
Abstract
Aims:
Sirtuins, a family of NAD+-dependent deacetylases, are recognized as nondispensable regulators of aging processes. Sirtuin 3 (SIRT3) is the main mitochondrial deacetylase that maintains mitochondrial bioenergetics, an essential prerequisite for healthy aging. In this study, using Sirt3 knockout (Sirt3−/− ) mice, we sought to establish whether Sirt3 deficiency affected life span, an endpoint that has never been tested formally in mammals, and uncover the mechanisms involved in organ damage associated with aging.
Results:
Sirt3−/− mice experienced a shorter life span than wild-type mice and severe cardiac damage, characterized by hypertrophy and fibrosis, as they aged. No alterations were found in organs other than the heart. Sirt3 deficiency altered cardiac mitochondrial bioenergetics and caused hyperacetylation of optic atrophy 1 (OPA1), a SIRT3 target. These changes were associated with aberrant alignment of trans-mitochondrial cristae in cardiomyocytes, and cardiac dysfunction. Gene transfer of deacetylated Opa1 restored cristae alignment in Sirt3−/− mice, ameliorated cardiac reserve capacity, and protected the heart against hypertrophy and fibrosis. The translational relevance of these findings is in the data showing that SIRT3 silencing in human-induced pluripotent stem cell-derived cardiomyocytes led to mitochondrial dysfunction and altered contractile phenotype, both rescued by Opa1 gene transfer.
Innovation:
Our findings indicate that future approaches to heart failure could include SIRT3 as a plausible therapeutic target.
Conclusion:
SIRT3 has a major role in regulating mammalian life span. Sirt3 deficiency leads to cardiac abnormalities, due to defective trans-mitochondrial cristae alignment and impaired mitochondrial bioenergetics. Correcting cardiac OPA1 hyperacetylation through gene transfer diminished heart failure in Sirt3−/− mice during aging. Antioxid. Redox Signal. 31, 1255–1271.
Sirtuin 3 (Sirt3) deficiency is associated with several diseases of aging, but whether the sequelae of Sirt3 deficiency translated into an effect on life span is unknown. We show that Sirt3−/− mice have shortened life span with cardiac abnormalities that include altered mitochondrial bioenergetics and hyperacetylation of optic atrophy 1 (OPA1), associated with aberrant alignment of trans-mitochondrial cristae in cardiomyocytes, and cardiac dysfunction. Given our finding that the correction of cardiac OPA1 hyperacetylation through gene transfer restored trans-mitochondrial cristae alignment and limited heart failure in aged Sirt3−/− mice, we envision future approaches to heart failure, including SIRT3 as a plausible therapeutic target.
Introduction
Sirtuins (SIRTs) are an evolutionary conserved family of NAD+-dependent deacetylases that regulate multiple cellular processes, including stress resistance, apoptosis, inflammation, and mitochondrial energy homeostasis, and are recognized as critical determinants in counteracting the aging process and age-related diseases (19, 27).
In mammals, three members of the SIRT family— sirtuin 3 (SIRT3), SIRT4, and SIRT5—are localized in the mitochondrial matrix and modulate mitochondrial metabolism, but it is SIRT3 that functions uniquely as a major regulator of the whole organelle acetylome (33, 42). In fact, in mice the deletion of Sirt3, but not of Sirt4 or Sirt5, resulted in striking global hyperacetylation of mitochondrial proteins (25), indicating that SIRT3 is central to mitochondrial deacetylase activity, which is an essential prerequisite for healthy aging (52). Indeed, almost 60% of mitochondrial proteins can be deacetylated quickly to meet cellular metabolic demands and contribute substantially to maintaining bioenergetic adaptation during aging (16).
In this context, it has been demonstrated that SIRT3 fine-tunes the process of energy production, due to its primary role in fatty acid oxidation through the deacetylation of long-chain acyl-CoA dehydrogenase (17), as well as the activation of mitochondrial enzymatic chain proteins, including complexes I (2), II (14), and V (39), which are involved in oxidative phosphorylation through which ATP is generated.
In addition to energy supply and nutrient oxidation, SIRT3 affects other major aspects of mitochondrial biology, including reactive oxygen species detoxification and mitochondrial dynamics (6, 31, 35). Sirt3 attenuation or ablation is known to be associated with the accelerated development of several diseases of aging, including cancer and cardiovascular, renal, hepatic, and neurodegenerative disorders (5, 18, 20, 29, 32, 34, 43). Features that are characteristic of aging are found in the myocardium of Sirt3 heart-specific knockout mice, such as mitochondrial protein dysfunction, enhanced oxidative stress, and impaired energy metabolism (24). Whether sequelae of SIRT3 deficiency translated into an effect on life span was not addressed.
In this study, we wanted (i) to investigate the impact that the lack of Sirt3 had on life span in mice, which, surprisingly, had not been studied in mammals yet; (ii) to establish which organ is primarily affected by Sirt3 deficiency during aging, and identify the molecular mechanisms underlying organ dysfunction in aged mice; (iii) to mitigate target organ dysfunction therapeutically; and (iv) to demonstrate the translational relevance of these findings by studying SIRT3-silenced human-induced pluripotent stem cell (iPSC)-derived cardiomyocytes in vitro.
Results
Sirt3 deficiency shortens life span and promotes cardiac dysfunction
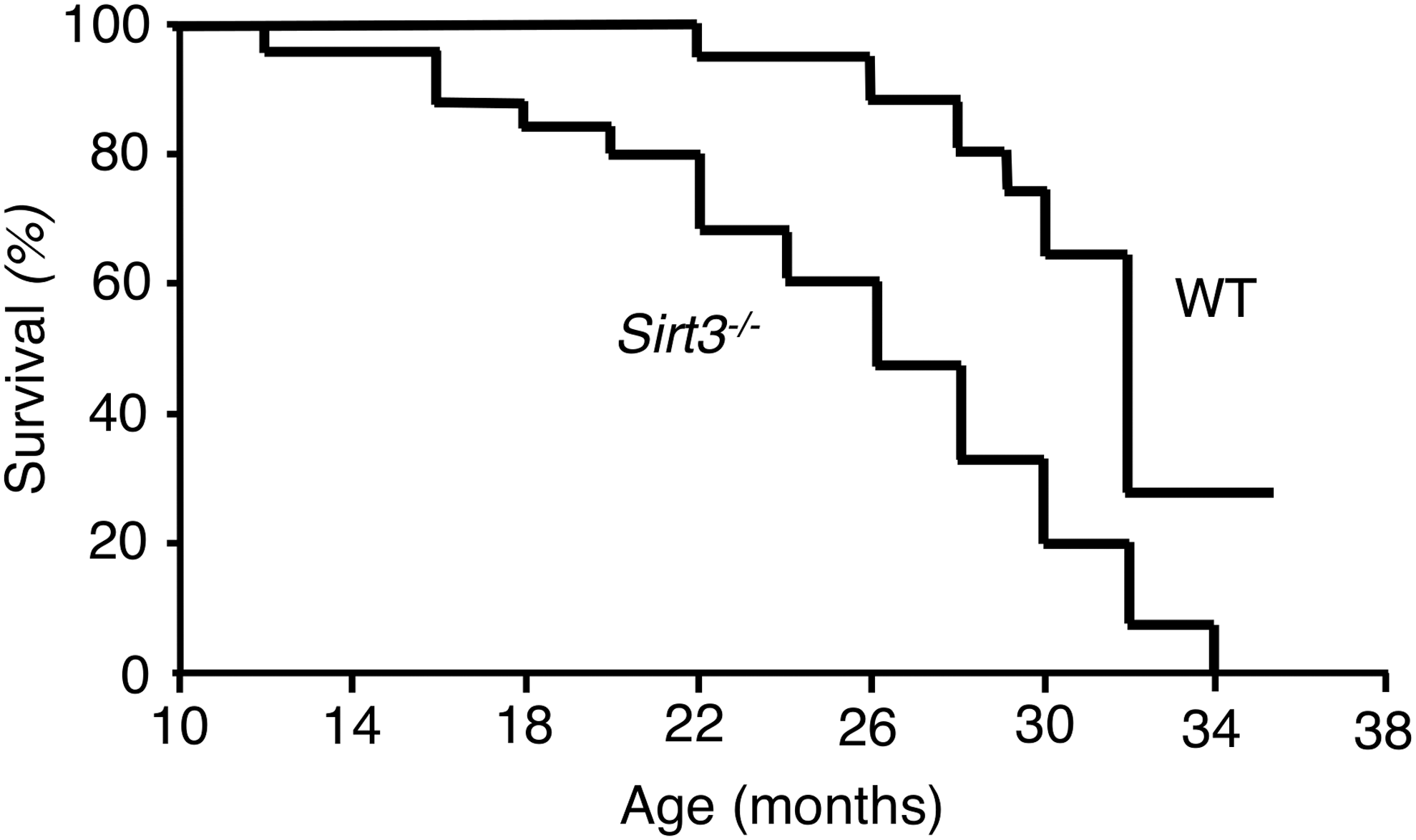
We set out to examine the impact of Sirt3 gene disruption in male mice compared with wild type (WT) control littermates. All Sirt3 knockout animals died within 34 months, at which time 28% of their control littermates were still alive (Fig. 1). Log-rank test analysis showed significant differences in the survival curves between Sirt3−/− and WT mice. Compared with WT littermates, the median life span of Sirt3−/− mice was 19% shorter (median = 50th percentile: from 32 to 26 months) (p-value of <0.0001, Fig. 1). Compared with WT littermates, the maximum life span of Sirt3−/− mice (i.e., the life span of the oldest 10% of a cohort to die) was 10% shorter (90th percentile: from 35.5 to 32 months), a p-value of 0.011.
