
Abstract
Significance:
In addition to their classical role in cellular ATP production, mitochondria are of key relevance in various (patho)physiological mechanisms including second messenger signaling, neuro-transduction, immune responses and death induction.
Recent Advances:
Within cells, mitochondria are motile and display temporal changes in internal and external structure (“mitochondrial dynamics”). During the last decade, substantial empirical and in silico evidence was presented demonstrating that mitochondrial dynamics impacts on mitochondrial function and vice versa.
Critical Issues:
However, a comprehensive and quantitative understanding of the bidirectional links between mitochondrial external shape, internal structure and function (“morphofunction”) is still lacking. The latter particularly hampers our understanding of the functional properties and behavior of individual mitochondrial within single living cells.
Future Directions:
In this review we discuss the concept of mitochondrial morphofunction in mammalian cells, primarily using experimental evidence obtained within the last decade. The topic is introduced by briefly presenting the central role of mitochondria in cell physiology and the importance of the mitochondrial electron transport chain (ETC) therein. Next, we summarize in detail how mitochondrial (ultra)structure is controlled and discuss empirical evidence regarding the equivalence of mitochondrial (ultra)structure and function. Finally, we provide a brief summary of how mitochondrial morphofunction can be quantified at the level of single cells and mitochondria, how mitochondrial ultrastructure/volume impacts on mitochondrial bioreactions and intramitochondrial protein diffusion, and how mitochondrial morphofunction can be targeted by small molecules.
Table Of Contents
I. Introduction
Virtually every mammalian cell contains mitochondria, which are classically recognized as key generators of cellular energy in the form of adenosine triphosphate (ATP) (99). In addition, mitochondria constitute an integral part of the mechanisms that control cell functioning and survival. This is exemplified by their established physiological role in cell differentiation (289), immune cell function (40, 252, 261), cell death regulation (20, 123), calcium homeostasis (77, 109, 110, 434), and neurogenesis (175). In addition, mitochondrial dysfunction is associated with a multitude of pathophysiological conditions, including metabolic disorders (196), cancer (6, 53), diabetes (336, 436), and neurodegeneration (188). Proper mitochondrial function in mammals requires ∼1200 genes [MitoCarta 2.0 (43) and MitoMiner 4.0 (369)].
Only a small fraction of these mitochondrial proteins is encoded by the mitochondrial DNA (mtDNA), of which multiple copies are present in each individual mitochondrion. The mtDNA consists of a light (L) and heavy (H) strand, has a size of 16,569 base pairs, and contains 37 genes (9 on the L-strand and 28 on the H-strand). These genes encode 13 proteins [all of which are subunits of the mitochondrial oxidative phosphorylation (OXPHOS) system; see section I.A], 22 transfer RNAs, and 2 subunits of the mitochondrial ribosome (135). The nuclear DNA (nDNA) encodes the large majority of mitochondrial proteins, which are synthesized in the cytosol and imported into the mitochondrion. This import is carried out by a dedicated machinery of protein translocases in the mitochondrial outer membrane (TOM) and inner membrane (TIM), as reviewed in detail elsewere (172, 422).
A. Mitochondrial adenosine triphosphate production
Mitochondrial adenosine triphosphate (ATP) is primarily generated by the action of the mitochondrial OXPHOS system (Fig. 1). This system is embedded in the mitochondrial inner membrane (MIM), consists of five multi-subunit complexes (CI–CV), and is fueled by NADH and FADH2 provided by the glycolysis pathway in the cytosol and the tricarboxylic acid (TCA) cycle in the mitochondrial matrix (234, 397). Functionally, the OXPHOS system consists of the electron transport chain (ETC; comprising CI-CIV) and the ATP-generating FoF1-ATP synthase (“CV”). ETC assembly from its nDNA- and mtDNA-encoded subunits requires assistance of at least 33 nDNA-encoded assembly factors (188). At a higher structural level, the ETC can be organized in respiratory supercomplexes (98, 260).

Within the ETC, CI and CII abstract electrons from NADH and FADH2, respectively, and donate them to Coenzyme Q10 (“Q”). The latter molecule transports electrons to CIII, from where they are conveyed to CIV by cytochrome-c (“c”). At CIV, the electrons are donated to molecular oxygen to form water. As an alternative to CI, CII, and CIII, various other MIM-associated enzymes can donate electrons to Q (241, 284). For instance, by metabolizing: (i) acetyl coenzyme A (acyl-CoA) (by electron transfer flavoprotein-ubiquinone oxidoreductase or ETFQ), (ii) glycerol-3-phosphate (s,n-Glycerol-3-phosphate dehydrogenase or G3PDH), (iii) proline (proline dehydrogenase or PRODH), (iv) dihydroorotate (dihydroorotate dehydrogenase or DHODH), and (v) hydrogen sulfide (succinate:quinone reductase or SQR). Moreover, electrons can be donated to cytochrome-c by Mo-pterin and B-type heme. In this sense, Q and cytochrome-c can be regarded as “junctions,” on which different electron-donating systems converge to feed electrons into the ETC (213). It appears that the “alternative” electron donors do not simultaneously supply electrons to the ETC. Moreover, these enzymes display tissue and species-specific expression (241). During electron transport, energy is gradually released and used (at CI, CIII, and CIV) to expel protons (H+) from the mitochondrial matrix across the MIM. As a consequence, an inward-directed trans-MIM proton-motive force (PMF) is generated, consisting of an electrical (Δψ) and chemical (ΔpH) component (448). The PMF is utilized by CV to catalyze the formation of ATP from adenosine diphosphate (ADP) and inorganic phosphate (Pi) by allowing the controlled re-entry of protons into the matrix (267, 410). This ATP generation requires Pi import in the form of PO4 3− by the Pi/H+ symporter (PiC) and the electrogenic exchange of ADP3− (import) against ATP4− (export) by the adenine nucleotide translocator (ANT; Fig. 1). This combined (forward) action of CV and ANT will depolarize Δψ, which is counterbalanced by ETC action. Under pathological conditions, CV can also hydrolyze ATP and expel protons from the mitochondrial matrix to sustain Δψ (285). This mechanism requires transport of ATP generated in the cytosol, for instance by the glycolysis pathway, into the mitochondrial matrix by ANT reverse mode action.
It is well established that the ETC plays a key role in the production of mitochondrial reactive oxygen species (ROS), particularly under pathological conditions. Information about how ETC-mediated ROS production relates to: (i) other sources of mitochondrial and cellular ROS, (ii) the spatial aspects of ROS action, (iii) oxidative stress induction, and (iv) ROS signaling is discussed in detail elsewhere (21, 89, 190, 241, 365, 425). Regarding the link between the ETC and redox metabolism, the mitochondrial nicotinamide nucleotide transhydrogenase (NNT) directly couples the trans-MIM influx of H+ to the transfer of electrons from NADH to NADP (Fig. 1). This coupling keeps the mitochondrial NADP/NADPH pool in a reduced state, which protects mitochondria against oxidative damage (273). The NNT can also operate in reverse mode, thereby oxidizing the NADP/NADPH pool and disrupting antioxidant defense (286). Both NAD+/NADH and NADP+/NADPH play important (regulatory) roles in mitochondrial/cellular metabolism and redox homeostasis. These roles, as well as their mechanistic connection and signaling function in health and disease are discussed in detail elsewhere (140, 145, 161, 435).
B. Cellular ATP production displays metabolic flexibility
In addition to mitochondrial OXPHOS, the glycolysis pathway also generates ATP by converting glucose (taken up by the cell via glucose transporters) into pyruvate. The latter is either converted into lactate (which can be released into the extracellular medium) or enters the mitochondrial matrix to form acyl-CoA as a TCA cycle substrate yielding additional ATP (Fig. 1). In addition, also fatty acids (FAs) and glutamine (Gln) can serve as TCA substrates (397). Cells display a substantial degree of metabolic flexibility, meaning that the balance between glycolysis- and OXPHOS-derived ATP generation is variable (394). This is exemplified by studies in C2C12 mouse myoblasts demonstrating that acute OXPHOS inhibition rapidly increases steady-state glucose uptake/consumption and that this increase fully compensates for the reduction in mitochondrial ATP production (219, 220). Alternatively, ATP can be generated by substrate-level phosphorylation within the mitochondrial matrix. In this process, a phosphorylated biomolecule directly transfers a PO3 2− (phosphoryl) group to ADP or guanosine diphosphate (GDP) to form ATP or guanosine triphosphate (GTP) (61).
C. ETC function is not only essential for mitochondrial ATP production
In addition to ATP generation, Δψ and/or ΔpH are crucial for the activity of transporters that exchange metabolites and ions between the cytosol, intermembrane space (IMS), and mitochondrial matrix compartment (Fig. 1) (302, 379). This exchange is of key importance, for instance to maintain Ca2+ homeostasis and viability in the heart (235). Further, Δψ plays a dual role in the trans-MIM import of mitochondrial preproteins via the TOM/TIM system (422). Given the fact that Δψ is negative on the matrix-facing side of the MIM, it exerts an electrophoretic effect on the N-terminal mitochondrial targeting sequence (or “presequence”), which often carries a positive charge (247, 392). In addition, Δψ directly activates a key TIM protein (TIM23), which translocates cleavable preproteins into the IMS or mitochondrial matrix (253).
A less negative (depolarized) Δψ has been associated with reduced mitochondrial ATP production and also connects to various other (patho) physiological phenomena by: (i) increasing cellular ROS levels (190, 192, 193), (ii) preventing fusion of the MIM but not of the mitochondrial outer membrane (MOM; see section III) (243), (iii) increasing the opening probability of the mitochondrial permeability transition pore (mPTP), ultimately leading to apoptosis induction (26), and (iv) stimulating mitochondrial degradation by organelle-specific autophagy (“mitophagy”) (137, 382, 393).
D. Mitochondria physically and functionally interact with other cell constituents
Further emphasizing their central role in cell physiology, mitochondria physically and functionally interact with various other cell components, including the cytoskeleton, plasma membrane (PM), endoplasmic reticulum (ER), Golgi apparatus, lipid droplets (LDs), peroxisomes, and lysosomes (316, 395, 428). With respect to peroxisomes, mitochondria are essential for their de novo biosynthesis from mitochondrial and ER-derived pre-peroxisomes (376). Mitochondria-ER interactions are currently the best studied and are mediated by mitochondria-associated membranes. The latter are at the center of intracellular signaling and other pathways, including cholesterol and phospholipid synthesis, ROS production, and mitochondrial calcium uptake/release. In this respect, various tethering proteins involved in mito-ER coupling have been identified in yeast and mammalian systems (Fig. 2) (118, 141, 224). Further detailed information about these protein–protein interactions, as well as their functional relevance, is presented elsewhere (50, 72, 118, 121, 141, 190, 224).

