
Abstract
Significance:
Cellular redox processes are highly interconnected, yet not in equilibrium, and governed by a wide range of biochemical parameters. Technological advances continue refining how specific redox processes are regulated, but broad understanding of the dynamic interconnectivity between cellular redox modules remains limited. Systems biology investigates multiple components in complex environments and can provide integrative insights into the multifaceted cellular redox state. This review describes the state of the art in redox systems biology as well as provides an updated perspective and practical guide for harnessing thousands of cysteine sensors in the redoxome for multiparameter characterization of cellular redox networks.
Recent Advances:
Redox systems biology has been applied to genome-scale models and large public datasets, challenged common conceptions, and provided new insights that complement reductionist approaches. Advances in public knowledge and user-friendly tools for proteome-wide annotation of cysteine sentinels can now leverage cysteine redox proteomics datasets to provide spatial, functional, and protein structural information.
Critical Issues:
Careful consideration of available analytical approaches is needed to broadly characterize the systems-level properties of redox signaling networks and be experimentally feasible. The cysteine redoxome is an informative focal point since it integrates many aspects of redox biology. The mechanisms and redox modules governing cysteine redox regulation, cysteine oxidation assays, proteome-wide annotation of the biophysical and biochemical properties of individual cysteines, and their clinical application are discussed.
Future Directions:
Investigating the cysteine redoxome at a systems level will uncover new insights into the mechanisms of selectivity and context dependence of redox signaling networks.
Redox Systems Biology: Goals
The goal of redox systems biology, defined here as integrative profiling or modeling aimed toward comprehensive, multiparameter understanding of the cellular redox state, is to address the dimensionality and reductionist challenges of redox biology. While systems biology often does not test clear hypotheses, it is best suited to answer many important redox questions that require an integrative solution. Is there a global redox setpoint or is each redox module (Figs. 1 and 2) independent (2)? How can bottlenecks of redox biochemistry be efficiently identified and confirmed in cells, such as contexts in which nicotinamide adenine dinucleotide phosphate (NADPH) or glutathione (GSH) becomes limiting (92)? What are the design principles of redox signaling and what distinguishes redox signaling from oxidative stress (76)? What specific roles does redox biology play as a hallmark of cancer (65) and other diseases? What sets redox homeostatic setpoints, what triggers their adaptation in cancer and other diseases (35), and how can therapeutic intervention restore them? How personalized or individualized are redox processes? What are the key buffers between redox modules enabling regulatory specificity and providing context dependence to redox signaling networks?
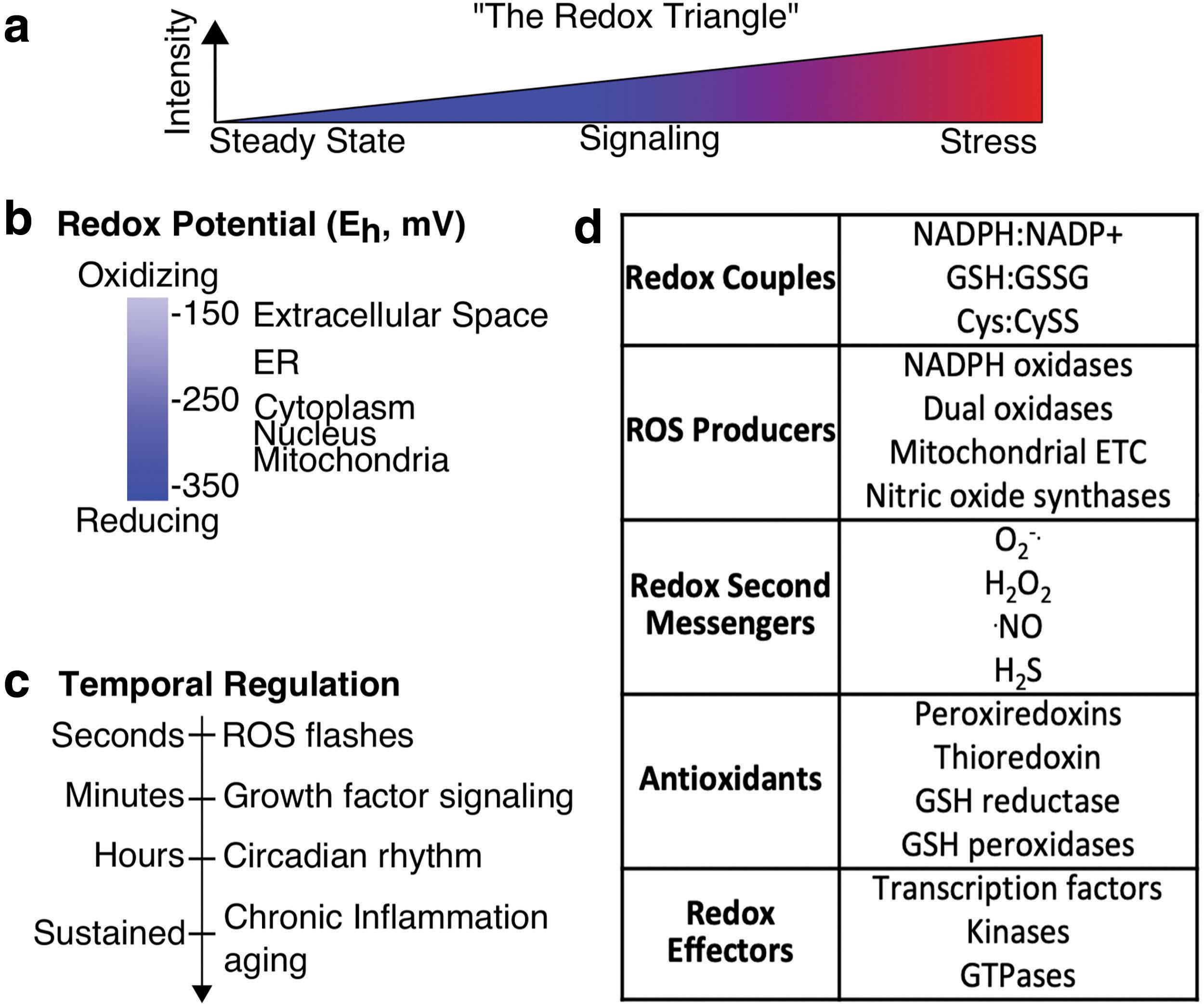
Broader goals of redox systems biology include generating new hypotheses, defining the design principles of redox signaling, and confronting common (mis)conceptions, which are discussed throughout. One essential challenge for redox systems biology is to supersede the ubiquitous “redox triangle” model (Fig. 1a). In the redox triangle, the primary driver of redox biology is the level of a vague redox stimulus, usually inferred as reactive oxygen species (ROS) that directs trinary outputs of cell steady state, signaling, and stress. While simple, the triangle representation of redox biology is misleading and puts a misplaced emphasis on ROS levels (20) instead of circuitry, network logic, and cellular location. Focusing on ROS levels, in turn, propagates the “antioxidant paradoxes” of cancer, aging, and diabetes (9, 25, 109, 143). Systems-level understanding of redox biology is a key to accurately simplify visualization of the essential network structures of redox biology (38) and to delineate and focus on the functional regulatory nodes and circuitry based on specific cellular contexts.
Redox Biology: Systems-Level Challenges
Redox reactions are essential for life as key transducers of cell signaling and metabolism. Redox processes are full of contradiction, however, defying easily interpretable cause–effect logic, and are often nonlinear, ultrasensitive, or hysteretic (55, 138, 163, 172). Discerning which redox module(s) are responsible for a specific function or phenotype and delineating how the module(s) function codependently with other cellular components remain challenging. The interconnectivity crucial to the structure of cellular redox networks taxes the reductionist model of breaking down systems into individual components to isolate a single root. Simplification is further hindered since many redox processes cannot be accurately modeled outside of a cell (137), yet within a cell there are many different redox processes that are not in thermodynamic equilibrium (80) and vary across organelles (60) (Fig. 1b). Kinetic competition among many reactions drives selectivity of redox processes, but adds another layer of complexity that is difficult to model as simply and intuitively as signal transduction and transcriptional networks (2, 183). In addition, redox cycles occur on diverse temporal scales since circadian rhythm, cell cycle, and light/dark cycles alter cellular redox state over the course of hours (39, 63, 167, 190), chronic diseases alter redox state over months and years (57), yet significant perturbation to redox homeostasis can be resolved in minutes or less to return to steady state (28, 139) (Fig. 1c). The relationship and interdependence of redox processes throughout the cell and across different time scales remain poorly understood.
