
Abstract
Significance:
Skeletal muscle is a crucial tissue to whole-body locomotion and metabolic health. Reactive oxygen species (ROS) have emerged as intracellular messengers participating in both physiological and pathological adaptations in skeletal muscle. A complex interplay between ROS-producing enzymes and antioxidant networks exists in different subcellular compartments of mature skeletal muscle. Recent evidence suggests that nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) are a major source of contraction- and insulin-stimulated oxidants production, but they may paradoxically also contribute to muscle insulin resistance and atrophy.
Recent Advances:
Pharmacological and molecular biological tools, including redox-sensitive probes and transgenic mouse models, have generated novel insights into compartmentalized redox signaling and suggested that NOX2 contributes to redox control of skeletal muscle metabolism.
Critical Issues:
Major outstanding questions in skeletal muscle include where NOX2 activation occurs under different conditions in health and disease, how NOX2 activation is regulated, how superoxide/hydrogen peroxide generated by NOX2 reaches the cytosol, what the signaling mediators are downstream of NOX2, and the role of NOX2 for different physiological and pathophysiological processes.
Future Directions:
Future research should utilize and expand the current redox-signaling toolbox to clarify the NOX2-dependent mechanisms in skeletal muscle and determine whether the proposed functions of NOX2 in cells and animal models are conserved into humans.
I. Introduction
Skeletal muscle constitutes ∼40% of total body mass and plays a crucial role in maintaining locomotion, metabolic health, and, thus, ultimately quality of life (386). External factors, such as diet and physical activity, as well as internal factors, such as gender, aging, and genetic muscle diseases, have been demonstrated to have a profound effect on both muscle function and whole-body homeostasis (55). Unravelling the molecular mechanisms responsible for the adaptive responses may allow us to identify novel pharmacological targets to treat muscle diseases (118).
Reactive oxygen species (ROS) are proposed to be molecular regulators of the physiological and pathological adaptation of skeletal muscle (286). ROS are continuously generated and buffered in skeletal muscle both at rest and in response to various stressors (286). A complex interplay between ROS-producing enzymes and antioxidant networks exists in different subcellular compartments of mature skeletal muscle (307), allowing for highly compartmentalized signaling by specific hydrogen peroxide (H2O2) and oxidation products. The resulting changes in the myofiber redox state are emerging as essential signal transduction elements linked to the regulation of, for example, muscle metabolism, repair, and growth (286).
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) complexes were first described in phagocytic cells, but have since been found to be expressed in many nonimmune cell types, including different brain cells, endothelial cells, hepatocytes, adipocytes, cardiomyocytes, skeletal muscle, and more (27). So far, NOXs are the only enzyme family known to produce ROS as their primary function, in contrast to other enzymes that generate ROS as byproducts of their catalytic functions. It has been shown that skeletal muscle expresses at least two isoforms, NOX2 and NOX4, in different subcellular compartments (144).
This review will focus on the many emerging connections between NOX2-dependent redox signaling and metabolic homeostasis in skeletal muscle and try to reconcile the several seemingly contradicting paradigms regarding NOX2 as being both a beneficial and a detrimental player in this context. First, we will provide an overview of fundamental mechanisms in general redox biochemistry to introduce emerging redox-signaling concepts to nonredox specialists, after which we will review the generic NOX2 complex expression and regulation, considering the relevance to skeletal muscle when possible. Then, the literature regarding the physiological regulation of NOX2 and its downstream effects in skeletal muscle will be discussed. Lastly, the involvement of NOX2 in various skeletal muscle disease states will be covered.
II. General Introduction to Redox Signaling
A. Regulation of signaling cascades by H2O2—reversible oxidation of cysteine residues
ROS clearly display signaling or regulatory functions, in addition to their described capacity to damage biomolecules and induce damage. Along with this idea, the redox biology community has developed the view that oxidants are responsible for oxidative damage, whereas signaling mechanisms involve H2O2 or other two-electron oxidants (383). Indeed, H2O2 has some features that make it a good signaling molecule; it is a robust two-electron oxidant, with a standard reduction potential (E′o) of 1.32 V at pH 7.0 (382), and it is, therefore, capable of oxidizing a wide range of substrates. However, in contrast to other two-electron reactive species, the reactions of H2O2 have to overcome a high activation energy barrier to release its oxidizing power and, therefore, they are kinetically rather than thermodynamically driven. This reactivity, restricted to very few biological molecules at an appreciable rate, for example, thiols in proteins, provides the basis for a highly selective messenger.
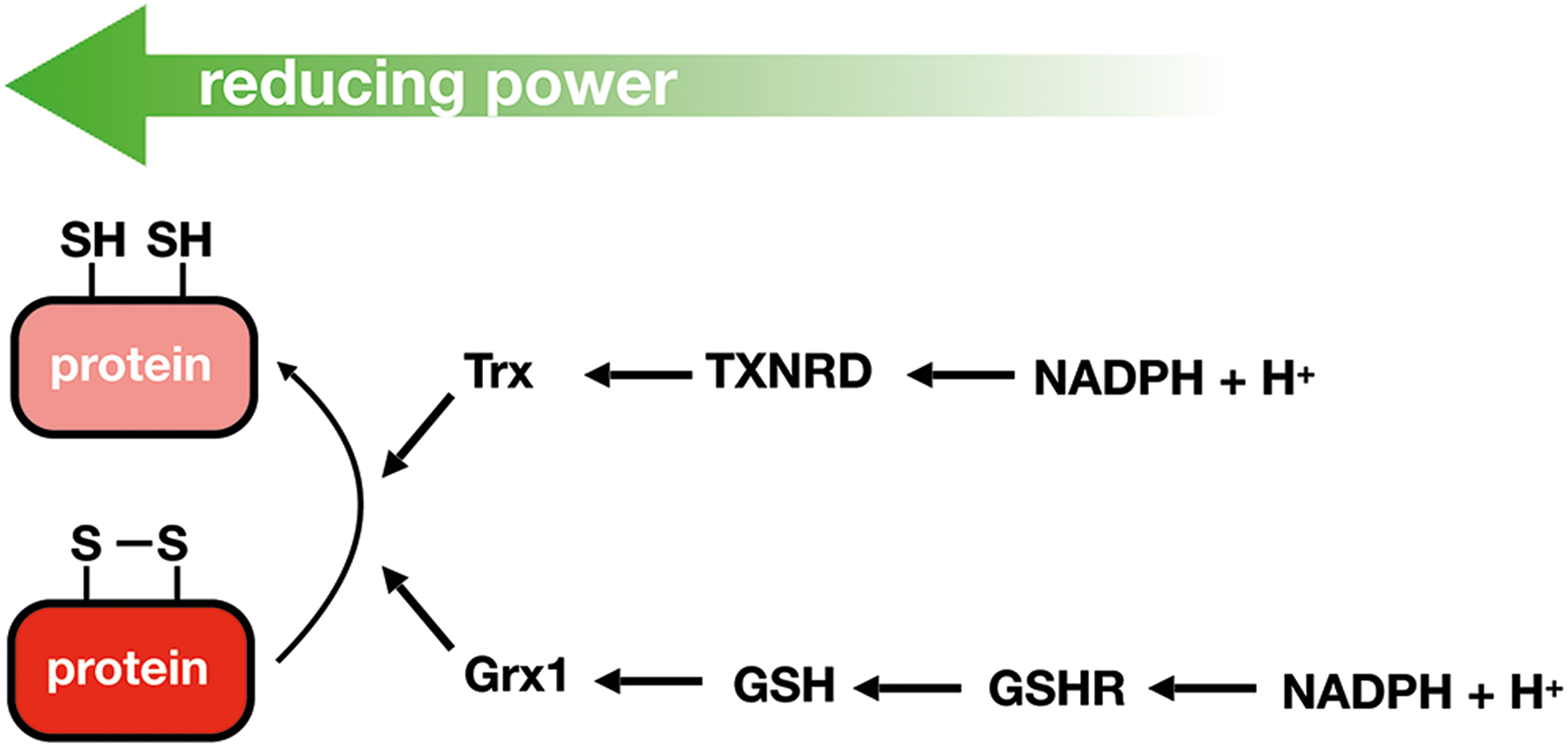
As proposed by Brigelius-Flohé and Flohé (45), a real redox signaling mechanism requires a primary signaling molecule, a sensor, transducers, effectors, and reversion reactions to terminate the signaling cascade. Thus, H2O2 will react in a specific, fast, and efficient manner with sensor proteins containing redox switches, such as metal centers or cysteine (Cys) residues (thiols); the reversible oxidation of these redox moieties controls the activity of the sensor proteins. The reactivity of Fe-S clusters toward redox-active molecules and its role in signaling has been recently reviewed (70). This section will only focus on H2O2 signaling mediated by reversible Cys oxidation, and on the reduction by the thioredoxin (Trx)/thioredoxin reductase (TXNRD) and glutaredoxin (Grx)/reduced glutathione (GSH)/GSH reductase pathways, which reduce proteins containing reversibly oxidized Cys using NADPH as the final electron donor (Fig. 1).
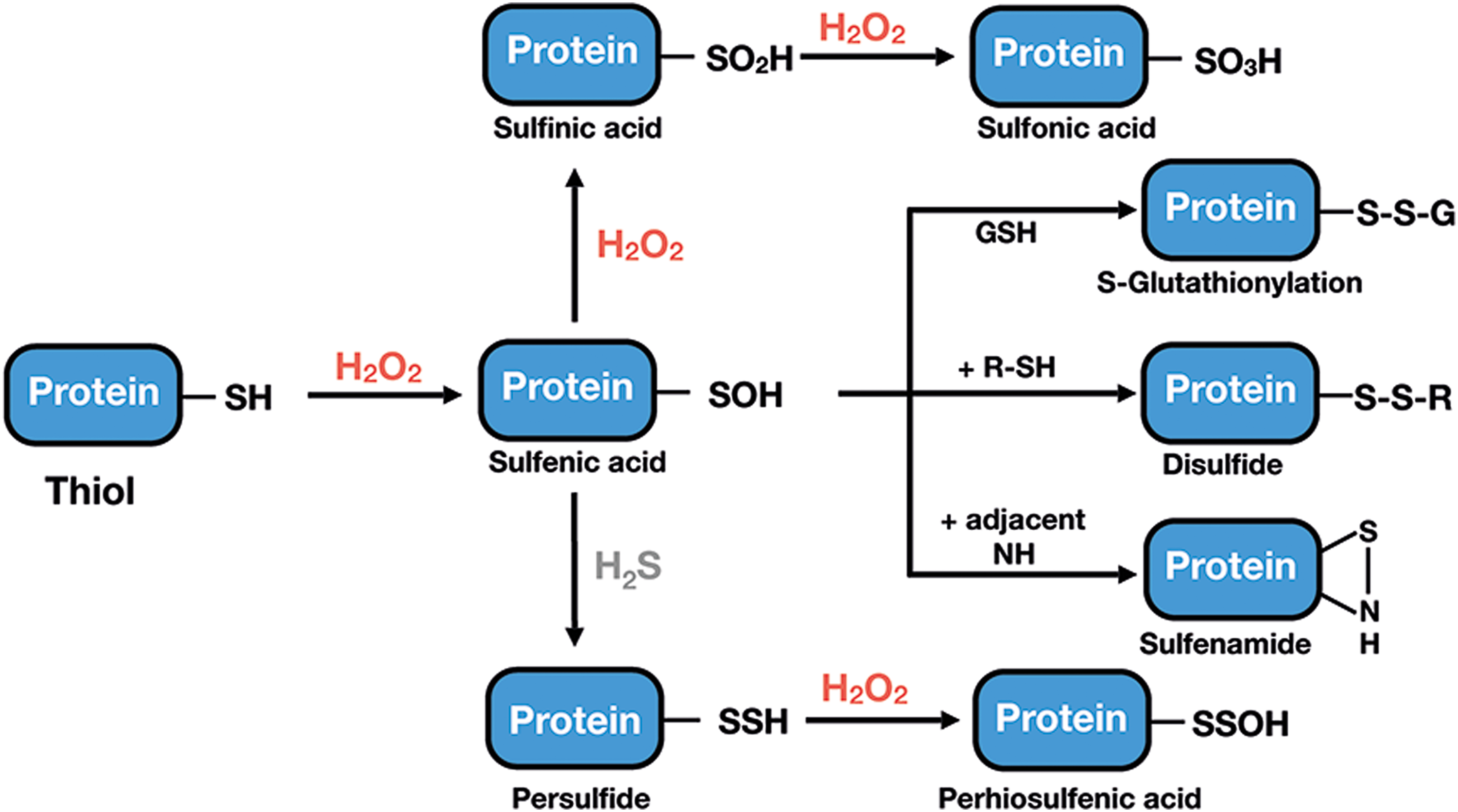
The oxidative modifications of Cys produced by H2O2 are summarized in Figure 2. Exposure of a Cys thiol (SH) to low levels of H2O2 results in the formation of a sulfenic acid (Cys-SOH) intermediate, which due to its high reactivity will rapidly react with GSH (glutathionylation; Cys-SSG), with a nearby Cys, to form an inter- or intramolecular disulfide bond (295), or with adjacent amino groups, to form a cyclic sulfenamide. Larger doses of H2O2 will result in the oxidation of Cys-SOH to sulfinic or sulfonic acid, modifications that are, in general, irreversible (Fig. 2). It is now becoming apparent that biological thiols can also exist in the form of hydropersulfide or polysulfide species (71, 157). They have a reduced pKa, which, in part, explains their enhanced nucleophilic properties and one-electron reductant activity compared with corresponding sulfhydryl species (34, 71, 157) and it has been proposed that they might represent important targets for H2O2, leading to the intermediate formation of perthiosulfenic acid species (143).
Laying aside the chemical nature of the thiol modification, key factors that account for sensitivity and specificity in the reaction of specific Cys residues with H2O2 and that, therefore, define a true thiol switch are accessibility, intermediate half-lives of disulfides, and sufficient reactivity of Cys thiols, the latter given by the rate constant and concentration (82). These parameters are determined by the protein environment, resulting in activated molecular geometries, particularly with regard to the transition state, as well as appropriate redox potential, pKa values of both the nucleophilic-Cys thiol and the leaving group (OH− in the case of H2O2), and complementary surfaces between signal molecules and interacting proteins (82, 108).
However, and despite theoretical predictions, there is a wide range of reactivity of thiol-containing proteins toward H2O2 spanning several orders of magnitude, from the low 20 M −1 s−1 for some protein tyrosine phosphatases, such as protein tyrosine phosphatase 1B (PTP1B) and SHP-2, to the high 107 M −1 s−1 for peroxiredoxin (Prx) 2 (384). More strikingly, the reactivity toward peroxides of proteins proposed to be redox regulated (such as phosphatases, kinases, and transcription factors that are activated or inactivated by thiol oxidation for signaling purposes) often falls in the range of mid-to-low intrinsic H2O2 reactivity (k ≈ 101–102 M −1 s−1) (229) and they are expressed at low levels. This moderate H2O2 reactivity should not be a problem as long as cells require a slow response, or if the H2O2 transient signal lasts for a long time (229). However, if these proteins are directly oxidized by H2O2, it is difficult to reconcile a fast oxidation of low reactive thiols by nanomolar concentrations of H2O2 in the presence of abundant high-affinity H2O2 scavenger proteins such as Prxs.
There are currently two models to explain the mechanism by which H2O2 is transduced to low-reactivity target proteins, which are not mutually exclusive, and the occurrence of one mechanism or the other could be influenced by the source of H2O2 generation, duration, and cellular location. The predictions, experimental evidence, and open questions of these two models have been recently reviewed (334), and they will be summarized here briefly (Fig. 3A, B).
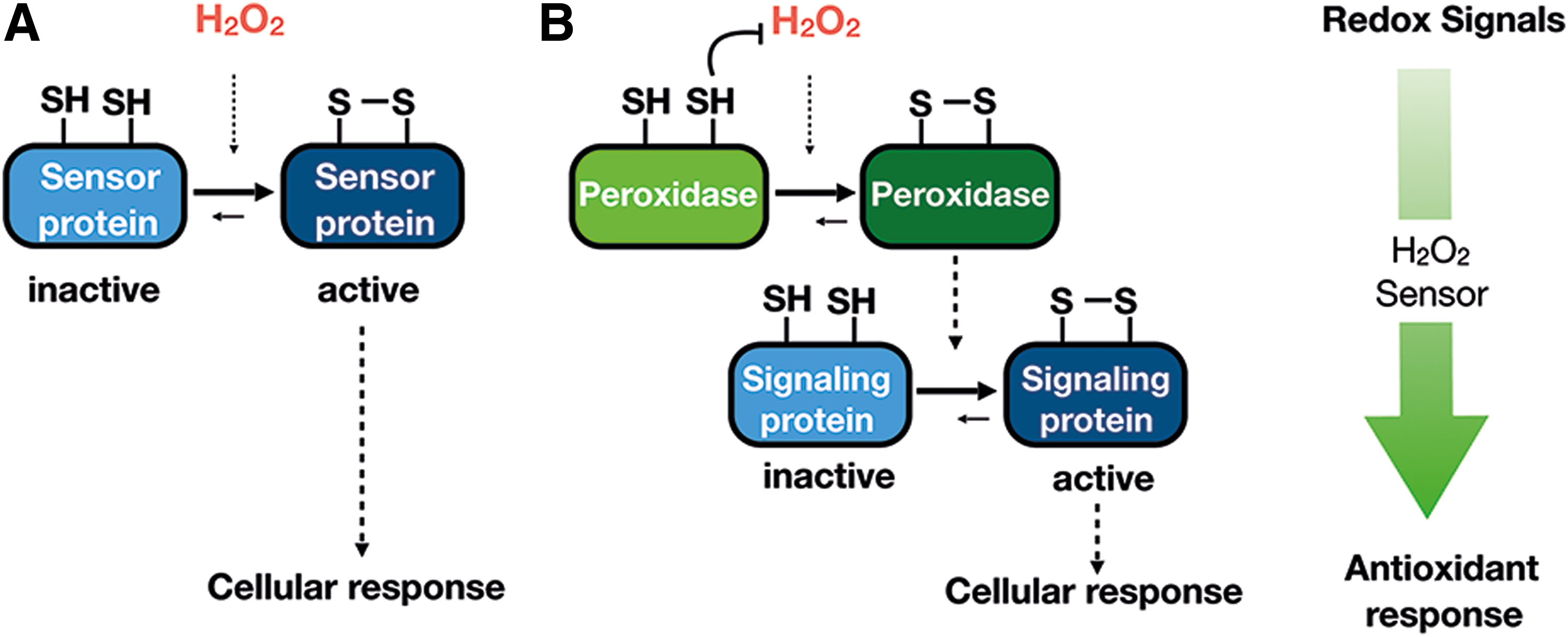
The first model consists of the direct oxidation of thiols in redox-regulated proteins by H2O2 present in the environment that has diffused from the original intra- or extracellular source (Fig. 3A). H2O2 is actually able to diffuse through lipid membranes, but aquaporins (AQPNs) facilitate the passage of H2O2 across membranes (246). As mentioned earlier, because of the higher H2O2 reactivity of Prxs and GSH peroxidases (Gpxs), these H2O2 scavengers are strong competitors of redox-regulated proteins, and they need to be transiently inactivated by post-translational modifications. Once the activity of Prxs has been temporally and locally blocked, the accumulation of H2O2 may reach concentrations in the micromolar range that now allow for the direct oxidation of protein thiols with low H2O2 reactivity (229). In support of this hypothesis, there are some 2-Cys-Prxs whose peroxidatic Cys are easily hyperoxidized to sulfinic acid, inactivating their peroxidase activity. The existence of sulfiredoxins, enzymes capable of specifically reducing sulfinic acid forms in Prxs, also supports the role of transient Prxs inactivation in H2O2 signaling (37). In addition to Prxs inactivation by hyperoxidation, inactivation by phosphorylation is an alternative mechanism described for Prx1. A comprehensive review regarding mammalian Prxs regulation has been recently published (296). An interesting concept related to Prxs inactivation is the localized accumulation of H2O2, providing an additional means of target selectivity. Only proteins that are recruited into high H2O2 microenvironments (e.g., in close proximity to H2O2 sources or to inactivated Prxs or confined in compartments lacking Prxs) would be oxidized. In favor of this idea, it is widely accepted that the spatial distribution of H2O2 in cells and tissues is not uniform; instead, substantial gradients exist both from extracellular to intercellular and between subcellular spaces (20, 228).
