
Abstract
Significance:
Type 2 diabetes development involves multiple changes in β-cells, related to the oxidative stress and impaired redox signaling, beginning frequently by sustained overfeeding due to the resulting lipotoxicity and glucotoxicity. Uncovering relationships among the dysregulated metabolism, impaired β-cell “well-being,” biogenesis, or cross talk with peripheral insulin resistance is required for elucidation of type 2 diabetes etiology.
Recent Advances:
It has been recognized that the oxidative stress, lipotoxicity, and glucotoxicity cannot be separated from numerous other cell pathology events, such as the attempted compensation of β-cell for the increased insulin demand and dynamics of β-cell biogenesis and its “reversal” at dedifferentiation, that is, from the concomitantly decreasing islet β-cell mass (also due to transdifferentiation) and low-grade islet or systemic inflammation.
Critical Issues:
At prediabetes, the compensation responses of β-cells, attempting to delay the pathology progression—when exaggerated—set a new state, in which a self-checking redox signaling related to the expression of Ins gene expression is impaired. The resulting altered redox signaling, diminished insulin secretion responses to various secretagogues including glucose, may lead to excretion of cytokines or chemokines by β-cells or excretion of endosomes. They could substantiate putative stress signals to the periphery. Subsequent changes and lasting glucolipotoxicity promote islet inflammatory responses and further pathology spiral.
Future Directions:
Should bring an understanding of the β-cell self-checking and related redox signaling, including the putative stress signal to periphery. Strategies to cure or prevent type 2 diabetes could be based on the substitution of the “wrong” signal by the “correct” self-checking signal.
Introduction
Type 2 diabetes etiology
Consensus on type 2 diabetes etiology
Traditionally, it is emphasized that insulin stimulates the disposal of glucose into the skeletal muscle and white adipose tissue (WAT), reduces glycogenolysis and gluconeogenesis in the liver, and suppresses lipolysis in WAT (30). Insulin resistance (IR) is consequently viewed as the inability of the above events or their reduction, such as insufficient glucose uptake into skeletal muscle, enhanced glucose release from the liver, and enhanced supply of free fatty acids (FAs) to the blood from WAT. Typically, during the glucose tolerance test, IR is manifested by the delayed response on glucose. For healthy individuals, after a glucose bolus, one can recognize the typical high and sharp first phase of insulin release, followed by the second phase with about a half amplitude and prolonged decay (up to 1 hour).
At the progressed type 2 diabetes, the first phase is often missing, whereas the second phase is enhanced and more prolonged. This is termed as hyperinsulinemia due to the fact that higher time-integrated insulin release exists in such a pathological state, despite the first phase being inhibited. In healthy individuals, about 75% of the insulin-induced glucose uptake is ensured into skeletal muscle, whereas this is substantially reduced in hyperinsulinemic and obese patients (21).
With the progressing molecular physiology research, it is recognized that numerous other factors contribute to the fine blood glucose regulations and insulin responses, namely nutritional signaling mediated by the metabotropic receptors (122), endocrine role of incretins, that is, glucagon-like peptide 1 (GLP-1) (97, 158) and gastric inhibitory polypeptide (GIP) (198) (and other gastrointestinal hormones), paracrine GLP-1 signaling (82), paracrine and endocrine secretion of other hormones (245), systemic control by brain (43), and immune system contribution. Concerning the type 2 diabetes development, emphasis predominates mostly in relation to WAT on the so-called low-grade inflammation causing IR (20, 130, 308). During prediabetes, at early type 2 diabetes stages, a compensation phase exists when β-cells respond by enhancing their mass and function.
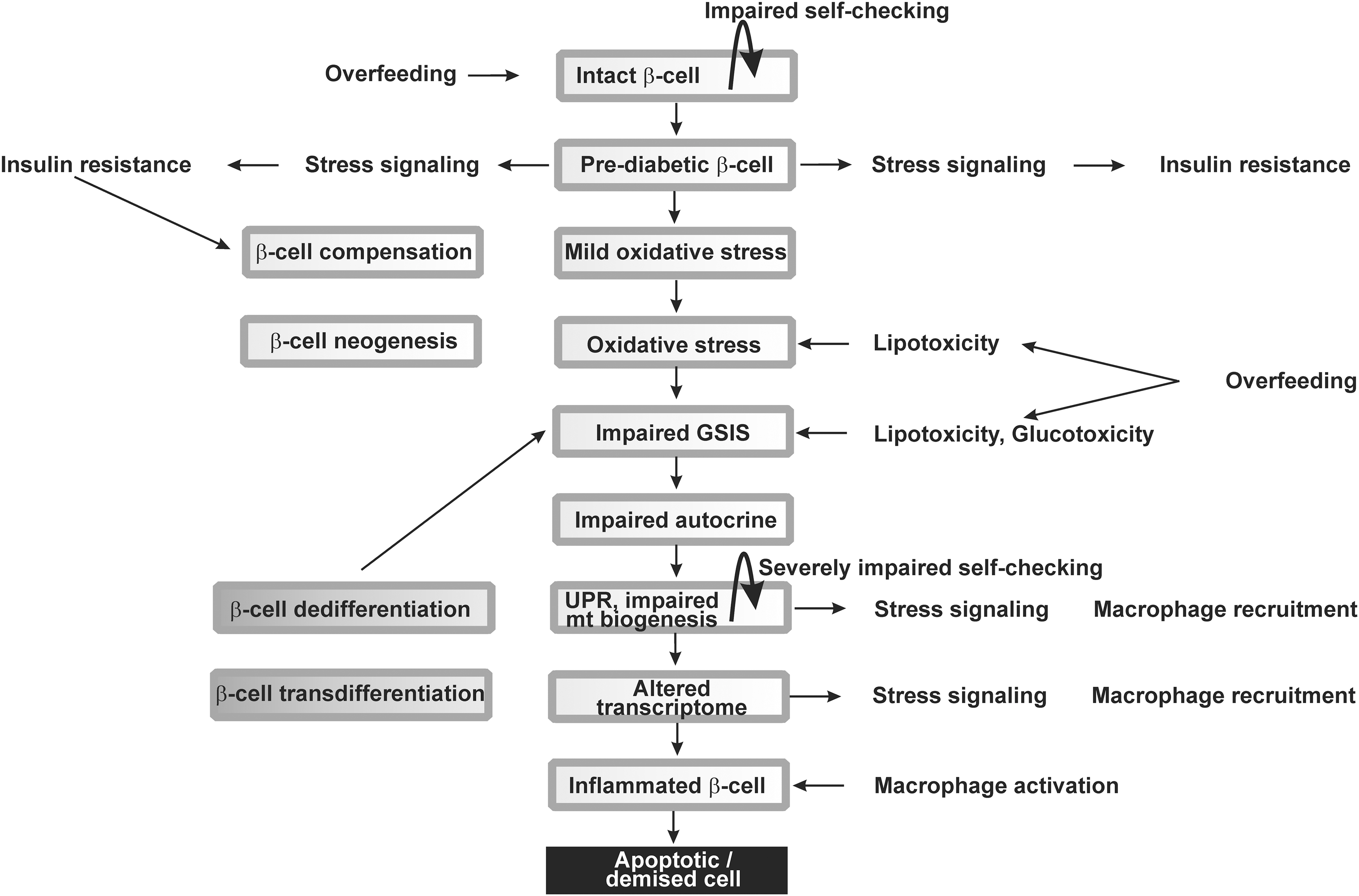
However, the overwhelming progress of such compensation induces further pathogenesis (7). Consequently, the onset of type 2 diabetes is accompanied by the inability of the existing functional β-cells to meet the altered, glucotoxic, metabolic demand (149) (Fig. 1).
In contrast, it is still a matter of debate whether the impaired autocrine factors such as autocrine insulin signaling participate in type 2 diabetes development. Two decades ago, the existence of the autocrine insulin secretion was considered plausible (14, 170), whereas it was later questioned and alternative explanations were elaborated explaining the apparent autocrine effects rather by the central nervous system regulation (241).
However, the impaired biogenesis and self-checking of pancreatic β-cells and islets, and specifically the loss of β-cell differentiated identity, are recently regarded as the most essential factors contributing to the origin of the type 2 diabetes (23, 270, 273), besides the low-grade inflammation in pancreatic islets, promoting β-cell oxidative stress, and cell death (Fig. 1). This is accompanied by a cross talk with the immune system and peripheral tissues. Nevertheless, this cross talk should be minimally substantiated by macrophage recruitment to the islets hypothetically initiated by the impaired “well-being” or self-checking and gradual loss of the β-cell identity. A common feature or outcome of some of these changes is either the oxidative stress or impaired redox signaling, and therefore, these aspects are reviewed here as the common theme.
A consensus exists for type 2 diabetes origin due to the overnutrition (252). The intensive overnutrition elevates the plasma levels of glucose and FAs, which cause metabolic stress at various organs and tissues including pancreatic β-cells. FAs and other metabolites act, for example, via numerous types of G-protein-coupled receptors (GPRs) family members (122). Sustained overnutrition not only progressively impairs the existing β-cells but also stimulates proliferation from their progenitors as a part of the adaptive responses. We should emphasize the compensatory role of β-cells in the initial phases of type 2 diabetes when increased β-cell biogenesis, and hence β-cell mass, elevates insulin gene expression attempting to compensate the insufficient insulin regulation. Despite the extensive description of this compensation, it is out of the scope of our review.
The involved phenomena of the increased antioxidant defense are within our scope. The compensation also involves a fine-tuned regulation to upgrade the endoplasmic reticulum (ER) capacity and quality control (237). Without such enhancement of ER capacity, the unfolded protein response (UPR) is initiated by the excessive unfolded proinsulin accumulation. All these changes include alternations of the identity checking and impaired “well-being” of β-cells.
Resulting tissue-specific metabolic stress thus also causes the release of cytokines and chemokines, which recruit immune cells and initiate the tissue-specific inflammation. Macrophages consist of a spectrum of cells with distinct functional plasticity. Among them, the two limiting classes are classified as M1 and M2 macrophages. One has to judge the M1/M2 macrophage proportion in diabetic pancreatic islets to discuss the molecular mechanism (77). Macrophages are activated by classical way, that is, M1 type are cytotoxic, produce superoxide (O2 •−) by activated NADPH oxidases (NOXs) or HOCl and HSCN by myeloperoxidase into extracellular environment, and produce proinflammatory cytokines.
All these events may induce apoptotic or necrotic type of death in β-cells or may alter their redox regulations and expression of housekeeping and β-cell-identity maintaining genes. Theoretically, this may induce even β-cell dedifferentiation or transdifferentiation. In contrast, M2 macrophages activated by alternative way resolve inflammation, and the pancreatic islets reparation is provided by phagocytosis of apoptotic cells and rebuilding of the extracellular matrix (77). The M1/M2 macrophage proportion in diabetic pancreatic islets has not yet been satisfactorily evaluated. Nevertheless, obviously, the M1 macrophage excess should cause a sufficient deterioration of β-cells.
Undoubtedly, the IR development in peripheral tissues also stems predominantly from the inflammatory responses (107). Surprisingly, skeletal muscle-specific insulin receptor knockout mice exhibited IR but normal fasting glucose, hence it has been evidenced that IR stems from multiorgan deficiency, including that of β-cells (44). Peripheral tissue inflammation is connected with expression of cytokines, chemokines, and adipocytokines in WAT due to transcriptional activation of their expression by oxidative and metabolic stress. At the initial stages, we can consider them as a low-grade inflammation. The ER stress, by means of UPR, is another mechanism that plays a crucial role in the development of IR in adipocytes and peripheral tissues.
Recently, a redox nature of certain β-cell-identity checks and maintenance has been predicted, giving further importance to redox biology of pancreatic β-cells. Receiving the “wrong” identity checking signals might subsequently lead to a release of putative stress signals, the nature of which has yet to be determined. Such β-cell stress signals may hypothetically cause IR in peripheral tissues and may be combined or followed with inflammatory responses in pancreatic islets. Besides inflammatory signals, exosomes can also carry out the stress signals from pancreatic β-cells to peripheral tissues, such as transduced by microRNAs (miRNAs) (150). Such an emerging novel mechanism of IR induction has to be studied further.
Cross talk between stressed β-cells and IR
Vice versa, the IR affects back pancreatic β-cells, by several lines of mechanism. The ultimate consequence is the accelerated islet inflammation. However, metabolic components stemming from nearly permanent hyperinsulinemia and glycemia induce a further turn of additional oxidative stress through the actions of oxidated or proinflammatory lipid species, advanced glycation end (AGE) products, and amyloid peptide. Consequently, the only partial β-cell impairment, at early stages, later contributes to a further turn of pathology spiral leading to irreversible insufficiency in insulin secretion. One can recognize that the oxidative stress can be regarded as a common pathogenic factor in inflammation and metabolic effects of the evolving pathology.
Metabolic, hormonal, and immune-related pathological factors thus “fight back” to pancreatic β-cells and promote their insufficiencies, apoptosis, ER stress, impaired autocrine functions, identity checking signaling of “wrong identity,” and dysregulated biogenesis of remaining progenitor cells. A common factor here is the insufficient INS gene expression and impaired secretion of insulin as responding to normal physiological stimuli.