II. Mitochondrial (Ultra)Structural Dynamics
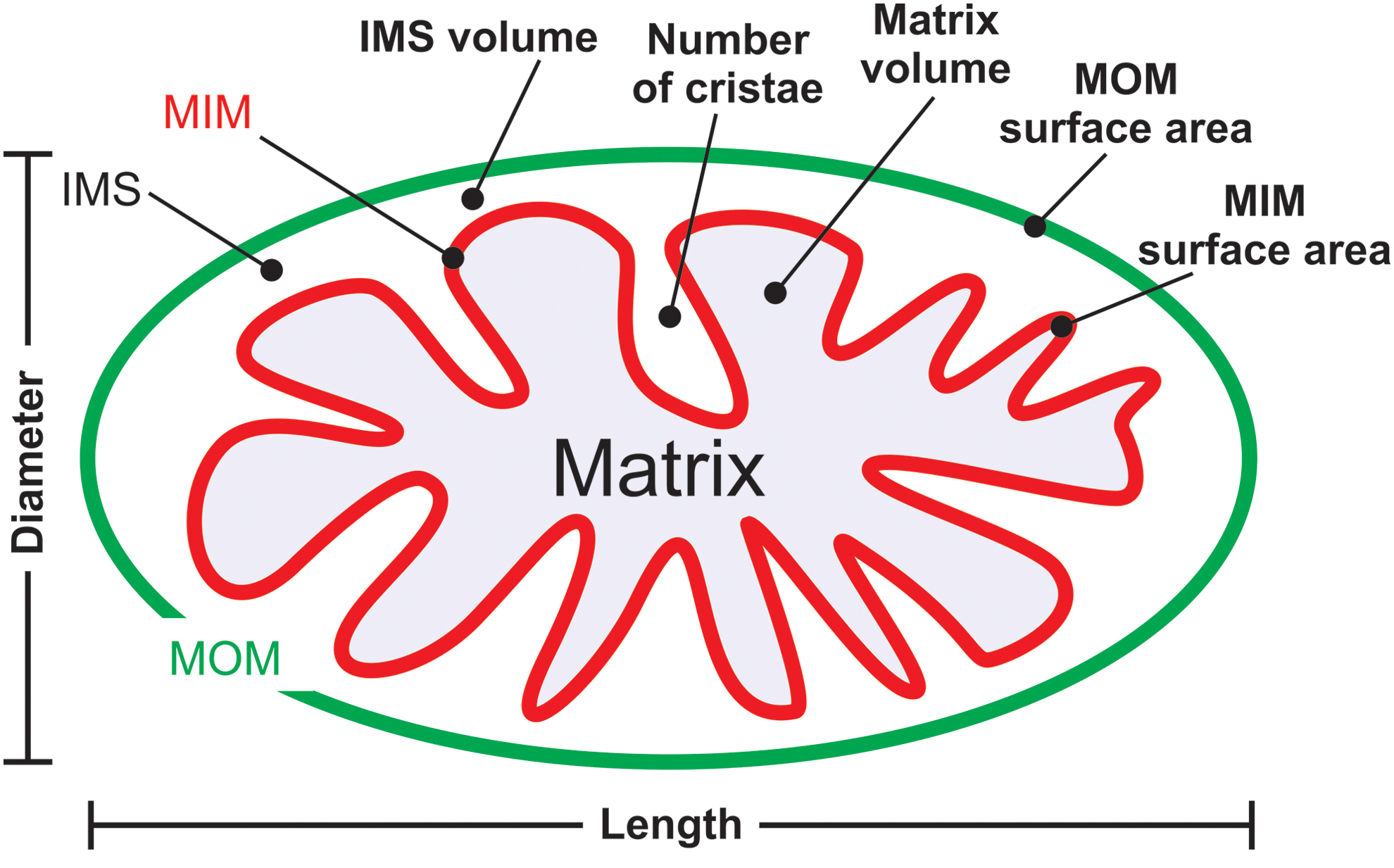
Mitochondria consist of a double membrane system, in which the MOM surrounds the MIM (Fig. 3A, B). The latter constitutes the boundary of the mitochondrial matrix compartment and contains many folds (cristae) that protrude into this compartment, thereby enlarging the MIM surface area. The MIM and MOM are separated by the mitochondrial IMS and are partially connected via contact sites that are involved in cristae organization (309). Important structural features of the mitochondrial matrix and cristae system include the inner boundary membrane (IBM), cristae junction (CJ), and cristae membrane (CM; Fig. 3C, D). Parameters describing mitochondrial (ultra)structure include the distance between the MIM and MOM (marked “a”), the intracristae space (marked “b”), and the distance between adjacent cristae (“inter-cristae space”) (403).

Maintaining CM integrity is of key importance for proper OXPHOS functioning (68, 149). The latter is illustrated by analyis of the yeast Saccharomyces cerevisiae using superresolution microscopy (see section IV.D) and cryo-immunogold electron microscopy (EM). This revealed that mtDNA-encoded subunits of CIII and CIV are inserted at different sites of the MIM when compared with CV subunits (374). In addition, it was found that early (but not late) steps in CIII and CIV assembly occur at the IBM, whereas CV assembly is carried out in the CM. This suggests that alterations in cristae structure directly but differentially affect the assembly of OXPHOS complexes. Net mitochondrial external/internal morphology and positioning greatly differs between cell types (39, 69, 201, 389) and dynamically changes over time (Fig. 4; “mitochondrial dynamics”). These changes require mitochondrial movement via mechanical interactions with the cytoskeleton, the functional impact and molecular mechanisms of which are discussed elsewhere (29, 223, 264, 362). In addition, mitochondrial external and internal morphology is affected by: (i) fission/fusion events (51, 297, 392, 420), (ii) chemiosmosis-related swelling/shrinking (160), (iii) the composition and physicochemical properties of the MOM and MIM (131), and (iv) the nature of the extracellular matrix (19).

As the integrated result of the mechanisms described earlier, mitochondrial external morphology ranges from elongated tubular structures (Fig. 5A–C) to donut-shaped organelles (Fig. 5D, arrows) and often displays heterogeneity within the same cell (Fig. 5C–E). It was proposed that this morphological plasticity allows: (i) mixing of mitochondrial content, (ii) redistribution of damaged proteins and lipids between mitochondria in stress reduction, (iii) local functioning of (subsets of) mitochondria within the cell, and (iv) mitophagy (179). Importantly, mitochondria cannot be generated de novo (363). This means that mitochondrial fission is crucial to allow their inheritance by both daughter cells during cell division.
In vivo mouse studies revealed that mtDNA-devoid tumor cells acquired mtDNA from host cells, suggesting transfer of whole mitochondria and/or their content between cells (381). In agreement with this idea, transcellular exchange of individual mitochondria via nanotubular structures (“nanotunneling”) has been demonstrated under certain conditions (255, 338, 404). On the other hand, it appears that mtDNA can be transferred between cells via exchange of extracellular vesicles (343). Also, the MIM displays heterogeneity and dynamic behavior (Fig. 6) and, although a rare event, cristae can disappear or form within a matter of seconds (87). Fission and fusion of the MOM and MIM are executed by a core machinery of nine currently identified proteins (Fig. 7A) (51, 308, 337). Directly below, we discuss these fission and fusion mechanisms in more detail.


A. Fission of the mitochondrial outer membrane
MOM fission is carried out by dynamin-related protein 1 (DRP1; Fig. 8), a cytosolic GTPase (314, 367). This protein also mediates peroxisome fission (see section II.F). The large majority of DRP1 protein appears to reside in the cytosol, from which it is recruited to the MOM by four currently unknown adaptor proteins (Fig. 8): (i) fission protein 1 (FIS1) (230, 300, 437), (ii) mitochondrial fission factor (MFF) (230, 299, 300), and (iii) mitochondrial elongation factor 1 (MIEF1/MID51) and mitochondrial elongation factor 2 (MIEF2/MID49) (229, 230, 300, 301, 331, 439, 445).

In principle, mitochondrial fission is a stochastic process (200) during which DRP1 is recruited to the MOM. Once recruited, DRP1 monomers oligomerize into a contractile ring-like structure. On GTP hydrolysis, this structure decreases its diameter, thereby inducing mitochondrial fission (41, 112, 250). Quantitative analysis of DRP1 protein distribution in HeLa cells expressing GFP-tagged DRP1 suggests that cytosolic GFP-DRP1 predominantly exists in a tetrameric form and constitutes ∼50% of the total GFP-DRP1 pool (258). The latter study estimated that a functional DRP1 fission complex contains ∼100 DRP1 molecules. A population of DRP1 oligomers was detected on ER membranes, being distinct from mitochondrial or peroxisome-associated oligomers (162). On MFF knockdown, DRP1 oligomer levels at the ER, mitochondria, and peroxisomes were reduced, suggesting that DRP1 can use these three organelle membranes as oligomerization platforms.
ER tubules are involved in marking the site of mitochondrial division (111) and mtDNA replication (216). Mitochondrial ER-mediated constriction further involves inverted formin 2, an ER-associated actin modulator that induced actin polymerization at the ER-mitochondrial contact site (197). Additional actin-binding proteins (e.g. SPIRE1C; 211, 245) and lysosome-mitochondrial contacts also play a role in marking DRP1- and ER-positive sites of mitochondrial fission (428). Mechanistically, these contacts were promoted by active (GTP-bound) RAB7, a lysosomal GTPase that is a member of the RAS oncogene family. Interestingly, disconnection of lysosome-mitochondria contacts involved FIS1-mediated recruitment of TBC1D15 (TBC1 Domain Family Member 15). The latter is a GAP (GTPase-accelerating protein) of RAB7, which deactivates RAB7 by promoting its GTP hydrolyzing activity (428). In yeast, FIS1 was identified as a factor involved in the mitochondrial recruitment of DRP1 (DNM1) (202). However, it appears that FIS1 is not essential for mitochondrial fission in mammals (296, 299) but binds to MFF on the MOM, which, subsequently, attaches to a complex containing FIS1 and ER proteins at the mitochondria-ER interface (360). This study also provided evidence that FIS1 is not strictly required for mitochondrial fission but is involved in mitophagy. In this sense, inactivation of FIS1 gives rise to formation of LGG-1/LC3 aggregates (360).
Taken together, the current mechanistic insights suggest that MFF is the main DRP1 adaptor protein that, together with MIEF1/MID51 and MIEF2/MID49, controls DRP1 ring formation and constriction (169, 200). It was suggested that MIEF1/MID51 and MIEF2/MID49 also regulate the association of DRP1 with MFF in a trimeric DRP1-MIEF/MID-MFF complex (439). In addition, evidence was provided that MFF can act as a biomechanical membrane-bound force sensor (139). This would allow recruitment of the fission machinery to mechanically strained sites of the MOM. Recently, a new member of the mitochondrial division machinery, dynamin 2 (DYN2; Fig. 8), was discovered (210). DYN2 is ubiquitously expressed and acts together with DRP1 in sequential constriction, ultimately leading to a final DYN2-mediated step in mitochondrial fission (210).
B. Fusion of the MOM
Mitochondrial fusion requires sequential merging of the MOM and MIM of the two precursor mitochondria (243, 372). MOM fusion is mediated by the GTPases mitofusin 1 and 2 (MFN1/MFN2) (46, 212, 334, 344, 431). This process is well studied (100, 211) and is mediated by homotypic and heterotypic interactions between MFN1 and MFN2 on opposing mitochondria (56). These interactions are believed to involve (indirect) formation of disulfide bridges (249, 364). Both mitofusins (Fig. 8) contain a GTPase (G) domain, a coiled-coil domain (heptad repeat domain 1 or HR1), an MOM-spanning transmembrane domain (TM), and a second IMS-protruding coiled-coil domain (heptad repeat domain 2) (116). It was recently demonstrated that mitofusins are single MOM-spanning proteins with their N-terminus facing outward into the cytosol and their C-terminus protruding into the IMS (249). Analysis of the MFN1 crystal structure suggests that it forms homodimers during mitochondrial fusion by conformational changes that promote GTPase-domain (G-domain) dimerization (46, 321). This is in contrast with previous evidence suggesting that the C-terminal antiparallel coiled-coil domain of MFN1 plays a key role in tethering (105, 199). It was further suggested that MFN1 might use a two-step tethering mechanism consisting of a nucleotide-regulated dimerization of the G domains and subsequent interaction between the coiled-coil domains (46). Using in situ and in vitro fusion assays, it was demonstrated that interaction with the surface of the lipid bilayer induces folding of a conserved amphipathic helix in HR1 (75). This suggests a mechanism in which HR1 destabilizes the MOM lipid bilayer, especially in membrane regions displaying defects in lipid packing, to facilitate MOM fusion.
C. Mitochondrial inner membrane fusion
MIM fusion requires the action of the GTPase optic atrophy protein 1 (OPA1) (108, 128, 293, 371). GTP is supplied to OPA1 by nucleosidediphosphate kinases, which produce GTP through ATP-driven conversion of GDP (32). The OPA1 protein exists in eight different isoforms (splice variants, Sp1–Sp8) due to differential splicing (Fig. 8) (51). Each OPA1 isoform contains various domains including an N-terminal mitochondrial targeting sequence (MTS), a TM, and a protease cleavage site (S1). Four of the eight OPA1 isoforms also contain an additional (S2) protease cleavage site (51). After mitochondrial import of the OPA1 precursor, its MTS is cleaved off by the matrix-soluble protein peptidase, yielding an MIM-anchored “Long” OPA1 form (“L-OPA1”; Fig. 9). Cleavage of the OPA1 precursor at S1 or S2 yields “Short” OPA1 forms (“S-OPA1”). Proteolytic processing of OPA1 at S1 and S2 constitutes a major regulatory mechanism (371) that is carried out by a complex proteolysis network (51). Two mitochondrial proteases, OMA1 (“overlapping activity with m-AAA protease”) and YME1L (“Human yme1-like protein”), are responsible for L-OPA1 cleavage at S1 and S2, respectively (Fig. 9) (8, 51, 265, 323). Because both L-OPA1 and S-OPA1 forms need to be present in a proper ratio for MIM fusion, it appears that some OPA1 processing is always required.

D. Regulation and maintenance of cristae structure
1. Role of OPA1
Experimental studies in HeLa cells revealed that OPA1 knockdown induced mitochondrial fragmentation, Δψ dissipation, and cristae disorganization, which were associated with cytochrome-c release and caspase-dependent apoptotic cell (AC) death (294). These observations are compatible with the observation that OPA1, independently of its role in MIM fusion and without interfering with activation of BAX and BAK, prevents mitochondrial cytochrome-c release, thereby protecting against apoptosis induction (108). Mechanistically, this is achieved by oligomerization of MIM-anchored L-OPA1 and soluble S-OPA1, which maintains tightness of the CJs during apoptosis (Fig. 10A).

