
Abstract
Significance:
Pulmonary arterial hypertension (PAH) is a progressive disease of the lung vasculature characterized by the proliferation of all vascular wall cell types, including endothelial, smooth muscle, and fibroblasts. The disease rapidly advances into a form with extensive pulmonary vascular remodeling, leading to a rapid increase in pulmonary vascular resistance, which results in right heart failure.
Recent Advances:
Most current research in the PAH field has been focused on the late stage of the disease, largely due to an urgent need for patient treatment options in clinics. Further, the pathobiology of PAH is multifaceted in the advanced disease, and there has been promising recent progress in identifying various pathological pathways related to the late clinical picture.
Critical Issues:
Early stage PAH still requires additional attention from the scientific community, and although the survival of patients with early diagnosis is comparatively higher, the disease develops in patients asymptomatically, making it difficult to identify and treat early.
Future Directions:
There are several reasons to focus on the early stage of PAH. First, the complexity of late stage disease, owing to multiple pathways being activated in a complex system with intra- and intercellular signaling, leads to an unclear picture of the key contributors to the pathobiology. Second, an understanding of early pathophysiological events can increase the ability to identify PAH patients earlier than what is currently possible. Third, the prompt diagnosis of PAH would allow for the therapy to start earlier, which has proved to be a more successful strategy, and it ensures better survival in PAH patients.
Introduction
Pulmonary arterial hypertension (PAH)/pulmonary hypertension (PH) is a fatal disease characterized by pathologic vascular remodeling, leading to right heart failure. Remodeling of the pulmonary arteries results in the thickening of the intima, media, and adventitia. Disease progression is associated with vessel lumen narrowing and the eventual occlusion, intimal fibrosis, and the development of the concentric and plexiform lesions. Despite the recent development of new therapeutics, survival remains poor. This is because at the advanced stage of the disease, the complexity of disturbed pathways is overwhelmingly complicated for delineation of causative or consequential mechanisms. Patients are usually diagnosed after the disease has had considerable progression and this is largely due to the consequence of the disease being asymptomatic early on. Thus, from a diagnostic standpoint, it would be of great benefit if more research efforts are focused on dissecting the pathways involved in the early initiation stages of the disease and the following chronological PAH progression.
Since this endeavor is clinically challenging, a critical approach would be to utilize preclinical animal models during the early and intermediate time-points in longitudinal studies (29) (Table 1; Fig. 1), rather than the endpoints on which the vast majority of literature is based. By focusing on the early disease development stages, it is highly probable that an understanding of the critical initiating disturbances in those signaling pathways can be attained. Moreover, it may be possible to study those disturbances in a more isolated environment that would alleviate some of the complexity and a vast number of cellular and molecular reprogramming that occurs in late-stage advanced disease. It is important to understand the early vital mechanisms that could help to treat familial cases of PAH and other predisposed PAH conditions such as connective tissue disease, hemolytic anemias, HIV, etc. There is also the importance of identifying early biomarkers that can move forward the diagnosis of PAH at an early, potentially reversible stage. This review is focused on the early mechanisms that were described in literature, and the potential early biomarkers of the disease. By discussing the current progress in understanding the mechanisms responsible for PAH initiation and early progression, we expect to highlight the importance to continue this line of research to advance the early PAH diagnostics and treatment.
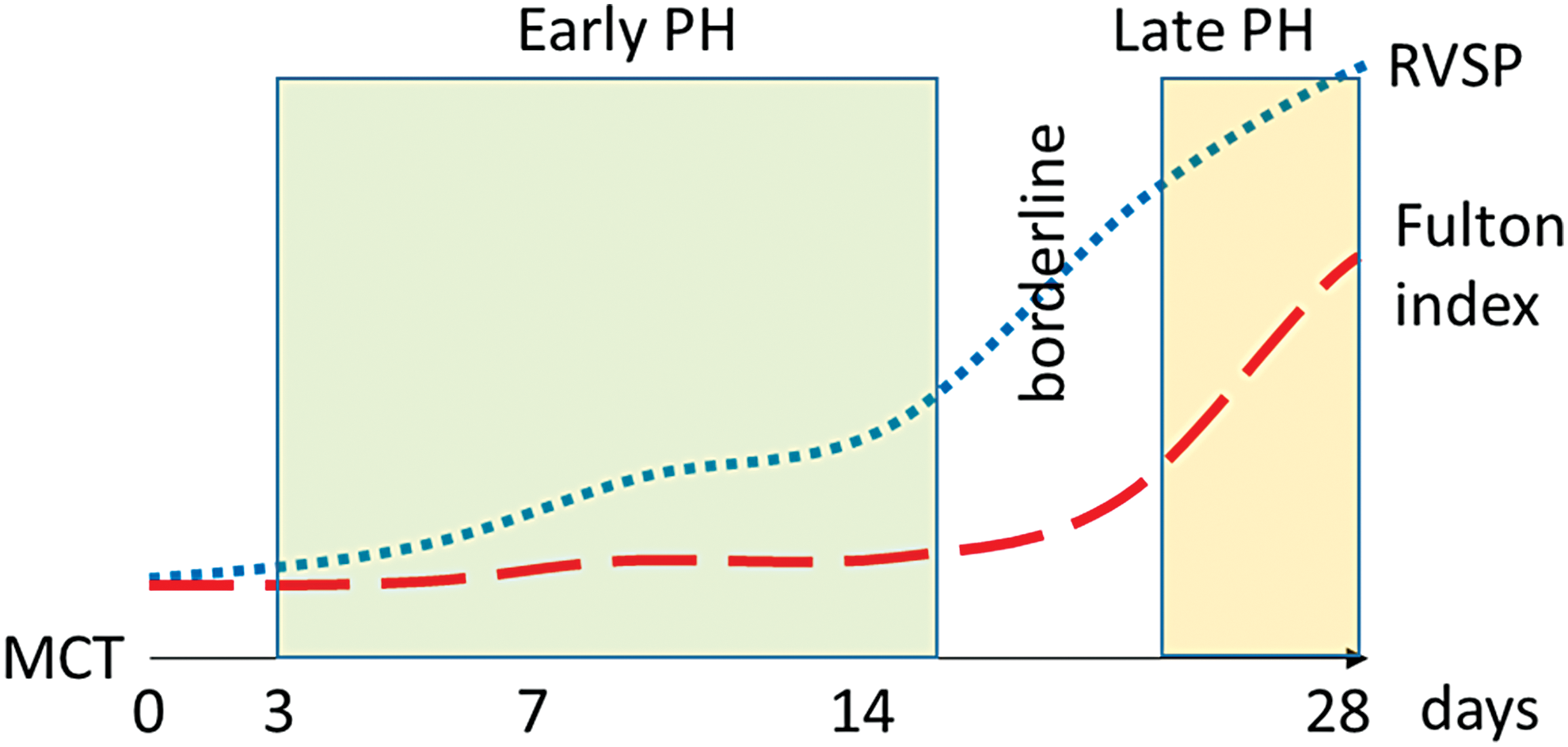
Preclinical Animal Models Utilized to Study Pulmonary Hypertension at Early and Intermediate Time-Points in the Longitudinal Studies
AA, antimycin A; i.p., intraperitoneal; , i.v., intravenous; MCT, monocrotaline; PH, pulmonary hypertension; RVSP, right ventricular systolic pressure; RVWT, right ventricle wall thickness; s.c., subcutaneous.
Metabolic Reprogramming
Recently, with the availability of untargeted metabolomic approaches, several reports were published that used metabolic profiling to evaluate possible biomarkers and shifts in metabolites in PAH patients (103, 116, 149, 207 –209), animal models of PH (72, 140, 144, 210), and cells (36, 86, 182). The cancer-like proliferation of pulmonary vascular cells requires metabolic adaptation for the heightened demand of dividing cells. Different vascular cell types can adapt differently. Metabolomic analyses of human pulmonary microvascular endothelial cells (ECs) with PAH-causing mutations in the bone morphogenetic protein receptor type 2 (BMPR2) showed the upregulation of glycolysis and pentose phosphate pathways (36). Glucose, ribose, and their phosphorylated intermediates were also significantly increased. On the other hand, these cells also have exhibited a decrease in carnitine homeostasis, fatty acid oxidation pathways, and tricarboxylic acid (TCA) cycle metabolites. Thus, the upregulation of cytosolic glycolysis in ECs was accompanied by a decrease in mitochondrial-based metabolic pathways. It was also demonstrated that in the model of increased pulmonary blood flow due to heart defects, carnitine homeostasis is downregulated via the inhibition of the carnitine acetylation/deacetylation system, which is responsible for the transport of fatty acid into the mitochondrion (178). Therefore, the altered transport of fatty acids could be a significant contributor to the decrease in fatty acid oxidation and subsequent reduction of TCA cycle activity as well as oxidative phosphorylation. On the contrary, smooth muscle cells (SMCs) from PAH patients instead exhibited decreased glucose metabolism and an increase in fatty acid biosynthesis (86). Other groups found that SMCs isolated from monocrotaline (MCT) animals showed reduced oxidative phosphorylation and an increased rate of glycolysis (107, 139). This discrepancy could be from many factors, such as stability of metabolic phenotype in relation to methods of isolation, cell growth, the influence of cell culture media, etc. The question remains, however, whether or not endothelial and SMC lineages have a similar metabolic shift in PAH. Our published data indicate that inhibition of mitochondrial respiration by antimycin A (AA) shifts ECs toward glycolysis more effectively than in SMCs (144).