The major metabolic feedback is mediated by chronically elevated plasma glucose and FA levels, whereas the cytokine signaling at low-grade inflammation stimulates expression of the toll-like receptor 4 (TLR4) (33). All these events promote oxidative stress of β-cells, their ER stress, dysfunctional mitochondria, all of which, in higher progression, lead to β-cell apoptosis (255).
Some human populations were faced to pressure for survival in extremely cold climates, leading to the selection of combination of specific genes or alleles. As a result, these genes predisposed certain today's ethnic groups living in North Polar regions to the decreased prevalence of type 2 diabetes at the simultaneous enhancement of thermogenesis.
Inflammation contribution to type 2 diabetes
Metabolic syndrome origin of diabetic complications
Obesity and lipodystrophy is substantiated by the abnormal mass of WAT, which in predominant cases results in the development of IR. Since WAT serves also as an endocrine organ regulating energy metabolism and insulin sensitivity by FAs and adipokines, its endocrine actions serve important homeostatic role. The adipokines include proinflammatory mediators such as leptin, tumor necrosis factor-α (TNF-α), interleukin (IL)-6, tissue inhibitor of metalloproteinases (TIMP-1), retinol-binding protein 4 (RBP-4), and monocyte chemotactic protein 1 (MCP-1), not only inducing local WAT inflammation but also systemic inflammation (215). The anti-inflammatory adiponectin, which declines in obesity, is also considered as an adipokine. Notably, an imbalance between leptin and adiponectin could also cause IR.
Origin of inflammatory responses
Proinflammatory cytokines contribute to IR development. Among them, interleukin-1β (IL-1β), IL-6, TNF-α, and the C-reactive protein (CRP) are taken as markers; however, the contribution of various other chemokines and adipocytokines may provide important responses. The master proinflammatory mediator is IL-1β since it induces production of other cytokines and chemokines. IL-1β binds to the interleukin-1 receptor type 1 (IL-1R1) and reduces the expression of insulin receptor substrate-1 (IRS-1) by the extracellular-related kinase- (ERK) dependent transcriptional regulation and also at post-transcriptional level (134). In this way, by reducing the ability to utilize insulin, the IR is developed in peripheral tissues.
IL-6 is known to be increased in plasma in aging. IL-6 activates the suppressor of cytokine signaling (SOCS) proteins blocking the signal transducer and activator of transcription 5B (STAT5B), a transcription factor of insulin receptor. As a result, IL-6 contributes to the IR development. Moreover, IL-6 suppresses the lipoprotein lipase, which leads to the increasing plasma levels of triglycerides (159). TNF-α, produced also by adipocytes, activates cascades of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and c-Jun N-terminal kinase (JNK), phosphorylating serine 307 in insulin receptor substrate IRS-1, thus contributing to IR (2).
Chemokines include, for example, MCP-1, MCP-2, MCP-3, MCP-4, CCL2, MIP-1α, and MIP-1β (224). Their involvement in IR development is nicely illustrated by the inability of high-fat diet to induce IR in MCP-1- and CCL2-deficient mice (152, 296). Chemokine receptors such as CCR2 and CCR5 in WAT are important for IR development. Besides inducing expression of other inflammatory genes, the activation of CCR2 inhibits insulin-dependent glucose uptake into adipocytes.
Pancreatic islet inflammation involving β-cells
Pancreatic islet inflammation is caused by the interaction of M1-like macrophages and β-cells producing inflammatory cytokines in vicious cycle. This, of course, predominates in type 1 diabetes. The number of macrophages increases in Langerhans islets during type 2 diabetes, thus elevating the production of proinflammatory cytokines (79). Macrophages also produce chemokines, which are essential for the recruitment of innate immune cells into pancreas along with promoting changes in signaling of resident leucocytes (59). It has been documented that M1 type of macrophages infiltration into islets of pancreas can induce a significant loss of β-cells (169).
Such an example of NLRP3 inflammasome activation (the third isoform of NLRP [NOD-like receptor containing pyrin domain]) in infiltrating macrophages is endocannabinoids, which contribute to lowering of β-cell mass in type 2 diabetes (144, 145). Classical activation of M1 macrophages induces inflammation-dependent superoxide and HOCl or HSCN and contributes to already existing oxidated stress in β-cells. Produced proinflammatory cytokines, such as IL-1β, clearly enhance the number of apoptotic pancreatic β-cells, manifested as cytochrome c release from mitochondria followed by downstream activation of caspases (56). Increased reactive oxygen species (ROS) production and subsequent redox regulations were reportedly shown as mediators of the cytokine-induced cell death since overexpression of antioxidant enzymes prevented this process (16). Cytokines were also found to induce ER stress (42).
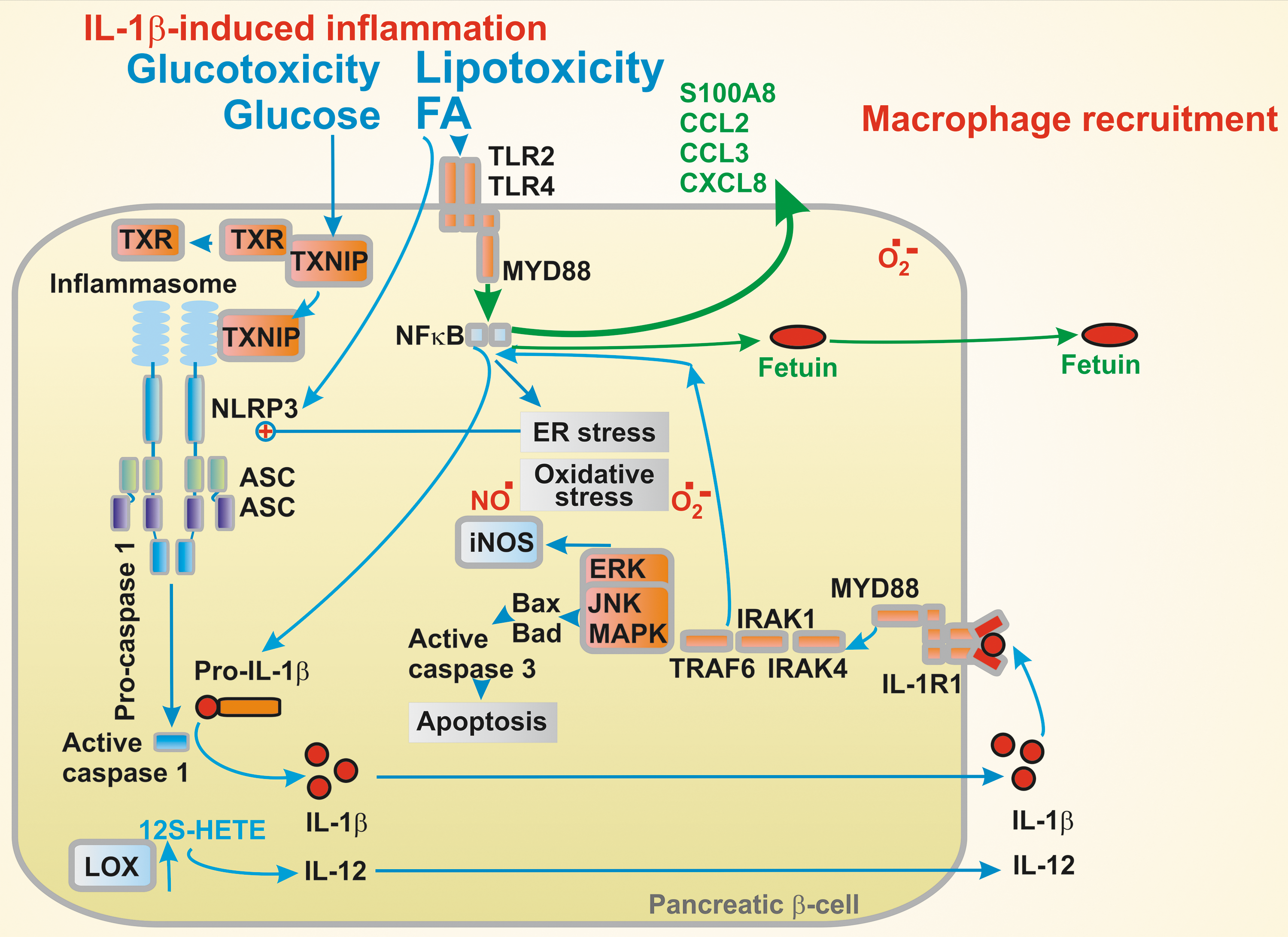
IL-1β induces inflammation in islets due to glucolipotoxicity, that is, prolonged exposure to FAs, glucose, or other nutrients. FAs seem to play the role of a main regulator of inflammation in β-cells. In β-cells, FAs act via GPRs, via the TLR2,4/MYD88/NF-κB pathway (where MYD88 [myeloid differentiation primary response 88 protein] is involved), and via the NLRP3 inflammasome/apoptosis-associated speck-like protein containing CARD (ASC)/caspase 1 pathway to increase IL-1β expression (312). Here, ASC is the ASC.
Glucose stimulates IL-1β expression by the thioredoxin-interacting protein (TXNIP) upstream of the NLRP3 inflammasome. Resulting IL-1β exerts an autocrine function by the activation of its receptor IL-1RI recruiting MYD88 and accelerating its own expression, whereas the activation of MYD88/NF-κB pathway stimulates the expression of CCL2, CCL3, and CXCL8 chemokines. The chemokines recruit macrophages also producing IL-1β (78). In later diabetes stages, islet amyloid stimulates inflammasome in the islet-recruited macrophages. Excessive palmitate was also found to mediate TXNIP downregulation (225).
Excess of saturated FAs (e.g., palmitate) induce chemokine and cytokine expression (CXCL1, CCL3, IL-6, and IL-8) within Langerhans islets in vitro (56, 123). Human islet donors with type 2 diabetes were shown to induce transcription of cytokine and chemokine (193, 274). Moreover, analyses of laser microdissected β-cells from type 2 diabetic donors have shown induced expression of cytokines IL-1β, IL-8, and IL-11, and chemokines CCL2, CCL11, and CCL13, plus downregulation of IL-1α cytokine (195). Notably, nondiabetic control samples have not uncovered IL-1β presence by in situ hybridization (34).
The S100 calcium-binding protein A8 (S100A8), a member of the damage-associated molecular pattern molecules, has been implicated in β-cell inflammation (128). Its expression was increased by TLR4 signaling in pancreatic islets induced by palmitate in the presence of a high glucose during their co-culturing with unstimulated peritoneal macrophages. S100A8 was suggested to mediate islet–macrophages interaction that results in β-cell apoptosis. TLR4 pathway inhibition avoided this interaction. Also, Fetuin-A secretion induced by palmitate acts via the NF-κB/MIF inflammatory pathway (MIF is the macrophage migration inhibitory factor). Fetuin-A is an α-2-HS-glycoprotein excreted by the liver. In pancreatic islets, Fetuin-A secretion sets “an inflammatory environment” leading to β-cell dysfunction (209).
Lipotoxicity can be ensured by major lipoxygenase in human pancreatic islets, such as by 12-LO-producing 12-S hydroxyeicosatetraenoic acid (12-S HETE) from arachidonic acid in β-cells. Elevated 12-S-HETE was found in islets and WAT of rodent diabetes models and diabetic patients. Proinflammatory 12-S-HETE induces cytokine production, such as IL-12, and activates JNK and oxidative stress pathways by p38 mitogen-activated protein kinase (MAPK), which induces the NOX1 activity. Consequently, the 12-S-HETE effects lead to the decreased β-cell viability and impaired insulin secretion (125).
Oxidative Stress and Antioxidant Protection in Pancreatic β-Cell
Damages to perfect glucose sensor
Glucose sensing in pancreatic β-cells
The current consensus on the glucose-sensing function of pancreatic β-cells lies in the increasing oxidative phosphorylation (OXPHOS) rate with the elevated glucose metabolism of incoming glucose (13, 164, 231, 246, 299). The resulting elevated ATP/ADP ratio closes the plasma membrane ATP-sensitive K+ channel (KATP), which then initiates orchestra of other channel events substantiating plasma membrane depolarization (72). This activates voltage-gated L-type Ca2+channels (CaL) leading to Ca2+ entry and hence Ca2+-dependent exocytosis of the insulin-containing secretory granules.
A large facilitation of insulin secretion exists due to the intestinal incretin hormone action. This includes GLP-1 stimulation via the GLP-1 receptor, Gs protein, adenylate kinase activation, and cAMP-dependent activation of the protein kinase A (PKA), and the exchange protein directly activated by cAMP 2 (EPAC2) (97). Both downstream pathways promote Ca2+-dependent exocytosis of insulin granules independent of KATP (Fig. 2). A similar stimulation exists with GIP (198).
