
Abstract
Aims:
Metabolic remodeling of cardiac muscles during pathological hypertrophy is characterized by downregulation of fatty acid oxidation (FAO) regulator, peroxisome proliferator-activated receptor alpha (PPARα). Thereby, we hypothesized that a cardiac-specific induction of PPARα might restore the FAO-related protein expression and resultant energy deficit. In the present study, consequences of PPARα augmentation were evaluated for amelioration of chronic oxidative stress, myocyte apoptosis, and cardiac function during pathological cardiac hypertrophy.
Results:
Nanotized PPARα overexpression targeted to myocardium was done by a stearic acid-modified carboxymethyl-chitosan (CMC) conjugated to a 20-mer myocyte-targeted peptide (CMCP). Overexpression of PPARα ameliorated pathological hypertrophy and improved cardiac function. Augmented PPARα in hypertrophied myocytes revealed downregulated p53 acetylation (lys 382), leading to reduced apoptosis. Such cells showed increased binding of PPARα with p53 that in turn reduced interaction of p53 with glycogen synthase kinase-3β (GSK3β), which upregulated inactive phospho-GSK3β (serine [Ser]9) expression within mitochondrial protein fraction. Altogether, the altered molecular milieu in PPARα-overexpressed hypertrophy groups restored mitochondrial structure and function both in vitro and in vivo.
Innovation:
Cardiomyocyte-targeted overexpression of a protein of interest (PPARα) by nanotized plasmid has been described for the first time in this study. Our data provide a novel insight towards regression of pathological hypertrophy by ameliorating mitochondrial oxidative stress in targeted PPARα-overexpressed myocardium.
Conclusion:
PPARα-overexpression during pathological hypertrophy showed substantial betterment of mitochondrial structure and function, along with downregulated apoptosis. Myocardium-targeted overexpression of PPARα during pathological cardiac hypertrophy led to an overall improvement of cardiac energy deficit and subsequent cardiac function, thereby, opening up a potential avenue for cardiac tissue engineering during hypertrophic cardiac pathophysiology.
Introduction
P
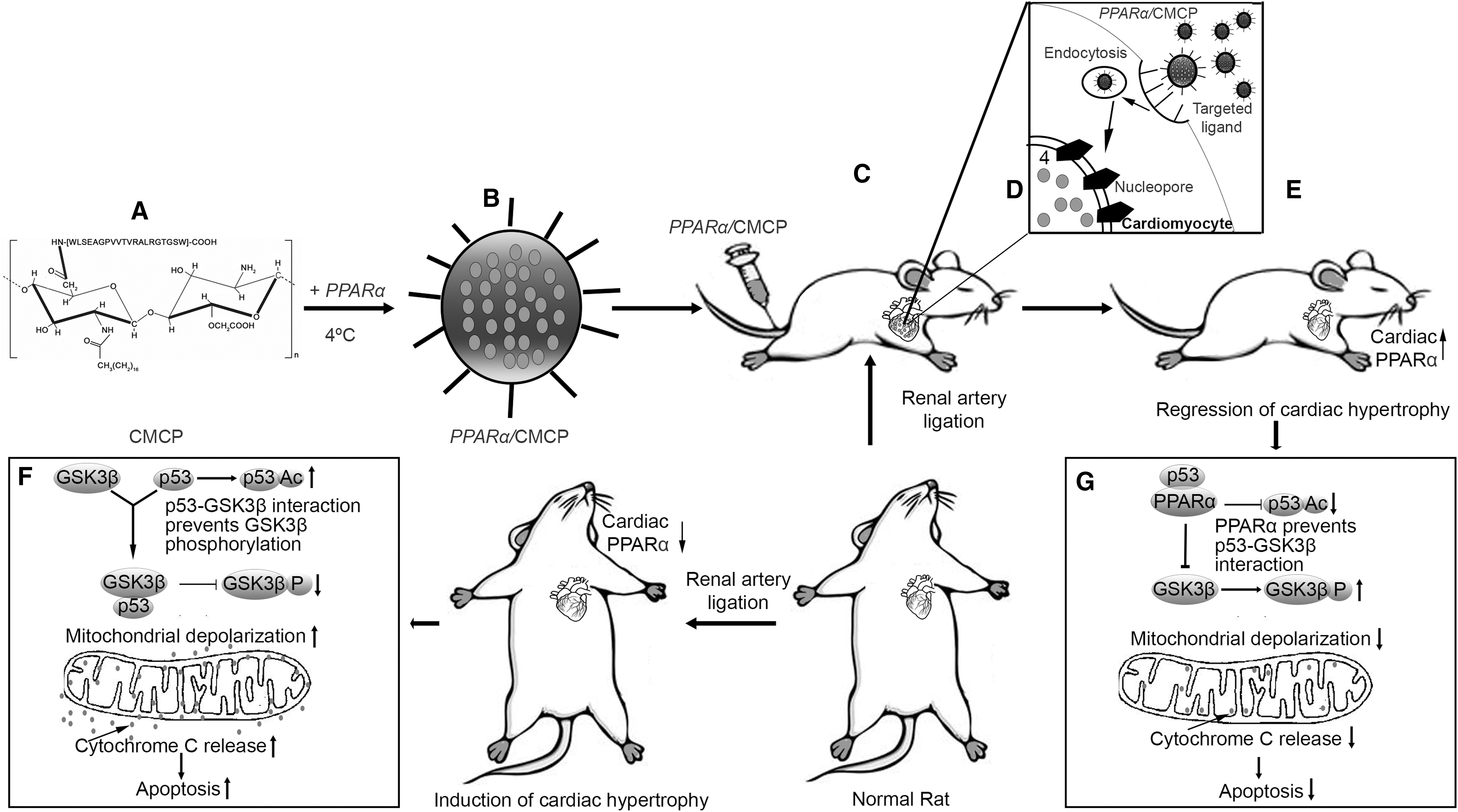
The study establishes welfare of fatty acid oxidation signaling preservation by targeted overexpression of nanotized peroxisome proliferator-activated receptor alpha (PPARα) during cardiac hypertrophy and points toward the role of PPARα in stabilization of myocyte mitochondria by moderating oxidative stress and resultant apoptotic load. Molecular exploration of such cells revealed that interaction of PPARα with p53 C-terminal domain inhibits activation of glycogen synthase kinase-3β (GSK3β); this contributes to improved structural integrity and functional quality of myocyte mitochondria during hypertrophy. Thus, targeted overexpression of PPARα arbitrates improvement of cardiac pathophysiology by resilience of myocyte mitochondria and precipitating further functional deterioration. Overall, the myocyte-targeted PPARα augmentation strategy translates to a novel therapeutic regimen in molecular engineering of the diseased myocardium.
The hypertrophic stress responses in cardiomyocytes are known to upregulate and acetylate p53 (16). Acetylated p53 has been associated with mitochondrial apoptotic pathways (40) and, along with glycogen synthase kinase-3β (GSK3β), plays a critical role in balancing pro- or antiapoptotic signals, integrating stress response to mitochondria for determination of cell fate (5). Concurrently, classical apoptotic signaling pathways lead to changes in mitochondrial structure and function (48). Basal (activated) state of GSK3β has been implicated in mitochondrial membrane depolarization and cytochrome-c release, whereas GSK3β (serine [Ser]9) phosphorylation (inactivated) stabilizes mitochondria and provides myocyte protection (30, 52). Inhibition of p53 and GSK3β activation thus emerges as an important target for stabilization of mitochondrial integrity and apoptosis in myocytes during cardiac hypertrophy.
This study reports a myocyte-specific delivery system for encapsulated overexpression vectors by a stearic acid-modified carboxymethyl-chitosan (CMC) nanopolymeric vector conjugated to a 20-mer myocyte-targeted peptide (CMCP) (57). Our study shows for the first time that myocardium-specific overexpression of PPARα during pathological hypertrophy binds the C-terminal domain of p53 leading to inactivation of p53 and GSK3β. Mitochondria of such overexpressed cells showed structural and functional improvement along with reduced apoptotic load. The study discerns the molecular mechanism of restoration of cardiac energy deficit, regression of cardiac hypertrophy, and consequent improvement of cardiac function in ligated rat hearts due to targeted overexpression of PPARα.