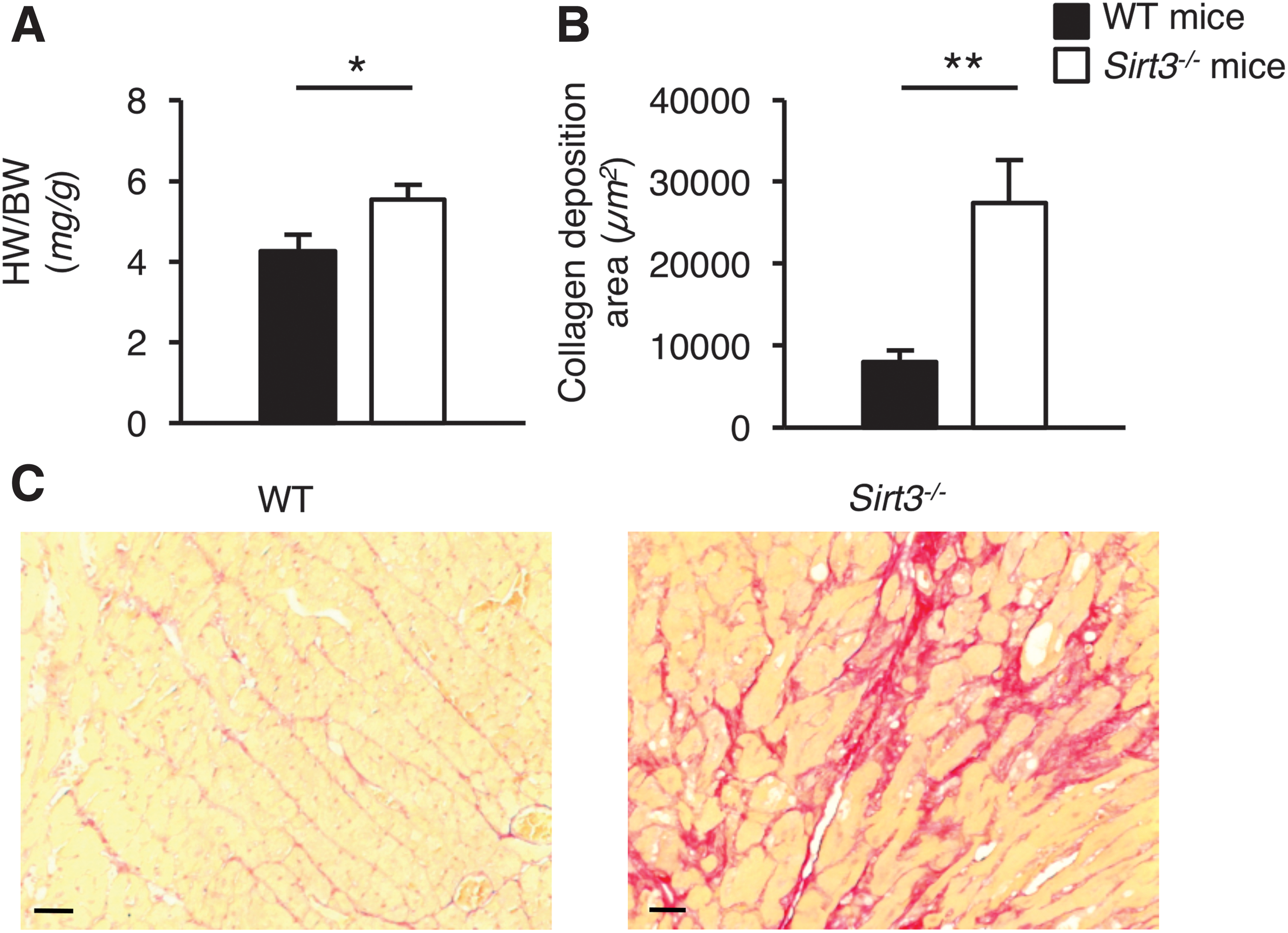
Of all the organs analyzed in aged mice in the present study, the heart was the main organ affected by Sirt3 deficiency. Aged Sirt3−/− mice developed severe cardiac damage, characterized by hypertrophy and fibrosis, which is in line with previous studies (15, 45, 46). An increased heart to body weight ratio (Fig. 2A) and the accumulation of collagen deposition in the interstitial space (Fig. 2B, C) were observed in aged Sirt3−/− but not in WT mice.
A similar number of immune cells were found in the cardiac tissue of Sirt3−/− mice and WT mice (neutrophils: 5.5 ± 0.4 vs. 4.4 ± 0.6; macrophages: 0.5 ± 0.2 vs. 0.8 ± 0.1; CD4+ T cells: 0.9 ± 0.1 vs. 0.6 ± 0.2; and CD8+ T cells: 0.8 ± 0.2 vs. 0.6 ± 0.2, expressed as number of cells/field in n = 6 mice/group), which ruled out the possibility that an immune or inflammatory reaction caused the cardiac changes in Sirt3−/− mice. Cardiac abnormalities in these animals were not associated with changes in lipid plasma levels or renal function impairment in terms of proteinuria (Supplementary Table S1).
As for the other organs, mild renal interstitial fibrosis developed in Sirt3−/− mice comparable with WT mice, as shown by stainings of the kidney tissue for collagen I and III fibers (Sirius Red, Supplementary Fig. S1) or for alpha-smooth muscle actin (α-SMA, Supplementary Fig. S1). However, these abnormalities did not translate into a pathological renal phenotype (Supplementary Table S1). As for the liver, we found modest abnormalities that did not translate into full-blown liver dysfunction, as demonstrated by normal levels of plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels (Supplementary Table S1). No histological alterations were found in the brain, nor was an increased incidence of tumors observed in Sirt3−/− mice compared with WT mice.
Sirt3 deficiency compromises mitochondrial connectivity and communication in the heart
The absolute mass of cardiac mitochondria was significantly reduced in Sirt3−/− compared with WT mice, which was demonstrated in the analysis of citrate synthase activity (Fig. 3A). In addition to mass reduction, the ATP synthesis rate was also reduced in Sirt3−/− hearts compared with those of WT mice, reflecting low energy production in the former group (Fig. 3B).
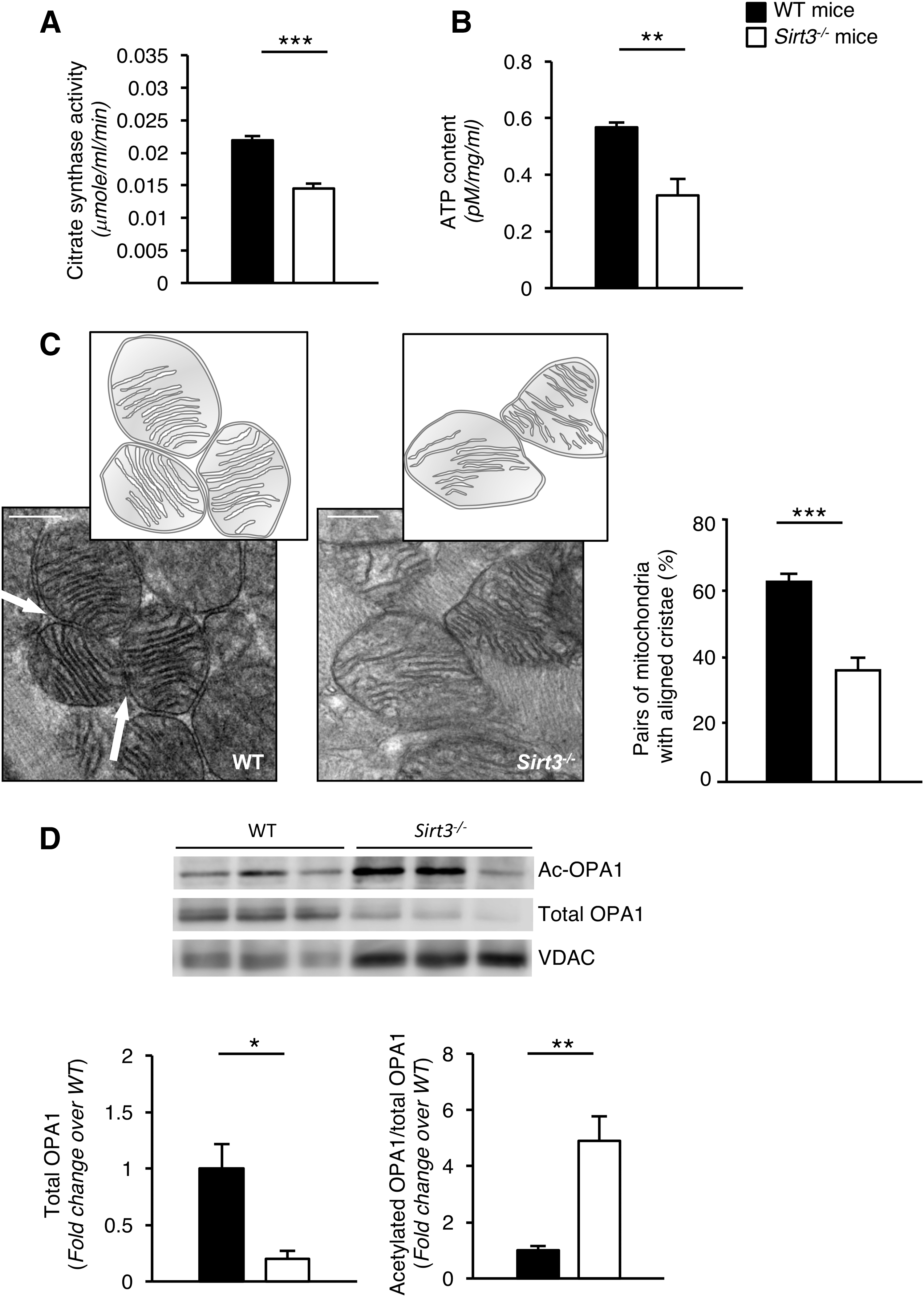
Next, we wanted to understand why the energetic state of Sirt3−/− mice was compromised. We thus turned our attention to mitochondrial connectivity and communication, a crucial factor in cellular homeostasis (21). We investigated the structural integrity of the mitochondrial network in Sirt3−/− and WT mice using transmission electron microscopy at a nanometer resolution range.
