
Abstract
Significance:
Macrophages are crucial for tissue homeostasis. Based on their activation, they might display classical/M1 or alternative/M2 phenotypes. M1 macrophages produce pro-inflammatory cytokines, reactive oxygen species (ROS), and nitric oxide (NO). M2 macrophages upregulate arginase-1 and reduce NO and ROS levels; they also release anti-inflammatory cytokines, growth factors, and polyamines, thus promoting angiogenesis and tissue healing. Moreover, M1 and M2 display key metabolic differences; M1 polarization is characterized by an enhancement in glycolysis and in the pentose phosphate pathway (PPP) along with a decreased oxidative phosphorylation (OxPhos), whereas M2 are characterized by an efficient OxPhos and reduced PPP.
Recent Advances:
The glutamine-related metabolism has been discovered as crucial for M2 polarization. Vice versa, flux discontinuities in the Krebs cycle are considered additional M1 features; they lead to increased levels of immunoresponsive gene 1 and itaconic acid, to isocitrate dehydrogenase 1-downregulation and to succinate, citrate, and isocitrate over-expression.
Critical Issues:
A macrophage classification problem, particularly in vivo, originating from a gap in the knowledge of the several intermediate polarization statuses between the M1 and M2 extremes, characterizes this field. Moreover, the detailed features of metabolic reprogramming crucial for macrophage polarization are largely unknown; in particular, the role of β-oxidation is highly controversial.
Future Directions:
Manipulating the metabolism to redirect macrophage polarization might be useful in various pathologies, including an efficient skeletal muscle regeneration. Unraveling the complexity pertaining to metabolic signatures that are specific for the different macrophage subsets is crucial for identifying new compounds that are able to trigger macrophage polarization and that might be used for therapeutical purposes.
I. Introduction
Macrophages (MΦ) are inflammatory cells with a high capacity for engulfing and digesting pathogens and cell debris. In addition, MΦ play increasingly defined roles in orchestrating the healing of various damaged tissues and show high heterogeneity, plasticity, and adaptation abilities. Based on their environment and on the activation of specific signaling pathways, they might display, schematically, a pro-inflammatory (M1) or an anti-inflammatory (M2) phenotype, which will hereby be described. Differentially activated MΦ also show diverse metabolic features, with a link between the metabolic pathways operating in MΦ and their pro-/anti-inflammatory status; this review will focus on the potentiality of manipulating metabolism to polarize MΦ.
Among the various tissues in which MΦ play a role, their effect on skeletal muscle will be emphasized; differentially activated MΦ are crucial for efficient muscle regeneration by means of a complex cross-talk with skeletal muscle resident cells. Specifically, MΦ polarization can be metabolically modulated to improve the regenerative process; this aspect will be highlighted, with special focus on therapeutical approaches.
II. MΦ: Origin and Fate
MΦ represent one of the body's first lines of defense against pathogens. First described in 1887 by Metchnikoff as a population of phagocytes with the capacity to engulf and kill pathogens, MΦ are long-living cells of the innate immune system that also play a central role in adaptive immunity (156). They are present in all organs and tissues and can assume different phenotypes and functions; their diversity depends both on their origin and on the tissue and extracellular mileu in which they are located.
A. Origin
Most tissue MΦ derive from circulating monocytes extravasating from the bloodstream into injured tissues where they differentiate into MΦ (8). MΦ are characterized by the expression of markers, including CD45, the colony-stimulating factor 1 (CSF1)-receptor (CSF1R), CD11b, and the GPCR F4/80, none of which are uniquely restricted to MΦ. Depending on the tissue, MΦ can express different markers associated to their function in that tissue and can also assume different names (Fig. 1).
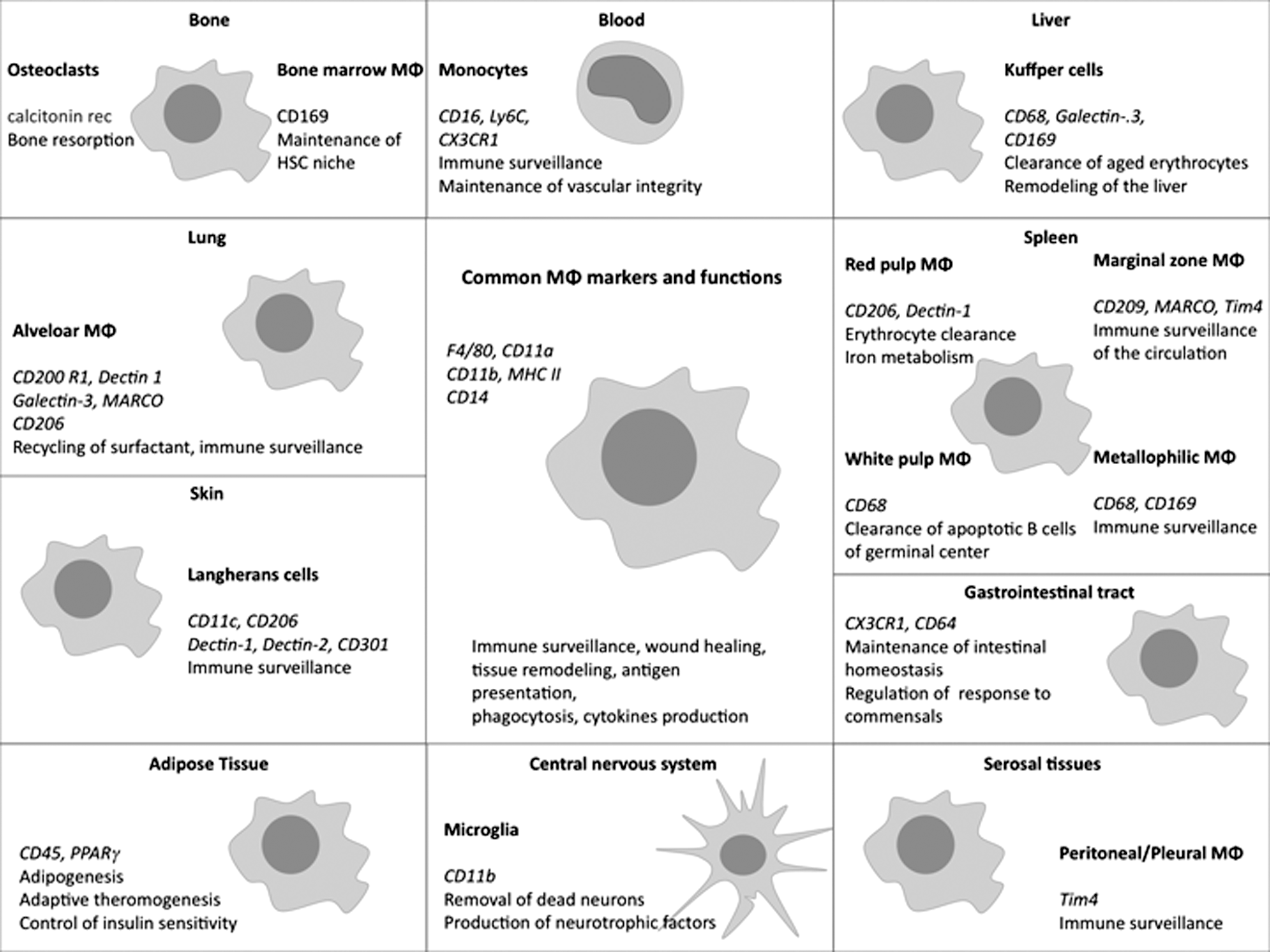
The study of MΦ ontogeny in mice showed that monocytes and MΦ derive from MΦ and dendritic cell progenitors (MDPs), which are bone marrow resident hematopoietic stem cells (c-kit+CX3CR1+Flt3+CD115+) (95). MDPs differentiate into a common monocyte precursor (c-kit+CX3CR1+Flt3−CD115+), which gives rise to the two main subsets of circulating monocytes, distinguished, in mice, by the expression of Ly6C and the chemokine receptor CX3CR1; Ly6C+CX3CR1low are considered pro-inflammatory monocytes homing to inflamed tissues, whereas Ly6ClowCX3CR1high monocytes are less abundant and home to noninflamed healthy tissues contributing to tissue repair (102).
Most MΦ derive from circulating monocytes, whereas some tissue-resident MΦ arise from a distinct mechanism of hematopoiesis. For example, microglia is ontogenetically distinct from bone marrow-derived MΦ (BMDM), as it derives from yolk sac mesenchymal progenitors, which early in mouse embryo development migrate to the neuroepithelium without passing through a monocytic stage (105). Indeed, during mammal embryogenesis, hematopoiesis takes place in different organs (mainly yolk sac and fetal liver) before the hematopoietic stem cells establish in the bone marrow.
The first hematopoietic cells arising in mice are mesenchymal progenitors appearing at E7.5 in the yolk sac. These precursors generate erythrocytes and MΦ but not lymphocytes. Around E8.5, progenitors from the yolk sac begin to seed into the fetal liver and start the first wave of hematopoiesis. A second wave starts at E10.5 and takes place in the major arterial vessels of the aorta-gonad-mesonephros region. Clusters of hematopoietic precursors with endothelial features, once they are passed through different stages, give rise to the definitive hematopoietic stem cells that are able to differentiate into multiple lineages (336). After E16.5, the transition to the bone marrow—the definitive site of hematopoiesis—takes place.
Besides microglia, long-lived embryonic precursors persist during adulthood and contribute to other resident MΦ in murine adult tissues—such as Kupffer cells in the liver or alveolar MΦ in the lung—whereas MΦ in other tissues are replaced. Thus, at least in mice, two ontogenetically distinct MΦ populations persist in adults, the ones derived from the bone marrow and the ones derived from the yolk sac. The latter population proliferate and sustain their presence in vivo independently of bone marrow-derived monocytes. Indirect evidence suggests that also in humans, some tissue MΦ are long-lived cells existing independently of circulating monocytes (269).
B. Activation/polarization
MΦ are phagocytes that respond to danger signals, sensing injury and infection; activated MΦ engulf and digest invading and damaged cells. They also present the antigen to lymphocytes and produce cytokines, thus further enhancing inflammation. In addition, MΦ are crucial for tissue homeostasis (232, 312); indeed, depending on their activation and on their microenvironment, MΦ might play pro-inflammatory or anti-inflammatory roles, thus leading to a gross classification into two groups: M1 and M2 (198, 213).
M1 are obtained by “classical” activation; they remove foreign and damaged cells thanks to their highly bactericidal and phagocytic capacity. Conversely, M2, obtained by “alternative” activation, resolve inflammation and mediate tissue regeneration and angiogenesis. MΦ are plastic cells that readily switch their phenotype. With a wide-ranging agreement among scientists, the acquisition of different phenotypes by MΦ is referred to as polarization (189, 196, 234).
1. M1 polarization
M1 polarization is elicited by interferon-γ (IFNγ) priming associated to pro-inflammatory cytokines, for example, tumor necrosis factor-α (TNFα), or by Toll-like receptor (TLR) ligands such as microbial products like pathogen-associated molecular patterns (PAMPs) or opsonins. PAMPs are molecules binding to TLRs that are highly conserved in different classes of pathogens, for example, lipopolysaccharide (LPS)—a component of the Gram-negative bacterial membrane—binding TLR4. LPS is commonly used to induce M1 activation in vitro. Other PAMPs are flagellin, peptidoglycans, and viral double-stranded RNA.
The activation of TLRs, together with NOD-like receptors, by PAMPs as well as by danger-associated molecular patterns (DAMPs) from the damaged tissue or by alarmins leads to M1 polarization. DAMPs are small molecules that are highly concentrated within healthy cells but absent or rare in the extracellular matrix (ECM), such as the high mobility group box 1 (HMGB1) protein regulating chromatin organization in healthy cells, nucleotides (ATP, ADP, UDP), oxidized phospholipids, heat shock proteins, and uric acid (224).
DAMPs are rapidly released after unprogrammed cell death, and they bind pattern recognition receptors (PRRs) on the immune cells' surface that become activated and start the inflammatory response. PRRs binding DAMPs might either be the same as those binding PAMPs (e.g., TLR) or be unique for DAMPs, such as RAGE (receptor for advanced glycation end products) binding HMGB1 or purinergic (P2) receptors sensing extracellular nucleotides, such as P2X7. MΦ activation by DAMPs occurs in case of sterile inflammation, meaning in the absence of pathogens like in muscle injury, where cellular debris triggers M1 polarization.
Differentially activated MΦ subsets might be identified by the spectrum of secreted soluble factors. Briefly, M1 produce antiviral proteins, pro-inflammatory cytokines, and chemokines (Fig. 2) [for review see Mortha and Burrows (223)]; high amounts of reactive oxygen species (ROS) and nitric oxide (NO), the latter mainly by inducible NO synthase (iNOS), which is considered among the most reliable markers of M1 activation (see section IV.C) (224, 280). M1 are characterized by an increased killing rate and antigen presentation ability (224). In addition, some cell surface molecules highly expressed in M1 compared with unstimulated MΦ (M0) or M2 are used as M1 polarization markers (Fig. 2) (373).
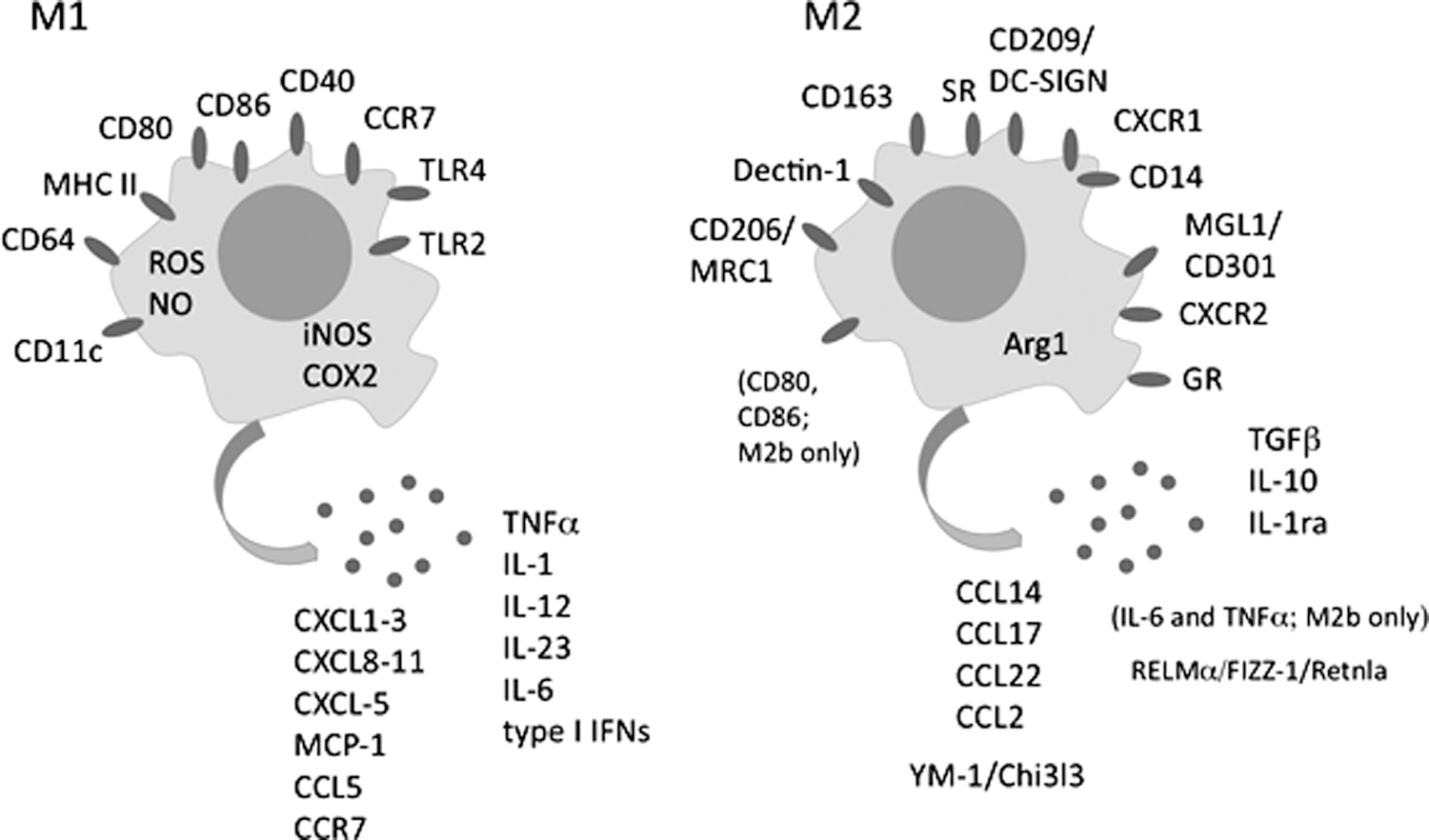
However, the high degree of MΦ plasticity and their ability to switch from a phenotype to another, through intermediate polarization status, makes the identification of unambiguous membrane markers distinguishing M1 from M2 difficult. Indeed, differently polarized MΦ can be better identified by also considering the pattern of cytokines and chemokines secreted, the effect exerted on surrounding tissues, and their metabolic status (see section IV) (313).
2. M2 polarization
On the other hand, M2 phenotypical activation is favored in normal tissue homeostasis and during recovery of tissues after damage, where M2 are necessary to resolve the inflammatory response and to allow tissue healing and remodeling. Anti-inflammatory and regenerative M2 produce interleukin (IL)-10, IL-1ra (IL-1 antagonist), growth factors, and polyamines (Fig. 2), which stimulate fibroblast growth, collagen, and production of ECM components, thus promoting fibrosis, angiogenesis, lymphangiogenesis, and damaged tissue healing.
M2 also play a role in parasite infection and tumor progression (114, 382). ROS production is reduced in M2 and arginase-1 (Arg1) is upregulated, thus also lowering NO production. By secreting IL-10, which inhibits T helper (Th)1 lymphocytes and inflammatory cytokine production, and by recruiting regulatory T cells in damaged sites, M2 directly contribute to dampen inflammation; in fact, inflammation is necessary for protection against infections, but if prolonged, it might cause tissue damage (148).
M2 activation is fostered by anti-inflammatory, pro-tolerogenic Th2 cytokines such as IL-10, IL-4, and IL-13; whereas it might be inhibited in a context in which Th1/Th17 cytokines dominate. Macrophage colony-stimulating factor (M-CSF), systemically expressed and crucial for MΦ differentiation from precursor cells, can also prime and support M2 polarization (although not acting by itself) by inducing the expression of M2-associated genes (Fig. 2) together with cell cycle regulatory genes (cyclins) that foster homeostatic proliferation (199, 287). The broad availability of M-CSF contributes to maintaining the pool of tissue resident M2 (67).
M2 upregulate Arg1 and activate the arginine pathways, producing ornithine and polyamines (see section IV.C.2). As NO and ornithine are directly involved in pathogen killing, angiogenesis, and tissue repair, these molecular signatures are considered the most typical features of M2 activation (212). Cell surface molecules over-expressed in M2 and used as their markers are indicated in Figure 2 and reviewed by Rőszer (198, 287). Notably, many of the reported M1 and M2 markers are not specific for MΦ but are also expressed by other cell types.
M2 have a high degree of heterogeneity and a further classification into three subgroups—M2a, M2b, and M2c—has been proposed, mainly on the basis of the inducing stimuli and on the panel of secreted factors (195). All three M2 subtypes are characterized by high IL-10 and low IL-12 levels.
M2a—where “a” stands for “alternative”—are induced by IL-4/IL-13 stimulation; they produce high levels of IL-10 and IL-1ra and express the mannose receptor CD206 and the receptor IL-1RII, a nonsignaling molecule that acts as a decoy receptor for IL-1. M2a are involved in Th2 responses, allergy, and the killing and encapsulation of parasites (195).
M2b are induced by exposure to immunocomplexes and agonists of IL-1R or TLRs such as LPS, thus displaying a pro-inflammatory cytokine profile, as they produce, together with IL-10, discrete amounts of IL-6 and TNFα but a very low amount of IL-12. As discussed later, similar to M1, M2b do not express high levels of Arg1, which is a marker for M2a and M2c (195).
M2c are induced by IL-10 and glucocorticoid hormones and are also referred to as “deactivated” MΦ. M2c express CD206, produce Arg1, and secrete IL-1ra, IL-10, and transforming growth factor-β (TGF-β). Moreover, they also express CD163, which is not expressed by other M2 subtypes (158). M2c are involved in tissue remodeling, ECM deposition, and immunoregulation (195). Interestingly, under the effect of IL-10, they express discrete amounts of CCR2 and CCR5 receptors for pro-inflammatory chemokines (MCP1/CCL2, RANTES/CCL5, macrophage inflammatory protein (MIP)1α/CCL3, and MIP1β/CCL4), which, in a mileu rich in IL-10, would serve as a scavenger receptor system to dampen inflammation (270).