The Presenilin-associated rhomboid-like (PARL) protease is involved in this OPA1-dependent protection, since Parl−/− mice displayed reduced levels of S-OPA1 and PARL −/− mitochondria exhibited accelerated cristae remodeling and cytochrome-c release during apoptosis (65). Knockdown of OPA1 in HeLa cells specifically reduced CM dynamics, whereas the IBM remained flexible (124). Recently, an elegant mechanistic framework was presented that integrates these earlier observations with the existence of OPA1 isoforms and OMA1/YME1L-mediated OPA1 processing (79 –81). In this model, all eight OPA1 isoforms (Fig. 9) are proposed to be involved in organization of OXPHOS supercomplexes, cristae shaping, and mtDNA maintenance. Independent of mitochondrial morphology, L-OPA1 and S-OPA1 isoforms support mitochondrial fusion and preserve mitochondrial bioenergetics, respectively. In addition to OMA1, YME1L, and PARL, various other biomolecules interact with or affect OPA1 function to regulate or maintain MIM structure. These include MFN1, MFN2, BAX/BAK, Paraplegin (SPG7), prohibitin 2 (PHB2), and mtDNA polymerase gamma. Detailed information on these interactions and their functional impact is provided in the Supplementary Table and elsewhere (67, 74, 447).
2. Role of mitochondrial contact site and cristae organization
In addition to OPA1, another key player in mitochondrial ultrastructural maintenance is the mitochondrial contact site and cristae organization (MICOS) system (Fig. 10B). Currently, this system is best understood in yeast (327, 398, 427). The MICOS complex impacts metabolite handling, cellular signaling, mitochondrial membrane architecture, and mitochondrial protein import. This coupling is mediated by MICOS interactions with: (i) the MOM protein Porin/VDAC (voltage-dependent anion channel; interaction with the MICOS subunit MIC60), (ii) the protein sorting and assembly machinery (SAM; MIC60 interaction), (iii) the protein translocase of the MOM (TOM; MIC60 interaction), (iv) the presequence translocase of the MIM (TIM23), (v) mtDNA, (vi) the ETC, and (vii) CV (352). Recent evidence suggests that MICOS also interacts with the MIM ATPase ATAD3A, and that the latter protein assists in stabilizing mitochondrial cristae, potentially via interactions with the MOM (307). Subunit MIC10 plays a central role in the yeast MICOS system (31), and MIC10 oligomerization induces MOM bending at CJs (17). Electron tomography analysis revealed that MIC19 and MIC60 were enriched inside cristae and localized at CJs (346). Moreover, the latter study provided evidence that MIC19 interacted with subunit IV of CIV. Various interactions between MICOS subunits and mitochondrial fission/fusion proteins have been reported (Supplementary Table). For instance, the core MICOS protein MIC60 binds to OPA1 and together they control the number and stability of CJs (119).
3. Role of CV
Another important determinant of mitochondrial ultrastructure is CV of the OXPHOS system, the dimerization of which induces MIM curvature (Fig. 10B) (133). Dissociation of CV dimers into monomers was paralleled by inversion of MOM curvature at the CV site, MOM vesiculation, and cristae structural aberrations during aging in the model organism Podospora anserina (76). Evidence was provided that CV dimerization is mediated by interactions between its Fo and F1 domains and promotes cristae formation (262). Alternatively, it was proposed that CV dimerization is mediated by connecting its two F1 moieties by a protein bridge formed by ATPase inhibitory factor 1 (IF1) (262). Compatible with this connecting function, IF1 overexpression and knockdown increased and decreased mitochondrial volume and mitochondrial cristae density, respectively (44, 45). IF1 limits mitochondrial ATP consumption when mitochondrial respiration is impaired by inhibiting CV reverse-mode action (44). Moreover, IF1 inhibits OMA1-mediated processing of OPA1, thereby impeding cristae remodeling during apoptosis (101). Evidence in yeast suggests that the extent of CV dimerization and/or clustering depends on the cellular demand for OXPHOS-generated ATP (164).
4. Role of MitoNEET
Interestingly, adjacent mitochondria display a coordinated cristae architecture at inter-mitochondrial junctions (IMJs), where cristae form parallel arrays that are perpendicular to the IMJ, suggesting a role in electrochemical coupling (311). This study also provided evidence that MFNs are not required for IMJ formation and that IMJs are dynamic structures, the amount and function of which are dynamically regulated by the bioenergetic state of the mitochondrion. It was recently demonstrated that knockdown of MitoNEET (CDGSH iron-sulfur domain-containing protein 1 or CISD1), a dimeric outer membrane protein, reduced IMJ frequency (402). This suggests that MitoNEET plays a role in the regulation of IMJ formation.
E. Role of “non-core” proteins in mitochondrial fission, fusion, and ultrastructure
In addition to the “well-studied” mechanisms described earlier, there is a large body of empirical evidence in higher eukaryotes reporting additional proteins and pathways that affect mitochondrial (ultra)structure. These effects are mediated not only by these proteins interacting with core fission/fusion proteins (Supplementary Table) but also by other mechanisms. For instance, the mitochondrial protein TMEM11 (transmembrane protein 11) regulated mitochondrial morphology by a mechanism that was independent of DRP1/MFN (332). TMEM11 is the human orthologue of Drosophila PM1 (pantagruelian mitochondrion 1), which controls cristae biogenesis and mitochondrial diameter in flies (236). Also, TMEM135 (transmembrane protein 135) localized to mitochondria and its overexpression and/or mutation disturbed mitochondrial dynamics (206).
Overexpression and knockdown of optic atrophy 3 (OPA3), an integral MOM protein of which the gene is mutated in hereditary optic neuropathies, induced mitochondrial fragmentation and elongation, respectively (340). OPA3-overexpressing cells did not display spontaneous AC death but were sensitized to apoptosis induction by staurosporine- and tumor necrosis factor-related apoptosis-inducing ligand. This suggests that OPA1 and OPA3 exert different functions in apoptosis. OPA3 gene mutations were associated with mitochondrial fragmentation in patient fibroblasts (125). Mitochondrial fragmentation and spontaneous apoptosis were also induced after overexpression of a pathological OPA3 mutant (G93S), suggesting that OPA3 plays a role in mitochondrial fission and links this process to optic atrophy (340).
Another protein involved in mitochondrial morphology is mitochondrial fission process protein 1 (MTFP1). Knockdown of MTFP1 inhibited DRP1-mediated mitochondrial fission, leading to mitochondrial hyperfusion (386, 387, 407). Translation of MTFP1, and thereby the mitochondrial recruitment of DRP1, is regulated by the nutrient-sensing complex mechanistic/mammalian target of rapamycin complex 1 (270). This mechanism provides a link between environmental/intracellular stimuli and mitochondrial morphofunction. With respect to calcium homeostasis, mitochondria can undergo “mitochondrial shape transition” (MiST), which is distinct from mitochondrial fission/swelling and mediated by the mitochondrial Rho GTPase 1 (MIRO1) (280). MiST was induced by increased calcium levels in the cytosol, but not by mitochondrial calcium uptake by the mitochondrial calcium uniporter (MCU). It was further demonstrated that autophagy/mitophagy induction requires MIRO1-dependent MiST.
F. Secondary functions of mitochondrial fission and fusion proteins
It is often overlooked that mitochondrial fission/fusion proteins also exert important “secondary” functions in cell physiology (412). For instance, the Mfn2 gene is also known as Hsg (hyperplasia suppressor gene) and is involved in the control of cell proliferation and apoptosis. The MFN2 protein exerts its antiproliferative effect by inhibiting the RAS-RAF-ERK signaling cascade by N-terminally interacting with RAF-1 and C-terminally with RAF (55). In brown adipose tissue (BAT), MFN2 was involved in the docking of mitochondria to LDs, allowing efficient FA transfer to mitochondria for β-oxidation (37). In mice, MFN2 in BAT was required for cold-induced thermogenesis and promoted insulin resistance in obese animals (240). On dysfunctional mitochondria, MFN2 can also act as a Parkin-receptor, an E3 ubiquitin ligase implicated in Parkinson's disease (57). Mechanistically, it was proposed that mitochondrial Δψ depolarization leads to stabilization of PTEN-induced putative kinase 1 (PINK1) at the MOM. After this stabilization, PINK1 phosphorylates MFN2, thereby increasing the binding of Parkin to stimulate mitophagy. In this way, absence of MFN2 interrupted the PINK1-Parkin mitochondrial quality control mechanism, leading to accumulation of dysfunctional mitochondria (57). In addition to the pathway just described, there is considerable additional crosstalk between mitochondrial fusion and Parkin-mediated mitophagy (92, 382). Evidence in adipocytes suggests that OPA1 also can reside outside mitochondria on LDs to act as an A-kinase anchoring protein (313). It was proposed that OPA1 is involved in lipolysis of neutral lipids stored in the LDs. However, this mechanism is still controversial (23).
Other “secondary” functions of mitochondrial fission/fusion proteins include (Table 1): (i) execution of peroxisome fission (DRP1, FIS1, MFF) (181, 353, 354), (ii) MOM protein distribution (MFNs) (418), (iii) ER morphology regulation and ER-mitochondria interactions (MFN2) (77, 102), (iv) the unfolded protein response (MFN2) (272), (v) apoptosis (DRP1, FIS1, OPA1) (108, 378, 416), (vi) metabolic regulation (MFN1, MFN2) (86, 312), (vii) cell cycle and division (hFIS1, OPA1, MFN1) (208), (viii) mPTP opening (hFIS1, DRP1) (184), (ix) endocytosis (DRP1) (217), (x) calcium homeostasis (MFN2, DRP1) (54, 77, 102, 378), (xi) antiviral signaling (MFN2) (433), (xii) melanosome biogenesis (MFN2) (73), and (xiii) cell senescence (hFIS1, OPA1) (207). This multi-functionality is possibly responsible for apparent discrepancies in experimental results between various studies. For example, stimulation of mitochondrial fusion in Drosophila dramatically reduced ROS levels (276), whereas stimulation of mitochondrial fission suppressed oxidative damage in murine postmitotic neurons (168). This means that similar alterations in mitochondrial morphology (e.g., fragmentation or filamentation; Fig. 7B, C) can serve a different purpose depending on the cell type, upstream signaling pathway, metabolic state, and physiological context. Obviously, the “secondary” functions of mitochondrial fission and fusion proteins need to be taken into account for proper interpretation of experimental results.
Other Selected Functions of Core Mitochondrial Fusion and Fission Proteins
BAP31, B cell receptor associated protein 31; BAT, brown adipose tissue; CRAC, calcium release-activated calcium channel; DRP1, dynamin-related protein 1; ER, endoplasmic reticulum; FIS1, fission protein 1; HSC, hematopoietic stem cell; LD, lipid droplet; MFF, mitochondrial fission factor; MFN1/MFN2, mitofusin 1 and 2; MIRO, mitochondrial RHO GTPase; MOM, mitochondrial outer membrane; NFAT, nuclear factor of activated T cells; OPA1, optic atrophy protein 1; PERK, protein kinase RNA (PKR)-like ER kinase; PKA, protein kinase A; PM, plasma membrane; STIM1, stromal interaction molecule 1; UPR, unfolded protein response.
III. Regulation of Mitochondrial (Ultra)Structure
The regulation of mitochondrial morphology is a subject of intense study. This control is primarily exerted by modifying the function of mitochondrial fission and fusion proteins at various levels. This includes microRNAs (Supplementary Table), which act as negative regulators of gene expression by inhibiting mRNA translation or promoting mRNA degradation (205). Here, we focus on the role of post-translational modifications (PTMs) and mitochondrial lipids.
A. Regulation by post-translationlal modifications of fission and fusion proteins
Mitochondrial fission and fusion proteins contain various (predicted) PTM sites, allowing their phosphorylation, S-nitrosilation, sumoylation, ubiquitination, tyrosine sulfation, acetylation, and S-palmitoylation (5, 70, 279, 425). Depending on the nature and location of the PTM, either mitochondrial fission or fusion is stimulated (51, 103, 289). These mechanisms are explained in more detail below (sections III.A.1–12).
1. Stimulating fission by DRP1 phosphorylation at S616 (human isoform 1)
S616 corresponds to S579 in rats and mice. In mitotic cells and neurons, DRP1 phosphorylation at S616 is stimulated by cyclin-dependent kinases (CDKs) (63, 375, 380) and protein kinase Cδ (319). This phosphorylation stimulates DRP1 activity and mitochondrial fission. To the best of our knowledge, no phosphatases are currently described for this phosphorylation site. Mammalian DRP1 genes include three alternative exons and evidence was provided that alternative splicing generates a cytoskeletal DRP1 “reserve pool,” which translocates to the MOM on CDK-induced phosphorylation (375). This means that alternative splicing and phosphorylation might function as an isoform-specific mechanism that (co)controls DRP1 subcellular localization and mitochondrial fission. Remarkably, S616 phosphorylation by CDK5 inhibited DRP1, thereby contributing to mitochondrial elongation during neuronal maturation under physiological conditions (63). It was proposed that CDK5 reduces the oligomerization activity of DRP1.
