted lipid metabolism in AD (330). ApoE and cholesterol colocalize with Aβ in the brains of transgenic AD mice and AD patients, and cholesterol is reportedly an early risk factor for AD (215, 302). ApoE binds to Aβ in a lipid-dependent manner, and Aβ deposition increases in ApoE mice (212). The lipid-binding region on ApoE4 mediates the neuro-mitochondrial toxicity of ApoE4 (50). The ApoE4 genotype is established in carriers ∼20–39 years of age, and these carriers have functional brain abnormalities as relatively young adults, several decades before the onset of dementia (330). Before the onset of AD, ApoE4 carriers supplemented with high-dose DHA showed a reduced risk of developing AD symptoms (450). Recent evidence suggests that single nucleotide polymorphisms in the genes St-AT-related lipid transfer domain 6 (STARD6) and near enoyl CoA-hydratase domain containing 3 (ECHDC3) are responsible for the lipid level and metabolism in AD, and variants in or near the STARD6 and ECHDC3 genes, especially ApoE4 carriers, are risk factors associated with AD (452). The role of ApoE4 in lipid metabolism and AD indicates a lipid-driven mechanism of AD pathogenesis.
B. Lipogenesis in AD
Lipogenesis is the biochemical process that converts acetyl CoA into fatty acids, which primarily occurs in the liver. The increased lipoprotein secretion in the periphery aggravates the Aβ burden in the AD brain. Lipoprotein receptor–related protein 2 (LRP2) clears Aβ40 rapidly across the BBB. Other lipid proteins, such as ApoE and ApoJ, mediate Aβ crosstalk between the brain and periphery. High ApoE-Aβ binding activity decreases Aβ brain efflux, whereas ApoJ-Aβ binding increases LRP2-mediated efflux. The poorly lipidated ApoE increases the Aβ deposition. Alternatively, the lipidation status of peripheral Aβ may also affect its clearance, which, in turn, might also be mediated by ApoE, ApoJ, and LRP1 (66). The ApoE4-driven disruption in fatty acid metabolism is proposed to be factor underlying why ApoE4 carriers are at a high risk for developing AD. ApoE4 in astrocyte exhibits an increase in the number of lipid droplet, which serves to sequester fatty acids depending on the saturation status of the fatty acid. The expression of the lipid droplet marker PLIN2 and the oxygen consumption rate from endogenous fatty acid oxidation are also enhanced in E4 astrocytes (102). Proteins involved in mitochondrial fatty acid oxidation, including PTEN-induced putative kinase 1 (PINK1) and PGC-1α, were shown to be cooperatively downregulated in AD and diabetic patient brains, indicating a role of mitochondria in linking AD and diabetes (57).
A high-fat diet (HFD) increases the risk of AD, a phenomenon that is related to dysfunctional lipid metabolism. Abnormal lipid accumulation leads to insulin resistance and vice versa. Depleting the negative insulin signaling regulator PTEN results in a fatty liver and induces lipogenesis (421). Our previous study compared C57 mice with APP/PS1 transgenic AD mice, showing that the HFD induced more severe body weight gain, hyperglycemia, and hepatic insulin resistance in AD mice than in C57 mice, potentially because an inactivated lipogenic pathway by inflammatory factors initiates the substrate flux for glucose production, leading to more severe hyperglycemia in AD mice (273, 387). Hepatic insulin resistance in diabetic db/db mice was intensified by hybridization with AD mice (384). Early inflammation induced by an HFD in APP/PS1 mice resulted in less hepatic steatosis than that in C57 mice that were also fed an HFD (387).
C. Lipolysis in AD
Under energy-demanding conditions, adipose tissue begins to undergo lipolysis, which activates the PKA and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathways that phosphorylate hormone-sensitive lipase, resulting in free fatty acid release (96). The activity of lipolytic enzymes, including monoacylglycerol and diacylglycerol, is stimulated in plasma membrane and synaptosomal plasma membrane fractions obtained from various regions of normal and AD brains (103).
APP can be processed to produce the Aβ peptide in adipose tissue, wherein the Aβ peptide stimulates lipolysis and adipokine secretion via PKA and ERK1/2-dependent pathways and stimulates the secretion of leptin and IL-6, resulting in the release of free fatty acids and proinflammatory adipokines (407). Elevated fatty acid levels trigger lipid deposition and insulin resistance, demonstrating a close relationship between energy metabolism and AD.
D. Ketogenesis in AD
In the ketogenesis pathway, acetoacetyl-CoA is generated by acetyl-CoA, which is mediated by thiolase, and β-hydroxy-β-methylglutaryl-CoA (HMG-CoA) is then generated by another acetyl-CoA that is mediated by HMG-CoA synthase. HMG-CoA generates acetyl-CoA, and acetoacetate is mediated by HMG-CoA lyase. The final product D-β-hydroxybutyrate (β-HB) derives from the reaction between acetoacetate and NADH, which is mediated by β-hydroxybutyrate dehydrogenase 1 (BHD1) (317). Therefore, acetoacetate and β-HB are two main ketone bodies. Ketone bodies are produced by the liver from fatty acids. Fatty acids, responsible for providing cellular energy to peripheral tissues, are oxidized in the liver, producing large amounts of acetyl-CoA, surpassing the ability of TCA to synthesize ketone bodies in liver mitochondria. Then, ketone bodies are transported to extrahepatic tissue through the blood for oxidation and the subsequent release of acetyl-CoA, which enters the TCA cycle, thereby generating energy (119, 205). Ketone bodies decrease the need for glycolysis and the mitochondrial NAD+/NADH ratio. Using label-free quantitative proteomics, SIRT3 was found to target each complex in the TCA cycle, as does every enzymatic step from fatty acids to ketone body β-HB. The mitochondrial deacetylase SIRT3 regulates acetylation via ketogenesis during trafficking to the mitochondrial matrix, maintaining the balance of energy homeostasis (323).
Although the brain mainly uses glucose as the sole energy substrate under normal conditions, under hypometabolic conditions in AD, ketone bodies, such as acetoacetate or β-HB, may be used as an alternative energy substrate for glucose. β-HB is the most abundant ketone, constituting ∼70% of ketone bodies primarily formed in liver mitochondria and then transported by blood to other tissues. β-HB-enriched ketogenic diets could be helpful for AD intervention, which will be discussed in part VI, titled “Mitochondrial Metabolic Dysfunction as Target for AD Prevention.”
In the AD mouse model (APP/PS1/Tau, 3 × Tg), the enzymes responsible for ketone body metabolism were significantly increased (447). In early AD, Aβ invades the mitochondria, initiating their production of superoxide radicals and the conversion of these molecules into hydrogen peroxide. These events lead to oxidative stress and activation of the C-Jun NH2-terminal kinase (JNK)-MAPK pathway and nuclear factor-kB (NF-κB), resulting in the release of proinflammatory interleukins and cytokines (8, 321). We recently reported that the hepatic proinflammatory cytokine IL-6 activates 3-hydroxy-3-methylglutaryl-CoA synthase 2, which then functions as a critical regulator via p38/NF-κB p65 signaling in inflammation-induced ketogenesis in the early stage of AD (359). This finding implies that as a self-adaptation mechanism, cerebral glucose metabolic dysfunction in early AD increases the ketone body supply from the liver to the brain, resulting in a shift to ketone body utilization (359). During the early stage of AD, the constant activation of ketogenesis could maintain mitochondrial function to prevent and/or delay further development of the disease. Enriched ketogenesis in the liver and ketogenic diets may help prevent or delay a bioenergetic decline in the early AD brain (449). It remains unclear which event in AD triggers a hepatic inflammatory response, such as IL-6 accumulation.
IV. Mitochondria at the Metabolic Core in AD
A. Mitochondrial dysfunction precedes metabolic disorder in AD
Mitochondria are crucial organelles for energy metabolism and signal cascade control. Metabolism of glucose, lipogenesis, and ketogenesis occurs in mitochondria, and mitochondrial dysfunction tends to be the cause or effect of metabolic dysfunction. The abnormalities in AD, including defective glucose metabolism and energy metabolism, suggest that mitochondrial dysfunction is involved in AD pathology and is an early prominent feature. Studies by ourselves and others have demonstrated reduced activities of enzymes mediating mitochondrial respiration and the TCA cycle in the AD brain and periphery, including succinate dehydrogenase (SDH) or complex II, COX or complex IV, pyruvate dehydrogenase complex (PDC), and α-ketoglutarate dehydrogenase complex (KGDH), confirming the link between energy metabolism dysfunction and mitochondrial dysfunction in AD (242, 377). Genetic mutations have been identified in mitochondrial-encoded cytochrome c oxidase subunits I, II, and III (COX-1, 2, and 3) in AD.
Mitochondria are very sensitive to extracellular glucose changes in both the brain and periphery, and changes in mitochondrial function precede AMPK/Akt signal fluctuations. In addition, studies by ourselves and others have also demonstrated that AMPK inactivation leads to PI3K-mediated insulin resistance and PGC-1α-mediated mitochondrial loss in both peripheral organs and the brain, resulting in disorders of glucose metabolism and neuronal damage that are ameliorated by normal glucose metabolism and mitochondrial activity, providing new insights into the interrelationship between hyperglycemia and cognitive impairment (140, 308). Embryonic neurons derived from the AD mouse hippocampus exhibited a significant decrease in mitochondrial respiration and an increase in glycolysis (448).
In addition, mitochondrial dysfunction is greater in ApoE4 carriers than in AD patients who do not carry ApoE4 (123). ApoE4 is also associated with decreased cerebral glucose metabolism in AD, indicating that a combination of cerebral metabolic rates and genetic risk factors offers a strategy for preclinical AD diagnoses (368). The level of α-ketoglutarate dehydrogenase, a mitochondrial enzyme in the TCA cycle, is decreased in the AD patient brain, particularly in those of ApoE4 carriers (123). The truncated ApoE4 elicits mitochondrial function and integrity, indicating that blockage of the interaction between mitochondria and ApoE4 is a potential treatment strategy (50).
As mitochondria are the site of the TCA cycle and respiration, mitochondrial dysfunction is a major intracellular event that contributes to AD pathology. This event occurs early in AD progression, even before the onset of Aβ pathology, making it critical to develop strategies aimed at mitochondrial function (257, 259, 327).
B. Mitochondria mediate pathological impairment during AD progression
Decreases in respiratory capacity, increases in mitochondrial fragmentation, and fractures in the mitochondrial cristae structure occurs in the AD brain, and the abnormalities in mitochondria appear before pathological Aβ plaque deposition (371, 438). Aβ accumulates exclusively in mitochondria. The mitochondrial membrane 40 import channel and mitochondrial membrane 23 channel were shown to bind to Aβ and cause mitochondrial dysfunction in the human AD brain but not in the age-matched controls (78). Aβ binds exclusively to ADH. The Aβ and ABAD complex promotes free-radical generation and mitochondrial dysfunction, interacts with mitochondrial components, impairs ATP production, and induces oxidative stress in AD, which could be the underlying mechanism of Aβ toxicity in mitochondria (248).
Cyclophilin D (CypD), an integral part of the mPTP, directly interacts with Aβ in mitochondria, promotes ROS generation, and mediates mPTP formation, resulting in mitochondrial stress-induced neuronal cell death in AD mice and patients (258). Moreover, the deficiency of CypD improves learning and memory in AD mice (84, 258). As the direct site of Aβ accumulation in AD neurons, mitochondria are responsible for ROS generation and oxidative damage. The Aβ peptide produced by APP splicing binds to ferroheme, resulting in the decreased activity of complex IV. Using the complex IV inhibitor NaN3, the accumulation of APP and the production of amyloid C-terminal fragments are greatly enhanced. In addition, the Aβ peptide stimulates GSK3β activity, phosphorylates pyruvate dehydrogenase, and finally inhibits energy metabolism. Hydrogen peroxide is increased and cytochrome oxidase activity is decreased before the appearance of Aβ plaques, suggesting that early mitochondria-targeted therapeutic interventions might be an effective strategy in delaying AD progression and treatment (257). Mitochondrial dysfunction also induces synaptic damage, impairs neurotransmission, and causes AD-related cognitive decline (225, 326, 348).