Cellular redox systems include a wide array of components that are cooperative (190), but hierarchical and have varying specificities and kinetics (Fig. 1d). These can be grouped into five primary categories: (i) small molecule redox couples (NADPH:NADP+, NADH:NAD+, GSH: oxidized glutathione [GSSG]) that are often catalytically coupled to the activity of (ii) antioxidant proteins such as peroxiredoxins (PRDXs), thioredoxin (TRX), and GSH-utilizing enzymes. (iii) Small metabolites often derived from oxygen such as superoxide (O2 −•), hydrogen peroxide (H2O2), and nitric oxide (•NO). Some of these play clear roles as signaling second messengers but others, such as the hydroxyl radical, lack the specificity or reversibility needed for signaling (48). Hydrogen sulfide (H2S) and cysteine are small molecule effectors that participate in redox processes but do not use oxygen (10, 17). (iv) The major endogenous sources of ROS for redox signaling include NADPH oxidases (NOXs), dual oxidases (DUOXs), nitric oxide synthases, and the mitochondrial electron transport chain. (v) The downstream effectors of redox biology, which may be directly or indirectly redox regulated, include transcription factors such as nuclear factor erythroid 2-related factor 2 (NRF2), hypoxia-inducible factor 1-alpha (HIF1α), aryl hydrocarbon receptor, and nuclear factor NF-kappa-B p100 subunit (NFκB), kinases such as MAP kinases (MAPKs), and GTPases (68).
Redox biology suffers from the “curse of dimensionality” due to the many integrated components and high spatiotemporal diversity. While new chemical and imaging technologies with increased chemical (4, 120, 124, 125, 137, 139, 140) or spatiotemporal specificity (103, 137, 166, 193) have unequivocally increased our knowledge of redox biology, building of integrative, cell-level, bottom-up models of redox biology with these narrowly focused techniques is experimentally untenable (20, 177, 193). For example, the current state of the art approach combining genetically encoded sensors for NADPH:NADP+, NADH:NAD+, H2O2 levels, and/or the thiol redox state in cells is limited by fluorescent spectral overlap for each fluorophore, and thus each parameter must be measured in nonoverlapping cellular compartments (193). Assaying additional spatial combinations of redox processes requires generating new cell lines with a different set of genetically encoded sensors. Using these tools to approach a full accounting of the cellular redox state is therefore experimentally challenging. ROS assays similarly suffer from spectral limitations that restrict parallel analysis of different ROS, localization, or other redox parameters. The ability to introduce fluorophores with specific redox readouts on demand would help alleviate these issues. Sallin et al. took a step toward this goal by utilizing Snifit tags, biosensors of cellular NAD+ levels, and the NADPH:NADP+ ratio, which are semisynthetic and can be flexibly conjugated to target proteins in live cells in real time similar to Halo- or SNAP tags (132).
Redox Systems Biology: Metabolism
Constraint-based mathematical modeling with flux balance analysis (FBA) is a common technique to simplify the complexity of biochemical networks and predict the steady state flux distribution of metabolites (116). FBA is especially useful to determine the capacity of metabolic networks (41, 42, 45, 191) and narrow down possible model topologies (2, 116, 172) when combined with experimental results, including public data. FBA coupled with carbon metabolite tracing revealed that, unexpectedly, serine-driven 1-carbon metabolism contributes as much to NADPH production as the pentose phosphate shunt (PPP) via the activity of methylenetetrahydrofolate dehydrogenase (42).
A hallmark of redox processes is their dynamic, yet robust mechanisms maintaining homeostasis (28, 66). Elucidating how the cellular redox system transitions to respond to perturbations, transduces adaptive signaling, and either restores or adjusts homeostasis remains an important challenge. To examine the mechanism of Escherichia coli's rapid response to stress, Christodoulou et al. quantified 30 metabolites at 10-s intervals up to 1 min after stimulation with high levels of H2O2 (28). FBA of glycolysis and the PPP that leveraged publicly available rate constants generated a candidate regulatory model focused on a negative interaction between glucose-6-phosphate dehydrogenase (G6PDH) and NADPH. The authors proposed, and subsequently verified, that H2O2 stress decreases NADPH levels within seconds, relieving NADPH-dependent inhibition of G6PDH to increase flux through the PPP to restore NADPH levels. This study highlights the robustness of the publicly available redox kinetic data and the power of computational modeling to in silico evaluate many potential redox components and narrow the set of possibilities down to be feasible for experimental testing. Kuehne et al. used time-resolved metabolic profiling to demonstrate that the same NADPH–G6PDH mechanism occurs in human fibroblasts in response to H2O2 or ultraviolet stress (88). This mechanism is distinct from oxidation of pyruvate kinase M2 C358, another redox-based mechanism inhibiting lower glycolysis to drive PPP flux and NADPH production (7) that likely works on a longer time scale to fine tune metabolism after it is restored initially by G6PDH activation. Notably, as these studies all focus on modeling oxidative stress, transitioning these systems-level approaches to nonstress conditions in which H2O2 drives redox signaling may reveal important new linkages between metabolic adaptation and sensors of redox alterations.
While the above studies focus on specific metabolic pathways, the ultimate goal of redox systems biology is large-scale prediction and characterization of redox modules (38). Toward this goal, Lewis et al. modeled the NADPH-dependent cycling of the chemotherapeutic β-lapachone in head and neck cancer cells at the genome level using transcriptomic results (92). After modifying the Recon 2 metabolite network to include all NADPH metabolic reactions, the authors performed FBA of a network encompassing >5000 metabolites and 7000 reactions. Utilizing transcriptomic data from either matched head and neck cancer cell lines or primary tumors in The Cancer Genome Atlas that were either sensitive or resistant to β-lapachone or radiation, the authors simulated the effect of gene knockdown on drug sensitivity and predicted that isocitrate dehydrogenase 1 (IDH1) played an especially important role in NADPH production that mediated drug resistance. These predictions were experimentally verified by IDH1 knockdown in radiation-resistant cells, which routed NADPH flux through glutamate dehydrogenase 1/2. This study demonstrates the potential for genome-scale modeling of redox reactions in cell lines and primary tumors using public data. It is especially exciting that specific proteins and redox reactions that are limited by, or tuned to, such a complex redox hub as NADPH can be predicted.
To date, most redox systems biology advances have been limited to metabolism and use of mathematical modeling. While FBA has clear utility, it has notable limitations, including the inability to predict the dynamic changes in flux to a stimulus since kinetic parameters are not incorporated (116). Therefore, FBA only has limited ability to study redox signaling, which is dynamic, or vastly altered metabolic states that are not included in a generic map of human metabolism such as those that occur during carcinogenesis. In addition, constraints lie at the heart of FBA, and thus FBA requires complete information for all reaction components without any missing data. Protein expression therefore has limited utility for FBA because proteomics datasets have substantial amounts of missing data. Kinetic modeling is the primary alternative to FBA for metabolic systems in applications where time-dependent dynamics are of interest (2, 181). Finally, since only the successfully verified mathematical models are published, it is not possible to unbiasedly gauge the overall accuracy of these approaches. Community-wide, crowdsourced predictions such as DREAM challenges (100) offer an important opportunity to gauge the effectiveness of current mathematical redox modeling approaches.