Currently, hypoxia-inducible factor (HIF) and RAC-alpha serine/threonine-protein kinase (Akt) are considered to play a regulatory role in the metabolic switch in PAH (15, 37, 107, 173). HIF upregulates expression of glycolysis genes such as hexokinase-2, Glut1,3, PFK, and growth factors that, in turn, can induce Akt (102, 129, 202). Akt activation can lead to cell proliferation, apoptosis resistance and further activation of glycolysis by the induction of HIF signaling, and the translocation of Glut4 and mammalian target of rapamycin (mTOR) phosphorylation. It has been shown that mTOR kinase is an important regulator of lung vasculature remodeling and right heart hypertrophy (51, 65, 124). Moreover, increased oxidative stress contributes to HIF and Akt activation (11, 53, 80, 138); this generates a feedforward loop. Besides, HIF-1α upregulates the expression of pyruvate dehydrogenase kinase (PDK), an inhibitor of the mitochondrial enzyme pyruvate dehydrogenase (PDH), and decreases intramitochondrial calcium, thus additionally impairing Ca2+-dependent activity of PDH (7, 28). Inactivation of PDH was previously found to be an underlying cause of the disease pathology as the inactivation of PDH limited pyruvate influx into the TCA cycle (179). By limiting the formation of oxidative phosphorylation substrates in the mitochondrial matrix, PDH insufficiency can contribute to a glycolytic switch. Indeed, ex vivo treatment of human PAH lungs with the PDK inhibitor dichloroacetate (DCA) induced the activation of PDH, increased mitochondrial respiration (112), and improved right ventricle (RV) function in animal models (127). DCA administrated to idiopathic PAH patients reduced mean pulmonary artery pressure and pulmonary vascular resistance and brought improvement in functional capacity, confirming the critical role of PDH activity in PAH pathogenesis, although showing a range of individual responses (112).
However, it is still not clear whether the role of mitochondrial dysfunction in the process of the glycolytic switch of vascular cells is a causative event or a consequence of the disease. We have recently demonstrated that the chronic inhibition of mitochondrial respiration by AA resulted in an increased pulmonary pressure and proliferation of the vascular wall (144). Interestingly, lung glucose and pulmonary pressure demonstrated a strong correlation in animals treated with AA. Metabolic assessment of the lung tissue from the AA model showed an upregulation of glycolytic intermediates with rising levels of glucose, ribose, and phosphorylated sugars, as previously seen in ECs with BMPR2 mutation. A recent study on the chronic hypoxia, Sugen SU5614 (SU)/hypoxia, and MCT rodent models also confirmed the upregulation of glycolysis and the downregulation of mitochondria-centered metabolism (63, 96, 98, 107). Studies showed that adventitia fibroblasts isolated from hypoxic animals exhibited an upregulation of glycolysis (173). Glycolytic fibroblasts, in turn, lead to the activation of inflammatory pathways via fibroblast–macrophage interactions, resulting in secretion of cytokines and chemokines such as interleukin (IL)-1β, IL-6, and vascular endothelial growth factor (VEGF)-A, which contribute to vascular remodeling. Therefore, we can conclude that the vasculature metabolic reprogramming could start both from ECs (inside) and from adventitia (outside).
Importantly, the metabolic shift in PAH discussed earlier occurs much earlier than the increase in pulmonary arterial pressure and right heart hypertrophy. By using rats just after 14 days of MCT injection, we found that before significantly increased pulmonary pressure and right ventricle hypertrophy, a complete metabolic reprogramming occurs in the lungs (140). Metabolic profiling indicated that at the early stage in disease development there were significant changes in glycolysis and fatty acids beta-oxidation as well as inflammatory, oxidative stress, and fibrosis biomarkers, which were previously described for developing PH (103, 116, 149, 208). Thus, metabolic reprogramming is an early event that takes place faster than the pathophysiologic changes in the pulmonary vasculature. Specifically, the accumulation of glycolytic intermediates and reductions in acyl-carnitine long-chain fatty acid metabolites were reported (140). Therefore, the increase in glycolysis with decreased fatty acid oxidation appears to occur very early in the disease. Kynurenine and kynurenate, metabolites of tryptophan degradation that are produced by activated indoleamine 2,3-dioxygenase during inflammation, could activate the production of inflammatory molecules such as IL-6 and could be involved in cell proliferation (175). Both pro- and anti-inflammatory prostaglandins, as well as omega-6 fatty acids, were increased during the development stage. Increased levels of glucosamine, its derivatives, and hydroxyproline indicate toward the predisposition to extracellular matrix (ECM) remodeling. Together with adventitial fibroblasts activation, this could involve profibrotic changes in the vasculature. Also, elevated levels of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase (NOS), and the upregulation of arginases (75, 174) could increase the consumption of arginine via the urea cycle with a 14-fold increase in urea, ornithine, and polyamines content. All these events indicate a disrupted arginine homeostasis and suggest the bypass of normal nitric oxide (NO) signaling. Profiling an early PH animal model may lead not only to finding the key pathways that modulate cells reprogramming but also to the discovery of metabolites that comprise the blueprint for early PAH diagnosis.
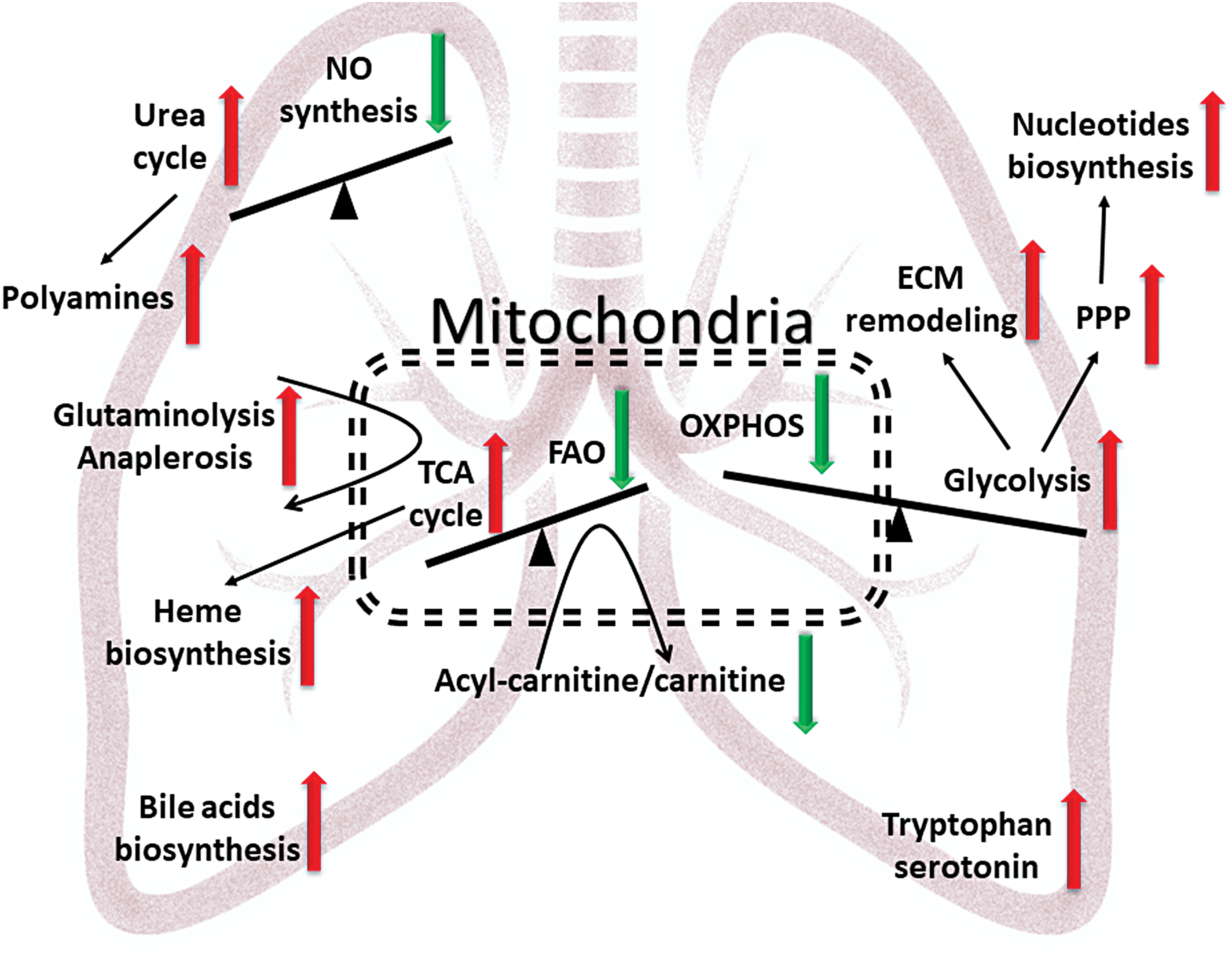
Early changes in metabolic footprints in the MCT model were mostly equal to the late stage changes found in patients with PAH. The plasma of patients with PAH show significantly increased TCA cycle intermediates, acyl-carnitines, urea, tryptophan, and polyamine metabolites, and they are similar to the early MCT model in the observed upregulation of these pathways (103, 140, 149). Interestingly, responders to vasorelaxation therapy, patients whose mean pulmonary artery pressure dropped >10 mmHg to <40 mmHg with preserved cardiac output in response to an acute pulmonary vasodilator challenge and remained stable on calcium channel blocker therapy alone, showed normalization of their metabolic profile back to healthy controls, providing the evidence of a strong association between the severity of PAH and metabolic alterations (149). Similar changes in arginine metabolism, ECM metabolites, polyamines, TCA intermediates, as well as increased heme degradation were found in lung tissues collected from PAH patients (207, 208). Common metabolites upregulated in PAH patients and the early model of MCT PH are indicated in Figure 2. This figure illustrates three main imbalances, which give rise to other metabolic disturbances. First, the shift from oxidative phosphorylation to glycolysis results in the activation of glycolysis-fed pathways involved in nucleotide biosynthesis and ECM remodeling. Second, the imbalance between the NO cycle and the urea cycle led to decreased NO production and increased polyamine synthesis, which contributed to vasoconstriction and proliferation. Third, decreased fatty acid oxidation and the demand for oxidative phosphorylation modify the TCA cycle by promoting anaplerotic reactions via glutaminolysis (12, 128), or pyruvate to oxaloacetate reactions, that are necessary for proliferating cells due to an increased need in building blocks for protein, fatty acids, and heme biosynthesis. Although those metabolic switches occur in the whole lung at the development stage of a preclinical model, they recapitulate metabolic disturbances in patients during the progressive phase of PAH. Metabolons of the whole lung are the most inclusive in terms of their variety of altered pathways; however, the plasma profile showed similar changes even with a reduced number of metabolites. Indeed, metabolites in plasma are well regulated by the liver, kidneys, and gut and do not reflect only lung status. However, availability of the plasma as well as difficulties to obtain lung biopsy should also be taken into account. Endothelial and SMCs isolated from animals or patients showed different results in metabolic profiling that could be explained by phenotype loss after isolation and cell culture. Thus, individual cell analyses are not so sensitive and could be cumbersome in clinical practice. Therefore, the profiling of the plasma samples could be important in the future diagnosis of PAH at the early stage.