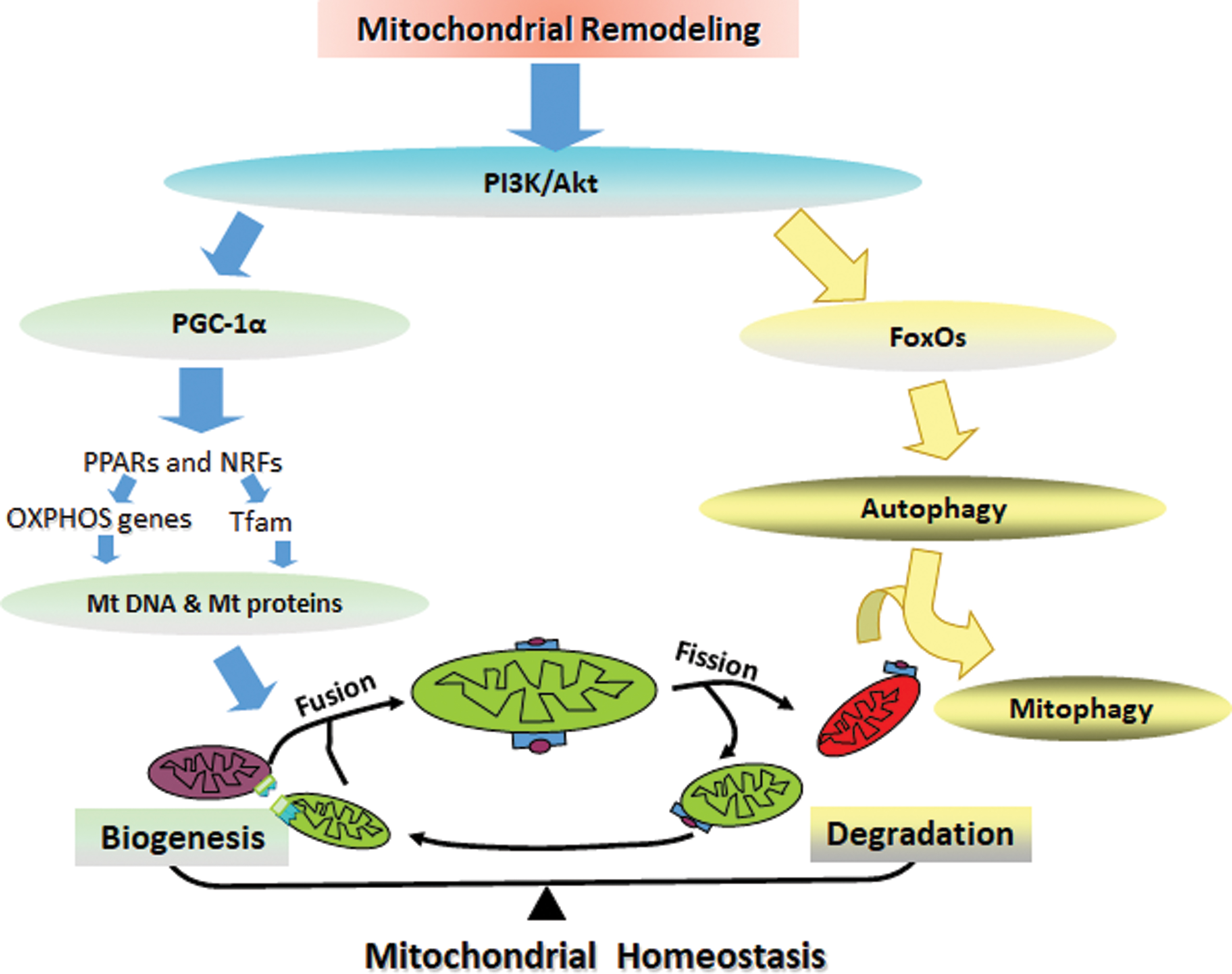
C. Mitochondrial remodeling in AD
Mitochondria are highly dynamic organelles that continuously, morphologically, and functionally undergo two opposing processes, namely, fission and fusion (15). Mitochondrial fission is controlled by the GTPase protein dynamin-related protein 1 (Drp1). Drp1 interacts with its receptors mitochondrial fission 1 (Fis1) and mitochondrial fission factor to stimulate mitochondrial fission (49). On the other hand, mitochondrial fusion is controlled by other GTPase proteins, optic dominant atrophy 1 (OPA1), mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) (48). It has been shown that excessive fission leads to mitochondrial fragmentation and the imbalance between fission and fusion compromises mitochondrial respiration function and ATP production (433). Besides, impaired mitophagy disturbs the clearance of defective mitochondria, which leads to the production of ROS and causes oxidative stress (198).
The levels of proteins governing mitochondrial fission, including Drp1 and Fis1, and mitochondrial fusion, including OPA1, Mfn1, and Mfn2, are significantly altered in AD mice and patients (258, 419). Besides the change in mitochondrial morphology, the distribution of mitochondria in neuron and axis is altered in AD patients (419). The distribution of mitochondria in neuron is a very complex topic. Inhibition of Drp1 in AD mouse neurons prevents cognitive decline, mitochondrial fragmentation, lipid peroxidation, BACE1 expression, and Aβ deposition (19). In addition to Aβ-related AD pathology, Drp1 reduction also protects against hyperphosphorylated Tau and phosphorylated Tau-induced mitochondrial dysfunction in AD (189). The S-nitrosylation of Drp1 at Cys644 causes Aβ-related mitochondrial fission and neuronal injury, leading to mitochondrial fragmentation in human AD neurons (56). Treatment of mitochondrial division inhibitor 1 (mdivi-1), a mitochondrial fission inhibitor, markedly improves mitochondrial dynamics and cognitive function in AD mouse model CRND8 early before the accumulation of amyloid pathology (417).
Similarly, manipulation of mitochondrial fusion also plays a role in AD. For example, OPA1 overexpression alleviates mitochondrial ROS overproduction in the AD brain (419). PS2 mutation in FAD promotes mitochondria and endoplasmic reticulum (ER) coupling in the presence of Mfn2. The PS2-Mfn2 binding is more efficient in mitochondria-associated membranes (107). Further, the activity of PS2 is affected by Mfn2 knockdown-increased mitochondria-ER coupling, leading to the decrease of Aβ accumulation (216).
Mitochondrial metabolic homeostasis is maintained by the balance between mitochondrial biogenesis and degradation. PGC-1α is a transcriptional coactivator that regulates mitochondrial biogenesis through the downstream factors peroxisome proliferator-activated receptors (PPARs), nuclear respiratory factor 1 (NRF1), and nuclear respiratory factor 2 (NRF2), which govern the nuclear genes encoding mitochondrial proteins, and mitochondrial transcription factor A (TFAM), which initiates mtDNA transcription and replication (434). The evolutionarily conserved form of degradation is autophagy, which is responsible for the degradation of accumulated and cytotoxic proteins. Autophagy facilitated in mitochondria is called mitophagy, and energy deficits initiate autophagic pathways. The molecular interaction between autophagy and AD pathology remains controversial. For example, the triggered autophagy pathway increases the degradation of the Tau protein and excessive Aβ accumulation in early AD. However, autophagosome accumulation exacerbates AD pathogenesis (140). More in-depth studies are needed to clarify the mechanisms by which molecules participate in autophagy pathway functions through AD progression, and the function of different molecules may be stage dependent in AD progression.
FOXO transcription factors are essential for memory and neurodegeneration, for the manipulation of glucose and lipid metabolism, and for autophagy in response to energy deprivation, providing a potential link between AD and energy deficits. FoxO3a, which induces LC3b, Beclin1, and Bnip3 for autophagy and PINK1 for mitophagy, is an important transcription factor for mitochondrial gene expression and ROS production. We found that AMPK, as an energy sensor, directly phosphorylates FoxO3 under energy deficient conditions (238). The decreases in FoxOs expression in the brain insulin resistance state are related to toxic Aβ formation and Tau hyperphosphorylation in diabetic transgenic mice and monkeys (340). However, other studies have demonstrated that the consecutive activation of FoxO proteins induces neuronal loss, Aβ deposition, and Tau hyperphosphorylation (263). In addition, in patients with AD, impaired glucose metabolism decreases PGC-1α levels and then increases FoxO3a levels, inhibiting α-secretase in the nonamyloidogenic processing of APP, thereby promoting amyloidogenic processing to produce excessive Aβ (318, 354).
Although more investigations are needed to determine the mechanism by which the dynamic state of mitochondria eliminates or aggravates AD, the current findings suggest that the steady state of mitochondrial remodeling in AD supports its early and fundamental role in AD-related pathological and cognitive impairments, suggesting abnormal mitochondrial remodeling as a possible therapeutic target for halting AD (Fig. 5). Nutrients targeting mitochondria-related signals may be a new strategy for AD prevention and treatment and will be discussed in part VI-A.
D. Mitochondria-induced inflammation and oxidative stress during the pathogenesis of AD
The brain is especially vulnerable to oxidative stress because of its high oxygen needs. Oxidative stress arises from excess ROS, most of which are generated from mitochondria by electron leakage from the OXPHOS and TCA cycle and are also generated from pathological AD conditions (110, 404, 420). Mitochondrial dysfunction in neurons leads to insufficient energy supply and the release of a large amount of ROS, which induces oxidative stress and the imbalance of calcium regulation, ultimately triggering neuronal apoptosis. Brain oxidative stress linked to mitochondrial dysfunction arises in early AD and contributes to AD pathogenesis, and the extent of oxidative damage is much higher in mtDNA than in nuclear DNA (276). In 3-month-old transgenic APP mice, mitochondria in the brain have shown decreased membrane potential, COX activity, and ATP levels. At this time, the Aβ levels in neurons are increased, whereas extracellular Aβ deposition does not yet occur, suggesting that mitochondrial dysfunction occurs earlier than Aβ pathological deposition. ROS-mediated Akt1 oxidative modification causes synaptic dysfunction, which occurs very early in the AD process (4).
Mitochondria are the hub of the redox system. The overall mitochondrial redox status can be elucidated by fluorescence imaging or HPLC. Free radical oxidation of polyunsaturated fatty acids, cholesterol, and glucose causes overload of toxic products, leading to the destruction of cellular redox homeostasis (55). Nrf2, a redox-sensitive transcriptional factor, is regulated by Keap1 and controls the expression of antioxidant genes through interactions with antioxidant response element (ARE) to balance the endogenous antioxidant stress defense system. Basically, Nrf2 binds to Keap1 with the E3 ubiquitin ligase Cullin 3-RING box1 for ubiquitination, resulting in proteasomal degradation. Without Keap1's target, Nrf2 binds to ARE to activate the transcription of the phase 2 response. Nrf2 and ARE levels decrease on increased Aβ plaque deposition in APP/PS1 transgenic mice. The Aβ toxicity could be decreased by enhancing Nrf2-ARE activity.
In addition, the activation of Nrf2 reduces Tau phosphorylation, suggesting that the activation of an Nrf2-ARE antioxidant defense has a neuroprotective effect and may prevent or delay the pathological changes in AD. A study in another AD mouse model confirmed that oxidative damage precedes Aβ and further demonstrated that oxidative damage increases the deposition of Aβ in the mouse brain. A mitochondrial superoxide dismutase (MnSOD) deletion in APP mice significantly increased Aβ levels and amyloid deposits in the brain (39, 247, 282). These findings suggest that oxidative stress exists before the characteristic features of AD pathology, such as Aβ deposition, and promotes Aβ deposition during AD progression.
Oxidative stress triggers the JNK-MAPK signaling pathway, leading to an inflammatory response and inflammasome activation (307). Aβ increases the production of the inflammatory cytokine TNF-α, causing insulin resistance by JNK in the AD mouse hippocampus (33). Activated JNK was found in the AD brain and periphery (33). Oxidative stress promotes the transcriptional activity of FoxO, which induces JNK activation. As JNK signaling pathways affect mitochondrial function and mitochondrial bioenergetics, it is reasonable to link mitochondria, ROS, and inflammation together with AD. In AD, excessive ROS contributes to insulin resistance and inflammasome activation mostly through mitochondria (360, 474). Oxidative stress and inflammation in early AD damage the neuronal membrane potential, mtDNA, TCA cycle, and electron transport chain in mitochondria, thereby reducing ATP generation and exacerbating oxidative damage (343, 448).
In AD, increased peripheral inflammation occurs early in the MCI phase and decreases as a function of the severity of AD. The release of inflammatory cytokines and chemokines improves the generation of fatty acids, lipid peroxidation products, and ROS. Our studies identified higher hepatic and serum TNF-α and IL-6 contents in the early stage of AD in transgenic mice before the activation of these inflammatory molecules in the brain (359, 387). Inflammatory-related genes are increasingly featured in AD genetic studies, and inflammatory cytokines even play a role as peripheral biomarkers to some extent. Thus, there appears to be an early response to abnormal protein deposition that develops into immune adaptation in chronic diseases, resulting in immune hyporesponsiveness. Accordingly, the early treatment of AD with anti-inflammatory drugs at the MCI stage may be beneficial (429).
The antidiabetic drug rosiglitazone, which also has a neuroprotective effect, has been demonstrated to exert anti-inflammatory effects by decreasing NF-κB levels in neurons (231). Blunting autophagy/mitophagy leads to the generation of ROS in mitochondria, which, in turn, activates the NLRP3 inflammasome (474). Inflammasome activation contributes to the onset and progression of AD. The NLRP3 inflammasome was first identified in AD by studying the IL-1β expression induced by Aβ (148). In addition, studies crossing NLRP−/− mice with APP/PS1 mice demonstrated the role of the NLRP3 inflammasome in the pathogenesis of AD (155). Peripheral insulin resistance exacerbates brain inflammation, accelerating the Alzheimer-like pathology in APP+-ob/ob mice obtained from a cross of Alzheimer transgenic APP23 mice and diabetic ob/ob mice (384). Activating GSK3β, a Tau phosphorylation kinase, could also induce inflammation via NF-κB and lead to apoptosis (62, 277).
The RAGE acts as an inflammatory mediator as well as an oxidative stress inducer in AD. RAGE belongs to the immunoglobulin superfamily and drives AD progression by mediating Aβ clearance, formation, and accumulation; NFT accumulation and neuronal degeneration. RAGE transports Aβ from the periphery to the brain, and transportation at the BBB disrupts BBB function (412). The RAGE-Aβ interaction inhibits the nuclear translocation of NF-κB and stimulates the release of proinflammatory cytokines, such as TNF-α and IL-6 and ROS. The interaction between RAGE and Aβ maintains the balance of Aβ via the regulation of β- and γ-secretase activities. Changes in fatty acid composition were present in AD human cases, with an increase in RAGE leading to a lipid peroxidation-derived protein modification in AD pathogenesis (300).
Mitochondrial uncoupling proteins (UCPs) located on the inner mitochondrial membrane function as regulators of mitochondrial membrane potential and ROS, thus protecting neurons. Studies have indicated that UCP2 is a novel mast cell function regulator with potential for the treatment of mast cell-mediated allergic and inflammation responses in neurodegenerative diseases (197).
Indeed, mitochondrial dysfunction induces inflammation and oxidative stress; in contrast, inflammation and oxidative stress induce mitochondrial dysfunction. Either way, inflammation, oxidative stress, and mitochondrial dysfunction coexist and exacerbate each other; therefore, approaches targeting mitochondrial dysfunction, inflammation, oxidative stress, or any two or all of these events could be beneficial for AD prevention and treatment.