Redox Systems Biology: Beyond Metabolism
Mathematical modeling and large-scale cysteine redox profiling have provided insight into the hierarchic interdependence of cellular antioxidant modules (83, 122, 123). Modeling H2O2 clearance in Jurkat cells, Adimora et al. discerned the relative contribution of various antioxidant modules to buffer H2O2 and the percentage of proteins that undergo S-glutathionylation versus disulfide formation (2). While catalase accounted for <1% of H2O2 catabolism in Jurkat cells, Benfeitas et al. modeled that catalase and PRDXs contribute similarly to H2O2 metabolism in red blood cells (16). Taking a different approach, Le Moan et al. and Go et al. performed proteomic analysis of either yeast or mammalian cells, respectively, in which the TRX or GSH systems were selectively impaired, both finding that each antioxidant module controls the redox state of different subsets of cysteines in the proteome (58, 107). Systems-level metabolic profiling also revealed a new metabolic pathway providing cellular antioxidant capacity in normal hepatocytes that, defying expectation, survive even in the absence of the activity of two critical antioxidants, TRX and glutathione reductase (GR) (41). Eriksson et al. discerned that methionine is able to serve as the precursor of cysteine in TRX/GR-null livers, in contrast to the canonical model in which cysteine is taken up as its oxidized form cystine that is then reduced by the TRX/GR system for use (41).
Redox systems biology techniques have facilitated development of biosensors for high-throughput, single cell analysis by identifying or directing evolution of NADPH-utilizing proteins. Siedler et al. developed an NADPH biosensor based on yellow fluorescent protein expression coupled to the SoxR protein promoter that is activated by increased NADPH (144). The authors performed a high-throughput, systematic screen of NADPH-utilizing enzymes in large E. coli-mutant libraries to identify novel variant proteins with increased NADPH utilization that have potential application in metabolic engineering. Zhang et al. similarly developed a growth selection platform capable of screening 108 variants per round of selection to identify NADPH-generating glyceraldehyde-3-phosphate dehydrogenases with improved activity as well as direct evolution of a Lactobacillus delbrueckii lactate dehydrogenase variant with NADPH specificity instead of NADH (192).
NRF2, NFκB, HIF1α, MAPK, and forkhead box proteins constitute the master regulators of redox-sensitive transcriptional networks (68). These proteins work in coordination to initiate a transcriptional response that pleiotropically reprogram the cell's redox homeostatic setpoint(s), not just a simple upregulation of antioxidant capacity (68). In addition, sirtuins are protein deacetylases that play a unique role in redox biology by being tuned to NAD+ levels, capable of inducing ROS production and regulating protein acetylation (147). Application of systems biology beyond redox biology has identified new regulatory architectures in which hyperconnected master regulator proteins, via post-translational regulation, serve as integrative controllers of vast transcriptional networks in cell homeostasis, cancer, drug resistance (3, 23, 174). These master regulatory proteins stably maintain complex phenotypes such as cancer despite the high genomic and proteomic heterogeneity across tumors, but notably are not frequently mutated or studied and difficult to identify using conventional experiments (3, 23, 174). Application of these computational techniques to redox biology may uncover new redox regulator effectors that are responsible for higher level organization and interconnection of redox modules.
Cysteines in the Proteome: Informative and Integrative Redox Sentinels
All redox parameters would ideally be exhaustively and quantitatively characterized in space, time, and abundance to make a full accounting of the cellular redox status. However, the requisite tools to do so are lacking, and the extensive amount of time it would take to acquire all results with quantitative accuracy limits feasibility. Tangible advancement of redox systems biology will require careful consideration of feature selection, data dimensionality reduction, and collection of minimally redundant, maximally orthogonal data. For example, how and when do ROS measurements provide value if the sources and targets of the ROS are characterized, especially given the continued technical challenges of measuring ROS and their limited mechanistic value (20, 40)? More generally, how is the cellular redox state best assessed, and what redox parameter(s) are most effective for modeling and most efficiently determined experimentally?
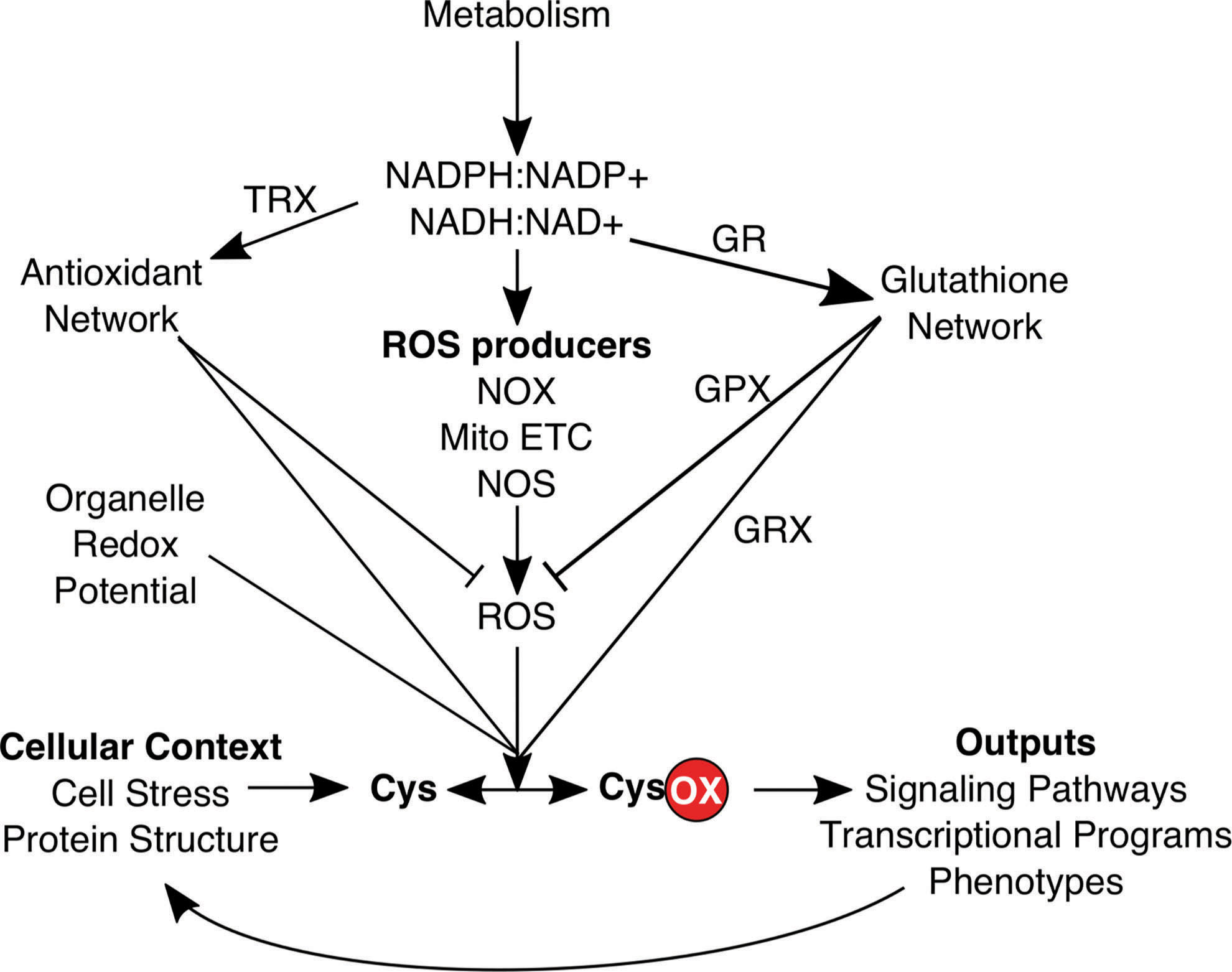
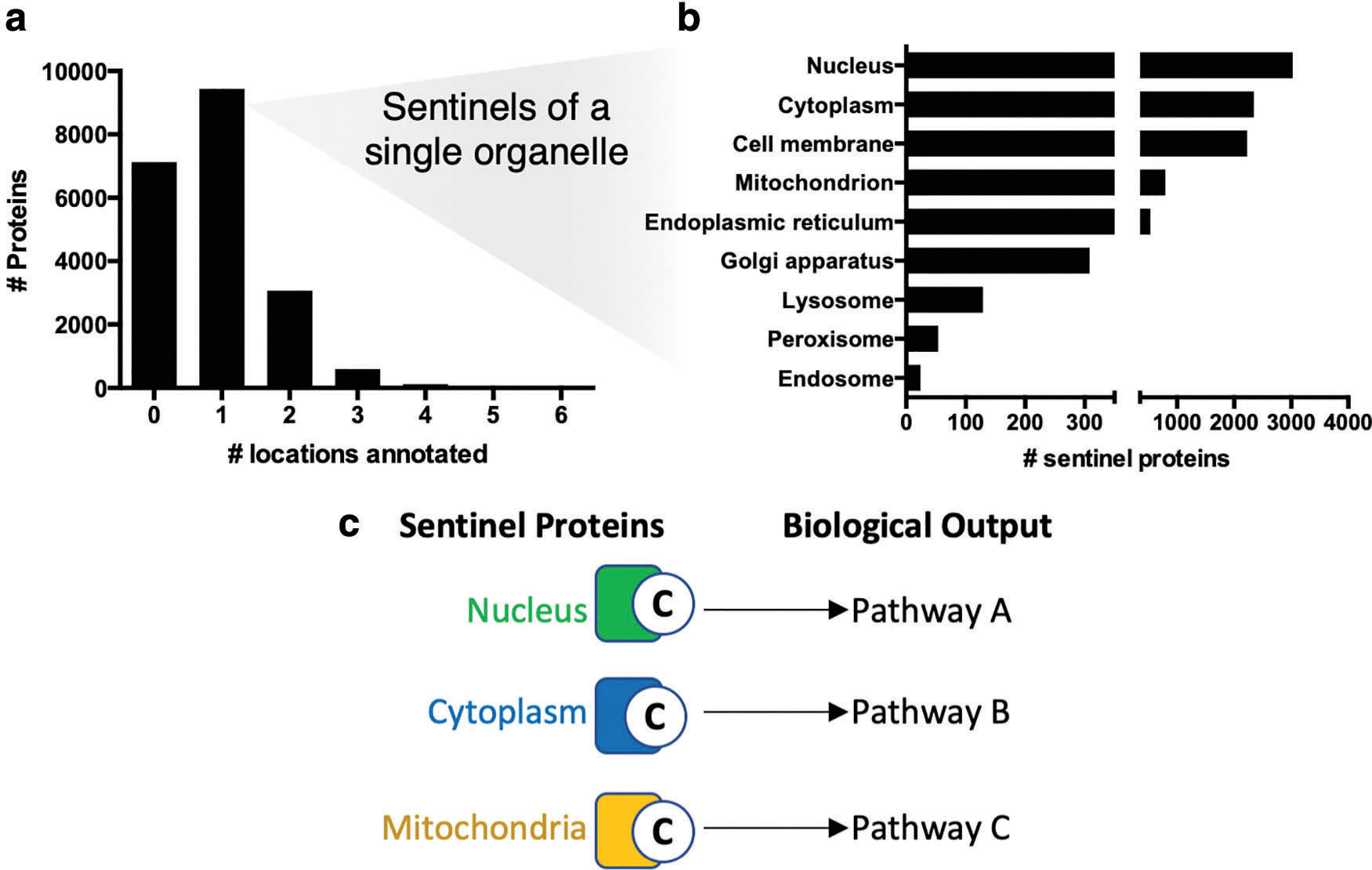