The second mechanism for H2O2 transduction postulated as a “redox relay”, proposes that Prxs and, to a lesser extent, Gpxs do outcompete all other less reactive protein thiols and transfer oxidized equivalents to redox-regulated target proteins, constituting a two-component sensor system (Fig. 3B). As opposed to the “direct oxidation” mechanism, Prxs are the “true” sensors in the signaling pathway and enable protein oxidation rather than compete (385). According to this model, protein and thiol specificity can be explained by protein
An additional level of complexity of the Prx (or Gpx)-based redox relay model is based on the fact that the reaction of H2O2 with peroxidases during the scavenging cycle requires the recycling by, and the concomitant oxidation of, Trxs. Oxidized Prxs would accumulate only when TXNRD or NADPH become transiently limiting or exhausted, transferring the oxidizing equivalents to target signaling proteins (53). Alternatively, the accumulation of oxidized Trxs could directly promote oxidation of the target signaling protein. Although no direct biochemical thiol-disulfide exchange between an oxidized Trx and a downstream target has been demonstrated, several signal transduction pathways have been proposed to be activated by oxidized Trx but not by reduced Trx [reviewed by Berndt et al. (31) and Netto and Antunes (262)].
B. Reversibility of H2O2 signaling: reduction of reversibly oxidized Cys residues in proteins
The two main reversible oxidative Cys modifications involved in H2O2 signaling are sulfenic acid and disulfides. The recycling/reduction of sensors and other oxidized proteins in H2O2 signaling cascades containing these modifications depends on the two major systems that cells are provided with to regulate the thiol-disulfide status of proteins: the Trx and Grx systems (Fig. 1). Trxs and Grxs are small oxidoreductases (9–15 kDa) originally identified as hydrogen donors for ribonucleotide reductase (150, 253). They are structurally similar and rely on a Cys-X-X-Cys/Ser active site motif (Trx fold) to reduce protein disulfides using a dithiol mechanism. In the Trx, the dimeric flavoenzyme TXNRD reduces oxidized Trx by using NADPH as the electron donor (397). Regarding the Grx pathway, disulfide bonds in proteins can be reduced by a dithiol mechanism, by which Grxs form mixed disulfides with the protein. In addition, Grxs can catalyze the reduction of mixed disulfides formed between protein Cys residues and GSH (deglutathionylation) by using a so-called monothiol mechanism that only requires the N-terminal Cys residue of the Cys-X-X-Cys/Ser motif (82). Both processes require a consequent series of reactions involving GSH, GSH reductase, and reduced cofactor (124). Crosstalk between these two electron flow pathways is exemplified by mammalian Grx2, characterized by the uncommon active site motif Cys–Ser–Tyr–Cys (instead of the regular dithiol consensus Cys–Pro–Tyr–Cys), that can receive electrons from TXNRD (167).
Although Grxs are the only oxidoreductases that are capable of reducing glutathionylated proteins, there is considerable overlap or redundancy between the Grx and Trx systems when recycling disulfides of redox proteins. The extent of redundancy might depend on the organism [reviewed in Garcia-Santamarina et al. (124)]. In Escherichia coli, the two systems seem largely redundant (at least one of the two systems is essential). In contrast, in eukaryotes such as yeast, they are not fully redundant and, actually, it seems that the GSH/Grx system does not have a significant function and GSH would only act as a backup for the Trx system. Despite this more or less significant redundancy, Trxs and Grxs have distinct substrate specificity, which is controlled by short- and long-range electrostatic interactions between oxidoreductases and their substrates as well as by geometric complementarity. Therefore, the amino acid composition of areas outside the active site or even outside the contact area is more important than the composition of the active site that would only affect the redox potential, not the determinant for specificity (32). In agreement with this, the surface of individual Grxs and Trxs is where they mostly differ.
C. Mammalian redox-regulated proteins
The development of in vivo thiol trapping methodologies combined with mass spectrometry has produced a growing body of data supporting the role of oxidative modifications of protein thiols in the regulation of the function of a diverse set of proteins involved in diverse physiological and pathological responses. Examples in mammals of these cellular processes include growth factor signaling, hypoxic signal transduction, autophagy, immune responses, cell proliferation, and differentiation or metabolic reprogramming, to name a few. It is not the purpose of this section to provide a comprehensive list of different types of mammalian redox-regulated proteins, but, instead, we suggest recent reviews that provide examples of redox-regulated kinases, phosphatases, and transcription factors involved in cancer and cell proliferation (121), hypoxia (327), inflammatory processes (219), blood pressure homeostasis (289), and glycemic control (273), among others.
Many of the redox-regulated proteins fall into the category of kinases, phosphatases, and transcription factors. However, there are also other types of proteins whose activity has been shown to be redox regulated, such as proteins involved in the cytoskeleton dynamics such as actin, tubulins, cofilin, semaphorins (125), GTPases (146, 184), protein quality control proteins (40, 389), or dehydrogenases such as GAPDH (282).
Despite a large number of identified redox-regulated proteins, the depth of the biochemical characterization of the signal transduction pathway (actual intra- or extracellular messenger, type of thiol modification, the potential role of other Cys in the same protein, transduction mechanism) is, with a few exceptions, quite low. A detailed biochemical characterization of the signaling mechanism is not only important per se, but also to properly design mutant thiol redox switches and animal knock-in models bearing these mutations into the native genomic loci to analyze the physiological relevance of a particular redox modification, which in mammalians is far from being established.
Regarding transcription factors, a number of them are believed to be redox regulated in vivo by H2O2. From a broad perspective, transcription factor regulation by H2O2 may depend on redox regulation of kinases and phosphatases whose final effector is a transcription factor. In a stricter sense, redox-regulated transcription factors refer to those bearing H2O2-sensitive Cys located at the DNA binding interface, or at the domains of interactions with either co-activators, co-repressors, or transport machinery. These transcription factors, on reversible oxidation, would modify their promoter recognition, recruitment of co-regulators, or subcellular localization. Many in vitro and cell culture studies support the identification of Cys residues as thiol switches in transcription factors, but, as mentioned earlier, studies showing the complete biochemical mechanism, or the real significance of these switches are still missing. We will discuss here two of the redox-regulated transcription factors that have been better characterized: Kelch-like ECH-associated protein-1 (Keap1) and STAT-3.
Keap1/nuclear factor erythroid 2–related factor 2 (Nrf2) is the Cys-based sensor of oxidative stress, reminiscent of the well-characterized Yap1 and Pap1 in budding and fission yeast (80, 363), respectively, that activates the transcription of antioxidant genes on induction by electrophiles and oxidants. This redox-stress sensing system has been thoroughly studied regarding its molecular mechanism and physiological significance (91). Keap1 interacts with the transcription factor Nrf2 through its degron domain and with Cullin 3 to form a ubiquitin E3 ligase complex that in basal conditions ubiquitinates Nrf2, which is then degraded by the proteasome. Under oxidative or electrophilic stress, including H2O2, specific Keap1 Cys residues are modified, Keap1-based E3 ubiquitin ligase activity is inhibited, and, as a consequence, de novo synthesized Nrf2 is stabilized and accumulates in the nucleus. Keap1 has at least 10 reactive Cys, among which Cys-151, Cys-273, and Cys-288 act individually and/or in cooperation as sensors of various electrophiles. In particular, the importance of Cys-151 as a sensor has been verified in transgenic mice expressing Keap1 with Cys-151 substituted with serine. The functional significance of the other Cys residues in Keap1 has also been examined by using site-directed mutagenesis. The diversity of response profiles depending on the inducers and modified Cys has led to the concept of “Cys code” by which the Keap1–Nrf2 sensor is unique in that it responds to a diverse array of chemicals and oxidative insults employing multiple Cys. Based on this code, Nrf2 inducers have been classified into at least five classes (306). In the particular case of H2O2, Nrf2 induction is independent of the three main Keap1 Cys: 151, 273, and 288. Thus, mouse embryonic fibroblasts established cell lines with Keap1C151S/C273W/C288E and showed Nrf2 levels similar to those of a wild-type (WT) cell line on treatment with H2O2 (306). The role of Cys-151 in H2O2 response seems to be restricted to the formation of an intermolecular bond between two Keap1 subunits (113), but how this redox switch is transduced into increased Nrf2 accumulation remains unknown. Other H2O2-sensitive Cys residues in Keap1 are Cys-226 and Cys613, which are involved in an intermolecular bond that is further increased after treatment with H2O2. Another unresolved question related to Keap1 H2O2 signaling is whether the specific Cys receive oxidant equivalents directly from H2O2 or through a redox relay.
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that responds to cytokines and growth factors by forming homo- and heterodimers and by nuclear translocation to the nucleus on phosphorylation by receptor-associated Janus kinases. In a screen for mixed disulfides with Prxs, the laboratory of Tobias Dick identified STAT3 as a mixed disulfide with Prx2 and demonstrated that Prx2 mediated the oxidation and oligomerization of STAT3 by both H2O2 and interleukins (ILs) (328). They could also identify that the STAT3 Cys was involved with the Prx2 interaction and suggested two independent pathways that led to the formation of either STAT3 dimers or tetramers. In both pathways, there was a concerted disulfide exchange that started from a Prx2-STAT3 monomer involving the C-terminal domain of STAT3 containing the trans-activation domain (AD). Interestingly, the Prx2-driven oxidation of STAT3 resulted in an inhibition of STAT3-driven transcription rather than activation of the STAT3 function. STAT3 represents the first example of a mammalian redox-regulated protein where a redox relay H2O2 transduction mechanism has been demonstrated.
III. Expression and Regulation of NOXs in Skeletal Muscle
Although ROS and other highly reactive molecules may be the result of environmental insults, they are continuously formed within cells from side reactions of normal metabolism. Under normal physiological conditions, ROS are usually produced at subtoxic levels, in a regulated way, and through different mechanisms. The cellular localization of the NOX family of proteins and the concomitant localized production of superoxide (O2 ·−)/H2O2 make these particularly relevant in terms of redox signaling, because transient and high local concentrations of H2O2 may be required to oxidize target proteins (see Section II.B). The following sections of this review will be devoted to a comprehensive revision of the NOX family biology and what it is known in muscle.
A. A brief history of the NOX family
The NOX family currently comprises seven mammalian homologs named NOX1 to 5 and DUOX1 and 2 (Fig. 4). For a detailed account of the differences in their structure/function, the reader is referred to several excellent reviews on this subject (27, 198). Here, only a brief account of the NOX2 discovery and of NOX isoforms will be given for reference, after which we will focus on the structure/function of NOX2 and its potential regulatory mechanisms in skeletal muscle.
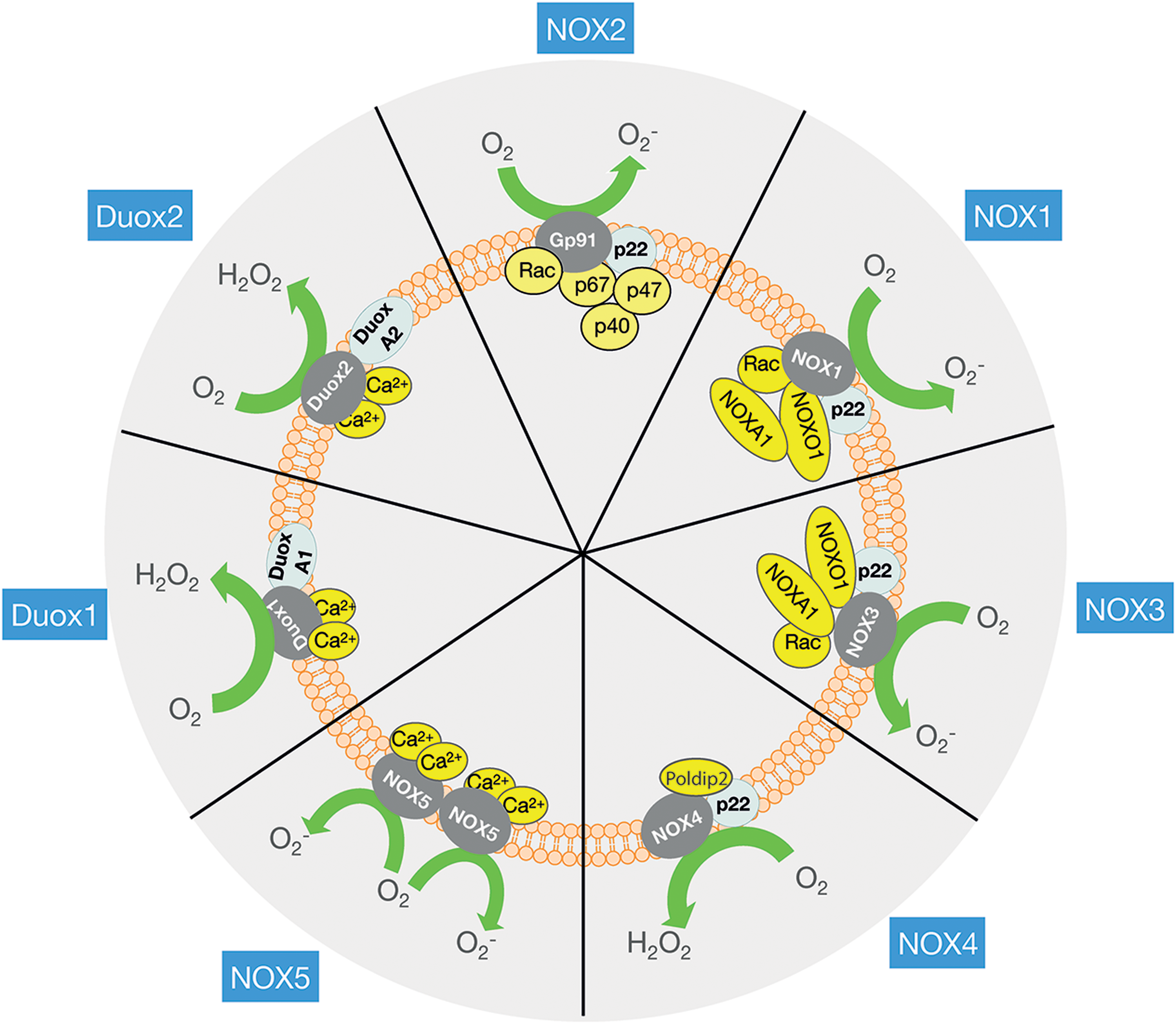
Historically, the heme-containing b558 flavocytochrome was described in 1978 as an integral membrane protein complex lacking in patients with the rare inherited human immune disorder chronic granulomatous disease (317), a condition characterized by an absent respiratory burst in phagocytes. Later, in 1986–1987, the catalytic gp91phox subunit of the b558 complex was cloned (304, 351), followed by the discovery of the remaining five current NOX2 subunits within the subsequent 7 years. These included the description of the second b558 integral membrane subunit p22phox in 1987 (90, 315), then the cytoplasmic subunits p47phox and p67phox (365), Rac GTPase isoforms (3), and, lastly, the nonessential modulatory subunit p40phox (379). Mutations in all of these subunits have now been linked to chronic granulomatous disease (303). The existence of additional nonessential subunits has been suggested but not firmly established.
Up through the 1990s, six other isoforms of the NOX superfamily of proteins sharing homology with the gp91phox catalytic subunit were discovered (Fig. 4). Gp91phox catalytic subunit was renamed NOX2 in the novel terminology and is, therefore, called NOX2 in the remainder of this review. The NOX family shows distinct tissue-selective expression patterns and functions. Thus, NOX1 is enriched in the colon epithelium, NOX2 in phagocytes, NOX3 in the inner ear, NOX4 in kidney epithelial cells, NOX5 in spleen and testis, and DUOX1 and 2 in the thyroid gland. Based on similarities in their regulation, the NOX family can be divided into three subfamilies: (i) NOX1–3, which requires p22phox and cytosolic subunits for activation; (ii) NOX4, which is p22phox- and Poldip2 dependent but active without cytosolic subunits (366); and (iii) the calcium (Ca2+)-dependent and p22phox and cytosolic subunit-independent NOX5 and DUOX1 and DUOX2. NOX1–3 and NOX5 release O2 −, NOX4 generates both O2 − and H2O2 (350), and DUOX isoforms generate predominantly H2O2 (153).
B. Expression of NOX isoforms in skeletal muscle
NOX isoform expression has been measured in different skeletal muscle model systems. For context, all NOX isoforms reported in skeletal muscle are included in this section.