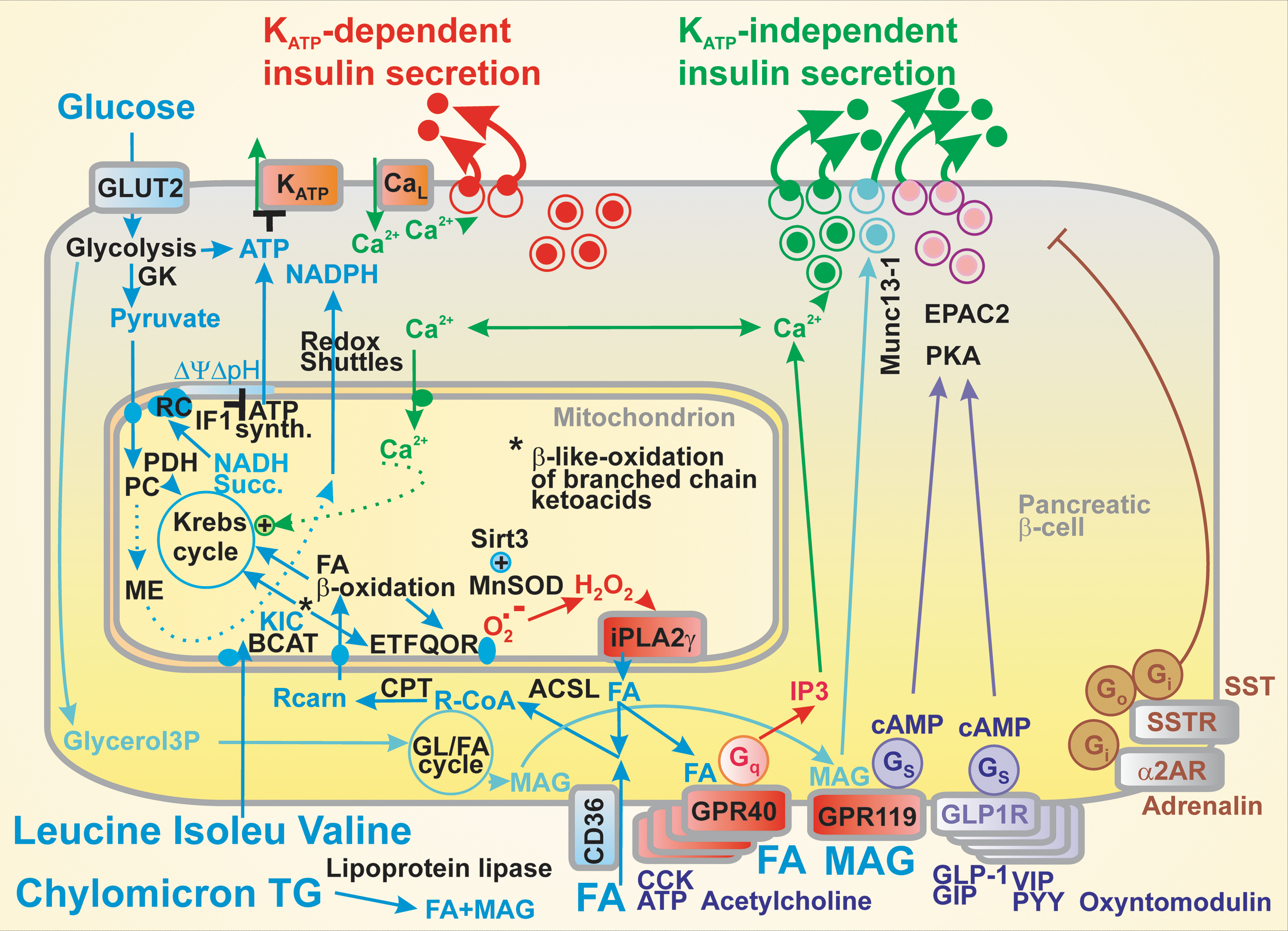
In addition, acetylcholine stimulates insulin secretion via muscarinic M3 receptors, extracellular ATP stimulates via purinoreceptors (246), and long-chain FAs via metabotropic receptors GPR40 (98, 113, 122, 129, 194). Activation of all these receptors is followed by the Gq pathway promoting the inositol trisphosphate- (IP3) mediated Ca2+ release, again independent of KATP. Other metabolites, such as aromatic amino acids, act via distinct GPRs (122). Also, the glycerol/FA cycle relevant for 2-monoacyl glycerol-mediated insulin secretion acts independent of KATP via Munc13-1 facilitation of exocytosis (231). Notably, insulin release can be inhibited by acting via the inhibitory Gi proteins upon the activation of α2-adrenergic receptors or somatostatin receptors via the inhibitory Gi and Go proteins (246).
Nature's engineering of the glucose sensor consists of several key points, without which the sensing mechanism would never work. At first, glucose transporter GLUT2 (or probably GLUT1 in humans) is constitutively active and is not dependent on the insulin receptor signaling cascade as, for example, in peripheral tissues. This allows glucose concentration in the β-cell cytosol being proportional to bloodstream glucose (151).
Second, the suppressed expression of lactate dehydrogenase (belonging to the so-called β-cell-disallowed genes) and the pattern of pyruvate dehydrogenase kinase (Pdk) genes are surely responsible for the fact that all glucose is metabolized by OXPHOS in β-cells.
Thus, β-cell PDK1 (pyruvate dehydrogenase kinase 1) and PDK3 are “constitutively blocked” (5), and PDK2 is “inefficient” so that it does not phosphorylate the E1α subunit of pyruvate dehydrogenase (PDH), hence does not inhibit its activity. At low basal glucose, PDH shows high activity, whereas at increased glucose, PDH is inhibited only by 22%.
The third aspect includes the fact that hexokinase IV (glucokinase) in β-cells is not inhibited by glucose-6-phosphate like other hexokinase isoforms (226). The missing feedback inhibition of glycolysis directly connects glycolysis to pyruvate. Consequently, the β-cell respiration and OXPHOS rates, leading to increased ATP/ADP ratio, are dependent on glucose availability, whereas in most other cell types, cell demand dictates respiration/metabolism rates and the ATP/ADP ratio.
Fourth, matching of physiological postprandial glucose concentrations with the sensitivity range of the glucose sensor (which pancreatic β-cells indeed represent) is ensured by the inhibitory factor IF1 of the mitochondrial ATP synthase (69). This aspect was derived from the IF1 ablation experiments, which allowed switching on the sensor, that is, elevation of respiration and OXPHOS, even at zero glucose concentration and shifted the insulin release dose–response to the range of 0–2 mM glucose. This demonstrated that IF1 ensures that mitochondrial respiration and resulting ATP synthesis and insulin release starts to increase sharply at ∼3 mM glucose levels due to a weak inhibition of the ATP synthase (and hence OXPHOS) by IF1.
Consequently, glucose metabolism in β-cells is finely adjusted to the blood glucose levels (201). Also, the above-mentioned complete repression of high-affinity type I–III hexokinases contributes to the prevention of insulin release at low plasma glucose concentrations (254).
At starvation with approximately 3–5 mM glucose levels, β-cell respiration as well as the intensity of ATP synthesis are low (263). The increase in glucose concentrations raises OXPHOS up to saturated concentration at approximately 12–15 mM glucose, when cells show maximum OXPHOS, maximum respiration and mitochondrial inner membrane potential ΔΨm.
Another peculiar aspect of the pancreatic β-cell glucose sensor dose–response, that is, glucose dependence of insulin response lies in its steepness, higher than would correspond to a Hill coefficient of 1. We have revealed that upon glucose-stimulated insulin secretion (GSIS), mitochondrial cristae become narrowed at glucose intake in model β-cells and, consequently, proton coupling localized into smaller intracristal space volume enables more efficient OXPHOS, thus contributing to the steepness of the glucose sensor (148). Overall, β-cells maintain a relatively high [ATP]/[ADP] value even in low glucose and glucose metabolism dramatically decreases free ADP while only modestly increases ATP (88). Since a high [ATP]/[ADP] ratio exists even at low glucose levels, the total adenine nucleotide content is unchanged during a glucose-induced elevation.
After a meal intake, blood glucose rises slowly and insulin release appears to be monophasic. Experimentally after a bolus of glucose, insulin release proceeds in two phases (287), which are also observed in perifusion experiments of the isolated pancreatic islets. Earlier considerations regarded the first phase as purely KATP-dependent, whereas the second phase was considered to occur due to the KATP-independent mechanism. However, this concept turned out to be incorrect since both classes of mechanisms contribute to both phases (13, 114, 231, 246).
Actually, Ca2+ elevations during GSIS have two phases, a sharp increase after glucose intake, which is followed by the prolonged oscillations (4). The first GSIS phase was also considered to be executed by a readily releasable pool of granules docked to the plasma membrane, whereas the second phase by a reserve pool of granules (13, 231, 246). Note that the insulin-degrading enzyme, an endopeptidase, was found in β-cells and α-cells (83), degrading insulin and glucagon. This should be the subject of further studies, whether it may also contribute to the phases, beside the insulin receptor internalization (see the Autocrine Insulin section).
Relationships between ROS homeostasis and glucose sensor
Redox homeostasis in cells and particularly in mitochondria and peroxisomes is physiologically regulated in multiple hierarchies and in pancreatic β-cells contributes to the mechanism(s) of insulin secretion. When these physiological relationships are dysregulated, an internal oxidative stress in pancreatic β-cells arises, which inhibits insulin secretion (178, 216). At the organism and islet levels, inflammation amends an additional component of the oxidative stress in β-cells.
Under physiological situation, GSIS in pancreatic β-cells is facilitated by the increase in cytosolic NADPH (13, 141, 143, 219, 231, 244, 246) or even by ROS (176, 228), such as resulting from the addition of mono-oleoyl-glycerol (247). The three major metabolic shuttles virtually export the reducing equivalents of NADH from the mitochondrial matrix to the cytosol during GSIS (141). They were named as the pyruvate/malate, pyruvate/citrate, and pyruvate/isocitrate shuttle (143). The two cytosolic enzymes produce NADPH, the cytosolic malic enzyme 1 (ME1) within the first two shuttles, whereas the isocitrate dehydrogenase 1 (IDH1) acts within the third one (141). As a result, the GSIS is facilitated by the NADPH increase, a mechanism that has yet to be revealed (Fig. 3).
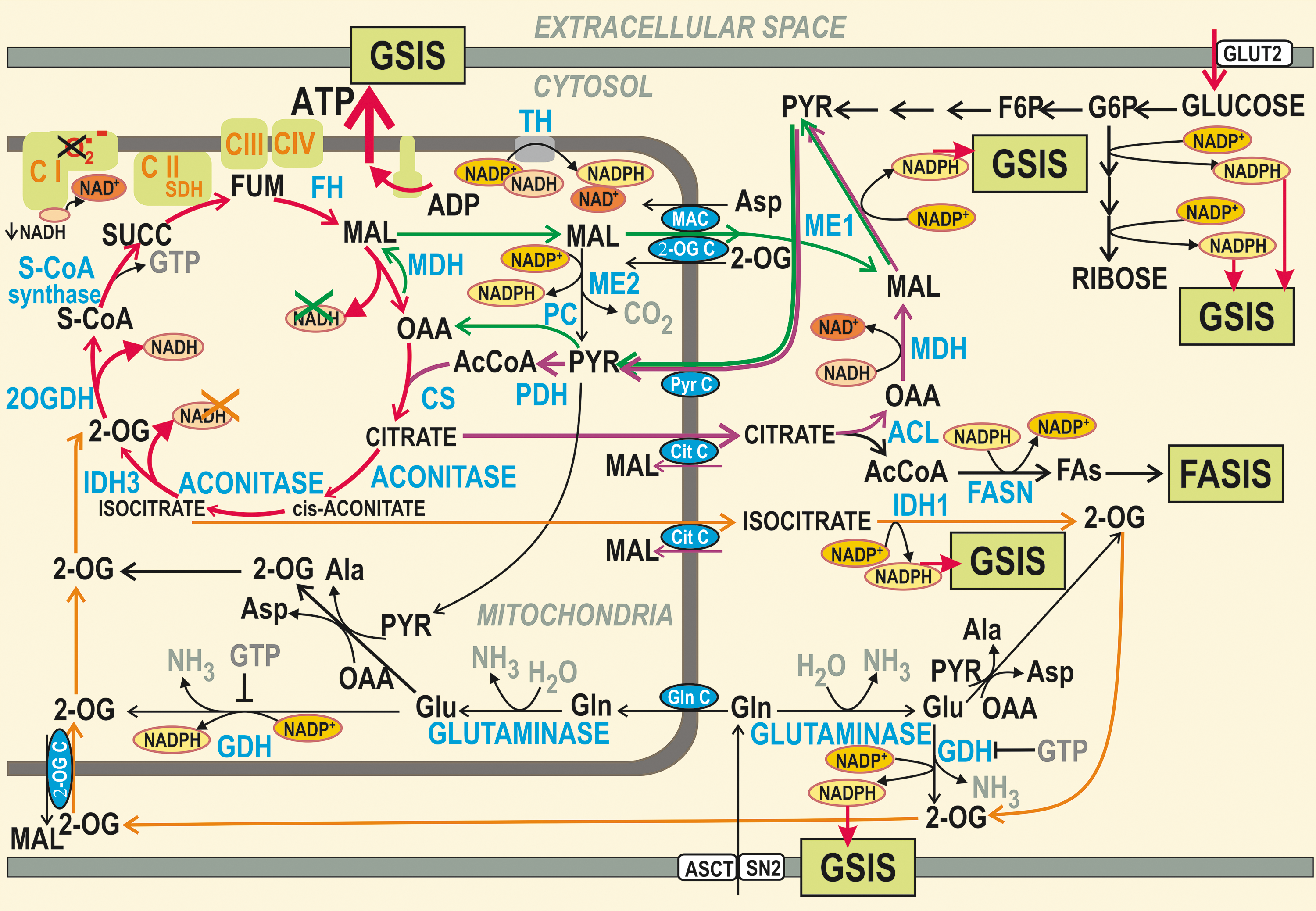
These redox shuttles also affect ROS homeostasis. For each molecule of malate or citrate/isocitrate exported from the mitochondrial matrix by the respective shuttle, one molecule of NADH is missing for the respiratory chain Complex II since the malate dehydrogenase (MDH) or IDH3, otherwise producing NADH, cannot use their substrates. Malate can be also cycled back after being converted to pyruvate by mitochondrial ME2. Consequently, the matrix NADH formation is predicted to be lower and NAD+ is assumed to accumulate in the matrix upon GSIS, specifically when compared with the situation when these shuttles are slowed down or blocked.
