
Abstract
Significance:
Heme binds to and serves as a cofactor for a myriad of proteins that are involved in diverse biological processes. Hemoproteins also exhibit varying modes of heme binding, suggesting that the protein environment contributes to the functional versatility of this prosthetic group. The subject of this review is a subset of hemoproteins that contain at least one heme regulatory motif (HRM), which is a short sequence containing a Cys-Pro core that, in many cases, binds heme with the Cys acting as an axial ligand.
Recent Advances:
As more details about HRM-containing proteins are uncovered, some underlying commonalities are emerging, including a role in regulating protein stability. Further, the cysteines of some HRMs have been shown to form disulfide bonds. Because the cysteines must be in the reduced, dithiol form to act as a heme axial ligand, heme binds at these sites in a redox-regulated manner, as demonstrated for heme oxygenase-2 (HO2) and Rev-erbβ.
Critical Issues:
HRM-containing proteins have wide variations in heme affinity, utilize different axial ligand schemes, and exhibit differences in the ability to act as a redox sensor—all while having a wide variety of biological functions. Here, we highlight HO2 and Rev-erbβ to illustrate the similarities and differences between two hemoproteins that contain HRMs acting as redox sensors.
Future Directions:
HRMs acting as redox sensors may be applicable to other HRM-containing proteins as many contain multiple HRMs and/or other cysteine residues, which may become more evident as the functional significance of HRMs is probed in additional proteins.
Introduction
The heme regulatory motif (HRM) is a short sequence containing a Cys-Pro core that, in many cases, binds heme with the Cys acting as an axial ligand. HRMs are found in a diverse set of proteins that have a wide variety of biological functions, but the study of HRMs began in earnest only relatively recently. In 1989, the DNA sequence of heme activator protein 1 (Hap-1), a yeast transcription activator, revealed a short sequence (Lys/Arg-Cys-Pro-Val/Ile-Asp-His) repeated six times over an ∼200 amino acid section of the protein that modulated DNA binding in response to heme (67). Then, in 1993, three copies of a similar sequence (Arg/Ser-Cys-Pro-Val/Ile-Leu-Ala) were identified by Lathrop and Timko in the precursor protein sequence of both the nonspecific (ALAS1) and erythroid-specific (ALAS2) forms of δ-aminolevulinic acid synthase. Two copies are located in the presequence for mitochondrial translocation that is proteolytically removed on import to form the mature protein, and mutation of both Cys residues to Ser in those copies eliminated the heme-dependent inhibition of mitochondrial transport of mouse ALAS2 (52). Thus, the name “heme regulatory motif” was bestowed on the short sequence by the authors of the report, and they additionally noted the presence of HRMs in sequences of catalase and hemopexin. Two years later, Zhang and Guarente also referred to the short sequences in Hap-1 as HRMs and identified HRMs in heme-regulated eIF2α kinase (HRI), heme lyase, and heme oxygenase-2 (HO2) (105).
The Zhang and Guarente report was much more defining to the field than simply cementing a name for the motif. First, an alignment of the sequences around the Cys-Pro dipeptide showed a tendency for a basic residue before the Cys and a hydrophobic residue after Pro (105), helping to define an HRM. Second, the report provided spectroscopic evidence of direct heme binding to a short peptide (Ala-Lys-Arg-Cys-Pro-Val-Asp-His-Thr-Met) corresponding to the consensus of the six HRMs of Hap-1 (105). Finally, based on mutational studies (Cys to Ala and Pro to Ala) of Hap-1, it was proposed that Cys acts as an axial ligand to Fe3+-heme and that Pro plays a structural role, possibly by exposing Cys for ligation (105). Thus, the study included several key factors in initially defining what an HRM is and how the motifs would be studied in the future.
The report also demonstrated that not all HRMs are created equal. The authors identified a seventh HRM (HRM7) in Hap-1 located near the activation domain closer to the C-terminus of the protein (the first six HRMs are located near the N-terminal DNA-binding domain) and demonstrated that it is HRM7 that modulates the activation of Hap-1 in response to heme (105), a finding that was confirmed by later studies that pointed to only a minor role of the other HRMs despite the ability of the peptide to bind heme (28, 33). Thus, given these findings on Hap-1 and ALAS2 demonstrating that HRMs even in the same protein can function differently, it became clear, and is one of the themes in this review, how important it is to characterize the function of the heme-binding event and to assess (as quantitatively as possible) heme binding to a designated HRM.
The functions assigned to HRMs
Assigning a function to a particular HRM is of great interest to those who study HRM-containing proteins. There does not appear to be one common function assignable to the HRMs—and it is believed that some HRMs have no functional relevance—so each HRM must be investigated thoroughly. Further, proteins that contain HRMs are varied, including transcription factors and enzymes involved in many different processes. Thus, the field includes a wide variety of approaches to study HRM-containing proteins, which has, in turn, led to the uncovering of a variety of effects of heme binding to an HRM (Fig. 1).