Results
PPARα modulation in cardiac hypertrophy
Western blot analyses showed significant downregulation of PPARα (in vitro: 2.73 ± 0.025-fold, p < 0.01 and in vivo: 3.05 ± 0.27-fold, p < 0.01) and ATGL (in vitro: 3.54 ± 0.27-fold and in vivo: 3.02 ± 0.32-fold, p < 0.001) during hypertrophy compared with respective control groups (Fig. 1A; Supplementary Fig. S1; Supplementary Data are available online at
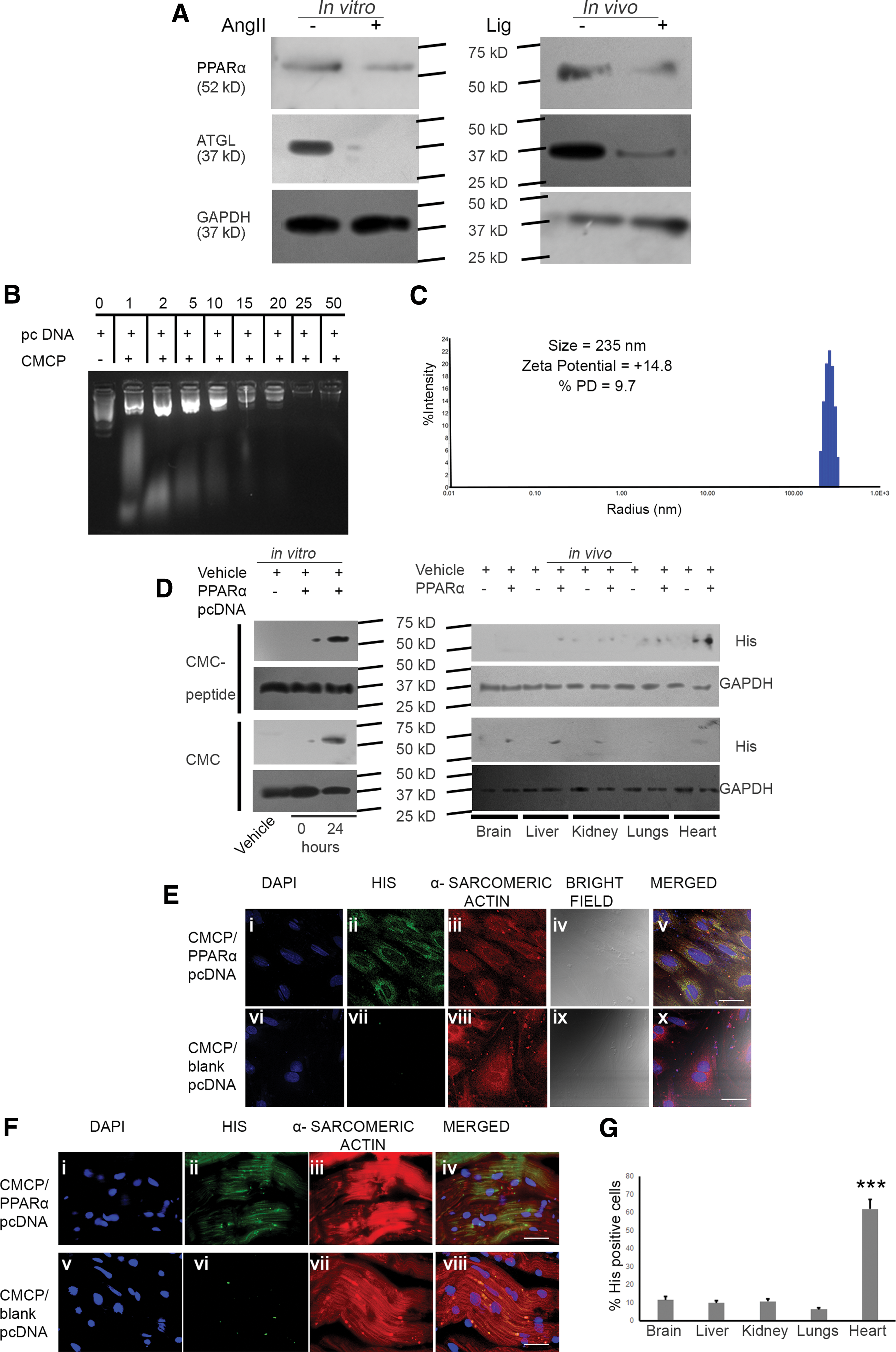
Characterization of nanopolymeric delivery system
Biophysical characterizations confirmed successful encapsulation of PPARα by CMCP and among the various encapsulated weight ratios, 1:50 (CMCP:pcDNA) showed significant retention and the encapsulation efficiency was 77.81% (±4.8). PPARα encapsulated within CMCP at 1:50 weight ratio showed a hydrodynamic diameter of 235 nm, % polydispersity of 9.7, and ζ potential of +14.8 mV (Fig. 1B, C). Scanning electron microscopic analysis of CMCP and CMCP-encapsulated PPARα showed homogenous size distribution of the nanopolymers (Supplementary Fig. S2). Moreover, serum incubation of CMCP-encapsulated PPARα at different time periods from 30 min to 24 h revealed substantial integrity of the plasmid DNA within CMCP encapsulation, whereas free pcDNA on serum incubation showed degradation within 1 h (Supplementary Fig. S2).
Myocyte-targeted overexpression of PPARα by nanopolymer
CMCP polyplex and CMC-mediated overexpression of PPARα in myocytes were estimated by Western blot analyses with anti-his antibody that showed plasmid transfection efficiency by CMCP is 1.91 ± 0.25-fold higher compared with CMC at 24 h. Furthermore, rats overexpressed with PPARα/CMCP showed significantly higher cardiac tissue-specific expression of histidine, “his,” compared with other organs, namely, the brain (5.42 ± 0.35-fold), liver (6.3 ± 0.24-fold), kidney (5.77 ± 0.42-fold), and lungs (9.92 ± 0.69-fold). CMC-mediated overexpression of PPARα showed no significant difference in “his” expression among different organs studied (Fig. 1D; Supplementary Fig. S1). Confocal microscopy confirmed targeted transfection by “his” positivity in vitro and in vivo from myocytes counterstained with α-sarcomeric actin. Moreover, CMCP-mediated overexpression revealed significantly higher percentage of “his-” positive myocytes in vivo the myocardium compared with other organs (Fig. 1E–G).
Overexpression of PPARα by CMCP encapsulation in ligated samples showed significant restoration of PGC1α (4.21 ± 0.36-fold), ATGL (4.9 ± 0.34-fold), and CPT1α (2.49 ± 0.16-fold) compared with nanotized empty plasmid-targeted ligated groups (Supplementary Fig. S3 and S4).
Regression of cardiac hypertrophy via targeted overexpression of PPARα
Overexpression of PPARα in angiotensin II (AngII)-treated hypertrophied myocytes showed significant downregulation of the hypertrophy marker gene expression in vitro ANF (2.14 ± 0.28-fold) and β-MHC (2.35 ± 0.26-fold) when compared with empty plasmid-transfected hypertrophied myocytes. Targeted overexpression of nanotized PPARα in ligated rat hearts also revealed significantly downregulated ANF (4.56 ± 0.21-fold) and β-MHC (4.34 ± 0.37-fold; Supplementary Fig. 2A) when compared with nanotized empty plasmid-targeted hypertrophied rat hearts.
Overexpression of nanotized PPARα in ligated rats resulted in significant reduction of myocyte cross-sectional area (518.4 ± 19.2 μm2) as well as heart weight (HW) to body weight (BW) ratio (3.32 ± 0.16) compared with nanotized empty plasmid-targeted ligated rats (678.6 ± 39.1 μm2 and 4.57 ± 0.09, respectively) (Fig. 2B, C; Supplementary Fig. S5).