At elevated glucose metabolism, these shuttles exporting redox equivalents should hypothetically decrease the substrate pressure NADH/NAD+ in the mitochondrial matrix, which would lead to the decreased superoxide formation in the flavin site IF of Complex I. Overall, this hypothetical mechanism would contribute to the diminished superoxide formation upon GSIS and should effectively prevent the ROS increase upon GSIS within mitochondria (Fig. 3).
Yet, previously, it has been reported that mitochondrial superoxide formation increases upon GSIS (32, 176, 228). In contrast, it has been found that low glucose levels induce ROS production in pancreatic β-cells (118, 251) and that ROS were suppressed by high glucose (196). The glucose decrease was also associated with the increased mitochondrial H2O2 (243).
Also, channel machinery has been related to ROS. Insulin granule exocytosis has been reported to be directly induced by H2O2, independent of Ca2+ (189). Plasma membrane depolarization may be hypothetically affected by direct or indirect inhibition of K+ ATP channel by H2O2 (305) or by direct or indirect inhibition of repolarizing K+ channels, such as KV (11, 87, 181, 190, 202, 303) or activation of TPRM2 (transient receptor potential melastatin 2) depolarizing channel (101). Indirect blockage would involve ROS-activated signaling pathways leading to KATP or KV closure. Also, the Ca2+-mobilizing messenger nicotinic adenine nucleotide diphosphate (NAADP) acts via endosomal and lysosomal two-pore channels (TPC) (12).
Physiological antioxidant mechanism in pancreatic β-cells
Next, let us discuss about the mechanisms that can be manifested in maintaining the physiological ROS homeostasis and their impaired shift to oxidative stress under pathological conditions. Nature engineered a plethora of native mechanisms to suppress the superoxide sources within the mitochondrial respiratory chain.
Recently, one of these mechanisms was found to be executed by the synergy of mitochondrial-specific calcium-independent phospholipase A2, isoform γ (iPLA2γ), and uncoupling protein UCP2 (136). Uncoupling cannot suppress superoxide formation when arising from the retarded electron transport pathway and proton pumps by mutations of subunits encoded by mitochondrial DNA (mtDNA) or when superoxide is formed due to the retarded cytochrome c shuttling (229). Otherwise, uncoupling by UCPs (UCP1–UCP5) is quite an effective way to suppress mitochondrial superoxide formation (138, 180).
Sole respiration and OXPHOS may increase mitochondrial formation of superoxide when a certain redox reaction is retarded and the input metabolic (e.g., NADH) flux keeps a metabolic pressure. Excessive superoxide is subsequently dismuted by the matrix-located SOD2/MnSOD. At sufficient H2O2 degradation, MnSOD thus has profound antioxidant effects. This substantiates the beneficial effects of Mn supplementation (174). MnSOD can be prooxidant when downstream degradation of H2O2 does not match its formation.
However, MnSOD can be inactivated by acetylation that is, in turn, rescued by the sirtuin-3-mediated deacetylation (67, 275, 276). Sirtuins in general promote an OXPHOS and OXPHOS-supporting pathways, affecting the involved proteins. Acetylation of numerous mitochondrial proteins inhibits their function, whereas their deacetylation by certain sirtuins abolishes such inhibition. Among mitochondrial sirtuins, sirtuin 3 is an NAD+-dependent deacetylase essential for OXPHOS (50, 160, 267, 313), whereas sirtuin 5 is a potent desuccinylase (309). Among others, subunits of the ATP synthase, OPA1, or MnSOD are regulated by the inhibitory acetylation and physiologically activated by sirtuin 3 (55), whereas enzymes of FA β-oxidation and ketogenesis are activated by desuccinylation mediated by sirtuin 5 (238). Mitochondrial sirtuin 4 removes three acyl moieties from lysine residues (9).
Numerous cases were found when sirtuin function has been linked to insulin secretion. Also, typically, sirtuin 3 expression is reduced upon high-fat diet suggesting the involvement of the oxidative stress arising from partly inactive MnSOD (313).
When H2O2 is formed, it diffuses out of mitochondria or from peroxisomes into the cytosol. Organelles and cytosolic milieu contain redox buffers and redox relaying systems providing redox signal propagation, besides a simple maintenance of the redox homeostasis. Thus, disulfide reductases, thioredoxins (TRXs), glutaredoxins (GRXs), peroxiredoxins (PRXs), and glutamate-cysteine ligase are also capable to propagate the redox signals to the targets.
TRX are disulfide reductases of protein sulfhydryl groups, maintaining proteins in the reduced state (17). The oxidized TRX are regenerated by the TRX reductase using NADPH for electrons. Similarly, GRX reductase-2 reduces H2O2 or hydroperoxy-FA lipid chains to water or hydroxy FA lipid chains, respectively, on the expense of conversion of reduced glutathione (GSH) to oxidized glutathione (GSSG) (240). Glutathione is regenerated by glutathione reductase.
PRX, as thiol peroxide reductases, use TRX or other thiol-containing proteins to clear H2O2 or lipid peroxides (311). The catalytic cysteine sulfhydryl group of PRXs is selectively oxidized by H2O2 to either sulfenic acid or disulfide intermediates. At the TRX shortage, PRX is inactivated to PRX-SO2, which can be reversed by sulfiredoxins, at the expense of ATP, yielding sulfenylated PRX (PRX-SOH) (302). PRX are classified into two major enzyme classes, differing by the mechanism of recycling of the sulfenic acid back to a thiol. 2-Cys PRXs are reduced by thiols such as of TRX, TRX-like proteins, or in certain cases by glutathione. The 1-Cys class of PRXs is reduced by ascorbic acid or glutathione in the presence of GST-π (206). Note that PRX3 is localized to mitochondria, where besides redox buffer function exerts its role in the redox H2O2 signaling.
Oxidative damage versus antioxidant defense in pancreatic β-cells
Oxidative stress is induced when redox homeostasis diverts to excessive ROS production, which exceeds the antioxidant mechanisms. A general oxidative stress in a cell arises when the function of redox buffers and antioxidant enzymes is diminished so that they no longer possess the ability to detoxify the produced ROS. A permanent character and the level of ROS amounts distinguish this stress from the physiological redox signaling. Oxidative stress possesses not only the direct pathological consequences, by increased appearance of oxidatively modified biological constituents, but also the induction novel cellular responses, that is, programmed cell death, such as apoptosis. Usually, these phenomena occur in parallel, after accumulation of the oxidative stress reached a certain threshold.
Oxidative stress can oxidatively modify DNA with more vulnerable mtDNA, lipids (by nonenzymatic lipid peroxidation), and proteins, such as carbonylation. Although cell owes physiological mechanisms for clearance of these oxidized constituents (repair mechanisms for DNA, glutathione peroxidase [GPX], and normal protein turnover), when their formation exceeds clearance capacity, some pathology can occur. Yet imbalance in identity check mechanisms in a cell can induce such stress even at mild oxidative stress. In this case, an impairment of normal autophagy and notably autophagic mechanisms dealing with mitochondria leads to the accumulation of products that were supposed to be cleared with serious consequences for the cell (157, 293).
A hallmark of β-cell ROS homeostasis is that redox buffers and antioxidant enzymes are rather weak when compared with other cell types (177), and consequently, the ROS homeostasis can be easily shifted to the oxidative stress conditions. Catalase, GPX, and superoxide dismutase (SOD1 or CuZnSOD) represent three of most important intracellular antioxidant enzymes of a primary defense system. However, pancreatic islets contain only 1% catalase, 2% GPX, and 29% SOD1 activities compared with the liver (100, 179, 203, 280). This makes them more susceptible to an oxidative insult. Human β-cells were suggested to be less prone to oxidative stress than rodent β-cells, possibly because they show enhanced catalase and SOD activity (297). But GPX activity is poorly detectable in human β-cells (281). Moreover, β-cells possess weak repair machinery for oxidatively damaged DNA (204). Besides small antioxidant molecules, that is, vitamin E (α-tocopherol), ascorbate, or uric acid, glutathione provides an important antioxidant protection of the β-cells against oxidative damage (167, 211). Glutathione, in millimolar concentrations, is kept in the reduced state (GSH) by glutathione reductase (272). GSH transfers its reducing equivalents to ascorbate, GPX, and GRX.
In turn, β-cells are rich in disulfide reductase-based antioxidant defenses, such as GRX and TRX (131). They were shown to be localized at periplasma membrane cytosol, and glutaredoxin GRX1 has been found to be implicated in the modulation of Ca2+-dependent insulin exocytosis [143]. NADPH was shown to stimulate the exocytotic machinery, which correlates with ∼30% inhibition in whole-cell Ca2+ currents. When GRX1 was silenced, NADPH was not amplifier of insulin release but still inhibited Ca2+ currents (240).
Although oxidative stress and metabolic dysregulation are considered as two of the most important pathogenic factors of impaired insulin secretion, the latter is usually regarded as dysregulated relationships between oxidation of glucose and FAs. Yet, there is still no consensus on what particular oxidants are responsive for oxidative stress and what exactly causes such a metabolic dysregulation. One type of the frequently overlooked products of oxidative stress is isoprostanes, which are prostaglandin-like compounds produced by nonenzymatic peroxidation of arachidonic acid (such as initiated by the hydroperoxyl radical, a conjugated acid of superoxide), together with a class of extremely reactive molecules that are generated as products of the isoprostane pathway (53). The levulgandins and isolevuglandins (currently referred to as isoketals) have been found to form covalent adducts with proteins and induce protein–protein and DNA–protein cross-links and thus are able to modify key biomolecules that are critical to normal cellular function and replication (249a). The levulgandins and isolevuglandins can accumulate over the lifetimes of proteins, and isolevuglandin–protein adducts represent a convenient dosimeter of oxidative stress central to many disease states (249a).
Unfortunately, despite increasing number of studies, which relate products of lipid peroxidation to the etiology of diabetes mellitus, the specific role of the isoprostane pathway of autoxidation of polyunsaturated fatty acids (PUFAs) in the regulation of insulin secretion has not been identified.
Kelch-like ECH-associated protein 1–nuclear factor erythroid 2-related factor system
Cells possess a protection against oxidative, electrophilic, or environmental stresses by activating genes via antioxidant-responsive elements facilitating the defense. Such a protective system includes the nuclear factor erythroid 2-related factor (Nrf2) together with a sensor Kelch-like ECH-associated protein 1 (Keap1). Nrf2 belongs to a family of transcriptional factors containing a unique basic-leucine-zipper motif, the cap-n-collar family [reviewed in Uruno et al. (284)]. Keap1 plays a substrate adaptor function and subjects Nrf2 to ubiquitination and degradation and thus suppress the transcriptional activity of Nrf2 under conditions of no oxidative stress. When cells are exposed to increased prooxidant conditions and oxidative stress, the cysteine residues of Keap1 are modified and Keap1 loses its ability to ubiquitinate Nrf2. Nrf2 then accumulates within the cell and induces the expression of target genes.
Nrf2-knockout islets and Nrf2-knockdown MIN6 cells were used to study the protective role of Nrf2 against acute oxidative stress-induced pancreatic β-cell damage (89). Both cells and islets expressed markedly lower amount of antioxidant enzymes in response to a variety of oxidative stressors, whereas pretreatment of β-cells with Nrf2 activators induced protection of the cells from H2O2-induced cell damage. Because it was shown that oxidants can amplify GSIS and persistent activation of Nrf2 decreases glucose-triggered ROS signaling insulin release, this study points out on distinct roles that Nrf2 may play in pancreatic β-cell dysfunction that accompanies different stages of diabetes.
The association of Nrf2 and inflammatory cytokines was found by a cross-sectional study involving healthy control subjects and subjects with newly diagnosed diabetes mellitus. Thus, while oxidative stress markers were significantly increased, the Nrf2 activity and its downstream targets were decreased in peripheral blood mononuclear cells of diabetic subjects in comparison with control. The circulatory increased levels of Nrf2 showed a positive correlation with anti-inflammatory Th2 type cytokines and negative correlation with proinflammatory Th1 type cytokines. Furthermore, the activation of Nrf2 restored the cytokine stress-induced impaired insulin secretion in pancreatic β-cells (259).