In immortalized mouse skeletal muscle C2C12 cell culture, NOX1, 2, 4, DUOX1, and DUOX2 are detectable at the messenger RNA (mRNA) level (105). In adult rat whole muscle lysates, NOX2, NOX4, and DUOX1 mRNA are expressed, whereas data on the expression of DUOX2 are conflicting (220, 339). NOX2 and NOX4 exhibit a fiber type-dependent mRNA expression, higher in slow-twitch oxidative compared with fast-twitch glycolytic muscles (220). At the protein level, the expression of all NOX2 subunits and NOX4 has been confirmed by immunoblotting in isolated mouse single muscle fibers (309). Consistent with the mRNA expression being higher in slow-twitch muscle, NOX2 protein expression by Western blotting was visually highest in adult rat diaphragm, intermediate in soleus, and lowest in the more glycolytic gastrocnemius muscle (162). Interestingly, however, the fiber-type dependence in rodents may only apply to some NOX2 complex regulatory subunits such as Rac1 (341) but not p22phox and p47phox (162). Whether the reported differences in subunit expression relate to intrinsic muscle fiber-type differences or to different fiber-type recruitment patterns (e.g., postural vs. nonpostural muscles) remains to be seen. DUOX1 mRNA did not differ significantly between muscle types (220). To our knowledge, DUOX1 protein expression has not been confirmed in skeletal muscle nor are data available on DUOX2 protein expression in muscle. In human muscle biopsies, p67phox (265) and Rac1 (341) protein expression was detected by immunoblotting, suggesting conserved expression of NOX2 complex in human muscle, but this requires further investigation.
C. Myocellular localization of NOX isoforms
At the subcellular level, NOX2 in phagocytes is known to reside in phagosomes, secretory vesicles, specific granules, and the surface membrane. Upon activation, NOX2 translocates to the plasma membrane via exocytosis to produce O2 ·− extracellularly and inside endocytosed phagosomes (263). Within the plasma membrane, NOX2 may be localized to lipid rafts and caveolae (275). A subset of endocytosed surface membranes containing NOX2 termed “redox-active endosomes” may produce O2 ·−/H2O2 at specific subcellular sites depending on their localization (275). In rat H9c2 cardiac muscle cells, NOX2 complex subunits p47phox and p22phox and oxidative stress measured as tyrosine nitrosylation increased at the nuclear pore complex in response to ischemia (131). In skeletal muscle, NOX2 has been reported at the protein level to be predominantly localized to not only T-tubules based on immunofluorescence microscopy and biochemical fractionation studies (144) but also close to or within the sarcolemma for NOX2, p67phox, p47phox, and p22phox in transverse cryosections (309). p40phox has been shown by immunofluorescence microscopy of single mouse flexor digitorum brevis (FDB) fibers to translocate to the sarcolemma, whereas no movement of p67phox could be detected (309). Whether this reflects a bonafide noncanonical movement pattern of some NOX2 regulatory subunits in skeletal muscle or a limitation of confocal microscopy to resolve movement in the molecularly packed muscle fiber is currently unclear. NOX4 was found in the surface membranes and in mitochondrial fractions (309) colocalizing with ryanodine receptor 1 (RyR1) in the sarcoplasmic reticulum (SR)-enriched fractions and by immunofluorescence microscopy (339). The localization of DUOX1 and 2 proteins has, to our knowledge, not been studied in skeletal muscle.
D. Molecular regulation of NOX2 activity and expression in skeletal muscle
As reviewed in detail in later parts of this review, NOX2 expression and/or activity is increased in skeletal muscle by a number of physiological and pathophysiological stimuli. Most of what is known about the regulation of NOX2 at the molecular level is inferred from studies in other cell types, in particular phagocytes.
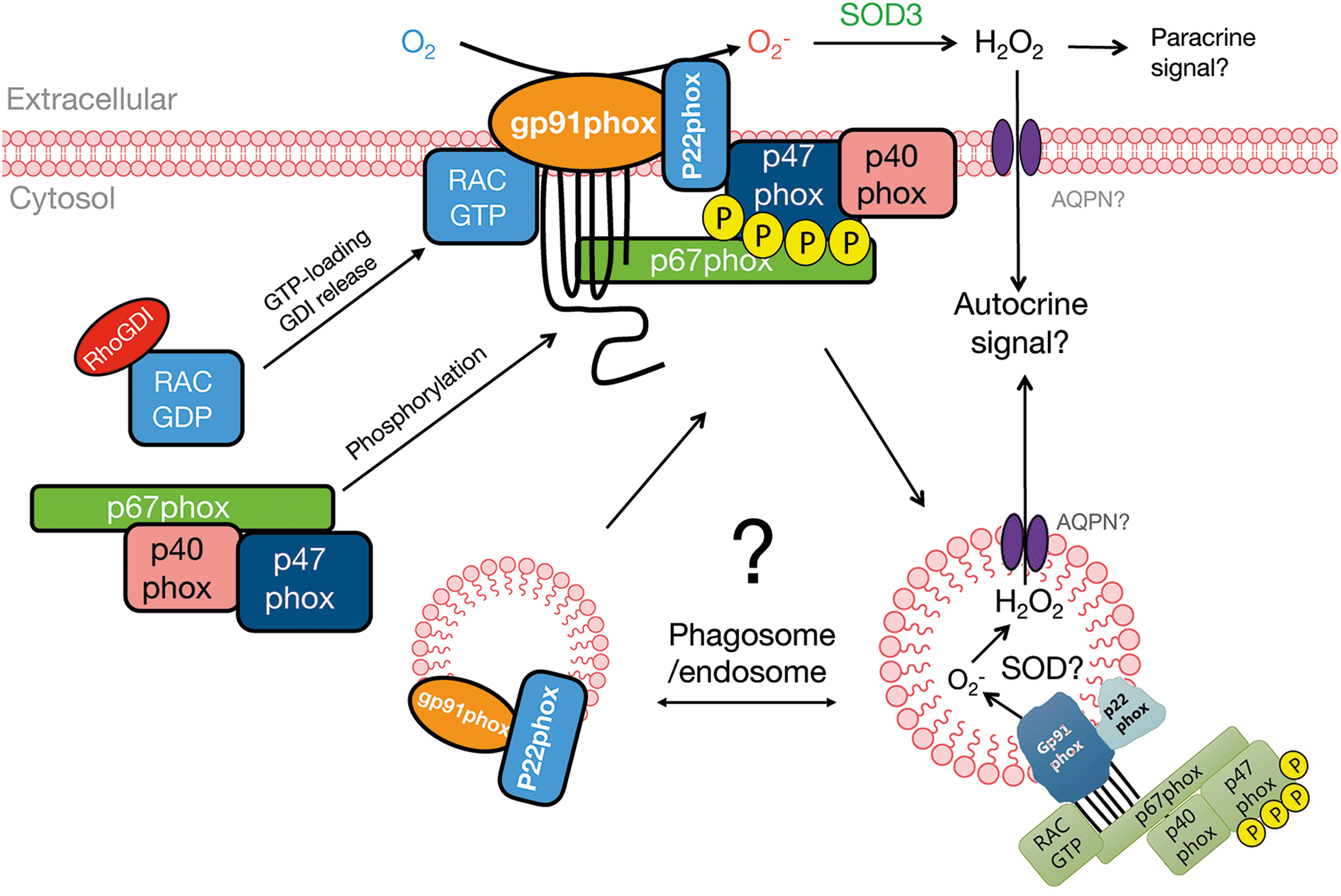
As illustrated in Figure 5, an increase in NOX2 activity requires the integral membrane protein heterodimer complex of NOX2 and p22phox to recruit a complex containing the cytosolic regulatory subunits, p47phox, p67phox, p40phox, and guanosine triphosphate (GTP)-loaded Rac GTPase (198). In the canonical model, p47phox forms a heterotrimer complex with p40phox and p67phox in resting cells, whereas inactive GDP-bound Rac is sequestered away from this complex by its cytosolic binding-partner Rho GDP-dissociation inhibitor (RhoGDI). On activation, GTP-bound Rac1 binds p67phox, and all four cytosolic subunits are recruited to the membrane-bound NOX2-p22phox heterodimer. In this process, p47phox is proposed to act as a molecular organizer that on phosphorylation becomes able to bind the cytosolic part of p22phox. p47phox also binds membrane phospholipids, assisted by the phospholipid-binding p40phox subunit. By interacting with p22phox, p47phox brings the NOX2-activating subunit p67phox in contact with the adjacent NOX2 subunit to activate the enzyme. Rac1 binding to p67phox further increases the recruitment of p67phox to the membrane (247) and is also proposed to cause a conformational change in p67phox that is necessary for NOX2 activation (285). Cell-free reconstitution studies have demonstrated that NOX2, p22phox, p47phox, p67phox, and Rac1 are sufficient for NOX2 activity in vitro (338); whereas p40phox is considered nonessential for NOX2 activity (379).
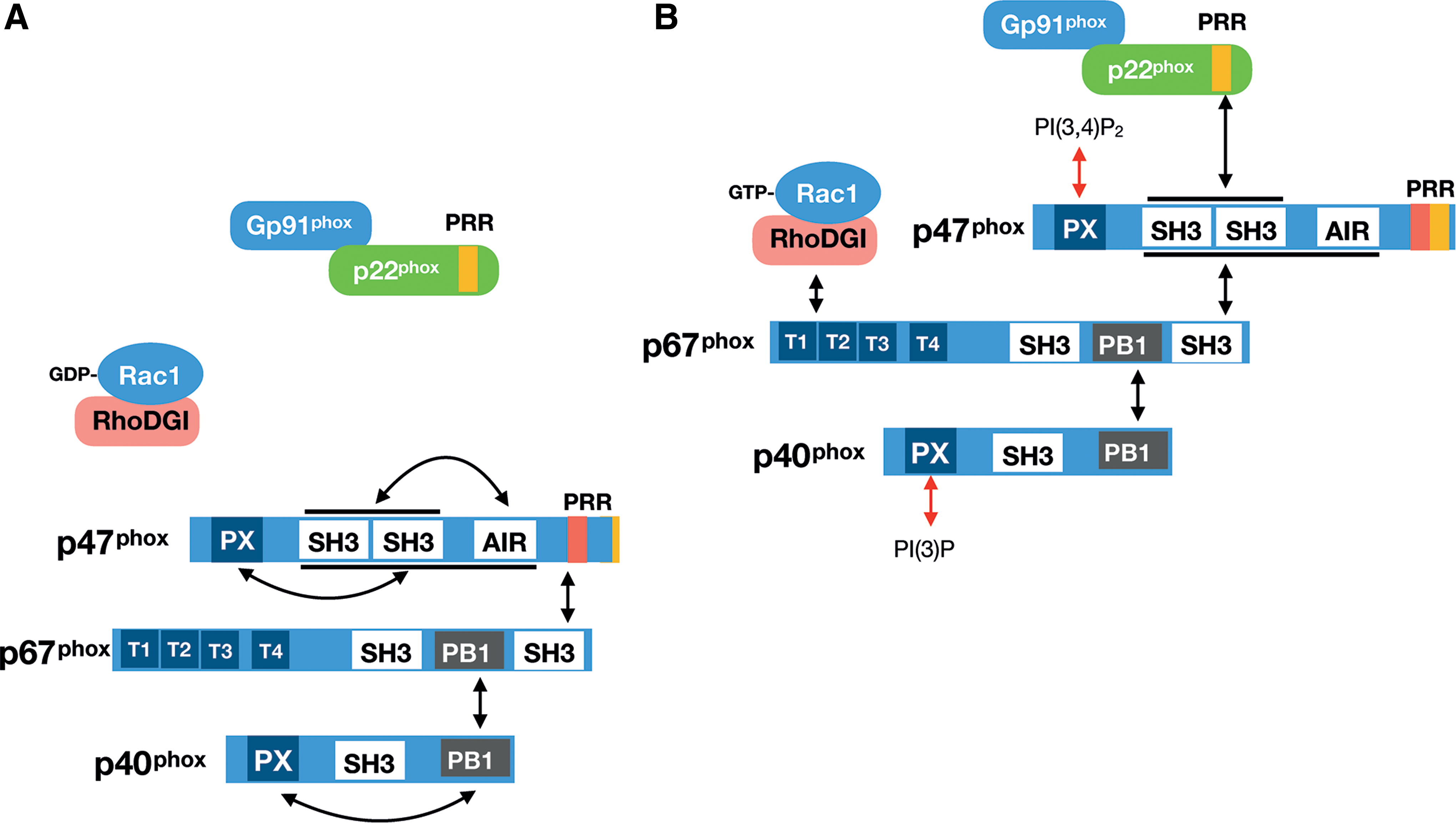
Later, the structure and function of the subunits of NOX2 and their role in NOX2 activation are detailed with the emphasis on possible skeletal muscle-relevant aspects. The connectivity between NOX2 subunits is further illustrated in Figure 6. For excellent reviews on generic NOX2 structure/function, see Lambeth (198) and Sumimoto (338).
1. NOX2/gp91phox
NOX2 is the catalytic subunit of the NOX2 holo-enzymatic complex. It has a predicted molecular weight of ∼65 kDa but runs as a broadband around ∼91 kDa on a Western blot due to variable N-terminal glycosylation (268, 369). Canonically, the NOX2 subunit generates O2
·− at the exofacial side of the phospholipid bilayer membrane by transferring electrons from NADPH on the cytosolic side via FAD and then via two stacked hemes to extracellular molecular oxygen. Structurally, the enzyme can be divided into two parts of roughly the same size, a C-terminal cytoplasmic domain containing NADPH and FAD-binding sites and the N-terminal membrane domain with six α-helical transmembrane parts and two distinct heme groups coordinated by four histidine residues in helix 3 and 5. NOX2 complex activity is proposed to be regulated by protein
It is worth noting that NOX2 activity is fairly specific to NADPH, and this enzyme works poorly with NADH as the electron donor (198). Seemingly in conflict with this view, a study in isolated rat diaphragm, and soleus and gastrocnemius muscles observed an approximately three- to six-fold higher extracellular H2O2 production by using the same molar concentration of exogenous NADH compared with NADPH when measured using Lucigenin-derived chemiluminescence (162). However, it should be noted that the 230 μM lucigenin concentration used in the study by Janiszewski et al. probably resulted in so-called “redox cycling” and overestimation of particularly NADH-driven oxidase activity (161). Case in point, this overestimation artifact—increasingly observable above ∼50 μM lucigenin concentrations—is shared by other cell types and is not unique to muscle (161, 209). Thus, we do not find compelling evidence to support the notion that skeletal muscle NOX2 has a unique preference for NADH over NADPH. This should be verified by other methods.
2. p22phox
p22phox is a shared constitutive binding partner of NOX1–4 (156). p22phox also co-immunoprecipitates with DUOX, although no functional role of this interaction has been established (370). p22phox is not glycosylated and runs around the predicted ∼22 kDa on a Western blot (90). p22phox is most likely a dual membrane-spanning protein with the N and C-terminals facing the cytoplasm (158). It has been ascribed the dual function of stabilizing the NOX2 subunit expression and acting as a linker to the cytoplasmic regulatory protein p47phox (81). In the absence of either p22phox or NOX2 subunit, the monomers are degraded by proteasomal degradation (81, 274). Similar to NOX2 protein, p22phox has long been known to be stimulation dependently phosphorylated (294). Site-directed mutagenesis studies of Thr147 near the C-terminal poly-proline helix and adjacent alpha helix region (residues 151–160) of p22phox critical for p47phox binding (204, 264) were shown to reduce O2 ·− production in CHO cells by >75% by blocking p22 binding to p47phox (205). Importantly, the reduction in activity occurred without effects on NOX2 subunit expression (205). PKC isoforms α and δ were able to phosphorylate this site in vitro but whether PKC is the bonafide kinase acting on this site in vivo remains uncertain.
3. p47phox
p47phox is a nonglycosylated protein running around the predicted ∼47 kDa on a Western blot. As is illustrated in Figure 6A, p47phox contains an N-terminal lipid-binding phagocyte oxidase (PX) domain, tandem SH3 domains, an autoinhibitory domain, and a C-terminal proline-rich region (PRR) (27). In the canonical model for NOX2 activation, this protein plays a critical role in the recruitment of cytosolic subunits to the membrane-inserted NOX2/p22phox heterodimer. p47phox is necessary for the activation of NOX2 in vivo, but its requirement can be overcome by high amounts of p67phox and Rac under cell-free conditions, supporting that it is an NOX2-organizing rather than an NOX2-activating protein (114, 192).
During NOX2 activation, p47phox becomes heavily phosphorylated on 10 sites between residues 303–379 in the C-terminal end (96, 316); for review see El-Benna et al. (98). Only the most phosphorylated form of p47phox translocates to the membrane (261). Phosphorylation of p47phox is proposed to relieve autoinhibition to allow the tandem SH3 domains to sandwich the PRR of p22phox (324). Early mutational analyses studies found that mutation of all 10 sites rendered p47phox incapable of activating NOX2 and suggested that Ser379 was most critical to NOX2 activation (104). However, the proposed role of Ser379 phosphorylation was challenged by later studies showing that Ser379 phosphorylation destabilized the interaction with p67phox and negatively regulated NOX2 activity (377). Rather, phosphorylation of Ser303, Ser304, and Ser328 was sufficient to relieve autoinhibition and allow p47phox to interact with p22phox (9). The conformational change elicited by phosphorylation of p47phox also allows the N-terminal PX domain to bind membrane phospho-inositides, presumably PtsIns (3, 4) P2, although the affinity and specificity of this interaction was reportedly low (174). It is worth noting that this region may also interact with phosphatidic acid (10), phosphatidylserine (333), and the actin cytoskeleton via Moesin (395). This may explain why p47phox can translocate independently of PtsIns (3, 4) P2 (8). p67phox binds with high affinity to the C-terminal PRR and two alpha-helixes in the tail-segment of p47phox (173).