Additional MΦ phenotypes have been more recently identified; some of them—namely Mox, M(Hb), Mhem, and M4—have been described in the atheresclerotic plaque, both in humans and in mice (22, 107, 154). These phenotypes are characterized by specific gene expression profiles and are elicited by different stimuli, such as oxidative stress (oxidized phospholipids) for Mox, hemoglobin–haptoglobin complexes for M(Hb), heme for Mhem, and CXC chemokine ligand (CXCL)4 for M4. Mox MΦ are believed to have a particular ability to deal with oxidative stress, as they express an increased glutathione/oxidized glutathione (GSH/GSSG) ratio (154) and the generation of the Mox phenotype is mediated by nuclear factor (erythroid-derived 2)-like 2 (Nrf2).
The various origins of MΦ (from circulating monocytes or from yolk sac), their polarization to pro-inflammatory or anti-inflammatory subsets, and their heterogeneity have been described. The nomenclature M1 and M2 emulates the T cells Th1/Th2 classification and underlines the functional cross-talk between lymphocytes and MΦ: Th1 lymphocytes produce INFγ, required for M1 polarization, and Th2 secrete IL-4 and IL-13, which drive M2 polarization. Notably, this classification is simplistic, due to the occurrence of several MΦ intermediate polarization statuses (234). The obstacles and lack of consensus in defining MΦ activation are described by the guidelines of Murray and coauthors (234). Based on such guidelines, describing MΦ-activation in vivo requires an explicit description of the populations under investigation, how they were isolated, from which tissue and conditions, and which marker combinations were used to ascertain MΦ activation.
III. Transcriptional Regulation of Polarization
A number of transcriptional factors participate in the differential activation of MΦ. In this section, we describe the most relevant, including the signal transducers and activators of transcription (STATs), the IFN-regulatory factors (IRFs), the nuclear factor κB (NF-κB), the hypoxia-inducible factors (HIFs), Krüppel-like factors (KLF), the peroxisome proliferator-activated receptors (PPARs), and several microRNAs (miRNAs) (170, 315).
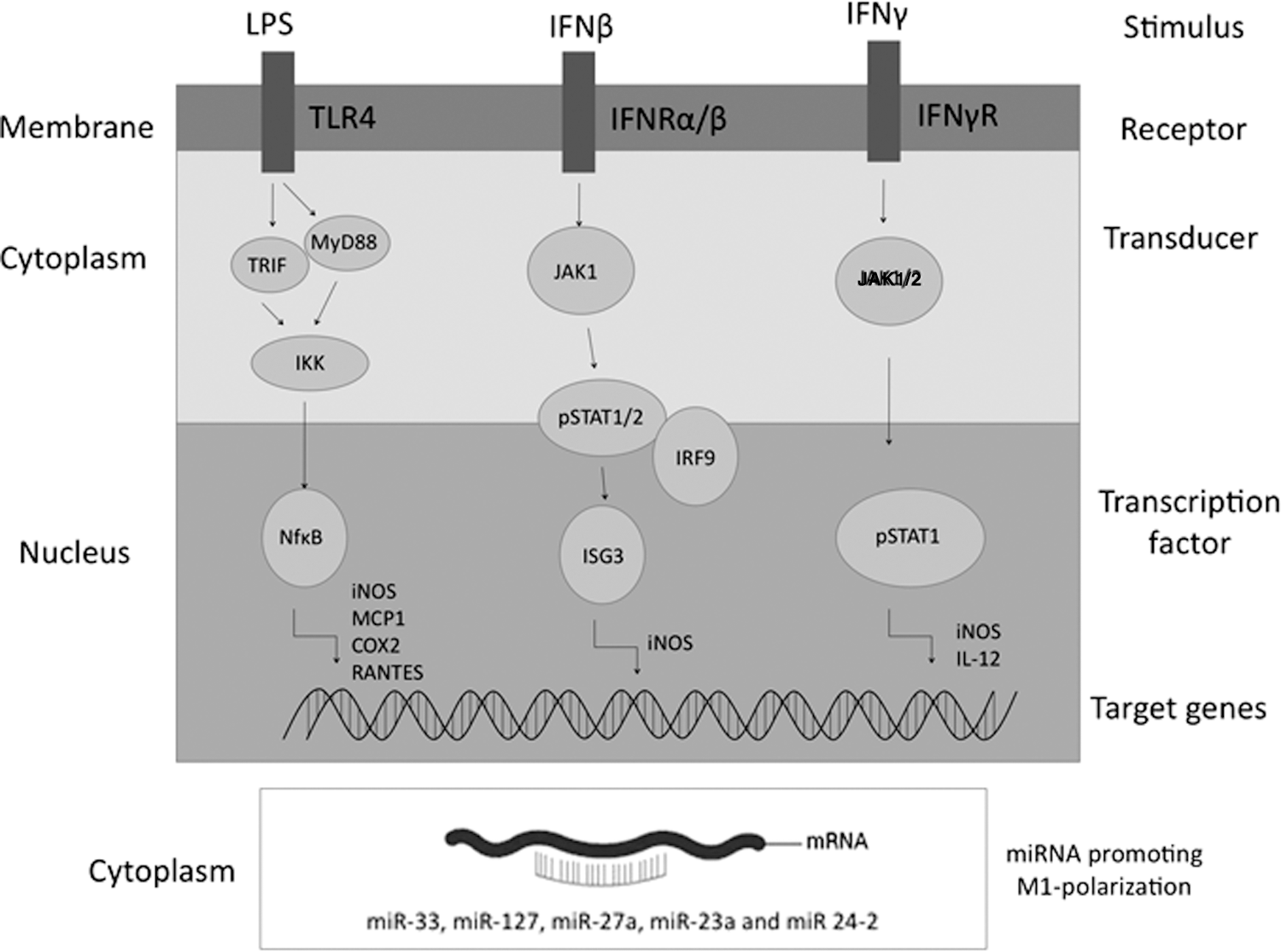
The members of the STAT family involved in MΦ polarization are STAT1, STAT2, STAT3, and STAT6. During M1 polarization, IFNγ binds to its receptor and triggers Janus kinase 1/2 (JAK1 and JAK2)-mediated phosphorylation of STAT1, which dimerizes and binds to the promoter region of target genes, including iNOS and IL-12 (Fig. 3) (66). STAT1 and STAT2 are also involved in the autocrine response to IFNβ, released by LPS-activated MΦ; IFNβ triggers the formation of the STAT1–STAT2 heterodimers that bind to IRF9 (Fig. 3) (see below within this same paragraph). In STAT1-knockout (KO) mice, MΦ fail to produce TNFα and NO, thus demonstrating that STAT1 is required for M1 polarization (179).
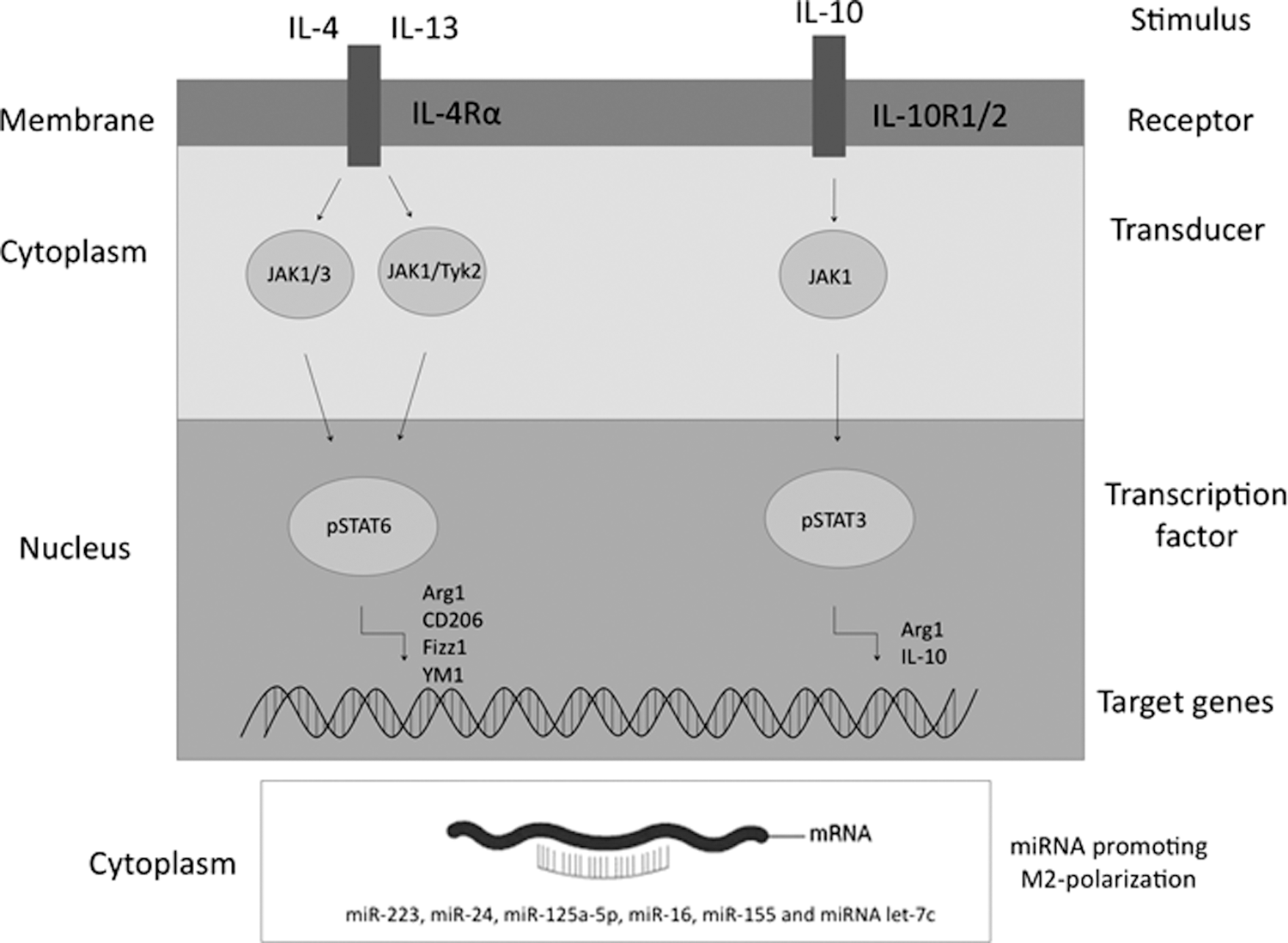
Conversely, STAT2 deficiency in mice does not block M1 polarization completely, but it impairs autocrine response to type I IFNs (309). STAT3 is considered one of the key signaling molecules in the induction of the M2 phenotype; STAT3 is activated by IL-10 and, in turn, enhances IL-10 expression while inhibiting TNFα, IL-1β, and IL-12 production. Notably, STAT3 activation by IL-6—considered a prototypic inflammatory cytokine—in a pro-M2 mileu (i.e., in the presence of IL-4/IL-13) enhances M2 polarization (90). Stimulation of MΦ by IL-4/IL-13, both binding the IL-4 receptor-α (IL-4Rα) and activating either JAKe1/JAK3 (by IL-4) or JAK1/Tyk2 (by IL-13), induces the phosphorylation and activation of STAT6, which dimerizes and induces the expression of most M2 genes, for example, Arg1, CD206, resistin-like molecule alpha (RELMα)/Fizz1/Retnla, and Ym1 (Fig. 4).
IRFs are also implicated in MΦ polarization. IRF1 (induced by type I IFNs) and IRF5 (induced by TLR) are required for proper M1 polarization and secretion of pro-inflammatory cytokines (327, 384); indeed, IRF5 polymorphisms are associated with immune-mediated diseases (115). IRF3 and IRF7 are activated in M1 and trigger type I IFNs production. IRF9 binds to the STAT1/STAT2 heterodimer to form the IFN-stimulated genes (ISG) factor 3, which stimulates the transcription of several M1-related genes such as iNOS (Fig. 3). On the other hand, IRF4 is specifically associated with M2 activation, as it is induced by IL-4-mediated activation of STAT6.
LPS binding to TLR4 and CD14 during M1 polarization fosters NF-κB activation, through myeloid differentiation primary response gene 88 (MyD88)-dependent and MyD88-independent but TIR-domain-containing adapter-inducing interferon-β (TRIF)-dependent pathways. NF-κB is normally present—but kept inactive by IkB—in the MΦ cytoplasm. TLR4 stimulation activates IkB kinase, which phosphorylates IkB and targets it for degradation; NF-κB is, therefore, released and migrates to the nucleus, where it contributes to its own activation by upregulating IL-1β and TNFα, these, in turn, stimulating NF-κB activation.
Other genes with NF-κB-binding sites in their promoters are iNOS, monocyte chemoattractant protein 1 (MCP1)/CCL2, cyclooxygenase-2 (COX-2), and regulated on activation, normal T cell expressed and secreted (RANTES)-CCL5, all related to M1 polarization. However, also M2 activation is partly regulated by NF-κB; in fact, KO mice for the NF-κB repressor, p50-NF-κB, display not only an exacerbated M1 response but also an impaired ability to establish a proper M2 response during parasitic infections (276). Also, the transcription factor activator protein 1 (AP1) is activated by TLRs.
A condition of hypoxia, occurring in damaged tissues and in case of bacterial infection—also due to high oxygen consumption by inflammatory cells—can influence MΦ polarization. The effect of low oxygen partial pressure (pO2) on cells is mediated by HIFs, two of which (HIF-1α and HIF-2α) are also expressed by MΦ (57, 64). Low pO2 reduces the prolyl hydroxylases (PHDs)-dependent degradation of HIF-1α and HIF-2α, which can dimerize with HIF-1β and become active gene regulators. HIF-1α and HIF-2α isoforms are directly involved in M1 and M2 polarization, due to their influence, in an antagonistic manner, on NO metabolism inducing the expression of iNOS and Arg1, respectively, although Arg1 seems to be a target of both HIF isoforms (28, 328).
HIF-1α plays a crucial role in orchestrating part of the M1 polarization, since it downregulates CD206, enhances IL-1β and the expression of other pro-inflammatory genes (324), and stimulates glycolysis as well as pyruvate dehydrogenase kinase-1 (PDK1) and glucose transporter type-4 (GLUT4) expression to allow ATP production in hypoxia; indeed, HIF-1α-KO (but not HIF-2α-KO) mice show alternative MΦ polarization and wound-healing improvement. In M1, HIF-1 and NF-κB are activated in parallel by hypoxia and ROS and act synergistically to induce common target genes such as iNOS; however, in some conditions, NF-κB seems to repress HIF-1 target genes, causing alternative polarization under hypoxia.
On the other hand, since NADPH-oxidases (NOX) and iNOS use oxygen to produce ROS or NO, a low pO2 might reduce their activity and M1 pro-inflammatory function. HIF-2α is barely detectable in M1, whereas it is upregulated in M2 where HIF-1α is downregulated. However, a decrease of the pro-inflammatory response has also been detected in HIF-2α-KO mice [for references see Brüne et al. (28)]. Besides Arg1, HIF-2 triggers anti-oxidant protein expression and ECM production. In M2, both HIF isoforms promote vascular endothelial growth factor (VEGF) signaling and angiogenesis, which are crucial for the regenerative process.
Interestingly, it has been found that PHD2 downregulation is critical for alternative polarization and promotes capillarization and arteriogenesis independently of hypoxia through the NF-κB pathway (120); activation of the angiopoietin receptor TIE2 in ischemia-associated MΦ, triggered by endothelium shear stress-released VEGF, induces PHD2 downregulation in normoxia, which activates the canonical NF-κB pathway, further upregulating the pro-arteriogenic TIE2. This is associated to M2 polarization and enhanced capillarization; indeed, arteriogenesis occurs in sites distant from the hypoxic area (120). It has more recently been observed that, in human MΦ under hypoxic conditions, a transcriptomic switch triggered by both HIF-1α and HIF-2α together (not by HIF-1α alone) occurs; this switch upregulates glycolytic enzymes, increasing anaerobic glycolysis (143).
HIF isoforms are differentially activated, HIF-1α being induced by Th1 cytokines and HIF-2α by Th2 cytokines (e.g., IL-4). HIF-1α clearly accumulates in M1 in normoxia conditions as well, paired with the contribution of NF-κB binding to its promoter. HIFs are also redox-sensitive transcription factors, since PHD is influenced not only by oxygen but also by NO and ROS. M1-produced ROS reduce PHD activity and promote HIF-1α expression also under an ambient oxygen level. Also, NO represses PHD and stabilizes HIF-1, which, in turn, induces iNOS in a positive feedback typical of M1 (see sections IV.C.1, IV.A.3.2, and V). However, NO becomes destabilizing under hypoxic conditions; by competing with oxygen for the binding to cytochrome c oxidase (COX), NO might leave more oxygen available for PHD activity (28).
Finally, LPS-induced TLR4 signaling triggers a cross-talk between HIF-1α and apoptosis signal-regulating kinase-1 (ASK1) pathways, both activated by ROS. ASK1 contributes to HIF-1α stabilization likely via p38 mitogen-activated protein kinase (MAPK), which is typically involved in M1 polarization (164, 322, 334). Even though HIF-1α levels mostly depend on PHD, also nuclear factor of activated T cells (NFAT) and STAT3—crucial for the M2 phenotype—seem to be required for HIF-1α expression as well as for the angiogenic role of MΦ; whereas transcriptional regulation of HIF-2α is barely known [for references see Brüne et al. (28)]. Through HIF-1α, hypoxia also upregulates some DAMP receptors such as RAGE and P2X7R, thus influencing MΦ polarization (325). HIFs promote stemness as well, including that of human embryonic stem cells, thus increasing their regenerative potential. Stem cells reside within hypoxic regions and HIFs are involved in their homeostasis also by decreasing their reliance on oxidative metabolism. HIFs maintain stemness also in cancer stem cells (203).
KLFs are zinc-finger transcriptional factors. Several members of the KLF family are involved in the MΦ-polarization process. KLF2, KLF10, and KLF13 attenuate M1 polarization through different pathways: (i) KLF2 inhibits NF-κB-mediated response to pro-inflammatory cytokines; (ii) KLF10 binds the promoter of TGF-βRII (257); and (iii) KLF13 mediates the anti-inflammatory effect of miR-125a-5p (11). On the other hand, KLF6 enhances the expression of pro-inflammatory genes in MΦ (159). As for KLF4, its function is controversial; it is upregulated in LPS/IFNγ-stimulated murine J774 MΦ, where it cooperates with NF-κB to enhance iNOS expression (89); whereas by contrast, it is over-expressed in murine peritoneal M2 and adipose tissue M2 MΦ from obese humans, where it interacts with STAT6 and inhibits NF-κB-mediated M1 response (182).
PPARγ is known to induce M2 activation; in fact, administration of rosiglitazone, a PPARγ agonist, leads to an increased expression of Arg1, found in inflammatory zone 1 (Fizz1), and IL-10 (127). On exposure to Th2 stimuli mediating M2 polarization, PPARγ deficiency downregulates Arg1 and IL-10 likely through the induction of miR-223 (392). Inhibition of pro-inflammatory cytokine signaling, for example, by suppressor of cytokine signaling (SOCS) proteins, also occurs in M2.
Several miRNAs have emerged as crucial players in MΦ polarization (Figs. 3 and 4) (148, 243, 253, 330, 391, 392). Along with miRNAs, other epigenetic mechanisms modulate MP polarization; just to give some examples of an extremely wide field, methylation of lysine 4 of histone 3 (H3K4) seems to be involved in TNFα transcriptional regulation (181). On the other hand, Xia et al. reported that methylation of H3K4 leads to TNFα and IL-6 downregulation in a murine model of sepsis (383). Moreover, the histone demethylase Jumonji domain-containing 3 seems to not only contribute to a fine regulation of LPS-dependent M1 gene expression (68) but also be crucial for M2 polarization (303). Further, histone acetylation represents another mechanism of transcriptional control; for example, histone deacetylases 3 (HDAC3) removes the acetylation on the enhancers of IL-4-induced genes, thus representing a brake for M2 polarization (148, 230).
IV. Metabolic Features of M1 and M2
In addition to the earlier described features characterizing M1 and M2, these two subsets might also be distinguished by key metabolic differences. Several years ago, Newsholme and collaborators proposed that MΦ polarization in mice is associated to a metabolic reprogramming; in synthesis, the switch toward a glycolytic or an oxidative metabolism is required for M1 and M2 polarization, respectively. Quiescent MΦ rely mostly on mitochondrial oxidative respiration to produce ATP, whereas M1 obtain energy mainly by glycolysis and M2 by enhanced oxidative metabolism (Fig. 5) (241, 242).