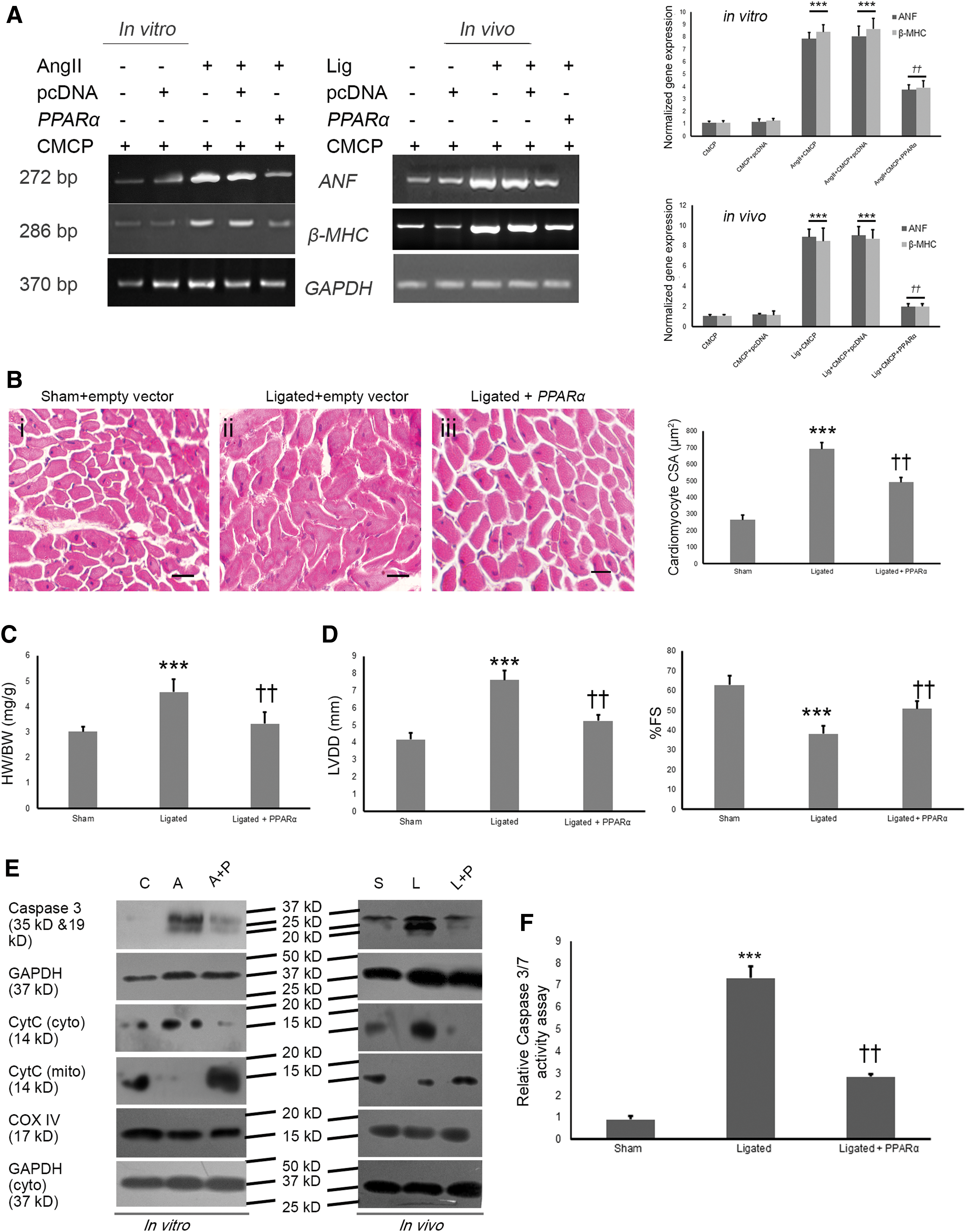
Myocyte-targeted overexpression of nanotized PPARα also showed improved cardiac function, as evidenced by a decreased left ventricular diastolic dimension (LVDd: 5.25 ± 0.32 mm), and restored the % fractional shortening (%FS: 50.74 ± 3.72%) during ligation compared with nanotized empty plasmid-targeted ligated rats (LVDd: 7.62 ± 0.53 mm; %FS: 38.16 ± 4.06%), as revealed by M-mode echocardiographic analyses (Fig. 2D). Other echocardiographic parameters, namely, left ventricular systolic internal dimensions (LVDs), left ventricular anterior wall diameter in diastole (LvAWDd), left ventricular anterior wall diameter in systole (LvAWDs), left ventricular posterior wall diameter in diastole (LvPWDd), and left ventricular posterior wall diameter in systole (LvPWDs), also support the previous findings (Supplementary Fig. S3B; Supplementary Table S1).
Overexpression of PPARα downregulates mitochondria-mediated apoptosis during hypertrophy
Nanotized PPARα-overexpressed hypertrophy samples showed reduced caspase-3 cleavage both in vitro and in vivo along with downregulated caspase-3 activity (in vivo: 2.57 ± 0.18-fold) compared with nanotized empty plasmid-targeted hypertrophy group (Fig. 2E, F). Targeted overexpression of PPARα in hypertrophy samples also showed significant downregulation of mitochondrial cytochrome-C release (in vitro: 2.67 ± 0.15-fold, p < 0.01 and in vivo: 3.79 ± 0.39-fold, p < 0.01) compared with respective hypertrophy groups (Fig. 2E; Supplementary Fig. S6B).
Overexpression of PPARα inhibits p53 acetylation during hypertrophy
Nanotized PPARα-overexpressed hypertrophy groups revealed significantly downregulated p53 acetylation (lys 382) (in vitro: 2.18 ± 0.19-fold, p < 0.01 and in vivo: 1.86 ± 0.22-fold, p < 0.01) compared with empty plasmid-treated hypertrophy groups (Fig. 3A). C646 treatment showed significant inhibition of p53 acetylation in vitro. Successful knockdown of p53 during hypertrophy by p53 siRNA treatment both in vitro and in vivo was used as negative control. C646-mediated inhibition of acetylated p53 or p53 knockdown by siRNA treatment in hypertrophy samples showed restoration of PPARα expression compared with hypertrophy groups (Fig. 3A; Supplementary Fig. S7).
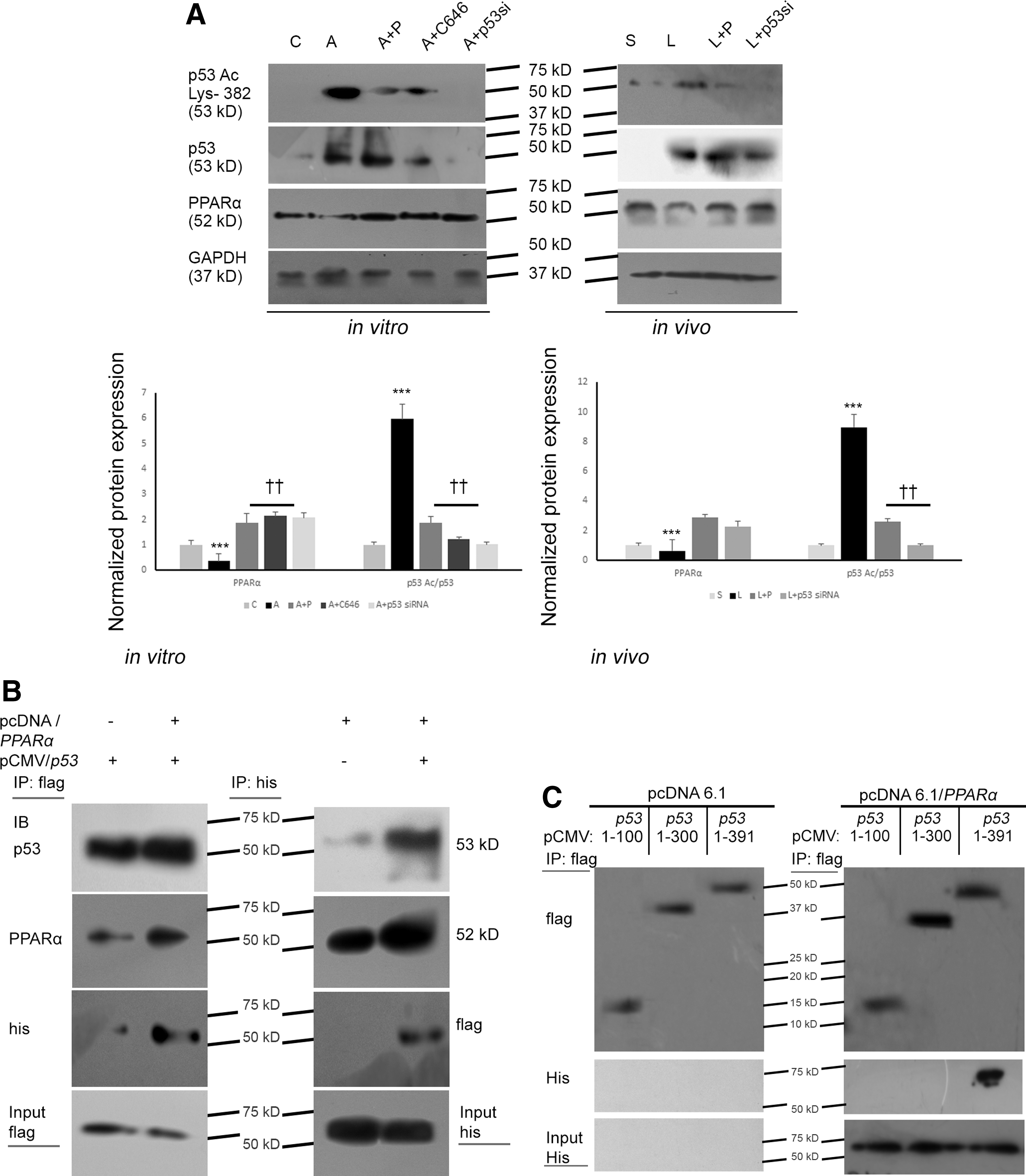
PPARα binds C-terminal domain of p53
Myocytes were transfected with p53-cytomegalovirus immediate-early (CMV) (flag tag) either in the presence or absence of PPARα-pcDNA (his-tag). Coimmunoprecipitation analyses, using the anti-flag antibody followed by Western blotting with anti-his antibody, showed substantial interaction between p53 and PPARα in myocytes (Fig. 3B; Supplementary Fig. S7). Reverse coimmunoprecipitation yielded similar results. Furthermore, interactions between the two proteins were mapped to the domains of p53. Myocytes were cotransfected with PPARα-pcDNA (his-tag) and the p53 domains 1–100, 1–300, and 1–391 (full length) cloned in CMV (flag tag). Flag tag pull down followed by immunoblotting with anti-his antibody revealed considerable interaction of 1-391 amino acid (aa) region of p53 with PPARα. However, the study did not reveal any substantial interaction of PPARα with 1-100 and 1-300 aa regions of p53, suggesting that C-terminal domain (300-391 aa) of p53 might be critical for physical interaction with PPARα (Fig. 3C; Supplementary Fig. S7).