Phorbol 12-myristate 13-acetate induces p47phox phosphorylation and is a classical activator of various PKC isoforms, and phosphorylation of particularly Ser303/304 and Ser328 has been ascribed to PKC isoforms (9, 74). Other kinases have also been suggested to phosphorylate p47phox in various cell types, including Src (61), extracellular signal-regulated protein kinase (ERK)1/2 and p38 mitogen-activated protein kinase (MAPK) (97), p21-activated kinases (PAK) (230), protein kinase B (Akt) (58), protein kinase A (28), and casein kinase II (271); whereas AMP-activated protein kinase (AMPK) was proposed to indirectly reduce p47phox phosphorylation via inhibition of PKC (329). A similar AMPK-induced inhibition of high glucose-induced NOX2 activity was observed in cultured adult rat cardiomyocytes exposed to GLP-1 (24). Lastly, arachidonic acid may potentiate phosphorylation-induced binding of p47phox to p22phox (324). All of the kinases and phospholipases mentioned earlier are present and activated in skeletal muscle by conditions such as exercise/contraction, adrenergic stimulation, insulin stimulation, and/or different forms of muscle disease (149, 343). To our knowledge, studies have yet to investigate the stimulus-dependent phosphorylation of p47phox in skeletal muscle. Teasing out which kinases target which sites with different stimuli in skeletal muscle will be a major challenge for the future.
4. p67phox
p67phox is a 526 amino acid nonglycosylated protein migrating around 67 kDa on a Western blot (predicted weight is ∼60 kDa). From N- to C-terminus, this protein contains four tetratricopeptide repeat (TPR): an AD, a Phox and Bem 1 (PB1) domain, flanked by two SH3 domains in the C-terminal half (27, 338). In the resting cell, p67 binds p40phox via the PB1 domain and p47phox at the C-terminal SH3 domain in the cytosol (Fig. 6B). On stimulation and recruitment of the cytosolic heterotrimer to the membrane NOX2/p22phox complex, p67phox additionally binds GTP-loaded Rac via its N-terminal TPR-containing domain (Fig. 6B). The general model is that p67phox is thereby put into contact with the NOX2 subunit via a direct protein
Not surprisingly, p67phox has also been reported to be phosphorylated on multiple sites on NOX2 complex stimulation via PKC (29, 112), p38 MAPK and ERK2 (75), an unidentified tyrosine kinase, MEK1/2, and phosphatase 1/2a-dependent mechanisms (76) although at a much lower magnitude and stoichiometry than p47phox (112). This phosphorylation occurs in the cytosol independent of p47phox (111). The functions of these phosphorylations have not been elucidated. In addition to phosphorylation, the stimulation-dependent binding of GTP-loaded Rac to p67phox is also required for NOX2 complex activation (Fig. 6).
5. Rac GTPases
Rac is a member of the Ras-like Rho family of small GTPases, best known for their canonical role in the regulation of the actin cytoskeleton (299). There are three highly homologous 192 amino acids and ∼21 kDa Rac isoforms, all of which can activate NOX2 (200, 248). Rac1 is ubiquitously expressed, including in skeletal muscle; Rac2 is restricted to hematopoietic cells; and Rac3 is mainly expressed in the central nervous system (147, 251). Key features of Rac GTPase is an N-terminal domain that binds GDP and GTP and contains the two effector-interacting switch regions, a middle insert domain specific to Rho but not to other Ras family GTPases, and a C-terminal polybasic tail containing a geranyl-geranyl lipid anchor (36). Activation of Rac GTPases occurs via a complicated series of molecular events. In the resting cell, Rac1 is sequestered by its molecular chaperone protein RhoGDI in the cytosol, covering the prenylated C-terminus of Rac1. GTP loading of Rac1 requires the release of Rac1 from RhoGDI, allowing the C-terminus of Rac1 to interact via electrostatic and lipophilic attraction with the negatively charged inner leaflet of the plasma membrane lipid bilayer. In a poorly understood sequence of events, Rac1 GTP loading presumably occurs somewhere in transit to the cell membrane, catalyzed by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins. The GTP loading of Rac GTPases acts as a binary on/off switch, allowing the N-terminal switch domain to interact with and modulate downstream effectors. In addition, phosphorylation by Akt on Rac1 Ser71 adjacent to switch II modulates the interaction between Rac1 and some substrates, including SRA-1, WASP, and PAK but not others (197, 396).
In relation to NOX2 activation, GTP-loaded Rac GTPases bind p67phox (87). Mutational analyses showed that this interaction required a functional N-terminal switch I region of Rac and a second region, residues 143–175, near the C-terminus for maximal binding (88). Rac1 binds p67phox in the N-terminal TPR repeat region (190). The ability to bind p67phox and activate NOX2 is a unique feature of Rac GTPases that cannot be performed by closely related Rho GTPases such as Cdc42, which lacks the critical residues for p67phox interaction. Rac has also been proposed to bind NOX2 subunit via its insert region (86), but this is controversial (285). Detailed analyses of a chimeric construct between Rac1 and p67phox revealed that Rac GTPases function to recruit p67phox to membranes but additionally may cause a conformational change in p67phox that is critical to its activation of the NOX2 subunit.
In skeletal muscle, Rac1 GTP loading and/or autophosphorylation of PAK1/2 Thr423 as a surrogate marker of Rac1 activation are increased by in vivo and ex vivo exercise/contraction, insulin, and ex vivo passive stretching in various experimental models in rodents and human skeletal muscle (340, 341, 355, 398).
As reviewed elsewhere (60), although a handful of GEFs have been linked to Rac1 activation in muscle cell culture with different stimuli, these have not been confirmed in adult skeletal muscle. No GTPase-activating proteins for Rac1 have been identified in skeletal muscle, and the role of RhoGDI in skeletal muscle has also not been investigated. Whether Ser71 phosphorylation of Rac1 modulates p67phox interaction has, to our knowledge, not been investigated (see Fig. 5 for subunits comparison and putative interactions).
Rac1 binds other effector proteins, including PAK and WASP, to regulate the actin cytoskeleton. As already mentioned, p47phox may be phosphorylated by PAK (230), and p47phox may interact with the actin cytoskeleton via moesin (380, 395), suggesting additional ways in which Rac activation might regulate NOX2 activity.
AMPK is a kinase activated in skeletal muscle by ATP-turnover during exercise/contraction, prolonged starvation, and pharmacological activators. Its classically ascribed function is to restore energy homeostasis by inhibiting anabolic signaling pathways and stimulating catabolic signaling pathways (187). As discussed in a previous section (see Section III.D.3), AMPK activation has been proposed to impair NOX2 activity. In skeletal muscle, AMPK activation by DNP also inhibits Rac GTP loading in L6 rat muscle cell culture (341). If reflective of the Rac1 pool regulating NOX2, this indicates that AMPK might reduce Rac1-dependent NOX2 activation in muscle. Another mechanism for AMPK inhibition of NOX2 activity might be speculated to involve inhibition of nucleoside diphosphate kinase (NDPK) by AMPK-dependent phosphorylation of Ser120 (267). NDPK catalyzes the ATP-dependent replenishment of GTP by using GDP as a substrate, and this has been suggested to regulate NOX2 activation (249, 285). Whether AMPK-dependent inhibition of Rac1 GTP loading occurs via NDPK deserves further study.
6. p40phox
This nonessential component of NOX2 is a 339 amino acid ∼40 kDa protein containing from N- to C-terminus a PX, SH3, and PB1 domain. It physically and stably binds p67phox and has been shown to enhance the recruitment of p67phox and p47phox to membranes (196). This enhanced recruitment is likely mediated by the N-terminal PX domain, which strongly binds phosphatidylinositol 3-phosphate (PI3P) (10, 174). The SH3 domain may, in addition, interact with p47phox (159, 231). A number of phosphorylation sites have been reported in p40phox, among which Thr154 and Ser315 are likely PKC sites that increase during NOX2 activation (42). The function of Thr154 is likely to decrease NOX2 activity (218).
Phosphorylation of phosphatidylinositol (PI) to generate PI3P is catalyzed by the class III phosphatidylinositol 3 kinase (PI3K; VPS34), an enzyme generally linked to membrane and vesicle trafficking events, including endocytic trafficking, phagocytosis, cytokinesis, and autophagy (22). VPS34 is present in skeletal muscle and is activated by acute resistance exercise in humans (222). VPS34 has been shown to exist in multiple complexes with distinct subunit composition, some of which regulate autophagy whereas others have been linked to general protein trafficking (183). Whole-body heterozygous kinase-dead VPS34 knock-in mice have increased insulin sensitivity, hyperactivated AMPK explained by lowered mitochondrial respiration (35). Whether modulation of NOX2 activity contributes to this phenotype is unclear.
E. The fate of generated O2 ·−
O2 ·− generated by skeletal muscle NOX2 is released extracellularly by NOX2 inserted into the sarcolemma or T-tubules (269). To be able to act as a second messenger within the cytosol, the O2 ·− generated by NOX2 must first reenter the muscle fiber. O2 ·− in solution rapidly and spontaneously dismutates to H2O2 at a rate of 105 M/s, and extracellular superoxide dismutase (SOD) 3 can increase this rate to 109 M/s (393). In contrast to O2 ·−, H2O2 is relatively stable in solution and is, therefore, often proposed as the more likely signaling molecule. It should be noted, however, that there is also evidence supporting a transmembrane signaling role for O2 ·− independent of conversion to H2O2 (110). H2O2 was long believed to cross the plasma membrane merely by passive diffusion. However, O2 ·− entry into cells is now known to occur via chloride channel 3 (ClC3) (110, 137). Similarly, AQPN isoforms 3, 8, 9, and 11 have been shown to facilitate H2O2 entry whereas AQPN1 and 4 have little to no H2O2 transport capacity (135, 235, 246). Skeletal muscle expresses multiple chloride channel isoforms including ClC3 (164) and AQPN isoforms 3,4,5, and likely 9 but not 11 (115, 368). There is evidence to suggest that these proteins are themselves regulated by post-translational modifications and subcellular trafficking (94, 206), adding further complexity to their potential role as H2O2 transporters.
NOX2 may also signal after endocytosis in “redox-active endosomes” (266, 331). In these organelles, O2 ·− is produced in the membrane-enclosed lumen (analogous to phagolysosomes) and must exit this compartment to initiate signal transduction cascades (Fig. 5). This presumably occurs by the same mechanisms as described earlier for the plasma membrane. Of note, redox-active endosomes appear to recruit SOD to their cytosolic surface to rapidly dismutate O2 ·− to H2O2 when exiting the organelle (258, 331). From thereon, the H2O2 signal may be then transduced via reversible Cys-oxidation as discussed in Section I.
In summary, ClC3 and muscle-relevant AQPN isoforms may contribute to the signal transduction by NOX2 by enabling O2 ·− and assisting H2O2 in movement across cellular membranes to initiate intracellular signaling. However, the requirement of these proteins needs to be confirmed experimentally in skeletal muscle. Further, both ClC3 and AQPNs have been shown to undergo stimulus-dependent protein trafficking (65, 226) and post-translational modifications (170), which may also modulate NOX2 signaling.
IV. NOX2 Signaling in Exercising Skeletal Muscle
A. Brief introduction to ROS signaling in the context of physical exercise
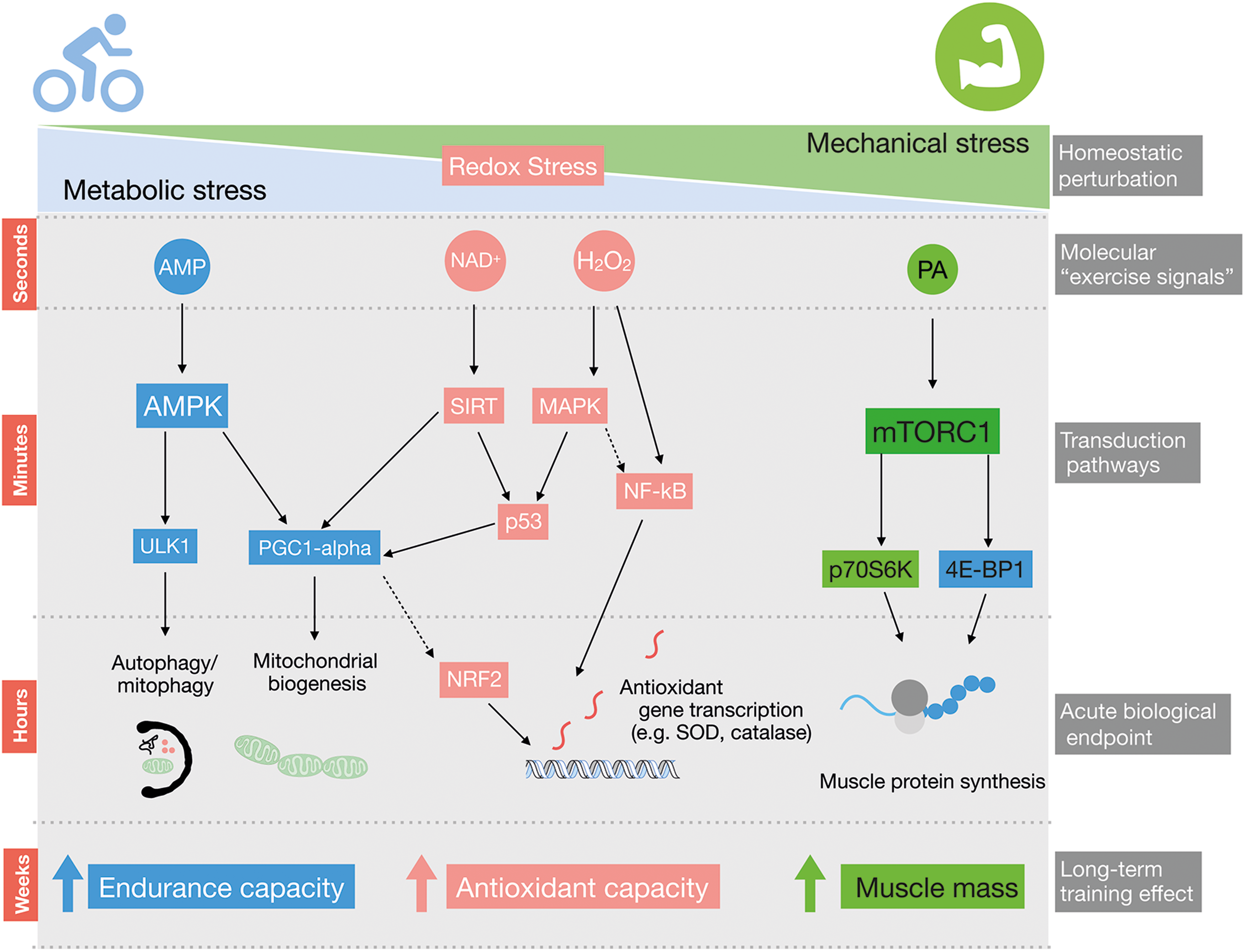
Exercise training improves the function of many organs in the human body, reducing the risk of developing diseases such as obesity, cancer, type II diabetes, and mental disorders (279). Skeletal muscle is highly responsive to both acute exercise and exercise training. Literally, thousands of post-translational intracellular signaling events are altered by a single bout of exercise in human skeletal muscle (149), acutely controlling, for example, metabolism, autophagy, protein trafficking, mRNA transcription, and protein translation (Fig. 7). The integration of molecular signaling from successive acute exercise bouts is further translated into chronic phenotypic changes produced by long-term training (283). Acute and chronic effects of exercise include enhanced skeletal muscle insulin sensitivity, mitochondrial biogenesis, and improved antioxidant defense (138). As is illustrated in Figure 7, several “exercise signals” have been proposed to relay these beneficial adaptations, including redox signals (286).
The first evidence suggesting increments in oxidants during exercise was obtained by measuring increased lipid peroxidation byproducts in expired air from exercising human subjects (89). Later, Davies et al. demonstrated an increase in free radicals elicited by treadmill running in rats by using electron spin resonance spectroscopy in muscle lysates (77). Currently, it is widely accepted that exercising muscle fibers may produce ROS from several sources, and the primary source of ROS has been a topic of controversy during the past decade (287). This controversy may, in part, relate to the technical challenge of measuring O2 ·−/H2O2 generation during in vivo exercise (160). This section will first briefly discuss studies investigating the role of different ROS in skeletal muscle exercise adaptations in general, and then focus on presenting the emerging evidence establishing NOX2 as a major source of ROS during physiological exercise conditions and linking NOX2 to both acute and chronic exercise adaptations.
B. NOX2 as a primary ROS source during exercise in vitro and in vivo
Since oxygen consumption is increased during exercise, several authors have assumed that ROS production during exercise is a byproduct of the elevated oxygen consumption by mitochondria to meet the increased myocellular energy demand (175, 177, 191). Early reports estimated O2 ·− production to account for 1%–2% of consumed oxygen by isolated mitochondria under particular experimental conditions (43). However, more recent studies with improved techniques showed that only ∼0.12%–0.15% of mitochondrial respiration is converted to H2O2 (194, 332, 349). Further, it was demonstrated that in vitro, electrical stimulation does not increase mitochondrial redox-sensitive GFP (roGFP) (244) or Mitosox oxidation in skeletal muscle fibers (309). We have recently found support for these observations during physiological exercise conditions by showing that oxidation of the mitochondrial matrix-targeted Orp1-roGFP is decreased, rather than increased, by moderate-intensity treadmill exercise in mouse skeletal muscle (141). There are several potential explanations for an exercise-stimulated drop in net mitochondrial H2O2. Overall, the net mitochondrial H2O2 production depends on the capacity for generation and removal of H2O2 (259). The production of H2O2 by mouse mitochondria is markedly decreased in the presence of high levels of ADP (259), which increases during exercise. Further, it has been shown that H2O2 consumption by rat skeletal muscle mitochondria is higher at lower pH (25), suggesting that intracellular acidification during muscle contraction shifts mitochondria toward a more antioxidant rather than pro-oxidant state. Together, available evidence suggests that mitochondria are not a significant contributor of oxidant generation during exercise.
In contrast to mitochondria, several cell-based studies in the past decade support the role of NOX2 in muscle contraction-induced O2 ·−/H2O2 generation. In primary rat myotubes, O2 ·− generation (276) and dichlorodihydrofluorescein diacetate (DCFH) oxidation (102) were first shown to be completely blocked by nonspecific NOX2 inhibitors diphenyleneiodonium (DPI) and apocynin (APO). These results, indicating that NOX2 is critical to contraction-stimulated ROS production in vitro, have since found increasing support by other studies in mouse primary myotubes and cultured FDB fibers using the more specific NOX2 inhibitor gp91ds-tat and genetic deletion of NOX2 complex subunits (Table 1).
Studies Addressing the Contribution of NOX2 in Contraction/Exercise Redox Signaling
APO, apocynin; CM-H2DCFDA, 5-(and-6) chloromethyl-2,7-dichloro dihydrofluorescein diacetate; DHE, dihydroethidium; DPI, diphenyleneiodonium; ES, electrical stimulation; FDB, flexor digitorum brevis; KO, knockout; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; Rac1 mKO, inducible muscle-specific Rac1 KO; roGFP, redox-sensitive GFP; TA, tibialis anterior.