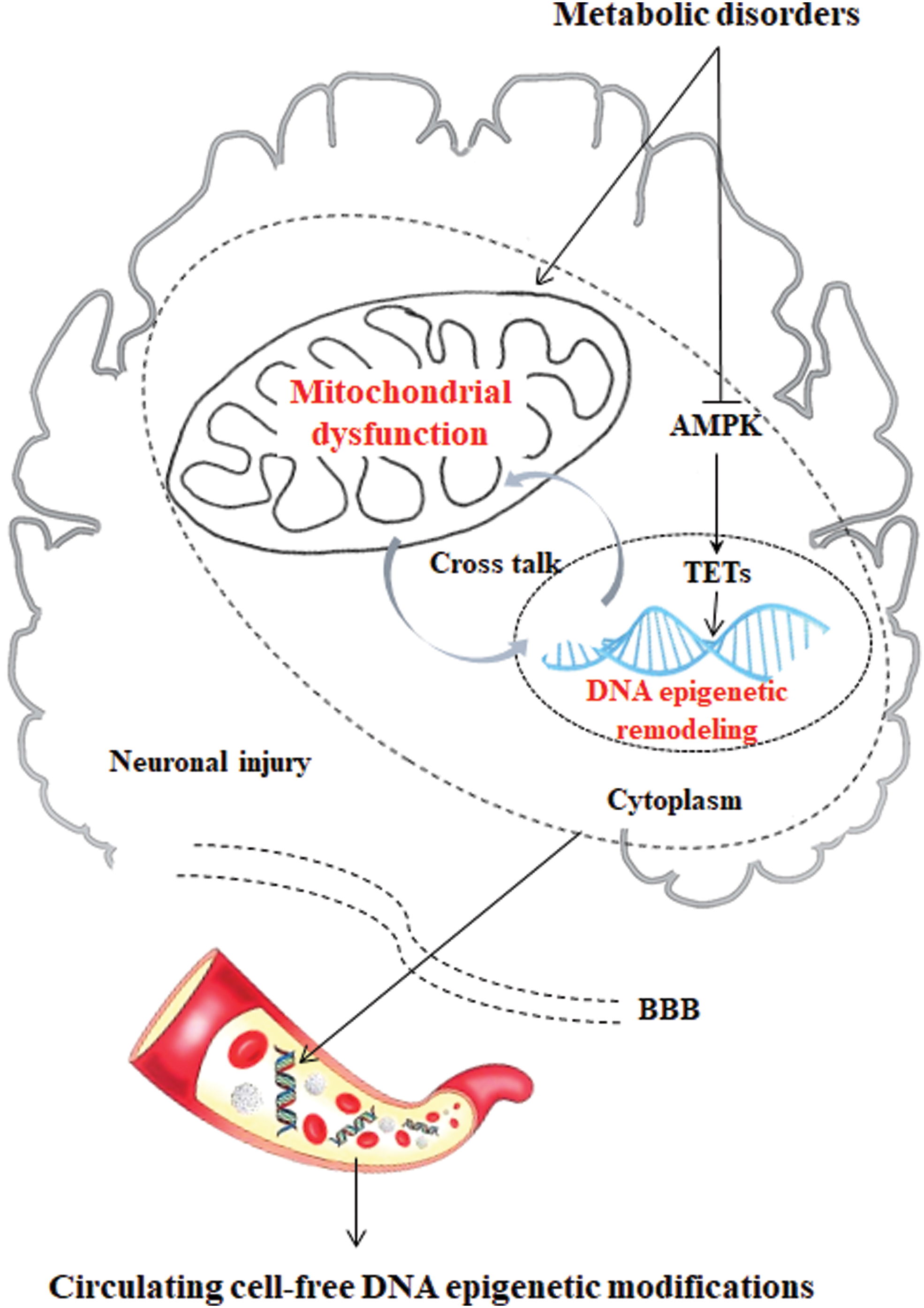
E. Mitochondrial metabolic disorder links nutritional imbalance to neural DNA epigenetic remodeling
The phenomenon of epigenetics has shown a new link between metabolic dysfunction and AD. Epigenetic changes, such as DNA methylation, have been observed in both metabolic disorders and AD. Metabolic disorders alter genome-wide DNA hydroxymethylation in two ways in association with mitochondrial dysfunction: (i) decreasing the stability of the TET2 protein by impeding AMPK phosphorylation and (ii) inhibiting ten-eleven translocation (TET) activity by increasing KGDH to competitively inhibit fumarate and succinate.
5-Hydroxymethylcytosine (5hmC) is a major and stable epigenetic marker generated from 5-methylcytosine (5mC) by the TET family of dioxygenases that is involved in a wide range of biological processes from development to various diseases (53). The epigenetic modulation that mtDNA methylation 5mC was found in the D-loop region of mtDNA in the entorhinal cortex in the human AD brain supported the epigenetic mtDNA regulation in AD human cases (30). 5hmC is regulated by multiple factors, and TETs play a central role in the generation and maintenance of 5hmC (208). TET-mediated oxidation reactions require oxygen and KGDH as substrates and Fe (II) as a cofactor to generate CO2 and succinate. As a result, 5hmC is directly affected by the availability of the substrates and cofactors (115).
Mitochondria are the regulatory centers of iron. The iron that is not used inside the cell is stored within the ferritin macromolecule to avoid the exertion of free iron toxicity in the cytosol (35). Multiple studies have shown that vitamin C can improve TET activity by improving the Fe (II) level (200). Therefore, iron homeostasis disorder induced by mitochondrial dysfunction can directly affect the 5hmC level in the genome. KGDH, as an intermediate in the TCA cycle for energy metabolism, is generated from isocitratevia isocitrate dehydrogenases, including IDH1, IDH2, and IDH3. Mutations in IDH1/IDH2 result in the simultaneous loss and gain of activities in the production of KGDH and 2-hydroxyglutarate (2-HG), respectively (208). These findings provide a biochemical basis for the hypermethylation observed in human glioma with IDH mutations and the mutually exclusive manner of IDH1/IDH2 and TET2 gene mutations in acute myeloid leukemia. In addition to IDH, two other TCA cycle genes, fumarate hydratase and SDH, are mutated in numerous human cancers, leading to the accumulation of fumarate and succinate. More importantly, 2-HG, fumarate, and succinate, which act as antagonists of KGDH, competitively inhibit the activity of TETs and the consequent alterations in genome-wide DNA hydroxymethylation (13, 143, 176, 436).
In addition to gene mutations, AMPK is impeded by high glucose levels, which results in the destabilization of TET2, followed by the dysregulation of 5hmC (135). Our recent work shows that the dysregulation of 5hmC in the CNS of mice with metabolic disorders induced by an HFD is closely related to the decreased stability of the TET2 protein and that the increased contents of TET enzyme inhibitors (fumarate and succinate) are induced by an HFD. Notably, knockdown of the TET2 gene leads to mitochondrial dysfunction and inflammation (unpublished) (Fig. 6).
V. Mitochondria-Centered Metabolic Markers in Early AD
There is an urgent need to identify alternative disease mechanisms and associated biomarkers that can help to diagnose AD in the preclinical and early clinical stages. Given that central and peripheral metabolism are remodeled in the early stage of AD and that mitochondria play a central role in the pathogenesis of AD, mitochondrial biomarkers in the CNS and periphery could be targeted for the early diagnosis of AD.
A. Biomarkers in the brain and CSF
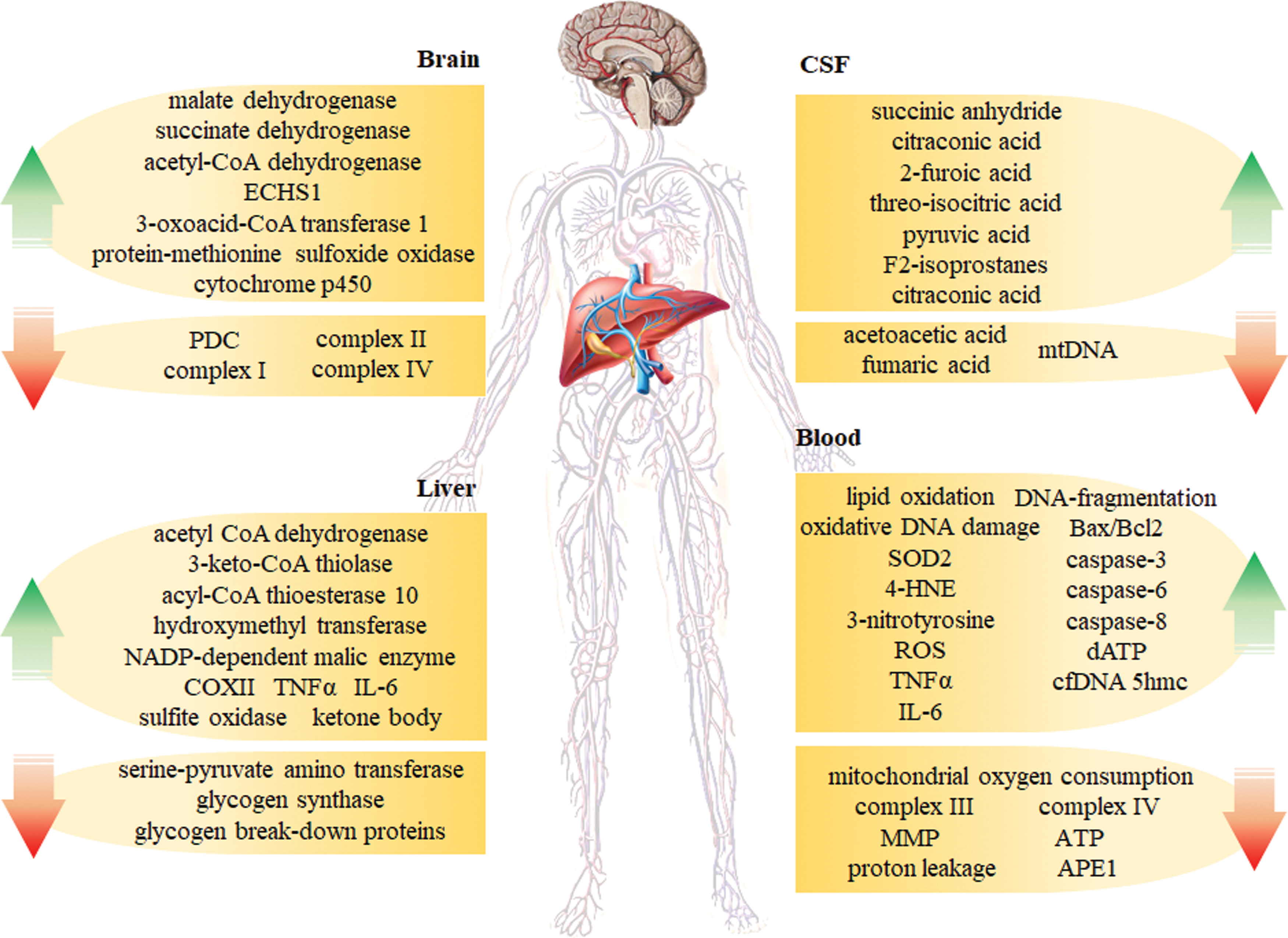
Another characteristic feature of AD is a reduced rate of brain metabolism, occurring before the development of significant amyloid plaques and NFTs (31, 166). The mitochondrial proteome in AD was analyzed by quantitative comparative proteomic profiling (59). Studies of transgenic AD mice have demonstrated that a wide variety of metabolic deficits, including the citric acid cycle, OXPHOS, pyruvate metabolism, glycolysis, oxidative stress, fatty acid oxidation, ketone body metabolism, ion transport, apoptosis, and mitochondrial protein synthesis, occur early in AD (99). COX (complex IV) of the electron transport chain shows decreased activity in both AD brains and platelets (124), as does the activity of the PDC, which connects glycolysis to the citric acid cycle in AD cortices (59, 250). Further, numerous mitochondrial proteins are reduced in AD cortices, including the subunits of complex I and complex IV in OXPHOS (59). Alternatively, other mitochondrial proteins, including malate dehydrogenase and SDH, show increased activity in AD cortices (447).
Moreover, some mitochondrial enzymes involved in energy production, such as acetyl-CoA dehydrogenase and enoyl-CoA hydratase (ECHS1), were upregulated in AD cortices (59). Increased levels of ketone body-producing enzymes have been previously observed in AD mouse models. The expression of 3-oxoacid-CoA transferase 1, which catabolizes ketone bodies to produce acetyl-CoA and thus generate ATP, was increased in 3 × TgAD mice (447). Protein-methionine sulfoxide oxidase is the enzymatic source of ROS production in neurons, and it could act as an increased biomarker in the AD brain (34). The removal of cholesterol from the brain is controlled by cytochrome P450 and was found to be genetically enhanced in 5 × FAD mice (126, 268). These alterations in the mitochondrial proteome of AD mouse cerebral cortices indicate that central mitochondrial protein alterations might be possible biomarkers for the early diagnosis of AD.
Metabolomics is a powerful tool for studying perturbations in the metabolome, reflecting alterations in multiple networks affected in AD. Previous studies have shown that many canonical pathways are significantly disturbed in MCI and AD patients. These pathways include energy metabolism, the Krebs cycle, mitochondrial function, neurotransmitters, amino acid metabolism, and lipid biosynthesis. Krebs cycle markers in the CSF and liver were significantly affected in patients with MCI compared with unaffected people. Succinic anhydride, citraconic acid, 2-furoic acid, threo-isocitric acid, citraconic acid, and pyruvic acid were increased in the CSF of MCI patients; however, acetoacetic acid and fumaric acid levels were decreased. Interestingly, most of the mitochondria-related metabolites affected early in MCI continue to be altered in AD, including succinic anhydride, citraconic acid, pyruvic acid, and acetoacetic acid (87). Moreover, F2-isoprostanes, which are stable products of lipid peroxidation, accumulate in the CSF of AD patients (126, 127). In addition, a new study reported reduced levels of cell-free mtDNA in the CSF, representing a possible novel biomarker of preclinical AD (83).
In 2018, the National Institute of Aging and Alzheimer's Association proposed a diagnostic standard for AD that is abbreviated as the ATN system (177), where A stands for β amyloid deposition, T stands for pathologic tau including total tau and phosphorylated tau, and N stands for neurodegeneration. Based on long-term clinical studies, this ATN classification system clusters different biomarkers through neuroimaging techniques, such as PET, thus providing standardized indicators for basic and clinical researchers to distinguish the cognitive impairment caused by AD pathology, even at an early stage (30–50 years old), thereby achieving early intervention. Many new mitochondria-related changes have been found in the CSF of transgenic AD mice and patients and are thus indicated as potential biomarkers. These mitochondria-related biomarkers should be further verified for their specificity in AD.
PET testing is expensive, as it requires demanding equipment and strict radioisotope operation. Further, the collection of CSF samples requires lumbar puncture, which is highly invasive. In addition, the contents of recent standard AD biomarkers found in the CSF are very low in the blood. Therefore, it is necessary to use peripheral body fluids, especially blood, to detect new biomarkers for the early diagnosis of AD.