Multiple properties of cysteines make them excellent and relatively specific redox sentinels due to their thiol side chain that can form seven stable oxidation states in vivo (56, 74, 127), which has been extensively reviewed (61, 70, 80, 127, 183). Experimental and bioinformatic analysis of the cysteine redoxome, defined as in Thamsen and Jakob (158) as the composite of all redox active cysteines, can integratively inform on many redox modules throughout the cell such as specific types of ROS, redox couples, ROS producers, and GSH-linked enzymes (80) (Fig. 2). For example, even in the absence of imaging data, cysteine redoxome analysis can spatially delineate redox alterations at the plasma membrane, mitochondria, and other organelles by examining which sentinel proteins localized to each organelle are redox regulated (Fig. 3).
Cysteines are oxidized by many ROS and redox effectors, including O2 −•, H2O2, •NO, H2S, Cys, and GSH, to form specific adducts that can be selectively analyzed (4, 79, 117, 120, 124, 125, 133, 154). Recently, disulfide linkage between cysteines and the sulfur in coenzyme A (S-CoAlation) directly linking cysteine oxidation to metabolism was found to be a new cysteine redox modification established by antibody-based assays and mass spectrometry (165). Analysis of specific cysteine oxoforms can rapidly or comprehensively assess global and spatial information on the state of these second messenger effectors when coupled with gel-based assays, microscopy, or proteomics. Cysteines in some proteins can provide multiple levels of regulation such as the PRDX family. Not only are different PRDX family members spatially localized to distinct organelles, but the catalytic and resolving cysteines are also highly tuned to H2O2. Thus, analysis of the redox state of PRDX family cysteines can inform on local H2O2 levels throughout the cell even without microscopy or ROS assays (129).
There is strong evolutionary selection on cysteines. On one hand, negative selection due to their intrinsic oxidizability and reactivity has led cysteine to be the rarest amino acid. Perhaps more insightfully, cysteine is also the most buried amino acid despite its relative polar nature (101, 102). On the other hand, when cysteines do arise during evolution they typically carry a clear functional benefit and are seldom lost later (171, 184). As cysteines are rare, buried, and highly selected for specific function(s) in proteins, it follows that each cysteine could be considered a unique sentinel tuned to a set of context-specific factors informing on one or more redox modules (18, 80) (Fig. 4). Conceptually, cysteines bridge the systems–reductionist divide (151) since analysis of individual cysteines provides detailed biological information, but the cysteine redoxome can provide a systems-level readout of the cellular redox state when viewed globally. Notably, cysteine redoxome analysis can link altered redox state with specific proteins and biological pathways, which adds considerable mechanistic value over other redox assays (20, 38).
Cysteine Redox Regulation: Properties and Mechanisms of Specificity in Redox Signaling Networks
Properties of a cysteine affecting its oxidizability are proximity to the source of ROS (183), low acid dissociation constant (pKa) (22, 96, 183), and solvent accessibility (102), which are necessary for direct interaction with a ROS. Redox regulation can also occur without direct interaction with ROS via disulfide relays (152, 161), transnitrosylation (64), or protein translocation to an organelle with a different redox setpoint (60). Notably, a subset of cysteines can be oxidized even in the absence of global changes to the antioxidant system (59) or detectable increase in ROS (20). This is due in part to the limited sensitivity of ROS assays and because the many endogenously redox-regulated cysteines are very reactive and highly tuned to the local redox milieu.
The oxidizability of a cysteine also depends on its neighbors. Kinetic competition, in which multiple potential reactions vie for a use of a reactant, is an important mechanism of redox selectivity in the complex cellular environment (2, 89, 183). For example, PRDXs are abundant proteins that are highly reactive with H2O2, potentially outcompeting local sulfenation of most other cysteines intracellularly (89, 149, 162, 170). Once oxidized, a PRDX can subsequently form a disulfide relay with neighboring proteins in a relatively specific interaction (150, 152, 161). Thus, protein–protein interactions also dictate redox regulation of a protein. Notably, kinetic modeling of the PRDX2-STAT3 disulfide relay suggests that its slow transfer kinetics are unfavorable in the context of potential competing reactions in the cell (89). The authors reconcile their results with published experimental findings (149) by re-evaluating the PRDX disulfide relay mechanism, and hypothesize that it may be facilitated by a third, scaffolding component. Transnitrosylation has also been reported to selectively relay a nitrosyl group from one cysteine to another in a protein (111).