2. Stimulating fission by DRP1 phosphorylation at S585
Analysis of a mouse model revealed that neuronal injury induced by N-methyl-D-aspartate was associated with DRP1 phosphorylation by CDK5 at S585 and mitochondrial fission (156).
3. Stimulating fission by DRP1 SUMOylation
SUMO (small ubiquitin-like modifier) proteins are attached to lysine residues of other proteins by the combined action of an E1 complex, a single E2, ubiquitin carrier protein 9, and several E3 enzymes (424). SUMOylation of DRP1 potentially stimulates its oligomerization at the MOM, leading to increased mitochondrial fission (416).
4. Stimulating fission by DRP1 glycosylation
In mouse cardiomyocytes, pharmacological inhibition of N-acetyl-glucosaminidase (OGA), which removes O-GlcNAc residues from Thr and Ser residues, resulted in increased O-linked-N-acetyl-glucosamine glycosylation of DRP1 at T585 and T586 (and possibly also S616 and S637) (115). Increased DRP1 glycosylation enhances GTP binding and MOM recruitment of this protein, paralleled by mitochondrial fragmentation and Δψ depolarization. Evidence was provided that increased O-GlcNAcylation decreases the phosphorylation of DRP1 at S637, compatible with stimulation of mitochondrial fission.
5. Stimulating fission by MFF phosphorylation at S155 and S172
MFF phosphorylation at S155 and S172 is mediated by AMP-activated protein kinase A (AMPK). This phosporylation stimulates mitochondrial fission by promoting DRP1 recruitment to the MOM (388).
6. Stimulating fission by MFN1 phosphorylation
ERK2 or MAPK1-mediated MFN1 phosphorylation inhibits MFN1 oligomerization, thereby stimulating mitochondrial fission (318).
7. Stimulating fission by MFN1 acetylation
MFN1 acetylation triggers its degradation via ubiquitination mediated by membrane-associated ring finger (C3HC4) 5 (MARCH5 or MITOL), thereby stimulating mitochondrial fission (304).
8. Stimulating fission by MFN2 phosphorylation at S27
During stress conditions (e.g., treatment with doxorubicin, cis-platinum, or tunicamycin), MFN2 is phosphorylated at S27 by c-JUN N-terminal kinase (JNK), which leads to recruitment of the ubiquitin ligase (E3) HUWE1/MULR/ARF-BP1/HECTH9/E3Histone/LASU1 to MFN2 (204). As a consequence, MFN2 is ubiquitinated and subsequently degraded, leading to mitochondrial fragmentation and enhanced AC death.
9. Stimulating fusion by DRP1 ubiquitination
Poly-ubiquitination of DRP1 leads to its degradation and is promoted by MARCH5 (278), the cytosolic E3 ubiquitin ligase Parkin (413), and anaphase-promoting complex/cyclosome and its coactivator CDH1 (APC/CCDH1) E3 ubiquitin ligase complex (144). MARCH5 is an integral MOM protein that interacts with ubiquitinated DRP1 and its overexpression stimulates mitochondrial filamentation (278). Parkin interacts with and ubiquitinates DRP1 to stimulate its proteasome-dependent degradation (413). During cell division, changes in mitochondrial morphology involve APC/CCDH1-driven DRP1 ubiquitination and its subsequent proteasomal degradation. Inhibition of this process prevents normal G1 phase regrowth of mitochondrial networks after cell division (144).
10. Stimulating fusion by DRP1 phosphorylation at S637 (human isoform 1)
S637 corresponds to S656 (in rat) and S600 (in mouse). This phosphorylation is stimulated by: (i) the proto-oncogene PIM1 (88), (ii) protein kinase A/A kinase anchor protein 1 (PKA/AKAP1) (52, 71), (iii) the rho-associated coiled coil-containing protein kinase 1 (ROCK1) (415), and (iv) calcium/calmodulin-dependent kinase 1α (CAMK1α) (136). The functional role of S637 phosphorylation is somewhat enigmatic. Under conditions of simulated ischemia, PIM1-induced S637 phosphorylation preserved mitochondrial filamentation by preventing DRP1 translocation to the MOM (88). Compatible with this result, PKA-induced phosphorylation of S637 decreased DRP1 GTPase activity (52, 71). In contrast, ROCK1-induced S637 phosphorylation stimulated DRP1 translocation to the MOM in a hyperglycemic mouse model (415). Similarly, Ca2+ influx through voltage-operated calcium channels blocked mitochondrial movement and stimulated CAMK1α-mediated S637 phosphorylation, associated with increased DRP1 recruitment to the MOM and mitochondrial fission in neurons (136). It was proposed that the functional consequences of S637 phosphorylation might be highly cell type and/or stimulus dependent (415).
Dephosphorylation of S637 involves: (i) phosphoglycerate mutase family member 5 (171), (ii) protein phosphatase 2A/Bβ2 (PP2A/Bβ2) (82), and (iii) calcineurin (CaN) (71). For these phosphatases, S637 dephosphorylation was associated with a stimulation of mitochondrial fission. Translocation of PPA2/Bβ2 to the MOM is regulated by its N-terminal phosphorylation (257). The Bβ2 regulatory subunit targets the scaffolding (A) and catalytic (C) subunits of PP2A to the MOM via transient association with receptor components of the TOM complex (426). In addition to PKA, AKAP1 also recruits other signaling enzymes to the MOM, including protein phosphatase 1 and CaN (426). Inducers of Δψ depolarization trigger Ca2+/CaN-dependent S637 dephosphorylation, which stimulates DRP1 translocation to the MOM and mitochondrial fission (49). Interestingly, it was recently demonstrated in mouse cells and brain lysates that the levels of S637-phosphorylated DRP1 displayed a 24-h rhythm. This was paralleled by rhythmic stimulation of mitochondrial fusion and circadian ATP production (349). This suggests that oxidative metabolism is regulated by the circadian rhythm in non-dividing cells mediated by cyclic DRP1 phosphorylation and inactivation.
11. Redox-dependent PTMs
Especially during pathological conditions, mitochondria are considered significant intracellular sources of ROS, which can not only potentially damage biomolecules but also play crucial signaling roles (188, 241, 357). Also, mitochondrial fission and fusion proteins constitute targets of various redox pathways, thereby allowing regulation of mitochondrial morphology (425): (i) nitration of Paraplegin and PKA/AKAP1 (potentially affecting DRP1/MFN2 activity and OPA1 processing); (ii) S-nitrosylation of DRP1 at C644 (potentially increasing its activity), OPA1 (potentially affecting its activity), Caspase 3 (inhibiting OPA1 processing), CDK5 (inhibiting DRP1 activity), Parkin (stimulating DRP1 and MFN2 activity), and JNK (stimulating MFN2 activity); (iii) disulfide bond alterations in amyloid-β (stimulating DRP1 activity), PKA/AKAP1 (inhibiting DRP1 activity), TIMM8A (potentially affecting DRP1 activity), and ROMO1 (stimulating OPA1 cleavage); and (iv) ROS-induced Δψ depolarization stimulating hydroxyl radical production, lipid peroxidation, protein carbonylation, and OPA1 cleavage.
Disulfide bond-mediated crosslinking between MFNs in a glutathione and GTP-dependent manner might contribute to mitochondrial hyperfusion that protects against oxidative stress (364). This protection might be exerted by sharing ROS-induced damage and/or mitochondrial antioxidants between individual mitochondria by stimulating fusion between “damaged” and “healthy” organelles (281). Recently, it was demonstrated that protein disulfide isomerase A1 (PDIA1) can function as a DRP1 thiol reductase (176). PDIA1 depletion stimulates DRP1 sulfenylation, thereby stimulating mitochondrial fragmentation.
12. Other PTMs
In case of MFN2, evidence was provided that its Parkin-mediated ubiquitination depends on phosphorylation of MFN2 residues T111/S442 by the mitochondrial kinase PINK1 (57). The same study demonstrated that MFN2 ablation prevents Parkin translocation to the MOM and suppresses mitophagy in mouse cardiac myocytes during Δψ depolarization. S442 can also be phosphorylated by PKA but this appears to play a role in MFN2-mediated suppression of vascular smooth muscle cell growth independent of mitochondrial morphology (446). MARCH5 binds and ubiquitinates mitochondrial MFN2 but not ER-associated MFN2, thereby regulating ER tethering to mitochondria by activating MFN2 via K192 ubiquitination (377). Polyubiquitinated MFN2 is then activated, leading to an increase in GTP-binding ability and is associated with the ER-localized MFN2, resulting in the oligomerization of MFN2 and attachment of mitochondria to the ER (277).
Evidence in yeast demonstrated the existence of two independent pathways for activation and degradation of MFNs to control MOM fusion (9, 423). In this mechanism, MOM fusion is activated by addition of stabilizing ubiquitin chains to MFN by an E3 ubiquitin ligase. Removal of these chains by a deubiquitylase (UBP12) then impairs MOM fusion and promotes mitochondrial fragmentation. In contrast, another E3 ligase attaches destabilizing ubiquitin chains to MFN, which are removed by a different deubiquitylase (UBP2) to support outer membrane fusion.
B. Role of mitochondrial lipids in mitochondrial (ultra)structure
Mitochondria display a distinct lipid composition when compared with other cell constituents (ER, PM, Golgi, and late endosomes) (399). More specifically, the MOM and MIM display a high content of non-bilayer phospholipids such as cardiolipin (CL) and phosphatidylethanolamine (PE) (384). Lipidomics analysis of mouse liver mitochondria revealed the following lipid composition: 25% PC (phosphatidylcholine), 18% CL, 14% PE, 10% phosphatidylglycerol, 10% phosphatidylinositol, 6% phosphatidylserine, 5% lysophosphatidylethanolamine, 5% sphingomyelin, 3% CER (ceramide), 3% lysocardiolipin, and 2% PA (phosphatidic acid) (13). In yeast, the MIM displayed a higher CL content than the MOM, whereas CL was virtually absent from the ER membrane (384). Mitochondrial membrane lipids are delivered by the ER or synthesized within mitochondria from ER-derived precursor lipids (347, 384). Inside the mitochondrion, phospholipids can be transported between the MIM and MOM and also be exchanged with the ER membrane. For instance, the lipid transfer protein STARD7 shuttles PC from the MOM to the MIM (342).
Various lipid-mediated signaling mechanisms are involved in the regulation of mitochondrial function. Examples include (254, 287): (i) mitophagy, where CL translocates from the MIM to the MOM to bind LC3 and LC3-B-II in collaboration with CER to induce autophagosomal mitochondrial recruitment; (ii) ETC assembly, where the chaperone PHB2 involved in CIV assembly interacts with sphingosine-1-phosphate (S1P); and (iii) apoptosis, where CER accumulates and acts together with Bax to induce pore formation, allowing cytochrome-c exit and apoptosis activation.
With respect to mitochondrial function, CL is the best studied mitochondrial lipid. CL has a dimeric structure, consists of four acyl chains and two phosphate groups, and plays a role in mitochondrial cristae organization. The latter is probably linked to the observation that the curvature of the MIM induces an asymmetric distribution of CL and PE (these segregate into the negatively curved monolayer leaflet facing the crista lumen) versus PC (this segregates into the positively curved monolayer leaflet facing the mitochondrial matrix (149). On CL incorporation, membrane bilayer deformability increases and CL becomes enriched in regions with a highly negative curvature (38).
Interestingly, specific proteins have been described that sense membrane curvature (membrane curvature sensing or MCS proteins) via their amphipathic helices (AHs) and Bin/Amphiphysin/Rcs BAR domains (239). Mechanistically, it was proposed that AHs detect and fill up lipid packing defects by inserting their hydrophobic part whereas BAR domains sense membrane curvature via electrostatic interactions (239). It is tempting to speculate that (CL-induced) changes in MIM curvature affect the stability and/or activity of integral membrane proteins (IMPs). Such an IMP-stabilizing effect might also be linked to the established role of CL in mitochondrial protein transport, MCU-mediated mitochondrial calcium uptake, and ETC functioning (35, 93). Indeed, CL-induced IMP stabilization has been described for individual ETC complexes in bovine and yeast as well as ETC supercomplexes and uncoupling protein 1 (274). Studying isolated CI revealed that its catalytic activity strongly depends on the phospholipid content of the preparation (359). CL was bound to CI, possibly playing a stabilizing role, and CI catalytic activity was influenced by binding of PE and PC to the complex (359).