We found that Sirt3 deficiency affected trans-mitochondrial cristae coordination to a significant extent. Specifically, we observed an aberrant alignment of trans-mitochondrial cristae in most mitochondria in the hearts of mice lacking Sirt3, compared with age-matched WT mice (Fig. 3C). Mitochondrial cristae architecture, although not completely disrupted, was predominantly abnormal in Sirt3−/− mice compared with the regular cristae distribution in WT mice (Fig. 3C). Specifically, mitochondrial cristae were rarely organized as parallel stacks, as is usually observed in WT animals (Fig. 3C).
Effect of Opa1 gene transfer on mitochondrial structure and function
There is evidence that SIRT3 regulates mitochondrial function and dynamics through the deacetylation and activation of optic atrophy 1 (OPA1), a pro-fusion protein of the inner mitochondrial membrane (31, 41). In this study, we wondered whether the lack of trans-mitochondrial cristae coordination in our model was caused by Sirt3 deficiency and the consequent hyperacetylation of OPA1. Total OPA1 was evaluated in isolated mitochondria of cardiac tissue by Western blot analysis. We found that total OPA1 was significantly reduced in Sirt3−/− compared with WT mice (Fig. 3D), and it was hyperacetylated (Fig. 3D).
We then investigated whether the impaired trans-mitochondrial cristae alignment found in Sirt3− /− mice could be corrected by inducing the expression of the deacetylated and active mutant (mut) form of OPA1. To this end, we generated an adeno-associated virus serotype 9 (AAV9)-mutOpa1-vector encoding for a deacetylation-mimetic version of murine OPA1, in which two conserved lysines, K944 and K949, were replaced with two arginins (R) to mimic the function of SIRT3. AAV9 was chosen for its uniquely exuberant cardiac tropism (55). An AAV9-LacZ vector served as a control.
Cardiomyocytes were efficiently transduced after the injection of 1.3 × 1012 viral genome/animal of AAV9-LacZ (Supplementary Fig. S2). Western blot analysis of mitochondrial extracts revealed that gene transfer of mutOpa1 increased total OPA1 protein levels, as expected, and effectively reduced the quantity of acetylated OPA1 in Sirt3−/− hearts (Fig. 4A). Two weeks after injection, the expression of the SIRT3-insensitive version of OPA1 did not have an impact on cardiac mitochondrial mass (citrate synthase activity, μmole/mL/min: 0.014 ± 0.003 in Sirt3−/− mice injected with AAV9-LacZ vs. 0.013 ± 0.002 in Sirt3−/− mice injected with AAV9-mutOpa1, n = 4 mice/group), but improved cardiac ATP content in Sirt3-deficient mice (Fig. 4B). Strikingly, mutOpa1 gene delivery restored the proper alignment of the cristae of adjacent mitochondria (Fig. 4C, D). Transfection with mutOpa1 also effectively reduced cardiac hypertrophy (Fig. 4E).
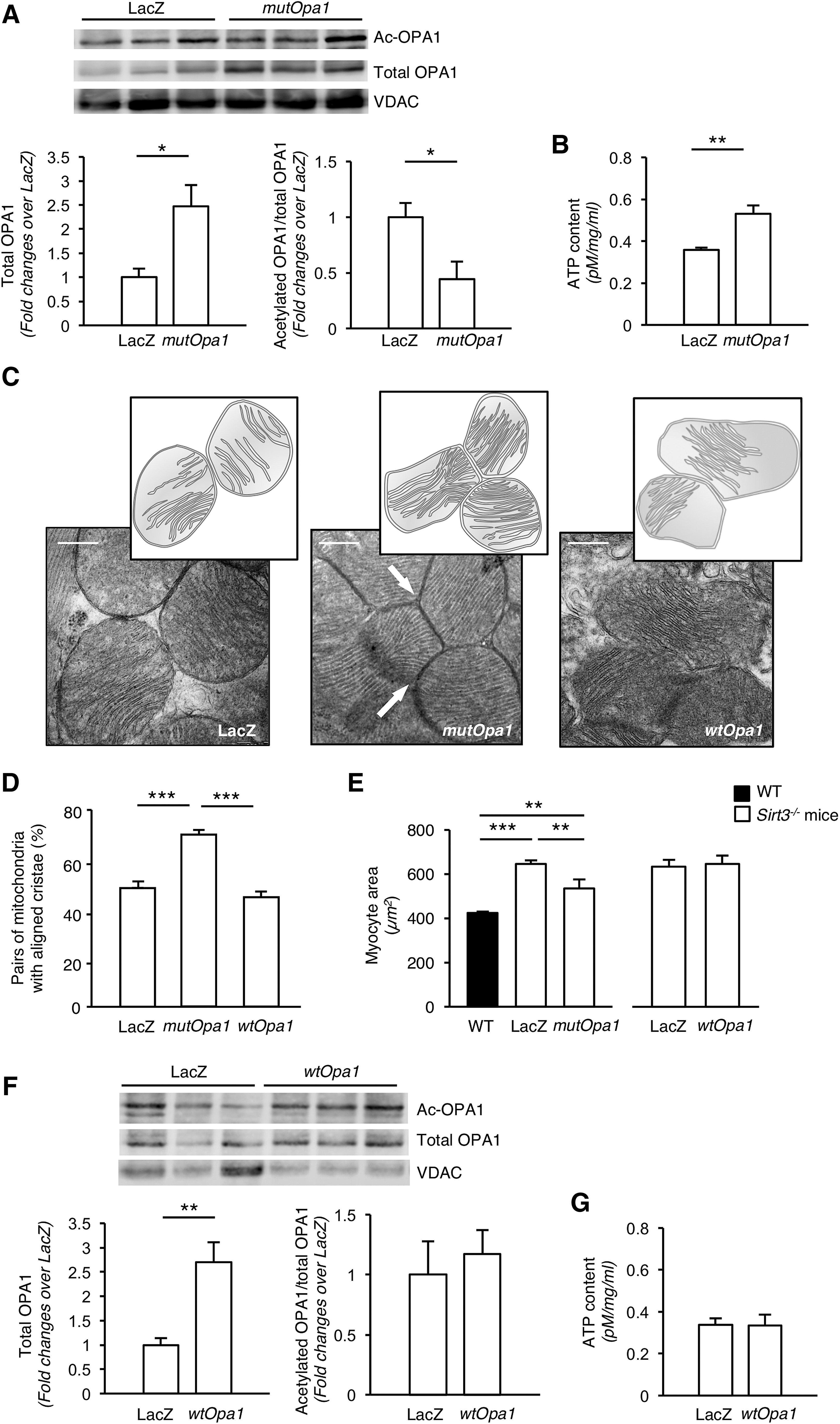
To confirm that the protective effect of gene transfer on cardiac abnormalities was fully dependent on OPA1's deacetylated status rather than on its expression levels, we performed gene transfer experiments of WT Opa1 (wtOpa1) in Sirt3−/− mice. Total OPA1 protein levels increased after gene delivery (Fig. 4F). Under Sirt3-deficient conditions, wtOpa1 could not be deacetylated as revealed by comparable levels of hyperacetylated OPA1 in mice injected with AAV9-wtOpa1 and AAV9-LacZ (Fig. 4F). Likewise, the transduction of wtOpa1 did not improve cardiac mitochondrial mass (citrate synthase activity, μmole/mL/min: 0.016 ± 0.001 in Sirt3−/− mice injected with AAV9-LacZ vs. 0.015 ± 0.001 in Sirt3−/− mice injected with AAV9-wtOpa1, n = 4 mice/group), or ATP content (Fig. 4G). Unlike mutOpa1, wtOpa1 did not promote trans-mitochondrial cristae alignment (Fig. 4C, D), nor did it limit cardiac hypertrophy (Fig. 4E).
Effect of Opa1 gene transfer on cardiac dysfunction in the long term
Next, we investigated whether the effects that the gene transfer of the mutOpa1 had on mitochondrial structure and function, observed earlier, translated into end-organ cardiac protection.
To determine the optimal timing for therapeutic intervention, we first evaluated α-SMA expression and collagen I deposition as markers of fibrosis in the cardiac tissue of Sirt3−/− mice (Supplementary Fig. S3A). α-SMA expression was already higher in Sirt3−/− mice compared with WT mice at 15 months (% of positive cells/field: 6.78 ± 0.29 vs. 3.60 ± 0.96, p-value of <0.05, n = 3 mice/group) and rose further at 20 months (15.27 ± 3.53 vs. 3.62 ± 0.19, p-value of <0.05, n = 3 mice/group) (Supplementary Fig. S3A). Collagen I deposition was similar in Sirt3−/− and WT mice at 15 months (score: 0.20 ± 0.03 vs. 0.17 ± 0.02, n = 3 mice/group), but rose significantly higher at 20 months in Sirt3−/− mice than in WT mice (0.56 ± 0.05 vs. 0.23 ± 0.03, p-value of <0.01, n = 3 mice/group) (Supplementary Fig. S3B). Based on α-SMA and collagen I expression in cardiac tissue, Sirt3−/− mice at 18–20 months of age were treated with AAV9-mutOpa1 or AAV9-LacZ and sacrificed after 3 months. The gene transfer approach proved capable of preserving in the long term an effective state of OPA1 deacetylation in Sirt3−/− mice (Fig. 5A).