The oxidizability of cysteines also depends on the type of ROS. For example, PRDXs are especially sensitive to H2O2, whereas aconitase and other iron sulfur (Fe–S) cluster containing proteins are more sensitive to O2 −• (169, 179). Global proteomic studies focused on analysis of specific cysteine oxoforms have established that the selectivity of certain cysteines for certain redox effectors is widespread, including •NO, GSH, and H2O2 (62).
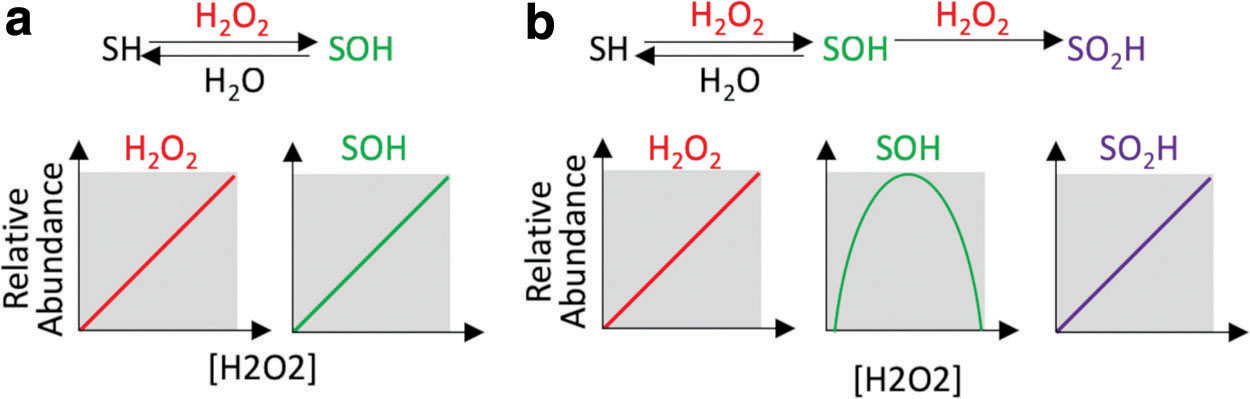
While the magnitude of cysteine oxidation is generally closely correlated with ROS levels, each cysteine oxoform has unique kinetics and selectivity for individual cysteine residues (138). Modeling the kinetics of cysteine sulfenic acid (SOH) formation serves as an example, which explains counterintuitive experimental results. Cysteine SOH is the direct oxidation product of cysteine with H2O2. Therefore, a priori, levels of H2O2 might be hypothesized to correlate directly with levels of cysteine SOH (Fig. 5a). However, empirically this is not observed; as increased exogenous H2O2 and epidermal growth factor (EGF) stimulation results in a biphasic change in SOH levels, increasing to a tipping point, beyond which increased H2O2 decreases SOH levels (120, 133). Kinetic modeling can explain these nonlinear, counterintuitive results. Inclusion of cysteine sulfinic acid (SO2H), the reaction product of SOH + H2O2, into the kinetic model fits the observed biphasic response of SOH to H2O2 treatment (Fig. 5b). This example underscores how simple network models can guide intuitive understanding of unexpected, but fundamentally important properties of redox biology as well as emphasizes that redox processes are rarely linear. Importantly, while SOH levels do not parallel H2O2 levels, assaying sulfenation of the cysteine redoxome output is potentially more meaningful as it indicates the transition point of sulfenation to sulfination (SO2H) and more accurately represents the oxidation of downstream H2O2 protein effectors.
The design principles of cysteine redox networks are most thoroughly understood by examining EGF signaling (Fig. 6), the best characterized model of redox signaling (120, 188) whose study was initiated by insulin receptor signaling decades earlier (21, 33, 104). Growth factor signaling highlights the importance of coordination to redox regulation, and includes both concerted oxidation of multiple cysteines in proteins and codependence with redox-independent processes to elicit specificity. The first principle is the importance of a spatially constrained source of ROS. In the case of EGF, this is activation of NOX or DUOXs at the plasma membrane (26, 60, 120). Second is the spatial specificity of the O2 −• and H2O2 produced by rapid intracellular degradation of ROS that limits their diffusion distance (95, 103, 120, 166). A third principle is the relatively short duration of altered redox state before returning to homeostasis (66), <60 min in the case of EGF stimulation (120). Fourth, oxidation of multiple cysteines in several proteins is critical to tipping the balance toward EGF-dependent phosphorylation. Oxidation of the catalytic cysteine in protein tyrosine phosphatases decreases the brake on phosphotyrosine signaling (46), and oxidation of cysteine in the kinase domain of epidermal growth factor receptor (EGFR) increases its activity up to a point, above which its kinase activity decreases (120). Notably, there is selectivity in which phosphatases are oxidized since EGF is a poor oxidizer of PTP1B yet oxidizes SHP-2 at very low EGF levels (120). While there is no clear mechanistic explanation for this, it is likely to be context-dependent changes in the phosphatases rather than an intrinsic property of the phosphatase(s) themselves since insulin preferentially oxidizes PTP1B (67). A fifth design principle is the interdependence of redox-dependent and -independent events (15). EGF-dependent phosphotyrosine signaling occurs more rapidly than redox processes, peaking ∼1 min after EGF stimulation (32, 115), whereas ROS production and cysteine SOH are delayed and maximal at 5 min (120). This delay is setup because EGF-dependent phosphorylation of NOX by SRC is required to trigger NOX activation and ROS production. SRC-dependent phosphorylation of Y194 of PRDX1 also locally deactivates it near the cell membrane (185), facilitating local buildup of H2O2 levels (97, 185). From a network perspective, redox signaling serves as an amplifier, rather than initiator, of EGF signaling, and the temporal delay of phosphatase and kinase regulation likely promotes ultrasensitivity of EGF signaling, sharpening the response to become more binary and less graded (72, 168).