B. Biomarkers in peripheral tissues and circulating body fluids
AD is a multifactorial disease that affects both the CNS and the periphery (127). Intriguingly, several recent studies investigating metabolites have reported significant metabolic changes in the liver and other peripheral organs (23, 325). β-oxidation, in liver mitochondria or peroxisomes (325), is the principal pathway for fatty acid metabolism. AD-related increases in long-chain specific acyl-CoA dehydrogenase, 3-keto-CoA thiolase, and acyl-CoA thioesterase 10 have been observed, suggesting elevated fatty acid β-oxidation and increased acetyl-CoA production (129). The increased fatty acid metabolism in the AD mouse liver may increase ketone body formation in blood, supported by the augmented ketone body production in AD mice (136). Pyruvate is an essential metabolite that fuels TCA and drives other biosynthetic pathways (87).
Other studies have shown that the levels of hydroxymethyl transferase and NADP-dependent malic enzyme are increased in the AD liver, while serine-pyruvate amino transferase levels are decreased, suggesting elevated pyruvate concentrations (129). Further, the decreased expression of glycogen synthase and glycogen breakdown proteins in the AD liver suggests reduced glucose storage (83). The observed proteomic changes suggest dysregulated glucose in the AD mouse liver (129). The enzymes involved in the electron transport chain can generate ROS by forming superoxide anions from molecular oxygen (396). COX-2 and sulfite oxidase were observed in the AD liver and thus potentially increase OXPHOS (129). Alterations in antioxidant regeneration rely on a continuous supply of NADH and NADPH and have been reported in the AD liver (136). The redox state in mitochondria depends on the ratios of the reduced and oxidized forms of several systems such as NADH/NAD+ (401). The changes observed in the liver proteome of AD mice provide further evidence that AD is a metabolic disorder and that the metabolites produced in mitochondrial metabolism might be liver biomarkers for early diagnosis and prevention.
Mitochondrial dysfunction has been demonstrated in the neurons of AD patients (396) and further shown to be linked to alterations in ROS production (462). These changes were observed in the brains of AD patients as well as in the periphery, that is, fibroblasts in humans and in mouse models (396, 462), and other tissues, including platelets and lymphocytes. Several studies showed that lipid oxidation and superoxide dismutase (SOD) activity were increased; whereas the activities of complex IV, complex III, mitochondrial membrane potential, and ATP were reduced in the platelets of AD patients compared with age-matched controls (167, 256, 275, 287).
Further, increased levels of oxidative DNA damage, Mn-superoxide-dismutase (SOD2) mRNA, 4-HNE, and 3-nitrotyrosine are observed in the lymphocytes of AD patients (358, 397), and ROS levels are higher in the lymphocytes of both AD patients and transgenic AD animals (74, 375). Moreover, apoptosis levels are elevated in lymphocytes from AD patients and transgenic mice as indicated by enhanced DNA fragmentation, SOD mRNA expression, Bax/Bcl2 ratios and caspase-3, caspase-6, and caspase-8 activity (93, 120, 220, 382).
In addition, Scott et al. reported that basal mitochondrial oxygen consumption, proton leakage, and AP endonuclease 1 (APE1) activity were reduced in peripheral blood mononuclear cells; whereas dATP levels were enhanced. MtDNA depletion is believed to be a typical pathophysiological factor of neurodegeneration in AD. A low cell-free mtDNA content in CSF may be a novel biomarker for the early detection of preclinical AD in both patients and transgenic mice. Further studies are needed to determine whether the CSF mtDNA concentration can be altered by disease-modifying treatments (315). Our laboratory recently identified cell-free DNA 5hmC as a potential biomarker in AD (unpublished). These studies highlight the relevance of mitochondrial function in blood cells as an early peripheral marker for the detection of AD.
Alterations in several types of mitochondrial enzymes and metabolites have recently been reported; however, selecting reliable and usable biomarkers from a large number of mitochondrial-related proteins is still a major challenge for the diagnosis, prevention, and control of AD. Detecting biomarkers in blood is the better method for diagnosing AD, but this has not yet been achieved. The identification of sensitive and easily detectable biomarkers should be a major goal in AD diagnosis, and the development of early diagnostic techniques has also advanced the study of early AD intervention programs (Fig. 7).
VI. Targeting Mitochondrial Metabolic Dysfunction for AD Prevention
There are currently more than 2000 registered clinical trials worldwide investigating anti-AD drugs that can be classified by their main mechanisms, including (i) targeting neurotransmission, (ii) preventing the accumulation of amyloid plaques or NFTs, and (iii) restoring mitochondrial function or energy metabolism; and other therapeutic approaches, such as anti-inflammation or rescuing nerve growth factor (ClinicalTrials.gov). Currently, the main therapeutic strategies and corresponding drug research and development include the following: (i) drugs that interfere with the formation and deposition of Aβ and amyloid cascade reaction; (ii) drugs that affect aggregated hyperphosphorylated Tau proteins; and (iii) drugs that regulate the CNS, including different types of neuronal receptors involved in neuronal plasticity and signal transduction.
The clinically approved drugs for AD treatment include (i) donepezil, an acetylcholinesterase inhibitor for all stages of AD; (ii) rivastigmine, an acetylcholinesterase inhibitor and butyryl cholinesterase inhibitor for mild to moderate AD; (iii) galantamine, a nicotinic receptor modulator for mild to moderate AD; (iv) memantine, an N-methyl
A. Mitochondrial nutrients
“Mitochondrial nutrients” refer to a group of micronutrients that, either directly or through metabolites, influence the structure and function of mitochondria (234, 239). These nutrients may also have other functions that are not necessarily related to mitochondria. The nutrients benefit mitochondria in four ways: by (i) ameliorating oxidative stress, for example, lipoic acid and hydoxytyrosol (234, 307); (ii) activating phase II enzymes that improve antioxidant defenses, for example, tocopherol and sulforaphane (106, 116); (iii) enhancing mitochondrial remodeling, including mitochondrial degradation, biogenesis, fission, and fusion, for example, acetyl-
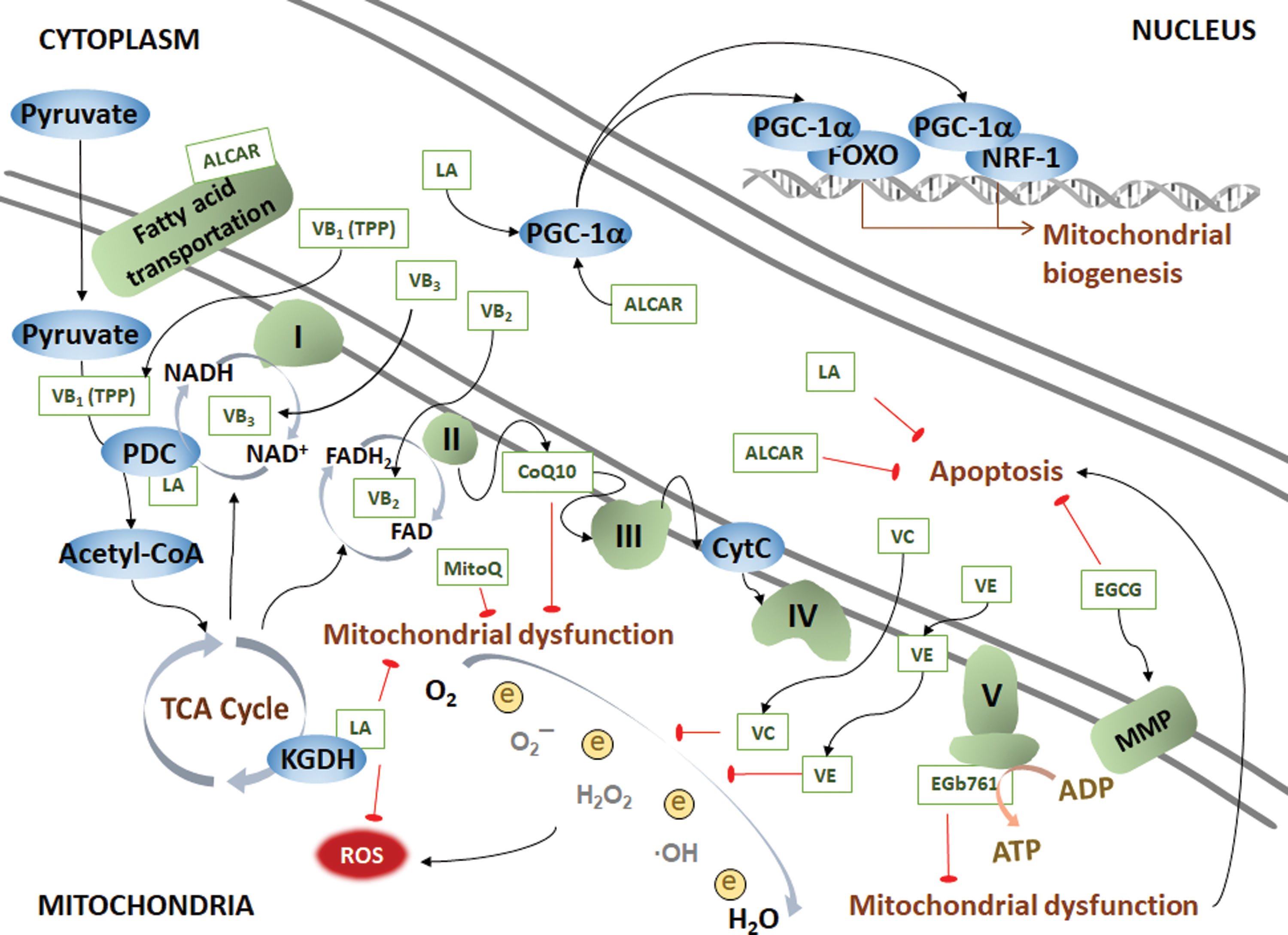
Clinical Trials Registered on
ADAS-cog, Alzheimer's Disease Assessment Scale-Cognitive Subscale; ADL, activities of daily living; DHA, docosahexaenoic acid; EGCG, epigallocatechin-3-gallate; EPA, eicosapentaenoic acid; MMSE, mini-mental state examination; MRI, magnetic resonance imaging.
1. Lipoic acid
α-Lipoic acid (LA), the in vivo cofactor for the PDC and KGDH in the TCA cycle, inhibits the formation of ROS, increases the levels of reduced glutathione, and scavenges lipid peroxidation products, such as hydroxynonenal and acrolein. LA crosses the BBB into nerve cells and protects against cell damage induced by Aβ or hydrogen peroxide (77, 235, 442). It also induces Akt expression, suggesting that the neuroprotective effect of the antioxidant LA is partially transmitted through PKB/Akt signaling. LA also exerts protective effects by activating phase 2 enzymes specifically by inducing the expression of Nrf2, which binds to ARE, in turn increasing the activity of the phase 2 enzyme system to reduce cellular oxidative stress, enhancing the intracellular antioxidant defense system, and alleviating mitochondrial oxidative stress. On the other hand, LA targets PGC-1α, which binds to FOXO or NRF1 to increase mitochondrial biogenesis, thereby improving cognitive function in AD (145, 374). A supplementation of LA and N-acetyl cysteine decreased mitochondrial-related oxidative damage in AD patient fibroblasts (275). A 48-month follow-up study in AD patients showed that treatment with LA might be a neuroprotective therapeutic option for AD. However, the phase II trial is needed (145).
2. Acetyl-l -carnitine
ALCAR is a natural product of the reaction of carnitine with acetyl-CoA. ALCAR is used to transport and activate fatty acids into the mitochondria, which is a rate-limiting regulator of fatty acid oxidation. ALCAR protects nerve cells from apoptosis and targets PGC-1α/FOXO or PGC-1α/NRF1signaling to improve mitochondrial biogenesis. ALCAR is especially enriched in muscle and brain tissues and can efficiently travel across the BBB to promote nerve cell regeneration, protect nerve cells from mitochondrial uncouplers and inhibitors, and enhance mitochondrial enzyme activity and mitochondrial function, thereby reducing brain damage in AD (144, 235, 341). After 6 months of the double-blind parallel design pilot study of ALCAR, the ALCAR group demonstrated significantly slowed deterioration in some cognitive areas among patients with AD (341). The perfusion of the precuneus was amplified in AD patients after ALCAR administration and their cognitive and neuropsychiatric symptoms were not worsened (180). Further clinical studies are warranted to determine the role of ALCAR treatment in AD.
3. Coenzyme Q10 and MitoQ
Coenzyme Q10 (CoQ10) is an integral part of the electron transport chain that acts as an electron acceptor that promotes ATP production and as an antioxidant in the mitochondrial matrix and its inner membrane. Several studies have identified CoQ10 as a potential target for AD, as stabilized mitochondria impaired by neurotoxins and oxidative stress and improved memory and behavioral performance in Tg19959 and APP/PS1 mouse models of AD (88, 286). The levels of the reduced and oxidized forms of CoQ10 in the CSF of AD patients and age-matched controls measured using HPLC showed that the oxidized/total CoQ10 level in AD was significantly higher than those in the controls, indicating that mitochondrial oxidative damage plays an important role in early AD pathogenesis (175). The mitochondria-targeting antioxidant MitoQ is a modified CoQ10 molecule. Compared with CoQ10, MitoQ is a much smaller molecule that is more easily absorbed by cells and is attracted to negatively charged mitochondria. MitoQ treatment was shown to prevent cognitive decline and oxidative stress in 3 × TgAD mice (269, 453) and to extend the lifespan, improve electron transport chain function, and protect the mitochondrial cardiolipin content in a Caenorhabditis elegans model of AD (292).
4. B vitamins
B vitamins have been found to reduce brain atrophy and slow the onset or progression of AD (82). B vitamins are components or precursors of corresponding mitochondrial coenzymes and participate in one-carbon metabolism (86, 395). Thiamine pyrophosphate is an active form of vitamin B1 that is responsible for glucose metabolism and the eventual production of acetyl-CoA. Vitamin B2 (riboflavin) is the precursor of FAD that participates in the transformation of FADH2-FAD in energy production and cellular respiration. Vitamin B3 is the precursor for NAD and NADP, which are responsible for DNA repair and mitochondrial respiration. However, vitamin B supplementation for AD remains controversial, because some trials failed to show its protective effect.
5. Hydroxytyrosol
Hydroxytyrosol (HT) is a phenyl ethanol with antioxidant activities. HT could supply a hydrogen atom from the phenolic hydroxyl group to free radicals. The second hydroxyl group in the ortho-position enhances antioxidation by generating the catechol ring, thereby increasing the rate of hydrogen transfer to the superoxide radical. The catechol ring is partially converted into o-benzoquinone and plays a direct role as an antioxidant.