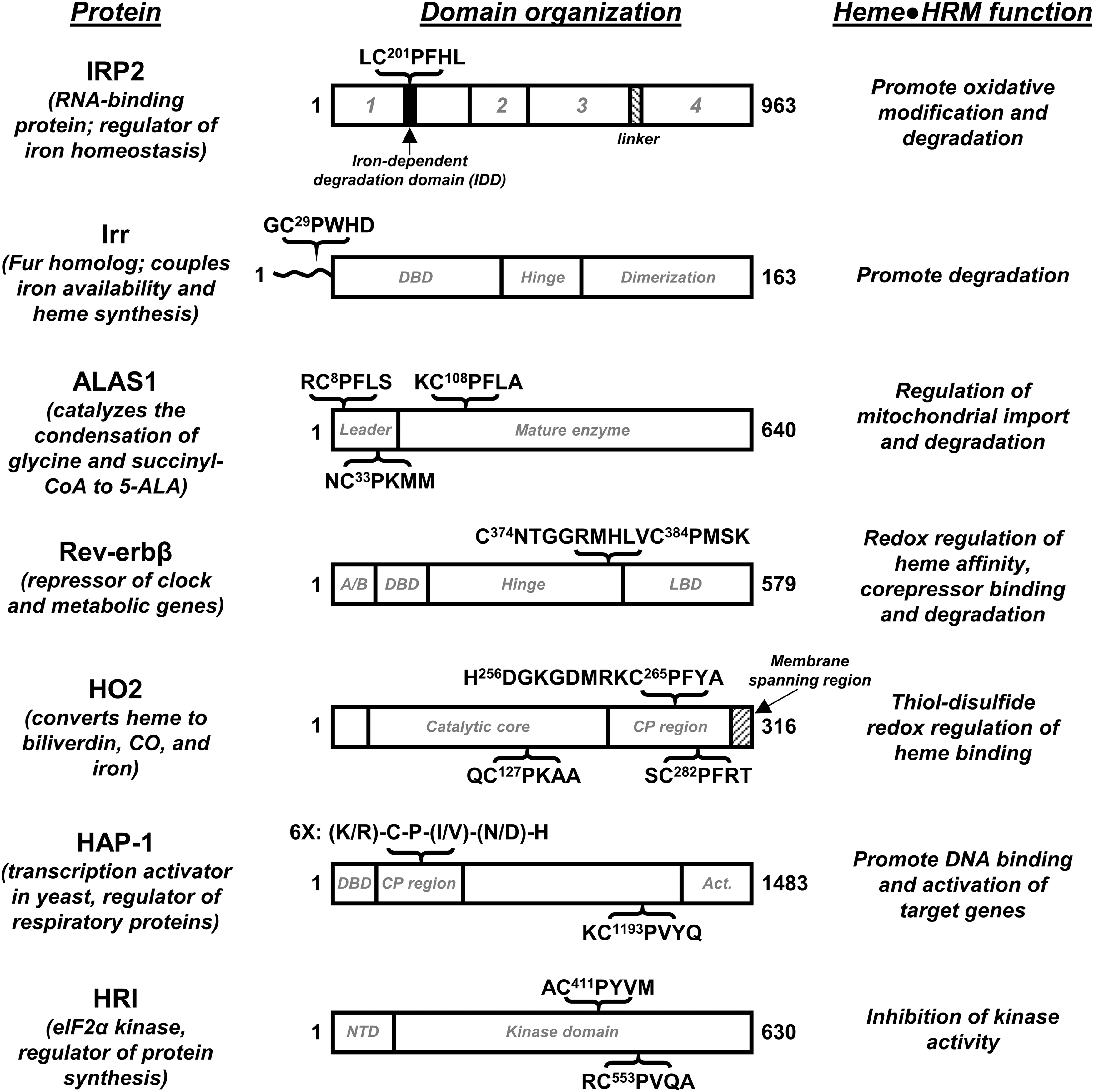
Despite a common function not having been assigned to HRMs, there are several examples of HRMs promoting heme-dependent protein degradation. The nuclear receptor (NR) Rev-erbβ, which represses the transcription of genes involved in regulating circadian rhythms and is described in detail later, is degraded in a heme-dependent manner (9). Heme binding to human Period-2 targets the protein for degradation, likely through the ubiquitin-proteasome pathway, and prevents formation of the heterodimeric complex between period-2 and human cryptochrome 1 that is involved in the regulation of circadian rhythms (94). Heme binding to tumor suppressor protein and metabolic regulator, p53, interferes with the ability of the protein to interact with DNA (14) (Fig. 2) and facilitates its nuclear export for cytosolic degradation (79). Similarly, the ability of Bach1, which regulates the expression of heme oxygenase-1 (HO1), to bind to DNA is significantly decreased on heme binding, which also targets Bach1 for nuclear export, polyubiquitination, and degradation (65, 82, 85, 103). Bradyrhizobium japonicum iron response regulator (Irr) regulates iron homeostasis and metabolism by acting as a repressor under low iron conditions and, under high iron conditions, is degraded on heme binding, allowing transcription to proceed (69, 97). Irr binds heme at both an HRM and a His-cluster site (His-His-His sequence) (39), and the protein appears to be degraded in a mechanism involving both sites that results in oxidative modification of the protein (46, 95, 96). ALAS1, the nonspecific form of δ-aminolevulinic acid synthase that catalyzes the first step of heme biosynthesis, contains three HRMs: two in the presequence involved in mitochondrial import (HRM1 and HRM2) and one near the N-terminus of the mature protein (HRM3) (52). HRM1 and HRM3 of rat ALAS1 were shown through mutagenesis studies to be critical for the heme-dependent inhibition of mitochondrial import (63). In addition, HRM3 of human ALAS1 was recently shown to be required for the heme-dependent formation of an ALAS1/ClpXP complex as well as for the oxidative modification of ALAS1 that triggers recruitment of LONP1, an ATP-dependent protease (48). Heme-dependent degradation on heme binding to HRMs has also been proposed for iron regulatory protein 2 (IRP2), a regulator of iron metabolism (38), and arginyl-transferase, which conjugates arginine to the N-terminus of proteins as a part of the N-end rule of protein degradation (35).