Overexpression of PPARα modulates p53 and GSK3β binding during hypertrophy
PPARα-overexpressed hypertrophied myocytes were found to be associated with increased binding of p53 with PPARα compared with hypertrophy group treated with empty vector. However, interaction of p53 with GSK3β was significantly reduced (2.43 ± 0.19-fold, p < 0.01) in such PPARα-overexpressed hypertrophied cells compared with hypertrophy alone, as revealed by coimmunoprecipitation analyses. Similarly, reduced interaction of p53 with GSK3β was also found in C646-treated hypertrophied myocytes (3.31 ± 0.34-fold, p < 0.01) compared with hypertrophied myocytes (Fig. 4A; Supplementary Fig. S8A).
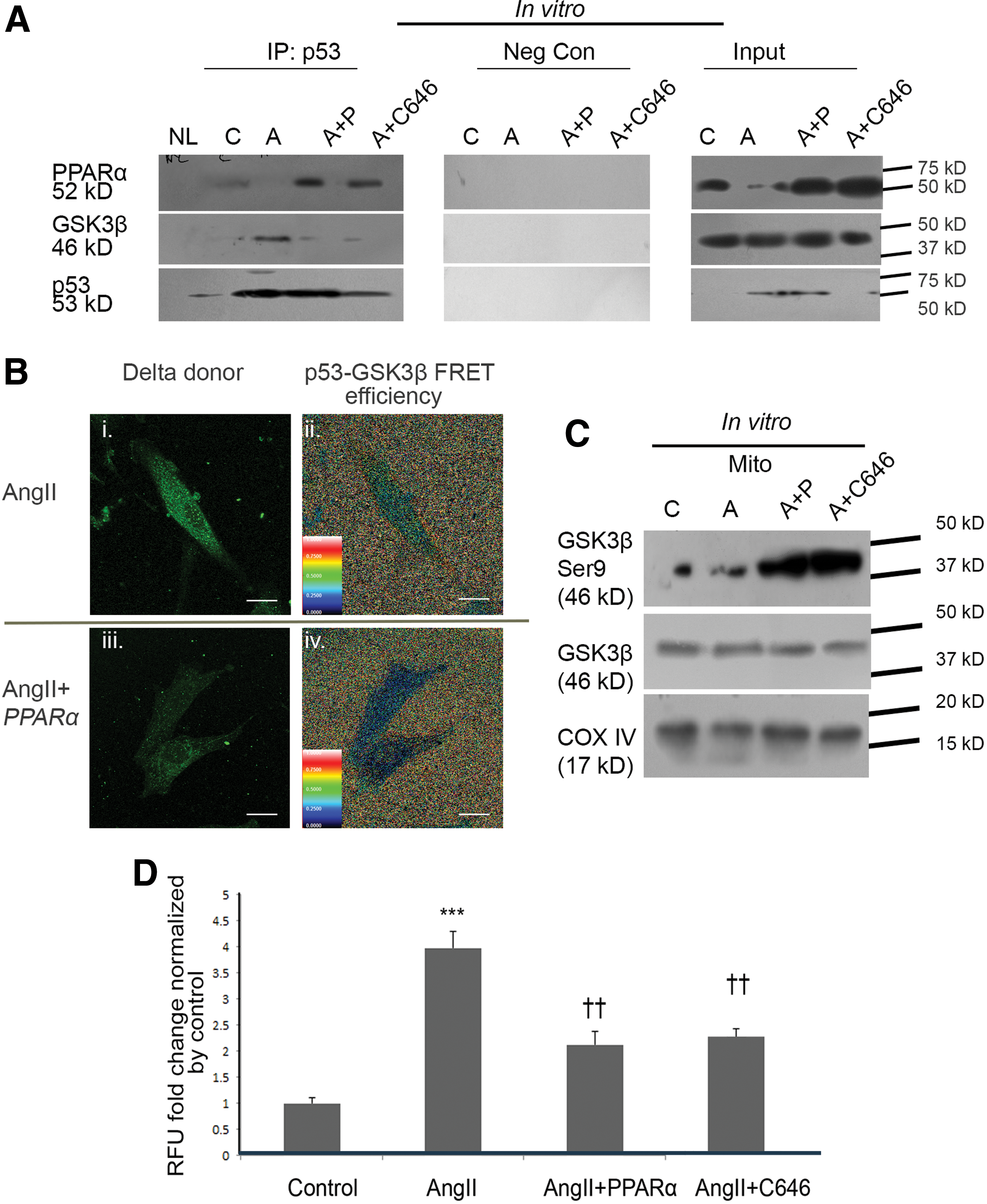
Comparative binding efficiency between p53 and GSK3β assessed by fluorescence recovery after photobleaching (FRAP) analyses showed a significantly lower fluorescence resonance energy transfer (FRET) efficiency (2.87 ± 0.42-fold, p < 0.01) in PPARα-overexpressed hypertrophied myocytes compared with empty vector-transfected hypertrophied cells, indicating reduced binding between p53 and GSK3β by increased PPARα-p53 interaction in hypertrophied myocytes (Fig. 4B).
Mitochondrial protein lysate from PPARα-overexpressed hypertrophied myocytes showed upregulated GSK3β (Ser9) expression levels (3.26 ± 0.34-fold, p < 0.01) compared with empty vector-transfected hypertrophied cells (Fig. 4C; Supplementary Fig. S8B).
PPARα moderates mitochondrial oxidative stress and associated protein expression during hypertrophy
PPARα-overexpressed hypertrophied myocytes also showed significant regression of cellular reactive oxygen species (ROS) level (3.27 ± 0.42-fold) compared with hypertrophied cells (Fig. 4D). Moreover, substantially upregulated mitochondrial superoxide (O2 −) in hypertrophied myocytes compared with control was significantly restored by overexpression of PPARα, as revealed by MitoSOX™ Red (Fig. 5A). Ratiometric analyses of 5, 5′, 6, 6′-tetrachloro-1, 1′, 3, 3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) aggregate to JC-1 monomer fluorescence intensity revealed substantially lower mitochondrial membrane potential (ΔΨm) in hypertrophied myocytes compared with control cells that was restored by overexpression of PPARα in hypertrophied myocytes; similar results were observed by C646 treatment of hypertrophied myocytes (Fig. 5B; Supplementary Fig. S9).
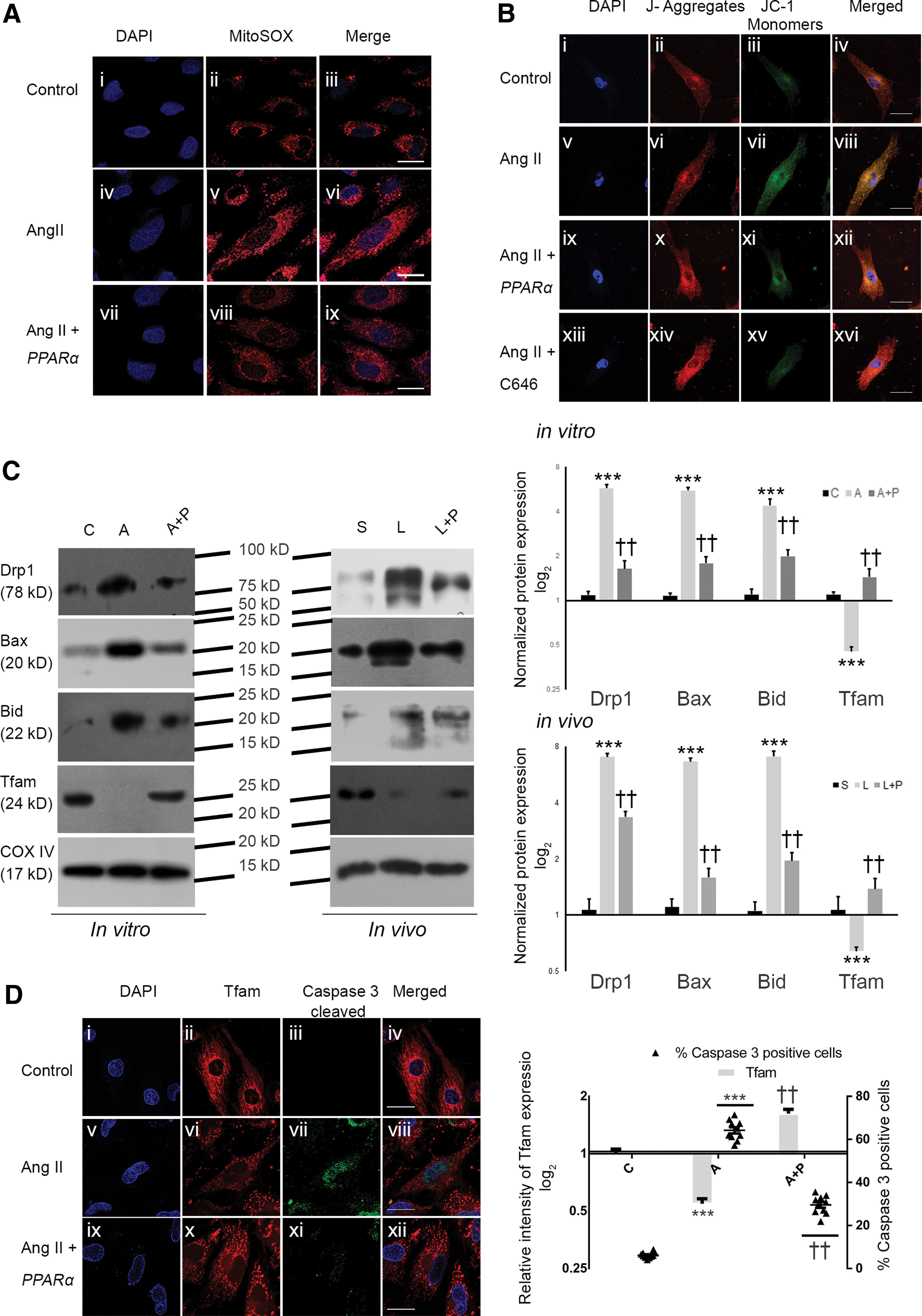
Mitochondrial protein lysates of PPARα-overexpressed hypertrophy samples revealed significantly downregulated expression of dynamin-related protein 1 (Drp1; in vitro: 3.5 ± 0.25-fold and in vivo: 2.09 ± 0.15-fold), BCL2-associated X protein (Bax; in vitro: 3.1 ± 0.21-fold and in vivo: 4.18 ± 0.29-fold), and BH3-interacting domain death agonist (Bid; in vitro: 2.19 ± 0.19-fold and in vivo: 3.61 ± 0.28-fold) compared with empty vector-treated hypertrophy group. Mitochondrial transcription factor of activated mitochondria (Tfam) expression showed significant downregulation in hypertrophy samples compared with control (in vitro: 2.43 ± 0.2-fold and in vivo: 1.65 ± 0.11-fold), which was again restored significantly by overexpression of PPARα during hypertrophy (in vitro: 3.17 ± 0.23-fold and in vivo: 2.15 ±0.19-fold, respectively; Fig. 5C; Supplementary Fig. S10).