Addressing the question of whether in vivo exercise increases NOX2 activity has been technically challenging. The first study indicating skeletal muscle NOX2 activation during endurance exercise was published in 2016 (140). One hour of swimming exercise increased the interaction between p47phox and the NOX2 subunit in mouse skeletal muscle and prevented by APO (140). Using the p47roGFP biosensor developed by Rodney's group (269), together with a redox histology technique in mice (116), we have provided in vivo evidence showing that moderate- (141) and high-intensity interval treadmill exercise (142) increased NOX2 activity in mouse skeletal muscle. Importantly, the oxidation of the cytosol-targeted Orp1-roGFP biosensor was observed in WT but not in ncf1* mice, suggesting that the NOX2 complex makes a substantial contribution to cytosolic O2 ·−/H2O2 generation during exercise.
DCFH oxidation induced by moderate-intensity exercise in human and rodent muscle measured in cryosections postmortem is completely absent in mice lacking either p47phox (ncf1* mouse) or Rac1 (141). This suggests that DCFH oxidation measures NOX2-dependent ROS production in mouse muscle and that exercise-stimulated NOX2 activation is likely conversed in humans (141). Together, these studies support a model where cytosolic O2 ·−/H2O2 production in myofibers during physiological requires an intact NOX2 complex.
C. NOX2 and exercise-stimulated myokines
In 1961, Goldstein suggested the possibility that muscle contraction stimulates the release of “factors” from muscles that communicate with other organs (127). Later, skeletal muscle was, indeed, discovered to be an endocrine organ releasing peptides dubbed myokines (272, 278), metabolites, and, more recently, small vesicles in response to exercise (378).
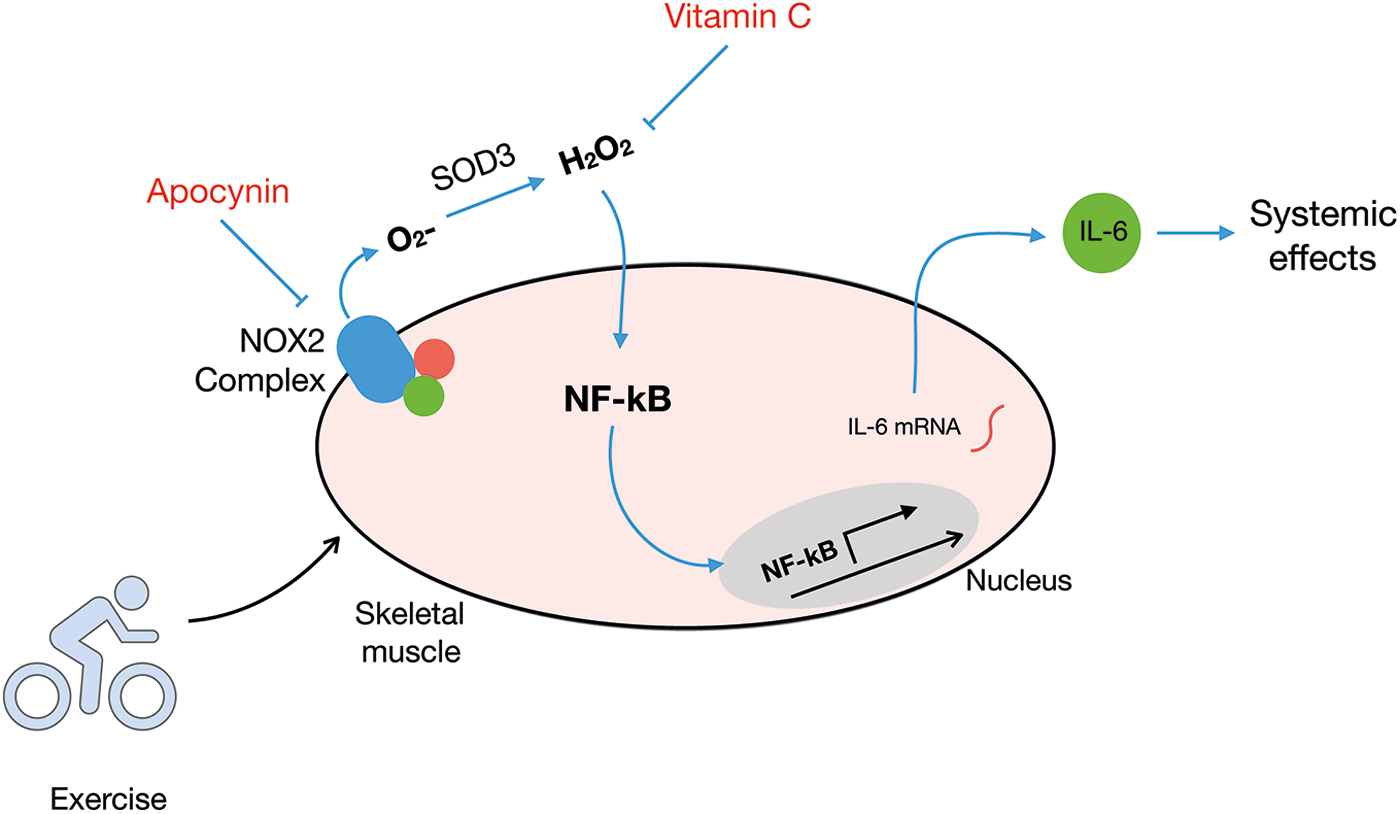
Interleukin 6 (IL-6) was the first identified myokine, and it is produced and released by skeletal muscle in response to diverse exercise modalities (278). The molecular mechanisms involved in IL-6 expression and release by skeletal muscle are still incompletely described. ROS have been shown to be sufficient to induce the expression of IL-6 in skeletal muscle cells (139, 193, 221). Interestingly, antioxidant supplementation was reported to lower the exercise-stimulated IL-6 expression in human subjects (109, 358, 390), suggesting that redox signaling regulates exercise-mediated IL-6 expression in muscle.
With regard to NOX2, Henríquez-Olguín et al. (140) demonstrated that IL-6 plasma levels were significantly lower in APO-treated versus saline-treated mice after 1 h of swimming exercise. This was accompanied by a reduction in IL-6 mRNA levels in muscle 2 h after in vivo exercise by both NOX2 and nuclear factor-κB (NF-κB) inhibitors. This suggests that NOX2 regulates exercise-stimulated IL-6 expression in skeletal muscle, possibly via NF-κB signaling (Fig. 8), which could be related to IL-6-mediated exercise benefits recently published in humans (199, 372). Studies testing the role of NOX2 directly in IL-6-mediated benefits after exercise training are highly required.
D. NOX2 as a regulator of acute exercise metabolism in muscle
Muscle contraction increases the demand for energy substrate metabolism and is well known to rapidly increase skeletal muscle glucose uptake by an insulin-independent mechanism (298). Key steps determining muscle glucose uptake during exercise are glucose delivery, glucose transport, and intracellular glucose metabolism (343).
Exercise-stimulated glucose transport requires the translocation of glucose transporter 4 (GLUT4) to the plasma membrane and transverse tubular system (202, 399). Among these, the muscle contraction-stimulated glucose transport across the plasma membrane of myofibers has been studied in great detail and is known to require the translocation of GLUT4 to the sarcolemma and transverse tubular system (343).
The first study linking ROS to glucose transport stimulation was by Cartee & Holloszy (1990), who observed that exogenous H2O2 (3 mM) could increase glucose transport into incubated rat epitrochlearis muscles (56). Later, an inverted U-shaped dose
In terms of necessity, glucose transport elicited by ex vivo contraction and passive stretch was partially blocked by preincubation with ROS scavengers (57, 242, 310), suggesting that ROS are required for the glucose transport response by these stimuli. Nevertheless, the infusion of N–acetylcysteine (NAC) in rats or humans did not affect in situ contraction- or exercise-mediated glucose transport in skeletal muscle (240, 243). Recently, Christiansen et al. demonstrated that intravenous NAC infusion attenuated exercise-stimulated glucose uptake in humans after blood flow restricted training in human volunteers (62). Methodological differences might explain these divergent results regarding the necessity of ROS for glucose transport in muscle, including the use of distinct exercise modalities, training state, and supplementation protocols.
During the past decade, the molecular mechanisms linking exercise-stimulated ROS generation and glucose metabolism have been a topic of research. The small GTPase and NOX2 complex subunit Rac1, previously linked to insulin-stimulated GLUT4 translocation via remodeling of the actin cytoskeleton, has emerged as a regulator of glucose transport by diverse stimulus in muscle cells (60) and mature skeletal muscle (345). Thus, both pharmacological inhibition of Rac1 and the actin-depolymerizing agent latrunculin B blunted passive stretch- and contraction-stimulated glucose transport in mouse soleus and EDL muscles ex vivo. Besides, incubated muscles from inducible muscle-specific Rac1 KO (Rac1 mKO) mice had impaired glucose transport responses to passive stretch (346) and contraction ex vivo (341). Strikingly, Rac1 mice seemed even more critical for the increases in glucose uptake and GLUT4 translocation in WT versus Rac1 mKO mice during physiological treadmill exercise in vivo (347, 348).
In parallel to the proposed actin-dependent mechanisms (60) for how Rac1 regulates exercise-stimulated glucose uptake, an NOX2-dependent mechanism has been recently proposed. The oxidation of p47roGFP and DCFH induced by exercise was shown to be absent in Rac1 mKO muscles (141). Interestingly, Rac1 and ncf1* mouse muscles presented striking phenotypic similarities, including greatly reduced muscle exercise-stimulated glucose uptake and GLUT4 translocation (141). The resemblance between these models suggests that Rac1 regulates exercise-stimulated GLUT4 translocation and glucose uptake via NOX2 (Fig. 9). The downstream mechanisms are unclear at present and may or may not involve modulation of the actin-cytoskeleton upstream, downstream, or in parallel to NOX2 activation.
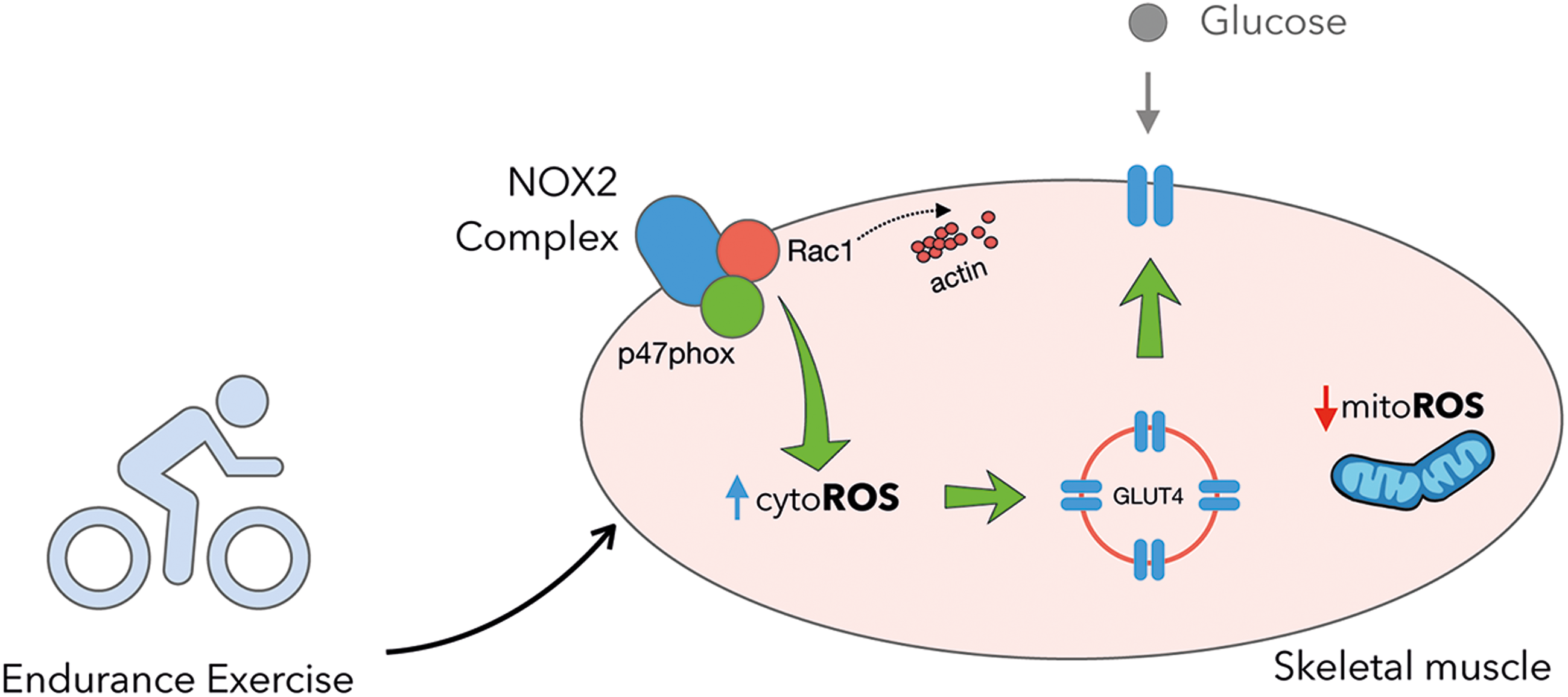
In summary, NOX2 activity is required to increase GLUT4 translocation and glucose uptake under physiological exercise conditions in mice (Fig. 9). The exact mechanism(s) downstream of NOX2 that control exercise-stimulated GLUT4 translocation require further investigation.
E. NOX2 and exercise training adaptations in skeletal muscle
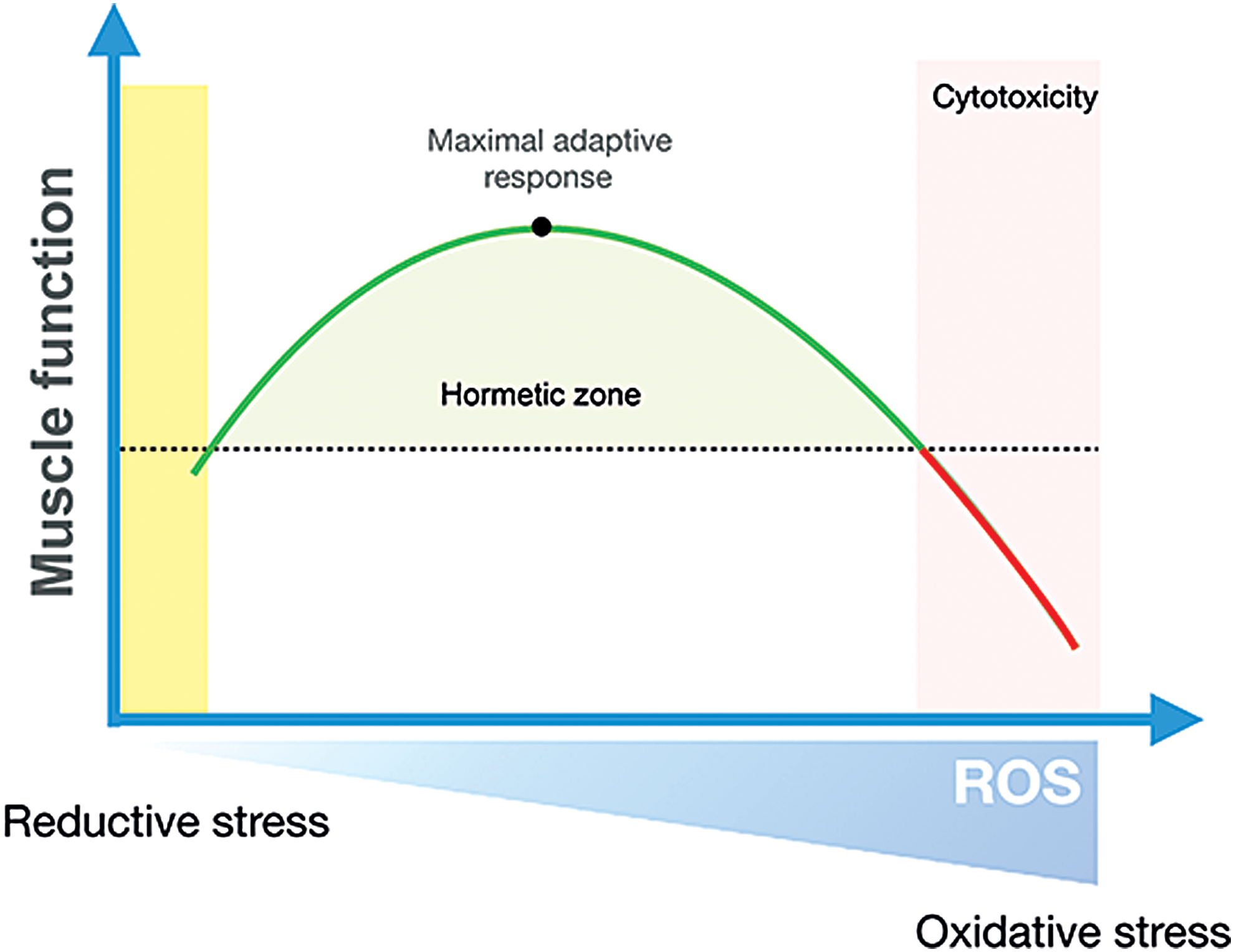
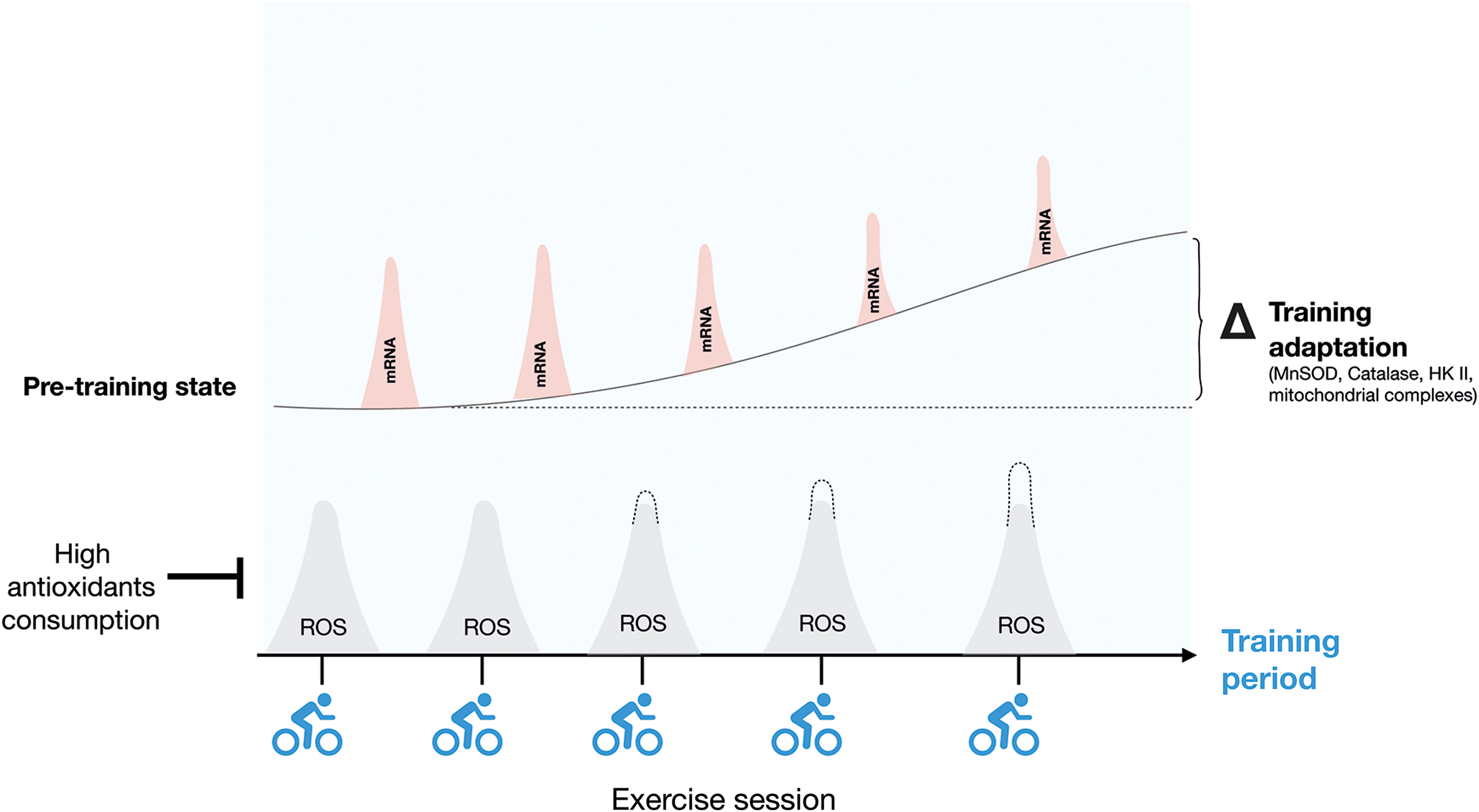
Exercise training is characterized by repeated transient exposures to metabolic, mechanical, and hypoxic stress, among others (277). These signals are translated into stress-protective adaptive responses, a concept referred to as exercise hormesis (300). The redox-signaling version of this paradigm is redox hormesis, where low-level exposure to ROS elicits beneficial stress adaptation, whereas too high ROS production relative to the antioxidant capacity promotes oxidative stress and cytotoxicity (Fig. 10). In the context of skeletal muscle, the hormetic response to nonharmful levels of oxidants during exercise has been proposed to contribute over time to the exercise-training response (Fig. 11). This hypothesis regarding the requirement of ROS for some training responses has mainly been investigated by providing general ROS scavengers/antioxidants during acute and long-term training periods [for review see Merry and Ristow (241)]. However, more recent studies have focused on identifying the specific ROS source(s) involved in these adaptations. The evidence for the involvement of first ROS, in general, and then NOX2, specifically, in some of the classical skeletal muscle training adaptations, including mitochondrial biogenesis, antioxidant defense, and increased insulin sensitivity, is reviewed later.