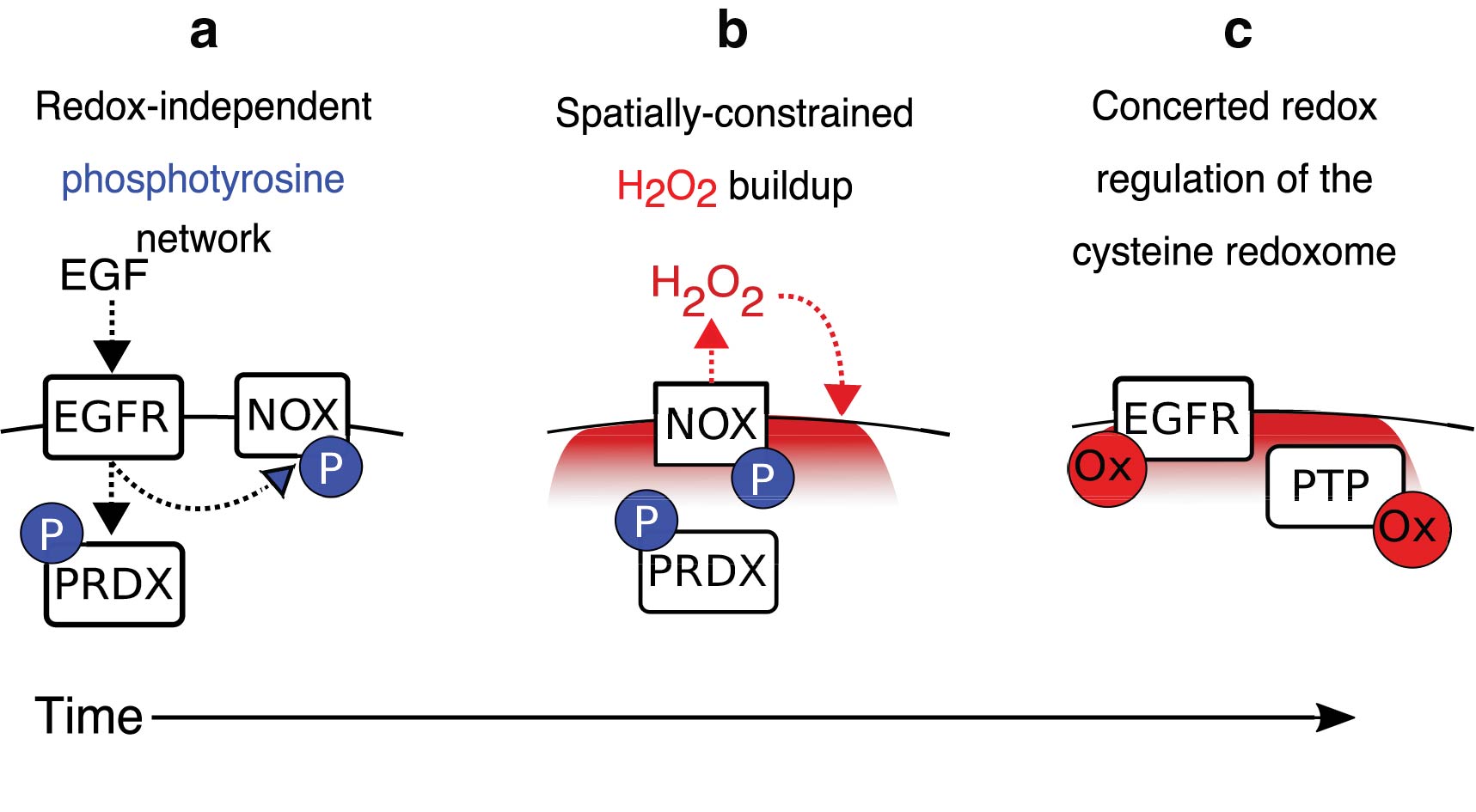
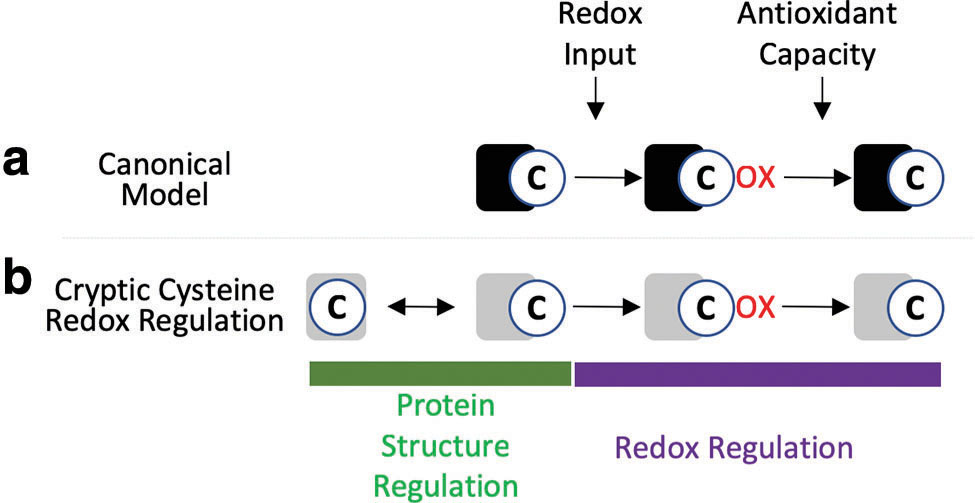
The temporal separation of redox-independent and redox-dependent events plays a key role in specifying which cysteines are oxidized by EGF due to an emerging relationship between protein structure and oxidation of cryptic cysteines, those that are only solvent exposed upon changes in protein conformation (6, 15, 75, 86). It has been estimated that 40%–70% of proteins are allosterically regulated and contain cryptic pockets (29, 114), and several studies have found that changes in protein folding can allosterically alter cysteine solvent accessibly (13, 53). Redox regulation of cryptic cysteines was conclusively demonstrated by Alegre-Cebollada in which cell stretching mechanically unfolded the elastic protein titin and allowed several cysteines to become solvent exposed and glutathionylated (6). Redox regulation inhibited titin refolding, stabilizing the extended structure to impart a mechanically weak state, which decreased cardiomyocytes stiffness (6). More broadly, Gould et al. performed a redox proteomic analysis of ∼3000 cysteines in mouse liver under steady state physiological conditions, and found that the cysteines oxidized at steady state had neither a low pKa nor were especially solvent accessible (62). A similar conclusion was reached in another large-scale profiling of redox-regulated cysteines (51). While this could be due to inaccurate pKa predictions, a similar prediction of the cysteines oxidized by H2O2 finds them to be largely solvent accessible in several studies (53, 188). A likely explanation is that endogenous ROS oxidizes cysteines that are solvent exposed at steady state, which is not reflected in crystal structures that are biased toward stable low-energy conformers (146). Once a cryptic cysteine is oxidized, the surrounding apolar environment may stabilize it in the oxidized state (163). Alternatively, it has been proposed that hydrophobic •NO may be able to tunnel to buried cysteines surrounded by hydrophobic residues (18). Since most cysteines are buried due to negative selection (101, 102), redox regulation of cryptic cysteines provides a plausible mechanism specifying the context-dependent selectivity of redox signaling pathways (15) (Fig. 4).
Additional connections between protein structure and cysteine oxidation are notable. Thiol/disulfide exchange in proteins can be regulated by mechanical force and can be influenced by protein folding (182). Second, forbidden disulfides with relatively high disulfide redox potential that introduces strain into a protein's structure occur in many protein crystal structures and can serve as redox switches (43, 186).
Together, the cysteine redoxome contains information integrating protein structural dynamics, redox-dependent and -independent regulation, as well as the location and levels of redox perturbations (Figs. 2–4). From the perspective of biological network design (168), cysteines serve inter alia as redox sniffers [transient responses, (149, 183, 185)], toggles [hysteretic switches, (33)], amplifiers (120), blinkers [oscillators, (38, 39)], and feedback loops (21, 68). Importantly, cysteine oxidation, as compared with other assays, is uniquely suited to leverage new insights into the context dependence of redox biology by directly linking to downstream pathways. The next section details cysteine oxidation assays and the tools available to maximally harness these results, especially those from cysteine redox proteomics datasets.
Techniques to Assay Cysteine Redox Regulation
Numerous probes can directly and covalently tag specific cysteine oxoforms such as SOH (120, 124, 125), SO2H (4), persulfide (117), and SNOs (139). Incorporating a clickable handle, such as an azide or alkyne, for one step conjugation to a wide array of detection modalities facilitates protein purification as well as in situ and in vivo imaging (187). Analysis of cysteine SOH oxoforms is the most mature, with SOH probes enabling imaging (78, 120, 140) and detection of >1000 sulfenated cysteines by proteomics (188). Proteomics has also been performed using direct SNO probes (139). While there is no probe to chemically label endogenous GSH, cells can utilize biotinylated GSH ethyl ester in place of GSH (Bio-GEE), as well as more flexible, clickable GSH analogs (44, 79). Unfortunately, no technique is capable of labeling disulfide bonds directly or indirectly, thus large-scale profiling of the disulfideome is not currently feasible. However, disulfide relay partners can be identified via catalytic mutants that prevent resolution of the disulfide (112). One limitation of cysteine oxoform trapping probes is their slow reactivity and efficiency that, coupled with low endogenous modification stoichiometry, often dictates long (30–60 min) incubation times (188) and limits their utility for temporal profiling or assays that require substantial protein input. In addition, the specificity of several oxoform-directed probes has been questioned (47, 130, 160).
While trapping cysteine oxoforms with probes can be coupled with mass spectrometry (62, 139, 188), most redox proteomics utilize differential alkylation to tag all reversibly oxidized cysteines (62, 70) or selectively reduce specific oxoforms such as SNO (50, 62), S-GSH (37, 62), or persulfides (36). In the differential alkylation procedure, free thiols are first covalently labeled with an alkylating reagent such as iodoacetamide or N-ethylmaleimide (NEM), reversibly oxidized cysteines are then reduced, followed by labeling the now free cysteine thiols with a different alkylating agent to distinguish previously oxidized thiols (70). While information about specific oxoforms is often lost, the lack of selectivity may be advantageous from a systems biology perspective by being more inclusive and integrative. Differential alkylation studies also enable detection of more cysteines and thus more sentinels, casting a wider net that can be more fully leveraged for functional analysis and spatial determination of cellular redox changes (15, 62, 128). Furthermore, since cells or tissues are rapidly lysed before differential labeling, a rapid snapshot of the redox state is acquired that enables assessment of cysteine oxidation dynamics with higher temporal precision compared with in situ trapping with covalent oxoform-specific tags (15).