Pulmonary Vascular Permeability
Inflammatory pathways in PAH are well accepted as important mechanisms of pathogenesis (21, 62, 134). PAH patients have increased infiltration of inflammatory cells (macrophages, lymphocytes, mast cells) in the perivascular region and an increased number of circulating cytokines in the plasma, such as monocyte chemoattractant protein 1 (MCP-1)/CCL2, regulated on activation normal T cell expressed and secreted/CCL5, tumor necrosis factor (TNF)-α, IL-1, and IL-6 (70, 91, 122, 130, 160). Importantly, the overexpression of IL-6 and TNF-α can induce a spontaneous development of PH in mice (43, 171). The mechanism behind cytokine-induced PH is based on studies that showed that a proliferative response could be induced in ECs, SMCs, and fibroblasts via yes associate protein/TAZ/SLUG, STAT3, mitogen-activated protein kinase (MAPK), protein kinase C (PKC), and AKT signaling cascades (24, 172, 192, 201). Thus, this promotes pulmonary vascular remodeling and subsequent increases in pulmonary arterial pressure. In the resolution phase of perivascular inflammation, ECM remodeling and the formation of fibrotic tissue can occur in the pulmonary vasculature (183). Interestingly, sex is an important component in the predisposition to a fibrotic phenotype of vascular remodeling. Our group found that the male sex is associated with increased inflammatory response and fibrosis of the pulmonary arteries (143). In a recent paper by Samokhin et al., it was found that important crosstalk exists between developmentally downregulated protein 9 (NEDD9) and mothers against decapentaplegic homolog 3 (SMAD3) within profibrotic transforming growth factor (TGF)β signaling (156). Oxidative stress-mediated post-translational modification in NEDD9 impairs its complex with SMAD3, leading to increased collagen production independent from ligand stimulation. Thus, this work established an important direct link between oxidative stress and vascular fibrosis. It is well accepted that female hormones mediate antioxidant responses (169, 206), and, thus, this mechanism may explain the predisposition of males to fibrotic changes in the vasculature. Reciprocally, connective tissue disease is known to be associated with a high risk of PH development (23, 58, 108). Fibrosis of the arteries increases vascular stiffness and results in an increased load to the heart through the shear stress of underlying fibrotic tissue vascular cells.
Although the complexity of inflammatory response in the pathogenesis of PAH is recognized (4, 17, 194), the initial trigger of inflammation that is involved in PAH development is not identified. It is still unclear whether initial damage to pulmonary vasculature activates an inflammatory response or whether pathogen/toxin-mediated inflammation damages the lung vasculature. However, both events must occur early in the development of PH. Initial damage to endothelial and SMCs induced via toxins or oxidative stress (1, 95, 196) can lead to damage-associated molecular patterns (DAMPs) activation, and a release that subsequently activates inflammatory cells via pattern recognition receptors (PRRs). Ruptured red blood cells (RBCs) can also be a source of DAMPs (77, 111). The pathogen can release pathogen-associated molecular patents (PAMPs) that also work by activating the inflammatory response via PRRs. The importance of one of the PRRs, receptor for advanced glycation endproducts, in PH was proven by elegant studies involving patient sample analysis and different animal models (31, 110). Therefore, a potentially feasible strategy is that PRRs and DAMPs/PAMPs inhibitors should be tested for clinical relevance in PAH management.
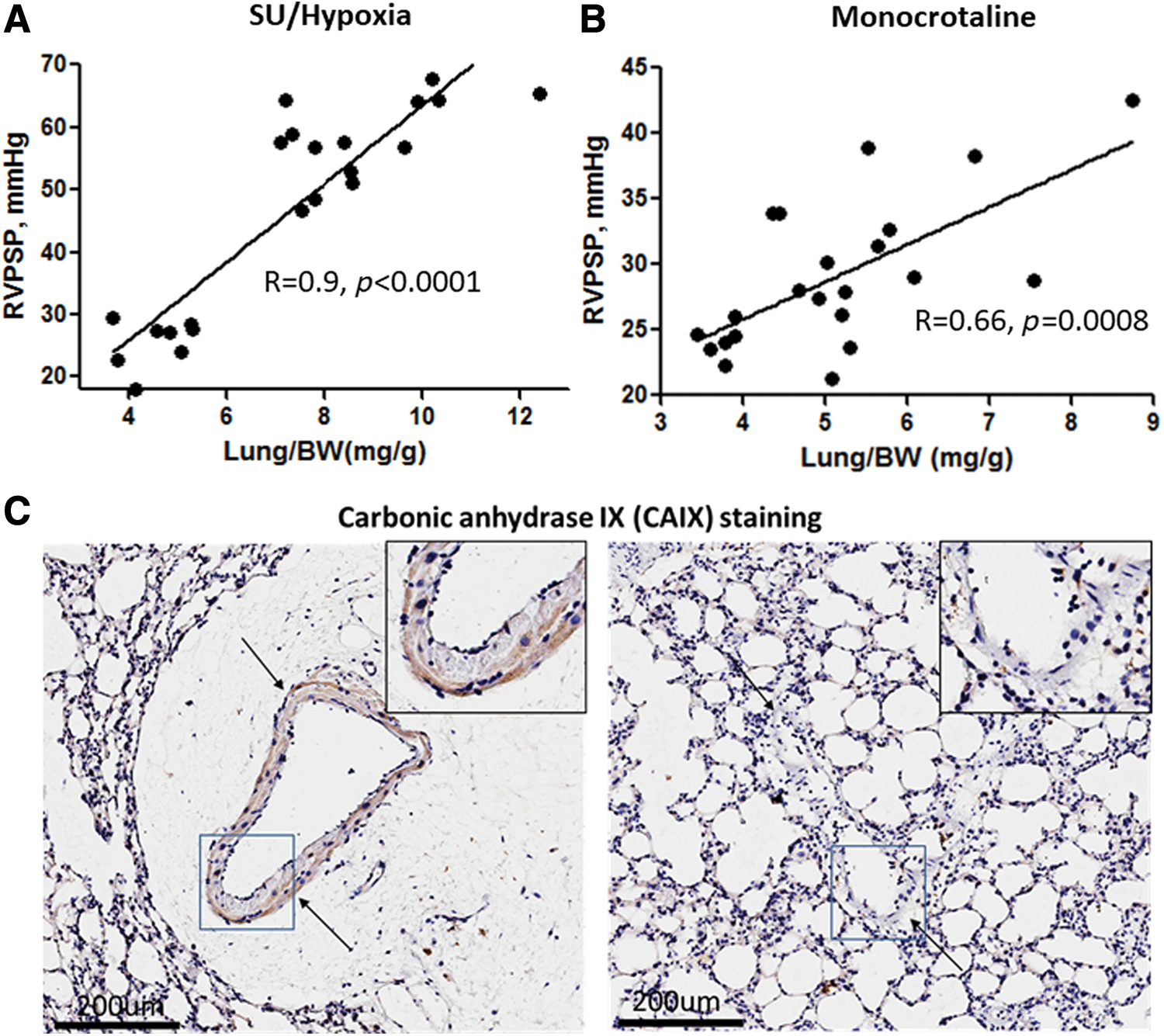
Another unanswered question in inflammation-mediated pathogenesis of PAH is why inflammatory cells have predominantly perivascular localization. This question may point to the increased infiltration of inflammatory cells through the vascular wall, which can be potentiated by the dysfunctional endothelial barrier. We and others have reported that chronic endothelial dysfunction due to different pathologies could be linked to PH (25, 106, 145, 196). In our study, we focused on the product of hemolysis, heme. Hemolysis leads to the accumulation of free hemoglobin in plasma, and this has correlated significantly with disease progression in PAH patients (145). Free hemoglobin can release free heme that, on the one hand, can damage the endothelium (77, 166), and, on the other hand, can induce disruptive endothelial barrier pathways leading to perivascular edema formation (145). Data indicated that the formation of perivascular edema and endothelial barrier dysfunction are early events in PH. In both SU/hypoxia and MCT models of PH, the increase in lung vascular leakage usually occurs during the first 2 weeks of disease induction and strongly correlates with the initial increase in pulmonary pressure (Fig. 3A, B). At an advanced stage of PAH, perivascular edema is diminished with highly increased thickness of the vasculature (145). Therefore, the perivascular inflammation due to infiltration of inflammatory cells into the perivascular area, crosstalk with adventitial fibroblasts, and the accumulation of cytokines in perivascular fluids could all be important contributors in the disease development phase. Moreover, fluid leakage in the perivascular area could impose local hypoxia to the flooded vasculature and induce vascular stiffness. Indeed, our data indicate the increased expression of carbonic anhydrase IX, the marker of hypoxic tissue, in the vasculature surrounded by perivascular fluid (Fig. 3C), but they showed no expression in the vessel without edema. This will induce HIF-mediated metabolic reprogramming in the lungs and increase the load on the right heart, leading to hypertrophy.