1. Training-induced mitochondrial biogenesis
Increases in mitochondrial content, function, and turnover in skeletal muscle are among the most well-described effects of endurance training on skeletal muscle (357). Muscle mitochondrial content is a product of synthesis (biogenesis) and degradation (mitophagy) (151). Apart from mitochondrial content, endurance exercise training is also known to influence mitochondrial morphology, by inducing an elongated mitochondrial network and decreasing mitochondrial fragmentation in trained muscles of rodents and humans (21, 133)
PCG-1α is a well-established regulator of mitochondrial biogenesis (44, 134) and mitophagy (132) in response to endurance training. Interestingly, administration of ROS scavengers reduced the expression of PCG-1α in response to endurance training in rat (128, 360), mouse (236, 335), and human muscle (301). Pharmacological inhibition of NOX2 decreased the exercise-stimulated mitochondrial gene expression (140). Recently, Baghersad et al. demonstrated that the increase of PCG-1α expression was blunted in NOX2 activity-deficient ncf1* compared with WT muscle in response to high-intensity interval training (23). Consistent with these observations, the high-intensity interval training-mediated increments of mitochondrial protein content and, in particular, changes in mitochondrial network morphology were markedly attenuated in ncf1* compared with WT muscle (142). Importantly, some exercise-responsive proteins such as GLUT4 responded similarly to exercise training in both WT and ncf1* mice, suggesting that NOX2 regulates a specific subset of training adaptations (142). Also worth noting, pharmacological inhibition of other myocellular ROS sources seemingly had no effect on training-related mitochondrial markers in mice (364, 367) and humans (323).
Taken together, this suggests that NOX2-derived O2 ·−/H2O2 production is required for a specific subset of exercise training adaptation in skeletal muscles related to mitochondrial content and morphology. Whether this requirement is specific to short-term HIIT training needs further investigation.
2. Training-induced changes in ROS production capacity and antioxidant defense
Gene expression of antioxidant defense proteins is increased in response to exercise training in mice and humans (165, 241). ROS seem to be necessary for induction of both manganese SOD (MnSOD) and GPx gene expression in rodents (128) and human skeletal muscle (284, 301), but the source (s) of ROS are again unclear. A recent study found that mouse skeletal muscle responded to a single bout of swimming exercise or electrical stimulation in isolated fibers by increasing MnSOD and GPx gene transcription (140). The exercise-induced MnSOD and GPx mRNA upregulation were prevented by the NOX2 inhibitors APO or gp91ds-tat. Later, catalase protein levels were found to increase in WT, but not in ncf1* muscles after 6 weeks of treadmill exercise training (139). This indicates that NOX2 is required to increase the expression of some antioxidant defense proteins in response to endurance exercise
Less attention has been given to the effect of exercise on ROS-producing enzymes. However, rats selectively bred for high running capacity show increased ROS production capacity without signs of oxidative damage (318, 354), suggesting that the capacity for both ROS production and clearance is increased by exercise training. In agreement with an increased capacity for ROS production, a rat study showed an increase in microsomal NOX2 activity and NOX2 mRNA levels after a 3-week treadmill exercise training program in gastrocnemius muscle (220). Hord et al. also found NOX2 expression to respond to exercise training in a rat study where 11 weeks of voluntary activity increased p67phox and p47phox protein levels in skeletal muscle (152). Interestingly, a 3-week training study also increased NOX4 mRNA in rat soleus but not gastrocnemius muscle, indicating a potential fiber/muscle-type dependence of NOX isoform adaptation to exercise training (220).
Overall, these data suggest that NOX2-dependent ROS production capacity is increased by exercise training (Fig. 11). However, whether the increased NOX2 expression with training actually increases maximal ROS production capacity and the physiological significance of this needs to be further tested. Further, the effect of different exercise training regimens on skeletal muscle NOX2 expression and activity, particularly in humans, is presently unclear.
V. NOX2 in Skeletal Muscle Pathophysiology
A. Insulin and NOX2 in healthy skeletal muscle
In the postprandial state, hyperglycemia is sensed by pancreatic β cells, resulting in the secretion of insulin. In insulin-responsive fat and muscle cells, insulin stimulates GLUT4 mobilization from intracellular compartments to the cellular surface to promote glucose uptake and it further stimulates the intracellular glucose storage and oxidation. Skeletal muscle plays a quantitatively large role in whole-body glucose homeostasis and accounts for ∼80% of whole-body glucose disposal under hyperinsulinemic-euglycemic clamp conditions (79). Interestingly, activation of different NOX isoforms has been suggested to modulate insulin action in muscle in both physiological and pathological contexts, as detailed later.
Studies in the 1970s explored the insulin-like effects of pro-oxidants in adipocytes. Czech and Fain demonstrated that thiols interacting with Cu2+ could transfer electrons to intracellular targets and increase glucose transport in rat adipocytes (73). Subsequent work by Livingston et al. showed that the insulin-mimetic effect of polyamines on glucose transport in adipocytes was mediated by H2O2 generation (213). Also, insulin was shown to generate endogenous H2O2 in adipocytes (257). Later, insulin-mediated H2O2 production was proposed as a second messenger mediating insulin-mediated glucose transport (232) and lipid synthesis (233) in rat adipocytes. Subsequently, Mahadev et al. (224) reported that genetic deletion of NOX4 reduced insulin-stimulated glucose uptake via upregulation of the activity of cellular protein-tyrosine phosphatases in 3T3-L1 adipocytes.
Although less studied in skeletal muscle models, insulin stimulation was also shown to increase H2O2 levels in L6 rat myotubes, and this increase was partially blocked by APO, p47phox siRNA, and gp91ds-tat (67, 101). This suggests that insulin increases NOX2-dependent H2O2 production in skeletal muscle, although this needs to be verified in adult muscle models.
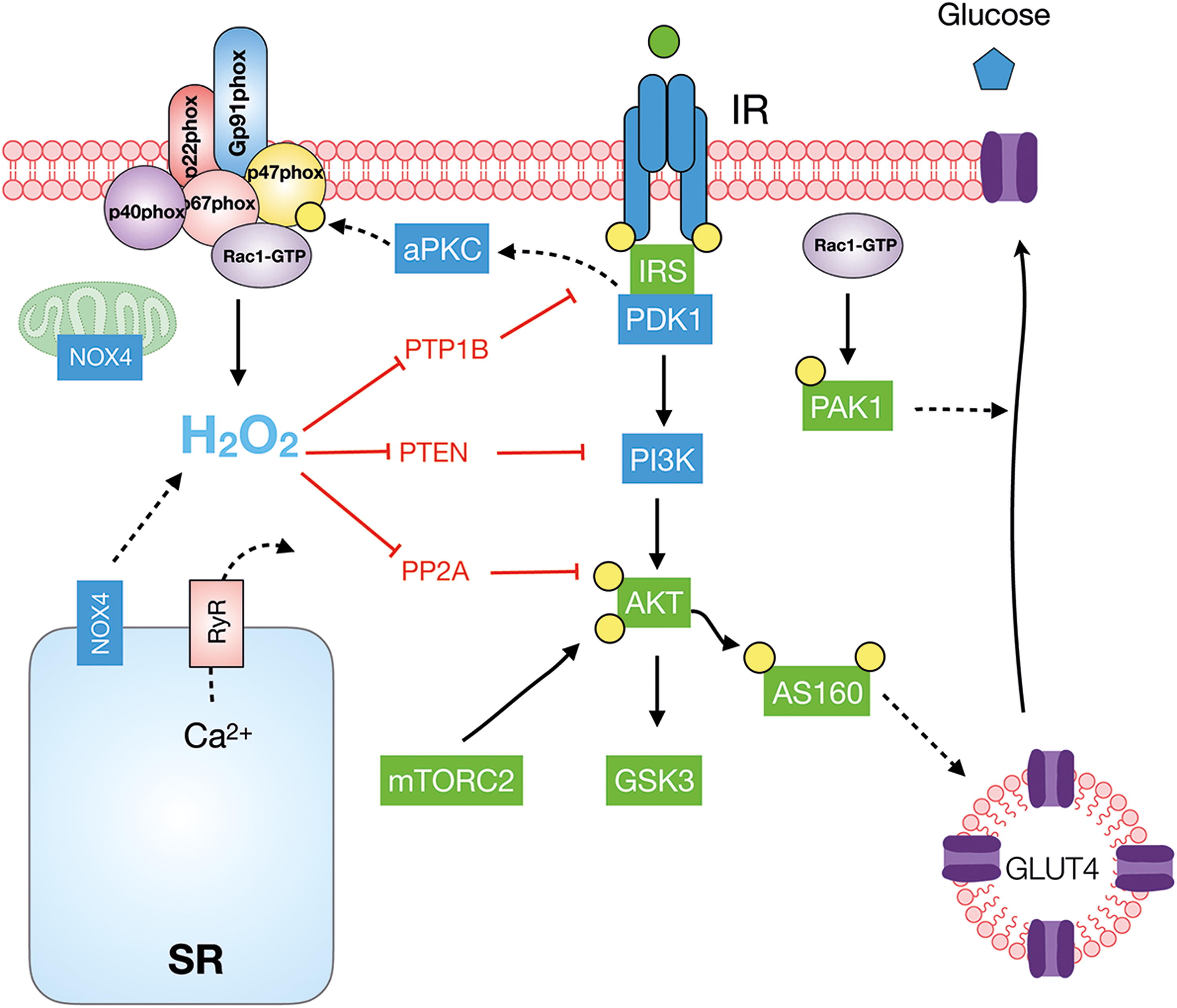
Downstream of insulin-stimulated NOX2, two potential mechanisms have been proposed to transduce the redox signal, reversible inactivation of phosphatases (120), and stimulation of Ca2+ transients (66).
The first possibility, tyrosine phosphatase inhibition, is proposed to lower the rate of de-phosphorylation of the insulin receptor and its substrates to enable/enhance insulin signaling, and it has yet to be directly tested in skeletal muscle. However, in 3T3-L1 adipocytes and HepG2 cells, insulin-stimulated H2O2 generation was reported to oxidize a conserved redox-sensitive Cys residue within the catalytic site of PTP1B to reversibly inhibit its activity (225). Skeletal muscle expresses several PTP1 isoforms (11). Further, overexpression and deletion of PTP1B in mouse muscle impairs and increases insulin signaling and insulin sensitivity, respectively (99, 392). Interestingly, human skeletal muscle from insulin-resistant subjects displayed increased PTP1B activity in skeletal muscle compared with insulin-sensitive controls (11). Thus, insulin-stimulated NOX2-dependent inactivation of PTP1 isoforms in skeletal muscle is an attractive hypothesis that deserves testing.
The second suggested mechanism involves insulin-stimulated Ca2+ signals in skeletal muscle triggered by H2O2 (314). Insulin is known to evoke transient Ca2+ waves in the proximity of the plasma membrane, but not globally, in mouse skeletal muscle (47). The insulin-stimulated Ca2+ spikes occur as a result of both extracellular Ca2+ entry (47) and SR Ca2+ release (67, 101). Interestingly, in cultured rat L6 myotubes, the knockdown of the p47phox subunit markedly reduced insulin-stimulated Ca2+ signals (101). Mechanistically, studies in cultured muscle cells have indicated that NOX2-derived H2O2 glutathionylates the RyR1 (144) to augment insulin-stimulated RyR-mediated Ca2+ release (67). However, whether the local insulin-stimulated Ca2+ transients depend on NOX2 in adult muscle requires further testing.
B. NOX2 and insulin-stimulated muscle glucose transport
In L6 myotubes, insulin-stimulated GLUT4 translocation was significantly reduced by gp91ds-tat and silencing of p47phox in a pathway requiring NOX2-stimulated Ca2+ release from RyR1 (67). Similarly, overexpression of a dominant-negative mutant of the NOX2 subunit Rac1 reduced insulin-stimulated GLUT4 translocation in L6 muscle cells (181). The muscle-specific deletion of Rac1 reduced insulin-stimulated GLUT4 translocation (355), induced mild whole-body glucose intolerance, and reduced muscle insulin-stimulated glucose transport ex vivo and in vivo (293, 340). The mechanism by which Rac1 regulates insulin-stimulated GLUT4 translocation and glucose transport has mainly been proposed to involve actin cytoskeleton remodeling (60). Consistent with the involvement of the actin cytoskeleton, pharmacological disruption of the actin cytoskeleton by Latrunculin B reduced insulin-stimulated glucose transport in rodents (234, 342). However, the cytoplasmic β- and γ-actin isoforms believed to undergo insulin-stimulated actin remodeling are downregulated during muscle differentiation (223). Further, muscle-specific KO of β-actin, likely a major isoform involved in actin remodeling in L6 myotubes, does not reduce insulin-stimulated glucose transport in adult mouse skeletal muscle ex vivo (223). Although actin polymerization is regulated by glutathionylation (371), the relative contribution of NOX2 versus actin remodeling and their potential crosstalk in mediating insulin-stimulated GLUT4 translocation, in particular in the context of adult skeletal muscle, needs to be systematically investigated. The various pathways proposed to involve NOX and ROS production in insulin signaling are depicted in Figure 12.
The proposition that ROS production could mediate the effect of acute exercise to increase muscle insulin sensitivity, defined as a greater insulin signaling or biological endpoint response at a given submaximal dose of insulin, has also been put forward. It is known that prior acute contractile activity increases the insulin sensitivity of the muscle in the hours after exercise (297). Antioxidant ingestion to attenuate ROS appears to blunt the acute postexercise increase in insulin sensitivity in humans (129). Some studies interfering with specific antioxidant defense enzymes such as Gpx1 also support a link between specific H2O2 and increased insulin signaling and glucose uptake into skeletal muscle (217). However, NOX2 does not appear to mediate acute insulin sensitization by exercise, since muscle-specific Rac1 KO mice were fully capable of improving their whole-body insulin sensitivity and insulin-stimulated glucose uptake measured 1h after a treadmill running bout (344). Rather, muscle insulin sensitization by contraction ex vivo and in situ in mice appears to require AMPK activity (188). How this relates to the reported dependence on ROS in humans (129) is currently unclear.
Exercise training also sensitizes skeletal muscle to insulin, but this is believed to rely on partly distinct mechanism involving increased capillarization, GLUT4, and hexokinase expression (343). It is unsettled whether ROS play a role in the exercise-training induced insulin sensitivity, since this effect was diminished by dietary antioxidants in some (301) but not other studies (390). NOX isoforms have, to our knowledge, not been studied in this context.