Mitochondrial lipids have also been implicated in the regulation of mitochondrial (ultra)structure (131). CL and PA coordinate mitochondrial dynamics by interacting with DRP1, MFN, and OPA1 (16, 170). Using cryo-EM analysis of DRP1 on nanotubes with a distinct lipid composition, evidence was provided that DRP1-CL interactions activate DRP1 oligomers (106). These authors further suggested that MIM-to-MOM translocation of CL enhances mitochondrial fission. PA inhibits DRP1 activity (2, 174, 405). A mitochondrial phospholipase D (mitoPLD) was detected on the MOM to promote transmitochondrial membrane adherence by a mechanism that was MFN-dependent and involved CL hydrolysis to form PA (64). It was suggested that PA could play the following roles during mitochondrial fusion: (i) to stimulate opposing membrane fusion by generating negative membrane curvature, (ii) to recruit additional biomolecules involved in fusion, and (iii) to be converted in another fusogenic lipid. A later study suggested that a PA-preferring phospholipase A1 (PA-PLA1) plays a regulatory role in mitochondrial morphology regulation since its overexpression and knockdown induced mitochondrial fragmentation and elongation, respectively (14). In this mechanism, increased PA-PLA1 activity (which leads to lower PA levels to inhibit mitochondrial fusion) counterbalances mitoPLD activity (which increases PA levels to stimulate mitochondrial fusion).
Evidence in a fly model and mammalian cells revealed that loss of mitoguardin (MIGA) induces mitochondrial fragmentation via its interaction with mitoPLD (443). This study provided evidence that MIGA functions downstream of MFN by stabilizing mitoPLD and stimulating MFN dimerization. Also, PE appears to play a role in this process since a moderate reduction (<30%) in mitochondrial PE levels induced mitochondrial fragmentation, impairment of mitochondrial function and reduced cell growth (383). Similarly, a role for lysophosphatidic acid (LPA) was proposed in mitochondrial fusion (292). This study revealed that mitochondrial GPAT (glycerol-3-phosphate acyltransferase; mitoGPAT) catalyzes the initial and rate-limiting step in LPA synthesis. Analyses in Caenorhabditis elegans and mammalian cells demonstrated that mitoGPAT depletion induced mitochondrial fragmentation, which was rescued by LPA. This suggests that mitoGPAT-mediated LPA-formation plays a role in mitochondrial fusion (292).
IV. The Concept of Mitochondrial Morphofunction
Changes in mitochondrial (ultra)structure and function are bidirectionally linked in many ways, and there are numerous studies in which (ultra)structural modulation induces functional changes and vice versa. The strong coupling between mitochondrial (internal) structure and function suggests that they are equivalent, thereby giving rise to the concept of mitochondrial “morphofunction.” This term was first proposed by Benard and Rossignol (24) when they defined “mitochondrial morphofunctional analysis” as: “the simultaneous quantification of mitochondrial morphological and functional readouts.” Later, we illustrate the importance of the mitochondrial morphofunctional concept by presenting results obtained in knockout (KO) animals and patients with mutations in mitochondrial fission/fusion proteins. Next, we discuss various other experimental studies that highlight the bidirectional link between mitochondrial (ultra)structure and function. Finally, we briefly summarize how mitochondrial morphofunction can be quantified in living cells, how mitochondrial (ultra)structure links to mitochondrial bioreactions, and how mitochondrial morphology can be targeted by small molecules.
A. Studies in knockout animals
Animal models have delivered valuable insights into the physiological and functional role of fission and fusion proteins. For instance, Drp1 gene KO mice display developmental abnormalities, particularly in the forebrain, and die after embryonic day 12.5 (152). The same study revealed that neural cell-specific Drp1-null animals die shortly after birth due to brain hypoplasia and apoptosis. Compatible with the dual role of DRP1 in mitochondrial and peroxisomal fission (see section III and Table 1), Drp1-null cells contain extensive mitochondrial networks and elongated peroxisomes, although a reduction in cellular ATP level was not detected (408). In postmitotic Purkinje cells of mouse cerebellum, Drp1 deletion induced mitochondrial elongation followed by formation of large spherical mitochondria due to oxidative stress (168). These mitochondrial spheres lost respiratory function, and accumulated ubiquitin and markers of mitophagy, leading to neurodegeneration. It was concluded that fission of mitochondria prevents oxidative damage, thereby contributing to neuronal survival. This is compatible with the idea presented earlier that fusion of individual mitochondria allows sharing of ROS-induced damage and/or antioxidants within the mitochondrial population (281).
MFN1 and MFN2 are of key importance for proper embryonic development, and KO mice die before mid-gestation (56). Loss of Mfn2 resulted in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit (310). The current experimental evidence in various pathological models suggests that increased levels of L-OPA1 are protective (238). Such a protection was induced by Oma1 KO (198, 429), increased OMA1 degradation (432), or OPA1 overexpression (400). Compatible with the latter, increased L-OPA1 processing was not detected during OPA1 overexpression associated with (mild) improvement of mitochondrial disease phenotypes in (CI-deficient) Ndufs4 KO mice and (CIV-deficient) Cox15 muscle-specific KO mice (66). In adult cardiomyocyte-specific Yme1l KO mice, L-OPA1-mediated mitochondrial fusion was required to preserve cardiac function whereas stress-induced OMA1-mediated processing of L-OPA1 and accompanying mitochondrial fragmentation induced dilated cardiomyopathy and heart failure (406). In this sense, Oma1 gene deletion and normalization of mitochondrial structure protected against cell death and heart failure. Interestingly, mitochondrial fragmentation triggered a switch in metabolism from FA to glucose utilization and reversing this switch preserved normal heart function even though mitochondrial fragmentation was not normalized (406). This demonstrates that in the absence of YME1L, heart function can be restored by preventing OMA1-mediated L-OPA1 processing and restoring mitochondrial morphology, or by normalizing metabolism that bypasses the deleterious effects of altered mitochondrial morphology on heart metabolic function (406).
B. Patients carrying mutations in mitochondrial fission and fusion proteins
Mutations in mitochondrial fission/fusion proteins induce human disease (11). This includes congenital microcephaly, lactic acidosis and sudden death (DRP1) (417), Leigh-like encephalopathy, optic atrophy and peripheral neuropathy (MFF) (182), Charcot-Marie-Tooth neuropathy type 2A (MFN2) (450), and optic atrophy (OPA1) (4). In addition, various alterations in mitochondrial fission and fusion proteins were demonstrated in experimental models of neurodegeneration (103, 180): (i) The levels/activity of DRP1 and mitochondrial fission were increased in a rat model of amyotrophic lateral sclerosis (various gene mutations), a mouse model of Alzheimer's disease (AD; various gene mutations), and human/mouse/rat models of Huntington's disease (HD; Huntingtin mutation); (ii) the levels/activity of DRP1 and mitochondrial fission were decreased in human and mouse models of autosomal-recessive spastic ataxia of Charlevoix-Sanguenay disease (SACS/Sacs gene mutation); (iii) the levels/activity of ganglioside-induced differentiation-associated protein 1 (GDAP1) and mitochondrial fission were reduced in human models of autosomal dominant Charcot–Marie–Tooth disease, axonal (CMT2K; GDAP1 gene mutation), autosomal recessive Charcot–Marie–Tooth disease, axonal, type 2K (ARCMT2K; GDAP1 gene mutation), and autosomal recessive Charcot–Marie–Tooth disease, type 4A, demyelinating (CMT4A; GDAP1 gene mutation); (iv) the levels/activity of MFN2 and mitochondrial fusion were reduced in human and rat models of autosomal dominant Charcot–Marie–Tooth disease, axonal, type 2A2 (CMT2A2; MFN2/Mfn2 gene mutation); and (v) the levels/activity of OPA1 and mitochondrial fusion were reduced in human and rat models of autosomal dominant optic atrophy (OPA1/Opa1 gene mutation).
With respect to CMT2A2, the consequences of its mutations on MFN2 activity and neuronal function were recently investigated in a Drosophila fly model (96). Analysis of four pathological mutations (R94Q, R364W, T105M, and L76P) revealed that they all increased mtDNA mutations, decreased oxidative metabolism, and induced mitochondrial depletion at neuromuscular junctions. R94Q and T105M mutations induced aggregation of nonfused mitochondria and their loss of function. Remarkably, R364W and L76P mutations stimulated mitochondrial fusion. It was proposed that both excessive mitochondrial aggregation and fusion can underlie CMT2A pathophysiology (96). Changes in mitochondrial (ultra)structure and mitochondrial function have also been described in human aging (356) as well as in a plethora of other human pathologies, including: (i) Parkinson's disease (339), (ii) AD (126), (iii) HD (130), (iv) ischemia/reperfusion injury (215), (v) mitochondrial metabolic disorders (196), (vi) cardiac disease (92), (vii) kidney disease (27), (viii) cell starvation (325), (ix) diabetes (336), (x) cancer (390), (xi) sepsis (122), and (xii) the effects of environmental toxins (163).
C. Bidirectional links between mitochondrial (ultra)structure and function
Next, we further elaborate on the concept of mitochondrial morphofunction by presenting experimental evidence demonstrating that: (i) mitochondrial (ultra)structure affects mitochondrial function, (ii) mitochondrial function affects mitochondrial (ultra)structure, (iii) mitochondrial internal structure affects mitochondrial external structure, (iv) mitochondrial external structure affects mitochondrial internal structure, (v) mitochondrial morphofunction affects cell function, and (vi) cell function affects mitochondrial morphofunction.
1. Mitochondrial (ultra)structure affects mitochondrial function
It is firmly established that MIM dynamics and topology impact on mitochondrial bioenergetics (244, 306 –308). For instance, changes in mitochondrial morphology and organization can enhance energy supply from OXPHOS in diabetic cardiomyopathy (159). Analysis of heteroplasmic human rhabdomyosarcoma cells harboring the pathological A3243G mtDNA mutation, associated with the neuromuscular syndrome MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), revealed that knockdown of DRP1 or FIS1 leads to increased levels of mutated mtDNA (242). This increase was not observed on knockdown of OPA1, suggesting that OPA1-dependent MIM fission and cristae organization are not involved in segregation of mutant and wild-type mtDNA (242). Supporting these results, MFN-mediated mitochondrial fusion protected against neurodegeneration in the cerebellum (58) and was required for mtDNA stability and tolerance of mtDNA mutations in skeletal muscle (59). MFN2-induced mitochondrial elongation protects against ROS-induced mPTP opening and represses staurosporin-induced BAX and cytochrome-c release (281).
During starvation-induced autophagy, the MOM participates in autophagosome formation by supplying membranes (134) and DRP1 was phosphorylated in a cAMP/PKA-dependent manner (120). This prevented DRP1 translocation to the MOM and induced formation of elongated mitochondria, which maintained their ATP production, contained more cristae, displayed increased CV levels/activity, and were not undergoing mitophagy. As a consequence, these elongated mitochondria were protected from autophagosomal degradation (326). Similarly, DRP1 knockdown and mitochondrial elongation protected melanoma cells from mitophagy-mediated death induced by CI inhibition (20).
Using a cell starvation model, evidence was provided that the autophagic process provides FAs for LDs, which transfer into mitochondria only if the latter have a tubulated morphology (325). This mechanism, which also required LD lipolysis, allows cells to survive during starvation by shifting their metabolism toward FA oxidation (409). Later studies argued that LDs are not required for FA delivery to mitochondria but instead function to prevent acylcarnitine accumulation and lipotoxic dysregulation of mitochondria (283). Further, a DRP1-independent mitophagy pathway was described (430). In this respect, inhibition and stimulation of the autophagy pathway were associated with increased and decreased DRP1 levels, respectively, suggesting that DRP1 is targeted by the autophagy process for lysosomal degradation (317).
Interestingly, both mitochondrial hyperfusion and hyperfragmentation protected cells against BAX-mediated apoptosis induction, suggesting that BAX requires a distinct mitochondrial shape to induce mitochondrial outer membrane permeabilization (330). The latter study proposed three factors that regulate BAX-mediated apoptosis: (i) a combination of stress-specific BH3-only proteins that induce apoptosis, (ii) an MOM composition that is regulated by lipid metabolic pathways, and (iii) a distinct net mitochondrial morphology resulting from the combined action of mitochondrial dynamics proteins that allows proper BAX integration and pore formation. In this sense, the large variety in mitochondrial morphology observed using different cell types and (patho)physiological conditions suggests that this structural diversity contributes to the differences in apoptosis sensitivity in these systems (330). The mitochondrial content of cells depends on cell size (177) and impacts mitochondrial function (259). In this context, single-cell analysis revealed that mitochondrial content affects the level of apoptotic proteins and thus cell sensitivity to apoptotic stimuli, meaning that cells with more mitochondria were more prone to cell death induction (246).
2. Mitochondrial function affects mitochondrial (ultra)structure
Mitochondrial Δψ regulates the configuration of the mitochondrial matrix and cytochrome-c release during apoptosis (123). Classical studies revealed that alterations in mitochondrial functional state, as typically observed in a wide range of (patho)physiological conditions and human diseases, are not only associated with altered mitochondrial dynamics but also affect the ultrastructure of the MIM. In response to low ADP concentrations, the structure of the MIM changed from a condition in which the mitochondrial matrix was contracted and dense (the “condensed state”), to a less dense and more expanded state (“orthodox”), associated with a more compact cristae compartment (132).