Vascular Damage and Inflammation
Injury of pulmonary vascular cells is one of the most recognized and established early mechanisms involved in PAH initiation and progression. In 2001, Voelkel and colleagues reported that inhibition of the primary endothelial prosurvival signaling molecule, VEGF, in combination with hypoxia induces apoptosis of pulmonary ECs and manifests as a severe angioproliferative PAH (184). This SU/hypoxia model now becomes a “gold standard” of experimental PAH that closely replicates the histological features seen in humans, including the formation of plexiform lesions composed of highly proliferative pulmonary vascular cells. It has been proposed that the initial apoptosis of pulmonary ECs induces a transition of and the selection for the remaining surviving cells. This causes a highly proliferative and apoptosis-resistant subpopulation, which, in turn, mediates PAH progression (154).
This conclusion may be viewed as paradoxical since it appears to require the initial inhibition of growth factors before it can promote the emergence of hyperproliferative cells. However, it seems likely that it is not growth inhibition itself, rather the induction of vascular cell damage, which is the main driving force in the stimulation of the proliferative potential of surviving cells. Indeed, the same apoptosis resistance could be achieved by exposure of naive cells directly to a conditioned medium from apoptotic cells (154). PAH could also be induced by the activation of apoptotic pathways (195). Apoptosis-inducing stimuli, including ultraviolet irradiation (49), drugs and toxins (46, 126), shear stress (155), mitochondrial dysfunction (203), oxidative/nitrative stress (186, 199), as well as genetic factors, such as loss of BMPR2 signaling (185), were shown to be directly involved in the pathogenesis of PAH.
In contrast, apoptosis inhibitors instead protect against PAH development. In two different studies, overexpression of VEGF in lungs blunts the onset of either hypoxia-induced PAH (35, 123) or PH associated with pulmonary fibrosis. Hameed et al. showed that the blockade or genetic deletion of a TNF-related apoptosis-inducing ligand that is responsible for endothelial apoptosis prevents the development of PAH in three independent rodent models (60). Another line of evidence comes from the research published by White et al. showing that attenuation of the programmed cell death 4/caspase-3/apoptotic pathway potently blocks SU/hypoxia PAH (195). Antagonizing the biosynthesis of macrophage-derived leukotriene B4 and directly inducing the apoptosis of pulmonary artery ECs reverses established PAH in animal models, as shown by Tian et al. (187). In contrast, overexpression of 5-lipoxygenase, which catalyzes leukotriene biosynthesis, was shown to have enhanced MCT-induced PAH (73). This last evidence supports the role of apoptotic cell death as not only in the initiation of PAH but also for its role in the continued progression during the developed stage. Indeed, it seems that PAH-associated damage is not a “hit and run” effect but rather an ongoing process continuously mediating disease progression. Thus, it was noticed that proapoptotic factors, such as caspase 3 and p53, remain elevated in the pulmonary arteries of patients with advanced irreversible disease (93). Another study reported that patients with a developed PAH continue to have circulating markers of vascular injury (167).
Although the causal role of initial vascular wall damage in PAH is widely accepted, the particular mechanisms responsible for the transformation of apoptosis into the proliferation of pulmonary vascular cells are still debated. In general, apoptosis serves as an essential regulator of tissue integrity and homeostasis that removes damaged or no longer functional cells. It represents a silent type of cellular damage with no detectable activation of the immune system. However, this silence is not complete; phagocytic clearance of apoptotic cells requires the production of chemoattractants that were described as “find me,” “eat me,” “listen to me,” or “stay away” signals that are critical for the recognition and the engulfment of the dying cell by the phagocytes (39, 55). This signaling along with the later discovered ability of apoptotic cells to regulate their local environment by releasing regulators of proapoptotic, antiapoptotic, and mitogenic pathways may explain the ultimate role of ongoing vascular apoptosis in the following proliferation of surrounding cells.
One of the examples of apoptotic cell communication is the releasing of long-range death factors. Thus, it was discovered that the induction of apoptosis in one part of the tissue stimulates an apoptotic death in remote cells through secretion of TNF-α (125). Other mechanisms inducing propagation of cell death require functional gap junctions. Two different research groups described that cell death signaling was propagated via gap junctions using two different systems. Krutovskikh et al. found that apoptotic cells use gap junction protein Connexin43 (Cx43) to couple with their nonapoptotic neighbors and propagate cell death (85). The formation of clusters of dying cells was inhibited in cells by expressing a dominant negative mutant of Cx43. Azzam et al. described that damage signals might be transmitted from irradiated to nonirradiated “bystander” cells that start expressing the genetic damage or changes in the expression of stress-induced genes through the same Cx43-mediated intercellular communication (8). Since gap junctions can typically pass molecules of up to 1000–1500 Da, the spreading of the damage signaling could occur through the exchange of metabolites: ions such as Ca2+, nucleotides, or peptides. Besides, these death messengers could be released from the cell itself. Lipid peroxide products, inosine nucleotides, and cytokines such as TNF-α, as well as reactive oxygen species (ROS), are the proposed candidates for transmission of damage signals from apoptotic to healthy cells (132). The same spread of the cell killing signaling cascade could coordinate apoptotic cell death in the pulmonary vasculature in response to various damaging stimuli discussed earlier. If confirmed, the propagation of death signaling could explain simultaneous apoptotic cell death in the remote parts of the pulmonary vascular tree.
Apoptotic cells are also known to mediate apoptosis-induced death resistance. The important survival signaling relevant to PAH is mediated through VEGF. Secreted from dying ECs (49), it induces apoptosis resistance in surviving ECs and vascular smooth muscle cells (VSMCs) (155). Another critical endothelial survival factor, angiopoietin-1 (121), is produced not only by apoptotic cells but also by monocytes interacting with apoptotic cells (161). Inflammatory cells are also a source of IL-6, a pleiotropic cytokine that serves to block apoptosis in different vascular cells exposed to the toxic environment (45). In contrast, apoptotic ECs promotes the survival of macrophages in a sphingosine-1-phosphate dependent manner, thus ensuring the rapid phagocytosis of dying cells (193). This double-sided protection represents a functional network between different types of cells that cooperatively stabilize the vascular wall against damage.
Such a combination of pro- and antiapoptotic stimuli does not fully explain the process of making the survival-versus-apoptosis decision for each affected cell. The current opinion tends to view each population of cells as a mixture of cells that are more sensitive or more resistant to apoptotic stimuli. That means that under the conditions of persistent self-perturbed apoptosis, the situation favors the selection of apoptosis-resistant cells due to the death of sensitive cells and the stimulation of apoptosis resistance in survivors. The secretion of prosurvival factors by apoptotic cells additionally shifts this balance toward survival, growth, and, eventually, vascular remodeling. Thus, TGF-1β released from the apoptotic ECs promotes intimal hyperplasia (105), SMC proliferation and migration (155), endothelial–mesenchymal transition with EC-derived SMC accumulation (94), and ECM deposition (64). The conditioned media were collected from the ECs exposed to hypoxia, a condition known to promote EC apoptosis (109), and stimulated proliferation of SMCs through the prostaglandin-mediated mechanism and the growth of fibroblasts via secretion on basic fibroblast growth factor (113). It was also reported that fibroblasts exposed to apoptotic media undergo myofibroblast differentiation in a connective tissue growth factor responsive manner (88). Endothelial injury impairs the secretion of NO, a main paracrine vasodilator with antimitogenic properties, and then stimulates the secretion of endothelin-1 (76), a potent vasoconstrictor and mitogen. Apoptosis is also associated with the increased production of ROS, which can initiate and control different aspects of apoptosis-induced proliferation (33).
Even in the context of immune system modulation, apoptotic cell death is now considered not “silent,” but rather anti-inflammatory (52). Inhibition of inflammation is achieved through a number of sequential steps. First, it requires a prompt and efficient engulfment of apoptotic cells by phagocytes to prevent an uncontrolled release of intracellular content. This is normally achieved by producing the “find me” and “eat me” factors. The examples of such attractants are lipid lysophosphatidylcholine, chemokine CX3CL1 (fractalkine [FKN]), the nucleotides adenosine triphosphate and uridine triphosphate, endothelial monocyte-activating polypeptide II, thrombospondin 1, TGF-β, annexin I, and oxidized phospholipids of apoptotic cells (55). Some of these attractants are also recognized as anti-inflammatory mediators. For example, apoptotic cell-derived TGF-β and FKN suppress macrophage proinflammatory responses (118).