Interestingly, Nrf2 nuclear translocation was elevated in embryonic development in UCP2 knockout mice, which revealed increased phosphorylation of protein kinase B (Akt) in their pancreata, suggesting that UCP2 controls pancreas development through ROS-Akt signaling (41). UCP2 was shown to mediate its antioxidant function by a synergy with mitochondrial phospholipase iPLA2γ (133), which leads to the prevention of lipotoxic oxidative stress in pancreatic β-cells (136), indicating that the oxidative stress observed in the pancreas by the ablation of Ucp2 gene is caused by the lack of iPLA2γ-UCP2 antioxidant synergy.
In addition, Nrf2 plays a role in the molecular mechanism of antioxidative effect induced by GLP-1. Results obtained with rat insulinoma cell line INS-1E suggest that exendin-4, a GLP-1 receptor agonist, activates and stabilizes Nrf2 through PKCδ activation, thus leading to the elevation of antioxidant gene expression, which then improves β-cell function during oxidative stress (161). GLP-1 activation of Nrf2 pathway with subsequent increase of antioxidant capacity signals via PKA-dependent ERK pathway, which induces Nrf2 nuclear translocation (84).
A chemokine C-X-C ligand 12 enhances survival and regeneration of pancreatic β-cells (217) and was shown to exert an antioxidant protection also via Nrf2 nuclear translocation through the activation of p38, ERK, and Akt kinases downstream of the chemokine receptor, thus contributing to the functional resistance of β-cells to prooxidant-induced stress (68).
Conditions for elevated ROS in pancreatic β-cells
Let us recapitulate the major events elevating ROS in β-cells. Mitochondria are traditionally reported as a profound source of oxidative stress in β-cell pathology (216). This can be deduced also from the effects of mitochondrial matrix-targeted antioxidants (126, 182).
As mentioned above, mitochondria may exhibit oxidative stress even in fasting conditions with low glucose levels (251). Mitochondrial respiration, especially when retarded at certain sites, provides the most upstream ROS, superoxide and its conjugated acid—hydroperoxyl radical, HO2 • (pKa 4.9) (137, 229, 268, 286). Superoxide is converted to H2O2 by SOD, cytosolic and intracristal space SOD1/ZnCuSOD (221, 291), and matrix SOD2/MnSOD. H2O2 may be converted by Fenton reaction in the presence of iron to the most reactive species—to the hydroxyl radical, •OH, acting locally. The hydroperoxyl radical, HO2 •, and hydroxyl radical, •OH, are capable to initiate nonenzymatic lipid peroxidation, a further amplifier of ROS (197).
Resulting hydroperoxy FAs, hydroxy FAs (converted from hydroperoxy FAs by GPX4), and numerous other derivatives of PUFAs coming from enzymatic lipid peroxidation are cleaved from oxidized lipids by phospholipases A2 (261). More frequently, the shorter lipid peroxidation products, for example, arachidonic acid metabolites act as proinflammatory, whereas resolvins coming from C22 ω-3 polyunsaturated fatty acids (PUFAs) act as anti-inflammatory (256).
Reports emphasized the function of succinate dehydrogenase as another ROS source (76). Other superoxide source is related to mitochondrial FA β-oxidation, when superoxide is produced at the EF site of electron-transferring flavoprotein:Q oxidoreductase (ETF:QOR) system (36). An ultimate MnSOD reaction in the matrix dismutes superoxide to H2O2, which activates iPLA2γ (136). Under conditions when concomitant activation of antioxidant synergy of UCP2 and iPLA2γ does not protect against these elevated, lipotoxic oxidative stress may arise (see the FA Sensing in Pancreatic β-Cells—Intact Versus Impaired section).
Cytosolic ROS are produced in pancreatic β-cells in association with the elevation of Ca2+ following the activation of protein kinase C (PKC), which promotes the assembly of and superoxide formation by NOXs (307a, 207).
Consequences of oxidative stress for glucose sensor
Let us discuss the consequences of oxidative stress for glucose sensing in pancreatic β-cells except for the stress given by FAs, which is described below in the FA Sensing in Pancreatic β-Cells—Intact Versus Impaired section (289). At first, the oxidative stress can affect mitochondria with the ultimate consequences manifested in diminished ATP synthesis by OXPHOS and thereby impairment of the KATP-dependent glucose sensing (85, 157, 191) (Fig. 4).
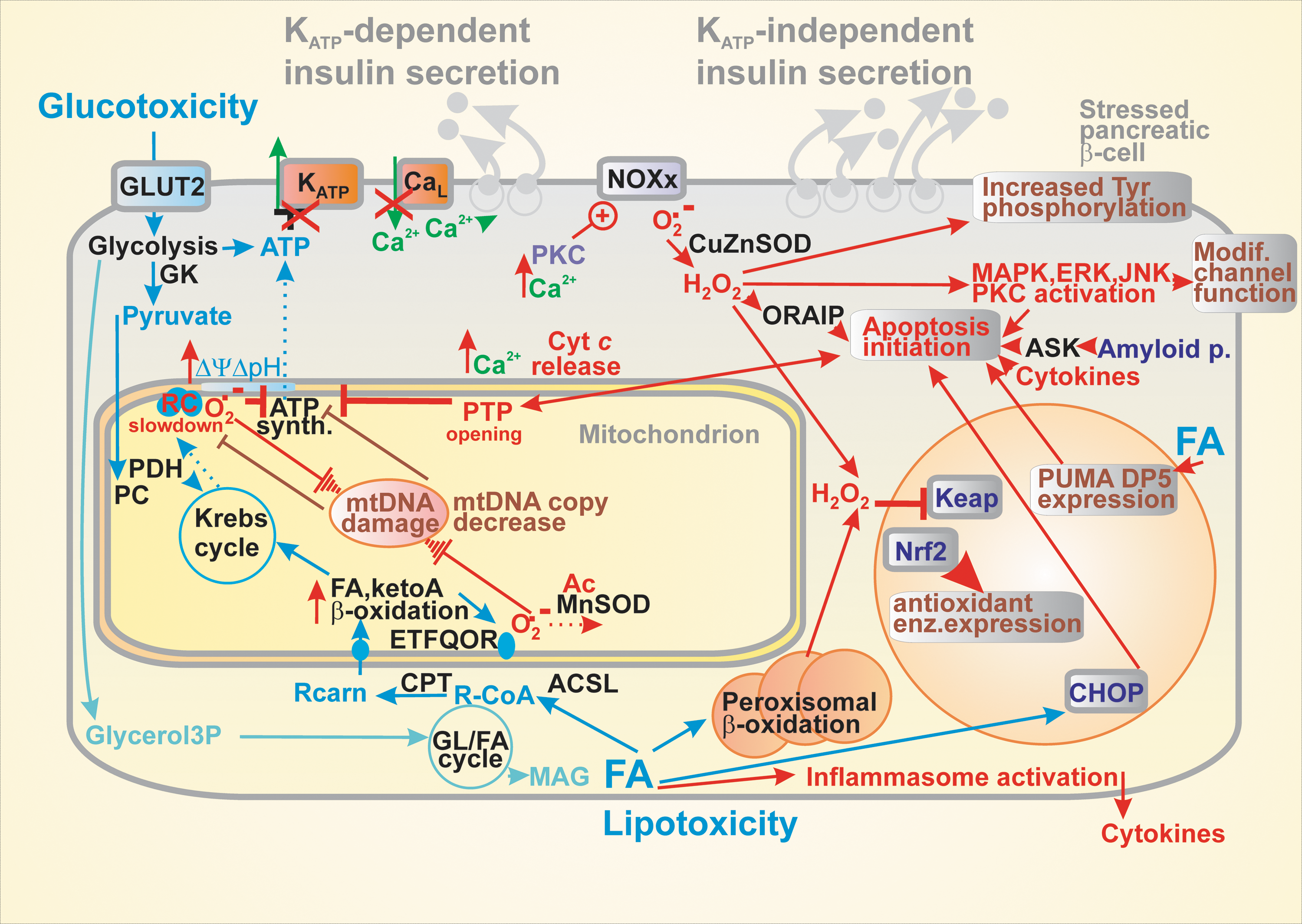
Mitochondrial dysfunction may result in induction of compensatory mechanisms such as the mitochondrial UPR, mitophagy, and autophagy. These systems, however, can be overcome by concomitant apoptosis and consequently reducing mass of β-cells and hence impairing insulin secretion and initiating the diabetic pathology. Nevertheless, the importance of autophagy can be illustrated by the effect of intermittent fasting, which preserves organelle quality via the autophagy–lysosome pathway and increases β-cell survival by stimulation of regeneration in obesity-induced diabetes (185). Thyroid hormones are also essential for β-cell maintenance as reflected by GSIS suppression, decrease in OXPHOS, and glucokinase activity in hypothyroid rats (249).
Oxidative stress is often interrelated with the disrupted Ca2+ homeostasis, in mitochondria further affecting OXPHOS and in exaggerated form leading to apoptosis accompanied by the so-called permeability transition opening (29, 95). Oxidative stress leads undoubtedly to impaired maintenance and biogenesis of pancreatic β-cells (see the Impaired Biogenesis of Pancreatic β-Cells section), including the putative “insulin resistance of β-cells,” that is, the dysregulation of autocrine insulin pathway, if it exists. This leads to decreased mtDNA copy number (262) and further GSIS deterioration by such a vicious cycle. Factors of mtDNA replication and transcription are essentially involved and their dysregulation or impairment leads to β-cell dysfunction including GSIS failure (214).
Also, the excessive generation of superoxide and H2O2 due to the elevated activity of NOXs results in impaired mitochondrial function and OXPHOS, consequently in impaired insulin secretion (207, 212). Elevated ROS also regulate signaling pathways by inhibiting activities of phosphatases and the consequent increase in protein tyrosine phosphorylation levels (22). In turn, redox signaling activates certain kinases, for example, p38 MAPK (234), JNK, PKC (166), and ERK (235). Such signaling often modifies ion channel function. Besides inducing Nrf2 signaling, oxidative stress also interferes with the inflammatory signaling of the NF-κB since the oxidized NF-κB exhibit reduced DNA binding (147). Also, a long-term activation of the 5′ AMP-activated protein kinase (AMPK) decreases GSIS (306).
Apoptosis of β-cells resulting from oxidative stress
Phylogenesis developed cell death mechanisms for situations when the above-described stresses would exceed the reparative capacity of tissue or organism. In contrast to the most investigated type of regulated cell death, termed apoptosis (118, 183, 199), necrosis was characterized as a nonspecific cell death. Recently, ferroptosis was classified as another type of cell death, arising from specific signals given by lipid hydroperoxides (266, 282, 307). It has been considered that not the apoptosis, but ferroptosis predominantly causes various neurodegenerative diseases. The role of ferroptosis in β-cells has yet to be investigated. Nevertheless, the disruption of iron homeostasis leads to β-cell death (19, 146).
The two main apoptotic signaling pathways have been described. First, the intrinsic pathway, also known as the mitochondrial pathway, which is activated by different types of cell stress such as hypoxia or oxidative stress, involves proteins of the Bcl-2 family and is manifested by the release of cytochrome c and other factors into the cell cytosol (106). The second one is the extrinsic pathway or the pathway mediated by cell death receptors, which involves ligands belonging to the TNF superfamily, such as TNF-α, Fas-L, and its respective receptors expressed on the surface of the pancreatic β-cells that transfer the apoptotic signal downstream through its cell death domain to other associated proteins (80, 242).
Apoptosis has been accepted as a major mechanism addressing β-cell failure in type 2 diabetes (Fig. 4). Oxidative stress and accompanying ER stress seem to be two major players upstream of the apoptotic cascade. Recently, an oxidative stress-responsive apoptosis-inducing protein (ORAIP) was found to induce apoptosis in pancreatic β-cells (304). Oxidative stress can induce β-cell apoptosis by MAPK plus JNK and ERK1/2 phosphorylation (119). The intracellular methylglyoxal accumulating at hyperglycemia was reported to induce the inositol-requiring protein-1 (IRE1)-JNK pathway of apoptosis initiation (184). Glycogen accumulation upon hyperglycemia is also setting conditions for apoptosis (40). In progressed diabetic states, amyloid polypeptide and NOXs are activated by the MAPK-JNK pathway when amyloid polypeptide activates apoptosis via the apoptosis signal-regulating kinase-1 (ASK1) (75, 102, 258).
A variety of apoptosis inducers exists. Notably, cytokines, and namely IL-1β, have been shown as important apoptosis mediators (310). Apoptosis as a response to nutrition overload involves the ER stress, but the distal effector mechanisms remain to be uncovered (31). The ER stress was described to be linked to apoptosis in β-cells chronically exposed to elevated FAs (155, 173). The main pathway axis includes the protein phosphatase 1 regulatory subunit 15A also known as growth arrest and DNA damage-inducible protein GADD34 (growth arrest and DNA damage-inducible protein 34); the eukaryotic initiation factor 2-α; the activating transcription factor 4 (ATF4); and protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK; EIF2AK3).