An inverse association was revealed between mitochondrial Tfam expression and nuclear cleaved caspase-3. PPARα-overexpressed hypertrophied myocytes showed a significantly reduced number of caspase-3-positive nuclei (2.16 ± 0.23-fold) and increased mitochondrial biogenesis (upregulated Tfam expression by 2.96 ± 0.18-fold) compared with hypertrophied cells (Fig. 5D).
PPARα improves myocyte mitochondrial number and structure during hypertrophy
The relative amount of mitochondrial DNA (mtDNA) copy number was significantly downregulated in vitro (4.12 ± 0.26-fold, p < 0.001) and in vivo (4.67 ± 0.34-fold, p < 0.001) in empty plasmid-targeted hypertrophy samples compared with respective control groups and was found to be reversed in PPARα-overexpressed hypertrophy groups (in vitro: 2.46 ± 0.32-fold, p < 0.01 and in vivo: 3.27 ± 0.31-fold, p < 0.01). Similar results were also observed either by C646 or p53 siRNA treatment during hypertrophy (Fig. 6A; Supplementary Fig. S11).
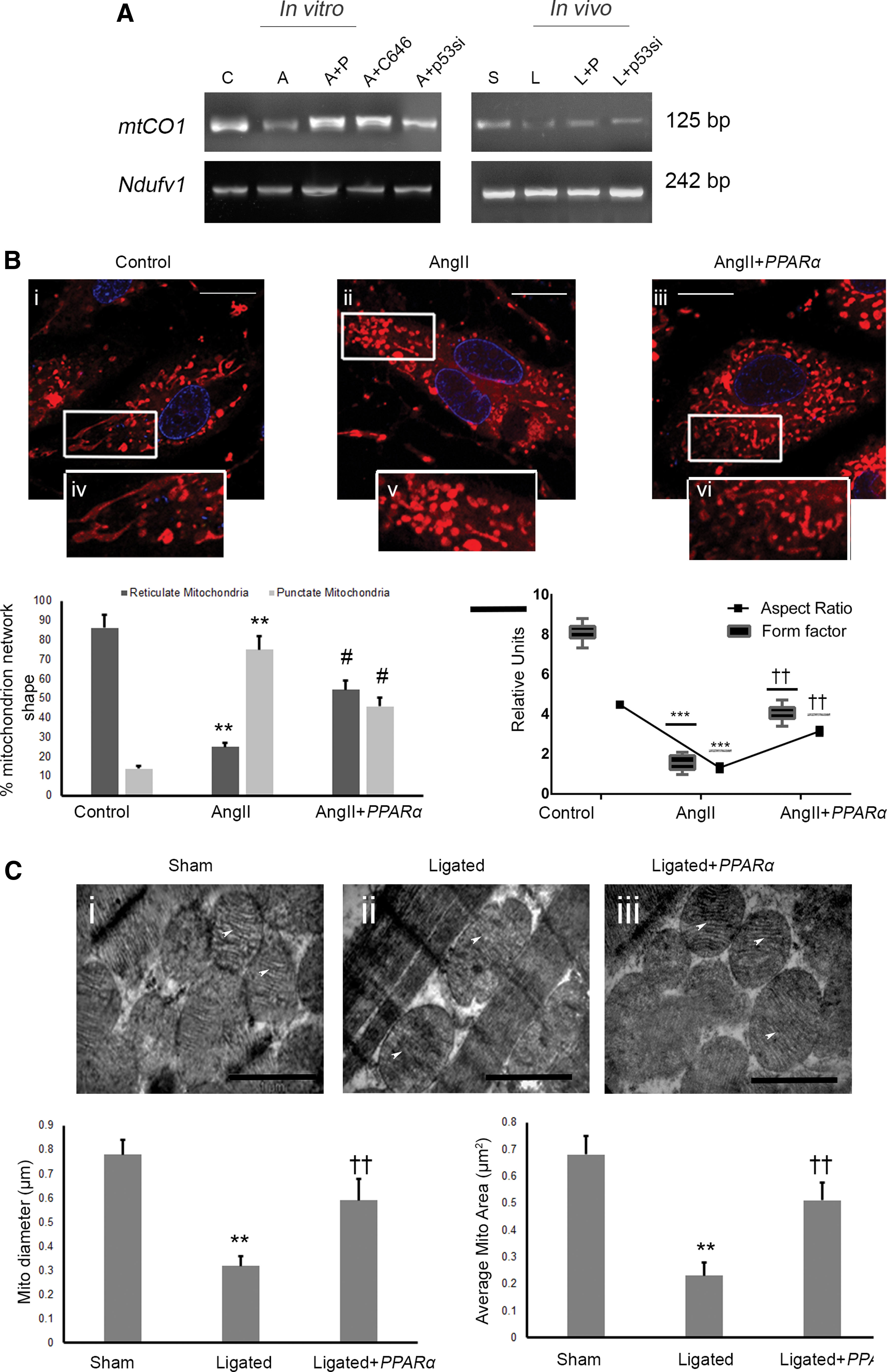
MitoTracker® Red-loaded control myocytes showed two different morphological structures of mitochondria, namely, reticulate or tubular and punctate networks (Fig. 6B). Hypertrophied myocytes revealed a decreased ratio of reticulate to punctate mitochondrial structures (7.05 ± 1.35-fold) compared with control myocytes that was significantly restored in PPARα-overexpressed hypertrophied myocytes (3.55 ± 0.38-fold; Fig. 6B). The aspect ratio (AR) and the form factor (F) revealed significant fragmentation of mitochondrial networks during hypertrophy (AR: 5.50 ± 0.47-fold and F: 4.86 ± 0.33-fold) with respect to control that was also restored significantly in PPARα-overexpressed hypertrophied myocytes (AR: 3.07 ± 0.22-fold and F: 2.17 ± 0.17-fold; Fig. 6B).
Targeted overexpression of nanotized PPARα in ligated cardiac tissue revealed substantial reduction of blurred cristae, mitochondrial swelling, and fragmentation (Fig. 6C), along with significant increase of mitochondrial diameter (1.85 ± 0.21-fold) and average mitochondrial area (2.21 ± 0.26-fold; Fig. 6C) compared with empty vector-targeted ligated group, as revealed by transmission electron microscopic analyses.
PPARα ameliorates myocyte oxygen consumption and adenosine triphosphate generation during hypertrophy
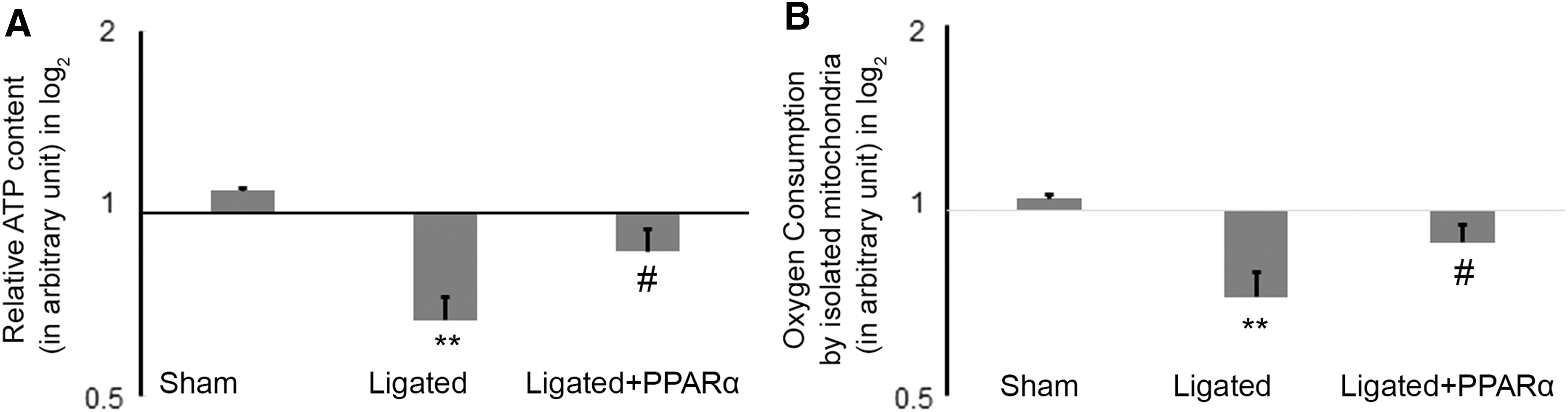
Ligated rat myocardium showed significant downregulation of baseline cellular oxygen consumption rate (OCR) (1.44 ± 0.11-fold) and adenosine triphosphate (ATP) generation (1.63 ± 0.14-fold) compared with sham control that was significantly restored by nanotized overexpression of PPARα in ligated rats (intracellular OCR: 1.22 ± 0.062-fold and ATP generation: 1.29 ± 0.15-fold) and compared with empty vector-treated ligated group (Fig. 7A, B).