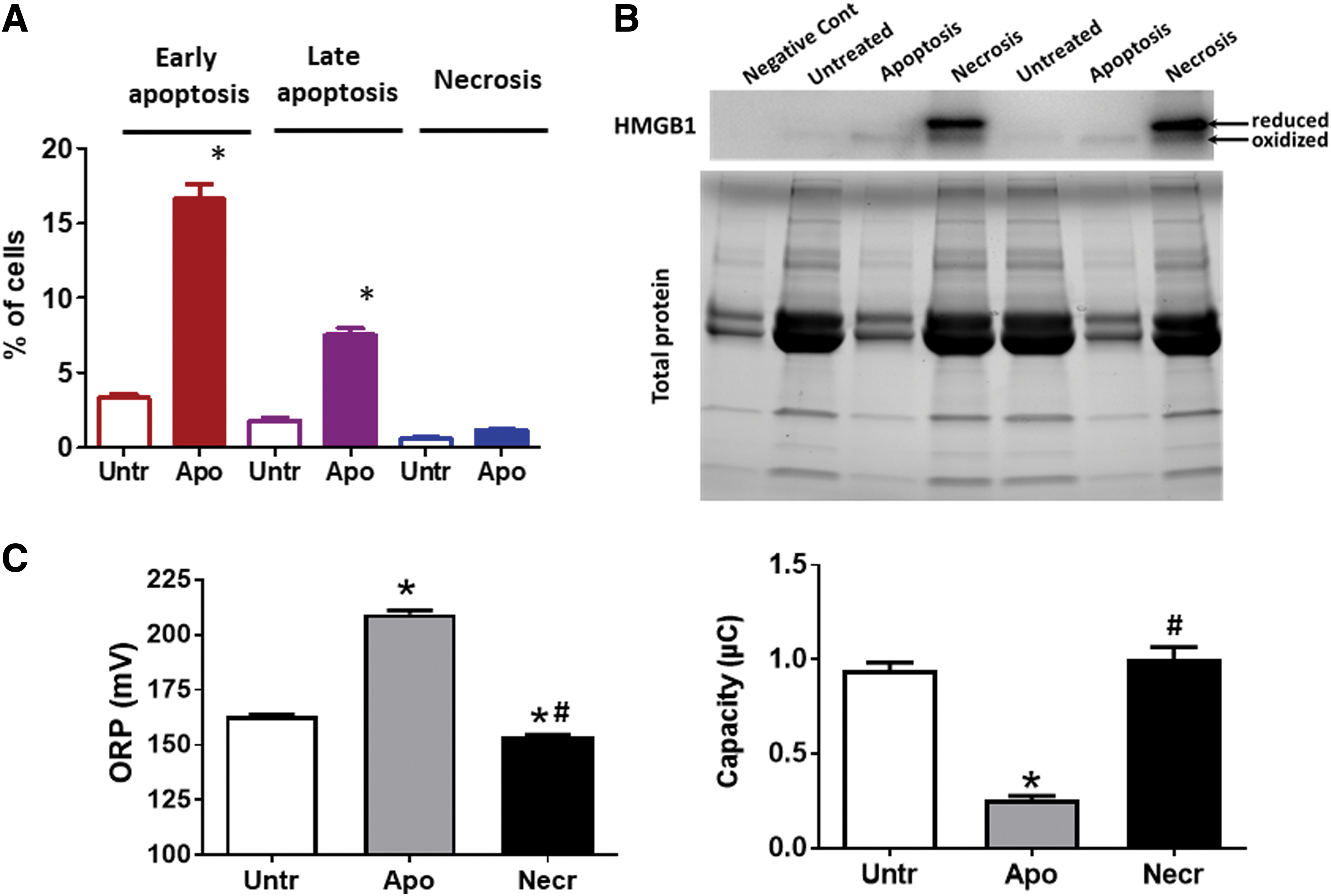
Dying cells are typically known to be a source of DAMPs, which initiate a cascade of inflammatory reactions. For example, high mobility group box 1 (HMGB1) stimulates inflammatory cell activation via the activation of PRRs. However, as opposed to necrosis, apoptotic cells were shown to produce HMGB1 in considerably lower amounts due to its ability to stay attached to the apoptotic chromatin (13). This is especially true for human pulmonary artery endothelial cells (HPAECs, Fig. 4A, B). ROS produced by apoptotic cells oxidize intracellular proteins (78) as well as proteins in the extracellular environment (Fig. 4C); therefore, HMGB1 and other redox-sensitive DAMPS would be likely secreted in an already preoxidized inactive form (78) or will be quickly inactivated by oxidation after secretion. Importantly, an oxidized HMGB1 is known to be responsible for the resolution of the inflammation due to its ability to initiate immune tolerance (78). Apoptotic cells are also capable of mediating innate immunosuppression via a mechanism uncoupled from paracrine effects or phagocytosis. This process consists of direct transcriptional inhibition of genes encoding inflammatory cytokines (14) and could be activated in macrophages, fibroblasts, and potentially all contacting neighbors of apoptotic cells.
It is also well established that once macrophages engulf the apoptotic cell, they resemble an alternative mode of activation and transform into the anti-inflammatory M2 phenotype macrophages, in contrast to the classical, proinflammatory M1 phenotype (193). Such M2 macrophages produce a spectrum of anti-inflammatory mediators including TGF-β, IL-10, and prostaglandins. Besides, the uptake of apoptotic cells by already activated M1 macrophages will suppress the production of pro-inflammatory cytokines such as TNF-α, IL-6, IL-1β, IL-8, and IL-12 (34, 55, 198). Thus, apoptosis could not only prevent the development of the inflammatory response but also cease any ongoing inflammation. Indeed, it was noticed that the addition of macrophages that ingest apoptotic cells into lipopolysaccharide-induced inflammatory cells reduces inflammatory pathways (52). Aside from anti-inflammatory activity, M2 macrophages also play an important role in tissue repair by secreting the ECM components, as well as angiogenic and chemotactic factors (151), and, thus, could directly contribute to vascular remodeling.
Despite the anti-inflammatory nature of apoptosis, PAH is associated with severe inflammatory changes in the pulmonary vascular wall (131, 135). Oddly, this activation of the innate and adaptive immune system in PAH is known to be directly connected to pulmonary vascular damage, which produces an apparent paradox. However, it is important to consider that apoptosis is not the only outcome for the damaged cell. The severe cell damage in PAH could also trigger necroptosis or necrosis. Surprisingly, the contribution of these types of cell death in the pathogenesis of PAH is almost unstudied. Both necroptotic and necrotic cells are the source of many DAMPs, known activators of the inflammatory response. Indeed, it was reported that the HMGB1 released into the extracellular space during the early stage of MCT-induced PAH contributes to PAH development (153, 200). Our group has also confirmed an accumulation of extranuclear HMGB1 in the pulmonary artery wall of male rats with PAH induced by SU/hypoxia (137). Apoptotic cells do not release HMGB1 even after they undergo secondary necrosis because of the hypoacetylation of one or more of the chromatin components that occurs during apoptosis and retains HMGB1 firmly bound to chromatin (158). Therefore, since the release of HMGB1 distinguishes the necrotic cells from apoptotic ones, it could be expected that MCT or SU/hypoxia induced PAH is associated with necrotic cell death. This necrosis is capable of initiating inflammatory signaling in pulmonary arteries through activation of TLR4/IL1β/E-Selectin axis (137) that activates ECs and increases inflammatory cell recruitment.
Activated monocytes and macrophages, in turn, are other cell types that could produce HMGB1, although not due to the passive release, but due to active secretion. This process requires HMGB1 hyperacetylation and, thus, is opposite to the hypoacetylation and nuclear retention that happens in apoptotic cells. However, the proinflammatory activation of phagocytes that should precede any cytokine production could not happen if the surrounding vascular cells died by apoptosis. In contrast, HMGB1 released from necrotic cells is an active facilitator of macrophage reprogramming toward the M1-like phenotype, whereas its neutralization reduces M1 macrophage infiltration (177). Therefore, necrosis, rather than apoptosis, appears to be responsible for the initiation of proinflammatory mechanisms (Fig. 5). By skewing macrophage differentiation toward M1-like phenotypes, HMGB1 also diminishes the amount of M2 macrophages and disturbs the normal clearance of apoptotic cells (159). Therefore, once released into the extracellular milieu, HMGB1 shifts the normally maintained balance between pro- and anti-inflammatory events toward inflammation, which, in turn, becomes self-perpetuating (204).