Inactivation of OPA1 by proteolytic cleavage mediated by OMA1 and YME1L is induced during cellular stress, leading to stimulation of mitochondrial fission (238). OPA1 cleavage also depends on mitochondrial ATP levels and bivalent metals (18). Both OMA1 and YME1L are activated by Δψ depolarization, thereby inducing OPA1 cleavage, mitochondrial fragmentation, and removal of damaged mitochondria by mitophagy (138, 238, 441). Inhibitory OMA1-mediated processing of OPA1 is also stimulated by knockdown of the m-AAA proteases AFG3L1 and AFG3L2 (95). Moreover, in the absence of the MIM PHB2, L-OPA1 is lost and mitochondrial fusion is defective (256). An MIM-specific effect on OPA1 cleavage was demonstrated in cells from patients with mtDNA mutations. This effect was mediated by YME1L, which cleaved OPA1 more efficiently under conditions at which OXPHOS activity was high (265). When Δψ drops below a certain threshold, DRP1-induced mitochondrial fission and OMA1-mediated cleavage of OPA1 are activated (165). OPA1 processing mediated by YME1L was modulated by Δψ, and OPA1 isoforms interact with MFN1 and MFN2 in a Δψ-independent manner (129). In combination with cellular ATP depletion, Δψ depolarization is paralleled by OMA1 activation and YME1L degradation (324). This differential degradation of OMA1 and YME1L affects the proteolytic processing of OPA1, which might allow cells to adjust mitochondrial morphology and function to different types of stress (324). Evidence was provided that Δψ-dependent processing of OPA1 impacts mitochondrial function (152, 294). In mouse muscle, age-associated loss of OPA1 can activate a signaling cascade starting from the ER that systemically affects metabolism and aging (385).
3. Mitochondrial internal structure affects mitochondrial external structure
A recent study in neuronal cells demonstrated that spontaneous and repetitive constrictions of the mitochondrial inner compartment (CoMIC) were closely associated with subsequent division of the mitochondrion (62). CoMIC was stimulated by increased matrix Ca2+ levels, mitoKCa-mediated ultrastructural changes, and Δψ depolarization and OMA1-catalyzed accumulation of S-OPA1, suggesting that these intra-mitochondrial constrictions prime mitochondrial division.
4. Mitochondrial external structure affects mitochondrial internal structure
Interestingly, cristae remodeling during apoptosis requires mitochondrial fission mediated by DRP1, MIEF1/MID51, and MIEF2/MID49 (298). This study revealed that during intrinsic apoptosis Mief1/MiD51-Mief2/Mid49-KO, and Drp1-KO cells did not exhibit cristae remodeling and cytochrome-c release, whereas this was not the case in Mff-KO cells.
5. Mitochondrial morphofunction affects cell function
Fis1 overexpression in HeLa cells induced fragmentation and perinuclear clustering of mitochondria that maintained a normal Δψ and normally sequestered Ca2+ released from the ER (110). In contrast, Ca2+ entering across the PM was taken up more slowly by these mitochondria, compatible with a role of subplasmalemmal mitochondria in modulating the activity of PM Ca2+ATPases (109). Supported by other studies (77, 434), this demonstrates that mitochondrial morphological aberrations affect (local) cellular Ca2+ homeostasis. Continued uptake of multiple ACs by phagocytes (a process known as efferocytosis) requires DRP1-dependent mitochondrial fragmentation in the phagocytes (414). The latter study demonstrated that mitochondrial fragmentation allows the release of Ca2+ from the ER into the cytosol to allow phagocytosis by stimulating Ca2+-mediated vesicle trafficking.
MFN2 KO in POMC (pro-opiomelanocortin) neurons (involved in the regulation of energy and glucose metabolism) induced defective POMC processing, loss of mitochondria-ER contacts, ER stress-induced leptin resistance, hyperphagia, reduced energy expenditure, and obesity (350). In agreement with these findings, DRP1-induced mitochondrial fission reduces the sensitivity of POMC neurons for leptin and glucose (345).
Mitochondrial structure affects cell cycle phase and vice versa (143). For instance, CDK5 inhibits DRP1 by phosphorylation (63) and mitochondria form a hyperfused network at the G1-to-S transition associated with buildup of cyclin E (268). The latter study also revealed that Δψ depolarization at early G1 prevents cell cycle entry into S phase. Perhaps related to this phenomenon, Δψ loss in HeLa cells was linked to homogenization and decompaction of chromatin structure, suggesting that the latter is linked to mitochondrial function (7).
6. Cell function affects mitochondrial morphofunction
Mitochondrial fusion appears to be ATP dependent (251), suggesting that mitochondria-generated or glycolytic ATP might (locally) affect mitochondrial fusion dynamics. In this context, high glucose induced mitochondrial fragmentation paralleled by increased ROS levels (391, 440) and inhibition of glycolysis impacted MIM fusion more than it did MOM fusion, whereas Δψ dissipation prevented MIM fusion but not MOM fusion (342).
In endothelial cells, mitochondrial morphology in situ was altered by Ca2+ overload (36). Using the same cell type, it was observed that KO of ROS-sensitive TRPM2 (transient receptor potential cation channel M2) channels prevented glucose-induced mitochondrial fission (1). The latter study proposed a mechanism in which: (i) TRPM2-mediated Ca2+ entry induces permeabilization of lysosomal membranes and lysosomal Zn2+ release, and (ii) the released Zn2+ stimulates mitochondrial DRP1 recruitment to induce fission (1).
AMPK is a key regulatory protein in cellular energy homeostasis (221). Mitochondrial morphofunction is linked to cellular bioenergetics via AMPK, which phosphorylates MFF to enhance DRP1 recruitment and induce mitochondrial fission (388). Interestingly, unphosphorylated MFF mutants inhibit mitophagy, connecting AMPK to mitochondrial fission and mitophagy regulation (388, 442).
Mitochondrial morphology changes were also induced via altered fusion dynamics during hypoxia-reoxygenation stress (222). These changes involved formation of donut-like (toroidal) mitochondrial structures during hypoxia in glucose-free media and reoxygenation-induced formation of these structures in glucose-containing medium. Formation of these donut-like morphologies involved partial mitochondrial detachment from the cytoskeleton and mitochondrial swelling, involving opening of the mPTP or mitochondrial K+ channels, allowing “autofusion” (222). Computational analysis suggested that an increase in osmotic potential during stress conditions induces donut formation and that this process is reversible (228).
D. Quantification of mitochondrial morphofunction in living cells
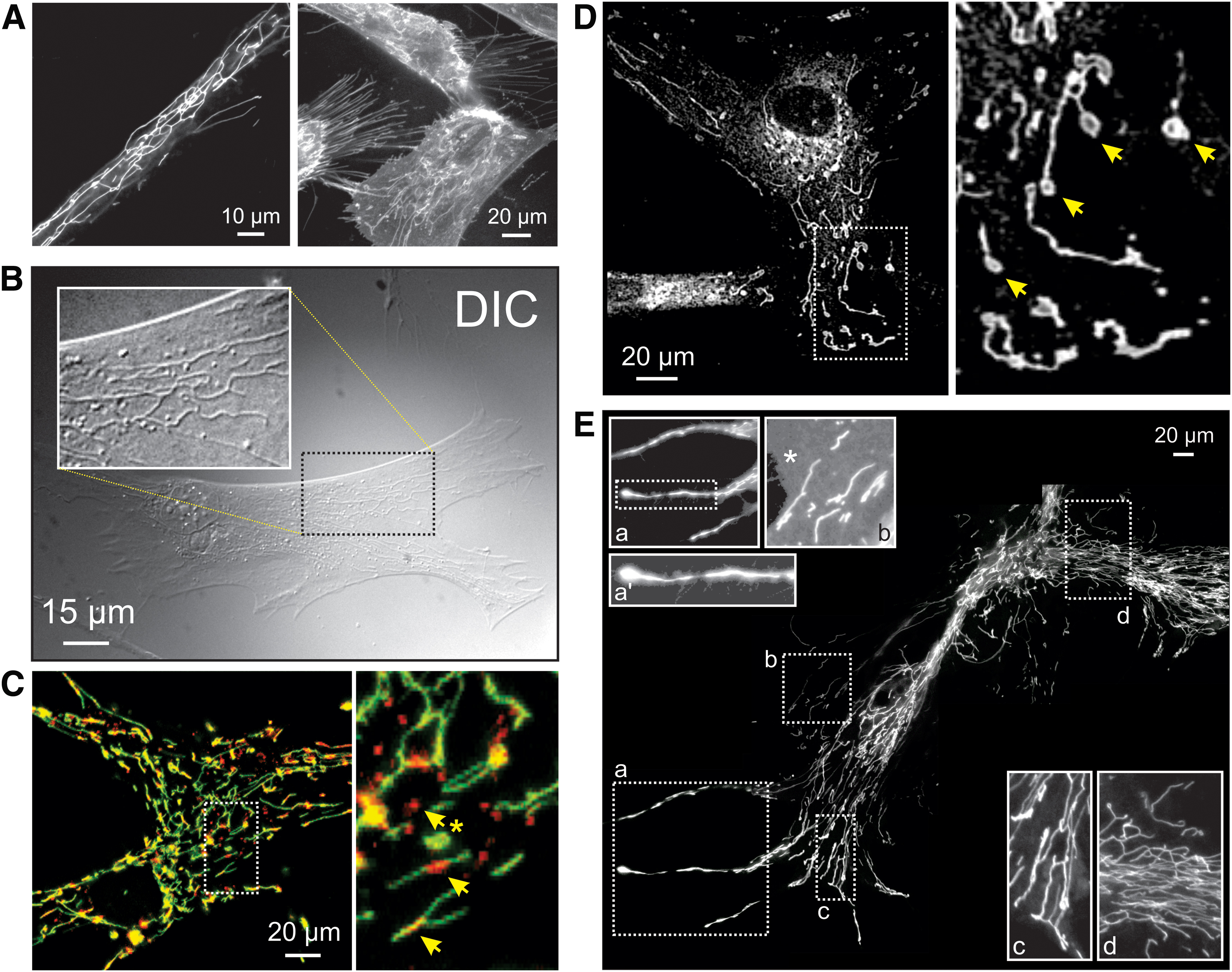
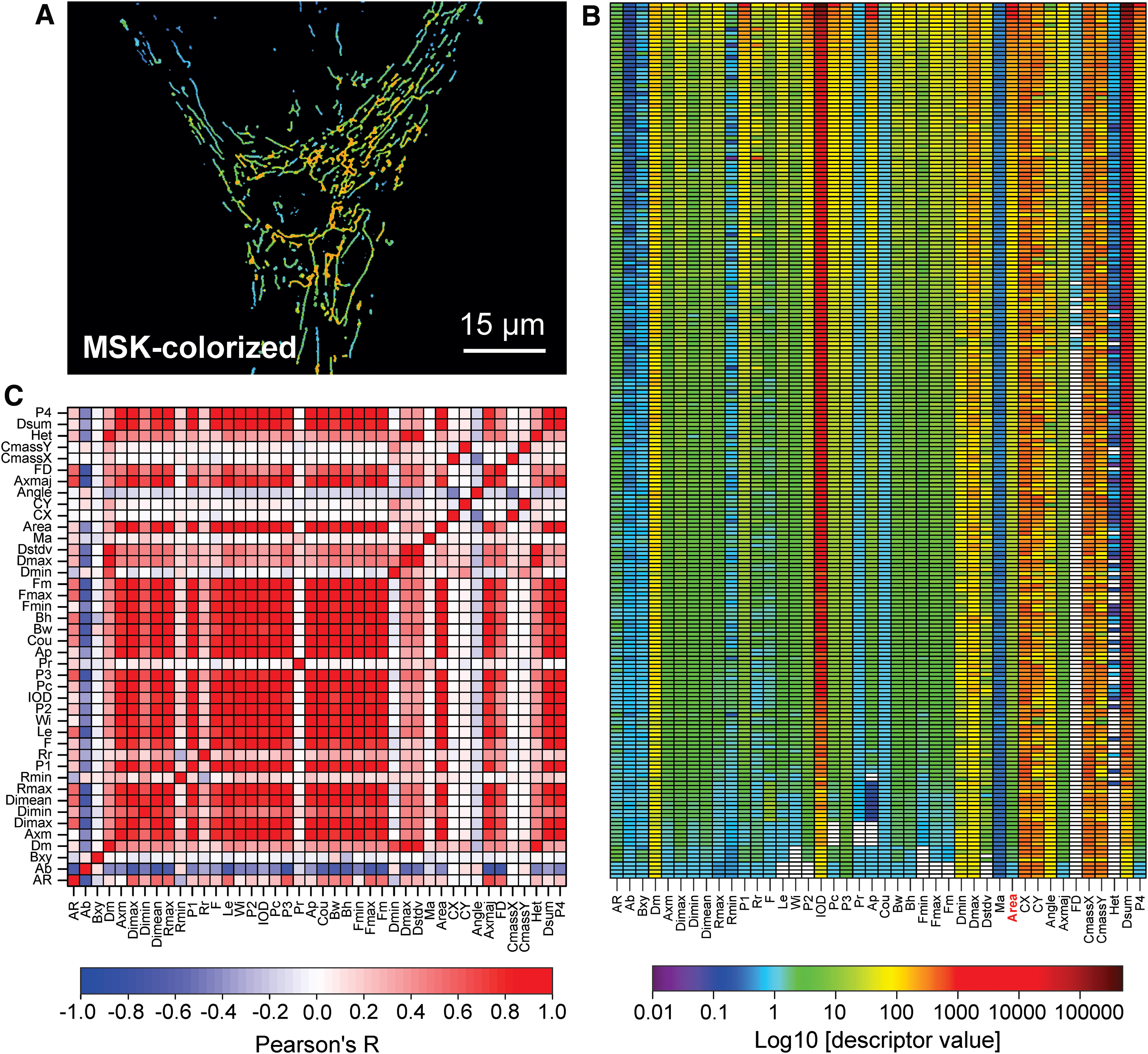
Although substantial progress has been made (24, 142, 266, 355, 407), we are still far from understanding how the morphofunctional heterogeneity of individual mitochondria within a single cell (Figs. 5, 7B, and 11) is maintained and how this heterogeneity connects to mitochondrial and cellular physiology. This lack of insight not only limits our understanding of mitochondrial morphofunction at the mechanistic level but also hampers the development and (large-scale) screening of morphofunction-targeting drugs. Given the tight connection between mitochondrial and cellular function, physiological analysis of mitochondrial morphofunction is ideally carried out within the context of a living cell (147, 148, 188, 196). If required, this can also entail analysis of mitochondrial fusion dynamics and/or motility using photoactivatable proteins (173, 231, 248). Micropatterned coverslips have been used to “standardize” cell size and shape and allow analysis of mitochondrial structure under controlled conditions (60). However, alterations in cell shape and cell-substrate adherence might affect mitochondrial mechanical interactions with the cytoskeleton and therefore mitochondrial (ultra)structure (19).