Discussion
The present study reports amelioration of apoptotic load and improvement of cardiac function by myocardium-targeted nanotized overexpression of PPARα during pathological hypertrophy. According to previous reports, MHC-PPARα transgenic animals showed diabetic cardiomyopathy accompanied with altered myocardial metabolic profile and lipid imbalance (21, 54), as well as high level of prenatal PPARα activation might lead to postnatal lethality (1). Our study aims to probe into the effect of the targeted transient overexpression strategy of PPARα in hypertrophied myocardium. In this study, we showed reduced bystander effect in other organs by the targeted nanopolymer. Myocyte-targeted overexpression of PPARα was achieved by a CMCP (Fig. 1B–G; Supplementary Figs. S1, S5B, C). M-mode echocardiographic analyses in such PPARα-overexpressed ligated rats showed significantly improved cardiac function (Fig. 2D; Supplementary Fig. S3; Supplementary Table S1) via effective inhibition of p53 acetylation (Fig. 3A), improved mitochondrial structure and function (Figs. 5 –7), and attenuation of myocyte apoptosis (Fig. 2E, F; Supplementary Fig. S6B). This report shows for the first time that targeted overexpression of PPARα in hypertrophied myocytes results in efficient amelioration of oxidative stress, thereby regressing hypertrophic stimuli and improving functional capacity of the diseased myocardium.
Studies have suggested that cardiac hypertrophy reduces ATP generation and oxygen consumption (59, 70), as has been reported in PPARα-deficient animals with blunted FAO gene expression (4, 9, 36). Previous works have also reported that maintenance of FAO during cardiac pathophysiology improved cardiac function (10, 32). Besides, active forms of PPARα and ATGL are reported to be mutual agonists in restoration of mitochondrial defects (12, 60) and PPARα agonists have been shown to attenuate stress-activated kinase-mediated apoptotic signaling pathways (19). Despite these findings, molecular mechanism of PPARα-mediated restoration of mitochondrial function in compromised heart still remains obscure.
In our study, significant downregulation of PPARα during cardiac hypertrophy supported the previous findings (Fig. 1A; Supplementary Fig. S5A). The pathophysiological outcome of PPARα transgenesis during cardiac hypertrophy emerged as a proposition toward empirical analyses. The gene therapy involved stable encapsulation of the overexpression vector by the myocardium-targeted CMCP nanopolymer. The targeted nanoconstruct (CMCP) showed stable encapsulation by gel retardation analysis, in which an encapsulation weight ratio of 1:50 (CMCP:pcDNA) showed significant plasmid retention and protection from serum-mediated nuclease degradation (Fig. 1B; Supplementary Fig. S1B). The plasmid-encapsulated CMCP revealed homogenous size distribution by dynamic light scattering (DLS) analysis and scanning electron microscopy, while positive charge was revealed by ζ potential analysis (Fig. 1C; Supplementary Fig. S1A).
Overall, the nanoconstruct signified an increased in vivo retention, while the cellular uptake of biomaterials possibly occurred via an endolysosomal pathway (57). Overexpression of PPARα by CMCP encapsulation showed stable expression of associated his-tag in vitro. CMCP-mediated PPARα gene delivery showed higher expression of his-tag in myocardium compared with bystander organs in vivo by Western blot analysis, thereby confirming the relative uptake and targeted bioavailability of the PPARα nanoconstruct delivery system (Fig. 1D, E; Supplementary Fig. S5C). Moreover, selective targeting by the 20-mer myocyte targeting peptide conjugated to the chitosan nanoconstruct was confirmed by colocalization of his-tag in myocytes counterstained with α-sarcomeric actin in vivo (Fig. 1F, G).
The CMCP nanoconstruct-mediated myocyte-targeted transgenesis and overexpression of PPARα led to concomitant upregulation of downstream signaling molecules, namely, PGC1α, ATGL, and CPT1α (Supplementary Fig. S2), involved in utilization of fatty acid and subsequent mitochondrial uptake of fatty acid for FAO (4, 27, 70). Interestingly, nanoconstruct-mediated targeted overexpression of PPARα during cardiac hypertrophy led to significant attenuation of hypertrophic marker genes, reduced cardiomyocyte cross-sectional area, and improved cardiac function compared with the respective hypertrophy group (Fig. 2A–D; Supplementary Fig. S3; Supplementary Table S1). Furthermore, overexpression of PPARα in hypertrophy samples showed significant downregulation of caspase-3 activity and reduced mitochondrial cytochrome-C release, suggesting reduced mitochondria-mediated apoptosis in the PPARα-overexpressed hypertrophied myocardium (Fig. 2E, F; Supplementary Fig. S6B).
Several reports have associated mitochondrial apoptosis with p53 acetylation during cardiac hypertrophy (40). Although our study revealed no significant change in p53 expression between myocyte-targeted PPARα-overexpressed hypertrophy groups compared with the empty plasmid-treated hypertrophy group, p53 acetylation (lys 382) was significantly downregulated in such samples compared with the hypertrophy group (Fig. 3A). Reportedly, acetylation of p53 protects it from mouse double minute 2 homolog (MDM2)-mediated ubiquitination (37). In hypertrophied myocytes, higher expression of MDM2 facilitates downregulation of PPARα (data not shown); moreover, MDM2-mediated proteasomal degradation of PPARα has also been associated in other cell types (7, 24). In line, p53 activates MDM2 transcription (3) and as a result knockdown of p53 during hypertrophy results in subsequent transcriptional downregulation of MDM2. Thereby, C646-mediated blockade to p53 acetylation and p53 knockdown showed an upregulated expression of PPARα. Next, the study focused on exploring the possible molecular mechanisms involved in downregulation of p53 acetylation during PPARα overexpression in hypertrophy groups.
To understand this conundrum, coimmunoprecipitation and reverse coimmunoprecipitation analyses with “his” and “flag” tag antibodies were performed in myocytes transfected with p53-CMV and PPARα-pcDNA, showing substantial interaction of PPARα with p53 (Fig. 3B). Furthermore, the interaction of PPARα to the C-terminal domain of p53 was revealed by the interaction of PPARα (his-tag) with different domains of p53 (flag-tag). Notably, flag-tag coimmunoprecipitation analyses followed by his-tag immunoblotting showed no interaction of 1-100 aa and 1-300 aa domains of p53 with PPARα, while full-length p53 (1-391 aa) showed significant interaction with PPARα compared with empty plasmid-treated myocytes (Fig. 3C), thereby indicating interaction of PPARα with 301-391 aa region of p53.
The specific region is also important for the interaction of p53 with its transcriptional machinery components, including TATA-binding protein and its associated factors ([TAFs] 49), and with its transcriptional coactivator p300 (64). This report brings us to the “masking model” hypothesis, through which the binding of PPARα to p53 conceals its C-terminal domain (53). The association of wild-type p53 in apoptosis is explicitly known (16, 25), moreover, studies have also implied the involvement of the p53 C-terminal domain in regulation of apoptosis (29, 65). Furthermore, interaction of GSK3β with p53 at 364-373 aa of the basic domain determines activity of both (67, 68) and is crucial in determining cell fate by modulating mitochondrial apoptosis (42). Moreover, myocyte-targeted overexpression of PPARα during cardiac hypertrophy showed reduced binding of GSK3β with p53 by coimmunoprecipitation analyses, as was observed by the C646 treatment. These data thus suggest the importance of p53 acetylation (lys 382) in GSK3β-p53 binding. Also, FRAP analyses confirmed that increased PPARα-p53 binding in hypertrophied myocytes abrogated the interaction of p53 with GSK3β (Fig. 4A, B; Supplementary Fig. S8A). Thus, interaction of PPARα with p53 at the C-terminal domain might inhibit the amino acid residues of that particular region for various post-translational modifications and interaction with other proteins (34, 56).