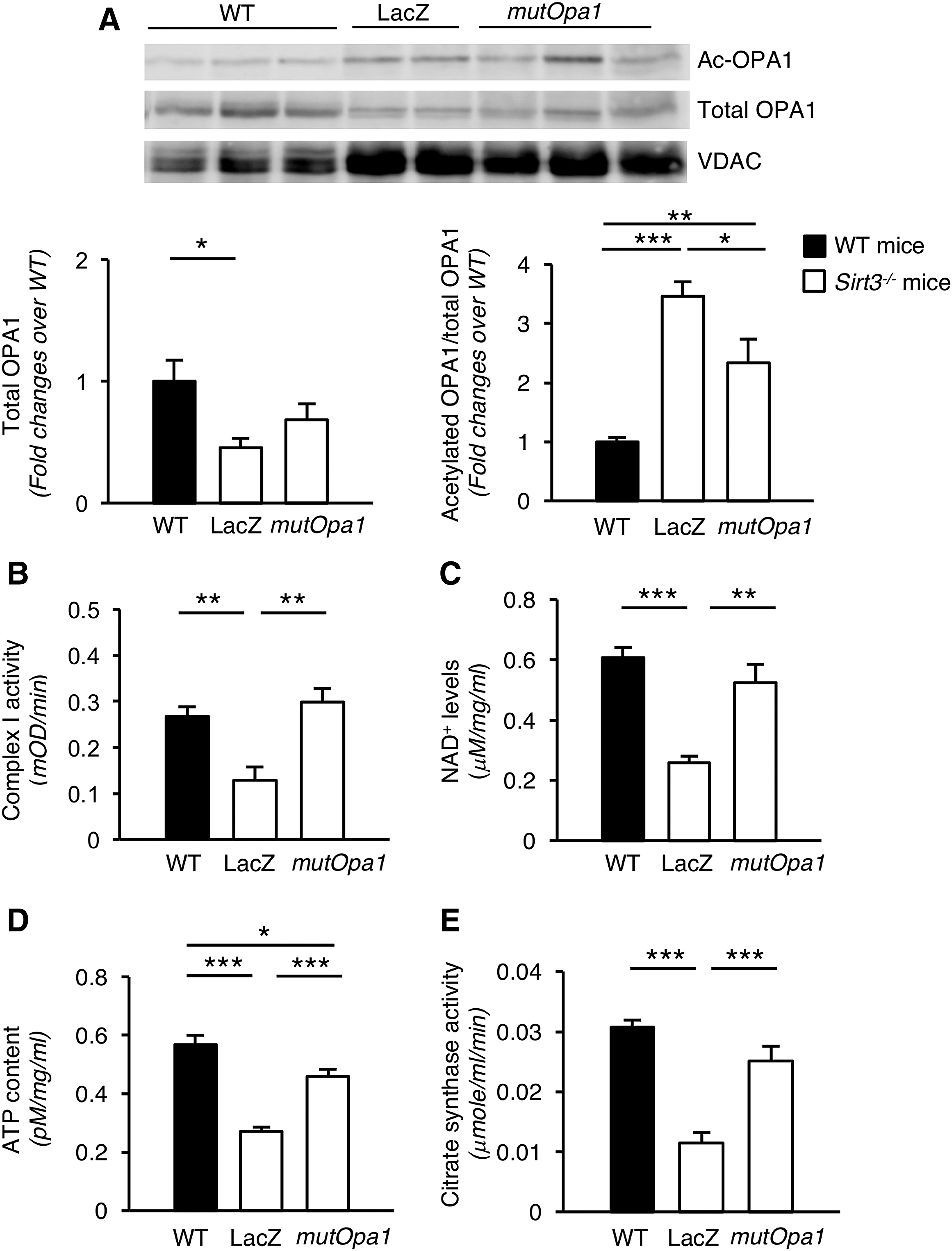
Since respiratory chain complexes are key structures for mitochondrial bioenergetics and reside on the cristae membrane, we evaluated the activity of the gatekeeper of the respiratory chain, Complex I, the formation and stabilization of which are dependent on OPA1. The activity of immunocaptured Complex I from cardiac tissue lysates was analyzed based on oxidation of NADH to NAD+, and found to be remarkably lower in Sirt3−/− mice receiving AAV9-LacZ vector compared with WT mice (Fig. 5B). MutOpa1 transduction restored the Complex I activity to levels that were indistinguishable from those of WT mice (Fig. 5B). As a consequence, NAD+ levels, which were reduced in AAV9-LacZ Sirt3−/− mice, were normalized after mutOpa1 overexpression (Fig. 5C).
The reduced respiratory capacity of AAV9-LacZ Sirt3−/− mice yielded lower ATP production, which was replenished to control levels by mutOpa1 (Fig. 5D). The improvement in mitochondrial bioenergetics by mutOpa1 was accompanied by an increase in mitochondrial mass, evaluated as citrate synthase activity (Fig. 5E).
Long-term transduction with mutOpa1 led to a reduction in hypertrophy and the heart-to-body weight ratio was normalized (Fig. 6A). The protective effect of mutOpa1 on cardiac hypertrophy was accompanied by reduced fibrosis, assessed through both α-SMA (Fig. 6B) and collagen I (Fig. 6C) immunostainings.
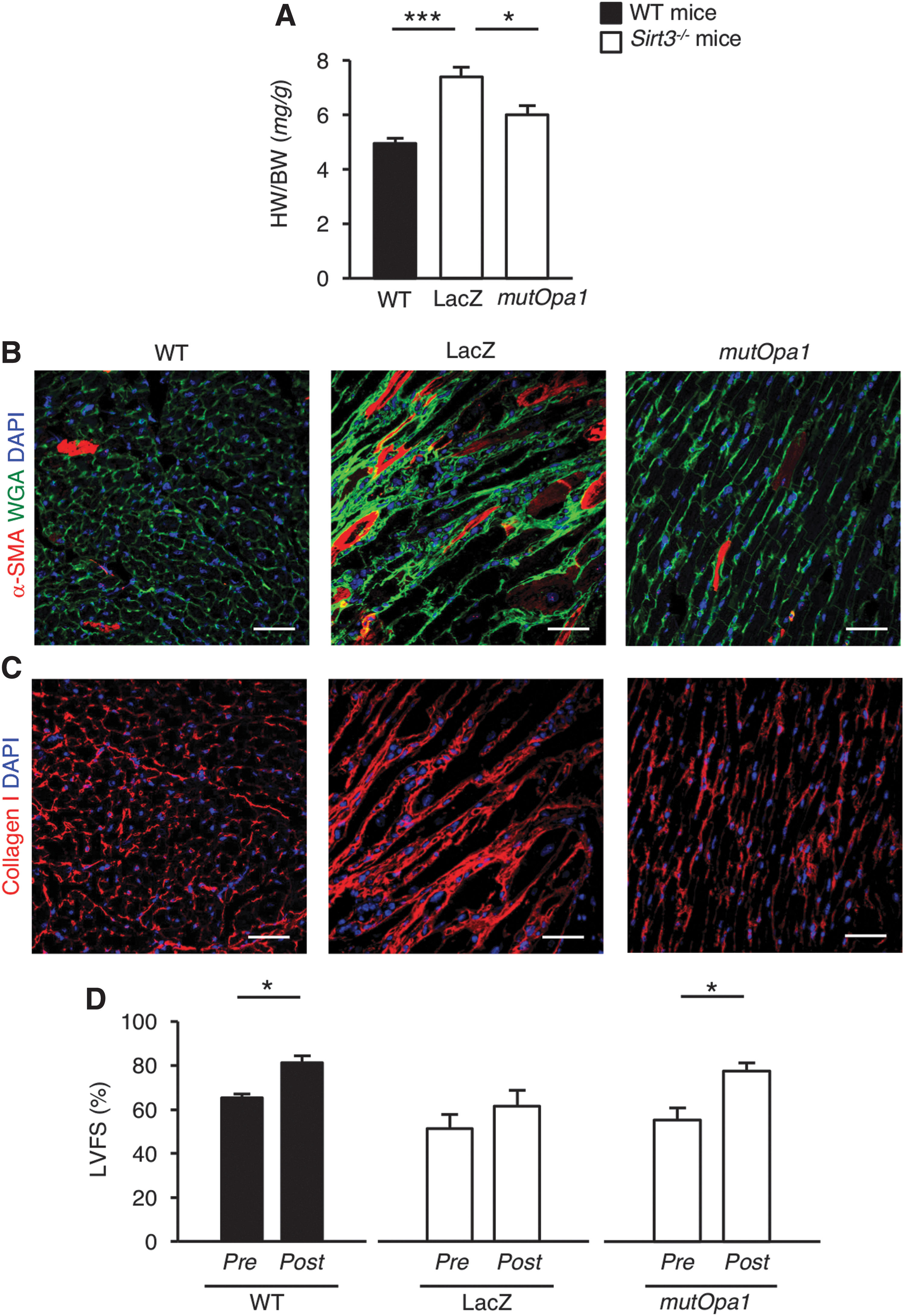
We then measured global left ventricular function using echocardiography at rest and during a low-dose dobutamine (LDD) stress test to evaluate myocardial viability and contractile performance in response to an acute increase in workload (48). LDD stress echocardiography was tolerated well in all animals and we did not encounter any complications. The dobutamine was effective at a low dose, as demonstrated by a statistically significant increase in left ventricle fractional shortening (LVFS), under stress, compared with baseline in WT mice (Fig. 6D). In Sirt3−/− mice receiving AAV9-LacZ vector, the LDD stress test did not increase the inotropic cardiac reserve, which instead increased significantly in Sirt3−/− mice transduced with mutOpa1 (Fig. 6D). There was no difference in chronotropic response to LDD between groups (AAV9-LacZ: pre 515.0 ± 34.1 beats per minute (bpm) vs. post 474.3 ± 34.7 bpm; AAV9-mutOpa1: pre 449.8 ± 8.8 bpm vs. post 459.0 ± 5.9 bpm; WT: pre 474.4 ± 40.3 bpm vs. post 536.6 ± 60.9 bpm). This is not surprising, since others have found that LDD did not induce changes in heart rate (8). All the above findings suggest a link between the replacement of trans-mitochondrial cristae alignment and recovery of cardiac structure and function.
That the effect of gene transfer actually depended on OPA1 deacetylation was supported by findings that hyperacetylation of total mitochondrial proteins, including the SIRT3 target superoxide dismutase 2 (SOD2), in Sirt3−/− mice (Supplementary Fig. S4A) was not affected by mutOpa1 gene delivery (Supplementary Fig. S4B).
SIRT3 silencing alters mitochondrial morphology and contractile behavior in human iPSC-derived cardiomyocytes
Having established that there is a strong correlation between SIRT3 and cardiac function in mice, we sought to investigate the effect of SIRT3 deficiency on spontaneously beating human iPSC-derived cardiomyocytes. To this end, human iPSC-derived cardiomyocytes were silenced for the SIRT3 gene through a specific small interfering RNA (siRNA). SIRT3 silencing resulted in 85% inhibition of SIRT3 mRNA expression, compared with cells transfected with small interfering RNA null (siNULL) (Fig. 7A).