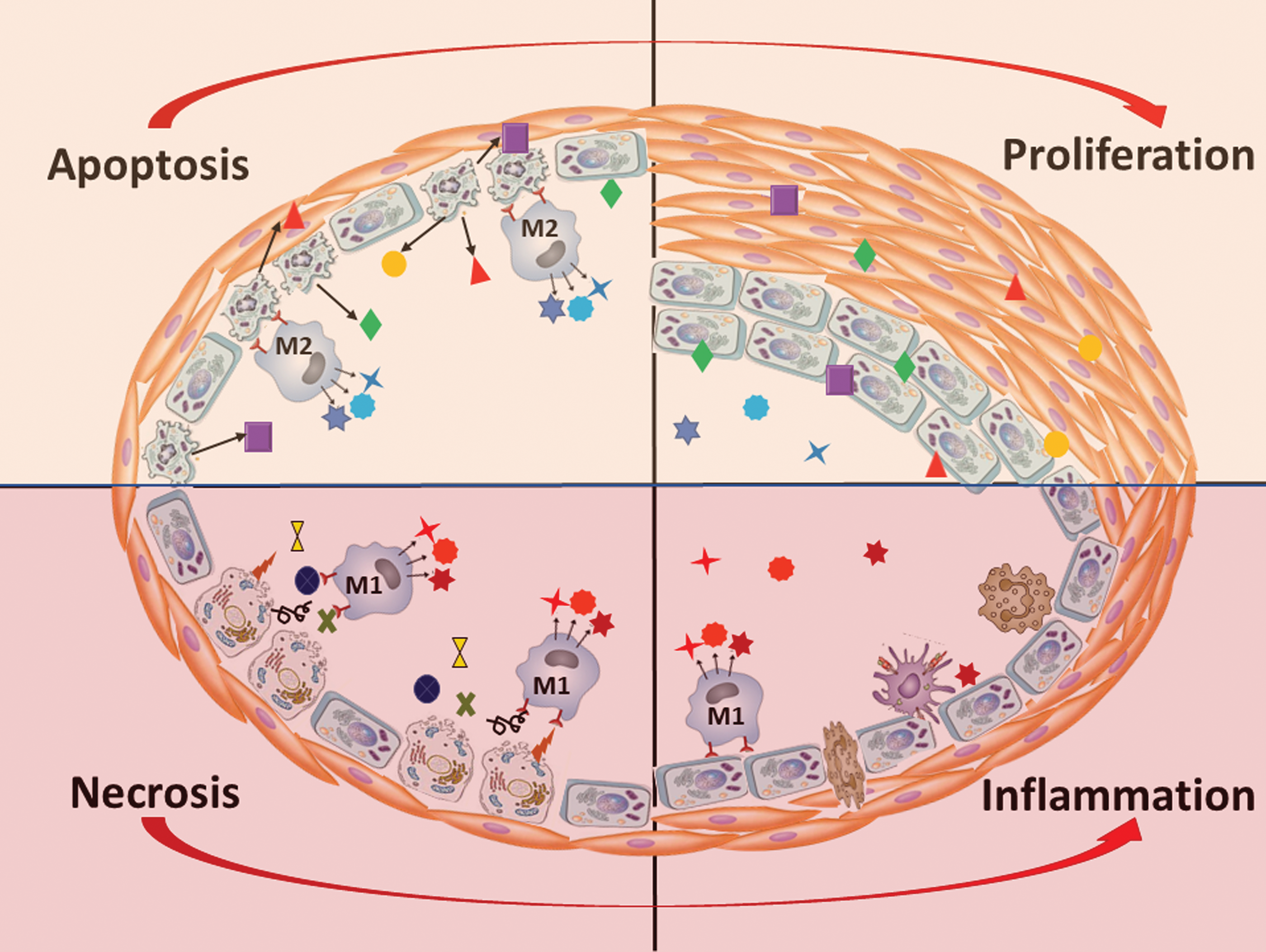
There are many other DAMPs produced by dying cells that have nucleic, cytosolic, or mitochondrial origin and could either participate in tissue healing through stimulation of prosurvival/proliferative pathways or induce inflammation (19, 30, 47, 100, 111, 120, 133, 147, 205). The particular contributions of all these factors are yet to be established. However, some of them have been already reported as promising biomarkers associated with PAH (74, 111, 137, 145). It, therefore, follows that further validation of such biomarkers that could be routinely screened, at least in the PAH patient's family members or in high-risk patients, could help to identify this deadly disease at the early, curable stage.
Oxidative/Nitrative Stress
ROS are a family of oxygen-based highly chemically active species that include the parent oxygen free-radical superoxide (O2 −•), its direct dismutation product hydrogen peroxide (H2O2), hydroxyl radical (OH−•), hypochlorite (OCl−), and hydroperoxyl radical (HO2 •) (3a). Cellular sources of ROS include the NADPH oxidase (Nox) family of enzymes, mitochondrial electron transport chain complexes (mainly I and III), xanthine oxidoreductase (XO), cytochrome P450, lipid oxygenases including cyclooxygenases and lipoxygenases, peroxidases, and uncoupled NOSs (3a, 10, 20, 189, 197). Nox proteins are considered the only “professional” ROS generators in that ROS are their main product rather than a byproduct of their chemical reactivity (3a). Nox1, 2, 4, and 5 are expressed in the lung vasculature (3a, 10, 48). To counterbalance the sources of ROS in the cell, a number of endogenous antioxidant systems exist. These include superoxide dismutases (SODs), which convert O2 −• to H2O2, catalase, which converts H2O2 to water, glutathione peroxidase, heme oxygenase, peroxiredoxins, glutaredoxin, and thioredoxin (41). Nonenzymatic antioxidant systems also contribute to the balance and include vitamins C and E, retinol, glutathione, and β-carotene (41). Similar to ROS, reactive nitrogen species (RNS) are chemical moieties derived from the free radical NO and include nitrogen dioxide (NO2), dinitrogen trioxide (N2O3), nitroxyl anion (NO−), and nitrosonium cation (NO+) (44, 181). The major enzymatic source of NO is the NOS enzymes, of which there are three isoforms: neuronal (nNOS or NOSI), inducible (iNOS or NOSII), and endothelial (eNOS or NOSIII) (44, 181). NO can also be produced through the reduction of nitrite by several nitrite reductases (81). NO can react with protein cysteine residues, heme iron in iron-containing proteins, and with lipids. Importantly, NO can also react with O2 −• to produce the highly reactive peroxynitrite (ONOO−), which can lead to protein nitration (tyrosine residues modification to nitrotyrosine) (181).
ROS, RNS, and their sources (e.g., Nox enzymes for ROS) have been implicated in PH and associated RV responses in both in vivo and in vitro studies, and there is mounting evidence that ROS levels are increased in PH patients (16, 38, 40, 42, 44, 57, 181). For example, systemic and pulmonary vascular EC, VSMCs, and adventitial fibroblasts express Nox1, 2, and 4 and these enzymes have all been linked to PH in multiple studies (9, 22, 26, 32, 38, 41, 42, 48, 56, 57, 61, 71). ROS scavenging molecules glutathione and vitamin E were observed to be low in PH patient plasma, whereas lipid peroxidation products such as malondialdehyde and lipid hydroperoxide were elevated (148). Levels of nitrotyrosine, 5-oxo-eicosatetraenoic acid, and hydroxyeicosatetraenoic acid were also elevated (16, 144). A role of NO and XO in PH has been shown in vivo and in pulmonary VSMCs (211). A global effect of ROS/RNS is the decrease in NO bioavailability through chemical scavenging, similar to what occurs when ONOO− is formed. This contributes to increased vasoconstriction and impaired vasorelaxation, and thereby to the development of PH (142, 181). NO is a major vasodilator and regulator of the basal vasomotor tone. Interruptions in NO signaling or its levels can lead to impaired vasorelaxation and are associated with PH (44, 81, 82, 181). Moreover, sources of ROS such as Nox enzymes and XO can interrupt NO production and signaling, further contributing to the decreased NO bioavailability (181). Indeed, it was demonstrated that the use of nitrite as an NO source has been associated with an attenuation of experimental PH (211). ROS/RNS have also been associated with increasing levels and signaling of the potent pulmonary vasoconstrictor endothelin 1, which is the main factor in PH development, further contributing to an overall imbalance in motor tone and PH (181).
ROS and RNS also have well-documented effects on cellular and molecular changes that promote vascular cell proliferation, migration, hypertrophy, and apoptosis, all processes linked to PH development (3a, 9, 10, 44, 57, 141, 144, 181), and have been implicated in promoting endothelial, smooth muscle, and fibroblast proliferation (9, 41, 48, 141). Interruption of Nox1 was associated with attenuation of pulmonary arterial EC proliferation, and targeting of Nox4 was linked to a reduction in pulmonary artery smooth muscle and fibroblast proliferation (9) as well as with increased fibroblast apoptosis. Both isoforms were upregulated in PH patient lung tissue (48, 97, 115). The Nox4 effect appears to be associated with activation of mTOR complex 2, mTORC2, and AMP-activated protein kinase (50). In models of persistent PH of newborn targeting ROS by the pharmacologic inhibitors, apocynin and N-acetyl-cysteine, was associated with improvement in angiogenesis in ECs (186). Generally, in addition to direct oxidation and free-radical damage of DNA, proteins, and lipids, ROS such as H2O2 affect key signaling pathways and transcription factors that drive the cellular pathophysiologic processes associated with PH and pulmonary vascular remodeling (141). These include the MAPKs pathways p38 MAPK and Erk1/2, the c-Jun N-terminal kinase pathway, the Akt/PKB pathway, the NF-κB transcription factor, p53, and AP1 (3a, 10, 44, 145, 181). Also, the cAMP-response element binding transcription factor, which is selectively activated in the lungs of hypoxic PAH mice and the hypoxic human lung microvascular EC (92), was implicated as an ROS-inducible transcription factor (157, 164, 165). Other examples include a role for Nox1 in driving pulmonary arterial EC proliferation via upregulation of sonic hedgehog-mediated expression of the bone morphogenetic protein (BMP)-signaling antagonist Gremlin1 (18, 48, 66, 212). BMP receptor and signaling impairments are pervasive in hereditary forms of PAH and are also implicated in a subset of idiopathic PAH (68, 152). In addition, Gremlin1 has been linked to HIF2α and the vascular smooth muscle proliferation, migration, and angiogenesis (18, 80a, 104, 170), further supporting a role for ROS in these processes and PH development. The link between ROS sources and cellular growth pathways is strengthened by studies that show a reduction in the hypoxia-induced pulmonary vascular endothelial and smooth muscle proliferation by Nox inhibitory drugs and a link of this reduction to reduction in TGFβ and rescue of peroxisome proliferator-activated receptor γ (54). Moreover, Nox1-mediated proliferation was linked to modulations of SOD2, Nrf2, cyclin D1, and cofilin in pulmonary arterial SMCs (191). Upstream, caveolin-1 (Cav-1) was associated with the inhibition of Nox enzymes through the modulation of NF-κB and the reduced expression of Cav-1 observed in PH models; this was linked to increased Nox activity and ROS elevation (22). Also upstream, evidence implicates TGFβ, which is a potent mediator of cellular phenotypes in PH, in upregulation of Nox, specifically Nox4, via activation of insulin-like growth factor binding protein 3 and Akt/PKB in pulmonary arterial SMCs (69).