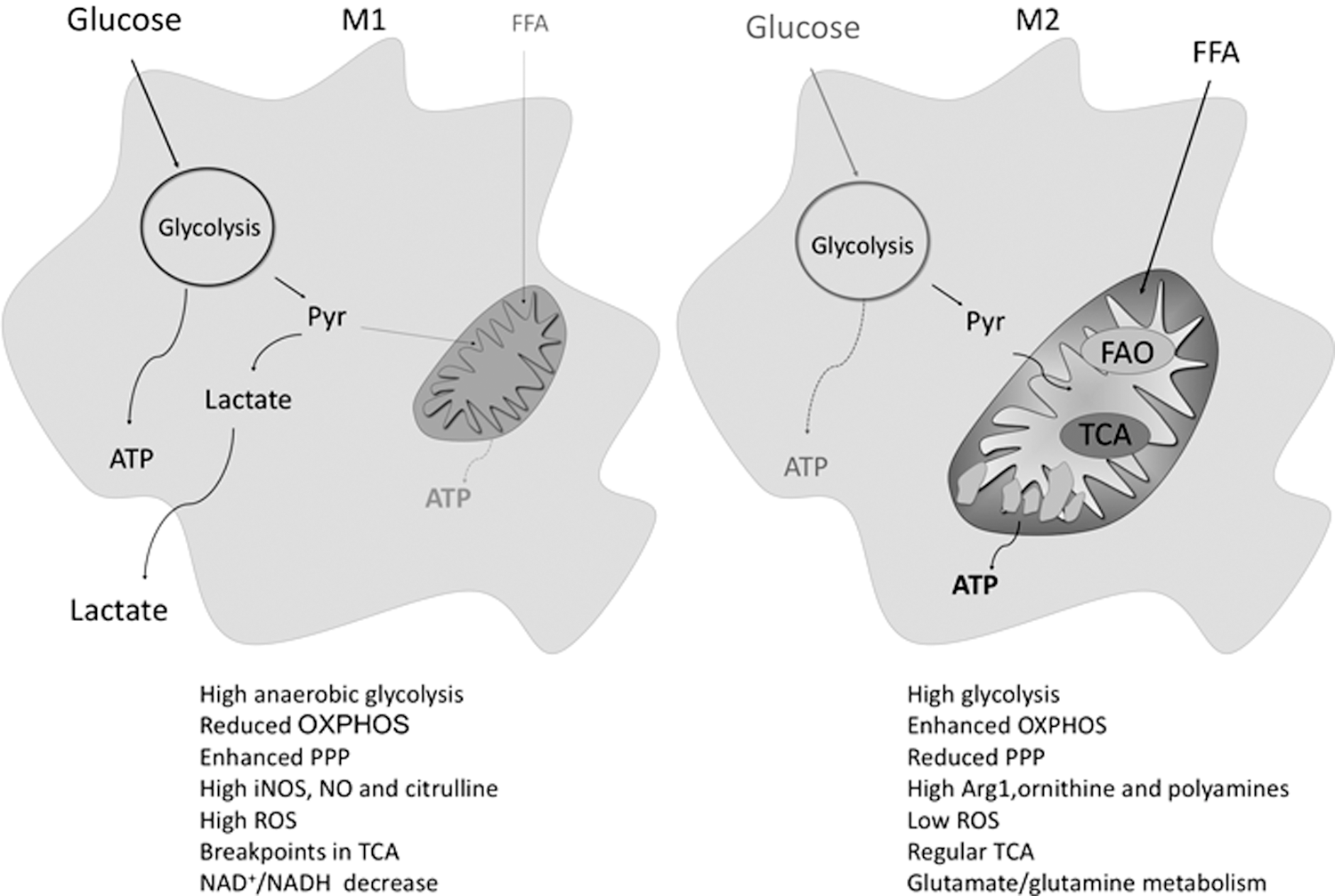
Importantly, the metabolic status characterizing differentially activated MΦ also affects their inflammatory state; therefore, different metabolic pathways not only are a different way to produce energy but also confer peculiar phenotypes and functions to MΦ subsets. Although a growing body of evidence emphasizes the crucial role of metabolic reprogramming in MΦ activation, the signaling allowing the metabolic switch leading to M1 or M2 activation is currently largely unknown.
A. M1-polarized MΦ
1. Enhanced glycolysis and reduced oxidative phosphorylation
M1 activation is associated with an oxidative phosphorylation (OxPhos) to glycolysis switch (Fig. 5). The first evidence of a higher glucose consumption in murine MΦ on pathogen stimulation was obtained in 1970 (124). This was confirmed in several manuscripts describing that, in LPS/IFNγ–polarized MΦ, an enhanced glycolysis fosters increased glucose uptake and conversion of pyruvate to lactate, the latter found in high concentrations both intra- and extracellularly; glucose is fermented to lactate, even though oxygen is sufficient to support OxPhos (241, 251). Vice versa, glucose metabolism remains unaltered in IL-4/IL-13-stimulated MΦ and dendritic cells (87, 164, 285, 355). In line with this, although not fully elucidated, endogenous GLUT4 seems to be crucial for M1 activation (238, 251).
In addition, metabolomic screenings and cDNA microarray gene expression analysis have shown that M1 upregulate glycolytic genes within 24 h after LPS stimulation, whereas they downregulate the mitochondrial ones. This is associated with a reduction of the respiratory chain activity evaluated by oxygen consumption rate (OCR), which is a measurement of cellular oxidative metabolism. It is also associated with an increased extracellular acidification rate, which is an indication of the glycolytic rate (238); in fact, extracellular H+ excretion derives both from anaerobic glycolysis-produced lactate (glucose is converted to lactate− and H+; glycolytic acidification) and from tricarboxylic acid (TCA) cycle-derived CO2 (exported CO2 is hydrated to H2CO3, which then dissociates to HCO3 − and H+; respiratory acidification).
Although the contribution of CO2 to extracellular acidification is often considered negligible, the proportions of glycolytic and respiratory acidification vary depending on the experimental conditions (221). To sum up, M1 polarization is characterized not only by enhanced glycolysis but also by repression of mitochondrial OxPhos (263, 264). M1 polarization also reduces the NAD+/NADH ratio in mice, this being in line with reduced oxidative respiration and NADH oxidation (Fig. 5) (214).
As recently reviewed by Van den Bossche et al. (355), glycolysis is also necessary for M1 activation given that it provides signals driving this polarization route, with glycolytic enzymes being crucial in supporting pro-inflammatory function. For example, the glycolytic activator 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) enhances the ability of murine MΦ to remove virus-infected cells (147). Moreover, when glycolysis is limited, glyceraldeyhe phosphate dehydrogenase (GAPDH) is not fully engaged in this cycle and might inhibit TNFα and IFNγ translation by binding their mRNA.
Therefore, glycolysis is a metabolically regulated signaling mechanism that is required to control cytokine production (38, 211). Moreover, α-enolase is expressed on the human M1 surface where it stimulates the production of pro-inflammatory cytokines (9), and pyruvate kinase M2 (PKM2) acts directly on HIF-1α and upregulates IL-1β. Further, through the activation of the eukaryotic translation initiation factor 2 alpha kinase 2—which modulates the inflammasome—PKM2 also indirectly promotes pro-IL-1β activation in mice (256). Finally, it has also been suggested that in coronary artery disease patients, over-utilization of glucose drives ROS production, leading to PKM2 nuclear translocation where it phosphorylates and activates STAT3 to boost the expression of IL-1β and IL-6 (310).
Glycolysis is required to induce and sustain a pro-inflammatory status also in other immune cells such as dendritic ones (86, 206, 263, 264). In fact, glycolysis produces low amounts of energy compared with OxPhos (2 ATP/glucose vs. around 30 ATP/glucose). However, it can be quickly activated, provides rapid energy, and reduces production of intermediates. As such, it has been suggested as crucial for acute bacterial killing in highly proliferating bacterial infection (164, 332).
2. Pentose phosphate pathway
Classical M1 activation also enhances the pentose phosphate pathway (PPP), branching from glycolysis and essential for NADPH production used to produce both ROS (by NOX) and NO (Fig. 6). Through the PPP, erythrose (precursor of amino acids) and ribose (nucleotide synthesis intermediate) are also synthetized by glucose (236, 355).
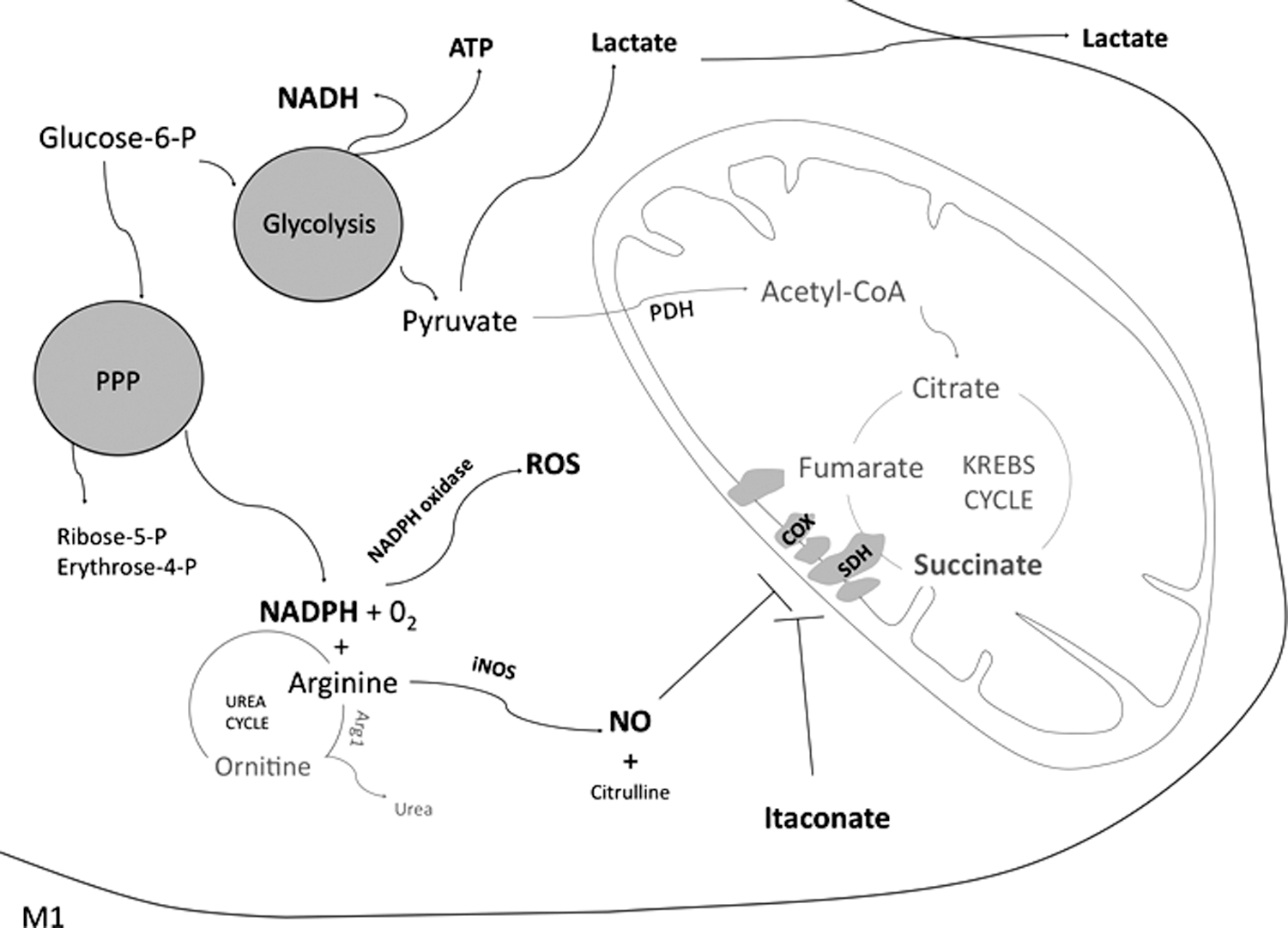
3. Breakpoints in the TCA cycle and TCA intermediates accumulation
Another typical consequence of the metabolic reprogramming characterizing M1 polarization is a flux discontinuity at several levels of the Krebs cycle, as demonstrated in murine MΦ (Figs. 6 and 7) (145). Such interruptions lead to the accumulation or reduction of some TCA intermediates, which influence the inflammatory response. However, the results of these studies are controversial due to the quick changes in metabolism and to the anaplerotic reactions feeding the TCA cycle. Notably, the TCA cycle fueled by both pyruvate and glutamine is globally maintained on LPS stimulation, whereas OxPhos decreases and NADH excess might possibly be converted into NADPH to support NOX activity during phagocytosis, as found in murine MΦ cell lines (207).
a. Citrate/isocitrate level
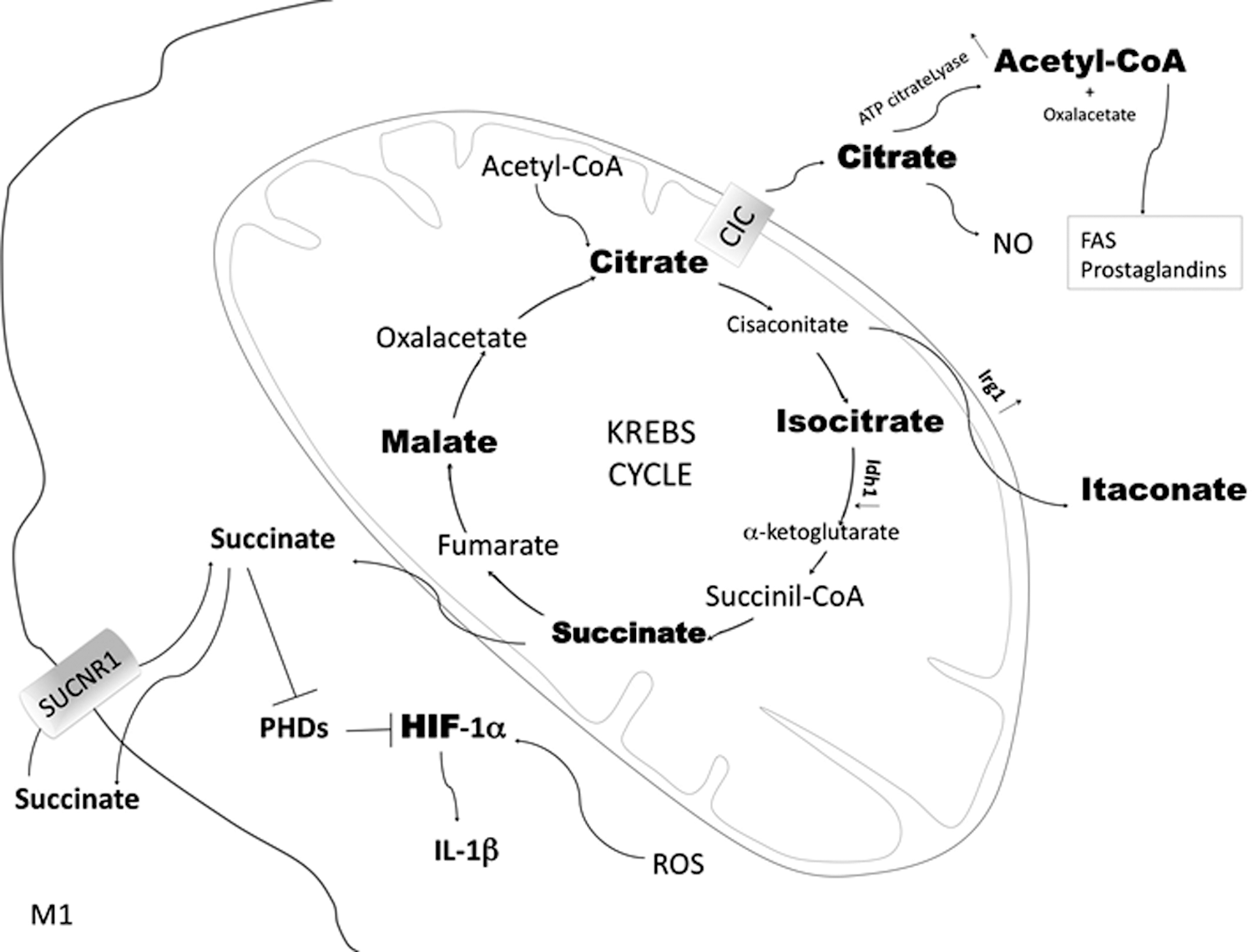
A robust increase of citrate occurs in M1 (Fig. 7). Citrate might translocate from the mitochondria to the cytosol by the citrate carrier (CIC; which is upregulated in M1) and be transformed into oxalacetate and acetyl coenzyme A (acetyl-CoA) by the ATP-citrate lyase (Fig. 7). Flux discontinuity at the citrate level might be responsible for the increased synthesis of acetyl-CoA, allowing higher synthesis of fatty acids (FA) and prostaglandins, characterizing the M1 subset.
FA are necessary for M1 to reorganize the plasma membrane and rapidly produce organelles that are crucial for cytokine synthesis and secretion, as demonstrated both in vivo and in vitro (86, 87, 208, 355, 378). FA synthesis occurring in M1 demonstrates that glycolysis is boosted and oxygen consumption is decreased yet not abrogated, as observed in vitro (214). Citrate is also critical for the production of pro-inflammatory mediators such as NO (140); if CIC or ATP-citrate lyase are inhibited, NO and ROS production by M1 is blocked (140, 141).
Deriving from the TCA cycle intermediate cis-aconitate (which, in turn, derives from the accumulated citrate), the itaconic acid is also highly concentrated in murine M1 (Fig. 7) (320). Pyruvate dehydrogenase (PDH) is necessary to obtain citrate (and, in turn, itaconate) from pyruvate (355). Itaconic acid acts as a microbicidal agent disrupting the glyoxylate cycle used by some pathogens (e.g., Salmonella enterica), but not by mammalian cells (209). Itaconic acid is secreted on M1 activation. In addition, Michelucci et al. have found that silencing the immunoresponsive gene 1 (Irg1)—the enzyme catalyzing the decarboxylation of cis-aconitate to itaconate—reduces MΦ antimicrobial activity (209, 331). Indeed, Irg1 is robustly upregulated in M1, thus linking the accumulation of citrate and succinate (Fig. 7) (61, 168).
More recently, disclosure of the metabolic rewiring typical of the M1- and M2-polarized states obtained by high-throughput metabolic and transcriptional data profiling (CoMBI-T analysis), besides confirming previous findings, has highlighted that M1 polarization is characterized by TCA cycle breakpoint at the isocitrate dehydrogenase 1 (Idh1) level (Fig. 7); M1 display higher levels of isocitrate compared with M0 MΦ and this was likely due to transcriptional downregulation of Idh1, the enzyme catalyzing the isocitrate to α-ketoglutarate reaction.
b. Succinate level
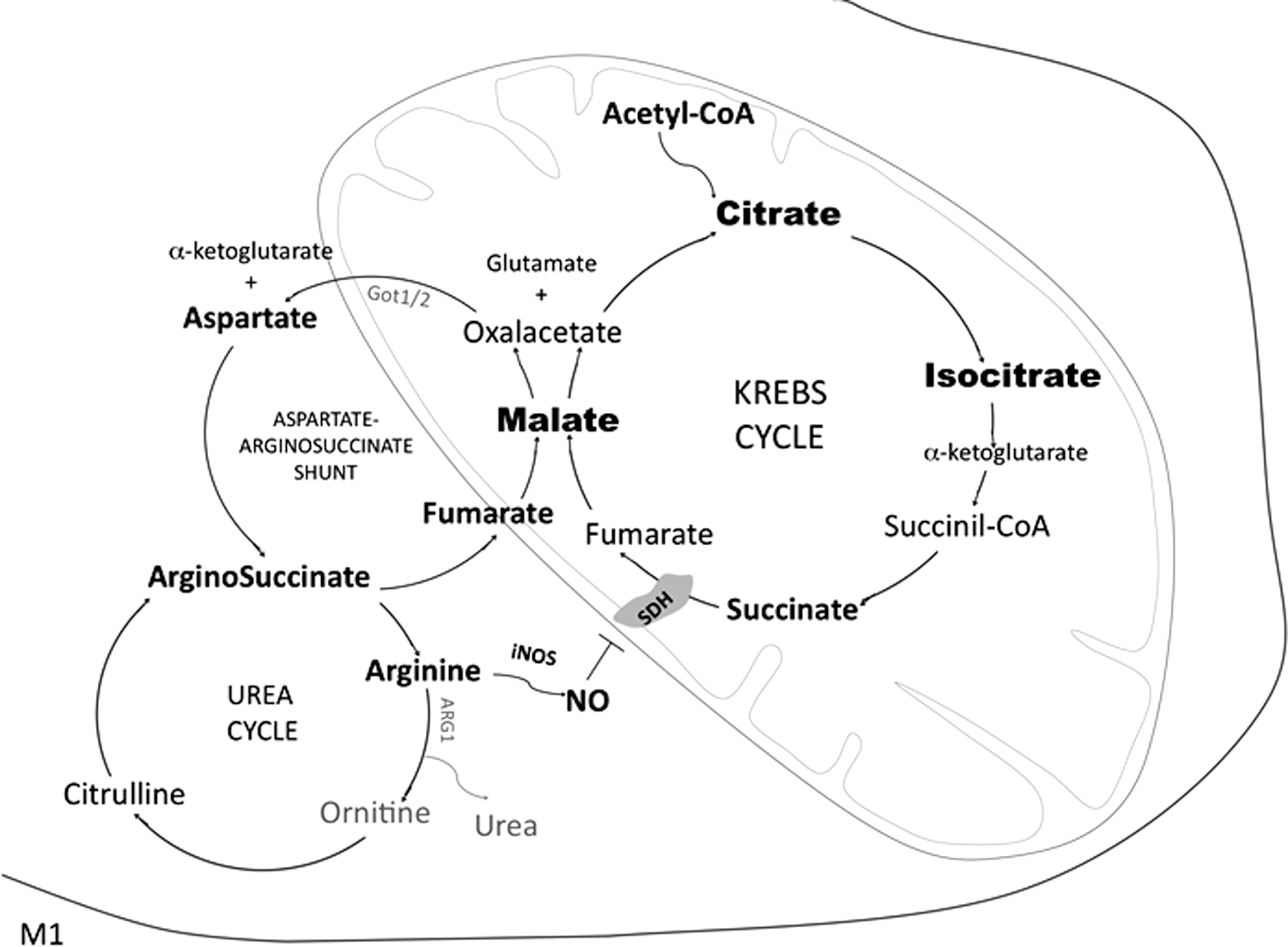
Another TCA flux discontinuity occurs at the succinate dehydrogenase (SDH) level; the succinate-to-fumarate transition is impaired, and the steady-state concentration of succinate and malate increases in M1. Data suggest that malate accumulates because it derives from the aspartate-arginosuccinate cycle, an anaplerotic set of reactions connecting the urea cycle (arginine-ornithine-citruline conversion) and NO production with the Krebs cycle (Fig. 8) (145). This was confirmed by inhibition of glutamic oxaloacetic transaminase 2-mediated aspartate production, which reduced NO production and iNOS expression, in turn decreasing IL-6 and blocking M1 conversion.