Markedly, the lipid binding property of p53 by the C-terminal domain is critical in mitochondrial p53 localization and resultant cell death pathways (38, 50). The active form of GSK3β within mitochondria induces oxidative stress injury and opening of mitochondrial permeability transition pore that leads to an upregulated cellular ROS generation (2). In our study, PPARα-overexpressed and C646-treated hypertrophied myocytes showed upregulated expression of GSK3β (Ser9) within mitochondrial compartments (Fig. 4C; Supplementary Fig. S8B). These data suggested that reduced acetylation of p53 promotes GSK3β inactivation, which renders improvement of mitochondria and subsequent cardioprotection during hypertrophic pathophysiology (30, 42, 63). The concomitantly increased expression of GSK3β (Ser9) phosphorylation and decreased expression of p53 acetylation were found to be associated with a downregulated ROS load in PPARα-overexpressed hypertrophied myocytes compared with untreated hypertrophied cells (Fig. 4D). Cellular activation of various protein kinases by redox-dependent mechanisms may be instrumental toward inactivation of GSK3β by phosphorylation at the Ser9 residue (12, 14, 20, 23, 30). Thus, the role of PPARα overexpression in the hypertrophied cells is instrumental toward lowering the apoptotic burden; however, the functional betterment of the myocytes required further exploration of the mitochondrial structure and function.
The failing heart has been associated with induction of O2 − flashes that transiently decrease myocyte ΔΨm, which further leads to qualitative and quantitative mitochondrial derangements such as energetic dysfunction, swelling, and rupture (26, 28, 35, 66), altogether reducing the capacity of heart to convert chemical energy into mechanical work (51). Our study revealed that PPARα-overexpressed hypertrophied myocytes reduced mitochondrial O2 − radical accumulation and restored ΔΨm compared with hypertrophied cells (Fig. 5A, B; Supplementary Fig. S4A). Myocyte-targeted overexpression of PPARα also reduced mitochondrial Drp1 expression that regulates cytochrome-C release during apoptosis (31), along with downregulated expression of Bax and Bid in hypertrophied myocardium (Fig. 5C). In addition, targeted overexpression of PPARα restored mitochondrial copy number and Tfam expression (Figs. 5D and 6A), reportedly downregulated during cardiac hypertrophy (15). Myocyte-targeted PPARα-overexpressed hypertrophy groups showed substantial restoration of mitochondrial structural integrity marked by increased ratio of reticulate to punctate mitochondrial networks, increased mitochondrial diameter and mitochondrial area, and partial restoration of mitochondrial AR and F compared with hypertrophy groups (Fig. 6B, C). This led to mitochondrial functional restoration as revealed by improved ATP generation and OCR (Fig. 7A, B) in myocyte-targeted PPARα-overexpressed ligated groups compared with ligated groups alone. Thus, restored energy generation stabilized mitochondrial oxidative metabolism and oxidative phosphorylation in the PPARα-overexpressed pathological cardiac hypertrophy groups.
Taken together, CMCP nanoconstruct-mediated myocyte-targeted overexpression of PPARα upregulated PPARα expression level during pathological hypertrophy that resulted in regressed apoptotic stress and altered the balance of apoptosis toward mitochondrial biogenesis by increased mitochondrial structural restitution against oxidative stress. Moreover, targeted delivery of PPARα ensures minimal bystander activity of this protein, thereby underlining the therapeutic benefit of myocyte-targeted overexpression of PPARα during cardiac pathophysiology. Altogether, this study for the first time delineates PPARα as a novel “tinkerer” in modulation of p53 and GSK3β axis toward prevention of mitochondrial apoptosis and restoration of cardiac function in ligated rats (Fig. 8).
Materials and Methods
Animals used
The 24-week-old male Wistar rats (Rattus norvegicus; n = 50) used in this study were procured from NIN, Hyderabad, India, and conform with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institute of Health (NIH Publication No. 85-23, revised 1996) and approved by the Institutional Animal Ethics Committee, University of Calcutta (Registration No. 885/ac/05/CPCSEA), registered under “Committee for the Purpose of Control and Supervision of Experiments on Laboratory Animals,” Ministry of Environment and Forest, Government of India.
Animal model and treatment
In vivo cardiac hypertrophy was generated by ligating the right renal artery of 24-week-old anesthetized rats (43). Animals were kept under optimum conditions for 14 days and sacrificed on the 15th day after surgery. Sham operated control rat groups underwent a similar surgical procedure without ligation. Heart, kidney, brain, liver, and lung tissues were collected for further experimentation. The HW/BW ratio, in mg/g, was calculated from heart tissue samples of different rat groups. Rats (n = 5) were used for each experimental group.
Isolation, culture, and treatment of myocytes
Hearts from 2- to 3-day-old neonatal rat pups were dissected out and minced in 10% fetal bovine serum medium. Collagenase media (80 U/mL) were added to minced hearts and stirred for 5 min at room temperature, this was repeated four times. The supernatants were centrifuged to pellet down the cells, which were resuspended in fresh media and incubated for 45 min. Finally, cells were plated in laminin (Sigma-Aldrich, St. Louis, MO)-coated plates and coverslips. Approximately 90% pure isolated cardiomyocytes were confirmed by staining with sarcomeric α-actin antibody. Myocytes were serum starved with Dulbecco's modified Eagle's medium (DMEM) for 12 h, before experimentation. Isolated serum-starved myocytes were treated for 24 h with 10−8 mol/L (Sar1)-AngII (Bachem, Torrance, CA) (52). Myocytes were treated with p300-HAT activity inhibitor C646 (10 μM; Sigma-Aldrich) during AngII treatment for blocking p300-mediated acetylation of p53 (58).
Plasmid construction
Full-length coding DNA sequence of PPARα (Reference Sequence: NM_013196.1) was cloned in pcDNA6/V5-His B mammalian expression vector (Thermo Fisher, Waltham, MA) with C-terminal his-tag as described previously (17). Full-length p53 (NM_030989.3) and its domains (1-100, 1-300, and 1-391 aa) were cloned in pFLAG-CMV™-3 expression vector (Sigma-Aldrich) with N-terminal flag-tag as described previously (6). All the clones were confirmed by sequencing (3730 DNA Analyzer; Applied Biosystems, Foster City, CA).
Preparation and biophysical characterization of targeted PPARα gene delivery system
The stearic acid-modified carboxymethyl-chitosan conjugated to the 20-mer peptide was synthesized according to Rana et al. (57), with minor modifications. Briefly, low-molecular-weight chitosan (Sigma-Aldrich) was used to produce CMC. The unreacted amine groups after modification with stearic acid were protected by di-tert-butyl dicarbonate (BOC2O; Sigma-Aldrich). A mixture of 1 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 1 mM N-hydroxysuccinimide (Thermo Fisher) was slowly added onto BOC2O-protected stearic acid-modified CMC along with dropwise addition of the peptide [H]-WLSEAGPVVTVRALRGTGSW-[OH] (Sigma-Aldrich) under continued stirring for 72 h at 4°C to ensure proper binding of peptide with CMC. The solution was centrifuged to precipitate peptide-conjugated CMC and then lyophilized. Finally, trifluoroacetic acid was added for BOC2O deprotection. PPARα cloned in pcDNA6/V5-His B mammalian expression vector was added to the CMCP at different weight ratios and incubated for 2 h at 4°C under constant shaking conditions (400 rpm), followed by centrifugation at 13,000 rpm for 20 min at 4°C, filtered, and lyophilized. PPARα-encapsulated CMCP at different weight ratios was electrophoresed.
PPARα-encapsulated CMCP dissolved in Na-acetate buffer was mildly sonicated and then centrifuged (4000 rpm at 4°C) for 5 min and characterized by DLS (DynaPro NanoStar™; Wyatt Technology, Santa Barbara, CA), ζ potential (Möbiuζ™ Mobility Instrument; Wyatt Technology), and scanning electron microscopy (Hitachi VP-SEM S-3400N).
The amount of DNA encapsulated within CMCP (DNA loading efficiency) was assessed by quantifying the concentration of plasmid DNA from a standard curve and encapsulation efficiency was calculated by dividing plasmid DNA entrapped within CMC peptide by plasmid DNA added for conjugation.
CMCP-entrapped pcDNA complex was incubated with 20% (v/v) rat serum diluted with DMEM (13). The complex was treated after regular time intervals in different tubes, with heparin sodium at a concentration of 30 IU/μg of pcDNA.
Treatment with targeted gene delivery system
Myocytes were transfected with 500 pmoles of p53 siRNA (FlexiTube siRNA; Qiagen, Germantown, MD) or negative control siRNA (AllStars Negative Control siRNA, Catalog No. 1027280; Qiagen) by a nanopolymeric encapsulation of CMCP (57). CMCP-encapsulated plasmids (2–4 μg) were transfected to myocytes. For in vivo experiments, plasmid and siRNA with CMCP encapsulation were intravenously injected via tail vein at a dose of 2 mg/kg of BW/day to renal artery ligated rats, on alternate days, starting from 8th day till 14th day of ligation.