ROS have been shown to be involved in both acute and chronic regulation of hypoxic pulmonary vasoconstriction, including associated processes such as vascular cell proliferation and medial hypertrophy, in part through modulation of calcium and potassium channels; however, the precise mechanisms remain elusive (41, 119, 162, 180, 190). For example, in pulmonary arterial SMCs from Nox1 null animals, there was a decrease in the Kv1.5 voltage-dependent potassium channel, an increase in intracellular potassium levels, and an associated reduction in apoptotic cells (71). On the other hand, interruption of ROS and Nox4 was shown to lift hypoxia-induced decreases in the potassium current via direct association with Kv1.5 (114). These discrepancies allude to the complexity of these processes but do support a clear link between ROS and potassium currents in PH. Genetic deletion of Nox2 or administration of SOD also reduced vasoconstriction in hypoxic mice, supporting the role of ROS and further supporting a potential interplay with the reduction in NO bioavailability as discussed earlier (99). Further, the mechanism by which ROS affects intracellular calcium and calcium channels, namely the Cav1.2 channel, may involve downregulation of the glycolytic pyruvate kinase M2 (59) and involve contributions from PKC signaling (146). One mechanism by which ROS carry out their second-messenger effects involves shifting the balance toward activation between the kinases and phosphatases of these major pathways by oxidizing and turning off phosphatases. This mostly occurs through oxidation of key cysteine residues of effector proteins. Also, the RNS ONOO− can induce direct protein tyrosine nitration, which inhibits and stimulates degradation of important enzymes involved in vasorelaxation, such as eNOS and protein kinase G (2, 181). Tyrosine nitration can also mimic phosphorylation and induce activation of Akt in ECs (138). Nitration of Akt increases as early as 1 week from SU/hypoxia model induction (Varghese VM, Niihori M, Eccles CA, Kurdyukov S, James J, Rafikova O, Rafikov R.). Nitrite treatment has been shown to effectively increase NO bioavailability and reduce manifestations of PH both in vivo and in vitro through a mechanism that involves inhibition of smooth muscle proliferation and upregulation of the cyclin-dependent kinase inhibitor p21/cip (211).
It can, therefore, be seen that ROS/RNS plays important roles in the development and progression of PH and PH-driving processes. They pose as viable therapeutic targets for future development. Indeed, some evidence supports this. For example, administration of recombinant SOD and SOD mimetics, eNOS inhibitor
Early Diagnosis/Biomarkers of PAH
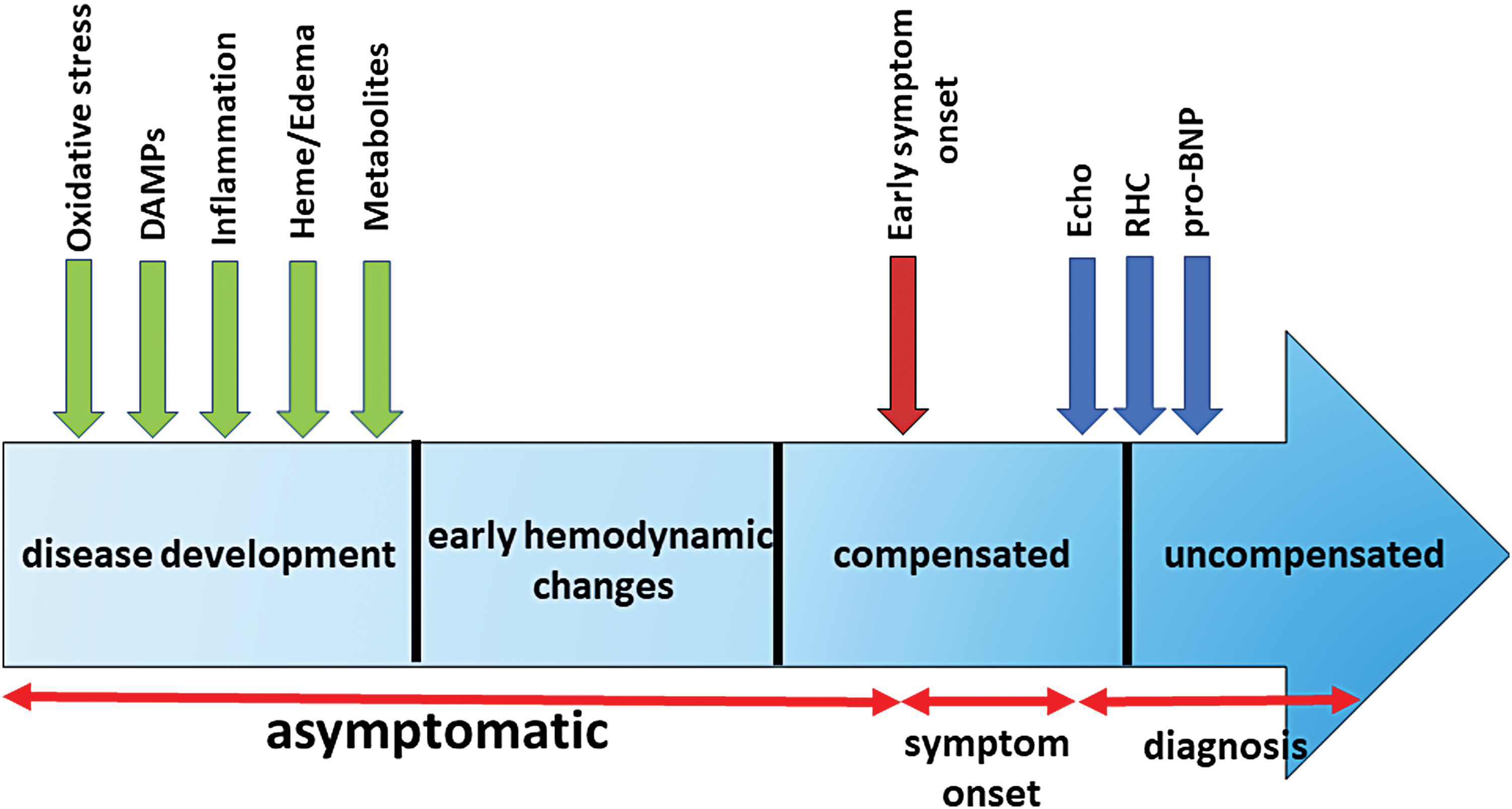
Initial diagnosis of PAH in patients is still problematic as patients experienced a very broad range of symptoms, and even at the earliest stage, PAH onset is asymptomatic (89, 90). The earliest symptom of PH, the dyspnea on exercise, is also a common symptom of asthma, chronic obstructive pulmonary disease, myocardial infarction, and pneumonia, among others. However, it is important to improve the early detection of PAH because functional class (FC) 1 and 2 patients with PAH have better survival rates than patients diagnosed in more severe conditions (67, 188). It can be argued that there is tremendous benefit in patients with risk factors such as familial PAH, sickle cell disease, thalassemia, glucose-6-phosphate dehydrogenase deficiency(87), portal hypertension, congenital heart disease, HIV infection, acute pulmonary embolism, and connective tissue disease(79), to undergo preventive assessment by echocardiography even in the absence of symptoms of PAH.
However, the accuracy of noninvasive echocardiography is not perfect, especially in mild conditions, and the gold standard of PAH diagnosis remains to be right heart catheterization (RHC). Nonetheless, RHC is an invasive procedure and monitoring even predisposed patients every year may be difficult and even unrealistic from a clinical perspective. Therefore, the importance of concerted efforts for the discovery of early biomarker “fingerprints” of PAH development at an early, asymptomatic phase becomes glaringly evident. The closest resemblance of a biomarker for PAH that exists today is the plasma levels of the probrain natriuretic peptide (pro-BNP). This is potentially a very useful, minimally invasive approach to assessing PAH that only requires plasma collection from patients (5). pro-BNP is a peptide that is released from heart tissue that is stressed by the extra load, similar to what is experienced by the right heart during PAH as a result of the rise in pulmonary vascular resistance and pressure. However, this approach presents important limitations. First, it can be a nonspecific marker of ventricular stress, be that from the right or left ventricles of the heart. Second, and more importantly, because an increase in pulmonary pressure is needed to induce RV stress, this biomarker cannot be used to assess early disease development that precedes pressure elevation.