In contrast to the examples provided earlier of HRMs participating in heme-dependent protein degradation are the HRM-containing proteins Hap-1 and HRI. Hap-1, briefly discussed earlier as one of the proteins in which HRMs were initially identified, activates the transcription of genes involved in respiration and the control of oxidative damage in yeast in response to heme. Heme binding to HRM7 of Hap-1 promotes the transition of a high-molecular-weight complex containing Hap-1 and several other proteins to a Hap-1 homodimer that binds DNA with a high affinity (106). Recently, it has been shown that Hap-1 represses transcription of its own gene and several others in the absence of heme (32, 34), making the HRM of Hap-1 an important sensor of heme levels in yeast. HRI also appears to be a sensor of intracellular heme levels. However, heme binding to HRI at an HRM promotes the transition from an active to an inactive state. It is when reticulocytes are heme deficient that HRI (without heme bound to the HRM) autophosphorylates and, subsequently, phosphorylates eIF2α to downregulate protein synthesis, particularly globin synthesis (12, 37, 62).
In addition to the examples listed earlier, there are many more HRM-containing proteins in which the function of heme binding is unclear. For example, in contrast to what Lanthrop and Timko reported on the function of HRMs in mouse ALAS2 (52), a subsequent study showed no effect of heme on mitochondrial import of rat ALAS2 (63); thus, the relevance of the HRM is questioned. In addition, there are examples of proteins for which the function of the HRM remains elusive: HO2, which will be described in detail later; stanniocalcin-2, a glycoprotein shown to interact with HO1 (42); and DiGeorge critical region 8 (DGCR8), a protein involved in the first step of microRNA processing that appears to bind heme at a Pro-Cys (rather than Cys-Pro) site (19). Lastly, using a combinatorial peptide library approach, several other proteins were recently identified as having potential HRMs that may bind heme (6, 49, 50, 77). The authors used high-throughput screening to identify heme-binding peptides, and their sequences were then searched against protein sequence databases to identify proteins that may bind heme. Dipeptidyl peptidase 8 [(DP8), a member of the serine proteases (104)], FeoB [a bacterial ferrous iron transport protein (44)], and interleukin-36α [(IL-36α), a cytokine that induces inflammatory responses (21)] were, among other proteins, identified. The results suggest that not all HRM-containing proteins have even been identified, so the full extent of neither the capabilities of HRMs nor the physiological links between heme levels and metabolic pathways is fully understood.