Protein isolation
After the experimental period, hearts were dissected out and perfused in chilled phosphate-buffered saline (PBS) for all in vivo experiments. For all in vitro studies, isolated myocytes after treatment were washed in chilled PBS. Protein from cells or tissues was isolated using protein extraction buffer [7 M urea, 4% (w/v) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate, 2 M thiourea, 1 × ethylenediaminetetraacetic acid-free protease inhibitor cocktail, and phosphatase inhibitor cocktail (Roche, San Francisco, CA)] as described previously (44). The protein supernatants were collected from each sample and concentrations were estimated by Bradford assay. Mitochondrial protein fractions from both cell and tissue samples were isolated by the differential centrifugation method as described previously (11, 44).
Western blotting and coimmunoprecipitation analyses
Proteins from ventricular tissues were extracted and separated by sodium dodecyl sulfate (SDS)/polyacrylamide gel electrophoresis before being transferred to polyvinylidene difluoride+ membrane. After blocking with 5% nonfat dry milk, membranes were incubated with primary antibodies in 5% bovine serum albumin solution at 4°C overnight. After washing, membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Thermo Fisher, Waltham, MA) at room temperature for 1 h. Finally, the immunoreactive bands were visualized by the enhanced chemiluminescence kit (Millipore, Billerica, MA) (11).
Coimmunoprecipitation was done following the manufacturer's protocol Pierce Coimmunoprecipitation Kit (Thermo Fisher). Briefly, proteins were incubated with fast flow protein G or protein A Sepharose beads and centrifuged to eliminate nonspecifically bound proteins (17). Concentration of the precleared proteins was estimated and 200 μg of protein was immunoprecipitated using primary antibodies of anti-p53, anti-his, and anti-flag. The immunoprotein complex was again incubated with protein G or protein A Sepharose beads. Attached proteins were then eluted from beads in 1% SDS buffer followed by immunoblotting with specific monoclonal antibodies (17). Normalization was done by immunoblotting using the same antibodies (Supplementary Table S2).
Reverse transcription-polymerase chain reaction
Total RNA and DNA from cells/tissues were isolated using TRIzol reagent (Thermo Fisher) following the manufacturer's protocol. Two micrograms of total RNA was used to make complementary DNA using cloned avian myeloblastosis virus First Strand cDNA Synthesis Kit (Thermo Fisher) and polymerase chain reaction (PCR) was done for the target genes using specific primers against ANF and β-MHC (11). Five hundred nanograms of total DNA was used for mtDNA copy number determination by PCR. Relative change in mtDNA copy number was determined by mtCO1 mitochondrial gene (forward: 5′-TGCTAGCCGCAGGCATTAC-3′; reverse: 5′-GGGTGCCCAAAGAATCAGAAC-3′) and single copy nuclear gene Ndufv1 (forward: 5′-CTTCCCCACTGGCCTCAAG-3′; reverse: 5′-CCAAAACCCAGTGATCCAGC-3′) (60).
Immunofluorescence analyses
Frozen ventricular tissue sections (4 μm) were prepared using CM 1850 cryostat (Leica, Germany). Tissue sections and cells plated on laminin-coated coverslips were stained with primary antibodies to anti-α-sarcomeric actin, anti-Tfam, anti-caspase-3 cleaved fragment, anti-GSK3β, anti p53, and anti-his followed by incubation with specific labeled secondary antibodies (Molecular Probes, Eugene, OR; Supplementary Table S2) (62). After mounting with Vectashield [with 4′,6-diamidino-2-phenylindole (DAPI)] (Vector Laboratories, Burlingame, CA), cells and tissues were visualized under FV1200 confocal microscope (Olympus, Japan).
FRAP analyses
Transfected and untransfected hypertrophied myocytes were stained with GSK3β-TRITC and p53-FITC. As TRITC and FITC work as a donor/acceptor in FRAP assay, TRITC was subjected to 50% bleaching, and FITC intensity in a single cell was measured. FRET efficiency was calculated from the difference between the postbleaching and prebleaching donor intensities (delta donor) using FV1200 confocal microscope (18).
Analysis of ΔΨm
Loss of ΔΨm was assessed on myocytes by incubating myocytes with tetraethyl benzimidazolyl carbocyanine iodide (JC-1; Thermo Fisher), a cationic dye that accumulates in energized mitochondria after treatment. Cells were incubated with 5 μM JC-1 for 15 min at 37°C to load the cells with dye. Myocytes after treatment were washed with 1 × PBS mounted on slides with Vectashield before scanning under confocal microscope (41). Stained myocytes were individually imaged using the 100 × objective; the monomeric form of JC-1 was excited using the 488-nm laser, and the aggregate form was excited using the 543-nm laser. Acquired images were analyzed using ImageJ, and the fluorescence intensity for each JC-1 form was determined for multiple regions of interest (ROIs).
MitoTracker labeling
Myocytes were treated with 500 nM MitoTracker Red FM (Thermo Fisher) for 15 min at 37°C, after which they were incubated in dye-free medium (39). The MitoTracker Red fluorophore molecules accumulated in mitochondria, which was determined by exciting with the 561-nm laser and the fluorescence intensity was determined from multiple ROIs per myocyte and analyzed with ImageJ software.
Mitochondrial morphological characteristics were quantified for AR (ratio between major and minor axes of an ellipse equivalent to the mitochondrion, measure of mitochondrial length) and F (perimeter2/4π·area, a combined measure of both mitochondrial length and degree of branching). AR is a measure of mitochondrial length and reflects the “length-to-width ratio”; F is a combined measure of both mitochondrial length and degree of branching and reflects the complexity and branching aspect of mitochondria. Both AR and F are independent of image magnification and have a minimal value of 1 (corresponding to a circular mitochondrion). Therefore, one was routinely subtracted from the experimental values obtained for AR and F (33, 55).
Assessment of ROS levels
Myocytes after respective treatments were incubated with 50 μM 2′,7′-dichlorofluorescin diacetate (DCFDA) (Abcam, Cambridge, MA) at 37°C in the dark for 45 min. The treatment media were completely removed before DCFDA staining. Cells were then washed with 1 × buffer and fluorescence intensities were acquired by fluorimetry at an excitation wavelength of 485 nm and emission wavelength of 535 nm. Tert-butyl hydrogen peroxide was used as positive control in ROS assay (57).
Mitochondrial ROS levels
MitoSOX Red mitochondrial O2 − indicator (Thermo Fisher) was used to monitor changes in mitochondrial O2 − production by confocal microscopy (69). Myocytes were loaded with MitoSOX Red (5 μm) for 30 min at room temperature, followed by 1 × PBS wash. MitoSOX Red was excited at 488 nm and measured at an emission of 585 nm.
Caspase-3 activity assay
Caspase-3 activity from ventricular tissue of all experimental groups was determined using the ApoAlert Caspase-3 Assay Kit (Clontech Q25 Laboratories, Mountain View, CA) following the manufacturer's protocol (11).
ATP assay and OCR in vivo
Ventricular tissue samples were lysed in ATP releasing buffer as described previously (39) and ATP levels were measured using the ATP Determination Kit (Molecular Probes®; Life Technologies, Camarillo, CA) as per the manufacturer's protocol. ATP content was calculated as nanomolars of ATP per microgram of protein.
Oxygen consumption by mitochondria isolated from cardiac muscle was measured in a Hansatech oxygraph (Hansatech, Norfolk, UK) as described previously (47). OCRs were calculated as nmol/mg tissue/min.
Histology (hematoxylin and eosin)
Heart tissue sections from animals of each group were fixed with acetone and methanol (1:1) and stained with hematoxylin and eosin for measurement of cardiomyocyte cross-sectional area (CSA) under microscope (Nikon Microscope, Singapore). Images were digitized and measured by Nikon NIS BR (57).
Electron microscopy
Cardiac tissue samples were fixed and embedded as described previously (45). Thin sections (80–90 nm) were briefly washed and stained with 2.5% uranyl acetate, and visualized under Morgagni 268D transmission electron microscope (Fei Company Q24, The Netherlands).
M-mode echocardiography
Cardiac function of lightly sedated animals from all groups was measured by M-mode analyses on a transthoracic study at the 15th day before euthanization. Digitized images were obtained using an ultrasound system (Vivid S5 System; GE Healthcare, Chicago, IL) for calculation of the following parameters, namely, LVDd, LVDs %FS, LvAWDd, LvAWDs, LvPWDd, and LvPWDs (44, 57).
Statistical analyses
All results are expressed as mean ± standard error of three independent experiments. Data were analyzed by Student's t-test (comparison of two groups) and one-way analysis of variance followed by Tukey's test (comparison of multiple groups) using GraphPad Prism (Version 6.01; GraphPad Software, La Jolla, CA). Results with p-value <0.05 were considered significant (18, 58).
Footnotes
Acknowledgments
The authors sincerely acknowledge Barbara Smith (Coventry University, Coventry, UK) for carefully editing the article. This work was funded by the Department of Biotechnology (DBT) (Grant Nos. BT-PR3709/BRB/10/980/2011 and BT/PR7016/NNT/28/641/2012) and Department of Science and Technology (DST) (Grant No. SB/SO/HS-148/2013), Government of India, to Dr. S. Sarkar and DST-FIST Program (Grant No. SR/FST/LS1-438/2000(G)).
Ethical Approval
This article does not contain any studies with human participants performed by any of the authors. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.