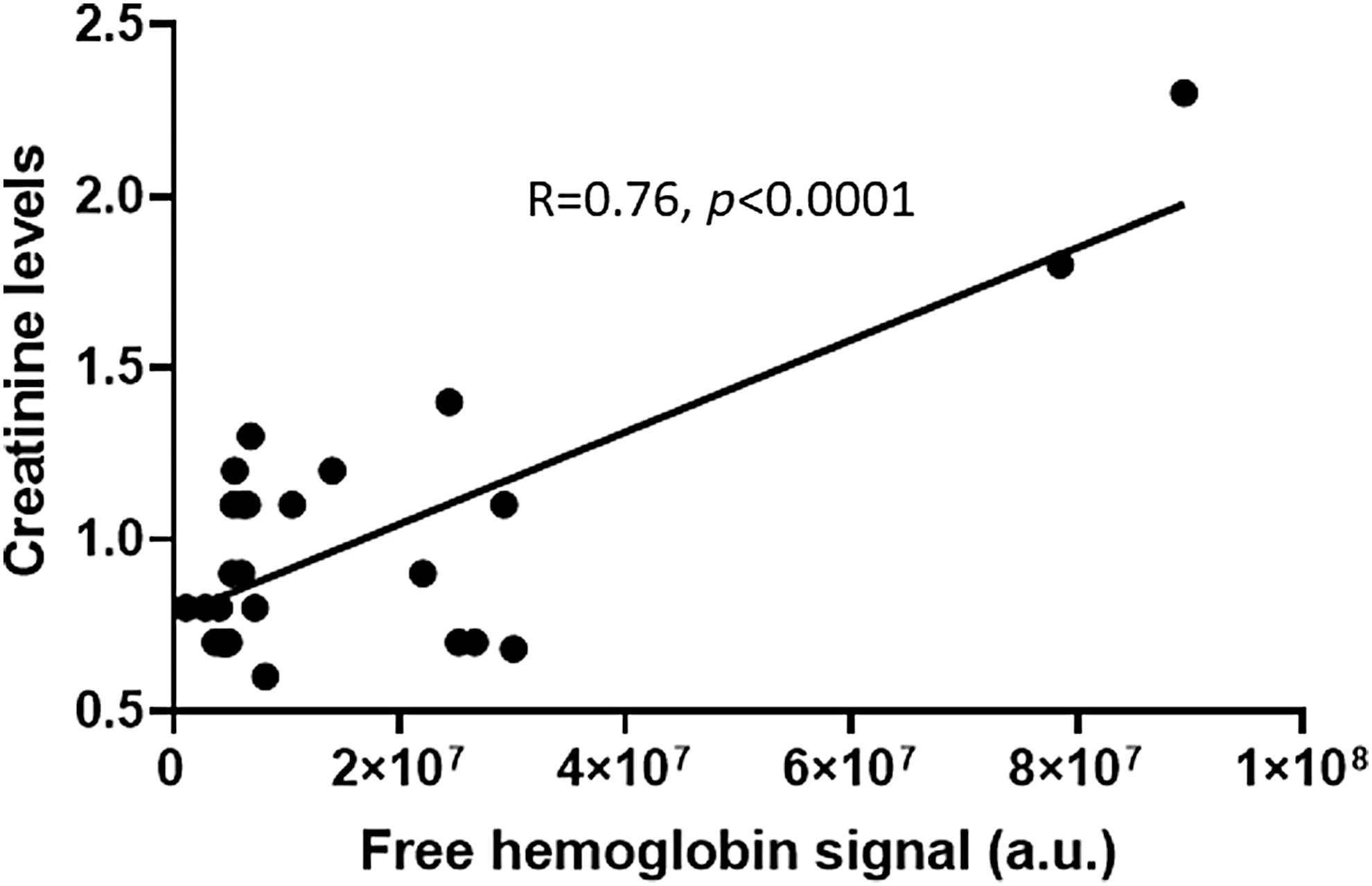
As discussed earlier, several conditions that initiate disease development could be used to assess early PAH. For example, several reports showed that metabolic changes in the lungs undergo significant disturbances. The importance of metabolic biomarkers in PAH is widely discussed. We found that even before hemodynamic changes occur in the pulmonary vasculature or right heart in the MCT model of PAH, more than 500 metabolites were significantly altered (140). Those metabolic changes could be used to generate the needed “fingerprint” of PAH for metabolic analysis. With the focus on the changes characteristic to PAH, these “fingerprints” could differentiate between other lung or heart diseases. PAH is accompanied with the glycolytic switch in metabolism, and indeed, our data indicate a more than 10-fold upregulation of glucose associated with the disease progression. Although a promising approach, the fact that these changes were assessed directly in lung tissue makes it infeasible due to the invasive nature of the needed procedures to collect lung biopsies. Building on these findings by examination of blood plasma for similar and specific metabolite changes, however, would be a favorable and feasible approach. Also, inflammatory markers, such as cytokines and DAMPs, could be used for early detection of PAH. The underlying hemolysis in PAH patients, which contributes to the inflammatory response can be assessed in plasma (111, 145). Therefore, erythroid-derived DAMPs may be promising targets for early detection. Indeed, signals from the free hemoglobin in plasma followed PAH progression, as demonstrated by a significant correlation with mean pulmonary arterial pressure (mPAP), pulmonary vascular resistance, and cardiac index. Free hemoglobin may not be an ideal reporter of hemolysis in PAH as it has a specific haptoglobin-mediated sequestering system; therefore, at the early phases of disease, it could be controlled by the detoxification system. Therefore, other markers of hemolysis may be utilized. Interestingly, an increase in plasma creatinine levels in PAH patients is common and correlates with disease progression; this is partially attributable to the worsening of renal function. Creatinine could also be found at a high level in RBCs and reticulocytes; thus, the release of creatinine from damaged RBCs could indicate ongoing hemolysis. Indeed, our data indicate that in PAH patients (N = 30) creatinine levels are strongly (p < 0.0001) correlated with free hemoglobin levels (Fig. 6). Two samples with increased free hemoglobin are probably producing the effect on correlation; thus, this required future studies with an analysis of the large cohort of patients.
It was also found that RBCs size distribution width (RDW) is an important biomarker of PAH progression and outcome (3, 168). RDW predicted survival of PAH patients independently of the pro-BNP and the 6-minute walk distance. Interestingly, increased RDW is common for iron deficit anemias and blood loss conditions, again pointing to the role of hemolysis in PAH. Other plasma biomarkers that we tested in the study that are related to the inflammatory response are IL-6 and the growth differentiation factor 15 (150). Both biomarkers demonstrated a significant correlation with hemodynamics and survival estimates. Therefore, early diagnosis of PAH could be based on circulating biomarkers that reflect the characteristic “blueprint” of PAH based on oxidative stress(117), inflammatory/DAMPs, hemolysis, and metabolic reprogramming markers (Fig. 7). Although the identification and initial validation of the promising biomarkers have to be done in a preclinical animal model, they could be further validated in the patients with exercise-induced PAH or patients with a mild borderline PH (mPAP 20–25 mmHg) that represent patient groups with early disease(84, 163). Although still uncommon compared with patients with more advanced PAH, such early cases are occasionally diagnosed, and their frequency has been increasing in the recent years.
Conclusion
The recent advances make PH more treatable than it was earlier. However, the early diagnosis of PH still remains the biggest challenge. The nonspecific symptoms of PH significantly delay its diagnosis. Currently, the time to diagnosis (TTD) was reported as being close to 4 years (176). On average, it takes a patient about 12 months after the initial symptom onset to initiate the first medical contact, five general practitioner visits, and three specialist visits before being referred to a PH specialist and before undergoing RV catheterization. During this period, the disease progresses from FC II (95%)/FC III (5%) at the time of symptom onset to FC II (5%)/FC III (90%)/FC IV (5%) at diagnosis (176). At the same time, a few national registry studies confirmed that FC I or II patients have significantly better long-term survival than patients with FC III/IV (67). Therefore, an approach that would allow decreasing TTD would help to change the distribution between early and advanced PAH and toward less progressive and more responsive PAH therapy. A further shift toward diagnosing PH at an asymptomatic stage could be expected to significantly improve the survival and prognosis for this deadly disease. The accumulated body of literature strongly supports the idea that such early diagnosis is highly possible. PH patients undergo significant changes in metabolism, redox status, chronic unrecognized hemolysis, cellular damage, and activation of inflammatory pathways that precede PH manifestation. All these early pathological events can be used to establish a combination of biomarkers that could serve as a blueprint of the early stage of PH and could be used for the prescreening of PH patients. Therefore, we believe that the effort of the research community has to be shifted from an attempt to treat the advanced stages of PH, which is hardly achievable due to nonreversible changes of a failing heart, toward getting a better understanding about mechanisms involved in PH initiation and early progression. Such an approach would not only advance our current knowledge regarding the early pathogenic events but also significantly advance PH diagnostics, increase therapy effectiveness, and improve patient survival.
Footnotes
Acknowledgments
This work was supported by NIH grants R01HL133085 (O.R.), R01HL132918 (R.R.), AHA grant 15SDG24910003, ALA grant RG-515656, and Gilead grant 0059493 (I.A.G.).