Based on these data, NO seems to be largely produced through the aspartate-arginosuccinate shunt. As shown in cultured murine dendritic cells, NO contributes to suppressing mitochondrial respiration by competing with oxygen and inhibiting SDH (87). To summarize, it seems that in M1, the aspartate-arginosuccinate shunt allows TCA cycle anaplerosis, which is useful in the context of the breakpoint at the SDH level. This shunt coordinates the NO cycle with TCA cycle anaplerosis (Fig. 8) (145).
Further, accumulated succinate, similar to NO, inhibits PHDs (Fig. 7). Usually, in the presence of oxygen, the α-ketoglutarate-dependent dioxygenases PHDs hydroxylate HIFs, targeting them for proteasomal degradation. At low pO2, PHDs are inhibited; HIF-1α is not degraded, thus stimulating glucose uptake, glycolytic genes, and IL-1β transcription while inhibiting pyruvate oxidation and favoring lactate reduction. PHDs inhibition by succinate stabilizes HIF-1α also in the presence of oxygen (334); succinate is, therefore, considered an inflammatory signal since HIF-1α plays a crucial role in orchestrating part of the M1 polarization by promoting glycolysis, GLUT4 and IL-1β expression, and MΦ migration.
Glycolysis is crucial for MΦ migration, whose inhibition suppresses systemic inflammation in vivo (308). Notably, in LPS-treated MΦ, inhibiting glycolysis by 2-deoxy-
Succinate might also be released by M1, acting extracellularly via the succinate receptor 1/G-protein coupled receptor-91 (SUCNR1/GPR91) expressed in many tissues. Recycling succinate induces a feed-forward loop of pro-inflammatory MΦ activation, which increases HIF-1α-dependent IL-1β expression. In fact, SUCNR1 synergizes with TLR on both human and murine dendritic cells to enhance the functions associated with antigen presentation (Fig. 7) (185, 290). Moreover, an LPS-dependent succinylation of numerous proteins—whose consequences are unknown—has been reported (335).
An increased succinate oxidation via SDH on M1 polarization, as observed in murine BMDM, has also been proposed (214). In addition to HIF-1α stabilization, succinate might have another inflammatory critical role; M1 polarization seems to drive mitochondrial membrane hyperpolarization (glycolysis supporting ATP generation), which, paired with SDH-mediated succinate oxidation to fumarate, leads to ROS generation (and, in turn, to IL-1β expression) via reverse electron transport through complex I rather than activating the conventional electron transport (214, 251, 317).
This hypothesis suggests a repurposing of mitochondria from ATP synthesis to ROS production, which promotes a pro-inflammatory state (214). This is accompanied by mitochondrial supercomplex destabilization; complexes I, II, and IV can accumulate as supercomplexes, improving coupling and reducing ROS formation. However, complex I, and, as a consequence, the whole supercomplex, is destabilized when MΦ are activated by bacteria; this activates SDH, which seems necessary for the control of bacteria (214), although the role of SDH and its link with IL-1β production is far from being clear. Succinate oxidation also leads to decreased anti-inflammatory gene expression; in line with this, inhibiting succinate oxidation by dimethyl malonate promotes an anti-inflammatory outcome.
To summarize, M1 polarization is characterized by enhanced glycolysis and PPP, whereas OxPhos decreases. Glycolysis is additionally necessary for M1 activation by providing signaling mediators driving it. Flux discontinuities on several levels of the Krebs cycle are also features of M1 polarization and lead to a robust increase of pro-inflammatory succinate and citrate, Irg1, isocitrate, and microbicidal itaconic acid and to the downregulation of Idh1. The variation of Irg1 and itaconate concentration represents a strong marker of M1 polarization similar to iNOS activation and NO over-production (see section IV.C). Specifically, PFKFB3, PMK2, α-enolase, citrate, succinate, and itaconate are not only consequences but also causal of M1 polarization.
B. M2-polarized MΦ
1. Enhanced OxPhos
Differently from M1, alternatively activated murine M2 have an intact TCA cycle and an efficient OxPhos supporting their energy demands and phenotype (Figs. 5 and 9) (357). Based on some authors' findings, glucose uptake also increases in M2 compared with untreated MΦ but is lower than in M1, and it is mainly oxidized by mitochondria; OxPhos accounts for higher but slower ATP generation, which can be sustained for a longer period compared with glycolysis (355). This is useful for the resolution of inflammation and also against prolonged parasite infections. OxPhos requirement for M2 polarization has been revealed by oligomycin-mediated inhibition of ATPsynthase and OCR, blocking IL-4-mediated M2 polarization. Also, OxPhos inhibition by rotenone impairs M2 polarization in murine BMDM (126).
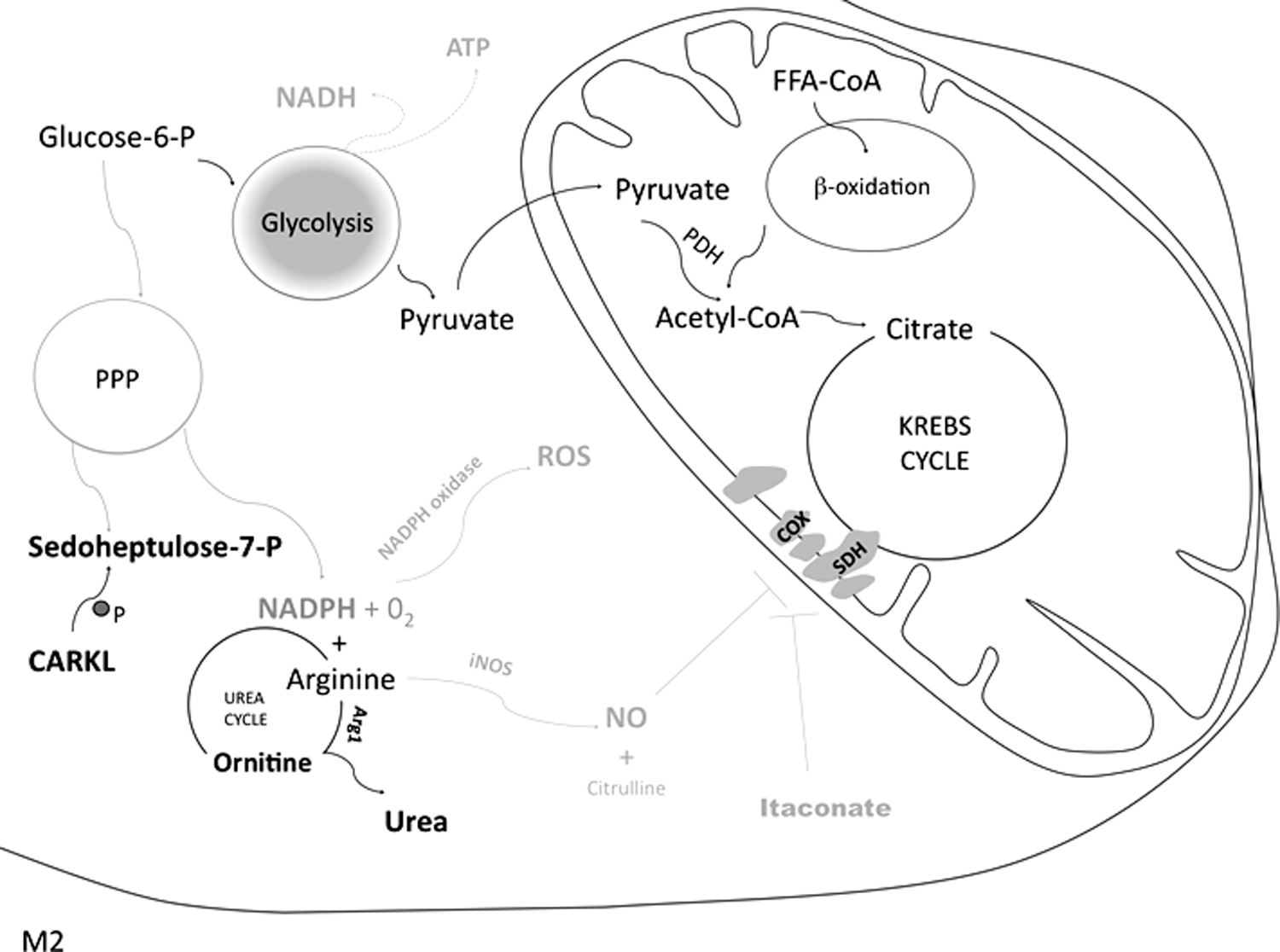
2. Reduced PPP
The carbohydrate kinase-like (CARKL) protein is a repressor of M1 activation both in human and in murine MΦ. In fact, CARKL phosphorylates the PPP intermediate sedoheptulose in sedoheptulose-7-P, thus stimulating the nonoxidative phase of this cycle; this reduces NADPH production and counteracts ROS production typical of M1. CARKL is critical for PPP regulation; its expression increases in M2 whereas it decreases in M1 (Fig. 9) (126).
3. Free fatty acid β-oxidation and glycolysis requirement in differentially polarized MΦ is controversial
The strict requirement of free fatty acid (FFA) β-oxidation (FAO) for M2 polarization has recently become a matter of debate (134, 237, 332, 355, 357). By using BMDM, Vats et al. suggested that FAO is crucial for M2 polarization; based on their data, FFA uptake, and FAO increase in M2 compared with untreated MΦ and M1; whereas IL-4-stimulated MΦ upregulate acyl-CoA dehydrogenases and enoyl-CoA hydratases involved in FAO (357). mRNAs of PPARγ-coactivator-1β (PGC-1β) and of genes involved in FFA uptake, transport, and oxidation—for example, lipoprotein lipase, fatty acid transporter (CD36/FAT), carnitine palmitoyltransferase 1 (CPT1), medium chain acyl-CoA dehydrogenase, long chain acyl-CoA dehydrogenase, and PPARγ—are also upregulated in M2 and downregulated in LPS/IFNγ-polarized M1 (357).
Triglycerides lysosomal lipolysis, after CD36-mediated lipoprotein uptake, fuels an intense OxPhos; lipolysis was found to be essential to allow oxidative metabolism and M2 polarization, both in humans and in mice (134). In line with this, and further corroborating the hypothesis of the key role of FAO in M2 polarization, PPARγ, necessary for mitochondrial function and FAO, enhances and is crucial for M2 activation and consequent reduction of inflammation; in fact, its inhibition impairs alternative polarization (163, 249).
Consistently, M2 activation requires PGC-1β and the induction of the transcription factor STAT6, enhancing mitochondrial respiration through the upregulation of PPARγ but also PGC-1α and PGC-1β, the master regulators of mitochondrial biogenesis (237). Accordingly, some authors have found that oxidative metabolism increases on M2 polarization and etomoxir-mediated FAO inhibition, similar to the inhibition of OxPhos by oligomycin or by the mitochondrial uncoupler, carbonyl-cyanide-4-trifluoromethoxy-phenylhydrazone, which completely abolishes IL-4-induced polarization as highlighted by the downregulation of the M2 polarization markers Arg1, Dectin-1, CD301, CD206, and RELMα (134, 238).
More recently, however, the strict FAO requirement for M2 polarization has become controversial (208); even though FAO and oxygen consumption increase in human M2, the global oxidative metabolism—not specifically FAO—might be crucial for M2 polarization, since glucose can be used to fuel OxPhos. In fact, although FAO decreases during M1 polarization, it has been demonstrated, in both murine and human MΦ, that etomoxir-mediated FAO inhibition does not block mitochondrial respiration and does not inhibit M2 polarization (237, 332, 355). Notably, suppression of glucose oxidation (not FAO suppression) would inhibit M2 polarization (332); this hypothesis is corroborated by the observation that 2DG-dependent glycolysis-inhibition—similar to mitochondrial ATPsynthase inhibition by oligomycin—blocks respiration and M2 polarization, as observed both in human and in murine MΦ both in vivo and in vitro (63, 246, 353, 397).
Glucose might fuel the TCA cycle for mitochondrial respiration in M2 and, as a consequence, glycolysis might be crucial not only for M1 but also for M2 polarization. In humans, FAO seems to be dispensable for M2 polarization; indeed, IL-4 leads to an unchanged expression of PGC-1α/β and to moderate changes in mitochondrial oxidative metabolism and FAO rate, thus highlighting a possible key difference between mice and humans (97, 332). Moreover, CPT2 knockdown and FAO disruption allows M2 activation, again demonstrating that FAO is dispensable for M2 activation (244); accordingly, some manuscripts report that FAO inhibition does not influence STAT6 phosphorylation and PGC-1β expression that are necessary for M2 polarization (238, 357).
Notably, the effect of etomoxir-mediated FAO inhibition is highly controversial not only because of differences among mice and humans but also because, as discussed in Namgaladze and Brüne (238), different concentrations of etomoxir trigger different effects. Low concentrations of etomoxir inhibit 90% of FAO whereas respiration is only slightly affected; this means that cells shift to another metabolism to fuel OxPhos. High etomoxir concentrations block FAO and also decrease respiration by 50%; they further reduce the expression of IL-4-target genes also in CPT2 −/− MΦ.
Moreover, genetic ablation of the FA transport protein 1 (FATP1) has been used to get insights into the role of FAO, without clarifying the issue. In fact, FATP1 deletion in murine MΦ triggers an FAO-to-glycolysis switch with iNOS upregulation and Arg1 downregulation, without altering the expression of M1 surface markers, including CD80, CD86, and major histocompatibility complex II, MHC-II (151). Also, the role of the adipocyte triglyceride lipase fueling FAO in MΦ is controversial (238).
Interestingly, it has also been proposed that IL-4 acts by activating AKT and, in turn, mammalian target of rapamycin (mTOR) complex-1 (TORC1), which stimulates glucose metabolism (63). Other authors reported that IL-4, in association with M-CSF, also acts through mTORC2 and IRF4 to increase glucose metabolism in murine MΦ, both in vitro and in vivo (135). This pathway resulted critical for alternative MΦ activation; in fact, deletion of Rictor (a component of mTORC2) reduces glycolysis and M2 activation. In conclusion, the necessity for glycolysis might not be typical of M1, but, vice versa, might be crucial for both inflammatory and anti-inflammatory responses. mTOR signaling regulation of MΦ polarization suggests that prolonged starvation might lead to an interesting interplay between mTOR signaling, metabolism, and MΦ polarization, which needs to be explored (30).
4. Glutamine-related metabolism
Recently, the glutamine/glutamate-related metabolism and the UDP-N-acetylglucosamine (UDP-GlcNAc) biosynthesis, through the hexosamine biosynthetic route, have been found to be enhanced and critical for M2 polarization in murine BMDM in vitro (145). Also, high levels of UDP-glucose and UDP-glucoronate characterize this polarization status (Fig. 10). Several studies confirm the crucial role of glutamine in the TCA cycle for M2 activation (124, 242, 285). The importance of glutamine-dependent pathways might also be associated with the requirement of UDP-GlcNAc as a sugar donor for N-glycosylation, possibly to properly fold and export cell surface or secretion proteins (145). N-glycosylation is crucial for M2 activation, since highly glycosylated lectin/mannose receptors are the most typical markers for M2 polarization; the N-glycosylation inhibitor tunicamycin inhibits the expression of the M2 markers RELMα, CD206, and CD301.
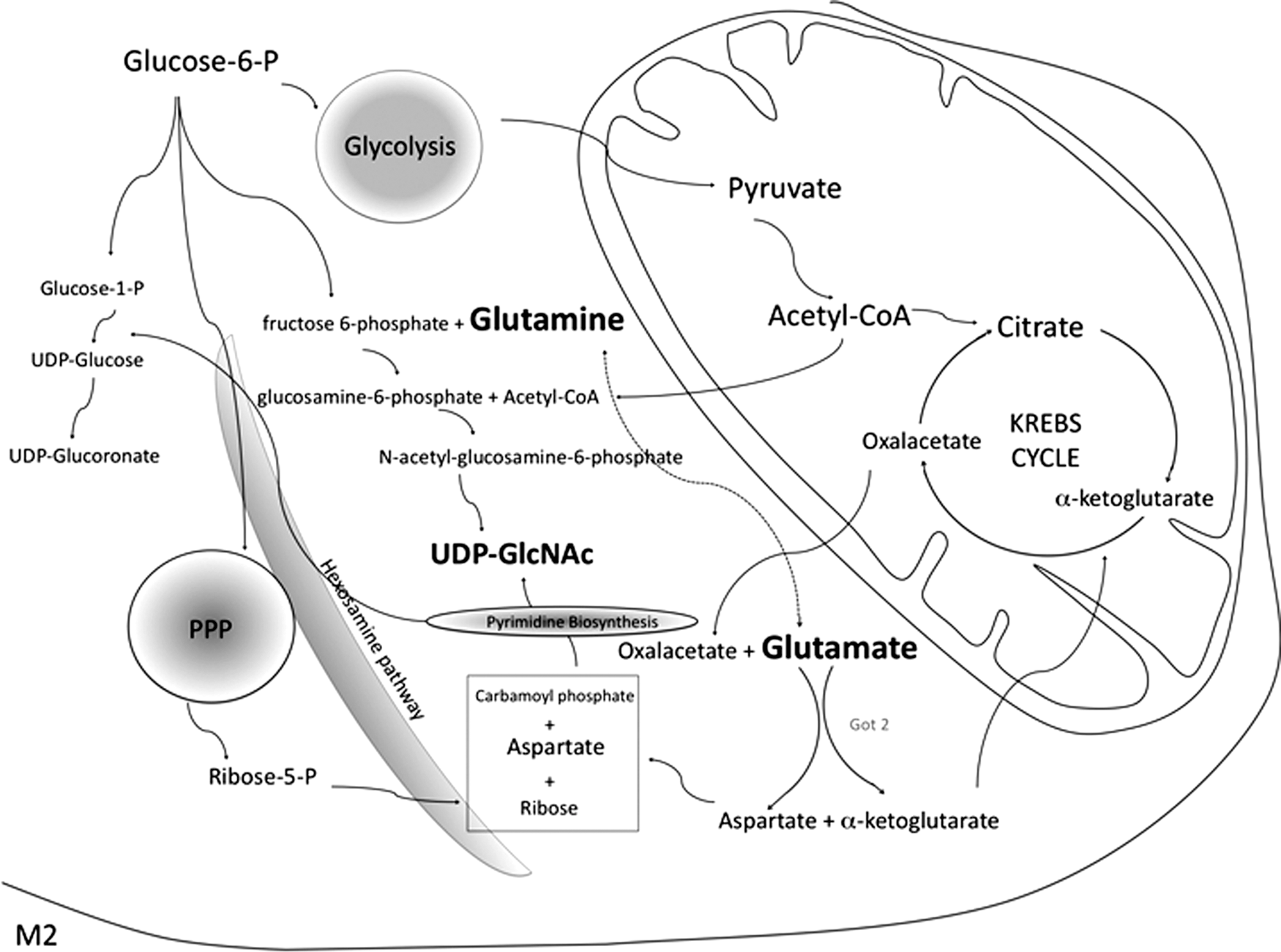
Moreover, UDP-GlcNAc might be used as a sugar donor for O-glycosylation, another pathway connecting cellular metabolism with signaling (145, 379). Finally, glutamate/glutamine support an active TCA (Fig. 10). In general, the availability of amino acids and nutrients in the microenvironment has a profound impact on metabolism and, by extension, on function. Metabolic intermediates are not just a source of energy, but they can also be directly implicated in the definition of a particular MΦ phenotype (117). For example, in lymphocytes, the inability to transport some amino acids (including glutamine) leads to inflammation (251). An extensive literature on the impact of amino acid deficiency on immune cells has been recently reviewed (192, 233).