There are two main sources of HT: (i) exogenous HT (oleuropein undergoes a double hydrolysis reaction to form HT, which is mainly found in olives, olive leaves, olive pulp, and olive oil in the Mediterranean diet); (ii) endogenous HT, the dopamine oxidation metabolite in the body. HT has now been identified by the Scientific Committee of the European Food Safety Authority as a polyphenol in olive oil that can help to protect lipids from oxidation. The pharmacokinetics of HT have been extensively studied, and one 14C-labeled HT distribution study showed that HT could exert its neuroprotective effect across the BBB (206). The cardioprotective, antitumor, antimicrobial, antidiabetic, and neuroprotective effects of HT have been investigated, due to its high antioxidant activity (337). Two major mechanisms involved in the regulation of HT antioxidant activity are: (i) the direct removal of ROS generated by oxidative stress and (ii) the activation of certain molecular targets to resist oxidative stress (Fig. 9).
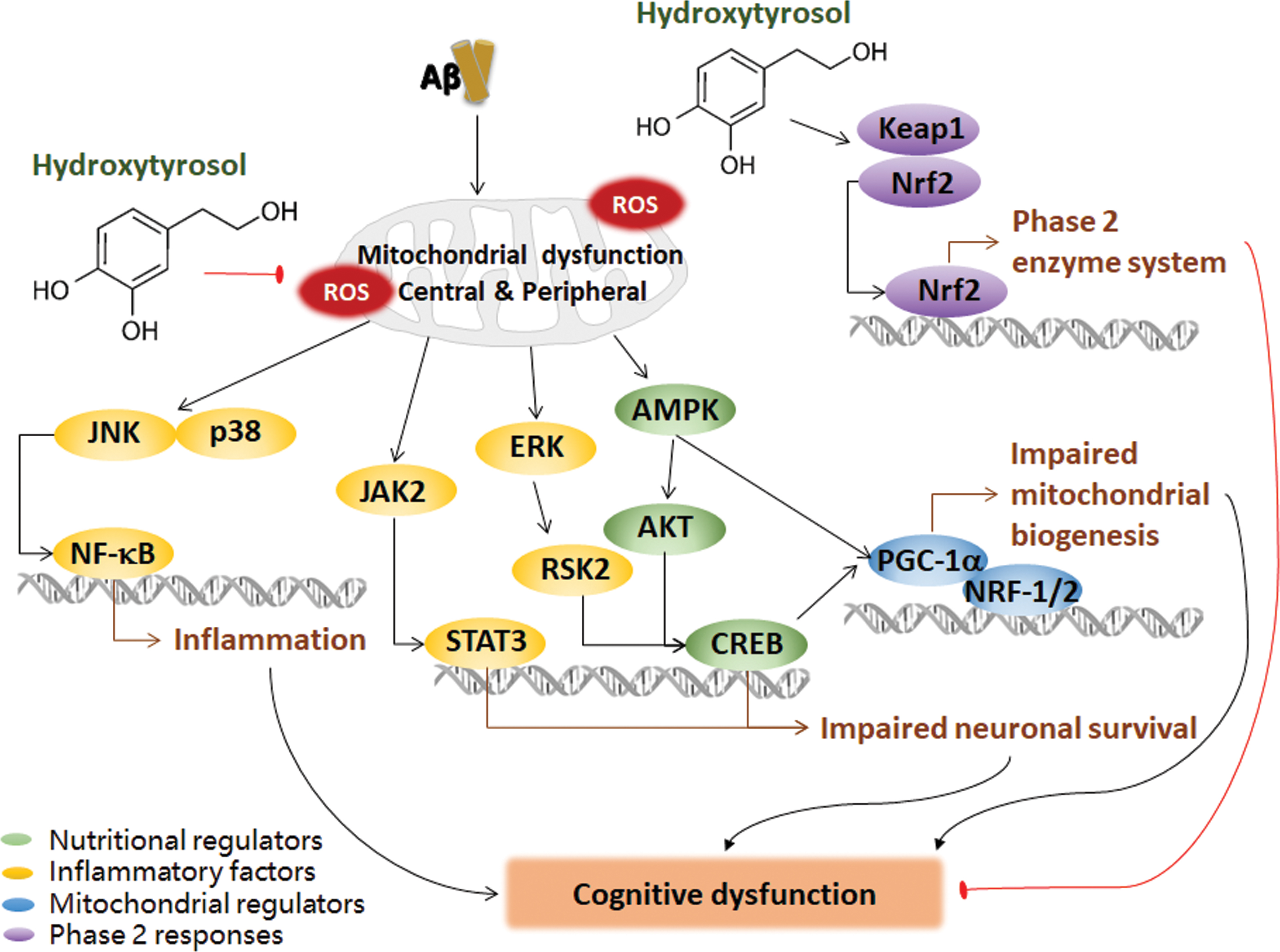
Adherence to the Mediterranean diet could reduce the risk of neurodegenerative diseases, such as MCI and AD, while helping to improve cognitive function (316, 335, 346, 366). The 2019 WHO guidelines for reducing the risk of cognitive decline and dementia recommended a Mediterranean-like diet to adults with normal cognition and MCI to reduce the risk of cognitive decline and/or dementia (425). One possible reason for the beneficial effects of the Mediterranean diet might be the functional ingredients, such as HT, which is the beneficial component of olive products. However, there are very few studies on the neuroprotective effect of HT. Oleuropein supplementation in TgCRND8 mice and aged mice resulted in a significant improvement in cognitive function; in addition, Aβ levels and plaque deposition in the brain were significantly reduced. These changes may be achieved through the mTOR pathway and, subsequently, activate neuronal autophagy (133). Another study showed that 14 days of 10 mg/kg HT treatment significantly enhanced the spatial cognitive abilities of C57BL/6 mice. The animals were injected with oligomeric Aβ1–42 plus isonicotinic acid and the improved spatial ability may have been due to the HT-induced improvements in the ERK-MAPK/RSK2, PI3K/Akt1, and JAK2/STAT3 signaling pathways in hippocampal neurons (16).
We identified mitochondrial dysfunction in AD and found that HT successfully reversed the mitochondrial respiratory chain enzyme activities in the brains of AD mice (307). A GSH reduction induces redox dysregulation and loss of mitochondrial membrane potential, and HT increases GSH levels via activating Nrf2. We also showed that HT significantly reduces mitochondrial protein carbonylation and repairs the phase 2 enzyme system by activating the Nrf2-Keap1 pathway and AMPK to constitute the energy-sensing protein network and trigger mitochondrial biogenesis (469, 476). Some vitagenes respond to the phase 2 enzyme system to protect against electrophiles and oxidants, and the Keap1-Nrf2/ARE pathway is critical for the phase 2 response. Vitagenes are protective genes that control stress and protein homeostasis with the health (40). Regulation of endogenous cellular defense mechanisms such as the vitagene network may open up new treatments for diseases related to tissue damage and cell death in AD.
Recent studies have reported that mitochondria may be a key target of HT in the amelioration of oxidative stress in AD. The oral administration of HT significantly improved the electroencephalogram activity of APP/PS1 mice as well as the learning and memory abilities and decreased mitochondrial oxidative stress, inflammation, and apoptosis in APP/PS1 mice. Our group recently identified an HT derivative HT-ac that has better bioavailability than HT and could remarkably improve cognitive function in APP/PS1 mice (unpublished). These findings suggest that HT may be a major functional factor in the Mediterranean diet to prevent and ameliorate AD (307) (Fig. 9).
6. β-hydroxybutyrate
Neuroprotective effects are observed with the appropriate concentrations of β-HB, as it provides mitochondrial protection (461). β-HB reduces ROS levels by targeting complex I in the mitochondrial respiration chain and induces ATP production in brain mitochondria (252, 393). Medium-chain triglycerides, such as β-HB, might improve memory in AD, which correlates positively with the plasma levels of β-HB. β-HB blocks Aβ entry and improves cognition in AD mice (72). Ketone bodies also have antiapoptotic activity by blocking ROS-activated PP2A, alleviating the inactivation of the antiapoptotic factor Bcl2 and thereby inhibiting apoptosis (251). In addition, β-HB may relieve rotenone-induced caspase 3 and caspase 9 activation (173). A recent study found that β-HB inhibited histone deacetylases (HDACs), releasing HDACs from closing the forkhead box O3α and metallothionein 2 genes and triggering cellular antioxidant responses accordingly (291). The activated ketone body β-HB in the liver activates Bdnf promoters via HDAC2 and HDAC3, leading to neurotransmitter release (367).
β-HB has also been found to protect hippocampal neurons against Aβ toxicity in Aβ-induced AD cell models by correcting defects in mitochondrial energy generation (193). These studies reveal that ketones, as an alternative energy source in the absence of glucose, may protect neurons by improving mitochondrial function, thereby relieving oxidative stress and inhibiting apoptosis. The detailed mechanisms by which β-HB and ketogenesis function in AD pathology are limited and worthy of further investigation (Fig. 10).
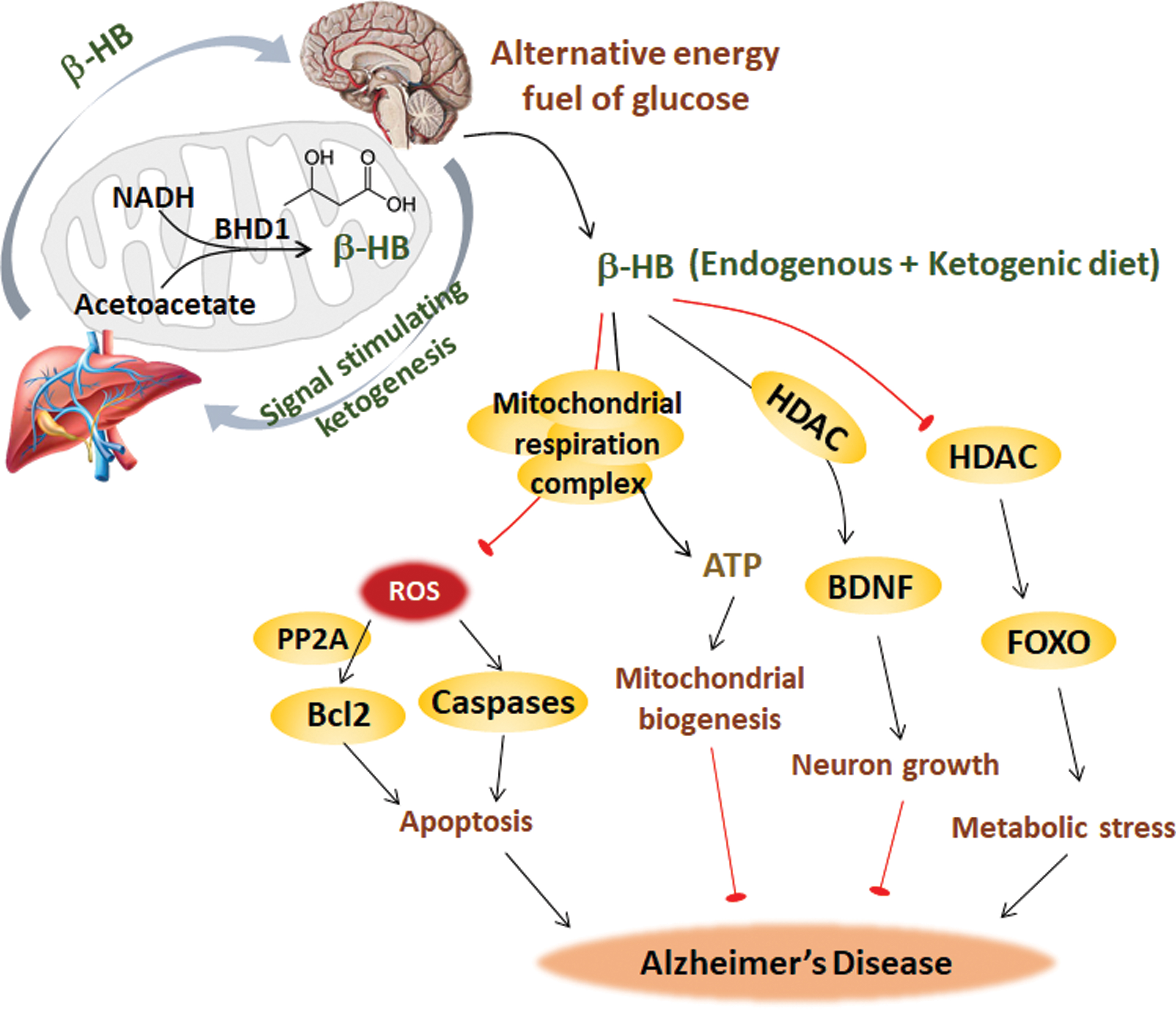
β-HB-enriched ketogenic diets, which account for 80% to 90% of high fat, are low in protein and low in carbohydrate diets that meet growth needs (399). Studies by ourselves and others have shown that unlike the HFD, which has certain negative effects (such as the risk of cardiovascular disease and cognitive impairment) despite the fact that the induced body weight gain improved bone health in healthy women and female AD mice (183, 309), ketogenic diets show potential for AD intervention based on the neuroprotective effect of ketone bodies. A ketogenic diet has been used for 80 years to cure epilepsy. The therapeutic effect might be related to mitochondrial bioenergy improvement, because the ketogenic diet enhances the genes and activities of enzymes in mitochondrial respiration, the TCA cycle, and mitochondrial biogenesis (267).
Microglia metabolize acetoacetate and β-HB produced by the ketogenic diet, and they respond to these ketones as metabolic signals (122). Studies have shown that the ketogenic diet has protective effects on neurodegenerative diseases, such as AD (461, 475). Low-carbohydrate diets were given to older adults with MCI, and the results showed that patient memory was improved and that the improvement was positively correlated with ketone body levels (72).
The increased risk of AD is associated with dietary habits of high levels of saturated fat and simple carbohydrates. A recent study modified a ketogenic diet with a Mediterranean diet, which makes up for the lack of higher carbohydrates in the ketogenic diet and adds intake of vegetables and fruits, while increasing fats and proteins derived from healthy sources such as olive oil and fish, and the resulting diet showed the potential to ameliorate AD (288).