Such signaling through the GADD34 protein-EIF2α-(ATF4)-PERK axis induces oxidative stress. This was reflected by the findings that knockout mice of db/db background, with the deleted CCAAT-enhancer binding protein homolog (CHOP), are protected against the β-cell oxidative stress. Similarly, other proapoptotic members, such as the p53-upregulated modulator of apoptosis (PUMA), or death protein 5 (DP5)/harakiri (Hrk), were found to be upregulated by palmitate independent of CHOP/ATF4 (64). The FA overload-induced apoptosis was described to be connected to permeability transition pore opening with the ADP/ATP carrier participation. These effects also correlated with the affected Ca2+ homeostasis, especially in mitochondria (29, 95). Also, ER stress is involved in parallel with apoptosis as induced by for example, palmitic acid (200).
Apoptosis is prevented by Bcl-2 and Bcl-xL promoting β-cell survival, but Bcl-2 was found also to modulate ROS signaling and suppress a redox-regulated mitochondrial proton leak in β-cells (3). Interestingly, insulin itself via elevated H2O2 promotes β-cell death (250). TXNIP is induced by strong ER stress leading to oxidative stress and inflammation. Interestingly, TXNIP is not induced by chronic lipotoxicity, however, is induced by chronic hyperglycemia (223).
It is well recognized that apoptotic mechanism accompanies cytokine-induced β-cell death mainly during type 1 diabetes but also in obese type 2 diabetic humans (27). However, the rate of β-cell apoptosis is relatively low and thus may not entirely explain the disappearance of β-cell mass in vivo during the type 2 diabetes development (142). This indicates that β-cell functional impairment rather than simply β-cell loss largely contributes to the insufficient insulin secretion and glycemic control in type 2 diabetes. Thus, the concept of β-cell de/transdifferentiation as an alternative mechanism of β-cell loss of function seems to be a preferential variant for explanation, as discussed in the β-Cell Biogenesis section.
Damages to sensing of non-glucose secretagogues
FA sensing in pancreatic β-cells—intact versus impaired
FAs were considered to only augment GSIS in pancreatic β-cells when exposed at low doses or for hours (49, 92, 99). The existence of FA-stimulated insulin secretion (FASIS) has not been widely recognized; despite it has been traditionally ascribed to the GPR40 pathway (96, 98, 248, 113, 122, 129, 139, 168, 171, 194, 235).
Since this pathway stimulates insulin granule exocytosis via the Gq protein or Gs protein plus arrestin routes, it is predominantly KATP-independent. That is why fatty acid-stimulated insulin secretion (FASIS) can exist even without raising glucose above the fasting levels (51, 139). Only the nonreceptor metabolic branch of FASIS involving the glycerol/FA cycle partly contributes to the canonical KATP-dependent pathway of insulin secretion by FA equivalents entering to the mitochondrial FA β-oxidation. Even the glycerol/FA cycle stimulates insulin secretion partly by a receptor pathway, in this case via the exocytosis-promoting protein Munc13-1 activated by the released 2/monoacyl glycerol (231).
Ježek et al. have identified FA β-oxidation, as one leg in the FASIS mechanism, the branch producing essential H2O2 from the ETF:QOR-formed superoxide (36, 136); but also revealed a new amplifying mechanism for the GPR40 branch of insulin secretion (providing ∼60% FASIS): amplification by FAs cleaved from mitochondrial phospholipids by the H2O2-activated iPLA2γ (136), recently confirmed in islets (138, 139, 175) (Fig. 2).
However, the acute lipotoxicity, due to the instantly exaggerated FA levels, is frequently found only in vitro (48, 165). It may result from the excessive mitochondrial uncoupling, inhibition of the respiratory chain, or permeability transition pore opening when imbalance with 2-monoacylglycerol metabolism and FA β-oxidation exists (10) (Fig. 4). Sirtuins via deacetylation of the forkhead box protein O1 (FoxO1) suppress expression of genes required for FA β-oxidation but do not affect GSIS (162).
Lipotoxicity is typically associated with the oxidative stress. For example, p47phox subunit of NOX was elevated by palmitate (207). The toxicity was exerted by long-chain FAs of total 500 μM concentrations with chain lengths >14, generating H2O2 reportedly in the peroxisomal β-oxidation (230). In contrast, in human studies, following oral ingestion of the three fat emulsions over 24 hours, plasma FAs were elevated by approximately twofold over the basal level and only ingestion of PUFAs resulted in absolute decrease in GSIS (300). Also, fasting serum FA levels correlated well with the impaired insulin secretion in type 2 diabetes patients (208). Simulations with a FA mixture mimicking in vivo situation in MIN6 cells showed decreased viability, reduced antioxidant capacity, and OXPHOS plus elevated lipid peroxidation (285).
Effects of various FAs on β-cell function are complex and pleiotropic, depending on concentration, the chemical nature, including length (62), exposure time, and interaction with other FAs and nutrients (28, 93, 94, 135). For example, high palmitic acid dose at high glucose, that is, under nutritional stress, induced ER stress and profoundly suppressed insulin secretion (57, 115). It can be recognized that most of the FA lipotoxic effects originate from the alternations of cell signaling pathways, often with involved redox signaling and hence accompanied by a parallel oxidative stress. Such altered signaling may subsequently transfer β-cells into more permanent oxidative stress states.
Palmitate impairs expression of the pancreatic and duodenal homeobox 1 (Pdx1) and GLP-1 receptors in β-cells by elevating a sterol regulatory element-binding protein SREBP-1C (210). Chronic palmitate treatment leads to GSIS suppression due to the decreasing activity of Ins promoter and binding of Pdx1 and MafA (a member of the basic-leucine-zipper family of transcription factors) to the preproinsulin gene-flanking sequence (108). The insulin exocytosis is also hampered by palmitate-induced dissociation of Ca2+ channels from secretory granules (117) and by the impaired cAMP generation (279). Impaired Ca2+ homeostasis leads to concerted effects with FAs in oxidative stress induction (188). Promotion of lipotoxicity exists also via miRNA, as mediated by PERK/p53-dependent pathway activated by miR-34a-5p upon stearic acid supplementation (187). A novel mechanism of glucolipotoxicity was revealed, which contributes to β-cell dysfunction and cell death. It is initiated by a small GTPase Rac1, which induces β-cell apoptosis by the activation of NOX consequently leading to the increased expression of FA transporter CD36 (81).
Sensing of other secretagogues—impaired versus intact
For insulin secretion stimulated by keto-acids, redox signaling similar to that existing during FA β-oxidation exists, which is linked to the respiratory chain by ETF:QOR, producing superoxide at the EF site (136). Mitochondrial H2O2 substantiating the redox signal originates from the MnSOD-converted superoxide. We hypothesize that this redox signaling also activates iPLA2γ and thus cleaves FAs for GPR40 pathway of insulin secretion (Fig. 2).
Leucine, isoleucine, and valine are transported to the mitochondrion where the branched-chain 2-ketoacid amino transferase converts them to 2-keto-isocaproate, 2-keto-3-methyl valerate, and 2-keto-isovalerate, respectively (58, 74). Next, the branched-chain 2-ketoacid dehydrogenase converts them to isovaleryl-CoA, 2-methylbutyryl-CoA, and isobutyryl-CoA, respectively. In the subsequent reaction series resembling β-oxidation, final products are made as acetyl-CoA (acetoacetate), propionyl-CoA (or again acetyl-CoA), and succinyl-CoA, respectively, entering the Krebs cycle and being oxidized by the ETF:QOR. Subsequently, the elevated OXPHOS results in the KATP-dependent insulin secretion.
The mitochondrial dependence of insulin secretion stimulated by keto-acids is reflected by its blockage due to ablation of mitochondrial transcription factor B2 (214). One can predict that interference between insulin secretion stimulated by keto-acids and FASIS exists. Also, the oxidative stress may result from the related metabolism, namely at the FA overload with severe consequences for insulin secretion.
Systemic oxidative stress affecting β-cells
Consequences of chronic high glucose, glucotoxicity
Concentrations out of the physiological glucose stimulatory range, a “GSIS range” (3–11 mM in INS-1E cells, approximately 5–10 mM in mice and similarly for humans), are harmful especially when exposed to β-cells at prolonged time. Thus, for example, AMPK activation by glucose can contribute to the oxidative stress (251). Physiologically, high glucose inhibits AMPK which increases MafA and activates Ins promoter (132 [retracted]). Glucose higher than 10 mM stimulates β-cell mass maintenance by stimulating proliferation, neogenesis, and hypertrophy (24), and, as previously interpreted, also via autocrine insulin signaling (38, 170, 186, 220).
The term glucotoxicity is referred to, when much higher chronic glucose levels, pathological effects that overwhelm the beneficial glucose “maintenance effects.” A hallmark of glucotoxicity is a profound oxidative stress due to inhibition of protein expression for antioxidant defense (124, 153) (Fig. 4). Thus, hyperglycemia activates JNK and impairs the function of Pdx1 and induces ER stress (223).
Another hallmark of glucotoxicity originates from the spontaneous reactions of glucose and other sugars with amine residues of proteins, lipids, and nucleic acids forming the so-called AGE (61, 121). Polyol pathway is activated when excess glucose is converted to sorbitol in the presence of aldose reductase, consuming NADPH and thus contributing to prooxidation state (109). Enhanced glucose influx via the so-called hexosamine pathway induces O-glycosylation of signaling molecules that then leads to dysregulation of a crucial survival pathway leading to oxidative stress. The crucial pathway is the insulin receptor and insulin receptor substrates/PI3 kinase pathway (154). Moreover, hyperglycemia increases diacylglycerol, which activates PKC inducing NOX activity and ROS production (127).
Finally, persistent hyperglycemia and accompanying enhanced ROS exposure activate glycosylation, leading to protein, lipids, and nucleic acids unfolding. Specifically, one polypeptide termed amylin is affected this way in islets in type 2 diabetes. The resulting antiparallel crossed β-pleated sheet structure amyloid is then sensitive to free radical polymerization and promotes significant β-cell cytotoxicity (1, 205, 298).
Cytotoxicity
The diverse molecular mechanisms involved in the pancreatic β-cell cytotoxicity and death include an above-described apoptotic process, which participates in the immune-mediated insulitis in type 1 diabetes mellitus. Among these cytotoxic mechanism, one can emphasize the action of proinflammatory cytokines, ROS production leading to oxidative stress, DNA fragmentation (typical of necroptosis in type 1 diabetic patients), excessive production of amyloid polypeptide, induction of ER stress, disruption in autophagy mechanisms, and initiation of the inflammasome formation [recently reviewed in Rojas et al. (242)].
A systemic source of proinflammatory cytokines may be WAT or immune cells. Proinflammatory cytokines are involved in provoking the pathogenesis of tissue-specific IR by interfering with the insulin signaling pathways (239). Endocrine and paracrine β-cell signaling responses on systemic cytokines are similar and have been frequently mimicked in vitro. The proinflammatory cytokines have been shown to cause a concentration and time-dependent effects disrupting pancreatic β-cell function and viability. To reproduce the acute regulatory effects of systemic inflammation on β-cell secretory responses, these cytokines were titrated starting at femtomolar concentrations and incubated for 14–16 hours (27).
In contrast, to illustrate the adverse effects of local chronic islet inflammation, either higher concentrations or longer times (more than 16 hours of exposure) were used (27). It has been concluded that the systemic (or paracrine or autocrine, i.e., extracellular for β-cell) inflammatory responses activate the β-cell transcription of various proinflammatory mediators, notably cytokines/chemokines, with involvement of various signaling pathways, oxidative and metabolic stress. The outcome lies also in the recruitment of macrophages to pancreatic islets (Fig. 5).
Consequently, if cytokines excreted by β-cells are low (drained away by the blood flow) that cannot contribute to the β-cell death, the acceleration of islet inflammation by the recruited macrophages increases the probability of the apoptosis. The most prominent apoptotic pathways involved are the NF-κB and MAPK pathways.
Using the human EndoC-βH1 β-cell line, a detailed analysis of cytokine toxicity was performed (105). In this study, the most prominent cytotoxic effect was observed with a mixture of three cytokines (IL-1β, TNF-α, and IFNγ), while any of single cytokine alone had either no or slight effect on the caspase-3 activation. The treatment of cytokines caused activation of NF-κB, Nrf1/2 gene expression, and ROS formation. However, the ER stress markers were not induced. In general, cytokines can induce the ER stress and further induce β-cell apoptosis. However, it is still unclear whether the ER stress is required to elicit the cytokine-induced β-cell apoptosis. In any case, the cytokine treatment caused a significant suppression of the insulin secretory response, as documented by significant reduction of ATP and the absence of ATP/ADP ratio elevations as responding to glucose.
Consequently, GSIS was abolished by the proinflammatory cytokines, but without induction of IL-1β expression. Cytokine-mediated caspase-3 activation was accompanied by the increased ROS formation while cytokines transiently increased the expression of UPR genes, without inducing ER stress-marker genes (105).