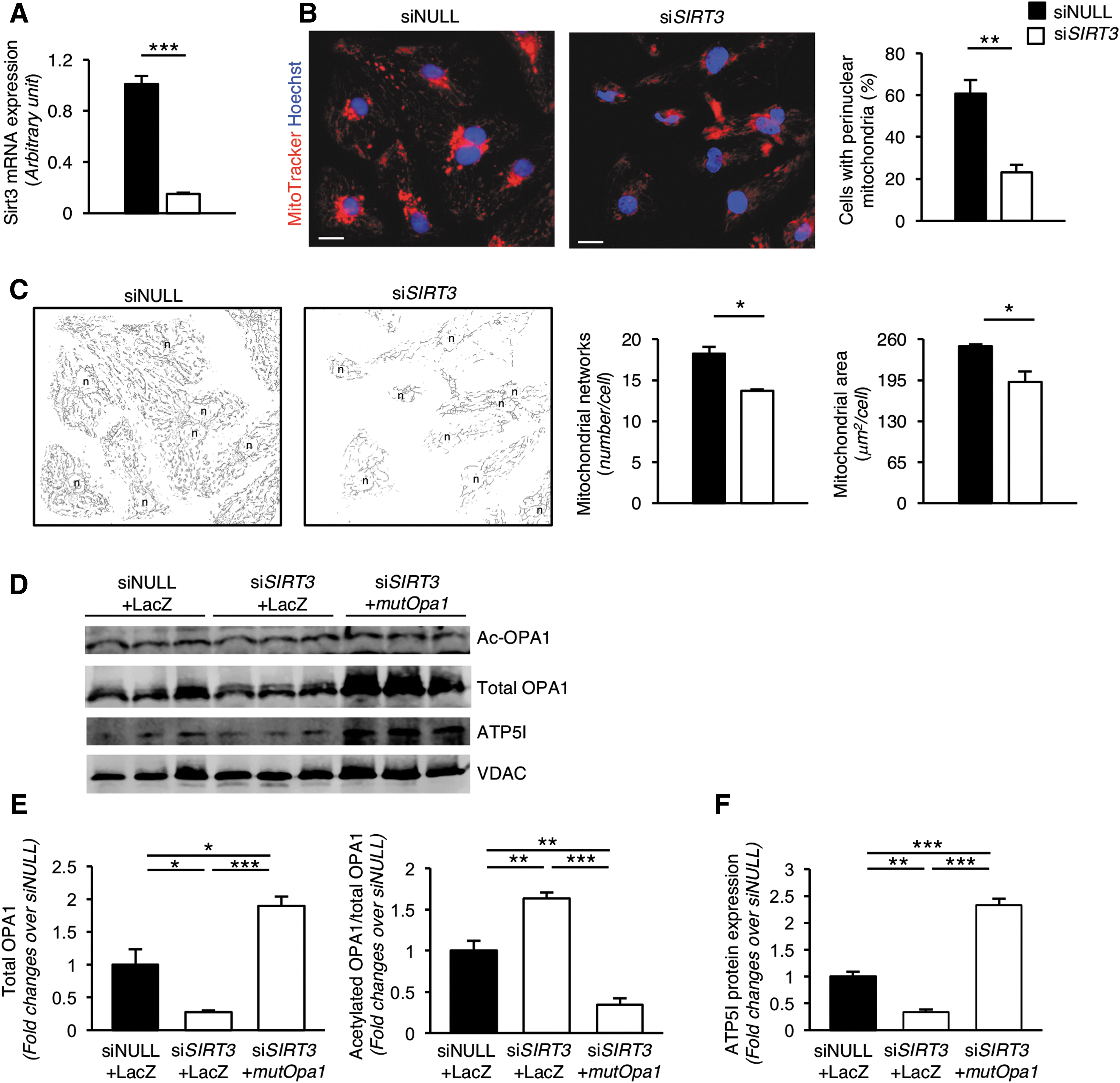
In this setting, we investigated the effect of SIRT3 deletion on mitochondrial structural integrity by using an image preprocessing tool to quantitatively capture morphological changes in the mitochondrial network (50). Using MitoTracker staining, we found that almost the 60% of siNULL iPSC-derived cardiomyocytes exhibited a subset of highly packed and round mitochondria near the perinuclear region, while highly packed mitochondria were found in only 20% of SIRT3-deficient iPSC-derived cardiomyocytes (Fig. 7B).
In addition, iPSC-derived cardiomyocytes exhibited a highly interconnected mitochondrial network, which extends through the whole cytoplasmic compartment of the cells following siNULL transfection (Fig. 7C). The deletion of SIRT3 markedly altered the overall structural architecture of the mitochondrial lattice in iPSC-derived cardiomyocytes, to the extent that the mitochondrial networks and area were significantly reduced compared with their SIRT3-competent counterpart (Fig. 7C). In-depth analysis revealed that the reduced mitochondrial network, which followed SIRT3 silencing, was the consequence of the specific loss of both mitochondrial branches (siNULL: 551 ± 29 vs. small interfering RNA for sirtuin 3 [siSIRT3]: 371 ± 20 number/cell, p-value of <0.01) and junctions (siNULL: 186 ± 9 vs. siSIRT3: 129 ± 8 number/cell, p-value of <0.01), as described by Valente et al. (50).
An extensive body of evidence has shown that both the shape and positioning of mitochondria tightly controls Ca2+-handling of cardiomyocytes (47). To investigate the mechanisms underlying the observed altered beating contraction, we analyzed the Ca2+-handling properties of iPSC-derived cardiomyocytes by using the Ca2+ fluorescent dye FLUO-4. The live-imaging recording showed that after siNULL transfection, high-intensity Ca2+ sparks emerged synchronously in both the nuclear and cytoplasmic regions of the iPSC-derived cardiomyocytes (Supplementary Movie S1). In contrast, SIRT3 silencing translated into irregular Ca2+ release that was not synchronized among iPSC-derived cardiomyocytes of a monolayer, without apparent changes in either intensity or duration of Ca2+ sparks (Supplementary Movie S2).
We subsequently examined how SIRT3 silencing affected cardiomyocyte contraction. Through Cell Observer analysis, phase-contrast monitoring of living human iPSC-derived cardiomyocytes treated with siNULL revealed a regular, autonomous beating rate with a frequency that remained almost constant during the temporal course of the measurements (Supplementary Movie S3). In the face of reduced SIRT3 expression, iPSC-derived cardiomyocytes exhibited uncontrolled spontaneous beating that led to the generation of ectopic foci of contraction (Supplementary Movie S4).
To evaluate whether the dysfunctioned phenotype of cardiomyocytes could be rescued by restoring functional OPA1, we transduced mutOpa1 in SIRT3-silenced iPSC-derived cardiomyocytes. First, we found that SIRT3-silenced iPSC-derived cardiomyocytes exhibited downregulation of OPA1 and increased protein acetylation compared with siNULL-transfected cells (Fig. 7D, E). By contrast, mutOpa1 gene transfer effectively increased total OPA1 protein expression and reduced its acetylation levels (Fig. 7D, E). To investigate whether the restoration of functional OPA1 translated into an amelioration of cardiomyocyte function, we assessed human iPSC-derived cardiomyocyte contraction. Cardiomyocytes transfected with siNULL and LacZ exhibited a regular, autonomous beating rate (Supplementary Movie S5), while SIRT3-silenced cardiomyocytes transfected with LacZ showed uncontrolled spontaneous beating (Supplementary Movie S6). Remarkably, transfection of mutOpa1 restored the spontaneous synchronous beating of SIRT3-silenced cardiomyocytes (Supplementary Movie S7).
To provide further functional data on mitochondrial activity in cardiomyocytes transfected with mutOpa1, we evaluated the expression of ATP synthase subunit e (ATP5I) that is required for ATP synthase dimerization and proper functioning (38). As shown in Figure 7D and F, we found that SIRT3 silencing in iPSC-derived cardiomyocytes significantly reduced ATP5I protein expression compared with siNULL-transfected cells. Levels of ATP5I protein were even higher than in siNULL-transfected cells by mutOpa1 gene transfer (Fig. 7D, F).
Discussion
In this study, we report for the first time that SIRT3 has a major role in regulating life span in mice. The limited survival time of Sirt3−/− animals is associated with severe heart structure damage and dysfunction. Heart damage parallels defective trans-mitochondrial cristae alignment and impaired bioenergetics, which are linked to suboptimal OPA1 deacetylation. The salient feature of the present study is the utilization of a targeted gene transfer involving mutOpa1 that reverses heart failure. Silencing SIRT3 in human iPSC-derived cardiomyocytes disrupts mitochondrial integrity and alters cell contractility, rescued by mutOpa1 gene delivery.
The first novel finding of this study is that Sirt3 deficiency shortens life span in mice. To the best of our knowledge, there are no other studies that document the effect of a specific mitochondrial SIRT on survival in mammals. A study that has been recently published (53) is worth mentioning here. In Drosophila melanogaster, life span is shortened by knockdown of Sirt4, encoding the sole predicted mitochondrial SIRT in this model organism, while flies overexpressing Sirt4 ubiquitously or in the fat body are long-lived. It is noteworthy that several phenotypes of Sirt4 knockout fruit fly are reminiscent of those found here in Sirt3−/− mice. As the authors of the above article have indicated, it is possible that Sirt4 in the fruit fly could be a counterpart of the mammalian SIRT3.
In this study, we have demonstrated that Sirt3-deficient animals primarily experience signs of severe cardiomyopathy as they age. While this finding may not be entirely new (15, 45), it is worth mentioning here that Sirt3−/− mice were followed up for a long period of time, long enough to demonstrate differences in heart pathologies, including hypertrophy and fibrosis. In the long run, these changes were associated with signs of heart failure.