In synthesis, M2 are characterized by an intact TCA cycle and a high and efficient OxPhos, which is required for M2 polarization; whereas PPP decreases and CARKL, a repressor of M1 activation, is upregulated. Glutamine is also crucial for M2 activation; moreover, PPARγ fosters M2 polarization, and GAPDH inhibits inflammation. Notably, the key role of FAO in M2 activation has recently been questioned, since it has been proposed that the global oxidative metabolism, also fueled by glycolysis, but not specifically FAO, is crucial for M2 polarization.
C. NO and ROS in M1- and M2-polarized MΦ
1. NO/iNOS and ROS in M1
Oxidative stress is associated to inflammation; classically activated M1 recognize invading microbes or cancer cells, engulf them into phagosomes, and destroy them on phagosomes-lysosomes fusion. M1 bactericidal action mainly relies on ROS production occurring in phagolysosomes, and on the production of the cytosolic diffusible NO, which reacts with phagolysosomal ROS to produce highly toxic species. NO and ROS account not only for M1 toxic, antimicrobial, and antitumor effects but also for the redox signaling that modulates many transcriptional events; this mainly occurs at low intracellular free radical levels, when changes elicited by ROS and NO are subtle and reversible.
Vice versa, high NO and ROS levels induce oxidative stress, which is a disturbance of the pro-oxidant/antioxidant balance occurring when the redox state redox systems shifts to the oxidized state. If prolonged, this condition is toxic and bactericidal and leads to protein, DNA, and lipid oxidation and damage. NO and ROS toxicity depends not only on their concentration but also on the type of oxidative species produced (see the subsequent paragraphs within this section).
A key effector molecule preferentially expressed in inflammatory M1 is the cytokine-inducible iNOS (NOS2); it produces NO from
Moreover, a mitochondrial NO synthase (mt-NOS) has been hypothesized; although it awaits characterization, it has been proposed that mt-NOS might directly interact with COX to block its activity in hypoxic conditions (104, 271). Notably, although production of NO by NOS is considered a coupled reaction, NOS might also produce ROS by uncoupled reactions, although their exact stoichiometry awaits further clarification (98, 302).
The NO moiety is added to thiols, secondary amines, or hydroxyl groups of cellular macromolecules by nitrosative reactions. In particular, S-nitrosylation mostly occurs at low NO concentrations and is the reversible addition of an NO moiety to sulfhydryl residues, generating S-nitrosothiol derivatives characterized mostly by regulative roles (220). NO binds heme and nonheme iron targets (Fe-nitrosylation) and inhibits the electron transport chain (ETC) both reversibly, by competing with oxygen for the COX heme, and irreversibly, by reacting (mostly as peroxynitrite) with iron-sulfur clusters in complexes I and II (229). In highly inflammatory conditions, NO might also impair the ETC of the cell in which it is produced, this being typical of M1.
Not only COX, SDH, and, as a consequence, the whole oxidative metabolism are directly inhibited by NO (possibly in concert with itaconate; see section IV.A.3.a), but also PHDs are inhibited by NO, thus leading to HIF-1α stabilization (87, 164, 321, 353) (see sections III and IV.A.3.a). iNOS may also generate N-hydroxyarginine (an inhibitor of Arg1) and the superoxide anion O2•−. In addition, oxidation and reduction of NO, as well as its reaction with oxygen or O2•−, produces several nitrogen species (among which are nitroxyl, nitrite, nitrate, nitrogen dioxide, peroxynitrite, and nitrosoperoxocarbonate), some of which are highly reactive (28). In particular, peroxynitrite and nitrogen dioxide, deriving from the reaction of NO with oxygen, are strong oxidants leading to the oxidation and nitration of proteins, lipids, and DNA; nitrotyrosines are a major marker of nitroxidative stress. iNOS levels and NO concentration have been suggested to be lower in humans compared with rodents, and the mix of TNFα and IFNγ used for rodents might not be sufficient to induce NO production in human MΦ in vitro (28).
Besides NO, M1 generate ROS. ROS production is associated with high MΦ phagocytic activity and inflammatory response, also leading to tissue damage along with cytokines and chemokines. NO and ROS combine to produce highly reactive species, thus increasing nitroxidative stress. Blocking ROS production inhibits the M1 inflammatory phenotype (288, 331). Although the mechanism by which MΦ generate ROS needs to be fully elucidated, ROS formation mainly derives from NOX, mitochondria, and also from NOS; indeed, as stated earlier, NOS participates in the elimination of pathogens also through direct generation of ROS (28). NOX1 to NOX5 and dual oxidases-1/2 (DUOX1 and DUOX2) are the primary sources of ROS.
NOX are transmembrane complexes that are able to transport electrons across membranes, thus leading to the reduction of oxygen into superoxide O2•−. When this occurs across the phagocytic membrane, O2•− is released into the vesicles. ROS production in MΦ on pathogen recognition is predominantly attributable to NOX2, whose subunits are assembled into phagolysosome membranes on stimuli such as IFNγ and TLR activation (31, 331). Interestingly, a direct interaction between TLR4 and NOX4 has been reported (331). NOX2 directly produces O2•−, which is converted to other ROS, for example, its dismutated product hydrogen peroxide (H2O2), the highly reactive hydroxyl radical (OH•) produced by the Fenton chemistry, and associated species.
As a consequence, the targets engulfed by MΦ become submerged into a mixture of toxic oxidants. NOX also contribute to lowering phagolysosome pH, thus favoring the activity of lysosomal proteolytic enzymes contributing to M1 antimicrobial action. TLR4 stimulation also leads to mitochondrial ROS generation from complex I through an unknown mechanism. Moreover, in M1, another mechanism dependent on succinate oxidation by SDH has been proposed for ROS production (see sections III and IV.A.3.b) (214); even the enzyme Irg1 and citrate induce ROS production (see sections III and IV.A.3.a) (118, 140, 141).
2. NO/Arg1 and ROS in M2
After the inflammatory phase, MΦ switch to the M2 phenotype and release IL-10 and TGF-β reducing inflammation; it also reduces ROS and NO production by upregulating, among other mechanisms, Arg1. Arg1 is inducible and competes with iNOS for their common substrate
Depending on the content of GSH, M1 and M2 have been defined as oxidative and reductive MΦ, respectively, confirming crucial and specific redox mechanisms in MΦ (331). Decreased ROS production, for example, by NOX2 inhibition, is necessary for M2 polarization. As stated earlier, NADPH and ROS production is reduced by CARKL, which contributes to M2 polarization. The interaction between MΦ and apoptotic bodies triggers the binding of the protein SYNCRIP to the NOX2 mRNA, thereby leading to its instability and favoring the M2 phenotype (166); this effect is also achieved by apocynin, an NOX inhibitor, and by mutation of the NOX subunit p47 phox (331). Moreover, differently from M1, extracellular ATP seems to block IL-1β in M2 by inhibiting inflammasome and ROS production (331).
3. Redox signaling in M1 and M2
Redox signaling in classical versus alternative MΦ polarization is far for being straightforward. Through transcription factor S-nitrosylation, NO regulates gene expression of not only pro-inflammatory but also respiratory chain and cell cycle genes (20). Although it is awaiting further clarification, along with NF-κB-induced iNOS expression there is also evidence of ROS and reactive nitrogen species (RNS)-mediated NF-κB activation and M1 polarization (28). NO activates NF-κB likely by S-nitrosylation; however, long-lasting NO exposure reduces NF-κB activity, contributing to M2 polarization (28, 262). NF-κB is also modulated by ROS, as well as by AP1 and p38 MAPK (262, 277, 331); also in this case, low levels of H2O2 enhance NF-κB activation, whereas high H2O2 levels inhibit it (28). Moreover, IkB oxidation reduces its degradation and, consequently, NF-κB activation, thus contributing to M2 polarization (262).
On the other hand, ROS also activate the inflammasome, and many redox-sensitive proteins are crucial in the signaling triggered by inflammatory mediators (14, 65, 289, 331); indeed, NOX inhibition favors M2 polarization (51). Although controversial [see below Lo Sasso et al. (188)], the NAD-dependent HDAC silent information regulator 2 (SIRT2) seems to be involved in LPS-induced ROS generation and NF-κB-dependent M1 gene expression, and H2O2 enhances M1 polarization by reducing TNFR1 shedding, which would reduce the inflammatory response [see references in Tan et al. (331)].
ROS and RNS are, therefore, crucial both for their cytotoxic effects and for signal transduction.
Notably, once generated in M1, they activate inflammatory genes but at the same time, also trigger protective mechanisms that might be necessary to allow the switch to the anti-inflammatory M2 phenotype; accordingly, a genetic defect in ROS production induces a hyper-inflammatory response (331). The potential role of ROS in M2 polarization is also suggested by the inhibition of monocyte-to-M2 differentiation caused by the antioxidant butylated hydroxyanisole (396); similarly, MCP1-induced protein-mediated ROS stimulation might be necessary for M2 polarization (155).
Moreover, the strong interaction of NO with the soluble guanylate cyclase heme is an Fe-nitrosylation, which produces cGMP, found to be protective and anti-inflammatory. NO also induces PPARγ, which antagonizes NOX2 assembly and attenuates NF-κB formation. This might be part of the protective mechanisms readily activated by MΦ against excessive inflammation and tissue damage. In this context, H2O2 produced by SOD1 has been found to promote M2 polarization by activating STAT6 and reducing TNFα and iNOS expression; H2O2 acts on a critical STAT6 cysteine leading to STAT6 nuclear translocation (128, 331). In addition, previous reports have shown with controversial results that SOD1 modulates pro-inflammatory genes such as TNFα, IL-1β, and iNOS and that, in other cell types, H2O2 might increase STAT6 phosphorylation via oxidative inactivation of the protein tyrosine phosphatase 1B (331).
Notably, free radicals produced by M1 MΦ might also be toxic for M1 themselves, which are, indeed, equipped to survive the bactericidal oxidative stress occurring during classical activation. A crucial role is played by the transcription factor Nrf2 activating antioxidant genes; M1 might, therefore, survive and persist at the sites of infection and, in principle, switch to an M2 phenotype to allow tissue remodeling. MΦ protect themselves from radical toxicity also by increasing the expression of DNA repair proteins and free radical scavengers, whereas the chromatin remodeling necessary to induce MΦ LPS tolerance is a redox-sensitive process (13, 31, 162, 279, 337).
Further, MΦ protect themselves by generating many toxic species into micro-compartments; indeed, O2•− formation mostly occurs into phagolysosomes where O2•− acts against pathogens but is separated from the rest of the cell, which is, therefore, protected. In phagolysosomes, ROS are also separated from cytosolic NO, thus preventing the production of highly reactive species deriving from the reaction of ROS with NO.
Moreover, oxidation and reduction of NO convert it into several nitrogen species whose ratio constantly changes during M1 activation and that might represent another form of protection from the high NO toxicity deriving by its reaction with ROS. Protection from NO is also achieved by S-nitrosylation of caspases, reducing the responsiveness to apoptotic signals. Over-production of ROS in MΦ during apoptotic cell phagocytosis is followed by attenuation of the oxidative burst by PPARγ activation as well as by resolvin D1 (derived from docosahexaenoic acid), which prevents MΦ death and ROS production by inactivating NOX2 (150, 174).
Although controversial, NO seems to accelerate the process of phagocytosis (304, 350). In particular, there is evidence that on FcγR stimulation of unprimed MΦ, nNOS and, to a lesser extent, eNOS produce low levels of NO that promotes phagocytosis by surrounding MΦ in a paracrine manner (137). NO produced by MΦ is necessary for PS externalization in dying cells through S-nitrosylation and inhibition of the aminophospholipid translocase (331, 351). Conversely, in nonactivated MΦ, NO stimulates NOX-dependent ROS generation by increasing mitochondrial ROS, but not phagocytosis; NO enhances mitochondrial ROS formation by inhibiting the ETC (see section IV.C.1) (229). Intracellular ROS are also able to increase MΦ phagocytic activity, and NOX2-deriving ROS seem to be necessary for apoptotic cell engulfment (but not for bacteria engulfment) (187, 229, 301, 331).
Extracellular ROS (mostly generated in the plasma membrane even by dying cells) have autocrine and paracrine signaling roles and are necessary for oxidative modification of dying cell surface molecules (e.g., oxidation of membrane proteins and lipids, such as phosphatidylserines), which are eat-me signals for MΦ (331). Phagocytosis and ROS production are closely linked by a common signaling pathway; in fact, phosphatidylinositol 3,4,5-trisphosphate is necessary not only for cytoskeleton reorganization and phagocytosis but also for NOX activation and ROS production (123). ROS and NO are also associated to high MΦ migration ability and to the consequent monocyte/MΦ recruitment (28, 331).
Mitochondrial function is linked to their morphology, which depends on mitochondrial membrane fusion and fission. For this reason, their shape is associated with metabolic homeostasis and changes rapidly in response to metabolic cues (370). Mitochondrial dynamics in MΦ polarization is far from being clear; in fact, it has been shown that mitochondrial fission promotes pro-inflammatory TLR-induced IL-12 expression in MΦ and inhibits IL-10 expression through IRF1 stabilization (99) and that, controversially, defective mitochondrial fission augments inflammasome activation (260).
Although most of the metabolic properties of polarized MΦ have been found to be a consequence of polarization, it is conceivable, although not always proven, that these metabolic features, if triggered in MΦ, would also be able to determine the direction of polarization. This is an important issue to be unraveled for future applications of immunometabolic therapies.
To sum it up, high ROS and NO production due to iNOS upregulation and Arg-1 upregulation-dependent ornithine and polyamine production are considered molecular signatures of M1- and M2 polarization, respectively. Decreased ROS production is also necessary for M2 polarization.
Although a dualistic MΦ classification in M1 and M2 subtypes with peculiar metabolic patterns is schematically useful, the link between MΦ polarization and metabolism, in particular the role of FAO, the differences between species and between in vitro and in vivo observations, awaits further elucidation. Indeed, as suggested earlier, polarized human MΦ might have, for some aspects, different metabolic features compared with murine MΦ, as observed by large-scale transcriptomic and proteomic analyses (200), raising the possibility of diverse immunometabolic therapeutical approaches between species. Moreover, the available information is mainly regarding the bipolar phenotypes deriving from in vitro LPS/IFNγ- and IL-4-induced MΦ, whereas the metabolic characterization of intermediate states is only at the beginning stage.
MΦ polarization requires a dramatic genetic and metabolic re-organization, going through several steps among the pro-inflammatory and the anti-inflammatory extremes. MΦ are extremely heterogeneous, and M1 and M2 categories are an over-simplification; in fact, M1 and M2 markers might also be expressed at the same time. Similar to other M1 and M2 features, even their glycolytic and oxidative metabolic features represent the extremes of a spectrum of several intermediate phenotypes (359).
V. Manipulating the Metabolism to Polarize MΦ
Resident MΦ show a high plasticity and adaptation to their microenvironment; MΦ polarization has been suggested to be reversible, so that differentiated MΦ might be reprogrammed to switch from one profile to another if transferred to a different environment (86, 169, 170, 335). However, evidence of in vivo M1-to-M2 repolarization is lacking; a recent in vivo study, performed with both murine and human MΦ, reported that M1 are unable to reconvert to M2, whereas M2 might be repolarized into M1 (353). The authors found that the inability to reconvert into an anti-inflammatory M2 phenotype is caused by an OxPhos inhibition occurring in inflammatory M1. More specifically, M1-produced NO impairs mitochondrial function; inhibiting iNOS during LPS/IFNγ-induced polarization recovers mitochondrial respiration and allows M2 repolarization. Notably, although both ROS and NO can inhibit mitochondria (87, 281), M2 polarization-inhibition has been proposed to be specifically caused by NO, since ROS inhibition by N-acetyl-cysteine (NAC) has no effect (353).
Cells are very flexible in relation to the type of substrate used; removal of a particular substrate or blocking a metabolic route is quickly compensated by others, to obtain energy. Metabolic changes imply a metabolic reprogramming due to epigenetic modifications, signal transduction, and transcriptional regulation; changes in metabolite levels also affect cell state. Since metabolic changes occur during MΦ polarization and metabolic intermediates are not just used to get energy but also directly contribute to drive specific MΦ phenotypes, targeting these metabolites to polarize MΦ and, possibly, to impact on several diseases is an exciting prospect.
Various metabolic strategies are able to redirect MΦ polarization, for example, enhancing the oxidative metabolism and protecting mitochondria allow M2 reprogramming (239, 240, 353). The metabolic sensor 5′ adenosine monophosphate-activated protein kinase (AMPK) activation by metformin and 5-aminoimidazole-4-carboxamide-1-b-4-ribofuranoside (AICAR) with the subsequent oxidative metabolism stimulation leads to M2 polarization also in vivo (21, 46, 239, 240, 399) and decreases MΦ infiltration into the central nervous system in multiple sclerosis mice models, thus reducing the progression of disease (239, 240). Interestingly, miR-33 drives M1 polarization by targeting AMPK (253). By contrast, it has been suggested that metformin inhibits M2 polarization of tumor-associated MΦ (TAM), thus preventing cancer metastasis both in vivo and in vitro, in murine and human MΦ (50, 71). It is conceivable that metformin's effect varies according to the microenvironmental changes (46, 139).
The NAD-dependent deacetylases sirtuins also modulate metabolism and impact MΦ polarization in vivo (138, 146, 153, 167, 237). By favoring oxidative metabolism, sirtuins inhibit M1 activation being anti-inflammatory during response against pathogens. In this context, the role of the nicotinamide phosphoribosyl transferase, an enzyme involved in NAD+ biosynthesis and sirtuin function, is crucial also in vivo and, possibly, linked to PPARγ (17, 335). SIRT3 modulates SDH activity and suppresses ROS by deacetylating and activating MnSOD (55). SIRT1 and SIRT6 favor the M2 phenotype by switching the metabolism from glycolysis to FA β-oxidation (186) and by inhibiting NF-κB and STAT3, as observed in human MΦ cell lines and in primary human and murine MΦ (48, 385). Also, SIRT2 and SIRT6 stimulate the anti-inflammatory pathway in murine MΦ (175, 188).
Accordingly, the flavonoid quercetin inhibits M1 polarization and inflammation in high fat diet (HFD)-fed mice through an AMPK/SIRT1-mediated mechanism (75), and the phytoalexin resveratrol activates SIRT1 and attenuates the inflammatory response in murine MΦ by inhibiting NF-κB/ROS-mediated IL-6 secretion along with TNFα and NO synthesis. Resveratrol promotes M1-to-M2 plasticity and M1 cell death in murine and human MΦ by a mechanism involving Arg1 activation (53, 177, 250, 292); however, it has controversially been reported that resveratrol inhibits TAM M2 polarization observed in vivo in mice (323). Synthetic SIRT1-activating compounds have been developed: They have a greater potency compared with resveratrol; among them, SRT1720 has an anti-inflammatory effect that acts by reducing the number of M1 in mice (44, 48). The SIRT3 activator Honokiol inhibits iNOS expression, NF-κB and TNFα secretion in LPS-stimulated murine MΦ (39).
Another molecule proposed as a metabolic modulator is the dimethylfumarate (DMF; Tecfidera), a fumaric acid methyl ester rapidly hydrolyzed to its active metabolite monomethyl fumarate (MMF). DMF reduced the progression of multiple sclerosis in a phase III clinical trial (110); since fumarate is a TCA cycle metabolite, it might act on MΦ metabolism favoring an M2 phenotype. In fact, DMF triggers a cytokine production shift from a pro-inflammatory to an anti-inflammatory pattern, inducing an M1-to-M2 phenotype shift in vitro. In particular, in LPS/IFNγ-treated human peripheral blood mononuclear cells, the expression of CXCL8, CXCL9, and CXCL10 chemokines is inhibited by DMF; whereas MMF-treated ones upregulate the anti-inflammatory IL-4, IL-5, IL-10, and IL1-ra (248, 335). Although controversial, DMF is categorized as anti-inflammatory by possibly activating the transcription factor Nrf2 inducing antioxidant genes (335).
Consistent with the earlier considerations, PDK1 plays a key role in in vitro murine MΦ polarization (332); it phosphorylates and inhibits some components (e.g., PDH-E1α) of the PDH complex, converting pyruvate derived from glycolysis to acetyl-CoA. Acetyl-CoA, in turn, enters the Krebs cycle, thus generating NADH and FADH2 fueling the ETC for OxPhos. PDK1 promotes M1 polarization, enhances glycolysis and lactate production, and inhibits M2 (332). Moreover, its downregulation decreases glycolysis and lactate production in response to TLR activation while increasing mitochondrial respiration; it also reduces IL-6 and iNOS mRNA levels as well as iNOS and COX-2 protein levels, therefore decreasing the induced M1-inflammatory response (332).