Fasting leads to the conversion of glucose from glycogen reserve to ketone from fatty acids. It has been reported that in an AD mouse model, intermittent fasting (IF) protects against GABAergic synaptic adaptations and cognitive deficits, which is mediated by the mitochondrial protein deacetylase SIRT3 (240). SIRT3 has been reported to regulate ketogenesis through activation of mitochondrial HMGCS2 in the fasting state (361). IF also protected against disturbances in energy, glucose, lipid, and bone metabolism in ovariectomized AD rats (362). However, IF could not ameliorate amyloid peptide deposition in 3 × AD mice (147). Further clinical trials of IF in the early stages of human AD are necessary.
7. Hydrogen
Hydrogen (H2) reacts with specific biomolecules at body temperature. Its high diffusivity allows it not only to rapidly penetrate the BBB but also to enter the cytoplasm, mitochondria, nucleus, ER, and other subcellular structures through the cell membrane structure to interact with target molecules.
The mild reducibility of H2 contributes to its selective reduction of hydroxyl radicals (·OH) and its low disturbance of redox balance in vivo (194, 299). Clinical trials have also observed no obvious side effects or H2 toxicity. H2-rich water, H2-rich saline, and H2 inhalation have antioxidant activities similar to those of molecular H2 (262). Our previous study applied coral calcium hydride (CCH), a new solid molecular H2 carrier made of coral calcium, to HFD-induced fatty liver rats and found that CCH could prevent glucose metabolic disorder via PI3K/Akt signaling and lipid metabolic disorder through ACC/FAS signaling and activate phase 2 enzymes and improve mitochondrial function (164).
In addition to antioxidant activity, the neuroprotective effects of H2 have been studied in recent years. Highly diffusible H2 gas inhalation protects against ischemia-reperfusion and stroke-induced oxidative stress by targeting intracellular sources of ROS (299). We found that small amounts of H2 inhalation ameliorated survival and neurological outcomes, including consciousness, cranial nerve reflexes, and sensory function, in the asphyxia rat model of cardiac arrest, and the beneficial effect was superior to that of mild hypothermia (414).
H2-enriched saline was found to ameliorate memory impairment in Aβ-induced AD mice (223) and to exert anti-inflammatory and antioxidant effects on the mouse brain (409). In the injured rat brain, H2-rich saline was found to reverse brain health by suppressing the inflammatory response involving the NF-κB pathway and NLRP3 inflammasome (352). This finding suggests that molecular H2 may play a beneficial role in protecting against the pathology of AD. Based on a previous H2 study, we speculated that H2 may ameliorate AD via the mitochondrial metabolic pathway and the anti-inflammatory response. Our recent study found that H2-enriched water significantly improved cognitive dysfunction in female mice via the estrogen-ERβ-BDNF signaling pathway and that H2-enriched water also exerted antioxidant and anti-inflammatory effects in APP/PS1 mice (163). As a newly identified small molecule playing a key role, the potential mechanism by which molecular hydrogen regulates the AD-related pathway needs to be elucidated (Fig. 11).
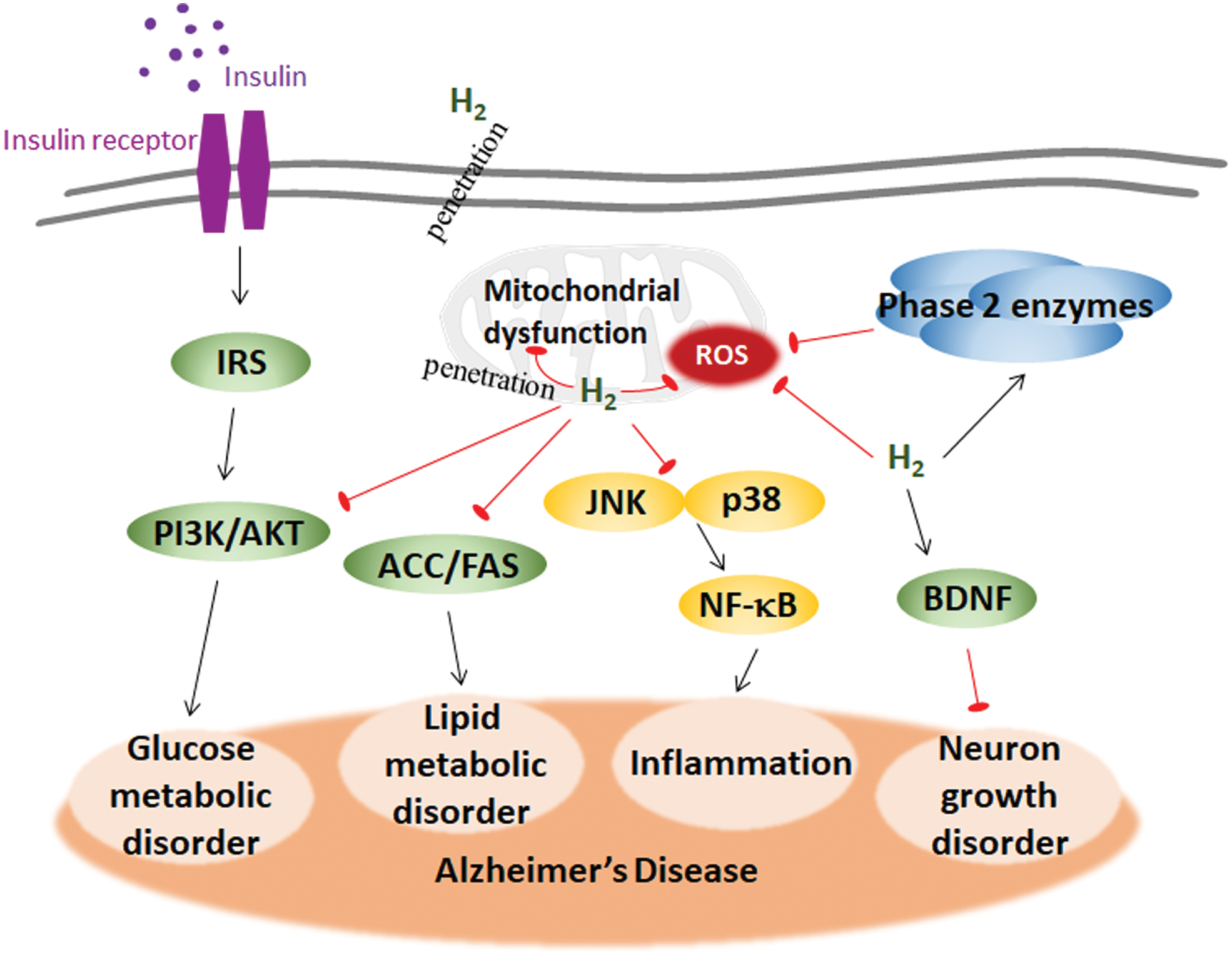
8. Other mitochondrial nutrients for AD
The nutrients administered to AD patients are typically antioxidants. The antioxidant vitamins A, C, and E reduced the risk of AD (455). The publications regarding the effects of vitamins on AD are mostly association studies in AD patients, whereas in-depth studies on the mechanism are rare. One possible reason underlying the effect of vitamins on AD is that they are involved in multiple metabolic processes. The excessive intake of vitamin A in the body causes poisoning, and β-carotene is a safe source of vitamin A because it can be converted to vitamin A after entering the body when needed. Studies have shown that β-carotene significantly improves behavioral abilities, learning, memory, and cognitive function in mice and rats through its antioxidant effect by scavenging free radicals (3, 457).
The first-line antioxidant vitamin C functions in reduction and hydroxylation reactions. Vitamin C deficiency in the brain accelerates amyloid pathogenesis and cognitive behavior in APP/PS1 mice, likely through oxidative stress pathways (81). Vitamin E also has the effect of scavenging free radicals, inhibiting lipid peroxidation, and maintaining the integrity and stability of the membrane structure. Vitamin E is especially abundant in the mitochondrial inner membrane and reacts rapidly with oxidants. Studies have shown that vitamin E significantly improves memory disorders and increased SOD activity (234). However, the clinical roles of vitamin A, C, and vitamin E in the prevention and treatment of AD are still under debate.
The standardized ginkgo biloba extract EGb761 was found to reverse the decrease in ATP production, thereby inhibiting mitochondrial dysfunction and oxidative damage in PC12 cells overexpressing the Aβ precursor protein (1, 92). Polyphenols could regulate the expression of antiapoptotic proteins in the Bcl-2 family to some extent and reduce apoptosis. Epigallocatechin-3-gallate (EGCG) has a certain protective effect on AD cell models and patients. EGCG intervention effectively reduces the decrease in mitochondrial membrane potential and cell apoptosis induced by Aβ (12, 464).
9. Combination of mitochondrial nutrients
The combination of mitochondrial nutrients ameliorates age-related cognitive dysfunction. An optimal combination of mitochondrial nutrients might be more effective than the individual nutrients in the prevention and treatment of AD. Combinations of multiple mitochondrial nutrients have been tested in different mitochondrial dysfunction-related disorders. For example, we initially reported that the combination of LA and ALCAR remarkably improved the cognition of aged rats. The combination of nutrients synergistically ameliorates mitochondrial enzyme activity and cerebral oxidative stress (235, 236). The efficiency of ApoE4 to transport cholesterol in neurons decreases under stress conditions. LA has anticholesterol activity, whereas ALCAR reduces cholesterol levels in the brain and blood; therefore, the combination of LA and ALCAR significantly improves cognitive behavior in transgenic ApoE4 mice (207, 356). The combination of ALCAR and donepezil or rivastigmine, cholinesterase inhibitors used for clinical AD treatment, could exert potent effects on the inhibitors of patients with mild AD who are not sensitive to cholinesterase inhibitors, indicating that a combination of anti-AD drugs and mitochondrial nutrients could be an effective strategy for AD treatment (28).
We found that the combination of LA, ALCAR, HT, and CoQ10 ameliorated skeletal muscle atrophy through the activation of mitochondrial biogenesis and reduction of oxidative stress (237). Previous studies have shown that the LA, ALCAR, nicotinamide, and biotin combination ameliorated the abnormal activities of mitochondrial complexes, mitochondrial biogenesis, oxidative stress, antioxidant defenses, and apoptosis, leading to the reversal of spleen, thymus, liver, and muscle dysfunctions in GK diabetic rats without causing weight gain (149, 150, 353). A combination of LA and ALCAR not only effectively protected cells against rotenone-induced mitochondrial dysfunction, oxidative damage, and the accumulation of α-synuclein and ubiquitin but also prevented mitochondrial oxidative damage in a cell culture model of Parkinson's disease in primary cells from the brains of aged rats.
ApoE-deficient mice fed a regimen that included vitamin E, ginkgo biloba, pycnogenol, and ascorbyl palmitate had significantly prolonged lifespans and reduced inclusion body histopathology in the hippocampus (398). Meta-analysis indicated that a dietary intake of β-carotene, vitamin C, and vitamin E lowered the risk of AD (222). Although vitamins A, C, and E are still controversial in the prevention and treatment of AD, clinical studies have shown that supplementation with vitamin E and vitamin C helps prevent vascular dementia, improves cognitive ability, and reduces the risk of AD; supplementation with vitamin E, vitamin C, and selenium contributes to AD prevention due to the antioxidative functions of these molecules (97). The findings from our studies and others support the hypothesis that the combination of mitochondrial nutrients or the combination of mitochondrial nutrients with other drugs is an effective strategy to prevent or treat neurodegenerative diseases, such as AD (235, 236, 241, 458).
B. Exercise
Similar to mitochondria-targeted nutritional intervention for AD, exercise advantageously has fewer side effects and better dependability than drug treatment; the key point is that exercise improves energy metabolism-centering mitochondria. Several studies in patients with AD and AD transgenic mice reported that exercise improved cognitive behaviors and ameliorated AD pathology (54, 98, 159, 232). In middle-aged healthy adults, exercise promotes cognitive function and reduces the risk of dementia in later life (76). Exercise targeting energy metabolism mimics the function of mitochondrial nutrients and vice versa. Consequently, exercise is considered an effective strategy for the prevention and adjuvant treatment of AD.
1. Exercise improves the metabolic profile in AD
Exercise improves the whole-body metabolic profile and brain health and, to some extent, decreases AD risk (290, 345), and several potential mechanisms have been found. BDNF regulates nerve cell survival, differentiation, and plasticity, improving cognitive function, learning, and memory, which are beneficial for AD. The release of BDNF enhances not only the function of nerve cells but also the cleavage of APP by α-secretase and inhibits β-secretase, reducing the production of toxic Aβ peptides (146, 296). Exercise was demonstrated to increase BDNF expression by activating the ketone body β-HB in the liver, inducing the activities of Bdnf promoters by HDAC2 and HDAC3 and leading to the neurotransmitter release (367). Blocking BDNF signaling by its receptor TrkB diminishes the positive effect of exercise, which confirms the role of BDNF signaling in the neuroprotective function of exercise (296, 367).
In addition to evidence showing that exercise slows AD-related pathological features in the brain, exercise could further prevent and alleviate cognitive function in AD by regulating blood pressure. Aerobic exercise induces endothelium-dependent vasodilatation by upregulating NO production and cardiovascular fitness, thereby promoting neuronal structure and function, mitigating cognitive impairment, and reducing brain atrophy in AD (37, 161). Exercise-derived circulating exosomes elevate miR-342-5p to afford cardioprotection against ischemia/reperfusion injury and regulate neural stem cell proliferation and astrocyte commitment (115, 165). Elevated miR-342-5p expression also downregulates the ankyrin G-induced axon initial segment in transgenic AD mice (376). Exercise reverses AD-related hippocampal decline through the induction of growth factors such as BDNF, IGF-I, and vascular endothelial growth factor (VEGF), in which IGF and VEGF from the periphery could pass through the BBB to promote neurogenesis and angiogenesis (65, 174). Exercise also increases glucose uptake in the hippocampus and enhances mitochondrial OXPHOS to produce ATP (400). PI3K/Akt signaling is hyperactivated in AD and contributes to Tau pathology. Treadmill exercise in AD mice increases PI3K/Akt phosphorylation, reduces downstream GSK-3β activity, reduces Tau protein hyperphosphorylation, and increases neuronal survival by elevating the Bcl-2/Bax ratio and inhibiting the activities of caspase 3 and COX-2, which are induced by Aβ (192, 203).