C. Redox signaling in muscle insulin resistance
Skeletal muscle insulin resistance, the inability of cells to efficiently respond to stimulation by insulin, is an early and causative event in the etiology of type 2 diabetes (T2D) (79). Left untreated, skeletal muscle insulin resistance is a major risk factor for T2D and cardiovascular disease (2). Excess weight is a well-established risk factor for T2D, with ∼80% of type 2 diabetics being overweight (171). Consequently, much effort has gone into mechanistically understanding how obesity causes insulin resistance in insulin-responsive tissues, including muscle. Currently, obesity is believed to drive the development of insulin resistance in skeletal muscle via a complex etiology involving multiple instigators, including gut-derived endotoxins and microbial products (130), low-grade inflammation (203), and ectopic lipid deposition causing lipotoxicity (326).
Increased cellular damage by ROS has long been linked to insulin resistance downstream of these insults (Fig. 13). For instance, humans in the early stages of the metabolic syndrome, that is, carrying a cluster of risk factors for macrovascular disease including impaired glucose tolerance, show elevated lipid peroxidation markers in plasma (178). A close relationship (r = 0.668, p < 0.01) between insulin resistance and oxidative stress markers has also been reported in obese subjects (356), suggesting maybe a causal relationship. Indeed, a highly pro-oxidative intracellular environment is well described as disrupting insulin signal transduction in multiple models of insulin-responsive tissues, including adipose cells (154), liver (123), and skeletal muscle cells (163).
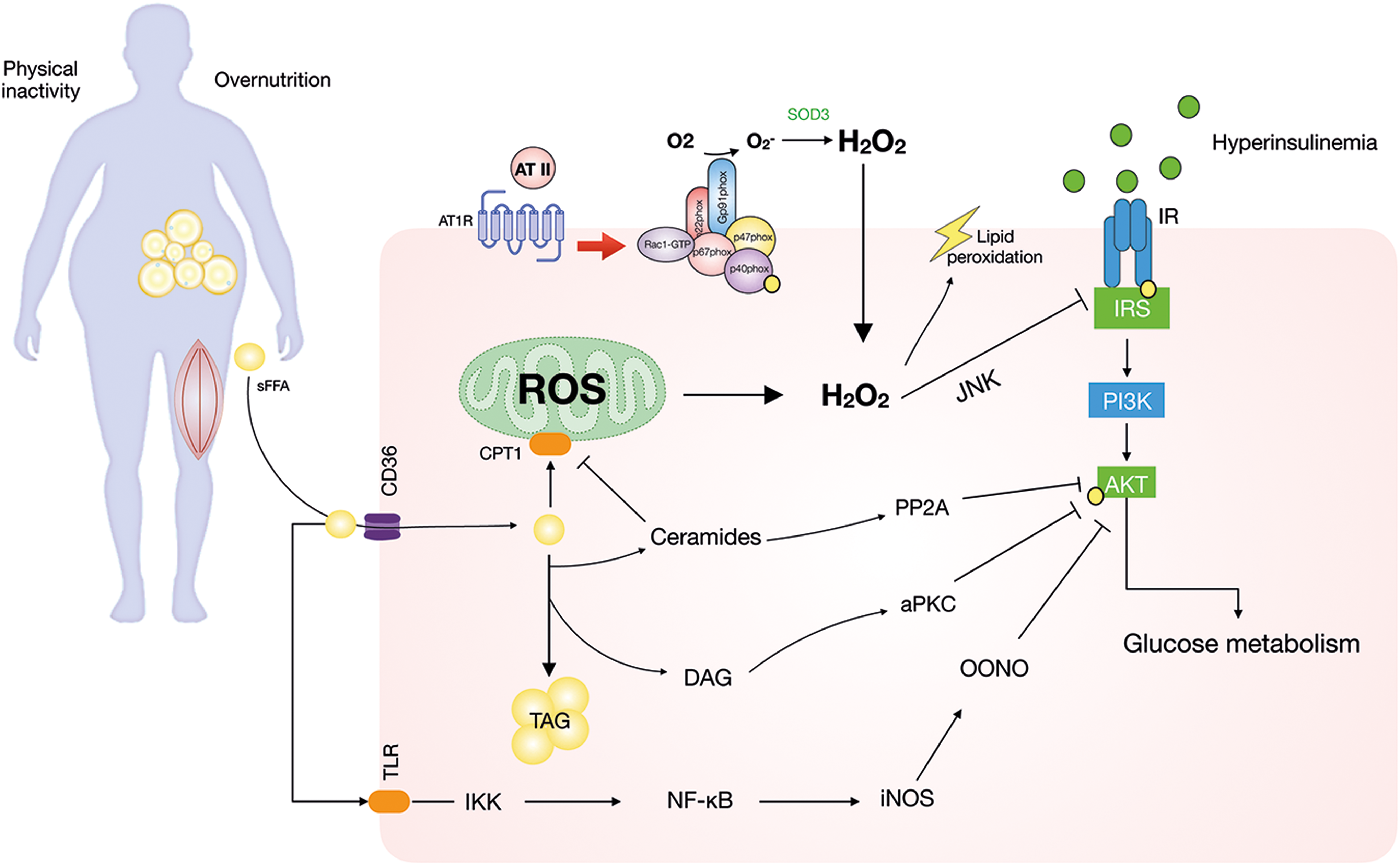
Analogous to exercise-induced ROS, mitochondria were first proposed as the major source of O2 ·−/H2O2 in relation to insulin resistance (17, 148). However, time course studies in rodents subsequently showed that insulin resistance occurred before mitochondrial dysfunction (39). Further, studies using pharmacological or genetically encoded antioxidants targeted at mitochondrial O2 ·−/H2O2 to prevent mitochondrial ROS-induced insulin resistance have generated mixed results (83). Consequently, mitochondrial ROS are increasingly believed to be modulators rather than initiators of insulin resistance (Fig. 13) (83).
Interestingly, obesity is characterized by increased NOX2 subunit expression in adipose tissue (117), skeletal muscle (100), and vasculature (166). For instance, Espinosa et al. (100) demonstrated that the p47phox protein expression was nearly 7-fold, and the NOX2 subunit was 1.6-fold higher in tibialis anterior (TA) muscle from high-fat diet-fed mice versus controls. This was accompanied by augmented insulin-stimulated cytosolic H2O2 measured by using the HyPer probe in FDB fibers. The mechanism of increasing NOX2 expression is unclear. However, NOX2 is well known to be increased by obesity-linked disease mediators, including endotoxin signaling via Toll-like receptor 4 (237, 238) and nucleotide-binding oligomerization domain (210), pro-inflammatory cytokines (208, 391), fatty acids (168), pro-fibrotic transforming growth factor type β1 (TGF-β1) (212), lysophosphatidic acid (359), and angiotensin II (AT II). It should be noted that many of these factors have also been linked to muscle atrophy and that obesity-associated muscle atrophy is known to exacerbate sarcopenia (172). For instance, Ang-II, suggested to induce muscle atrophy via NOX2 (4, 63), also caused insulin resistance in incubated rat soleus muscles via a mechanism partially alleviated by the antioxidant tempol (84). Thus, a contribution of NOX2-mediated oxidative stress might be speculated to be a contributor to both skeletal muscle insulin resistance and atrophy.
The role of NOX2 in muscle insulin resistance is unclear since no studies have inhibited skeletal muscle NOX2 alone and measured this endpoint (Table 2). Most studies in whole-body NOX2 KO mice and pharmacological inhibition generally report a protective phenotype against whole-body glucose intolerance and insulin resistance (Table 2), with one notable exception (68). Since myeloid-specific NOX2 KO mice present a similar protective phenotype (280), the protective phenotype in many of the whole-body NOX2 KO studies probably involves decreased inflammatory signaling, consistent with NOX2-dependent oxidative stress contributing to muscle insulin resistance. It is worth noting that the C57BL/6J-m obese diabetic mouse model and db/+ carry a point mutation in the p47phox gene, which results in aberrant splicing of the p47phox transcripts and, consequently, loss of NOX2 function (155). This might contribute to the phenotype in these mice, otherwise attributed to leptin receptor deficiency (394).
Studies Addressing the Contribution of NOX2 to Insulin Resistance
Akt, protein kinase B; EDL, extensor digitorum longus; GLUT4, glucose transporter 4; GTT, glucose tolerance test; HFD, high-fat diet; ITT, insulin tolerance test; vWAT, visceral WAT; WAT, white adipose tissue; WT, wild-type.
It should be evident from what has been mentioned that the study of ROS-mediated muscle pathology has moved from a simplified redox-hormetic concept, where global ROS dosage is the only determinant of detrimental effects (Fig. 14A), to an increasingly nuanced working model, considering also the specific subcellular ROS sources that play an important role (Fig. 14B), with H2O2 generation in specific compartments being either beneficial or detrimental. In line with the latter, NOX2 has gained increasing attention as a potential compartment-specific mediator of insulin resistance. Studies in whole-body NOX2 KO studies are generally supportive of the concept of NOX2-dependent oxidative stress as a mediator of insulin resistance, but they make firm conclusions about the role of NOX2 in muscle impossible. Currently, the only muscle-specific NOX2-deficient mouse model is the Rac1 KO mouse, but Rac1 has many NOX2-independent signaling functions, notably regulation of insulin-stimulated actin remodeling and GLUT4 translocation (60), making it hard to evaluate the role of skeletal muscle NOX2 as a driver of insulin resistance in this tissue. Future studies need to generate and characterize other muscle-specific NOX2 subunit KO mouse models to clarify this.
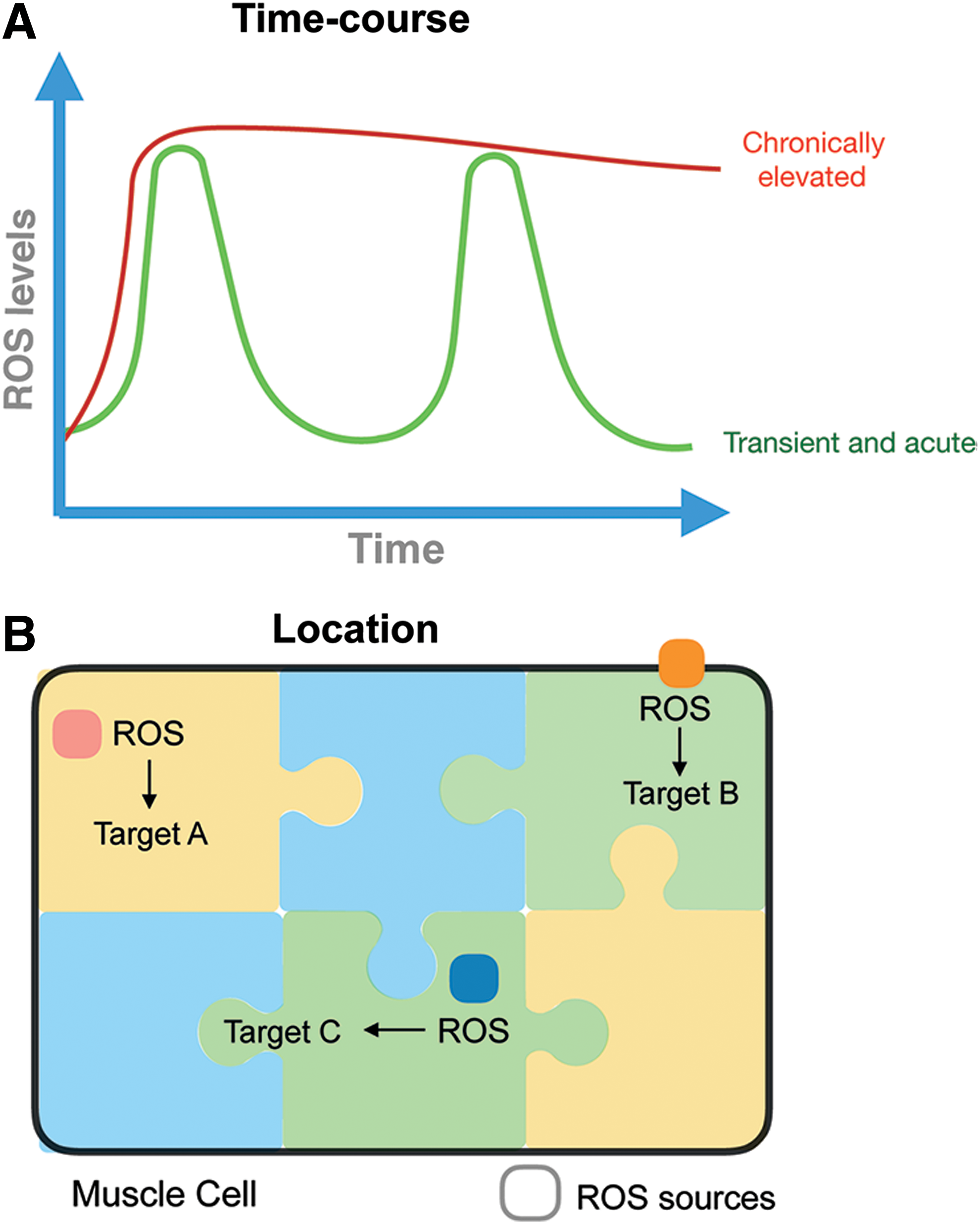
D. Altered redox signaling in muscular dystrophies
Muscular dystrophies represent a heterogeneous group of genetic diseases affecting skeletal muscle in both children and adults and are characterized by skeletal muscle weakness, wasting, and degeneration, with Duchenne muscular dystrophy (DMD) being the most severe (322). DMD is an X-linked disease that affects between one in 3600 and 6000 live male births (48). At the molecular level, DMD is characterized by a severe reduction or absence of the protein dystrophin (189) and this absence causes the rupture of the sarcolemma of the muscle fiber during contraction (12). At the cellular level, muscle tissue of DMD patients shows evidence of degeneration, regeneration, myofiber atrophy, fatty accumulation, necrosis of muscle fibers, inflammation, and fibrosis (16, 321, 361).
The most studied animal model of DMD is the mdx mouse, which presents a less severe phenotype than human DMD patients (1). NOX2 is upregulated in the mdx mouse, and this is a potential contributor to oxidative stress and DMD pathology (179, 375, 381). In this context, an increase in the complex subunits NOX2, p67phox, and Rac1 was reported in TA muscle from mdx mice compared with WT (377). Interestingly, the increased expression of NOX2 subunits preceded muscle necrosis and inflammation, suggesting that NOX2 complex may be a crucial source of oxidative stress triggering these events in mdx mice (1).
One of the main occurrences related to the deleterious effects observed in dystrophic muscle is Ca2+ influx to elevate cytosolic Ca2+ (119, 376). It has been demonstrated that Ca2+ influx through either nonspecific plasma membrane damage (250) or increased facilitated transport by stretch-activated channels (SACs) such as transient receptor potential channel 1 (TRPC1) (13) induces muscle-cell damage in the mdx mice. Another candidate for mediating the chronic elevation in intracellular Ca2+ concentrations in dystrophic skeletal muscle is a store-operated Ca2+ channel (95, 201). Meanwhile, basal Ca2+ release from the SR also appears to be increased (14). Both IP3 receptors and ryanodine receptors seem to be involved in this SR Ca2+ release, which has been suggested to increase NF-κB transcriptional activity, inducible nitric oxide synthase expression, and reactive nitrogen species. In fact, when the basal Ca2+ levels were restored by nifedipine injection, mdx muscles recovered their functional capacity (15). Regarding the downstream mechanism involved in the damage induced by the Ca2+ influx, the participation of TRPC1, caveolin-3, and Src-kinase has been documented. Thus, TRPC1, caveolin-3, and Src-kinase protein levels were increased in mdx muscle compared with WT (126). This is relevant, because caveolin-3 colocalized and coimmunoprecipitated with TRPC1, suggesting an interaction between these proteins that is dependent on NOX2-derived H2O2 with the participation of Src-kinase (126). Moreover, the activation and localization of TRPC1 is caveolin-3-dependent (126). Also, NOX2 may show increased sensitivity to mechanical stress in dystrophic muscles (376), and recent evidence indicates that NOX2 is activated by stretch and contributes to muscle damage in the TA of mdx mice (377). It has been described that in dystrophic single muscle fibers the excessive O2 ·− production derived from NOX2, and the simultaneous activation of abnormal Ca2+ signals amplify each other to produce a vicious cycle of damaging events, which may contribute to the abnormal mechanical stress sensitivity (325). Interestingly, nonspecific pharmacological inhibition of NOX2 by DPI significantly reduced the intracellular Ca2+ rise after stretched contractions in mdx single fibers and attenuated the loss of muscle force (377). In agreement, cross-breeding of p47phox KO with the dystrophic mdx mice was protective against pathological Ca2+ influx in skeletal muscle and reduced fibrosis and stiffness (215, 216). The microtubule cytoskeleton was also less disorganized in p47phox−/−/mdx muscles (216), with post-translational detyrosination of alpha-tubulin proposed as a mechanism that increases stiffness and thereby stretch-stimulated NOX2 activation in dystrophic muscle (180).
Along the same lines, it has been demonstrated that in double transgenic p47phox−/−/mdx mice the elimination of NOX2-dependent O2 ·−/H2O2 production protected against force decrements, probably by decreasing the aberrant Ca2+ influx through the sarcolemma (270). The disorganization of microtubules seemed to participate in the increased ROS production mechanism observed in dystrophic muscles (216) since stimulation by passive stretch increased microtubule-dependent activation of NOX2 activity, which ultimately produced the augmented Ca2+ influx through SACs in mdx muscle (180).
NOX2 activity and increased H2O2 levels have been linked to increased NF-κB activity (14, 54) (Fig. 15). The presence of an activated form of NF-κB has, thus, been demonstrated in both regenerating and necrotic fibers from DMD muscle by Western blotting and electrophoretic mobility shift assay analysis (252). In the mdx mouse, a skeletal muscle-specific activation of NF-κB has been demonstrated even before the onset of dystrophic damage (195). The pharmacological inactivation of NF-κB activity by pyrrolidine dithiocarbamate decreased the damage in dystrophic muscles elicited by oxidative stress, with consequent improvements in functional, morphological, and biochemical parameters. Studies in vitro indicated using APO or gp91ds-tat that electrical stimulation of primary mdx myotubes compared with WT-controls induced an increase of NOX2-dependent O2 ·−/H2O2 production and NF-κB activation (139). Further, increased MMP-9 expression and activity in mdx mice was required for NF-κB activity, contributing to the pathogenesis of the dystrophic muscle and the regulation of regeneration (207), with MMP-9 expression also being linked to chronic activation of NOX2 and NF-κB activation in multiple cell types (182, 353).