As reported earlier, PPARγ is crucial for alternative MΦ activation (47, 249, 258, 259). Also, PGC-1β promotes M2 polarization through IL-4 and STAT6 in BMDM, while stimulating FAO and mitochondrial biogenesis (357). The energy production coordinator PGC-1α promotes oxidative metabolism; it is upregulated in M2, and its over-expression stimulates M2 polarization (72).
MΦ polarization is also influenced by FA-induced metabolic reprogramming; different FAs exert different effects on MΦ polarization, for example, human MΦ incubated with the saturated FA palmitate, but not with oleate, stimulate inflammatory cytokines and ROS production inducing M1 polarization and reducing OxPhos dependency through ceramide-dependent PPARγ inhibition (259). Vice versa, the omega-3 FA eicosapentaenoic acid (EPA) increases IL-10 and reduces INF-γ levels, thus promoting an M1-to-M2 shift in mice (33). Similarly, other lipid derivatives such as lipoxins and palmitoleic acid have anti-inflammatory effects (287).
In obesity, adipose tissue is characterized by infiltrating pro-inflammatory MΦ; high levels of circulating FA might trigger M1 polarization in the adipose tissue of obese individuals. The effect of modulating FAO in the presence of high levels of FA is still unclear; activating FAO in MΦ might be beneficial since it metabolizes FA, an anti-inflammatory effect. On the other hand, it has been shown that palmitate oxidation fuels OxPhos and produces ROS, thereby activating the inflammasome and pro-inflammatory cytokines production.
As discussed earlier, hypoxia-activated HIF-1α induces iNOS and IL-1β; moreover, the effect of HIF-1α on glucose uptake potentiates glycolysis and pro-inflammatory activity, as observed in cultured human and murine MΦ and in a murine model of atherosclerosis (143, 338). More recently, it has been found that HIF-1α leads to increased mitophagy, which, in turn, induces a metabolic reconfiguration toward glycolysis, causing M1 polarization (85). Notably, intramuscular pO2 is lower compared with atmospheric oxygen levels even in physiological conditions; since pO2 influences cell physiology, data obtained in vitro might not fully reproduce in vivo processes also for this reason, even though M1 and M2 metabolic features occur in different tissue pO2 conditions.
In conclusion, these data emphasize the potentiality of metabolic therapies in directing MΦ activation.
VI. MΦ and Skeletal Muscle Regeneration
Besides their role in innate immunity, MΦ are also involved in tissue repair (108, 180, 201). In particular, we will hereby describe the crucial role of MΦ in allowing efficient skeletal muscle regeneration (245); the muscular regenerative program is first illustrated, followed by the analysis of the role of MΦ in regeneration on acute injury and chronic muscle diseases. In particular, the relevance of the M1-to-M2 switch and the interaction between MΦ subsets, satellite cells (SCs), and other muscle resident or infiltrating cells is discussed.
A. Skeletal muscle regeneration
In response to skeletal muscle damage, a finely regulated regeneration program is rapidly activated to allow muscle recovery (339). Muscle regeneration relies on proliferation and differentiation of SCs, the muscle resident stem cells localized under myofiber basal lamina usually quiescent during homeostasis (205, 389). In response to muscle damage, in both acute and chronic conditions, SCs exit from quiescence and undergo proliferation followed by either asymmetric division-mediated commitment to terminal muscle differentiation or return to quiescence as part of the SC pool (40).
SC asymmetric division guarantees that SC number does not change on multiple regenerative events, thus maintaining the muscles' regenerative potential (374). The ability of SCs to stay in a quiescent state is essential for SC self-renewal and maintenance of muscle stem cells pool (19, 227). Quiescence is a regulated condition, characterized by the expression of negative cell cycle regulators such as p27Kip1 and retinoblastoma tumor suppressor protein (Rb) (36).
Several markers for SCs and regenerating myofibers have been identified. Quiescent SCs specifically express Pax7 (306); SC activation is induced by mitogenic factors released in the damaged muscle area, such as insulin-like growth factor-1 (IGF-1), which downregulate p27Kip1, allowing cell cycle entry (37). Activated SCs (Pax7+, Myf5+) are also characterized by MyoD expression, typical of proliferating myoblasts that are able to undergo both self-renewal and differentiation.
Myoblast commitment to terminal muscle differentiation—involving Wnt and Notch pathways—is characterized by Pax7 downregulation and induction of early and late muscle regulatory factors (MRFs), specifically Myogenin and MRF4/Myf6 (25, 58). At this stage, differentiating myocytes fuse with pre-existing myofibers or with each other, to generate multinucleated myotubes expressing embryonic myosin heavy chain (MyH3/eMyHC) and undergoing further fusion with neighboring myotubes to produce myofibers (339).
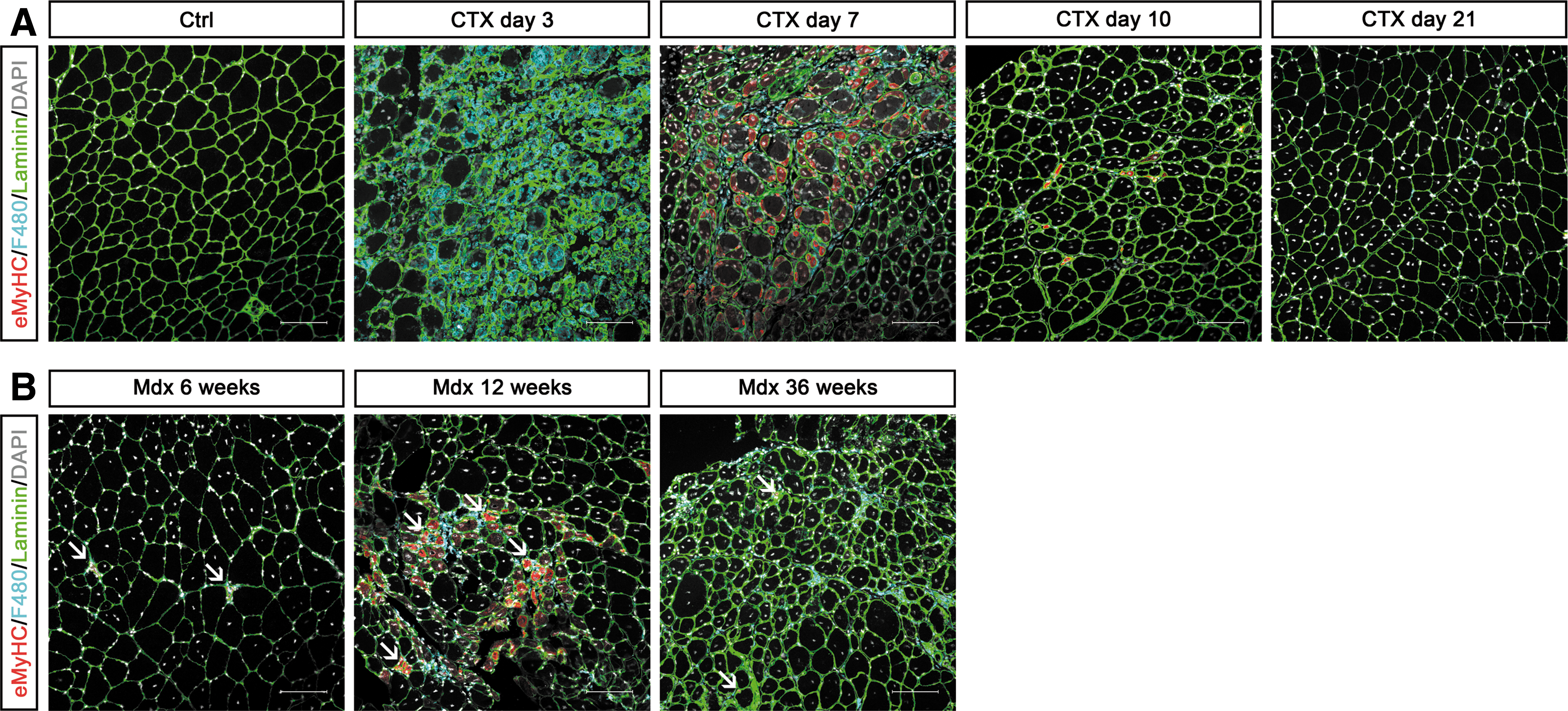
Newly formed myofibers can be distinguished by their small caliber, by eMyHC expression, and by centrally located myonuclei; whereas mature myofibers have bigger diameters, express different isoforms of adult MyHC (Myh1/fastMyHC2x, Myh2/fastMyHC2a, Myh4/fastMyHC2b, Myh7/slowMyHC1, Myh8/perinatal MyHC) (305) along with other late differentiation markers (e.g., muscle creatine kinase) and their nuclei are peripherally located (Fig. 11) (54, 393). Regenerating myofibers express the same MyHC isoforms that are characteristic of the specific damaged muscle, in both acute and chronic injury (305).
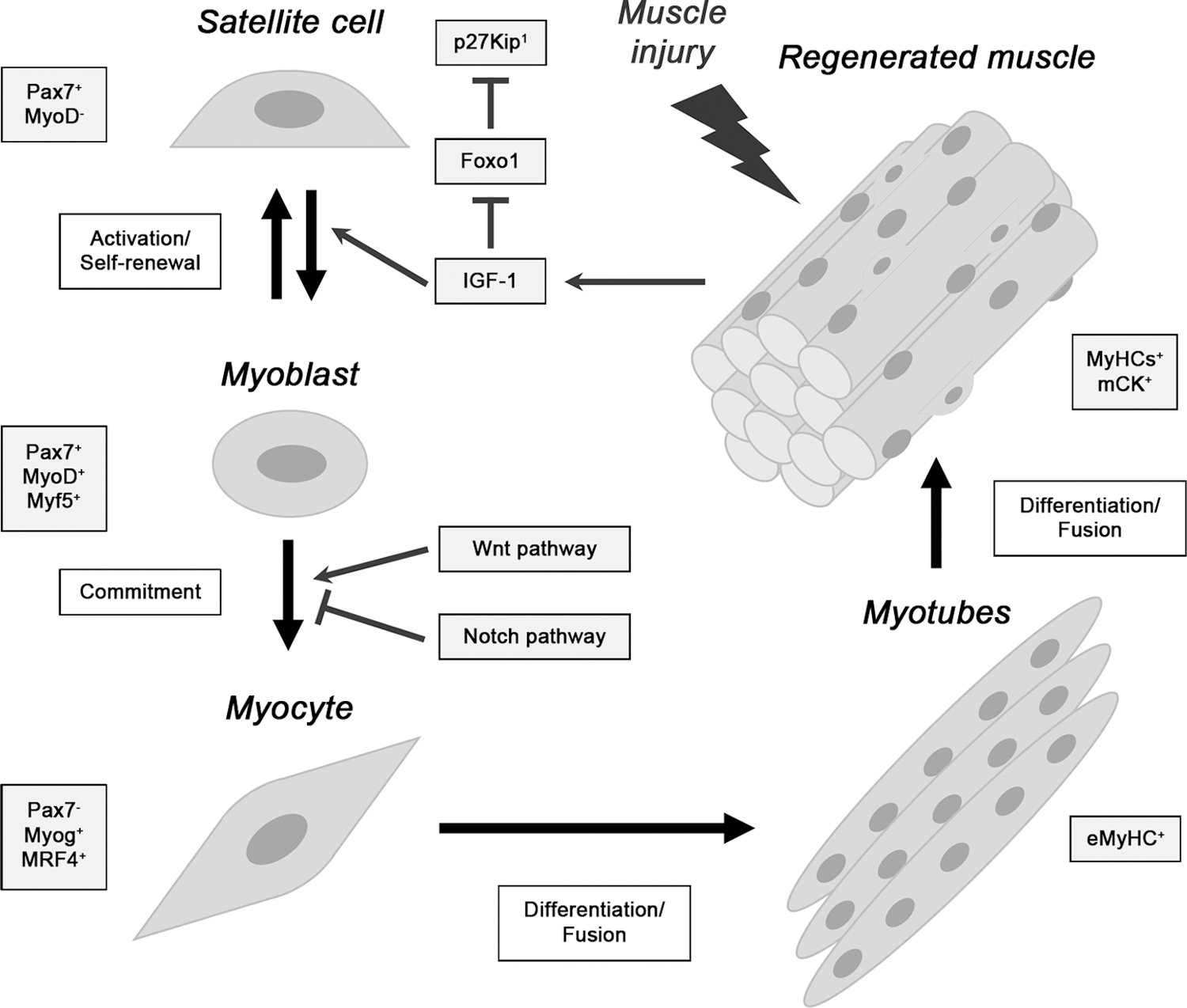
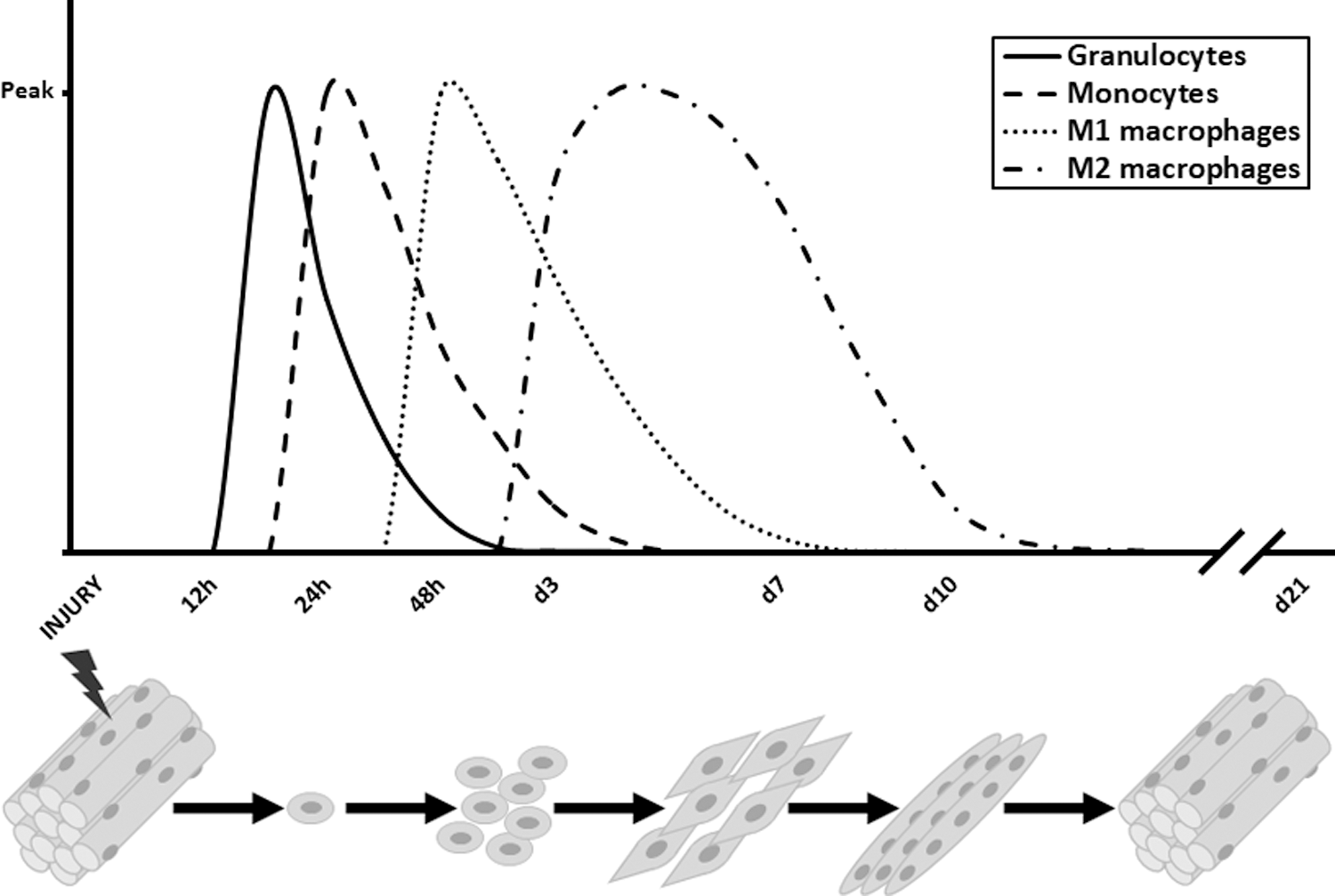
A detailed histopathological analysis of several regenerative stages on acute injury is shown in Figure 12A, whereas immunofluorescence detection of MΦ and newly formed myofibers is shown in Figure 13A. The scheme in Figure 14 shows the timing and trend of MΦ recruitment after muscle injury in parallel with the different muscle repair stages in which they are involved (125).
B. Role of MΦ in skeletal muscle regeneration
Muscle infiltrating immune cells play a critical role in skeletal muscle regeneration; neutrophils and MΦ are very abundant in damaged muscles, as observed in human biopsies and in mice (6, 93, 340, 342), whereas eosinophils (129) and lymphoid cells (CD4+, CD8+, and regulatory T cells/Treg) are rare although relevant for successful regeneration (29, 34, 367). However, a definitive understanding of the complex temporally coordinated MΦ roles in acute injury and in muscle degenerative diseases, and of the mechanisms regulating the crucial MΦ polarization occurring in regeneration, is still lacking (158, 296, 366).
As stated earlier, MΦ are able to assume, in response to a large variety of stimuli, a wide spectrum of polarization statuses corresponding to defined transcriptome signatures (386). Although the gross in vitro M1/M2 classification, including M2 subtypes, is schematically useful, it represents a conventional description that does not exhaustively recapitulate the in vivo process and the MΦ milieu in regenerating muscle, characterized by cell-to-cell interactions and by several uncharacterized MΦ phenotypes intermediate between M1 and M2 subtypes (142).
1. On acute injury
After injury, the number of MΦ within the skeletal muscle (located in the interstitial space of regenerating muscle) exponentially increases; damaged myofibers undergo necrosis; and release of normally muscle-compartmentalized factors recognized by TLRs triggers the production of pro-inflammatory cytokines and chemokines, including CCL2, which recruit bone marrow-derived monocytes (Ly6C+CCR2+CX3CR1low) into damaged muscles. In murine models of muscle injury, monocytes differentiate into highly plastic MΦ (Figs. 1 and 2) that, in response to cues from a damaged environment, polarize toward M1/Ly6C+ and M2/Ly6C− phenotypes mounting and resolving the inflammatory response, respectively (6, 224, 319).
Blocking MΦ recruitment to damaged muscles in the first 24 h after injury impairs muscle regeneration, increasing necrotic fibers and fat deposition, as revealed in transgenic mice expressing the diphtheria toxin receptor (DTR) under the CD11b promoter control (CD11b-DTR mice), so that CD11b+ cells (mainly MΦ) might be depleted on diphtheria toxin injection (6, 374). Moreover, studies in mice highlighted the crucial role of MΦ infiltration in muscle regeneration, which is impaired by genetic deletion of CCR2 (CCL2 receptor) or CCL2, both causing lower MΦ recruitment (191, 197, 311, 376).
Experiments in mice suggest that monocyte recruitment occurs as a result of an early activation of resident MΦ (Ly6C+CX3CR1−) releasing chemoattractants CXCL1 and CCL2, determining a massive neutrophil influx, in turn responsible for Ly6C+CCR2+CX3CR1low circulating monocytes extravasation toward damaged skeletal muscle (26, 340). The involvement of muscle-resident MΦ in regeneration is still obscure; transplantation of WT DT-insensitive bone marrow cells in CD11b-DTR mice—used to deplete MΦ and to discriminate between muscle-resident and recruited MΦ—demonstrated that muscle-resident MΦ are involved in monocyte recruitment after muscle damage (42).
On the other hand, if MΦ recruitment to the muscle is blocked (for example in CCR2 −/− mice), the muscle fails to regenerate, suggesting that resident MΦ are only poorly involved in regeneration (60, 376), possibly due to the low number of MΦ residing in skeletal muscles in physiological conditions (6).
Besides neutrophils, T cells have also been implicated in monocyte/MΦ recruitment via the CCL2/CCR2 axis; they contribute to CCL2 production and proper regeneration (394). Moreover, Treg rapidly accumulates in acutely injured and dystrophic muscles, ameliorating muscle repair by suppressing inflammation and enhancing SC expansion (29, 34, 367).
a. M1-to-M2 switch in muscle regeneration
The early steps of muscle regeneration are associated with pro-inflammatory M1 deriving from recruited monocytes (Ly6C+CCR2+CX3CR1low), whereas later on, the intermediate-late steps of regeneration, in mice, are associated with Ly6C−CX3CR1high anti-inflammatory M2. M1 promote phagocytosis of necrotic fibers and debris, sustain inflammation, and support activation and proliferation of SCs during early regeneration; whereas M2 promote the resolution of inflammation and enhance muscle regeneration by inducing muscle progenitor cells (MPCs) differentiation (194, 291).