2. Exercise improves mitochondrial remodeling in AD
Exercise improves mitochondrial remodeling and induces mitochondrial biogenesis through the hippocampal PGC-1α/FNDC5/BDNF pathway to stimulate Bdnf gene expression (431). Exercise enhances neurological health through SIRT1/PGC-1α, increases citrate synthase, the rate-limiting enzyme in the TCA cycle, and induces mtDNA production, leading to mitochondrial biogenesis (373). Exercise also improves electron transport chain function by increasing complex I activity and improves mitochondrial dynamics by upregulating Drp1 levels (142). Swim training regulates mitochondrial quality control by inhibiting LC-3b turnover-mediated autophagy and PINK1/Parkin-mediated mitophagy in aged mice (465). Regular maternal exercise during pregnancy has long-lasting effects on metabolic programming in the brains of offspring and increases BDNF levels, specifically in mitochondrial function as indicated by increased mitochondrial membrane potential and mass, activates α-ketoglutarate dehydrogenase and complex IV, and enhances Mfn1 and Drp1 levels, which then protect against Aβ-induced neurotoxicity and the cognitive impairment of the offspring in their lifetime (202).
3. Exercise decreases inflammation in AD
Exercise also systemically decreases inflammatory markers. The proinflammatory mediators, COX-2 and iNOS and cytokines TNF-α, IL-1β, and IL-6 are attenuated by exercise via the MAPK (p-p38 and p-ERK1/2)-dependent NF-κB pathway in transgenic AD mice with Tau abnormalities (219). The p38-MAPK pathway, TNF-α, and IL-1α are activated by exercise in PS1 transgenic mice (191) (Fig. 12).
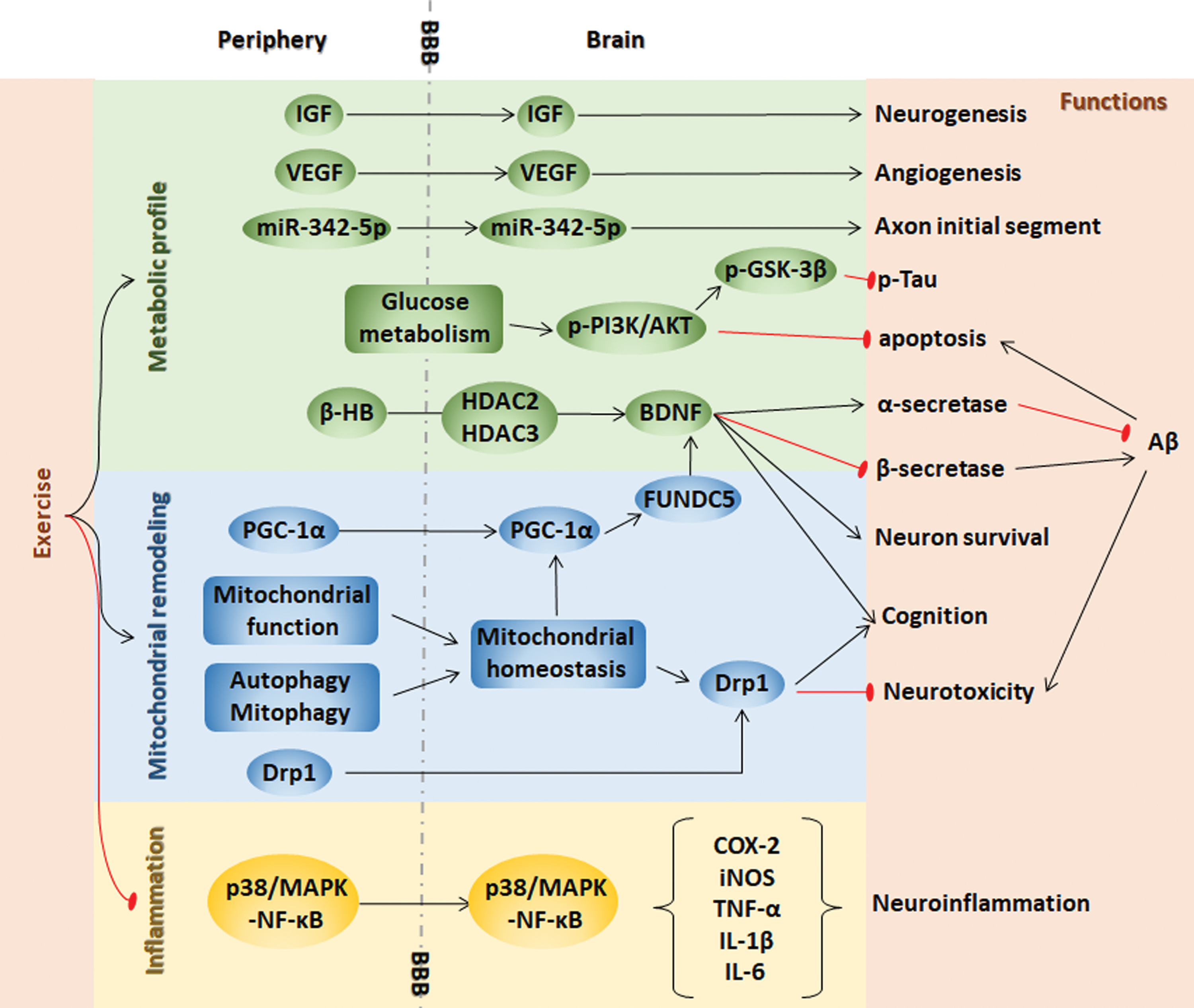
Although several clues and experimental evidence indicate that exercise reduces the pathological features associated with AD, the current research on AD in the field of sports science and medicine is still rare and not yet in-depth. Because exercise and mitochondrial nutrients have similar benefits for AD, the study of mitochondrial nutrients may provide some clue regarding the mechanism. The specific molecular mechanism by which exercise regulates energy metabolism and further prevents, delays, or slows AD may help to fully understand the occurrence and development of AD systemically.
C. Natural compounds from traditional Chinese medicines
As AD is a multifactorial systemic disease with metabolic disturbance in central and peripheral tissues involving insulin resistance, inflammation, and oxidative stress, approaches aiming at either single molecular targets, such as Aβ and the p-Tau protein, might not be effective in treating AD, and numerous clinical trials have provided fruitless evidence of this notion.
Traditional Chinese medicines (TCMs) undergo a trial-error paradigm, and the effective compounds are active in maintaining bioenergy and metabolic homeostasis and are more likely to maintain mitochondrial energic metabolism and function, for example, agents that may partially rescue the entirety of the metabolic disorders may prevent the progression of AD pathogenesis. Therefore, the alternative strategy for AD prevention and treatment may lie in TCMs.
Douglas Wallace proposed mitochondria as chi, which can be translated as “vital force or energy,” and emphasized that the Asian herbal medications that target the mitochondria could be used to treat age-related and neurodegenerative diseases, such as AD (406). Therefore, based on the mitochondria-chi theory, we have associated “chi” with the mitochondrial studies in our laboratory. In addition, according to the traditional concept, Yin and Yang represent the basic opposition of everything. In TCM, all physical structures of the human body can be divided into the corresponding parts of Yin/Yang. Normal life activities are the result of the coordination between Yin and Yang. Once existence Yin/Yang is abnormally strong or weak, it will cause the occurrence of disease.
As shown in Figure 13, when mitochondria are damaged, chi are diminished, resulting in a lack of energy supply to the body; therefore, AD continually worsens. Using TCMs targeting mitochondria as well as mitochondria-induced oxidative stress, inflammation, insulin resistance, and apoptosis constitute a new idea for slowing down or treating AD.
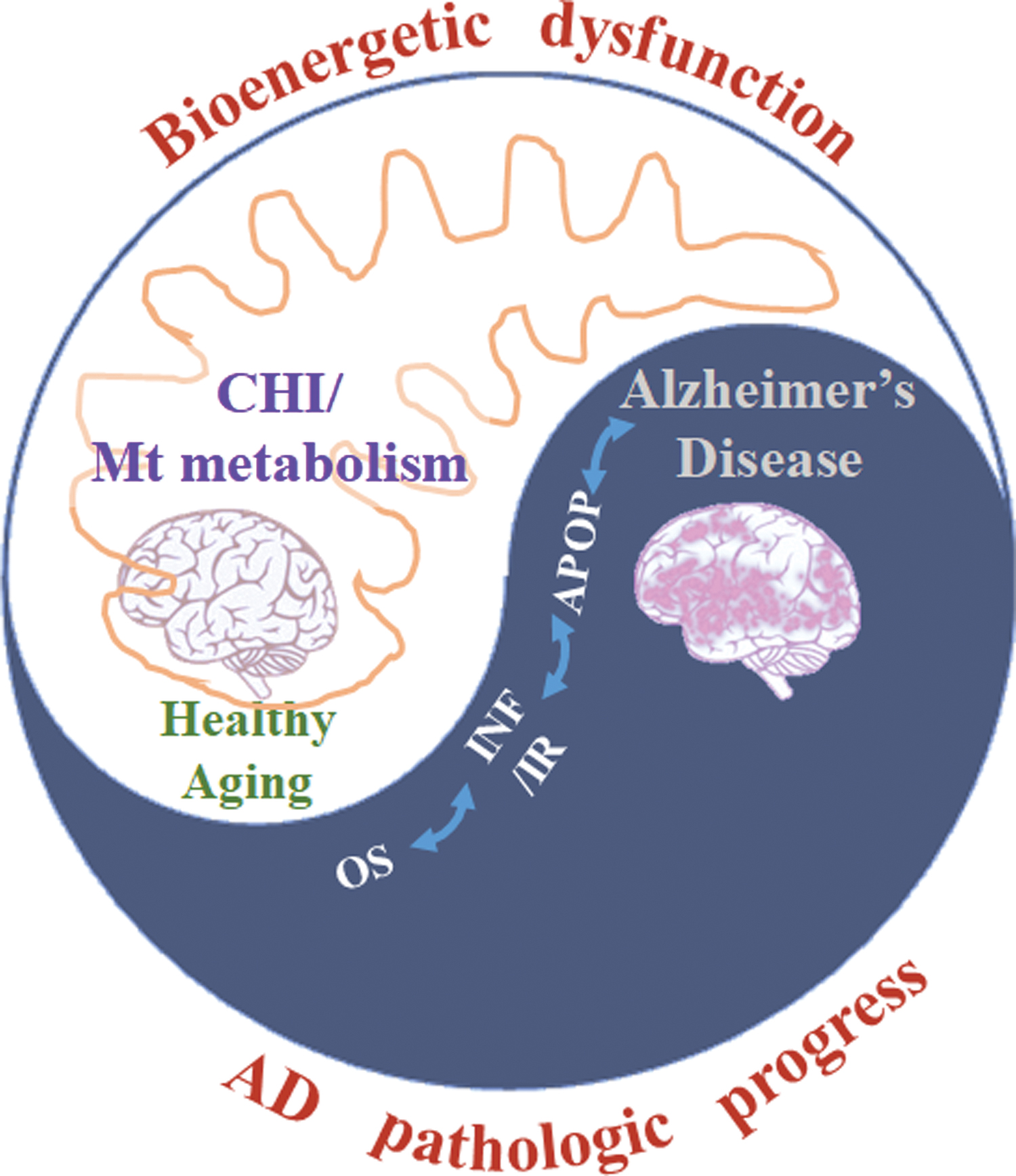
According to numerous scientific articles on Chinese medicine and a long history of clinical practice, several TCMs have shown certain protective effects in treating metabolic disorders and cognitive impairments. Some of these studies have recently been verified, greatly facilitating the screening of promising compounds for AD prevention. A list of TCM nutraceuticals with mitochondria function-related readouts is presented in this section (Table 2). We listed mitochondrial dysfunction, oxidative stress, inflammatory disorder, insulin resistance, and hepatic metabolic dysfunction as representative mitochondrial function-related readouts (Fig. 13).
Potential Nutraceuticals from Traditional Chinese Medicines for Alzheimer's Disease Prevention and Treatment
Aβ, beta amyloid; AD, Alzheimer's disease; IDE, insulin degrading enzyme; IR, insulin receptor; RAGE, receptor for advanced glycation end products; TCM, traditional Chinese medicine.
1. TCMs ameliorate insulin resistance and hepatic metabolic dysfunction
Ginseng is the most commonly mentioned TCM with broad beneficial effects on the CNS and the cardiovascular, endocrine, and immune systems (17, 199). Ginsenoside Rg1, the major active component in ginseng, is effective in recovering insulin sensitivity and reducing blood glucose levels in diabetic db/db mice and ob/ob mice, and the mechanism was also investigated in insulin-resistant cell models in which ginsenoside Rg1 activates the AMPK pathway (218, 439, 440). In addition, ginsenoside Rg1 also increases insulin-degrading enzymes, decreases Aβ levels, and improves learning and memory in a rat model injected with Aβ1–42 (320). Curcuma longa L (turmeric) is another TCM that is mainly used to treat abdominal pains (14). Curcumin, a polyphenolic compound derived from turmeric, ameliorates insulin resistance and glucose homeostasis in diabetic db/db mice by activating glycolysis and inhibiting gluconeogenic and lipid metabolic enzymes in the liver (350). Further, curcumin effectively reduces the plaque burden and insoluble Aβ accumulation in Tg2576/APPsw mice and protects against Aβ-induced mitochondrial dysfunction in neurons by inhibiting GSK-3β (25, 169). These results suggest that TCM nutraceuticals that ameliorate insulin resistance, such as ginsenoside Rg1 and curcumin, have the potential to prevent AD.
As reviewed earlier, compromised hepatic metabolic function is an early event that plays a key role in the pathogenesis of AD, and nutraceuticals/drugs that preserve normal hepatic function should be another type of candidate for treating AD. Salvia miltiorrhiza Bunge in TCM promotes blood circulation to remove blood stasis (301). Cryptotanshinone (CTS), a major constituent extracted from S. miltiorrhiza Bunge, decreases apoptosis and inflammation and ameliorates reduced liver failure (182). In addition, CTS prevents cognitive decline in APP/PS1 mice (271).
2. TCMs ameliorate mitochondrial dysfunction and oxidative stress
The TCM Huperziaserrata has been used for more than 1000 years in China for the treatment of numerous of ailments, including contusions, schizophrenia, swelling, myasthenia gravis, and, most recently, organophosphate poisoning (422). Huperzine-A, a component derived from Huperziaserrata, protects mitochondria from Aβ-induced damage, at least in part by preserving membrane integrity, inhibiting oxidative stress and improving energy metabolism (117, 118). Huperzine-A inhibits the neurotoxicity of Aβ and improves memory in patients with AD (437, 459, 460).