Quantification of mitochondrial internal and external structural parameters (Fig. 12) is not trivial, given the small dimensions of this organelle (diameter typically <1 μm). For this reason, mitochondrial ultrastructure has primarily been visualized by using EM-based approaches, although only in fixed cell specimens (107, 113, 233). In addition, various superresolution microscopy techniques (104, 396) have been applied to mitochondrial (ultra)structural analysis (157). These include: structured illumination microscopy (124, 358), 4Pi-confocal microscopy (94), STimulated Emission Depletion microscopy (158), Gated STimulated Emission Depletion microscopy (28), stochastic optical reconstruction microscopy (STORM), 3D-STORM, and direct STORM (146, 178) and the “Zernike Optimized Localisation approach in 3D” (12). Unfortunately, the superresolution approaches that are currently available, have not been optimized for live-cell imaging and lack a sufficiently high acquisition rate (i.e., with low subsecond time resolution) and axial imaging depth to allow real-time analysis of mitochondrial ultrastructural dynamics in living cells (104).
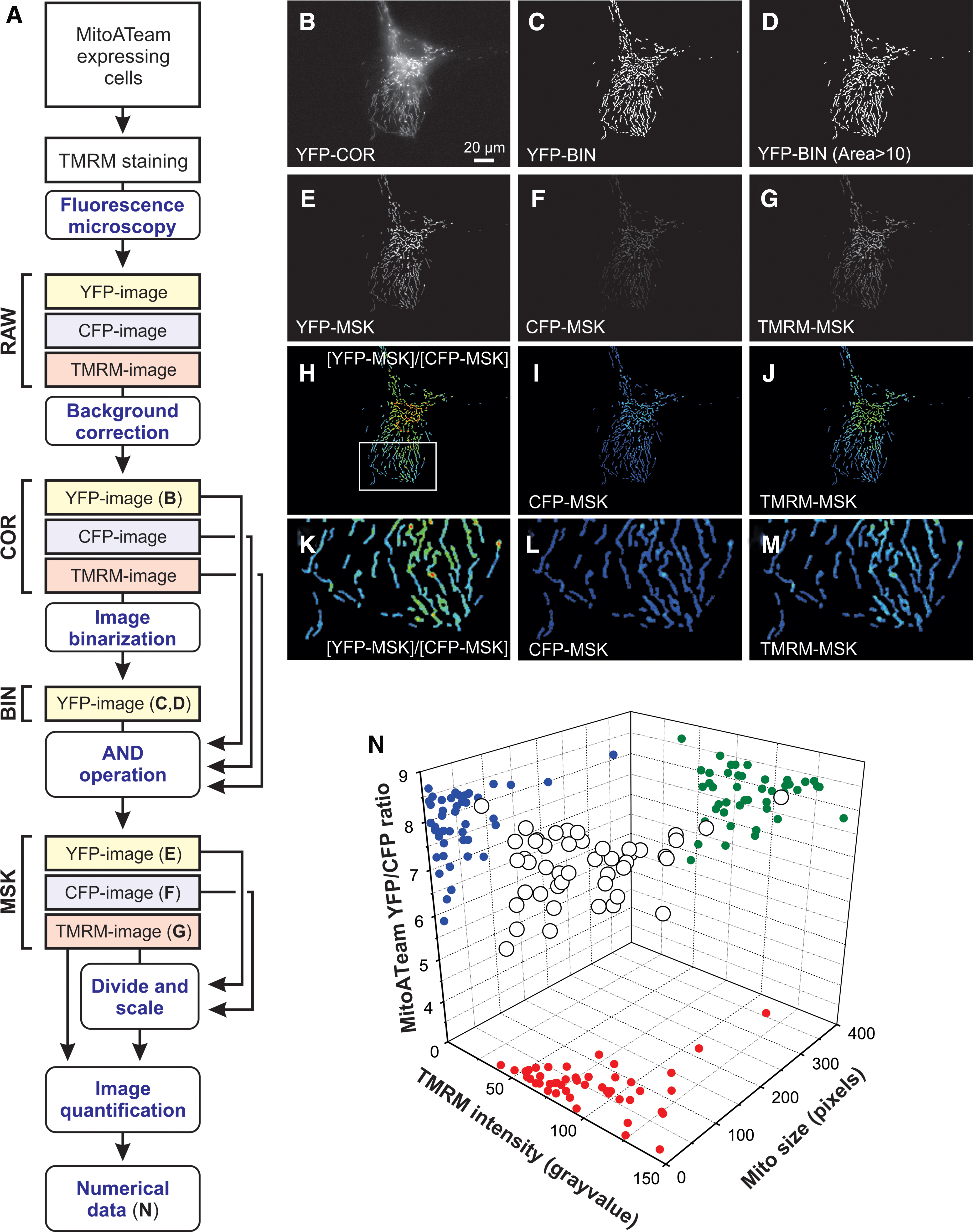
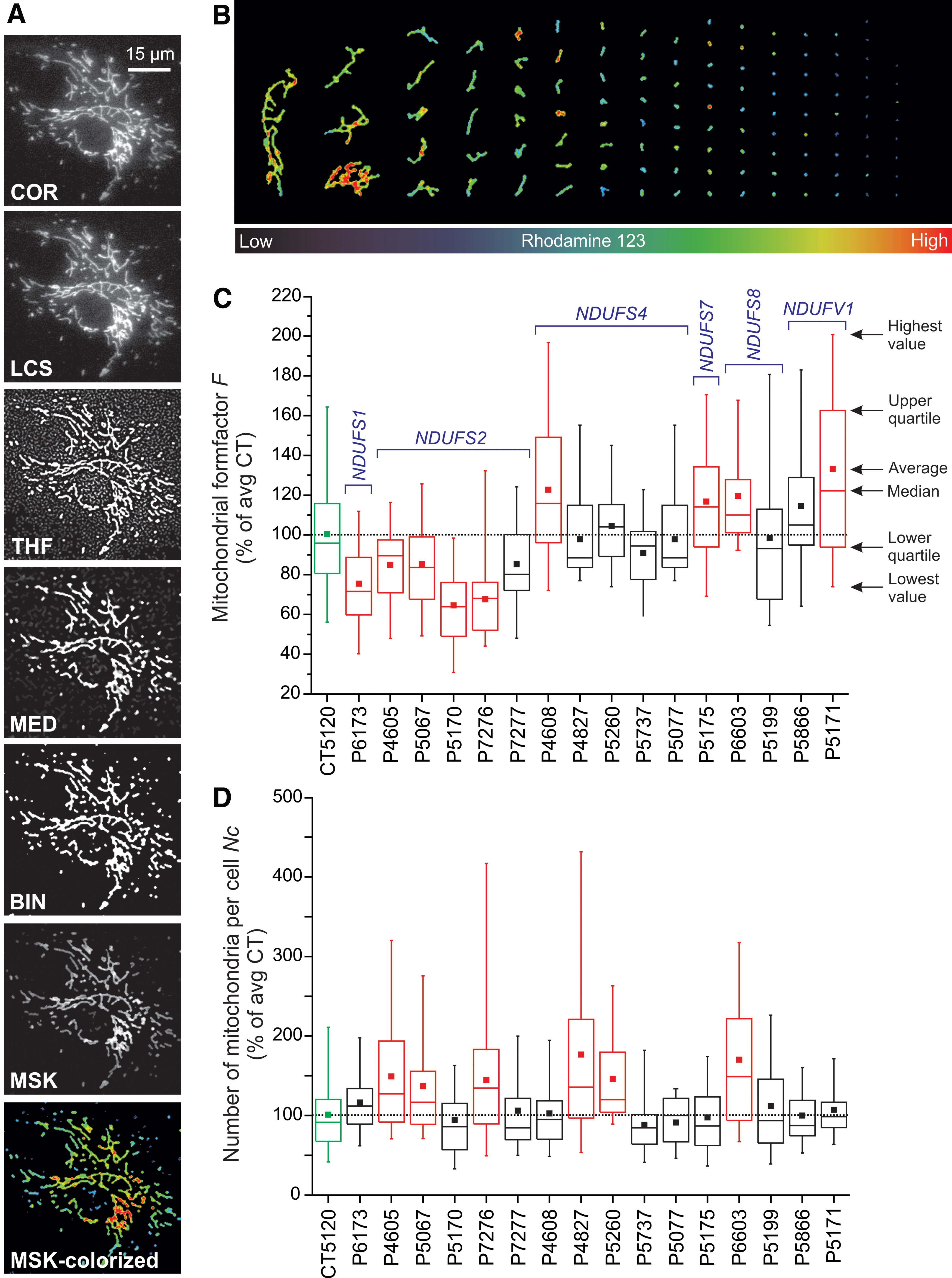
Currently, live-cell visualization of mitochondrial morphology and function is primarily carried out by combining fluorescent cations and/or mitochondria-targeted fluorescent proteins (FPs) with confocal laser scanning microscopy (CLSM) or epifluorescence microscopy. During the past decade, we and others had developed integrated experimental and computational strategies for semiautomatic quantification of mitochondrial morphology (Fig. 13) (78, 192, 194, 389). Our approach is optimized for analysis of relatively large flat cells such as primary human skin fibroblasts (PHSFs) but, after optimization, is also applicable to other cell types (147). As an alternative, (fixed) non-flat cell types and/or tissues can be analyzed by using 3D-CLSM (22, 288). In case of PHSFs, high-content microscopy images are acquired by using cells co-stained with: (i) tetramethylrhodamine (TMRM), which accumulates in mitochondria in a Δψ-dependent fashion; (ii) Calcein-AM, which accumulates in the cytosol; and (iii) Hoechst 33258, to stain nucleic acids (dsDNA). In a collaborative effort, we also developed high-content protocols for cells co-stained with TMRM and 5-(and-6)-chloromethyl-2’,7’-dichlorodihydrofluorescein (CM-H2DCFDA), a reporter of cellular oxidant levels (365). The acquired images are then processed (Fig. 14A) to allow extraction of various descriptors of mitochondrial morphofunction (e.g., number of mitochondria per cell, mitochondrial size, TMRM staining intensity; see Table 2). These descriptors then can be used for quality control and further evaluated by principal component analysis, univariate/bivariate/multivariate methods, and machine-learning strategies (Fig. 14) (3, 22, 30, 214, 329). For example, quantitative analysis and machine learning were applied to determine whether certain mitochondrial morphologies pre-disposed individual mitochondria to undergo subsequent fission or fusion (421). This revealed that mitochondrial perimeter and solidity (i.e., the compactness of its shape) positively and negatively correlated with the occurrence of a mitochondrial fission or fusion event, respectively. It was concluded that the mechanical properties of the MOM might influence mitochondrial fission and fusion probability (421).
Descriptors of Mitochondrial Morphofunction for Tetramethylrhodamine
Adapted from the definitions in the Image Pro Plus software. Whenever the measurement unit is not specified between brackets, it is arbitrary units.