As the main mitochondrial SIRT, SIRT3 deacetylates many proteins in the organelles and promotes diverse aspects of health span, but the role of SIRT3 goes beyond deacetylating specific mitochondrial substrates. It also regulates mitochondrial function at many other levels (26). Given that cardiac tissue has the highest number of mitochondria and the highest levels of oxygen consumption of all organs, Sirt3 deletion primarily affects the heart. Indeed, Sirt3−/− mice exhibited abnormal cardiac mitochondrial clustering and reduced mass associated with a lower than normal ATP synthesis rate, which would suggest an altered bioenergetic state.
Actually, bioenergetics in mitochondria depends on the reciprocal connectivity, which is guaranteed by specific junctions acting as unique electrochemical systems (3, 7, 36). While we confirmed the elegant findings of Picard et al., which found that adjacent mitochondria finely coordinate the curvature of their cristae with each other (37), we also found that in Sirt3−/− mice, this parallel alignment with the cristae of neighboring mitochondria was lost. This could explain the compromised mitochondrial energetics in Sirt3-deficient animals in the present study. To date, no other instance of aberrant trans-mitochondrial cristae alignment in a disease model has been reported. It is tempting to speculate that shortened life span and heart abnormalities could indeed be direct consequences of abnormal intermitochondrial interaction.
Since mitochondrial dysfunction in the hypertrophic heart is dependent on the state of deacetylation of OPA1 (41), we chose to investigate whether the lack of coordination of mitochondrial cristae was due to suboptimal OPA1 deacetylation of Sirt3−/− animals. Gene transfer experiments with constitutively deacetylated mutOpa1 fully reconstituted mitochondrial cristae coordination with the appropriate curvature within mitochondria in Sirt3−/− mice. This unique property of OPA1 has never been reported before, to the best of our knowledge.
We also found that the restoration of mitochondrial connectivity translated functionally into recovery of ATP content, which can be taken as an indication of restored mitochondrial bioenergetics. The possible role that mutOpa1 has in restoring energy production in Sirt3−/− mice could be due to the capability of OPA1 to maintain cristae shape, a prerequisite for the assembly and stability of respiratory chain supercomplexes and hence mitochondrial respiratory efficiency (11). Finding that gene transfer of wtOpa1 did not affect mitochondrial structure and function, further corroborated the hypothesis that SIRT3 has a critical role in activating OPA1. SIRT3-dependent deacetylation of OPA1 is the only reported posttranslational modification known to promote the activity of OPA1. Our data therefore provide unique proof of principle that modulating the expression of a single gene can have a profound influence on reduced organelle efficiency in Sirt3− /− mice. The finding that the correction of OPA1 hyperacetylation via gene transfer sufficed to limit the abnormal myocyte size of Sirt3−/− mice 2 weeks after mutOpa1 injection was already an indication that this maneuvre could theoretically preserve cardiac dysfunction.
The next step was to explore the potential of mutOpa1 gene delivery to mitigate accelerated cardiac hypertrophy, which develops in Sirt3−/− mice with age. To actually determine whether this was the case, we performed two sets of experiments based on echocardiography and LDD stress to assess cardiac inotropic reserve (48), given that there is evidence that cardiac inotropic response is a function of mitochondrial integrity and energy production (40). While global systolic function in Sirt3−/− and WT mice under resting conditions was the same, the dobutamine challenge unmasked an insufficient contractile response that can reasonably be attributed to the impairment of mitochondrial energetics production in Sirt3−/− mice. Indeed, we found that Sirt3−/− mice exhibited reduced Complex I activity in the mitochondrial electron transport chain, with a consequent decrease in NAD+ and ATP generation.
These findings may underlie the close link between the SIRT3-dependent OPA1 deacetylation and the preserved spatial distribution of cristae membranes. There is evidence showing that improvement of mitochondrial cristae shape induced by OPA1 ameliorates mitochondrial energetic efficiency (51), as it occurs here with the restoration of Complex I activity after mutOpa1 gene transfer. This interpretation was confirmed by the finding that these abnormalities were completely overturned by long-term mutOpa1 gene transfer experiments, which also restored the inotropic response of the heart. It is worth mentioning that deacetylation of OPA1—but not of other mitochondrial proteins including SOD2—maintained in the long term in Sirt3−/− animals, corrected cardiac dysfunction and fibrosis, demonstrating this maneuvre's previously unanticipated potential for cardiac protection. Studies have shown that mitochondrial dysfunction in cardiomyocytes critically contributes to the development of age-associated cardiac fibrosis by impairing bioenergetics and reducing cell survival (1, 28, 49). Furthermore, based on the recent evidence that Complex I inhibition is sufficient to promote the development of fibrosis in the lung, it is tempting to infer that the normalization of Complex I activity through mutOpa1 gene transfer contributes to the amelioration of cardiac fibrosis (30).
In search of a potential proof-of-concept that SIRT3 deficiency induced cardiomyocyte dysfunction in human specimens as it occurred in mice lacking Sirt3, we sought to study human iPSC-derived cardiomyocytes and used these cells to abrogate SIRT3 expression via specific siRNA. SIRT3 silencing led to altered mitochondrial morphology by reducing the subset of perinuclear mitochondria in iPSC-derived cardiomyocytes. This is significant, given that this mitochondrial clustering is an important factor in ATP generation and the formation of direct energetic channeling between mitochondria and the nucleus (9, 13, 22), possibly explaining the energetic defect observed in Sirt3−/− mice.
On the contrary, SIRT3 also affected the overall cytoplasmic mitochondrial network, which contributes to Ca2+-handling and sustains the proper cardiac contraction (12). In this context, our findings revealed impairment in the iPSC-derived cardiomyocyte contractile phenotype associated with altered Ca2+ signaling in the face of reduced SIRT3 expression. We know that human iPSC-derived cardiomyocytes may exhibit several limitations mainly due to their immature phenotype compared with adult cardiomyocytes. However, the novel finding of the present study is that SIRT3 silencing impairs cardiomyocyte contraction and that this phenomenon is fully rescued by restoring OPA1 activity. It is conceivable that our present results explain recent evidence that DNA sequence variants, which actually decrease SIRT3 transcription, play a role in promoting myocardial infarction in humans (54).
Given the theoretical potential for targeting SIRT3 pathways for healthy aging and cardiovascular fitness, this study provides an interesting opportunity to look at gene transfer technology as a therapeutic strategy for the clinical arena in addition to present pharmaceutical treatments. A gene modifying strategy targeting SIRT3 should be especially welcome, in view of the fact that attempts at identifying a specific SIRT3 activator have repeatedly failed.
Materials and Methods
Animal studies
Sirt3−/− mice generated in a mixed genetic background (provided by Professor Frederick W. Alt, Harvard Medical School, Boston, MA) (25) and their C57BL/6x129 WT littermates were used. Mice were maintained in a specific pathogen-free facility with a 12-h dark/12-h light cycle, in a constant temperature room with free access to standard diet and water. Only male mice were used. Animals were followed up throughout their life span (n = 25 for each group). Mice were sacrificed when death appeared imminent, and heart, kidneys, liver, and brain were harvested for tissue analysis. Tumor incidence was determined through histopathological examination of hematoxylin- and eosin-stained slides from the heart, liver, brain and aorta. The oldest WT mice (4/25) were sacrificed at 35.5 months of age for organ collection. Additional WT and Sirt3−/− mice were sacrificed at 15 months of age for analysis in cardiac tissues. AAV9-LacZ, AAV9-mutOpa1, and AAV9-wtOpa1 gene delivery experiments were performed in Sirt3−/− mice at 15 months of age (n = 3/group), and mice sacrificed 2 weeks after AAV9 injection for cardiac analysis. Additional AAV9-LacZ and AAV9-mutOpa1 gene delivery experiments were performed in Sirt3−/− mice at 18–20 months (n = 4/group), and mice were sacrificed 3 months after AAV9 injection for cardiac analysis. WT age-matched mice were studied as controls.
All procedures involving animals were performed in accordance with institutional guidelines in compliance with national (D.L.n.26, March 4, 2014) and international laws and policies (Directive 2010/63/EU on the protection of animals used for scientific purposes).
Laboratory parameters
Plasma levels of cholesterol, triglycerides, ALT, and AST were measured by the Reflotron test (Roche Diagnostic Corporation, Indianapolis). Urinary protein excretion was measured through the Comassie method using Cobas Mira autoanalyzer (Roche Diagnostics Systems, Risch, Switzerland).
Cardiac tissue analysis
Cardiomyocyte cross-sectional areas were evaluated on light microscopy images at × 400 magnification, from at least 50 cells in each group, and measured by manually tracing the cell contour using the ImageJ analysis program (ImageJ 1.42q) (4). Cardiac tissues were formalin fixed, paraffin embedded, and sectioned at 5 μm for hematoxylin/eosin and at 10 μm for collagen staining (0.1% Sirius Red in picric acid). Collagen content was quantified in digitally acquired images. The nature of Sirius Red-stained collagen deposits was confirmed by examination under a microscope equipped with polarized light that renders collagen fibers birefringent.
α-SMA and collagen I expression was evaluated through immunofluorescence analysis. Frozen heart sections (3 μm) were fixed with cold acetone and incubated with 1% bovine serum albumin (BSA) to block nonspecific sites. Sections were then incubated with cy3-conjugated mouse anti-α-SMA (clone 1A4; Sigma-Aldrich, St. Louis, MO) or rabbit anti-collagen I (ab34710; Abcam, Cambridge, United Kingdom), the latter followed by Cy3-conjugated goat anti-rabbit immunoglobulin G (IgG, 111-165-144; Jackson Immunoresearch Laboratories, West Grove, PA). Nuclei were stained with 4′,6-diamidino-2-phenylindole, while fibrotic tissue was labeled with fluorescein isothiocyanate wheat germ agglutinin (Vector Laboratories, Burlingame, CA). The expression of α-SMA was quantified as percentage of positive cardiomyocytes, and collagen I signal was graded on a scale of 0–3 (0: no signal, 1: weak, 2: mild, 3: diffuse), in at least 10–15 randomly selected high-power fields for each animal.