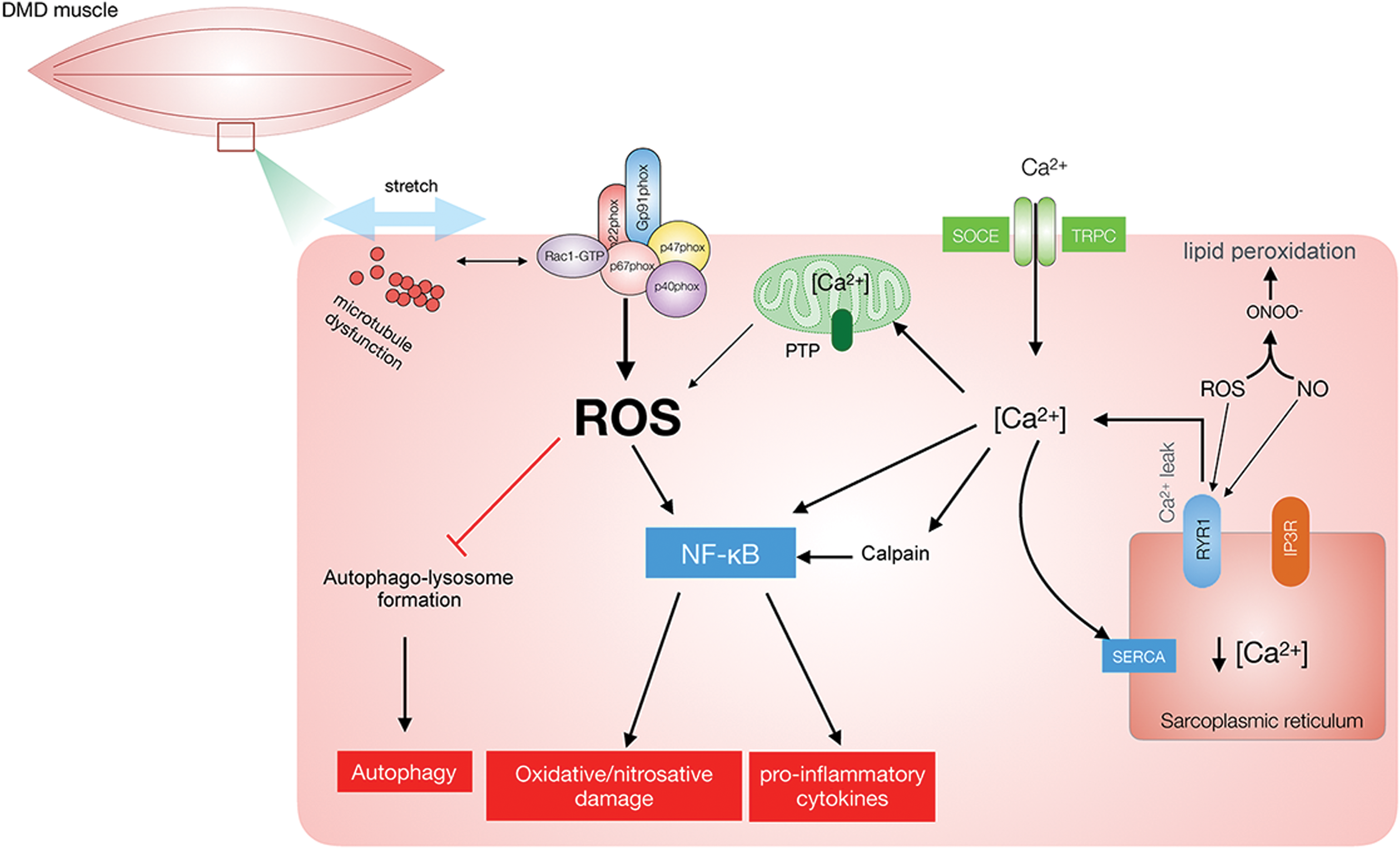
Another NOX2-dependent process proposed to contribute to damage in dystrophic skeletal muscle is autophagy (78). Hence, a recent study showed that autophagy in mdx mouse muscle was decreased via an NOX2-dependent mechanism (374). The decreased autophagy could be restored to normal levels by the use of statins, which reduced NOX2 expression and are also known to inhibit Rac GTPase function by blocking the synthesis of isoprenoid intermediates (19, 292). Overall, this resulted in lower muscle damage, inflammation, fibrosis, and an increase in muscle force production (373, 374).
One of the main features of dystrophic muscles is the development of fibrosis. Pathophysiological fibrosis is characterized by an excessive accumulation of extracellular matrix (ECM) components. Many pro-fibrogenic factors are produced within the affected tissue, leading to the activation of fibroblasts and the expression of ECM components and pro-fibrogenic factors. Among them are TGF-β and AT II (49, 50, 388). TGF-β is a potent pro-fibrotic cytokine that contributes to the pathogenesis of several muscular dystrophies (6, 30). Interestingly, it has been reported that TGF-β induces its own expression in C2C12 mouse muscle cell culture by a mechanism inhibited by APO, suggesting a dependence on NOX2-induced ROS production (4). Thus, TGF-β may activate an autocrine positive feedback loop dependent on NOX2 that perpetuates the development of fibrosis (6). This would be consistent with the observed reduced fibrosis and stiffness in mdx mouse muscle lacking p47phox (216).
AT II is the main peptide of the classical axis of Ras (51). AT II is formed by the action of the angiotensin-converting enzyme, and its main pathophysiological effects are mediated by the angiotensin receptor type 1, which is expressed in skeletal muscle (51). The fibrotic effects induced by AT II, such as increased expression of fibronectin, collagen III, and connective tissue growth factor, were dependent on the NOX2-induced ROS production in C2C12 muscle cells (49). Also, AT II mediates its pro-fibrotic effects via crosstalk with signaling pathways activated by TGF-β, suggesting that several pro-fibrotic factors shown to influence NOX2-induced O2 ·−/H2O2 production may interact (4, 256).
Recent studies in mice suggested that decreasing muscle fibrosis is a potential therapy for DMD (7, 52, 255). Based on the studies just cited, reducing NOX2-dependent O2 ·−/H2O2 production may improve pro-fibrotic signaling in dystrophic skeletal muscle and be a potential treatment strategy for this disease.
E. Potential role of NOX2 in muscle wasting and contractile dysfunction conditions
Atrophy is a typical pathological condition observed in skeletal muscle as a consequence of aging, disuse, starvation, sepsis, or denervation, or secondary to different chronic diseases such as diabetes, cancer, and kidney, lung, heart, and liver diseases (352, 362).
The main feature of atrophy in skeletal muscle is the loss of muscle mass due to a reduction in the myofibrillar protein content, resulting in decreased contractile capacity and force generation (38, 103). Relevant to this review, skeletal muscle wasting has been mechanistically linked to oxidative stress caused by excessive ROS production and/or decreased antioxidant defense (286). This section will highlight some of the emerging roles of NOX2 in different conditions where skeletal muscle wasting/dysfunction occurs.
1. Denervation and disuse-related atrophy
As discussed in several excellent reviews, a number of studies have demonstrated that lowering ROS production by using general or mitochondrial-targeted antioxidants inhibited the upregulation of proteolytic pathways and muscle atrophy induced by disuse or denervation (122, 288). Similarly, denervation atrophy in mice and fasting in C2C12 myotubes is associated with increased ROS production in mice and is suggested to be sensitive to antioxidant-treatment (290). Given the established role of NOX2 in other kinds of muscle wasting, it is tempting to speculate that NOX2 is a shared contributing ROS source in response to denervation, disuse, or starvation, despite these atrophy paradigms eliciting partially distinct molecular signaling (46, 245).
The specific role of NOX2 remains largely uninvestigated in these contexts. However, Scalabrin et al. (312) studied the effect of prolonged surgical denervation on peroxide release and proteins that regulate redox homeostasis in murine muscle fibers. Together with increased mitochondrial H2O2 production, denervation upregulated NOX subunits such as p67phox, p40phox, and NOX2 within a 21-day period. In addition, APO reduced mitochondrial H2O2 in denervated skeletal muscle fibers, which might imply communication between mitochondrial and cytosolic ROS (i.e., NOX2) sources (Fig. 16). Confirming these findings, a recent study showed increased protein levels of NOX2 and NOX4 after spinal cord injury in human patients. The upregulation of NOX2 and NOX2 was accompanied with increased procaspase 3 protein levels, suggesting that altered redox state could drive myocelullar apoptosis in human muscle (311).
2. Aging-related sarcopenia
In most mammalian species, including humans, aging is associated with a reduction in skeletal muscle mass and function termed sarcopenia. Sarcopenia is characterized by atrophy of the constituent muscle fibers, displaying larger atrophy of fast-twitch type 2 compared with slow-twitch type 1 fibers, with increased heterogeneity in fiber size, and an increase in noncontractile (adipose and connective) tissues within the muscles (302).
Animal studies suggest the involvement of ROS in the development of sarcopenia. Similar to other models in which atrophy occurs, ROS production measured as dihydroethidium (DHE) fluorescence, and H2O2 in skeletal muscle homogenates of a mouse sarcopenia model were increased with aging, concurrent with increased mRNA expression of NOX2 subunits and NOX4 and changes in antioxidant defense enzymes (337). However, a recent series of studies comparing KO mice with specific deletion of SOD1 in nerves, muscle fibers, or globally demonstrated that the loss of SOD1 in either motor neurons or muscle fibers alone produced only mild phenotypic changes and did not recapitulate the sarcopenic phenotype of global SOD1 KO mice (308). In contrast, the crossing of the global SOD1 KO with a transgenic synapsin-1 promotor-driven SOD1 overexpression mouse to partially restore nerve SOD1 expression was sufficient to preserve both NMJ and skeletal muscle morphology and function (309). Taken together, these studies suggest a critical role of redox status in motor neurons in the development of sarcopenia, which subsequently affects skeletal muscle to further perpetuate NMJ damage in a negative feedback loop. Notably, sarcopenia in the global SOD1 KO model occurred without changes in markers of oxidative stress such as protein carbonyls and 4-hydroxy-2-nonenal in peripheral nerves (308), suggesting that disturbances in specific redox signaling pathways rather than unspecific oxidative stress resulted in the sarcopenic phenotype.
The role of NOX2 in aging-induced sarcopenia is currently controversial. NOX2 is expressed in both neurons and muscle fibers. Interestingly, SOD1, the protein deleted in the sarcopenia model just cited (308), is the most commonly mutated protein in familial amyotrophic lateral sclerosis (ALS), a disease associated with fatal degeneration of cortical and spinal motor neurons. In a mouse model of ALS expressing mutated SOD1 G93A, deletion of NOX1 and, in particular, NOX2 impeded disease progression and enhanced survival (227). In a follow-up study, SOD1 was proposed to directly bind Rac1 and inhibit its GTPase activity in a manner disrupted by oxidation of Rac1 by H2O2 (136). Mutations in SOD1 were proposed to decrease this redox-sensitive uncoupling to increase NOX2 activity in ALS neurons (136). Consistent with the role of NOX2, pharmacological blockade of NOX2 by APO increased the average life-span of the SOD1 G93A mutant mouse ALS model (136). However, an independent follow-up study found no effect of either NOX1 or NOX2 KO or broad-spectrum pharmacological NOX inhibition by perphenazine and thioridazine on the survival of the SOD1 G93A ALS mouse model, despite an evident reduction in O2 ·− levels in vivo (320). This makes the role of NOX2 in ALS unclear and hard to generate a clear hypothesis regarding the role of NOX2 in aging neurons.
Apart from a possible role in motoneurons, NOX2 might play a role in the secondary development of sarcopenia-associated muscle atrophy. Studies of wheel-running exercise training and 8% calorie restriction in young versus aged rats suggest that NOX2 subunit expression in skeletal muscle decreased with aging and that either calorie restriction alone or calorie restriction + exercise training prevented this decrease and aging-associated muscle atrophy (152). Although this does not seem to fit the generic NOX2-oxidative stress-muscle dystrophy/atrophy model, decreased NOX2 subunit expression in conjunction with decreased antioxidant defense may result in an increased net oxidative stress, as reported in a cancer cachexia mouse model (336). Consistent with such a scenario, previous measurements of total hydroperoxides in the rat wheel-running exercise and 8% calorie-restriction sarcopenia study suggested increased oxidative stress with increasing age, which was attenuated by combined caloric restriction and exercise (185). More studies are needed to verify the decreased NOX2 expression in aging skeletal muscle and tease out the role of NOX2 activity, if any, in the development of sarcopenia.
3. Circulating factors mediating muscle atrophy and contractile dysfunction
There is some evidence suggesting that NOX2 activity can be upregulated by blood-borne factors linked to muscle atrophy (Fig. 16). For instance, a recent study of cardiac cachexia in a rat model of heart failure (HF) demonstrated increased NOX2 and p47phox protein expression and NOX2 activity in isolated membrane fractions accompanied by NF-κB activation and increased p38 phosphorylation and atrophy of glycolytic plantaris muscle. In this model, the muscle wasting and the NOX2 complex activity were reduced by 8 weeks of treadmill exercise training, suggesting that NOX2 may be a good candidate drug target for treating cardiac cachexia and perhaps other types of muscle wasting (72). In contrast, a study investigating mouse cancer cachexia reported decreased mRNA expression of multiple NOX2 subunits and antioxidant defense enzymes commensurate with increased DHE fluorescence in skeletal muscle (336), suggesting that decreased antioxidant defense rather than increased NOX2 activity caused oxidative stress in that model.
Among the potential atrophic factors, a term used for extracellular soluble factors eliciting skeletal muscle atrophy (51), is AT II. This hormone has been implicated in the pathology of conditions such as cardiac failure and kidney disease-related muscle atrophy (18). The relationship between AT II and NOX2 in skeletal muscle has been evaluated by different researchers (69, 169). One study suggested using pharmacological inhibitors that AT II signals via NOX2 to increase intracellular Ca2+ in mouse EDL skeletal muscle fibers (69). Experiments in vitro using C2C12 myotubes indicated that AT II increased the ROS production by a DPI-inhibitable NOX2-dependent mechanism (305). In mouse models of AT II-dependent muscle atrophy, elevated p47phox mRNA expression was observed in parallel to decrease in SOD1 and catalase mRNA (254). In experiments infusing Ang-II via osmotic minipumps for 7 days, an NOX2-dependent increase in ROS levels and muscle atrophy was observed in p47phox KO mice (319), demonstrating the necessity of NOX2 activity in the atrophic process induced by AT II (319). This is consistent with another recent study in NOX2 KO mice where the KO mice compared with WT were strongly protected against 4 weeks of AT II infusion-induced skeletal muscle atrophy and associated changes in anabolic and catabolic signaling (169).
TGF-β1, in addition to inducing fibrosis in skeletal muscle, also promotes muscle fiber atrophy (239). It was recently demonstrated that TGF-β, through its receptor ALK5, activated both canonical and noncanonical TGF-β signaling pathways in mouse skeletal muscle (6), and likely induced muscle atrophy by an NOX2-dependent mechanism, evidenced by its inhibition by APO (4, 5). Besides, TGF-β-stimulated NOX2-dependent ROS production was upstream of Smad, ERK, and c-Jun NH2-terminal protein kinase pathways that were necessary for the atrophic effect mediated by TGF-β (6).
The association between inflammation and contractile dysfunction has been linked to elevated sphingomyelinase activity in patients with chronic HF (92). Exogenous sphingomyelinase stimulation results in an increase in ceramide generation in skeletal muscle cells and mouse diaphragm muscle in a redox-dependent manner (106). Moreover, sphingomyelinase-induced ROS is involved in the reduction in muscle force in mouse muscle fibers (107). Loehr et al. studied the subcellular source contributing to sphingomyelinase-mediated ROS production and contractile dysfunction (214). Using intact FDB muscle fibers, this study showed that sphingomyelinase increased NOX2-dependent O2 ·−/H2O2 generation and had no effect on mitochondrial oxidants. Moreover, the incubation with gp91ds-tat or genetic deletion of either NOX2 or p47phox conferred protection against decreased force production induced by sphingomyelinase (41, 214). Together, these studies demonstrated that NOX2 plays a central role in sphingomyelinase-mediated ROS production and contractile dysfunction associated with inflammatory states.
VI. Conclusions and Future Directions
In this review, we have attempted to provide a comprehensive overview of what is currently known about the regulation and function of NOX2 in skeletal muscle and areas where our knowledge is still highly limited. In general, the working models regarding the various functions of ROS in muscle have expanded from a simplified redox-hormetic concept, where global ROS dosage is the sole determinant of biological effects (Fig. 14A), to an increasingly nuanced working model, where specific subcellular redox-signaling events determine biological effects (Fig. 14B), with H2O2 generation in specific compartments being either beneficial or detrimental. In line with the latter, NOX2 has gained increasing attention as a potential compartment-specific mediator of insulin resistance. Studies in whole-body NOX2 KO studies are generally supportive of the concept of NOX2-dependent oxidative stress as a mediator of insulin resistance but they make firm conclusions about the role of NOX2 in muscle impossible. It should be evident that the knowledge gaps are distributed across many aspects of skeletal muscle NOX2 signal transduction. Major outstanding questions in skeletal muscle include where NOX2 complex assembly occurs, how NOX2 is regulated, how ROS generated by NOX2 reach the cytosol, what signals are downstream of NOX2, whether the signal transduction differs between health and disease, the requirement of NOX2 for different physiological and pathophysiological processes, and perhaps, most importantly, whether the proposed functions of NOX2 in cells and animal models are conserved into humans. Already, however, it appears from the literature that NOX2-mediated ROS production is involved in a large number of processes in skeletal muscle fibers, including exercise metabolism, gene regulation, insulin action, and muscle mass regulation. Uncovering the exact underlying mechanisms might allow the development of novel treatment and prevention strategies against major lifestyle and aging-associated muscle diseases such as insulin resistance and atrophy.
Footnotes
Acknowledgments
The authors apologize to those authors whose works they have not cited because of space limitations.
Funding Information
C.H.-O. was supported by Chilean National Commission for Scientific and Technological Research (CONICYT) and a postdoctoral research grant from the Danish Diabetes Academy, funded by the Novo Nordisk Foundation (Grant no. NNF17SA0031406). T.E.J. was supported by a Novo Nordisk Foundation Excellence project grant (no. 15182) and a Research Council grant (no. 9039-00029B). C.C.-V. was supported by CONICYT (FONDECYT 1161646), Millennium Institute on Immunology and Immunotherapy (P09-016-F), Programa de Cooperación Científica ECOS-CONICYT (C16S02), and BASAL Grant CEDENNA (FB0807).