Recently, a gene expression analysis on murine MΦ, sorted as GFP-positive cells from CX3CR1GFP/+ mice at four subsequent time points after cardiotoxin (CTX)-induced muscle damage, revealed highly dynamic changes in MΦ expression profile in response to injury (356) at different steps of regeneration: (i) Ly6C+-MΦ show an IFNγ-independent inflammatory profile; (ii) a glycolytic-to-oxidative metabolism switch sustains the anti-inflammatory M2 polarization; (iii) Ly6C−-proliferating MΦ highly express M-CSF, and the administration of anti-M-CSF-receptor antibody during muscle regeneration suppresses MΦ—but not neutrophil—infiltration and compromises muscle repair, affecting SC proliferation and differentiation and increasing fibrosis (307); and (iv) Ly6C−-MΦ produce secretory ECM-related molecules involved in intercellular communication and regeneration, such as matrix metalloproteinase-2 (MMP2).
A sequentially synchronized recruitment and activity of different MΦ subtypes, with an M1-to-M2 shift, is required for successful muscle regeneration (6, 372); in fact, chronic M1 activation exacerbates muscle injury by releasing mediators of cell damage and cytokines amplifying inflammation. For this reason, in parallel or soon after any inflammatory response, mechanisms aimed at reducing inflammation are activated; M2 anti-inflammatory cytokines, such as IL-10, inactivate pro-inflammatory genes and further drive MΦ-alternative activation (364).
The transition from the early M1 immune response to the intermediate-late M2 immune response is, therefore, required and, functionally, corresponds to the progression of the myogenic regenerative program, since M2 stimulate myogenic differentiation and promote myocyte fusion (297). Interference with M2 polarization is detrimental on muscle regeneration, and in vitro polarized MΦ and FACS-isolated Ly6C−-MΦ are beneficial when transplanted into injured skeletal muscle (294, 344, 372).
However, the cause of the sequential presence of M1 and M2 in the damaged area of skeletal muscle is still a matter of debate; it is not clear whether skeletal muscle M1 are able to repolarize to M2, due to changes in the damaged microenvironment, or whether new Ly6C−CCR2−CX3CR1+ monocytes are recruited, at later stages, to a repair-promoting environment (e.g., when CX3CL1/fractalkine attracting M2 is produced by resident MΦ due to interaction with dying cells) where they differentiate to M2, as further suggested by selective depletion of Ly6C− monocytes enhancing M1 generation (102, 103, 116, 121).
By contrast, a pivotal paper, first describing the M1-to-M2 polarization within muscle as dependent on necrotic muscle phagocytosis, supports the hypothesis of MΦ phenotypic transition within muscle by which injured skeletal muscle recruits only pro-inflammatory monocytes (Ly6C+CCR2+CX3CR1low) that, within muscle, switch phenotype to become proliferating anti-inflammatory Ly6C−CCR2+CX3CR1high cells, further differentiating into F4/80+ MΦ (6). Moreover, the MAPK-phosphatase-1 (MKP-1)-p38-Akt axis is crucial for sequential M1-to-M2 transition, and deregulation of MΦ skewing by MKP-1-loss, in mice, results in impaired SC activity and defective regeneration (268).
Also, AMPKα1 is crucial for phagocytosis-induced pro-to-anti-inflammatory MΦ skewing, at the time of inflammatory resolution; indeed, damaged muscles of AMPKα1 −/− mice show decreased M2 and increased M1 subsets, along with regenerative impairment (226). In addition, CCAAT enhancer-binding protein (C/EBP)β in infiltrating MΦ is required for the upregulation of M2-specific genes and for M1-to-M2 switch and muscle regeneration (210, 291). The critical role of the MΦ-phenotypic switch for regeneration is also confirmed by the recent finding that miRNA-155 expression in myeloid cells modulates MΦ activation in vivo in mice and is necessary for proper muscle regeneration; during the initial inflammatory phase, miRNA-155 suppresses SOCS1 (a negative regulator of the JAK/STAT pathway) (243).
Also, in human skeletal muscle regeneration, different MΦ subsets modulate MPC fate; specifically, pro-inflammatory M1 are mainly associated with regenerating areas containing proliferating myogenic cells, whereas M2 are preferentially associated with differentiating myogenic cells (297). Among the different anti-inflammatory M2 subtypes, M2a are associated with halted muscle regeneration, collagen accumulation, fibrogenesis, and angiogenesis, preceded by anti-inflammatory M2c involved in tissue remodeling and regeneration, ECM deposition, and immunoregulation. As reported earlier, under the effect of IL-10, M2c express receptors for pro-inflammatory chemokines, which would possibly serve as a scavenger receptor system to dampen inflammation (270).
2. In chronic muscle diseases: for example, Duchenne muscular dystrophy
Similar to acute injury, also at early stages of neuromuscular disorders, such as Duchenne muscular dystrophy (DMD), muscle regeneration compensates chronic degeneration. However, differently from acute injury, chronic muscle diseases are characterized by overlapping and asynchronous cycles of degeneration and regeneration, associated with continuous MΦ infiltration producing, at each stage of disease, a mixed MΦ population containing variable levels of MΦ subtypes. The balance between different MΦ subpopulations is determinant for the progression of dystrophy in mdx mice, the commonly used mouse model of DMD disease (364).
For instance, the competition between iNOS-producing M1 and Arg1-producing M2a influences the extent of myofiber lysis by MΦ. In fact, M2a reduces the cytolytic activity of M1 (363 –365). Both M1 and M2a are present in muscles of 4-week-old mdx mice, the acute necrotic stage of the pathology (364). Arg1 expression in M2 increases in mdx muscles as dystrophy proceeds, and Arg1 metabolism might contribute to fibrosis driven by M2 in response to Th2 cytokines; indeed, ablation of Arg1 reduces fibrosis (377).
In DMD patients, the dystrophic pathology onset coincides with the onset of muscle inflammation, suggesting a key detrimental role of inflammatory cells in the progression of DMD and other skeletal muscle degenerative diseases characterized by chronic inflammation (366). The inflammatory progression in DMD patients is similar to what is observed in mdx mice. To study the role of MΦ in DMD, we recently generated an MΦ-depleted dystrophic mouse model by crossing the CD11b-DTR mouse with the mdx one, and we have confirmed the crucial beneficial role of MΦ in muscular dystrophies (unpublished observations).
A detailed histopathological analysis of various stages of skeletal muscle regeneration on chronic injury is shown in Figure 12B. Specific markers, identified by immunofluorescence, show that 12-week-old mdx mice muscles are similar to CTX-injured muscles at the intermediate phase of regeneration (day-7 CTX injection) (Fig. 13A, B) [reviewed in Rigamonti et al. (282)].
C. Cross-talk between metabolically diverse MΦ subpopulations and muscle resident cells
MΦ affect myogenesis and muscle fibrosis, after acute damage and in chronic diseases, by producing cytokines, chemokines, soluble factors, and cross-talking with muscle resident cells, for example, fibroadipogenic progenitor cells (FAPs), fibroblasts, SCs, and endothelial cells (218, 275). A reciprocal cross-talk between muscle cells and infiltrating immune cells also occurs, with muscle cells responsible for modulating the MΦ phenotypic shift during repair (261). The proper interplay among different cell types ensures phagocytosis of necrotic myofibers, activation of SCs, execution of the myogenic program (6), angiogenesis (247), and SC self-renewal to restore stem cell pool (225), thus forming new functional myofibers and allowing muscle homeostasis.
MΦ participate in the final stages of skeletal muscle repair by releasing not only anti-inflammatory mediators, such as TGF-β and IL-10, but also pro-regenerative and pro-angiogenic growth factors such as IGF-1 and VEGF-A, fibroblast growth factor (FGF), placental growth factor, and hepatocyte growth factor (HGF). Also, SDF-1 is necessary for collagen deposition into the damaged muscle and for regeneration (287), and through polyamine and collagen synthesis M2-produced ornithine promotes cell proliferation, fibrosis, and tissue remodeling (272). MΦ-produced growth factors act on the ECM, triggering angiogenesis; MΦ also release MMP2 and urokinase-type plasminogen involved in ECM protein degradation and proteolytic activation of HGF [reviewed in Rybalko et al. (294)], whereas collagen VI is also critical for MΦ migration and M2 polarization in mice (47).
Apoptotic cell clearance also contributes to triggering M2 activation and wound healing. Moreover, in response to sterile injury, vessel-associated stem cells (mesoangioblasts) specifically upregulate genes involved in the clearance of apoptotic cells and in M2 polarization—such as CD163—in MΦ (23). Notably, MΦ-secreted complement C1q impairs regeneration; blocking the angiotensin-II type 1 receptor by irbesartan induces M2 polarization and reduces MΦ-associated C1q expression, thus ameliorating regeneration and reducing fibrosis (387).
Immune system-produced ROS also participates in tissue repair key signaling and could be beneficial or detrimental depending on their levels. For example, ROS modulate the release of pro-inflammatory cytokines by modulating the balance between reduced and oxidized HMGB1 forms, involved in SC activation and differentiation in damaged skeletal muscle (358). However, excessive ROS levels can hyper-activate the inflammatory response, thus impairing regeneration. It has been suggested that MΦ-derived ROS are not involved in skeletal muscle regeneration (171); vice versa, iNOS seems to be crucial in acute muscle damage in vivo, indeed NO stimulates muscle repair (59), and M2 express factors such as SOD1 and thioredoxin that are implicated in HMGB1 reduction (360). A comprehensive analysis of redox regulation in resident and infiltrating cells during skeletal muscle regeneration is reviewed in Le Moal et al. (172).
The cross-talk between MΦ and SCs and between MΦ and FAPs will be discussed (Fig. 15).
1. Cross-talk MΦ: SCs
At early stages of regeneration, the cross-talk between SCs and MΦ is mainly modulated by M1-produced pro-inflammatory cytokines, which further recruit immune cells that are responsible for fiber debris and necrotic cell phagocytosis regulating, in an autocrine manner, the balance between MΦ subpopulations. Pro-inflammatory mediators also exert paracrine effects on muscle cells, for example, activating quiescent SCs that start proliferating, as observed in mice and in humans, and regulating FAP fate (43, 297, 343). In fact, SCs/MPCs proliferation was found to be associated with M1, both in vivo and in vitro; M1 migrate toward SCs/MPCs, where they stimulate their proliferation and prevent their premature differentiation (Fig. 16) (297).
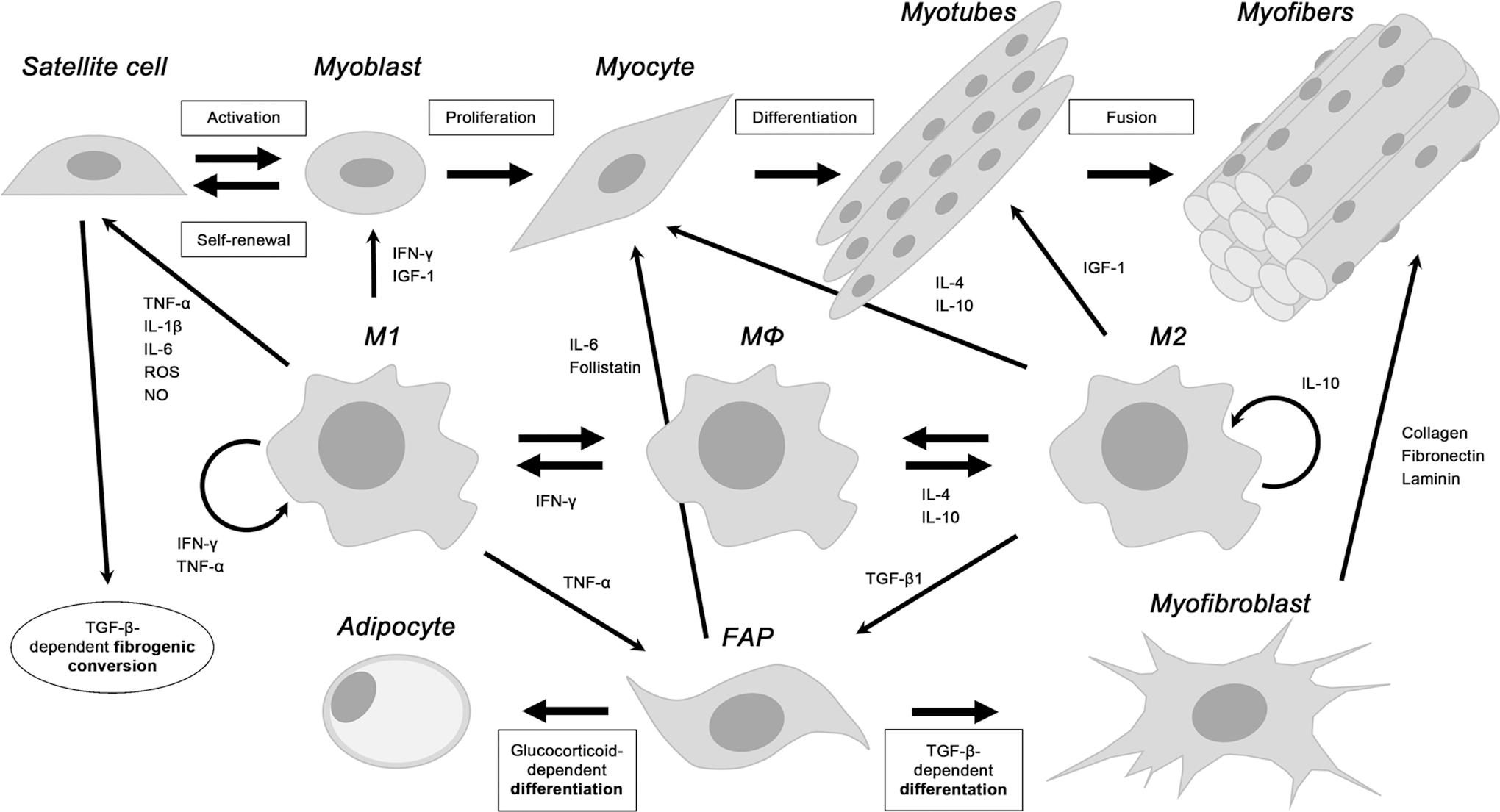
As described in mice, after the phagocytic phase, an M1-to-M2 switch occurs, with M2 inhibiting M1 and reducing pro-inflammatory cytokines-mediated MPCs proliferation while stimulating their differentiation (70).
IFNγ rapidly increases in injured muscle; it binds to its receptor expressed by both M1 and SCs/MPCs and activates target genes via JAK-STAT1 (49, 190, 222). As observed in mice, IFNγ promotes M1 polarization and represses SCs/MPCs differentiation by inhibiting myogenic genes— for example, Myogenin—through recruitment of Jarid2 and polycomb repressive complex-2 to their promoters (190, 381). Early in regeneration, IFNγ signaling maintains the M1 phenotype, allowing MPCs expansion; however, at intermediate-late stages of regeneration, IFNγ signaling must be switched off to avoid terminal muscle differentiation impairment (Fig. 16) (190). IFNγ ablation in mdx mice modulates the M1/M2 balance, this having different consequences at the inflammatory (3–4 weeks of age) and regenerative stages (6–12 weeks of age) of disease. The early inflammatory stage was not affected by IFNγ ablation, suggesting that M1 polarization is not strictly dependent on IFNγ.
By contrast, at the regenerative stage, IFNγ deletion is beneficial in that it promotes pro-regenerative M2 polarization and increases MyoD expression (363). Similar to IFNγ, TNFα acts both on MΦ, promoting M1 polarization, and on MPCs, affecting regeneration (Fig. 4). TNFα induces transcriptional repression of specific muscle genes (Pax7, MyoD, Myogenin, and MEF2C) in MPCs by Ezh2 recruitment on promoters (1, 58, 255). IL-6, TNFα, IL-1β, and G-CSF are all known to enhance myogenic proliferation (41, 122).
The anti-inflammatory cytokine IL-10 is crucial for the M1-to-M2 switch, improving muscle regeneration in both acutely injured and dystrophic muscles (Fig. 4) (70, 365). IL-10 effect on M2 polarization depends on IL-10-mediated AMPK activation, critical for the glycolytic (predominant in M1) to the oxidative metabolism (predominant in M2) switch, as observed in vitro (399). Indeed, AMPKα1 deletion in myeloid cells delays muscle repair, likely by influencing MΦ polarization (226). Although demonstrated only in vitro, IL-10-expressing M2 might also promote myoblast proliferation (70).
Moreover, M2-dependent IL-10 production is essential to support mesoangioblast survival and function in vivo (24). IL-4, mainly produced by eosinophils and Th2 cells, contributes to promoting M2 polarization, thus creating a pro-regenerative environment (Fig. 16). IL-4 is also expressed by muscle cells and controls myoblast/myotube fusion; it is secreted by myotubes and binds to myoblast-expressed IL-4R, therefore recruiting myoblasts to myotubes. IL-4 is regulated by the transcription factor NFATc2, crucial for myoblast fusion (132).
Produced by several resident cell types or ones infiltrating the regenerative muscle, TGF-β, crucial for muscle regeneration, is abundant in acutely injured muscles and in muscles of mdx mice and DMD patients [reviewed in Duffield et al. (78)]. In dystrophic muscles, anti-inflammatory TGF-β is mostly produced by M2 (273, 398). M2-secreted TGF-β supports the formation of myotubes (297). However, excessive levels of TGF-β2, induced by elevated canonical Wnt signaling in dystrophic muscles, affect SC fate, which undergoes fibrogenic conversion (Fig. 16) (18). TGF-β1 can cause fibrosis and neutralizing it significantly promotes muscle regeneration, enhances angiogenesis, prolongs SC activation, and recruits a greater number of M2. If MΦ infiltration is compromised, endothelial-derived progenitors undergo an endothelial-to-mesenchymal transition, possibly triggered by TGF-β, collagen accumulates, and the muscle is replaced by fibrotic tissue (400).
In regenerating muscle, both M1 and M2 produce IGF-1, although it is more prominently expressed by M2 (191). IGF-1 is a potent regeneration enhancer and is upregulated during the inflammation-to-repair transition phase (12, 235). MΦ-derived IGF-1 influences muscle regeneration by a double action: by acting on myogenesis, increasing MPCs proliferation and boosting their terminal differentiation, and in an autocrine manner, by inducing a pro-regenerative M2 polarization and contributing to resolve inflammation (346). Indeed, IGF-1 deletion in myeloid cells impairs M2 accumulation, thus compromising in vivo regeneration (346).
By means of released factors such as IGF-1, MΦ also act on intra-myocellular processes that are crucial during regeneration, such as protein synthesis; IGF-1 promotes myofiber protein synthesis predominantly by activating the phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway and phosphorylating 4E-BP1 and S6K1 (12, 235). Notably, the PI3K/Akt/mTOR pathway also mediates autophagy, which might, therefore, in principle, be modulated by MΦ-produced IGF-1 (286). IGF-1 is also produced by muscle cells on exercise; it might have a trophic action, possibly even physiologically, independently from the occurrence of inflammatory conditions (265). Also, IL-6 might act on the Akt/mTOR pathway, influencing protein synthesis and, presumably, autophagy; however, controversial results suggest opposite roles for IL-6 in the regulation of muscle fiber size and regeneration (10).
Notably, after muscle injury, MΦ secrete the metalloproteinase Adamts1, which targets and impairs Notch signaling, thus strongly increasing SC activation (77). Moreover, in mice, damaged myofibers and infiltrating MΦ release the protein S100B, which expands the myoblast population and promotes M2 polarization modulating collagen deposition. However, prolonged high levels of S100B compromise regeneration by delaying the M1-to-M2 transition and promoting fibrotic tissue deposition (283). Interestingly, anti-inflammatory M2-expressed CD163 is a receptor and scavenger for the cytokine TNF-like weak inducer of apoptosis (TWEAK). A soluble portion of CD163 functions as a decoy receptor for TWEAK, regulating its ability to activate Notch signaling and stimulate MPCs proliferation and tissue regeneration (3).
SCs recruit monocytes/MΦ using various chemotactic systems that are also useful to reduce apoptosis [reviewed by Rybalko et al. (294)]. The chemotactic factor CX3CL1 is produced by resident MΦ on interaction with dying cells and is also expressed by myoblasts; CX3CL1 attracts M2 and also induces the expression of pro-angiogenic factors, thus promoting a microenvironmental shift toward a more regenerative milieu. CX3CL1 also increases in human skeletal muscle after exercise. Notably, an interaction between CCL2/CCR2 and CX3CL1/CX3CR1 chemokine systems in modulating MΦ function during regeneration exists; the impaired MΦ infiltration in CCL2 −/− mice is rescued by CX3CR1 deficiency through enhanced MΦ-dependent ApoE production, improving MΦ phagocytic activity and compensating for defective monocyte recruitment (7).
2. Cross-talk MΦ: FAPs
FAPs are multipotent mesenchymal progenitors of fibroblasts and adipocytes, residing in the skeletal muscle interstitium (Fig. 15) and expressing the surface markers platelet-derived growth factor receptor-alpha and Sca1. Muscle regeneration is associated with an early increase of FAPs that provide signals promoting MPCs proliferation and differentiation, as observed in vivo (129, 149, 295, 352). For instance, IL-6 is upregulated in FAPs from day 2 to 5 on muscle damage in mice and enhances MPCs commitment (149). FAPs-derived Follistatin, a TGF-β-superfamily “bio-neutralizer,” is crucial for proper myotube formation, also improving muscle repair by increasing MΦ and Pax7-positive cell density (228, 388). FAPs are also responsible for ECM deposition during muscle regeneration and, once muscle injury is fully repaired, FAPs return to quiescence (16, 352).