Cysteine redox proteomics has been applied to many models, including oxidation analysis of ∼170 peptides in Caenorhabditis elegans during development and aging (87), ∼1200 cysteines in Drosophila melanogaster under conditions of fasting and aging (105), ∼2100 cysteines in Synechocystis spp. PCC 6803 in light versus dark cycles (63), ∼2500 sites in mouse liver at steady state (62), and as many as ∼4500 and ∼9500 cysteines in yeast and mouse cells, respectively, upon oxidative stress (128). The magnitude of oxidation of the cysteine redoxome by endogenous perturbations is surprisingly widespread with ∼49% of cysteines detected having altered redox regulation by endogenous perturbation, including EGF stimulation, light–dark cycling, or fasting (15, 63, 93, 188). Despite the utility of redox proteomics to assess redox signaling, many redox proteomic studies continue to focus on models of oxidative stress (8, 27, 128, 173).
The cysteine proteome also contains reactivity and solvent accessibility information that is complementary to redox status. Cysteine protection assays have long been utilized to determine protein structural changes (1). Bar-Peled et al. adapted this approach to profile solvent accessible cysteines proteome wide in cultured cells using high levels of alkylating agents to fully label cysteines (13). Alternatively, Weerapana et al. utilized limiting doses of the alkylating reagent iodoacetamide to quantify the nucleophilicity of cysteine in the proteome (180). Nucleophilicity is correlated with pKa but is nonlinearly related to oxidizability (163). Petrova et al. profiled the reactivity of ∼6000 cysteines in drosophila in the presence or absence of the ovary-specific TRX Deadhead using a similar alkylation approach (121).
Redox systems biology, like other systems-level analyses, is currently limited in practice more by the lack of data availability than computational power (172). Unfortunately, redox proteomics assays have limited throughput due to the multiple steps in sample preparation and data analysis. Therefore, generation of proteomic data is the primary bottleneck to proteome-level characterization of cysteine sentinels in the redoxome. Analysis of multiple oxoforms (62) expands the number of samples to analyzed, restricting the number of conditions that can be profiled and limiting characterization of the dynamics of the cysteine redoxome (176). Isotope encoded, cysteine-reactive iodoTMT reagents capable of six-plex sample analysis can improve throughput of sample analysis (8, 14, 118, 119, 141) and can be optionally paired with N-terminally labeling TMT reagents to quantify protein expression changes in parallel (14, 119). Pairing matched stable-isotope encoded variants of alkylating reagents before and after reduction can estimate the oxidation stoichiometry of each cysteine detected (63, 69, 70, 91, 145); however, absolute percentage oxidation has limited biological inference, and thus relative changes in oxidation levels are typically the focus of analysis.
Large-scale mass spectrometry-based proteomics are also limited by the dynamic range, roughly three orders of magnitude, which can be overcome by targeted mass spectrometry assays to improve sensitivity (34, 69). Multiple reaction monitoring mass spectrometry has been applied to quantify targeted cysteines in many proteins (34, 69). While a downside of targeted assays is quantitation of fewer analytes, new approaches using data-independent acquisition (DIA), also known as SWATH, and retention time scheduling analysis of targeted analytes across the liquid chromatography (LC) profile dramatically increase the number of analytes that can be measured in a single mass spectrometry run (15, 134). Notably, proteomic datasets using isobaric tags such as TMT often have up to 50% of quantitative data points missing. DIA mass spectrometry can quantify nearly all analytes detected in a sample, but can be more challenging to analyze (30, 71).
Bottom-up mass spectrometry-based proteomics often does not detect key cysteines. For example, the catalytic cysteines in protein tyrosine phosphatases and PRDXs are rarely detected due to their placement in poor tryptic peptides, low abundance, or low ionizability. Despite the progress of top-down proteomics (49), translating cysteine redox proteomics assays to a pure top-down format has many technologies hurdles. However, integration of top-down and bottom-up proteomics proved useful for post-translational modification (PTM) analysis of cancer samples and may increase the completeness of redox proteomics datasets (113). Alternatives to mass spectrometry-based proteomics, such as immobilized antibody arrays, have been used to rapidly profile the glutathionylation of 1000 proteins, many of them typically below the detection limit of mass spectrometry as an additional benefit (110). However, this approach is not able to determine which specific cysteine residues are oxidized.
Feature selection is an important challenge in redox biology given its high data dimensionality. While orthogonal assays, for example, temporal characterization and proteomics, may be an ideal pair, integrating disparate datasets can be challenging. Self-organizing maps have potential to visualize and classify high dimensional data (52). Network inference to conceptualize and simplify biological reality using coexpression or transcription regulatory networks has also shown promise (148), but for redox analyses these need continued advancements to include information about redox couples, PTMs, metabolites, and/or ROS. Thus, the most efficient approach to understand the redox landscapes and the network architecture of redox modules remains elusive.
Redox-Regulated Cysteines: Predicting and Annotating Biochemical Properties
The large-scale, site-specific information provided by redox proteomic analysis is advantageous for translating information about the cysteine redoxome to integrative knowledge of the cellular redox state. While cysteine redox profiling does not provide a full account of redox biology, each analysis generates an enormous amount of data that can be harnessed for multiparameter assessment of the redox state of cells using relatively user-friendly tools. Characterizing the properties of cysteine redox sentinels can be used to infer spatial information or ROS regulation independent of microscopy or ROS assays, and is valuable for feature selection and hypothesis generation. Available resources to annotate or predict the properties of individual cysteines and redox-regulated proteins are detailed here to maximize the information content of each cysteine and utilize it as an individual redox sensor. In some cases, this information is empirically determined and publicly available, and in other cases the properties require computational prediction. Tools that are open source are prioritized whenever possible as are those that are suitable for proteome-scale analysis, rather than analysis of a single cysteine or protein at a time.
Quantitative redox proteomics methods universally proteolyze proteins into peptides with trypsin before LC-mass spectrometry analysis. Thus, the first step of proteomic data analysis is assigning each peptide to a protein or proteins that include the peptide amino acid sequence. In many cases, peptides can be assigned to a single protein, but in some cases multiple proteins share a single peptide sequence. Peptide sequences shared between homologous genes, such as AKT isoforms, may offer valuable biological information similar to PTM-specific antibodies that cannot distinguish between highly conserved regulatory sites within gene families. However, if the peptide is shared between two unrelated proteins the peptide has far less utility. Most mass spectrometry-based proteomic analysis software, such as the open source and well-supported MaxQuant (31), will indicate all proteins that can be assigned to a given peptide as well as the residue number of the modified cysteine that can be carried forward for further bioinformatic analysis.
Solvent accessibility and local protein domain information are important components of cysteine redox regulation. NetSurfP-2.0 uses deep learning to predict the relative surface accessibility, secondary structure, and disorder of all amino acids in a protein, and can process large batches of protein inputs (84). UniProtKB is a large, curated, database that contains and links to the naturally occurring and disease-relevant variants of specific amino acids, known PTMs or role in metal binding, as well as the protein domain containing each cysteine. Since the entire UniProt database can be downloaded and readily parsed, it is amenable to proteome-wide annotation.