Immunofluorescence analysis of leukocytes in cardiac tissues
Intragraft leukocytes were analyzed in situ using an indirect immunofluorescence technique on frozen tissue sections (3 μm). The sections were incubated with the following antibodies: rat anti-mouse granulocytes (Gr-1, clone RB6-8C5; Biolegend, San Diego, CA) or rat anti-mouse F4/80 (clone BM8; Invitrogen, Carlsbad, CA) for macrophages, both followed by Cy3-conjugated goat anti-rat IgG (112-166-062; Jackson Immunoresearch Laboratories); rat anti-mouse CD4 (clone RM4–5; Biolegend); or rat anti-mouse CD8a (clone 53-6.7; BioLegend) both followed by AF488-conjugated goat anti-rat IgG (A-11006; Invitrogen). For each marker, the number of total positive cells was counted in at least 15–20 randomly selected high-power fields for each animal.
Transmission electron microscopy
Immediately after excision, cardiac tissue samples were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) overnight at 4°C and postfixed in 1% osmium tetroxide for 1 h. Then, specimens were dehydrated through ascending grades of alcohol and embedded in Epon resin. Semithin sections (1 μm) were stained with toluidine blue in borax and examined by light microscopy. Ultrathin sections (60–100 nm) were cut on an ultramicrotome (LKB Instruments, Milan, Italy), collected on copper grids, and stained with uranyl acetate and lead citrate. The morphological analysis of ultrastructure was performed using transmission electron microscopy (Morgagni 268D; Philips, Brno, Czech Republic). In total, at least 100 pairs of adjacent mitochondria were analyzed for WT and Sirt3−/− mice, and mitochondria showing coordination of cristae were counted. In Sirt3−/− mice injected with AAV9-mutOpa1, or AAV9-wtOpa1, a total of 288 pairs of adjacent mitochondria were studied, for a total of 576 mitochondria.
AAV production and delivery
A previous study has described that the deacetylation of two Sirt3-targeted lysines (K926, K931) in the C-terminal GTPase effector domain of the human OPA1 sequence (NP_056375.2) results in the activation of OPA1 (41). These two lysines are conserved between human and mouse, and they correspond to the K944 and K949 lysines in the Mus musculus OPA1 dynamin-like 120 kDa protein mitochondrial isoform 1 precursor (NP_001186106.1). The cDNA for the deacetylated form of mouse Opa1 was synthetized from GenScript. It differs from the wt Opa1 (GI: 312836757) in two base pair substitutions, c.A2831 > G and c.A2846 > G, and codifies for a protein containing two arginins instead of the conserved lysines K944 and K949 (K to R mutant plasmid). The cDNA was cloned in the AAV vector backbone pZac2.1 (pZac2.1-mutOpa1; Gene Therapy Program, Penn Vector core, University of Pennsylvania). To obtain the cDNA for the WT mouse Opa1 (pZac2.1-wtOpa1), site-directed mutagenesis on pZac2.1-mutOpa1 with the QuickChange II XL Site-Directed Mutagenesis Kit (#200521; Agilent Technologies, Santa Clara, CA) was performed according to the manufacturer's instructions. The following mutagenic primers were used: fwd tgagaagaaggttaaattgctcactggcaaacgagttcagctggc; rev gccagctgaactcgtttgccagtgagcaatttaaccttcttctca. The sequence of the full-length wtOpa1 was verified by Sanger sequencing on the 3730 DNA Analyzer (Applied Biosystem, Foster City, CA).
pZac2.1-mutOpa1 and pZac2.1-wtOpa1 were packaged into AAV capsid serotype 9 (AAV9-mutOpa1 and AAV9-wtOpa1, respectively). Infectious vector stock was prepared by the ICGEB AAV Vector Unit. Briefly, pZac2.1-mutOpa1 and pZac2.1-wtOpa1 were cotransfected together with a packaging/helper plasmid into HEK293 cells. Viral stocks were obtained by CsCl2 gradient centrifugation. Titration of AAV viral particles was performed by real-time PCR quantification of the number of viral genomes, measured as cytomegalovirus copy number.
For animal studies, AAV viral vector constructs (1.3 × 1012 vector genomes per animal) were diluted in saline to a final volume of 200 μL and administered to Sirt3−/− mice at 15 or at 18–20 months of age via a single tail vein injection, and animals were sacrificed 2 weeks or 3 months after AAV9 injection, respectively.
X-Gal staining
Fresh heart specimens were frozen in optimum cutting temperature compound and stored at −80°C. Three-micrometer cryosections were fixed with acetone for 10 min at 4°C. X-Gal staining was performed according to the manufacturer's instructions (200384, in situ Beta-Galactosidase Staininig Kit; Stratagene, San Diego, CA). Sections were counterstained with Harris's hematoxylin and mounted in Faramount acqueous mounting medium (S3025; Dako, Santa Clara, CA). Transduction efficiency was expressed as percentage of X-Gal-positive cardiomyocytes on the total number of cardiomyocytes, evaluated in 38 random fields from three different areas of the heart.
Citrate synthase activity
Mitochondrial content in cardiac tissues was evaluated by assessing citrate synthase activity. Citrate synthase activity is an accurate surrogate marker for detecting mitochondrial content in tissues, and compared with other mitochondrial mass indicators, it has the highest positive correlation with morphometric analysis of mitochondria performed with transmission electron microscopy (23). Hearts were washed twice in 0.9% (w/v) sodium chloride solution and homogenized in CelLytic MT buffer (C3228; Sigma-Aldrich) supplemented with protease inhibitor cocktail (P8340; Sigma-Aldrich), and tissue disruption was completed by using a blunt-ended needle and a syringe. As previously described (35), the sample lysates were then centrifuged 16,000 g for 10 min at 4°C to remove detergent-insoluble material. Supernatants were collected and total protein concentration was determined using DC™ assay (5000112; Bio-Rad Laboratories, Hercules, CA). Equal amounts of proteins (10 μg) were analyzed by citrate synthase activity assay (CS072; Sigma-Aldrich) according to the manufacturer's protocol. The citrate synthase activity was determined by the multimode microplate reader TECAN Infinite M200® PRO (Tecan Group Ltd., Mannedorf, Switzerland) at 412 nm under temperature controlled on a kinetic program for 1.5 min every 10 s.
ATP content assay
Excised cardiac tissues were washed twice in 0.9% (w/v) sodium chloride solution and homogenized in ATP assay buffer (#K354; BioVision, Milpitas, CA), and tissue disruption was completed by using a blunt-ended needle and a syringe. As previously described (35), the lysates were centrifuged 12,000 g for 10 min at 4°C to remove detergent-insoluble material. Supernatants were harvested and total protein concentration was determined using the DC assay (5000112; Bio-Rad Laboratories). Then, supernatants were deprived of proteins by using a deproteinizing preparation kit (K808; BioVision) according to the manufacturer's protocol. ATP content assay (K354; BioVision) was then performed on deproteinized samples according to the manufacturer's protocol. The fluorescence intensities were determined by the multimode microplate reader TECAN Infinite M200 PRO (Tecan Group Ltd.) at an excitation wavelength of Ex 535 nm and emission wavelength of Em 587 nm. Results were normalized for the total protein concentration of each sample.
NAD+ content assay
Cardiac tissues were washed twice in 0.9% (w/v) sodium chloride solution, homogenized in extraction buffer for NAD+ evaluation (KA1657; Abnova, Taipei City, Taiwan), and total protein concentration was determined using DC assay (Bio-Rad Laboratories). As previously described (35), samples were then processed for the determination of the NAD+ content by the multimode microplate reader TECAN Infinite M200 PRO (Tecan Group Ltd.) at 565 nm at a controlled temperature for 15 min according to the manufacturer's protocol. Results were normalized for total protein concentration of each sample.
Complex I activity assay
Cardiac tissues were washed twice in 0.9% (w/v) sodium chloride solution, homogenized in detergent solution according to the manufacturer's instructions (ab109721 Assay Kit; Abcam), and the total protein concentration was determined using DC assay (Bio-Rad Laboratories). A total of 20 μg of heart extracts was then processed for the determination of Complex I activity according to the manufacturer's instructions (ab109721 Assay Kit; Abcam). Complex I activity in each sample was determined by the multimode microplate reader TECAN Infinite M200 PRO (Tecan Group Ltd.) at 450 nm under a kinetic program at a controlled temperature for 30 min according to the manufacturer's instructions.
Cardiac function and dobutamine stress test
Cardiac function in mice anesthetized with 2 vol% isoflurane in 0.5 L/min O2 was assessed by transthoracic echocardiography using a commercially available echocardiographer equipped with a 12-MHz probe (MyLab30; Esaote, Geneva, Italy), as previously described (44). Warmed echo gel was placed on the shaved chest and each parameter was measured using B-mode-guided M-mode imaging in the parasternal short-axis view at the level of the papillary muscles. Images were analyzed off-line in a blinded manner. The global cardiac function was calculated as the percentage of fractional shortening (LVFS %) of the left ventricle. Heart rate and rhythm were simultaneously monitored by three-lead electrocardiogram (ECG). The cardiac function was evaluated at rest and at peak after intraperitoneal injection of a single bolus of dobutamine (1.5 μg/g of body weight), an established method for assessing the myocardial contractile reserve (10, 48). Echocardiographic scans and ECG recordings were repeated immediately after a single bolus injection and then periodically over 10 min, until the peak heart rate response was reached and heart rate began to decline again. ECG was recorded for 3 min at rest and at the peak of dobutamine stress.
Human iPSC-derived cardiomyocyte culture
Human iPSC-derived cardiomyocytes (C1006; Cellular Dynamics International, Madison, WI) were seeded on 0.1% gelatin-coated wells and cultured in a plating medium (M1001; Cellular Dynamics International) at a density of 45,000 viable cells/cm2. At day 2, the medium was replaced with maintenance medium (M1003; Cellular Dynamics International) and changed at day 4 and 6.