Heme binding to HRMs
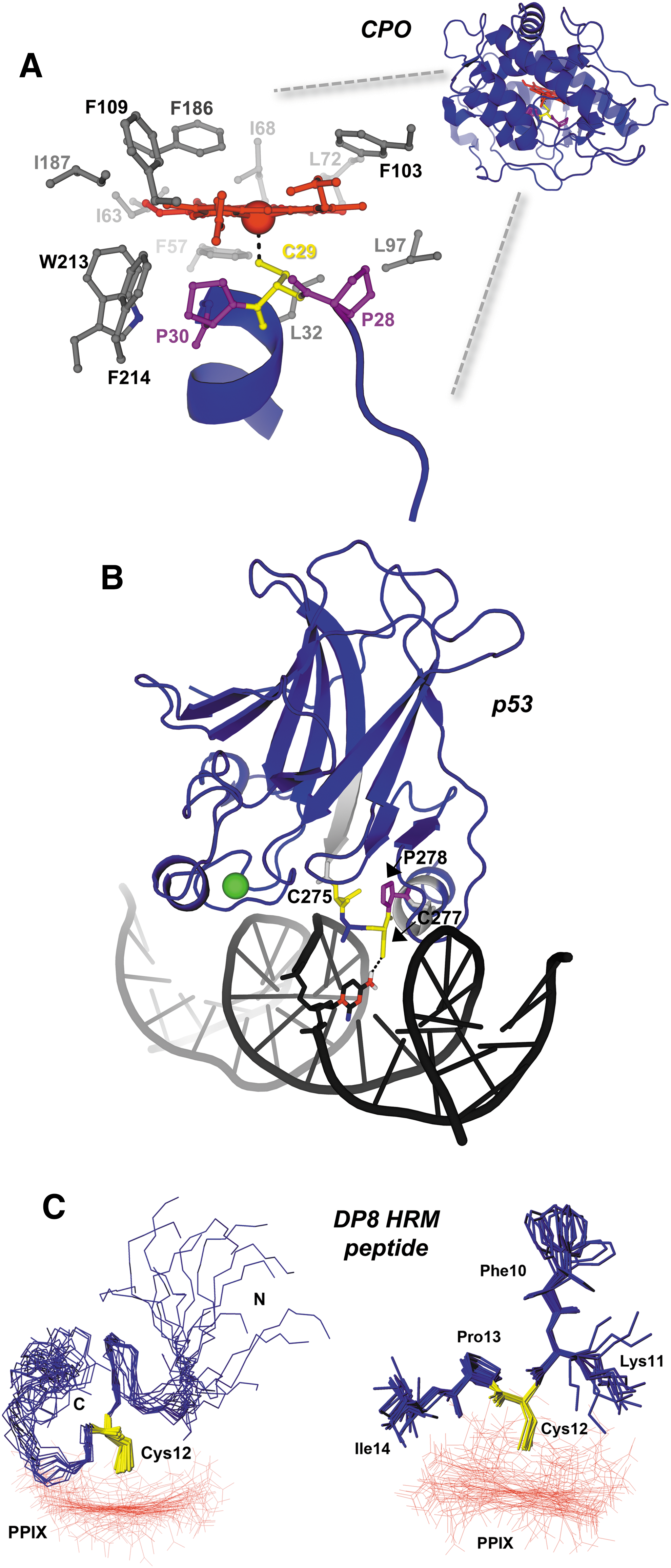
A critical point in assigning a function to an HRM-containing protein is to show direct heme binding to the HRM. Providing evidence of this HRM-heme interaction subsequently eliminates the possibility of indirect heme regulation and, particularly in the case of proteins such as Hap-1 (105) and Bach1 (65) that contain multiple HRMs, helps to distinguish responsive from unresponsive HRMs. Although X-ray crystal structures of only a few of the HRM-containing proteins (and their heme-bound forms) have been reported (Fig. 2), heme and heme-bound species lend themselves well to a variety of spectroscopic methods.
The classic model of the heme-HRM interaction includes Fe3+ of the heme in a penta-coordinated state with Cys acting an axial ligand. A shift in the absorbance of Fe3+-heme in aqueous solution from ∼385 nm to ∼370 nm in the presence of the HRM-containing peptide or protein is characteristic of Fe3+-heme binding to an HRM in this manner (105). Such complexes have an electron paramagnetic resonance (EPR) signal at g ∼ 6, although the signal is often broadened as compared with the signal for free Fe3+-heme in solution (39, 77), as shown later in the UV-vis and EPR spectra of some specific HRMs. Other spectroscopic methods such as resonance Raman (39, 50, 77), electron nuclear double resonance (ENDOR) (2), extended X-ray absorption fine structure (EXAFS) (2), and X-ray absorption (XAS) (2) have demonstrated Cys ligation of heme at HRMs and assisted in the characterization of the heme-HRM interaction.