Protein structures can also inform about the potential role of cysteines. While visual analysis is necessarily low throughput, protein crystal and solution state nuclear magnetic resonance structures can be found in the protein data bank. Chimera is an open source and powerful visualization tool for protein structure analysis that is integrated with MODELLER to build homology models if no protein structure is available (189). Interactive, virtual reality visualization of redox PTMs on proteins has recently been reported, and is an exciting new option for protein visualization (98). Finally, predicting the structural consequence of cysteine oxidation using an existing crystal structure as a starting point can be performed with Forcefield_PTM, a web-based tool that includes many cysteine oxoforms (81). An important caveat of all protein structure analyses, however, is that the full extent of protein dynamics and modifications are not represented.
Cysteine pKa, especially lowered pKa that can predispose a cysteine for oxidation (22, 96, 183), is a key physicochemical property of each cysteine. A comparison of four methods to predict pKa benchmarked to experimentally determined values was recently evaluated, including the commonly used algorithm PROPKA (11). Unfortunately, all methods were found to be unreliable with pH deviations averaging 2.5–3 U from empirically observed values. Adding to the challenge of pKa prediction, all currently available tools are limited to analysis of a single protein structure. As an alternative approach, pKa is correlated with nucleophilicity and reactivity (101, 102), which can be empirically determined on a large scale at the level of specific cysteines. The reactivity of 1082 cysteines in breast cancer cell extracts has been profiled (180), providing a large-scale, empirically determined dataset as an alternative to pKa. These data, along with other parameters, have facilitated sequence-based prediction algorithms of cysteine reactivity and oxidizability using machine learning (155, 175).
For protein localization annotation, UniProtKB references the COMPARTMENTS database, which combines experimental data, informatic predictions, and text mining (19). Over 9400 canonical human proteins, accounting for 46% of the human proteome, are annotated to a single major subcellular organelle in the UniProtKB that can be used as location sentinels of subcellular changes in redox state (Fig. 3a). These sentinels are mostly localized to the nucleus and cytoplasm, but numerous sentinels of even the peroxisome and endosome are annotated (Fig. 3b). The Human Protein Atlas also provides localization information based on antibody staining (159).
Numerous resources are available to assign the biological role(s) and function(s) of redox-regulated proteins. UniProtKB offers a curated description of protein function. The dependency map is generating genome-scale drug inhibition and gene deletion datasets to assess the functional dependency of genes in cancer models (164). Large-scale CRISPR screens coupled with high content imaging and reporter gene assays are being used to examine gene-dependent phenotypic changes at the organelle level (142). Protein interaction partners often provide orthogonal insight into the life of protein. The STRING database includes protein–protein interactions for ∼10 million proteins that are based on a combination of in silico prediction, high-throughput experiments, text mining, and coexpression information (156). Alternatively, BioPlex is using high-throughput protein affinity purification mass spectrometry to empirically determine protein–protein interaction partners of >2000 bait proteins to generate a list of 70,000 experimentally verified protein–protein interactions (73). Full functional assessment of redox-regulated cysteines often includes site-directed mutagenesis, but this is a low-throughput approach that is importantly not applicable to enzymes in which the cysteine residue is directly involved in their activity (82, 85).
Pathway-level annotation of proteins can be performed with protein set enrichment analysis (90). Alternatively, peptides can be assigned to the gene level (178), and the more commonly utilized gene set enrichment analysis (153) can be used. Notably, since identification by proteomic technologies is biased toward abundant proteins, such as metabolic and ribosomal proteins, it is critical to include all detected proteins in over-representation analysis. Specifically, inputting list of significantly regulated proteins identified by proteomics to investigate their enrichment compared with the human proteome will provide minimally useful biological insight. In addition, since proteomics datasets contain many proteins, multiple-hypothesis correction of p-values by Benjamini-Hochberg or another method should be considered for differential expression analysis. While protein interaction maps can be difficult to meaningfully interpret, network inference algorithms are a systems biology tool to infer important subnetworks and focus analysis (99). Since cysteine oxidation is a protein PTM event at the level of a specific cysteine rather than a gene, pathway mapping of the modified residues remains challenging though several tools are pushing PTM pathway analysis forward (126, 131). While there is no complete database of known redox-regulated sites, ProteomeScout (108) and OXID (53) are large protein PTM databases that include oxidized cysteine sites that can be downloaded to determine which redox-regulated cysteines identified may be novel. Other tools are available to predict sites of cysteine sulfenation (5) and S-nitrosation (24).
Cysteine Oxidation in the Clinic
Antioxidant therapies have had mixed results in patients due to many factors, including the relatively poor reactivity of most antioxidants, a focus on ROS levels rather than redox circuitry, and the lack of good biomarkers of drug activity (25). Next generation redox therapeutic approaches targeting the source of ROS, such as NOX (157), or targeting a specific redox vulnerability of cancer cells with relatively nonspecific antioxidants, such as ascorbate (135), provide continued optimism for redox-based therapies.
Assaying the cysteine redoxome in vivo could provide information on the mechanism of drug action, identify biomarkers of drug efficacy, and delineate the altered redox networks in individual patients to tailor redox-based therapeutic strategies (25). Key questions are whether the redox state of a disease is personalized and whether it can be drugged to restore homeostasis. The Cys:CySS ratio can serve as a redox biomarker of cells, tissues, and plasma (61), and served as a peripheral biomarker of treatment response in epileptic rats treated with kainic acid or pilocarpine (94). For in vivo analysis of Cys:CySS or GSH:GSSG, plasma samples must be immediately alkylated to preserve the redox state and to prevent artifacts during sample analysis (77). Oxidation of serum albumin C34, its lone nondisulfide-linked cysteine that is the major plasma oxidant sink, has also demonstrated potential as a biomarker (12, 41). While only minimally invasive, plasma analysis of redox couples or a single cysteine is unlikely to provide much specificity about the underlying disease biology or drug action. In comparison, cysteine oxoform trapping probes coupled with positron emission tomography imaging would allow noninvasive, in vivo analysis of the cysteine redoxome with more information content; however, this has not been reported to date.
Tissue biopsies offer more flexible options for cysteine redox analysis though best results require consistent sample processing after harvesting to minimize differential effects due to hypoxia (106). The in vivo redox state of the thiol-based roGFP, representative of the cysteine redoxome generally, has been shown to be accurately preserved after snap freezing, cryosectioning, and immersion if the fast acting alkylating agent NEM is added immediately to prevent ex vivo oxidation by atmospheric oxygen or fixation (54). As NEM alkylation is often the initial step of differential alkylation, downstream processing of tissue biopsies for biochemical assays or proteomics is similarly technically sound (70). While the dynamic nature of cysteine redox regulation may limit their utility as biomarkers, the availability of techniques to preserve their redox state likely makes them at least as robust assay targets for in vivo analysis in the clinic as ROS assays or redox couples. Development of new cysteine analysis tools for patient samples is needed, such as antibodies that recognize unique oxoforms localized to specific cysteine residues in target proteins that could be used for immunohistochemistry or enzyme-linked immunosorbent assay.
Drugs containing irreversible thiol reactive groups are being incorporated into the next generation of targeted therapies toward kinases EGFR or Bruton's tyrosine kinase to minimize drug resistance (136). Notably, it is estimated that up to 20% of EGFR C797 is sulfenated (120). Since oxidation of these cysteines blocks reactivity, the efficacy of these new inhibitors may depend on the redox state of the cysteines and assaying it may inform on drug efficacy.
Concluding Remarks
Redox signaling via cysteine sentinels plays a multifaceted role in biology despite the biochemical complexity, transience, and likelihood for deleterious stress that a priori seems insufficiently robust to be selected for critical cellular tasks (190). The prevalence of cysteine redox regulation may simply be an ancient, anachronistic holdover from the great oxidation event (39) that persists as an inevitable consequence of life or, rather, is sustained due to robust design principles that are still being elucidated. New insights into redox signaling and regulation of the cellular redox state can be gleaned by harnessing the network of individual cysteines in the redoxome as sensors of cellular redox state that bridges reductionist and systems-level perspectives.
Funding Information
The Held lab acknowledges funding and support from the CA200893, CA138308, CA202852, CA179452, CA200893, AB SCIEX, and the Diabetes Research Center at Washington University (DK020579).