NOX1 plays a key role in cytokine-induced β-cell dysfunction (295). Plasmid-driven elevation of NOX1 resulted in elevated ROS, loss of GSIS, and increased apoptosis, with outcomes analogous to cytokine treatment. In contrast, the lowering of NOX1 expression conferred protection to β-cells and islets from the damaging effects of inflammatory cytokines (295).
Serum levels of the alpha-cytoplasmic isoform of heat shock protein 90 (hsp90) were shown to be elevated in individuals with new onset type 1 diabetes (294), and a detailed study showed that human β-cell lines and cadaveric islets released hsp90α in response to stress induced by treatment with a combination of proinflammatory cytokines, including IL-1β, TNF-α, and interferon-γ (218). The hsp90α release was found to be driven by cytokine-induced ER stress mediated by JNK, a pathway that can eventually lead to β-cell apoptosis (218).
The proinflammatory signaling also include TLR4, which has been directly associated with β-cell dysfunction (63). The TLR4 receptor is activated by exposure to lipopolysaccharides (LPS), which are a major component of the outer membrane of gram-negative bacteria. LPS have been shown to trigger the upregulation of inflammatory cytokines, increase ROS production, and decrease insulin secretion in INS-1 pancreatic β-cells (73). The inhibition of TLR4 partially prevented LPS-induced dysfunction of rat islets (290).
Impaired Biogenesis of Pancreatic β-Cells
Heterogeneity and plasticity of pancreatic islets
Is there only a single β-cells phenotype?
In type 2 diabetes development, a significant decrease in the β-cell mass occurs not only due to the cell death but also due to β-cell dedifferentiation/transdifferentiation. Upon the dedifferentiation, β-cells regress to a less differentiated state or even a precursor-like state, whereas the other hormonal cell type is established during transdifferentiation. Transdifferentiation of β-cells to α-cells occurs in diabetic animals and patients (39, 54, 264, 265).
The dedifferentiation is characterized by downregulation of gene expression of Mafa, Neurod1, Nkx6.1, and Foxo1 genes encoding β-cell-specifying transcription factors, plus downregulation of insulin expression and expression of genes encoding processing and secretory pathway proteins (Hspa8, Dnaja2, Ssr1, Sec13, Copb2). In contrast, expression of atypical or “disallowed” genes for β-cells is enhanced by removing their suppression. For example, lactate dehydrogenase, hexokinase-1, monocarboxylate transporter MCT1, or progenitor cell genes (Neurog3, Pax4, Sox9) are induced. These alterations lead to phenotype changes, undoubtedly causing the deterioration or the lack of insulin secretion.
Although the mechanism of initiation is not fully understood, one of the stimuli is the glucotoxicity as such (24). In contrast, normal glucose metabolism is a major regulator of the β-cell differentiated phenotype due to the identity checking mechanism(s) (see the β-cell biogenesis section). Practically, model β-cell cultures have optimum maintenance at about 10 mM glucose (201), which increases messenger RNAs (mRNAs) of critical genes for insulin secretion and preceding metabolic pathways, including Glut2, Kir6.2, and Sur1 (25).
The question arises, how to really define the β-cell? Is it a cell with the active INS promoter, independent of transcribing other genes in different pattern? Alternatively, is there only a single ideal transcriptome pattern that defines the real β-cells having intact and presumably maximum GSIS? Swisa et al. defined the β-cell as a cell “capable of synthesizing, processing and secreting mature insulin in response to metabolic, hormonal and neurologic stimuli, or on a molecular level as a cell that expresses the full complement of genes associated with normal, regulated insulin secretion” (270).
Recent findings rather show that a spectrum of β-cells exists not only during ontogenesis but even after finished islet biogenesis. Such “flexible” biogenesis results in the existence of different states of maturation for β-cells. Undoubtedly, further remodeling of β-cells exists during the pathogenesis development. One could classify this as the initiating compensation responses, and after their failure, escape responses leading either to dedifferentiation or to transdifferentiation. We will briefly describe this in the next sections, leaving out the insulin promoter interactions and insulin expression that would deserve a self-standing review.
β-cell biogenesis
Neonatal β-cells are functionally incompetitive, whereas in adult β-cells, the replication is hindered but GSIS is fully functional. The transition between these limiting states can be reversed by the physiological non-oncogenic levels of c-Myc (233). Importantly, redox regulations are also involved in β-cell biogenesis.
Indeed, decreasing ROS levels were correlated with the diminished β-cell differentiation. In pancreatic explants cultures, increased ROS stimulated differentiation from progenitors by affecting ERK1/2 signal pathways (116). In turn, manipulations leading to decreases in cellular ROS levels prevented normal β-cell differentiation. Moderate increases in cytosolic H2O2 were found to promote β-cell proliferation in zebrafish, whereas high H2O2 levels can stimulate differentiation of new β-cells from progenitors (8). It has been hypothesized that the moderate H2O2 levels are necessary for β-cell proliferation. This may happen also during overnutrition in compensation by the increased β-cell mass.
Also miRNAs significantly regulate β-cell development, proliferation, insulin biosynthesis, and secretory processes, in addition to dedifferentiation or transdifferentiation (66). In contrast a different miRNA set (miR-7a, miR-132, miR-212) is involved in β-cell dysfunction by hyperglycemia and type 2 diabetes development. Interestingly, some of the so-called β-cell-disallowed genes are targets of miRNA and their expression is thus excluded. Thus, for example, miR-29a/b targets Slc16a1 gene encoding the monocarboxylate transporter MCT1 (70). Also, miR-328 and miR-384 were found specific for β-cells (46).
Regulation of insulin expression
The INS gene enhancer region is not only the site enhancing INS expression but also provides an important identity check for β-cells (see the β-Cell Identity Checking section). Sequence elements A, C1, C2, E1, E2, Z, and the cAMP-response element (CRE) have been identified in the INS gene enhancer, containing ∼400 bp-spanning binding sites essential for transcription factors (90).
The INS gene encodes a 110-amino acid precursor preproinsulin. Cytosolic ribonucleoprotein signal recognition particles enable translocation across the rough ER into the lumen upon cleavage by the signal peptidase, thus yielding proinsulin. Further ER processing involves the formation of three disulfide bonds to yield insulin (90).
Interestingly, the profound regulation of insulin exists at the translation level. The preproinsulin translation is controlled by glucose (283). Glucose added to primary β-cells thus initially recruits polyribosome complexes with preproinsulin mRNA, which causes up to 30 minutes lag when studied experimentally. However, subsequently after the lag, proinsulin biosynthesis can be increased up to 10-fold. Note that in humans resulting 51 amino acid insulin (5.8 kDa) is associated with the granules with ∼50 other secretory granule proteins. Insulin stored in β-cells is packed in these granules in an insoluble crystalline hexameric form of ∼40 mM concentration.
β-cell identity checking
The direct transcriptional activation of β-cell-specific genes and repression of β-cell-disallowed genes substantiate the basic mechanism of checking the identity of pancreatic β-cells. Thus, for example, Pdx1 represses MafB preventing (trans)differentiation into α-cells (91). Nkx6.1 represses numerous genes including somatostatin gene (277). Nkx2.2 mRNA levels were found to be increased at 10 mM glucose versus nonstimulating 2–5 mM glucose (25). However, the master regulator of β-cell-specific genes is Pax6 (269) (Fig. 6). Interestingly, transdifferentiation into β-cells also exists from the other islet cell types. Specific epigenetic pattern is most likely locking these cells as endocrine islet cells, which never transdifferentiate beyond their lineage (270).
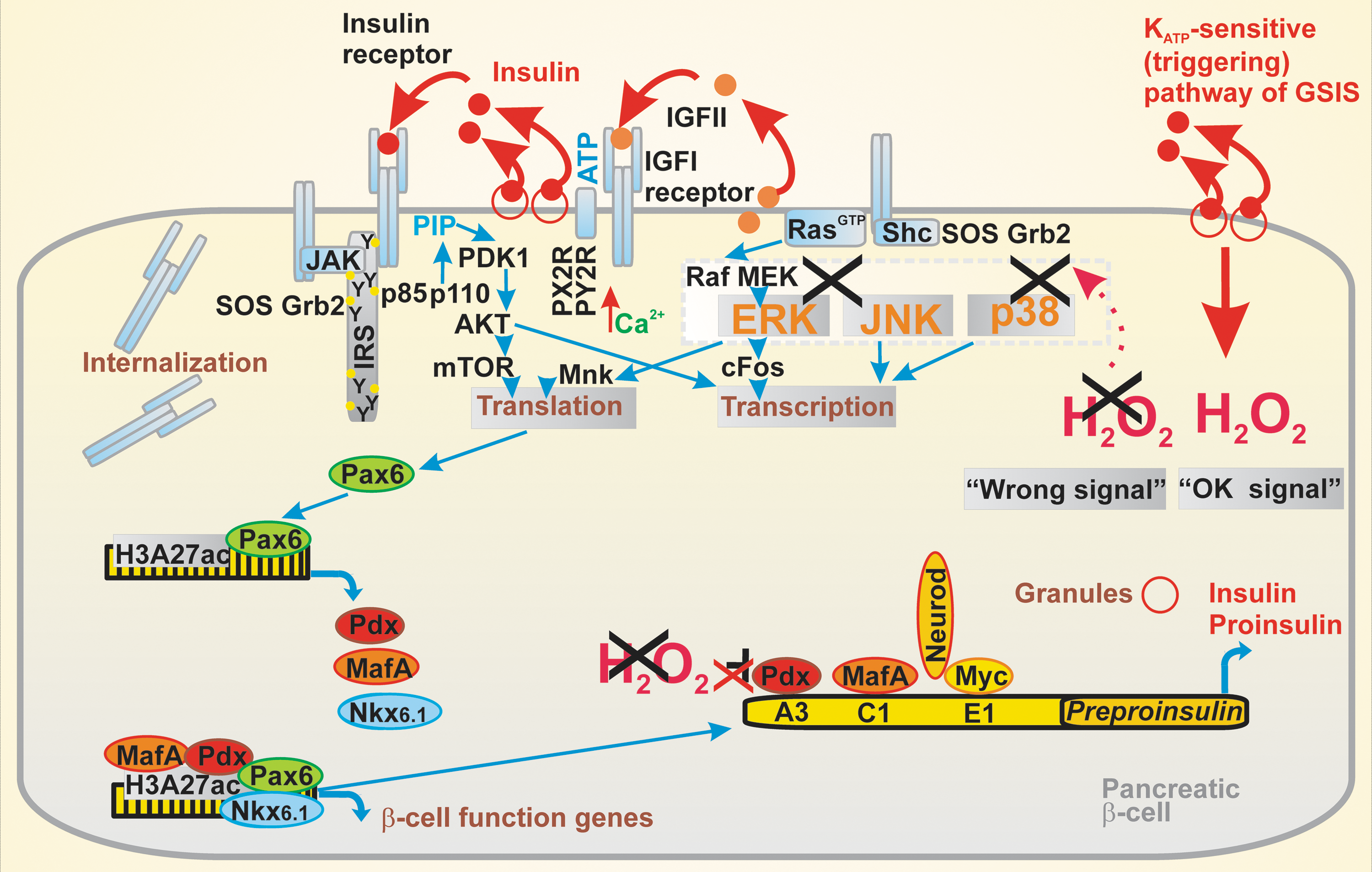
We may introduce a wider concept of the β-cell identity checking, which by homeostatic coordination of external factors and autocrine factors maintains optimum insulin gene expression, epigenetics, and insulin secretion on nutritional stimuli. When self-checking receives signals of the functional impairment, for example, an insufficient autocrine function, the consequences usually lead to cell death initiation or escape from it by de/transdifferentiation.
Certain modes of the β-cell identity checking also involve redox signaling or redox regulations. For example, Pdx and MafA as the main β-cell differentiation regulators are redox-sensitive (104). Pdx acts in a redox-sensitive manner during ontogenesis and MafA in mature β-cells. Together with the transcription factor NeuroD, Pdx and MafA bind to the Ins promoter. Pdx cooperates with the basic helix-loop-helix (bHLH) proteins such as β-2 and E47 that bind to the E1 site of insulin promoter (236).
Thus, both high glucose and ROS induce proinsulin expression as well as GLUT2 expression. The continuous activity of Pdx1-maintained gene expression is essential for mature β-cells (91). Also, Nkx6.1 serves as one line of identity checking, although being formerly considered merely as ontogenic factor important for β-cell differentiation in embryonic stage. Loss of Nkx6.1 leads to transdifferentiation into δ-cells (277). Importantly, hyperglycemia sets conditions when a “wrong” identity signals are received by the identity checking mechanisms (54).
Recently, the canonical pathway of GSIS including KATP, CaL, and being triggered by ATP rise due to OXPHOS elevation has been recognized (and hypothesized) as providing the major redox identity checking signal (270). The ongoing ROS (superoxide and H2O2) cytosolic release is assumed to substantiate the physiological redox identity checking signal (Fig. 6). Upon its loss (decline of redox signaling), the “wrong” identity signal is imposed (Fig. 6). The molecular nature of this is, however, unknown.