Structural data available for HRM-containing peptides and proteins provide insights into how heme binds at these sites. Chloroperoxidase (CPO) is a versatile enzyme that uses heme to catalyze a variety of oxidative reactions: halogenation, dehydrogenation (heme peroxidase-like activity), hydrogen peroxide decomposition (catalase-like activity), and oxygen insertion (cytochrome P450-like activity). The structure of CPO shows the heme bound within an 8-helix bundle; the proximal side resembles cytochrome P450 and provides an axial thiolate (cysteine) ligand, whereas the distal side contains polar residues that form the peroxide-binding site, like other peroxidases (Fig. 2) (84). The Cys ligand in CPO resides within an unusual Pro-Cys-Pro motif on a helix that is nearly perpendicular to the heme plane, and the two prolines position the Cys for ligation to the heme. The only access to the Fe3+-heme is in the distal face, allowing only small organic substrates to interact with an iron-linked oxygen atom, accounting for its P450-like reaction. Another example of an HRM in a structured region is the Cys275-X-Cys277-Pro motif of the transcription factor, p53, which lies at the interface of the protein and its cognate DNA element (14) (Fig. 2). Cys277 of the HRM accepts a hydrogen bond from a nearby cytosine, but it is also responsible for binding heme along with neighboring C275 (14, 79). High heme levels drive binding to Cys275 and Cys277 and cause p53 dissociation from DNA, likely by disrupting the DNA:p53 hydrogen bond network.
In contrast, a number of HRMs are within disordered regions, as represented by the nuclear magnetic resonance (NMR) structures of two peptides spanning the HRM-containing regions of DP8 (Fig. 2) and IL-36α (6, 50). UV-vis, EPR, and resonance Raman spectroscopies demonstrated that both a 23-mer peptide (SGGLPAPSDFK
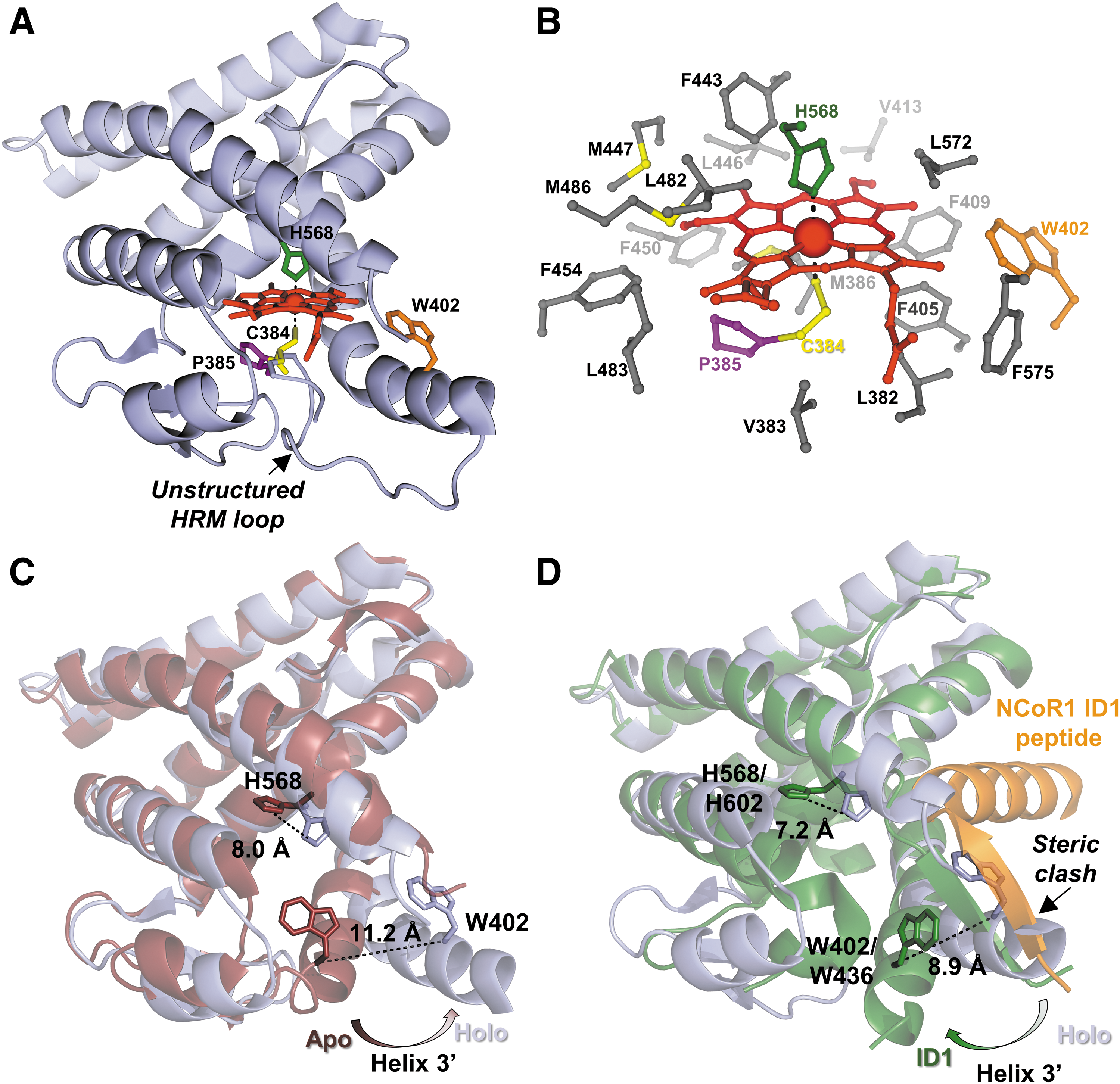
The HRM of Rev-erbβ is also in a flexible unstructured loop, but, in contrast to the structures of CPO and the DP8 peptide, His and Cys act as axial ligands to Fe3+-heme (Fig. 3) (66). Although the structure will be discussed in greater detail later, it demonstrates that Fe3+-heme bound to HRMs can also be hexa-coordinated. In fact, a growing number of proteins have been shown to bind Fe3+-heme at their HRM as a hexa-coordinated complex with Cys and an additional protein residue, usually His, acting as axial ligands. In addition to Rev-erbβ, HRI (37, 62) and HO2 (20) also bind Fe3+-heme with Cys and His ligands. These proteins have characteristic absorbance at ∼420 nm and a low-spin rhombic EPR signal with g-values of 2.5, 2.3, and 1.9. Resonance Raman has also been shown to be useful in distinguishing penta- and hexa-coordination (39, 50).