SIRT3 silencing and deacetylated Opa1 transfer
On day 7 postplating, iPSC-derived cardiomyocytes were transfected with 50 nM Silencer Select predesigned siRNA human SIRT3 (siSIRT3, s23768; Life Technologies, Carlsbad, CA) or control nontarget siRNA (siNULL, Silencer Select Negative Control #2 siRNA; Ambion, Carlsbad, CA) using the TransIT-KO Transfection Reagent (MIR2150; Mirus Bio, Madison, WI) according to the Cellular Dynamics International instructions. After 48 h of transfection, cells were used for subsequent experiments.
In selected experiments, iPSC-derived cardiomyocytes were transduced with 2 × 106 multiplicity of infection/cells of AAV9-mutOpa1 and 2 days later, SIRT3 silencing was performed as above. AAV9-LacZ- transduced iPSC-derived cardiomyocytes transfected with siNULL were used as control. On day 5 posttransduction, iPSC-derived cardiomyocytes were used for the analysis of cell contractility and total protein extraction.
Real-time PCR
Human iPSC-derived cardiomyocytes were harvested in TRIzol reagent (15596026; Invitrogen, Life Technologies), and total RNA was extracted according to the manufacturer's instructions. Contaminating genomic DNA was removed by RNase-free DNase (M6101; Promega, Madison, WI) for 1 h at 37°C. The first-strand cDNA (2.5 μg) was produced using a SuperScript VILO cDNA Synthesis Kit (11754050; Life Technologies) according to the manufacturer's instruction. To amplify cDNA of human SIRT3, we used the TaqMan Universal PCR Master Mix (4304437; Applied Biosystems) and inventoried TaqMan assays of the SIRT3 gene (FAM-MBG probe Hs00202030_m1), and of the human HPRT endogenous control (FAM-MGB probe, Hs99999909_m1) according to the manufacturer's instructions. PCR was performed on the Viia7 Real-Time PCR System (Applied Biosystems). Data were analyzed using the 2−ΔΔCT method and presented as fold changes relative to siNULL cells.
MitoTracker analysis
To evaluate mitochondrial morphology, living iPSC-derived cardiomyocytes were incubated with a 250 nM fluorescent probe MitoTracker® Red (M7512; Thermo Fisher, Invitrogen) for 30 min, at 37°C, 5% CO2. Nuclei were counterstained with NucBlue® Live ReadyProbes® (Hoechst; R37605; Thermo Fisher, Invitrogen) according to the manufacturer's protocol. At the end of the probe incubations, living cells were examined using the Cell Observer system (Axio Observer.Z1; Zeiss Oberkochen, Germania) and mitochondrial morphology was evaluated using the Mitochondrial Network Analysis toolset on Fiji software (50). Data were normalized for the number of Hoechst-positive cells in 10 random fields per sample.
Cell contractility and FLUO-4 analysis
To detect cell contractility, living iPSC-derived cardiomyocytes were seeded on 35-mm glass-bottomed dishes, monitored under phase contrast using the Cell Observer system (Axio Observer.Z1; Zeiss), and stably recorded for at least 2 min in different fields of the dish. For calcium analysis, the nonratiometric Fluo-4 dye Calcium Imaging Kit was used (F10489; Thermo Fisher, Invitrogen), according to the manufacturer's protocol. Briefly, living iPSC-derived cardiomyocytes were incubated with Fluo-4 AM in Live Cell Imaging Solution (Thermo Fisher, Invitrogen) supplemented with 20 mM glucose (Sigma-Aldrich) for 30 min at 37°C, 5% CO2. At the end of probe incubations, living cells were rinsed with the iCell Cardiomyocytes Maintenance Medium (M1003; Cellular Dynamics International) and recorded for at least 2 min in different fields of the dish by the Cell Observer system (Axio Observer.Z1; Zeiss).
Mitochondrial isolation from cardiac tissue
Mitochondria were isolated from mouse cardiac tissue using the Qproteome Mitochondria Isolation Kit (37612; Qiagen S.r.l., Milan, Italy) according to the manufacturer's protocol. Isolated mitochondria were solubilized in mitochondrial storage buffer (37612; Qiagen S.r.l.) and total protein concentration was determined using the DC assay (5000112; Bio-Rad Laboratories).
Protein extraction from human iPSC-derived cardiomyocytes
For total protein extraction, human iPSC-derived cardiomyocytes were washed twice in 0.9% (w/v) sodium chloride solution and lysed by sonication in mammalian CelLytic M lysis buffer (C2978; Sigma-Aldrich) supplemented with protease inhibitor cocktail (P8340; Sigma-Aldrich). After sonication, the sample lysates were centrifuged 16,000 g for 10 min at 4°C, and protein concentration was determined using DC assay (Bio-Rad Laboratories).
Evaluation of OPA1 and SOD2 acetylation
OPA1 and SOD2 acetylation was determined by standard Western blot analysis. Briefly, equal amounts of mitochondrial proteins from cardiac tissue (30 μg) or total extracts of iPSC-derived cardiomyocytes (20 μg) were separated on 12% sodium dodecyl sulfate (SDS)/polyacrylamide gel electrophoresis (PAGE) under reducing conditions and transferred to nitrocellulose membranes (Bio-Rad Laboratories). After blocking with 5% BSA in Tris-buffered saline (TBS) supplemented with 0.1% Tween-20, membranes were incubated overnight at 4°C with the following antibodies: rabbit anti-acetylated-lysine (1:1000, BK9441; Cell Signaling) and mouse anti-OPA1 antibody (1:1000, 612606; BD Bioscience) or sheep anti-SOD2 (1:1000, 574596; Merck Millipore). The signals were visualized on an Odyssey®FC Imaging System (LiCor, Lincoln, NE) by infrared (IR) fluorescence using a secondary goat anti-rabbit IRDye 680LT antibody (1:1000 FE3680210; LiCor) and a goat anti-mouse IRDye 800CW (1:1000FE30926210; LiCor) or by enhanced chemiluminescence-Western blotting detection reagent (Pierce, Thermo Fisher Scientific, Inc.) using donkey anti-sheep (1:10,000 A3415; Sigma-Aldrich) horseradish peroxidase secondary antibodies. Mouse anti-VDAC (voltage-dependent anion channel [VDAC], 1:2000ab186321; Abcam) was used as sample-loading controls in isolated mitochondria of the same membranes. Bands were quantified by densitometry using the Image Studio Lite 5.0 (LiCor) software. OPA1 and SOD2 acetylation was expressed as the ratio between the band of acetylated lysine colocalizing with the band corresponding to total OPA1 or SOD2. Total OPA1 protein level was expressed as the ratio between the bands of OPA1 and VDAC. For any cropped Western blot shown in the main body of the article, the full uncut gels are found in Supplementary Figure S5.
Western blot analysis of ATP5I
Equal amounts of total extracts of iPSC-derived cardiomyocytes (20 μg) were separated on 12% SDS-PAGE under reducing conditions and transferred to nitrocellulose membranes (Bio-Rad Laboratories). After blocking with 5% BSA in TBS supplemented with 0.1% Tween-20, membranes were incubated with a rabbit anti-ATP5I antibody (1:1000 HPA035010; Sigma-Aldrich). Mouse anti-VDAC (1:2000ab186321; Abcam) was used as sample-loading controls in isolated mitochondria of the same membranes. The signals were visualized on an Odyssey®FC Imaging System (LiCor) by IR fluorescence using a secondary goat anti-rabbit IRDye 680LT antibody (FE3680210; LiCor) and a goat anti-mouse IRDye 800CW (FE30926210; LiCor), as appropriate. Bands were quantified by densitometry using the Image Studio Lite (LiCor) 5.0 software. Total ATP5I protein level was expressed as the ratio between the bands of ATP5I and VDAC. For any cropped Western blot shown in the main body of the article, the full uncut gels are found in Supplementary Figure S5.
Statistics
The significance level of difference between individual groups was analyzed by the unpaired or paired Student's t-test, or analysis of variance with Tukey post hoc test comparisons, and the statistical significance was defined as p-value of <0.05. Results are given as mean ± standard error of the mean (SEM).
Statistics for life span comparison between Sirt3−/− and WT mice was evaluated through log-rank calculations. Maximum life span of Sirt3− /− and WT mice was compared by means of a Laplace regression, considering the time by which 90% of the mice had died (90th percentile of survival). The ‘Laplace’ package for STATA version 11 was retrieved for this purpose.
For the analysis of risk of cristae alignment alterations, odds ratios with their 95% confidence intervals were estimated taking into account familial dependence between observations with generalized estimated equations using the GENMOD procedure in SAS®, allowing the extension of traditional linear model theory to generalized linear models by allowing the mean of a population to depend on a linear predictor through a nonlinear link function. In this article, we investigated a binary outcome modeling approach using PROC GENMOD with the link functions and aggregate, with post hoc Bonferroni correction. Results are given as mean ± SEM, and the statistical significance was adjusted for multiple comparisons and was defined as p-value of <0.005.
Footnotes
Acknowledgments
The authors thank Sebastian Villa and Fabrizio Giordano for the animal study, Paola Rizzo for technical assistance with immunostaining, and Prof. Gino Villetti for histologic analysis. They thank Manuela Passera for helping to prepare the article and Kerstin Mierke for English language editing. L.P. is the recipient of a fellowship from Fondazione Aiuti per la Ricerca sulle Malattie Rare (ARMR), Bergamo, Italy. Do.C. is the recipient of a fellowship from Fondazione Istituti Educativi di Bergamo, Bergamo, Italy.
Authors' Contributions
A.B. and G.R. designed the study, discussed the data, and wrote the article; P.C. and S.C. designed the study, performed the experiments, discussed the data, and contributed to writing the article; L.P. performed the experiments, discussed the data, and contributed to writing the article; Do.C. and Da.C. performed the in vivo experiments; L.Z. generated the AAV9 vectors; P.T. performed the experiments; C.Z. discussed the data and contributed to writing the article; A.P. performed statistical analysis; V.L. discussed the data and performed measurements of cardiac function; M.G. discussed the data and contributed to writing the article; S.T. contributed to the research plan, discussed the data, and contributed to writing the article. All authors commented on the article.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Movie S1
Supplementary Movie S2
Supplementary Movie S3
Supplementary Movie S4
Supplementary Movie S5
Supplementary Movie S6
Supplementary Movie S7
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.