TMRM, tetramethylrhodamine.
E. A brief primer on the role of mitochondrial (ultra)structure in mitochondrial bioreactions
Understanding the dynamics and design principles of biochemical reactions within an inhomogeneous cell compartment such as the mitochondrion still represents a major challenge (127, 373). This is due to the fact that mitochondrial bioreactions are spatially compartmentalized, meaning that they generally involve the conversion of (im)mobile substrates by (im)mobile enzymes into (im)mobile products. As a consequence, these reaction systems require biomolecule transport and/or diffusion within or between mitochondria and, therefore, constitute “reaction-diffusion” systems (226, 419).
It is higly likely that changes in mitochondrial (ultra)structure affect mitochondrial function by modulating mitochondrial bioreactions, for instance by: (i) changes in MOM surface area and matrix volume, and (ii) changes in the extent of MIM folding, which affects cristae number/morphology/dimensions/topology. In addition, these bioreactions will be affected by alterations in matrix solvent properties, including ionic strength, viscosity, and macromolecular crowding. In this manner, changes in mitochondrial (ultra)structure and matrix physicochemical parameters could affect many diffusion-influenced mitochondrial reactions, including: (i) metabolic reactions (ATP, metabolites), (ii) protein folding, (iii) protein–protein interactions (signaling and macromolecular assembly), and (iv) mtDNA replication/translation. In addition, physicochemical differences between individual mitochondria could be underlying Δψ and functional inhomogeneities between and within single mitochondria.
Although the general properties of reaction-diffusion systems are relatively well understood, detailed quantitative information on solute diffusion and biomolecular interactions in the mitochondrial matrix is currently lacking (25, 91, 97, 155, 295, 351). The analysis of protein diffusion in the mitochondrial matrix generally involves expression of an FP with favorable spectral and biophysical properties. FP mobility then can be determined by using fluorescence recovery after photobleaching (FRAP), fluorescence loss in photobleaching (FLIP; Fig. 15), and/or fluorescence correlation spectroscopy (FCS) analysis (10, 85, 187, 189). Given the complex nature of FP diffusion within the matrix (i.e., it is affected by the molecular weight (MW) of the FP, cristae diffusion barriers, mitochondrial length and diameter, the size of the bleach region, and solvent viscosity), one will always measure an “apparent” FP fluorescence recovery time (FRAP/FLIP) or diffusion constant (FCS). This necessitates the use of a diffusion model in which various parameters are experimentally constrained (i.e., mitochondrial dimensions, number of cristae, FP concentration, and FRAP region size) (84). Applying this strategy in HEK293 cells, we demonstrated that, as expected (303), FP diffusion in the mitochondrial matrix is MW-dependent. Unexpectedly (305, 401), our experiments also revealed that cristae severely hinder FP diffusion. This suggests that differences in mitochondrial ultrastructure, as observed between cell types and between healthy and pathological conditions (403, 447) affect diffusion-limited reactions in the mitochondrial matrix. Moreover, it is highly probable that changes in mitochondrial metabolic state accompanied by orthodox-to-condensed transitions (132, 335) will also affect intra-matrix solute diffusion. This implies that alterations in mitochondrial nanostructure, as observed during numerous (patho)physiological conditions, can affect the properties of intramatrix reaction-diffusion systems and therefore mitochondrial and cellular function (84).

In contrast to FPs in the matrix, early evidence suggested that MIM-embedded ETC complexes exhibit restricted diffusion (117). Indeed, after artificial cell fusion, FP-labeled ETC complexes and CV displayed a patterned arrangement, possibly as a result of their incorporation in supercomplexes within cristae (275). This suggests that after mitochondrial fusion complete equilibration of ETC complexes, being MIM-embedded, is slow relative to FP diffusion in the mitochondrial matrix.
The volume of the reaction compartment is a key parameter in chemical reaction dynamics. Changes in mitochondrial volume (Fig. 7A) were induced by alterations in OXPHOS function, ischemia/reperfusion, anoxia, and apoptotic signaling. Evidence was provided that these volume changes (co)control mitochondrial function (48, 225, 291). Mitochondrial volume depends on the osmotic balance between the cytosol and the mitochondrion (167) and is mainly regulated by potassium (K+) ions (Fig. 1), which enter and leave the mitochondrial matrix in a Δψ-dependent manner (341). The latter study revealed that Δψ depolarization induced mitochondrial swelling and impaired mitochondrial motility in neuronal cells. Together with K+, also Ca2+ ions are important in triggering mitochondrial swelling, especially when the matrix influx of these ions is increased and/or their matrix efflux is decreased (160). Under these conditions, the osmotic pressure and water accumulation in the mitochondrial matrix is increased, leading to mitochondrial swelling. Importantly, the rate at which water enters the mitochondrial matrix is dependent not only on the osmotic gradient with the cytosol but also on the water permeability of the MIM (167). In addition, it was suggested that metabolic water produced by the OXPHOS system is involved in the regulation of mitochondrial volume (48). A water channel (aquaporin-8/AQP8; Fig. 1) was detected in the MIM of rat liver mitochondria and proposed to mediate rapid matrix expansion, thereby regulating the activity of the ETC and CV (42). Whether this AQP8-mediated influx is a general mechanism across tissues and whether AQP8 also participates in the trans-MIM transport of other molecules still remains to be determined (160, 209).
Physiologically relevant changes in matrix volume (Fig. 7A) are moderate, reversible, and linked to regulation of the ETC and mitochondrial ATP production (160). It appears that mitochondrial osmotic pressure regulates mPTP opening (15) and that massive swelling of the mitochondrial matrix associated with long-term (Ca2+-induced) mPTP opening leads to cell death by compromising mitochondrial function (291). Mechanistically, evidence in rat liver mitochondria suggests that this mPTP regulation involves volume-induced changes in matrix concentration of mPTP regulatory factors (290). In this context, the latter study revealed that mitochondrial shrinkage stimulated mPTP inhibition by Mg2+ and ubiquinone 0 (Ub0) and decreased the effect of the mPTP-inhibitor Cyclosporin A. It is currently unknown how the transition from physiological to pathological volume changes (Fig. 7A) is exactly regulated, although a role for ROS and metabolic stress was proposed (160).
F. Small-molecule targeting of mitochondrial morphology
The concept of mitochondrial morphofunction strongly suggests that mitochondrial function can be modulated by rational manipulation of mitochondrial (ultra)structure and vice versa. This is of great relevance for the large number of pathological conditions in humans where the mitochondrial structure-function relationship is disturbed (11, 328).
Various small molecules have been developed that inhibit DRP1 activity. These include Dynasore (237), Mdivi1 (47), and the P110-TAT peptide (130, 166, 320). All three of these molecules inhibited mitochondrial fission in various studies, suggesting that they might be useful to reduce excessive mitochondrial fission and oxidative stress in neurons affected in AD, Parkinson's disease, and HD (328). However, in case of Mdivi1, off-target effects have been described, including inhibition of K+ channels and membrane potential in HL-1 murine atrial cardiomyocytes (370) as well as CI inhibition and modulation of mitochondrial ROS production (34, 322). The pros and cons of the use of Mdivi1 as a specific DRP1 inhibitor were recently discussed (368). It was concluded that although the current evidence supports the previously described Mdivi1 bioactivity, use of this molecule requires inclusion of stringent positive controls that directly demonstrate its effect on mitochondrial fission. Importantly, DRP1 also mediates peroxisomal fission (353, 354) and exerts various other functions (Table 1). This means that mitochondrial fission cannot be specifically modulated by targeting of DRP1.
In addition to DRP1-targeting molecules, a cell-permeant fusogenic peptide (TAM-MP1Gly) was developed that destabilized the fusion-constrained configuration of MFN2, thereby promoting the fusion-permissive conformation (105). Interestingly, a small-molecule mimetic of the MFN2 peptide-peptide interface (“Chimera B-A/long”) allosterically activated MFN2, thereby promoting mitochondrial fusion (333). This small molecule mitigated mitochondrial clumping, Δψ depolarization, fragmentation, and dysmotility in CMT2A mutant neurons. The strategies described earlier focus on inhibition and stimulation of mitochondrial fission and fusion, respectively, to improve mitochondrial function. However, altering mitochondrial (ultra)structure by exogenous means also affects the cell's susceptibility to death-inducing agents. The latter is likely to be cell type- and condition-dependent. For example, mitochondrial fragmentation induced by DRP1 overexpression protected against the efficacy of CER-induced, Ca2+-dependent, apoptosis (378). In contrast, DRP1 knockdown induced mitochondrial filamentation and protected against necroptosis and apoptosis induced by CI inhibition in melanoma cell models (20).
In our own studies, we observed that Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid), a water-soluble vitamin E-derived antioxidant, increased the levels of fully assembled and active CI in primary skin fibroblasts of children with isolated CI deficiency (191). In these patient cells, Trolox displayed pleiotropic effects since it: (i) reduced ROS levels, (ii) normalized aberrant cytosolic Ca2+/ATP handling, and (iii) restored Δψ depolarization (90, 191). Mechanistic studies in healthy primary skin fibroblasts revealed that Trolox: (i) reduced ROS-induced formation of chloromethyl 2′,7′-dichlorofluorescein, (ii) did not affect oxidation of the ROS sensor HEt (hydroethidium), (iii) did not affect mitochondrial NAD(P)H levels or Δψ, (iv) reduced lipid peroxidation, (v) increased the expression and activity of all OXPHOS complexes, (vi) stimulated mitochondrial routine and maximal oxygen consumption, and (vii) prevented hydrogen peroxide-induced aberrations in cytosolic Ca2+ handling (89). Within the context of this review, it is important that the latter study demonstrated that Trolox stimulated the protein levels of MFN2 but not of DRP1 and FIS1 in mitochondria-enriched fractions from PHSFs and Chinese Hamster Ovary cells. Moreover, Trolox-induced stimulaton of mitochondrial filamentation was glutathione-dependent and absent in Mfn1 −/−, Mfn2 −/−, and Mfn1 −/− Mfn2 −/− cells. Given the fact that the Trolox effects were observed in healthy fibroblasts, we proposed that Trolox reduces the levels of endogenous CM-H2DCF oxidizing signaling ROS, which increases the levels of mitochondria-attached mitofusins and therefore induces mitochondrial filamentation (89). Functionally, this filamentation is associated with increased mitochondrial function as exemplified by increased activity of OXPHOS complexes and higher mitochondrial oxygen consumption. Given these pleiotropic effects, it remains to be determined whether Trolox and other antioxidants can exert positive effects by targeting aberrant mitochondrial (ultra)structure in human disease (21, 185, 348).
V. Conclusions
In this review, we highlighted the basic mechanisms and experimental evidence, demonstrating a tight and multidirectional connection between mitochondrial internal structure, external structure, and function. This supports the novel concept of “mitochondrial morphofunction.” Currently, a comprehensive understanding of mitochondrial morphofunction is still lacking, and obtaining this information requires quantitative information at the level of individual mitochondria within single living cells. In addition, more detailed insights are required into how the volume and complex nanoarchitecture of the mitochondrion impacts its biochemistry. It is expected that a detailed understanding of mitochondrial morphofunction will significantly contribute to deciphering: (i) why individual mitochondria display heterogeneous (ultra)structural and functional properties between and within cells, (ii) how mitochondrial (ultra)structure affects mitochondrial and cellular bioreactions and therefore mitochondrial and cellular functioning, (iii) how exogenous manipulation of mitochondrial (ultra)structure can be used to “control” mitochondrial function, and (iv) whether such manipulation is therapeutically relevant in human diseases with disturbed mitochondrial morphofunction.
Footnotes
Acknowledgments
The authors apologize to those authors whose articles they were unable to cite because of space limitations. This research was supported by an RIMLS junior researcher project granted to W.J.H.K. The authors are grateful to Dr. J. Esseling (Department of Biochemistry) for Differential Interference Contrast microscopy (supported by the Dutch Ministry of Economic Affairs; IOP Grant No. IGE05003), Ing. H.G. Swarts (Department of Biochemistry) for generating baculoviruses, Dr. J.A. Fransen and Ing. M.M. Kea-te Lindert (Department of Cell Biology) for electron microscopy analysis, and Dr. C.E.J. Dieteren (Department of Biochemistry) for creating inducible HEK293 cell lines and performing photobleaching experiments (supported by a joint grant of the “Nijmegen Center for Mitochondrial Disorders” to W.J.H.K. and Dr. L.G.J. Nijtmans from the Department of Pediatrics).
Author Disclosure Statement
W.J.H.K. and P.H.G.M.W. are scientific advisors of Khondrion B.V. (Nijmegen, The Netherlands). W.J.H.K. is scientific advisor of Mitoconix Bio., Ltd. (Ness Ziona, Israel) and of Fortify Therapeutics (Palo Alto, California). These SMEs had no involvement in the data collection, analysis and interpretation, writing of the article, and in the decision to submit the article for publication.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.