On acute muscle injury, ECM deposition is a transient and beneficial event supporting regeneration by providing a scaffold for regenerating myofibers. In pathological conditions, for example, muscular dystrophies, connective tissue production is not reversible and leads to myofiber replacement with fibrotic scars and fat deposition (Fig. 13A, Sirius) (228, 284). The persistence of activated FAPs (also induced by chronic inflammation) in the damaged muscle is associated with increased collagen and pro-fibrotic factor production, impairing the niche for proper SC activation and differentiation and exacerbating the degenerative phenotype (59, 267). Moreover, in injured muscles, FAPs are able to differentiate into intramuscular adipocytes by undefined mechanisms (76). Based on these considerations, the balance between FAPs activation and FAPs apoptosis is crucial for determining the extent of fibrosis, and the modulation of signaling pathways that fine-tune this balance represents a potential therapeutic approach (178).
FAPs are regulated by signals deriving from other resident muscle cells whose behavior is also influenced by FAPs, in a complex network of reciprocal functional interactions (16). Chronic inflammation is a driving force of fibrosis, with several cell types interplaying with inflammatory MΦ and contributing to ECM accumulation (194); if M1 were to persist in damaged muscles, this would contribute to fibrosis. As an example, fibrinogen, an ECM factor accumulated in muscles of mdx and DMD patients, binds Mac-1 on the MΦ surface, thus upregulating IL-1β and other pro-inflammatory cytokines. IL-1β induces MΦ-dependent TGF-β expression, which increases muscle fibroblasts collagen production (Fig. 16) (362); compromising fibrinogen-Mac1 interaction in mdx mice decreases inflammation and improves muscle regeneration (361).
Recently, Lemos et al. demonstrated that MΦ-produced soluble factors modulate ECM production and FAPs apoptosis balance in murine models of acute and chronic muscle damage (178). M1-produced TNFα mediates FAPs apoptosis that is necessary to reduce the number of FAPs and to limit fibrosis. M2-produced TGF-β counteracts FAPs apoptosis, triggering FAP differentiation in matrix-producing cells (often referred to as α-SMA+ myofibroblasts) providing ECM components such as collagen isoforms, fibronectin, and laminin (16). Accordingly, blocking MΦ infiltration in regenerative murine muscles, by CCR2 ablation or by TNFα expression inhibition, decreases FAPs apoptosis and increases fibrotic tissue deposition (178).
Moreover, the tyrosine kinase inhibitors nilotinib and imatinib induce FAPs apoptosis, thereby reducing muscle fibrosis in mdx mice (136, 178). However, nilotinib-mediated FAPs expansion inhibition also impairs proper muscle regeneration in a murine model of acute muscle damage, since it negatively affects SC expansion. This underlines the relevance of the FAPs trophic supportive function exerted on SCs (94). Besides MΦ, other inflammatory cells affect FAPs activation during muscle regeneration; in particular, eosinophil-secreted IL-4 induces FAPs proliferation, supporting SC myogenesis and inhibiting FAPs differentiation toward adipocytes (129).
Moreover, additional cell types other than FAPs are involved in collagen deposition during the progression of chronic muscle diseases in mice, for example, pericytes (81), SCs undergoing fibrogenic conversion (18, 273), and MΦ-dependent endothelial-mesenchymal transition of endothelium-derived progenitors (400).
We illustrated how MΦ recruitment to damaged muscles is crucial for proper regeneration; in particular, a sequential recruitment of different MΦ subtypes, with M1 followed by M2, is necessary, since at each stage of regeneration, a reciprocal cross-talk between muscle cells and differently activated MΦ subsets mediated by soluble factors is required. However, the origin of M1 and M2 in the damaged area of the skeletal muscle is still unclear.
VII. Metabolic Reprogramming of MΦ as a Potential Therapeutical Approach to Improve Skeletal Muscle Regeneration
The concept of MΦ reprogramming to promote anti-inflammatory/regenerative M2 polarization that is able to reduce inflammation is emerging as a new therapeutic immunometabolic approach to promote tissue healing and reduce inflammation in chronic inflammatory diseases such as atherosclerosis, rheumatoid arthritis or multiple sclerosis, as well as type 2 diabetes and obesity while improving aging health. Vice versa, a reprogramming from tumor-promoting M2 to anti-tumor M1 has been hypothesized as a therapy against cancer [for references see Ref. (335)].
The study of the contribution of metabolic pathways and the integration of local and systemic metabolism at the cellular level in regulating immune cell development and function is often referred to as immunometabolism; it further includes studying the role of immune cells in metabolic homeostasis. Manipulating immune cell metabolism can beneficially enhance or temper the immune response, including driving MΦ polarization and function, useful for the potential treatment of several diseases.
Compared with other strategies such as anti-inflammatory, stem cells-based or antioxidant therapies, approaches based on immunometabolism might have the unique advantage to both reduce inflammation and, at the same time, enhance the tissues' regenerative potential, thus being particularly attractive for chronic inflammatory, degenerative, and metabolic diseases [reviewed in Refs. (133, 152)]. We will discuss how this is particularly evident in skeletal muscle pathologies, including degenerative myopathies, acute injury, and cachexia, as well as in aging and regenerative medicine, where immunometabolism modulation could treat inflammation as well as improve regeneration. In particular, as mentioned earlier, MΦ effect on muscle resident cells strongly depends on their polarization status, which might be oriented by immunometabolic strategies (268).
A. Aging
M1-to-M2 polarization is influenced by a metabolic reprogramming that is induced by circulating FFA. In particular, aged individuals' FFA profile (characterized by an overall circulating FFA increase) impairs M2 activation. Also, circulating TNFα and IL-6 concentrations increase with age, whereas IL-10 and TGF-β1 levels decrease, thus negatively affecting skeletal muscle regeneration; therefore, acting on MΦ metabolism might be beneficial (259).
In fact, some nutraceuticals can play a prominent role in modulating skeletal muscle physiology (56); among them, resveratrol, green tea, catechins, and β-hydroxy-β-methylbutyrate improve SC function and regeneration in muscles of aged mice after disuse, therefore contributing to reducing sarcopenia (4, 15). In particular, resveratrol, also considered an “exercise mimetic,” promotes murine M2 polarization in vitro; however, whether this mediates the beneficial effect of resveratrol on skeletal muscle regeneration or not needs to be clarified (53, 177, 292).
B. Duchenne muscular dystrophy
The balance between different MΦ subpopulations can determine the progression of dystrophy in mdx mice; drugs that are able to modify this balance might attenuate symptoms of muscular dystrophies. Anti-cytokine drugs favoring a pro-regenerative M2 phenotype might be used, as observed in murine models (333, 363). Also, corticosteroids are used with some success to treat DMD, as observed in mice and humans; however, the side effects of these drugs often outweigh their benefits. Interestingly, HDAC inhibitors counteract disease progression in mice by directly acting on muscle cells and also by modulating MΦ polarization toward M2 (216). Moreover, the FA EPA increases IL-10 expression and reduces IFNγ levels, thus promoting M2 polarization, which decreases inflammation in muscles of dystrophic mice (33). Similarly, the SIRT1-activator resveratrol promotes M2 polarization, decreases inflammation, and increases utrophin expression in mdx mice (112).
C. Regenerative medicine
Since regeneration of injured skeletal muscle depends on M1-to-M2 phenotypic transition, in a tissue engineering context, after implantation of the artificial device, the control of M1-to-M2 progression might ensure a proper and timely coordinated transition from the inflammatory to the healing stage. Notably, it has recently been shown that degradation products from the mammalian ECM biologic scaffolds used for skeletal muscle reconstruction in regenerative medicine promote alternative M2 polarization in in vitro mechanically loaded murine MΦ, thus facilitating migration and myogenesis of MPCs in vivo and, ultimately, stimulating tissue repair (82, 298, 314).
This IL-4-dependent pro-regenerative response is characterized by cycolooxygenases-1 and -2 (COX-1/COX-2) activity, which seems to be crucial to allow myogenesis and collagen deposition in the damaged area (69). Resveratrol pro-M2 activity has been suggested to promote vascularization in tissue engineering applications (292, 349, 371). Also, transplantation of acellular ECM scaffolds improves performance of diseased muscles by promoting M2 polarization and activating MPCs (274, 278); however, other authors showed that, conversely from vital grafts, devitalized grafts were unable to promote an anti-inflammatory phenotype and regeneration in vivo (100).
D. Obesity-insulin-resistance
Infiltrating pro-inflammatory MΦ characterize the adipose tissue of obese individuals. Pro-inflammatory cytokines impact on the surrounding tissue, which is associated to insulin resistance. Moreover, HFD—mainly saturated FA—also upregulates pro-inflammatory cytokines in skeletal muscle and in skeletal muscle-infiltrating fat, which increase M1 polarization, further contributing to insulin resistance. Therefore, metabolic modulators promoting M2 polarization might be beneficial against insulin resistance, reducing aberrant glucose metabolism; palmitate-treated MΦ, as well as myoblasts, downregulate miR-16, which is known to impair M1 polarization.
Interestingly, MΦ over-expression of miR-16 enhances insulin sensitivity in co-cultured murine myoblasts (330). In addition, a major contributor to the insulin-resistance onset might be an altered and pro-oxidant cellular redox state, which decreases NADPH levels that, in turn, activate glucose-6-phosphate dehydrogenase (G6PDH), which is crucial for NADPH maintenance. G6PDH activity is elevated in adipose tissue and its over-expression causes insulin resistance (176). In such a context, the anti-inflammatory and antioxidant effects of M2 polarization might also help in reducing G6PDH expression.
E. Cachexia
Some diseases, such as cancer and chronic heart failure (CHF), are characterized by weight and skeletal muscle loss that is referred to as cachexia (219, 368). In cachexia, an increased systemic inflammatory state is associated with impaired myogenesis. In this context, the MΦ-reprogramming approach might be useful to promote anti-inflammatory M2 polarization. Notably, anti-inflammatory therapies have failed in counteracting cachexia (368). Therefore, the MΦ-immunometabolic approach, which does not merely reduce inflammation but also increases M2 activation, might potentially promote muscle regeneration, limiting muscle loss in cachexia. In this regard, we have recently found that the metabolic modulator trimetazidine (TMZ) enhances myogenesis in cancer cachexia in mice (101), along with stimulating M2 reprogramming in vitro (unpublished observations).
The MΦ-reprogramming approach against cachexia of cardiac origin is supported by preclinical studies demonstrating that M2 polarization (e.g., by EPA) is beneficial for the myocardium, by reducing inflammation and promoting cardiac healing after myocardial infarction (52, 111, 160, 326, 341). M2 polarization also counteracts systemic and plaque inflammation in atherosclerotic disorders, thus reducing CHF-associated factors (119, 299). As far as cancer-induced cachexia is concerned, however, it must be taken into account that, although being controversial, the most widely proposed immunometabolic therapy against cancer implies achieving M1 polarization to potentiate elimination of cancer cells by TAM (335). Nevertheless, MΦ balance might have different effects at different stages of tumor progression; therefore, different time points in the reprogramming therapy targeting either cancer or cachexia might also be considered.
F. Stimuli leading to metabolic reprogramming-mediated MΦ polarization
Based on the earlier considerations, metabolic reprogramming stimuli modulating the M1/M2 balance might interfere with skeletal muscle regeneration (300). In this paragraph, we will be considering exercise, calorie restriction (CR), and nutrients. Notably, it must be taken into account that the chosen immunometabolic strategy must be selective to MΦ without affecting metabolism of other cells. In fact, besides MΦ, other resident muscle cells such as SCs undergo metabolic remodeling during regeneration, switching from FAO to glycolysis during the transition from quiescence to proliferation. Moreover, metabolic reprogramming and metabolic substrate utilization shift occur during cell differentiation, when mitochondrial activity increases in most cell types, including SCs.
1. Exercise
Moderate training modulates MΦ activation by stimulating M1-to-M2 polarization and exerting a global anti-inflammatory effect in multiple organs [reviewed in Refs. (109, 315)]. For example, in HFD-induced adipose tissue of obese mice, exercise inhibits inflammation by accelerating M1-to-M2 polarization (157, 184, 193, 252). HFD-driven M1 accumulation in white adipose tissue correlates with insulin resistance in obese individuals (380); thanks to its anti-inflammatory effect, exercise improves insulin sensitivity. Also, in rats with nonalcoholic fatty liver disease, moderate exercise increases hepatic M2 polarization (183). Exercise suppresses IL-12 production, a stimulator of IFNγ, and reduces β2-adrenergic receptors in monocytes and MΦ by modulating TLR4 signaling (315).
In skeletal muscle, physical activity stimulates the release of myokines, among which cytokines are related to M1/M2 ratio regulation (e.g., IL-6, TNFα, and IL-10) and are involved in skeletal muscle regeneration. Exercise also triggers skeletal muscle PGC-1α over-expression; PGC-1α coordinates energy production, modulates myofiber metabolism, and regulates exercise-induced phenotypic adaptation (91). As discussed earlier, PGC-1α is upregulated in M2. PGC-1α also stimulates M2 polarization by accelerating necrotic resolution and counteracting fibrosis and muscle wasting in regenerating murine skeletal muscle (72). Muscle regeneration is characterized by a PGC-1α-dependent increased mitochondrial biogenesis and activity. PGC-1α also contributes to the fast-to-slow myofiber conversion after muscle injury in mice, which also influences damaged muscle recovery (80, 204, 369).
An anti-inflammatory role for PGC-1α has been reported in cultured muscle cells with PGC-1α downregulating NF-κB (72). Interestingly, exercise-induced PGC-1α upregulation also plays an immunomodulatory role in skeletal muscle by influencing cytokine expression; a PGC-1α-dependent B-type natriuretic peptide (BNP) production in myofibers induces M2 polarization, playing an anti-inflammatory and pro-repair role. Therefore, BNP might be considered a novel PGC-1α-dependent myokine mediating the cross-talk between tissue resident MΦ and skeletal muscle, as observed in mice (96). In addition, the myokine irisin—upregulated by exercise-induced PGC-1α—also suppresses inflammation and stimulates M2 polarization in vitro (74).
Gordon et al. showed that resistance exercise enhances M2-associated gene expression in human skeletal muscle (113) and also reduces stress response, improves glucose metabolism, mitochondrial activity, and OxPhos, thus being protective for skeletal muscle (113). MΦ activation seems to be relevant in maintaining skeletal muscle energy metabolism, and the transcription factor C/EBPα controls both M1- and M2 polarization; indeed, in the skeletal muscle of C/EBPα–KO mice, mitochondrial respiration and FAO are reduced, consistently with an overall decreased exercise capacity of the animal (173).
The effect of exercise strongly depends on its modality, intensity, and timing. Even when over-vigorous exercise results in damage, it still promotes M2 polarization and increased myogenesis in skeletal muscle despite TNFα increase, as observed in rats (215). Moreover, pO2 decreases in skeletal muscle also on intense exercise, activating HIF, which promotes capillarization and also acts on MΦ. M1-to-M2 polarization influences myogenesis in vitro in co-cultures of myoblasts with murine MΦ, whereas few data are available regarding the effect of MΦ polarization on myogenic cells in vivo as a consequence of physical exercise.
2. Calorie restriction
CR implies a negative energy balance stimulating adaptive metabolic changes with many positive effects such as lifespan extension, delayed age-associated disease onset, and improvement of metabolic health. Studies of CR effects on MΦ polarization in mice mainly concern white adipose tissue, where it has been found that CR leads, by an IL-4Rα- and STAT6-dependent signaling, to M2 polarization and metabolic improvement (88). Mild CR contributes to transient M2 accumulation in inflamed adipose tissue of obese subjects, where M2 supports remodeling of altered adipose tissue and enhances the formation of healthy metabolically flexible adipocytes that are able to control tissue FA levels, triglycerides/FA cycling, and OxPhos (202).
Therefore, M1-to-M2 switching supports healthy adipose tissue via the maintenance of metabolically beneficial MΦ. It has also been found that CR promotes M2 activation in adipose tissue, in both mice and humans, by increasing the appetite-reducing neuropeptide-FF (NPFF) plasma levels. NPFF upregulates IL-4R-α, Arg1, IL-10, and alkylglycerol monooxygenase; enhances p-STAT6 stability and, thus, M2 polarization (375).
3. Nutrients
MΦ-specific PPARγ deletion leads to OxPhos gene downregulation in skeletal muscle and also in the liver, indicating that resident MΦ have a beneficial role in regulating nutrient homeostasis (249). Besides resveratrol (see sections V and VII), other nutrients impinging on metabolism, such as amino acids, n-3 polyunsaturated FA (e.g., EPA or DHA), polyphenols, and vitamin D, improve skeletal muscle regeneration by targeting immune and muscle cells (73, 329). Indeed, the bioactive form of vitamin D3, 1,25(OH)2D3, inhibits M1 activation and promotes M2 activation, an effect also observed in vivo (390) and being mediated by PPARγ (395). A critical role for the MΦ vitamin D receptor in the inflammatory response to injury has also been reported in mice (316). Moreover, although controversial, a beneficial role for vitamin D in human skeletal muscle regeneration has been proposed (32, 254, 266, 318).
Cocoa polyphenolic extract influences MΦ metabolism by promoting oxidative pathways and M2 polarization in human MΦ in vitro (79). Pomegranate and its polyphenols promote M1-to-M2 switch in murine MΦ (2), and grape seed-derived polyphenols (proanthocyanidolic oligomers) accelerate muscle regeneration in rats by activating SCs and by promoting an anti-inflammatory switch (165). Interestingly, polyamines are able to drive M2 polarization in murine MΦ (354) and also to promote repair, fibrosis, and tissue remodeling in mice (272).
Sirtuins play a crucial role in skeletal muscle physiology; indeed, SIRT1 inactivation in skeletal muscle impairs muscle regeneration (293, 348). Based on the ability of some sirtuins to modulate metabolism, promoting an M1-to-M2 transition in vivo (see section V) (17, 75, 138, 153), a potential MΦ-mediated effect of these deacetylases on skeletal muscle regeneration might be worth further investigation. SIRT1 activators are considered “exercise mimetics” and, as reported earlier, other “exercise mimetics”—that is, resveratrol and AICAR—as well as metformin might drive M2 polarization also in vivo (21, 44, 48, 239, 240).
More recently, the metabolic modulator TMZ (144) has also been found to act as an “exercise mimetic”; TMZ increases oxidative metabolism while enhancing skeletal muscle myogenesis (92, 101, 219). Moreover, TMZ reduces the expression of pro-inflammatory cytokines in LPS-stimulated MΦ and in a murine model of myocardial dysfunction (5, 45, 84), stimulating M2 reprogramming in vitro (unpublished observations). This makes this drug and, possibly, other metabolic modulators (161, 345) promising candidates for further investigation in the attempt to find new treatments that are able to reprogram MΦ metabolism.
VIII. Conclusions
In conclusion, MΦ might assume different features and play different roles following specific activation routes, leading to the acquisition of two extreme phenotypes that are referred to as pro-inflammatory M1 and anti-inflammmatory/regenerative M2, which, simplistically, resume a much higher degree of heterogeneity. M1 and M2 subsets are characterized by a differential expression of cytokines, chemokines, and surface markers, by different molecular signatures and transcriptional regulation, and also by different metabolic features specifically illustrated here. Importantly, differently polarized MΦ play different roles, and MΦ are extremely plastic cells that are able to readily switch their phenotype and function if properly stimulated.
In particular, the role of differentially polarized MΦ in skeletal muscle regeneration has been discussed here; coordinated MΦ-phenotype transition is crucial to allow successful muscle regeneration, and promoting M2 polarization potentiates the tissues' regenerative phase. Specifically, the modulation of MΦ metabolism by immunometabolic strategies might be an appealing tool, which is possibly able to drive specific MΦ polarization routes and to open up a new scenario in terms of rehabilitative protocol design since it represents a potential therapeutic approach for several skeletal muscle conditions that might benefit from reduced inflammation and enhanced regeneration.
In the future, in-depth studies will hopefully disclose MΦ heterogeneity and the cellular metabolic pathways associated to specific MΦ polarization, thus identifying novel immunometabolic molecular targets and bolstering therapeutic interventions for degenerative and immunometabolic diseases.
Footnotes
Acknowledgments
The authors are grateful to M. Bennet and V.N. Di Robilant for their valuable editorial work. This work was supported by the Italian Ministry of Health (RF-2010-2318508) to E.F., by AFM-Telethon (#16772), by NUTRAMED (PON03PE_00078_2) by Duchenne Parent Project Netherlands (#DeSanta-DPP-NL), and by the Italian Ministry of Education, Universities and Research (MIUR) (IRMI:CTN01_00177_888744) to F.D.S.