Stress-induced dedifferentiation and transdifferentiation of β-cells
A consensus has been established, pointing out that the β-cell functional impairment due to metabolic stress and involving de/transdifferentiation predominantly contributes to type 2 diabetes, rather than outright β-cell loss. Indeed, β-cell de/transdifferentiation occurs, leading to, for example, that 20% of the islet cells found not to express any of five hormonal transcripts and only 34% to be monohormonal (278). The dedifferentiation causes the loss of functional β-cell mass with ultimately defective insulin secretion in type 2 diabetes (273). Notably, dedifferentiation of β-cell is reversible when exposed to normal glucose levels (292). In any case, the β-cell dedifferentiation represents a partial or complete reversible loss of the β-cell identity, such that, for example, gastrin is expressed in certain dedifferentiated cells and can be therefore used as a dedifferentiation marker (54, 65).
However, not only late but also earlier events include cytokine and chemokine secretion by β-cells, recruiting macrophages by stress signaling (77). They reflect the impaired self-maintenance of β-cells, which might be hypothetically considered as the initial event in prediabetes development (Fig. 1). Such β-cell decompensation can also be regulated by miRNAs (66). The β-cell dedifferentiation can also result from the hyperglycemia-induced zinc depletion due to excessive losses of zinc in the secreted insulin granules (172).
When β-cells are dedifferentiated, they become invisible for many common methods of detection. Moreover, cellular differentiation does not need to be unidirectional. In the case of sustained hyperglycemia, β-cells can lose their differentiated phenotype in the process of dedifferentiation, whereas in the late stage of type 2 diabetes, they may even transdifferentiate into α-cells (71) or δ-cells (6). It has been demonstrated for the isolated human pancreatic islets that 20% of the cells do not express any of five hormonal transcripts and only 34% were found to be positive monohormonal (213).
Similarly, the other group demonstrated that 45% of rodent β-cells express multihormonal transcripts (156). Consequently, dedifferentiation in type 2 diabetes causes the effective loss of functional β-cell mass (“true β-cells”) with the ultimately defective insulin secretion. However, there is a period during the earlier development of diabetes, when dedifferentiated β-cells can fix their identity through resumption of expression of β-cell-specific genes if unfavorable conditions of glucose saturation are released.
It is recognized that high glucose levels besides duration of hyperglycemia are the two major upstream regulators of this process. Thus, it has been shown that rat islets and purified β-cells are optimally preserved by ∼10 mM glucose, which is above blood glucose levels at fasting, whereas either low (2–5 mM) or high (30 mM) concentrations cause glucotoxicity that can lead in long term to β-cell dedifferentiation (25, 253).
Also, the monogenic form of type 2 diabetes due to mutations in the KATP Kir6.2 subunit as well as gene polymorphism resulting in the in-excitable β-cells, thereby affecting insulin secretion, cause the increase of “empty” β-cells lacking the mature β-cell markers and instead expressing multipotency markers reflecting dedifferentiation. Interestingly, attempting to achieve euglycemia, the exogenous insulin supplementation, causes regaining of insulin and re-expression of identity genes in β-cells (292).
The signaling pathway(s) translating conditions of chronically increased glucose to gene transcription leading to de/transdifferentiation also include ER. The strong metabolic load inducing insulin-producing machinery occurring during obesity in compensation for IR might cause severe ER stress and insufficient cellular adaptation to ER stress. Consequently, the concomitant UPR can drastically reduce GSIS.
The ER stress arises from the accumulation of misfolded proteins often caused by the protein overload when the rate of synthesis exceeds the capacity of ER and further protein exit from ER into the secretory pathway. Alternatively, the ER stress results from alterations in the ER milieu that comprises the folding efficiency. We can easily explain why pancreatic β-cells are vulnerable to the ER stress, considering that the proinsulin represents ∼20% of all transcribed mRNA in the β-cell and insulin production occupies around 70% of the protein synthetic machinery.
The failure of adaptive UPR is associated with diabetes progression and affected β-cell differentiation. The adaptive UPR genes (Hspa5, Fkbp11, Dnajc3) are upregulated in prediabetic db/db mouse islets (age of 6 weeks), but their expression will decrease in diabetic mice (age of 14 weeks). Numerous UPR, ER-Golgi transport, ER quality control, endoplasmic reticulum-associated degradation (ERAD), and retro-translocon-related genes were significantly downregulated in islets of type 2 diabetic donors (45). The defective ER protein folding machinery manifested mainly by major signaling arms of PERK, ATF6, and the serine/threonine–protein kinase/endoribonuclease IRE1 leading to subsequent accumulation of unfolded proteins in ER has been shown to trigger oxidative stress, which induces altered gene expression (18, 110).
Redox status was shown to have an impact on cellular localization of Pax6 in MIA-PaCa2 cells representing the cells from the pancreatic origin. While under low oxidative stress, Pax6 diffused from nucleus to the cytoplasm, under moderate stress, Pax6 moved back to nucleus (257). H2O2 treatment of β-cell line and isolated islets altered the nuclear expression and DNA binding activity of MafA and Pdx1 (103, 112). Their altered expression was found in the islets of diabetic ZDF rats. The expression was normalized by treatment with GPX mimetic ebselen, thus reflecting the participation of redox signaling (192).
Many redox signaling pathways act through various miR regulations of β-cell identity genes (301). Other genes under redox regulation include Myc, Foxo1, or an inhibitor of differentiation proteins (26). Under elevated ROS levels, FoxO1 translocates to the nucleus where it enhances Neurod1 and Mafa gene expression (163).
In addition to high glucose, hypoxia and low-grade inflammation were also shown to downregulate UPR genes (52). At the same time, cytokine- and glucotoxicity-induced perturbations in ER calcium luminal content that compromise the overall folding capacity might cause β-cell failure as well (47, 111). Lipotoxicity mainly induced by saturated FAs was shown to disrupt ER-to-Golgi protein trafficking (232). This can lead to ER stress resulting in impaired proinsulin maturation and loss of insulin content. In conclusion, dedifferentiation can serve as the potential adaptive mechanism to escape cell death under chronic hyperglycemia at the price of altered identity and function.
Impaired β-cell homeostasis and autocrine or paracrine functions
Autocrine insulin
Two decades ago, studies with β-cell-specific insulin receptor knockout (βIRKO) mice emphasized the possible role of autocrine insulin on fitness, housekeeping and identity, and correct biogenesis of β-cells (170, 186). In contrast, skeletal muscle-specific insulin receptor knockout mice exhibited IR but normal fasting glucose. This suggested that IR originates from multiorgan deficiency and inflammation cross talk, including that of β-cells (44).
Later, an influential report considered the autocrine insulin function as unlikely and theoretically redundant (241). The previously observed effects such as in βIRKO mice were explained rather by the temporal negative feedback of insulin on stimulated insulin secretion mediated via the central nervous system. The arguments against the internalization of insulin receptor have been emphasized if local insulin concentrations were higher (at slow vein drainage). The alternative argument against the autocrine insulin function is a probable very low (picomolar) insulin concentration in the pancreatic artery.
Despite the fact that this controversy has not yet been solved, pancreatic β-cells contain active insulin receptor (14, 15, 38, 170, 186, 220, 271). Consequently, an acute autocrine insulin signaling was suggested to have similar impact as in skeletal muscle and the liver, besides positive effects on β-cell proliferation (38). If exists, the autocrine insulin secretion would be beneficial for the regulation of adult β-cell mass, but the acute effects on insulin secretion itself would be even less probable (37, 120).
βIRKO mice showed increased apoptosis, low proliferative rate, and decreased β-cell mass (170). The insulin receptor was found to be required for islet compensatory growth response to IR (220). Nevertheless, the important role is suggested for downstream insulin receptor substrate IRS-1 (14, 15, 222), emphasizing that another receptor and not the insulin receptor mediates the observed beneficial effects. Consequently, insulin-like growth factor 1 (IGF) may be the true autocrine modulator since its receptor is acting upstream.
There are two parts of insulin signaling through the insulin receptor, the Shc-RasGTP-Raf-1 kinase pathway and the Akt kinase pathway, interlinked by the growth factor receptor-bound protein 2 plus son of sevenless homolog 1 (Grb2-SOS) kinase signaling initiated upon tyrosine phosphorylation of IRS-1 (Fig. 6) (107). Akt is linked to IRS-1 via p85 p110 phosphatidyl inositol stimulation of PDK1 and subsequently Akt. Insulin would stimulate primary β-cell proliferation via the Shc-RasGTP-Raf-1 kinase pathway and simultaneously suppress apoptosis.
The Akt pathway would enhance β-cell mass and improve glucose tolerance. A signalosome complex of glucokinase, proapoptotic protein, Bcl-2-associated death promoter, BADS, and PKA has been reduced in βIRKO, thus linking the lack of autocrine insulin with the development of type-2 diabetes. The INS-1 cells with stable insulin receptor knockdown after reducing glucose and its subsequent elevation decreased insulin gene expression as well as Pdx1 and GLUT2, suggesting rather a positive role of autocrine insulin (288).
Oxidative stress due to altered autocrine function of insulin and other factors
Controversially, studies of mice with mutated insulin receptor have indicated that oxidative stress induced by high-fat diet ceased at impaired insulin receptor signaling, evidencing that such autocrine signaling evokes a certain redox signaling (271). In healthy humans, presumable autocrine insulin has acute (4 hours) effects on GSIS (35). It has been discussed, however, whether central nervous system effects are involved (241).
Also, insulin signaling was found to activate mitochondrial nitric oxide synthase (mtNOS), although the Akt-2/protein-kinase-B mediated phosphorylation in skeletal muscle (86). Released freely permeable nitric oxide, NO•, having a half-life of 1–10 seconds, causes a mild oxidative and nitrosative stress accompanied by transiently diminished respiration. NO• can also facilitate glucose to glycogen conversion in the skeletal muscle and liver. Nonpeptidyl mimetic L-783,281-activated insulin receptor through Akt-2/phosphatidylinositol-3-kinase (PI3K) signaling causing inhibition of GSIS and basal insulin secretion in human pancreatic islets (227). Also, recent report on isolated mitochondria showed that insulin signaling regulates mitochondrial function in β-cells (186).
Stress signals of pancreatic β-cells
We hypothesize that self-checking or identity checking of β-cell is the most essential event regulating the correct housekeeping and phenotype of the “true” β-cell. When such self-checking senses that the less intensive or less frequent autocrine signals occur (37, 120), the absence of beneficial controls by, for example, IRS-1 downstream signaling or GLP-1 (60) probiogenic pathways leads to the release of stress signal. Note that this is the sensing of the absence of self-checking, hence it might be executed as, for example, the lack of previously existing redox signals with consequent termination of downstream redox-activate kinase signaling. Alternatively, the absence of elements promoting INS gene expression may substantiate the initiation of putative stress signals (Fig. 6).
Thus, a link may be hypothetically predicted between the deteriorated autocrine signals including that of insulin and low expression of β-cell-specifying factors and hypothetically also between deteriorated endocrine or paracrine incretin action and increased cytokine and chemokine expression in pancreatic β-cells. Since incretins also stimulate ERK, the absence of certain ERK-dependent gene expression of, for example, repressor of the stress gene might substantiate the stress signal. The primary stress signal can be identical to IL-1β-signaling and downstream cytokine and chemokine expression and release from the “unchecked” β-cell. Besides inflammatory signals, exosomes can also carry out the stress signals from pancreatic β-cells to peripheral tissues, such as transduced by miRNAs (150).
Islet inflammation has already been described above. Inflammation is expected to amplify the stress signaling by pancreatic β-cells. Moreover, a cross talk is inevitable between already weak inflammatory states of WAT and skeletal muscle and incorrectly checked β-cells or dedifferentiated β-cells.
Conclusions
Oxidative stress is traditionally considered as an umbrella for phenomena of multiple changes in β-cell during the type 2 diabetes development. Oxidative stress cannot be separated from numerous other cell pathology events, such as the attempted compensation of β-cell for the increased insulin demand and dynamics of β-cell biogenesis and its “reversal” at dedifferentiation and from changing islet β-cell mass (also due to transdifferentiation) and inflammation.
However, at the initiated pathology, the compensation responses of β-cells despite the delaying pathology progression set a new state when a strong self-checking function might hypothetically send the putative stress signals to the periphery. Subsequent changes together with lasting glucolipotoxicity undoubtedly promote islet inflammatory responses and further spiral of pathology.
Future research should bring an understanding of the β-cell self-checking and putative stress signals since substitution of the “wrong” or “incorrect” signal by the “correct” self-checking signal might help in type 2 diabetes prevention.
Overfeeding, namely the excessive glucose intake, likewise other nutrients acting in a synergy with glucose, has been identified as a major causative origin of numerous events leading to the progressive onset of type 2 diabetes. Both lead to metabolic dysregulations, frequently being related to the oxidative stress in periphery as well as in pancreatic β-cells. All such dysregulations may progress in type 2 diabetes development, being interrelated with the metabolic syndrome.
Footnotes
Acknowledgment
L.P.-H. was supported by the Grant Agency of the Czech Republic, grant No. 16-06700S, while P.J. by No. 17-01813S.