Although spectroscopy may simplify the assignment of the ligands as Cys and His, actually identifying the His residue can be a challenge. In each of the three examples—Rev-erbβ, HRI, and HO2—the His is located far outside of the Cys-Pro sequence motif of the HRM. Further, likely due to the surface binding of Fe3+-heme to some HRMs rather than in a more defined binding pocket, Fe3+-heme binds to some HRM-containing proteins in multiple conformations. For example, Fe3+-heme bound to the HRM of Irr is in a mixture of penta- and hexa-coordinated states (39), complicating the assignment of ligands. A mixture of states was also observed for several peptides used in recent combinatorial peptide library studies (6, 49, 50, 77). For instance, an Fe3+-heme-bound peptide, TPIL
Although most of the HRMs that bind Fe3+-heme in the hexa-coordinated state use Cys and His as axial ligands, one example of a protein that does not is DGCR8. As mentioned earlier, the sequence of the HRM of DGCR8 is Pro-Cys, rather than Cys-Pro. Spectroscopic studies have concluded that DGCR8 binds Fe3+-heme with an unusual ligand arrangement of two Cys axial ligands (3). Fe3+-heme is postulated to bind at a dimer interface with the Cys of an HRM from each monomer acting as an axial ligand. Two Cys ligands are not likely to be a common ligation environment for heme, but the example of DGCR8 is given here to emphasize that since not all HRM-containing proteins have been characterized, protein residues other than His can be the sixth ligand to hexa-coordinated Fe3+-heme bound to HRMs.
HRMs and redox chemistry
This review focuses on Rev-erbβ and HO2, which have been extensively studied in our laboratory, leading to characterization of the function and the various modes of heme binding to HRMs. In both Rev-erbβ and HO2, the HRMs act as redox sensors in which the Cys residues form disulfide bonds, altering the affinity of the HRM for heme. Although this has thus far only been noted for Rev-erbβ and HO2, many HRM-containing proteins contain multiple HRMs and/or other Cys that may be available to form disulfide bonds. Therefore, by highlighting these two very different proteins, we hope to contribute to a more general understanding of HRMs and their potential link to the redox state of the cell.
Rev-Erb
Regulation of circadian rhythms by heme
Mammals are entrained to the diurnal cycle through light perception, a signal that is transduced via the optic nerve to the suprachiasmatic nucleus (SCN), a small region of the brain located at the optic chiasm (73, 76). Within the SCN, a set of core molecular clock proteins synchronize cellular processes through activation and repression of gene transcription. At the apex of the molecular clock are the Bmal1 and CLOCK genes, whose protein products form a heterodimeric complex that binds to the promoters of clock-controlled genes to activate transcription. Among those genes activated are Period 1, 2, and 3, Cryptochrome 1 and 2, and Rev-erbα and Rev-erbβ. Period (Per) and Cryptochrome (Cry) proteins form a complex that inhibits the activator function of Bmal1•CLOCK, whereas Rev-erbs repress the transcription of Bmal1 and CLOCK genes. Together, Per, Cry, and Rev-erbs comprise the negative limb of the molecular clock that completes the feedback loop required for circadian rhythm maintenance (47).