Gynostemma pentaphyllum has various pharmacological effects, such as detoxification and heart palpitation clearance abilities (324). Gypenoside, a main bioactive ingredient in G. pentaphyllum, was reported to improve the cognitive function of an Aβ-induced AD rat model, probably by scavenging free radicals, inhibiting oxidative damage, and maintaining normal mitochondrial ultrastructure (446, 470). The TCM Moutan Cortex is typically used to treat diseases, such as atherosclerosis, infection, and inflammation, among many others (432). Paeonol, isolated from the bark of Moutan Cortex, possesses anti-inflammatory and antioxidant properties and has been reported to increase COX-1 expression and improve the cognitive function of Aβ-induced AD rats (471).
3. TCMs ameliorate inflammation
Central and peripheral inflammatory activation is featured during the progression of AD. Some TCM nutraceuticals have demonstrated robust anti-inflammatory activity. Forsythia suspensa (Thunb.) Vahl is a climbing plant, the fruit of which is a famous TCM commonly used in clinical treatments for gonorrhea, erysipelas, inflammation, pyrexia, and ulcer (435). Forsythiaside, isolated from F. suspensa, has been reported to improve cognition and memory in accelerated aging mice and SAMP8 mice, by suppressing oxidative stress and decreasing inflammatory cytokines (411). The CTS mentioned earlier also has an anti-inflammatory effect (293, 386). These studies imply that the constituents with anti-inflammatory or antioxidant properties may partially relieve symptoms of AD by improving oxidative stress or decreasing inflammation.
4. A variety of TCMs with mitochondria-related functional readouts may reveal a new therapeutic strategy in AD treatment
Given that complex metabolic disorders interact and progress from early to late stages of AD, the TCMs that rescue more than one AD-related critical metabolic disorder may be better candidates and be more helpful for AD treatment (Fig. 12).
In addition to the TCMs mentioned earlier, other TCMs, such as Coptis chinensis Franch, berberine, and Sophora flavescens Ait, also show benefits in both metabolic dysfunction and AD treatment (71, 170). Berberine, a major isoquinoline alkaloid in C. chinensis Franch, an important TCM herb that has been widely used (444), has multiple pharmacological functions, including anti-inflammatory, antioxidant, antidiabetic, liver-protective, and neuroprotective activities. Berberine has been reported to efficiently reduce cerebral Aβ levels and glial activation and significantly ameliorate cognitive impairment in TgCRND8 mice (90). Other recent studies have also found that berberine can additionally treat senile dementia by improving antioxidative stress, metabolism, and other multitarget pathways (170).
Matrine, the major active component of S. flavescens, which is typically used to treat dementia and exerts multiple pharmacological functions similar to berberine, has been found to be efficient in improving cognitive deficits in APP/PS1 mice by inhibiting Aβ aggregation in vivo and blocking the Aβ/RAGE axis (71). These two components not only recover the metabolic symptoms associated with AD but also reduce Aβ levels.
Although most of the studies described earlier were conducted in cellular and animal AD models, a systemic survey of TCMs using a variety of mitochondrial functional readouts may reveal new therapeutic strategies (406) based on clues accumulating from Chinese medicine practices. TCMs and some other herbal medicines are occasionally challenged due to a lack of rigorous scientific studies. It has been proposed that TCMs may act through hormetic dose
VII. Concluding Remarks
AD and metabolic syndrome are persistent challenges to morbidity, mortality, lifestyles, and human life expectancy. AD is almost incurable at this stage, and there are almost no effective interventions or treatments. The long-term use of the existing drugs is often accompanied by serious adverse side effects. Currently, treatment is aimed at improving cognitive function, working ability, and quality of life. However, the current treatment can hardly delay or reverse the course of this disease, let alone cure it. AD is a complex disease involving many signaling molecules, and overlapping genetic and environmental factors are involved in the development of diseases. Various factors have made the treatment of AD a challenging and urgent research hotspot.
The pathogenesis of AD is still unclear. The association between AD and metabolic syndrome from the evidence of clinical, epidemiological genetic, and molecular discoveries has inspired scientists to investigate the underlying mechanisms. Increasing evidence implies that central and peripheral mitochondrial metabolic deficiency occurs even earlier than the preclinical phase of AD (Fig. 14). The early diagnosis of AD, indicated by biomarkers, and the early intervention may provide a new strategy for AD treatment. As an important organelle for regulating the “life” and “death” of cells, mitochondria play an important role in the occurrence and development of AD. Therefore, targeting mitochondria will become a new strategy and an important means for the treatment of AD. Mitochondria are susceptible to the earliest signs of AD pathogenesis; remodeled mitochondrial metabolites represent a potential group of novel peripheral diagnostic markers of AD and are more readily detected than CNS-oriented markers in bodily fluids.
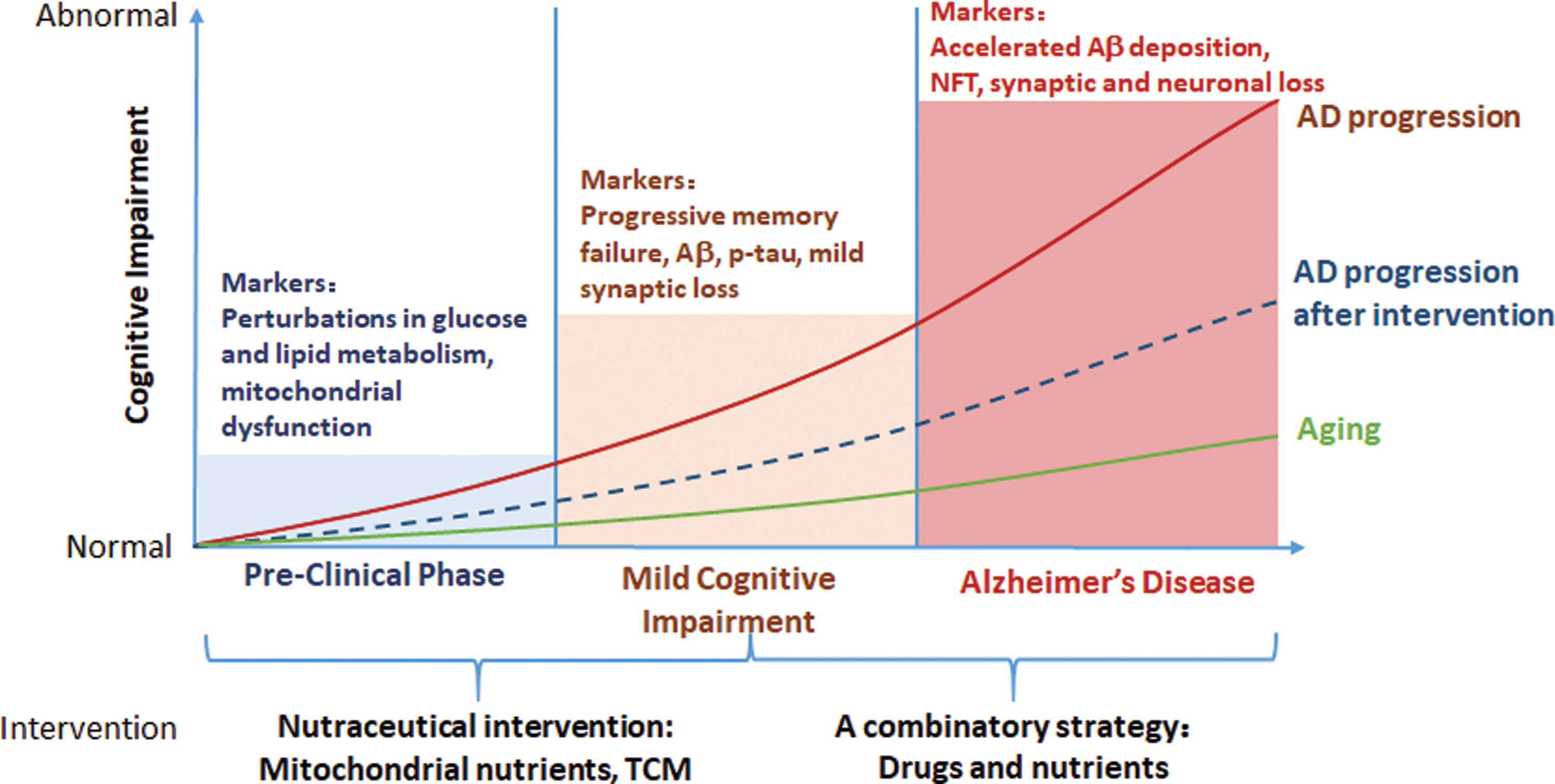
Mitochondrial nutrients, as well as some compounds from Chinese traditional medicine, ameliorate some metabolic disorders, including mitochondrial metabolic dysfunction, insulin resistance, and inflammatory provocation in AD. Moreover, the crosstalk between the brain and periphery links metabolic regulation and brain function, which implies a new aspect for identifying therapeutic targets for AD. Therefore, treating AD by targeting mitochondria provides a promising approach to preventing and delaying AD progression (Fig. 15). Mitochondrial-targeted therapy is still at the exploratory stage. Although many studies have shown that this treatment has a certain therapeutic effect, significant clinical improvement has not yet been confirmed. Thus, both clinical experimental and basic research on the pathogenesis of this disease is warranted.
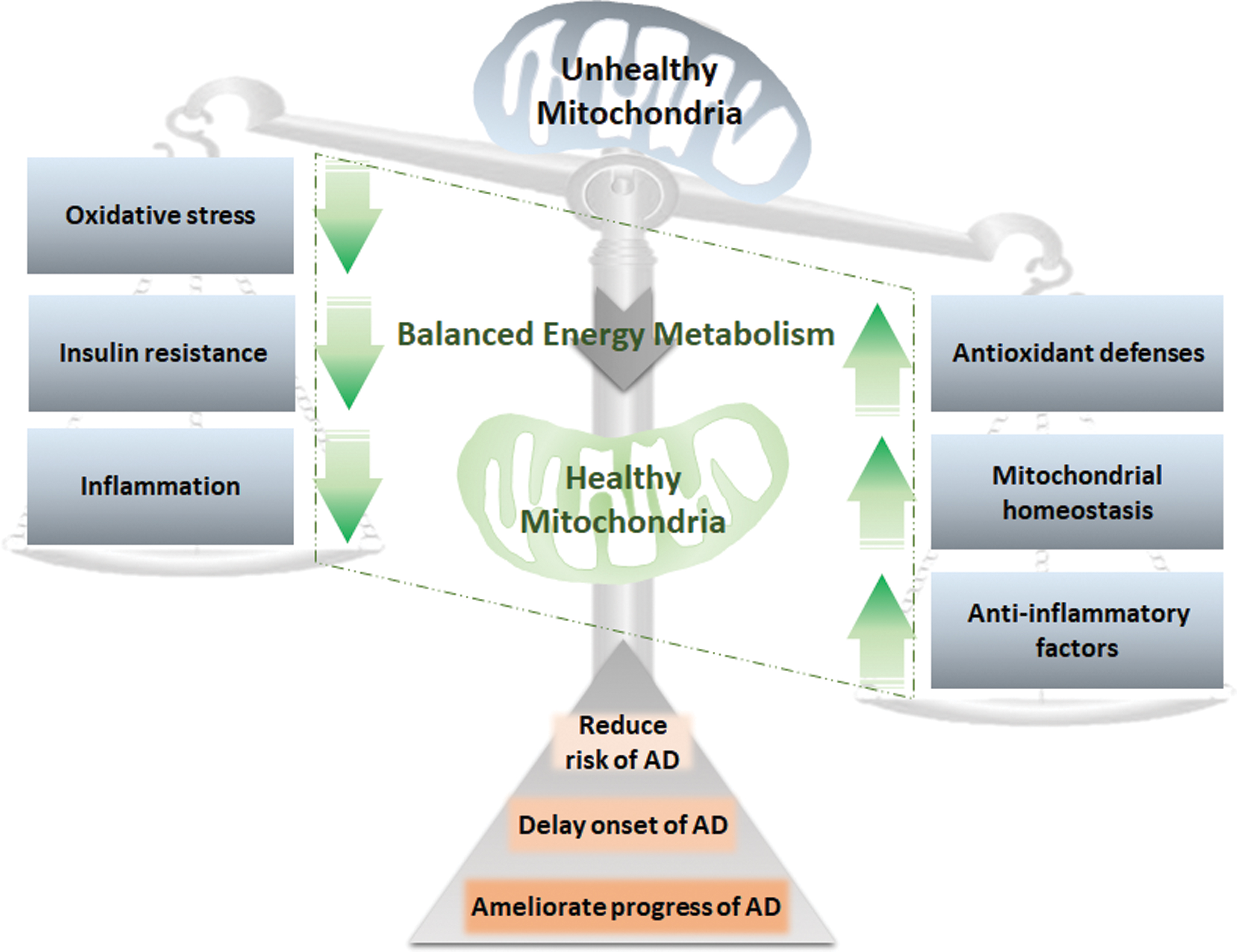
Treatments aimed at a single target or single pathology of AD are less efficient in AD medicine development. In contrast, the combined therapy of mitochondrial nutrients or TCMs and exercise synergistically ameliorates mitochondrial-induced oxidative stress, metabolic dysfunction, and inflammation in both the CNS and the periphery and could be more successful than monotherapy.
To date, numerous publications have shown that dietary or lifestyle interventions are associated with the risk of AD or reduce the progression of AD; however, scientific evidence is limited, and further studies and clinical trials are needed. The mechanism of AD is still the most important issue to investigate and remains highly prioritized, and the exact treatments needed to cure AD cannot be discovered until the mechanism has been elucidated.
Footnotes
Acknowledgment
The authors thank Dr. Byron C. Jones, University of Tennessee for the critical comments and editing.
Funding Information
This work was supported by the National Basic Research Program of China [973 program nos. 2015CB856302 and 2015CB553602], the National Natural Science Foundation of China [Nos. 81741110, 31770917, 31570777, 31870848, and 81802787], the China Postdoctoral Science Foundation [no. 2018M643673], and the opening foundation of the State Key Laboratory of Space Medicine Fundamentals and Application, Chinese Astronaut Research and Training Center [no. SMFA15K01].