The interplay of heme with the molecular clock is emerging as an important signaling axis. For example, heme biosynthesis and intracellular heme levels are entrained to the clock through regulation of ALAS1 by the Bmal1•NPAS2 (a forebrain CLOCK homolog) heterodimer (43). Significantly, NPAS2 is also a hemoprotein that binds carbon monoxide (CO), which further regulates its DNA-binding activity (17). On the negative limb of the circadian oscillator, Per2 contains an HRM that binds Fe3+-heme and promotes its degradation (94). Further, Per2 expression is regulated by p53, another HRM-containing transcription factor and metabolic regulator as discussed earlier (61). Rev-erbα and β isoforms also contain a redox-sensitive HRM that regulates heme binding, corepressor recruitment, and degradation (9, 11, 27). Because heme plays an integral role in energy production and metabolism, controlled degradation of Per2, Rev-erbs, and p53 through heme binding to their HRMs effectively couples those processes to the molecular clock. In the next section, we will discuss in detail the heme-dependent activities of Rev-erbs, and how they relate to circadian rhythm maintenance and entrainment of metabolic, inflammatory, and differentiation pathways.
Heme binding to Rev-erbα and Rev-erbβ indirectly facilitates corepressor recruitment
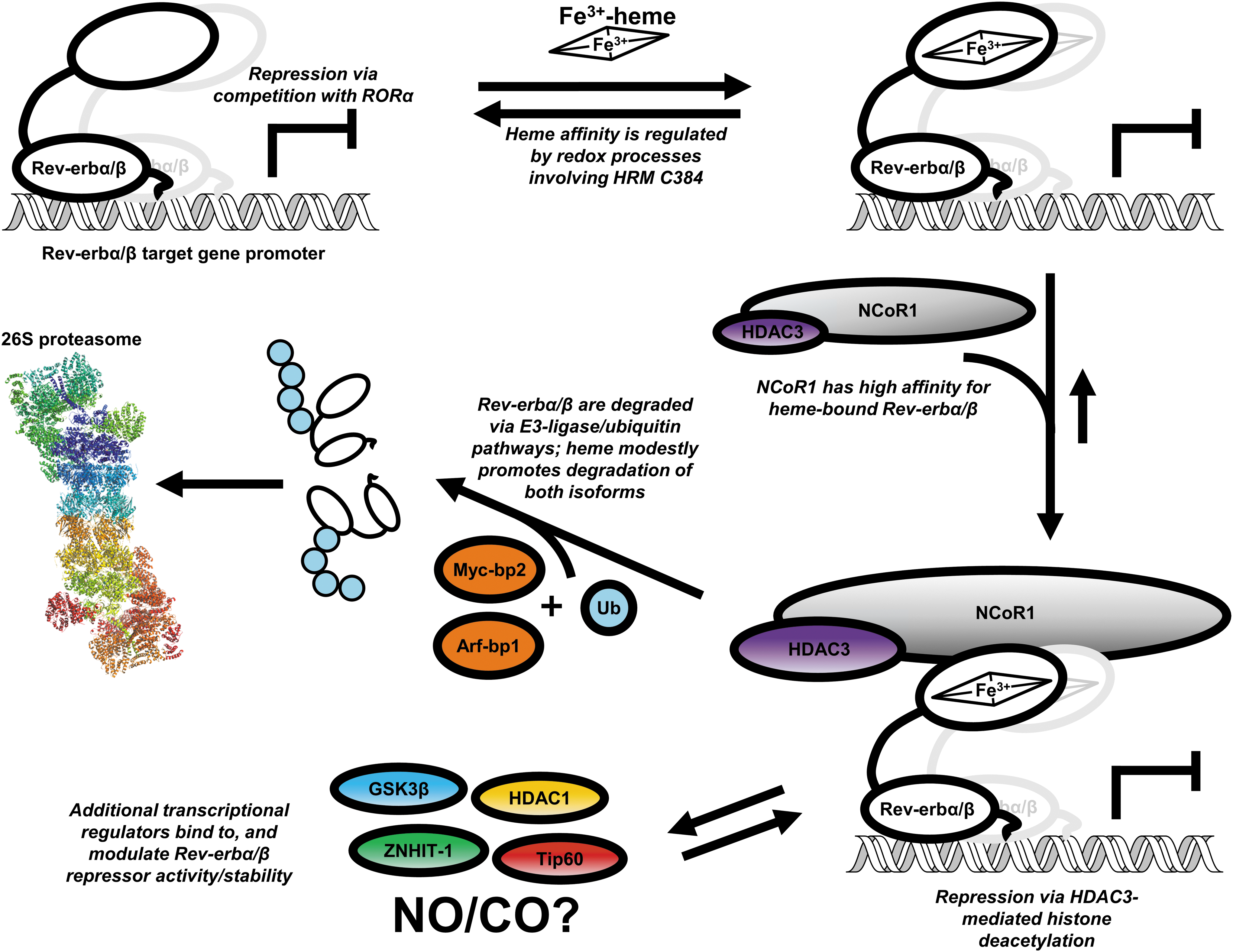
Rev-erbs are transcription factors that belong to the NR superfamily, which includes receptors of thyroid, estrogen, and testosterone hormones. In the ligand-bound state, many NRs recruit transcription coactivator complexes with histone acetyltransferase activity that ultimately leads to target gene activation (25). Rev-erbs are unique to the superfamily in that they lack a structural determinant required for coactivator binding, rendering them as repressors of transcription (93). The α and β isoforms share >70% amino acid identity in their DNA- and ligand-binding domains (LBD), and they are highly redundant in terms of circadian expression patterns and their target genes (8, 13, 18). Rev-erbs repress transcription either by direct competition for promoter elements with the transcription activator, RAR-related orphan receptor (RORα), or by interacting with nuclear receptor corepressor (NCoR1) (1, 8, 13). NCoR1 is a 270 kDa protein that acts as a scaffold bringing together NRs and histone deacetylase complexes that enhance repression (40). As discussed earlier, Rev-erbs have been well studied in their role as circadian factors, but they also repress the transcription of key genes involved in glucose and lipid metabolism, inflammatory responses, differentiation, and body temperature regulation, entraining these processes to the clock (23, 24, 72, 88, 91, 102).
In 2007, two independent groups identified heme as the endogenous ligand for Rev-erbs (70, 102). Estimates of Rev-erbα/β affinity for Fe3+-heme using isothermal titration calorimetry (ITC) were between 0.35 and 3.52 μM (59, 70). Diminution of cellular heme levels with the ALA-dehydratase inhibitor, succinylacetone, reversed Rev-erbα-dependent repression of target genes by disrupting the interaction of Rev-erbα with NCoR1 (70, 102). There is also evidence that heme binding to both α and β isoforms promotes their degradation likely through a ubiquitin-dependent pathway involving the E3-ligases, Arf-bp1, and Myc-bp2 (9, 51, 100). Based on these observations, a model was proposed in which Rev-erbs rapidly equilibrate with the regulatory heme (RH) pool so that when heme levels are high, Rev-erbs exist as holoproteins bound to NCoR1 and repress target gene transcription in a fine balance with their own degradation; controlling transcription output via degradation has also been observed with the thyroid hormone receptor (15). Presumably, low heme levels occlude binding to Rev-erbs based on the high K d of the complex, leading to NCoR1 dissociation and relief of repression (Fig. 4). A potential problem with this model is that the apparent weak affinity for heme (μM K d) coupled with very low free (or exchangeable) cellular heme levels (<100 nM) would be inconsistent with the proposed (70) role of Rev-erbs as sensors of intracellular heme. The results of other in vitro studies indicated that the simple heme-based recruitment model was incomplete.
Despite congruity among the cellular results described earlier, structural and in vitro studies provided evidence contrary to the heme-dependent NCoR1 recruitment model. The structure of the Fe3+-heme•Rev-erbβ complex shows heme bound in a six-coordinate system with axial ligands H568 and C384; the latter is provided by an HRM found on a flexible loop that is not present in the structure of the apoprotein (Fig. 3A, C) (66). The heme pocket is highly hydrophobic, stabilizing the nonpolar character of the porphyrin ring (Fig. 3B). Significant conformational changes are associated with heme binding to the apoprotein, namely a large shift of helix 3′ that opens the pocket to accommodate heme, a pivot of H568 to assume its position as a heme axial ligand, and the appearance of the disordered HRM loop that provides C384 (Fig. 3C) (93). More importantly, comparing the structures of the Rev-erbβ holoprotein to apoRev-erbα in complex with an NCoR1 peptide reveals a significant steric clash between helix 3 in the holoprotein structure and an anti-parallel beta sheet formed between helix 11 of Rev-erbα and the NCoR1 peptide (Fig. 3D) (68). The axial His residue, H602 in Rev-erbα, also swings out and away from the heme pocket in the NCoR1 structure, suggesting that heme and NCoR1 would not bind simultaneously to the LBD. Several in vitro studies supported the structural data and demonstrated that heme and cobalt-PPIX effectively compete with NCoR1 peptides for binding to purified Rev-erbα/β LBDs (59, 70, 102).
A potential caveat to the structural and in vitro studies was the use of truncated Rev-erbα/β LBDs and NCoR1 peptides that may not accurately represent the interactions between full-length proteins. For instance, the C-terminus of NCoR1 contains three NR interaction domains (IDs), two of which are known to associate with Rev-erbα (45); thus, the use of a single ID peptide for binding and structural studies may not be sufficient to promote cooperative interactions between different IDs on a single polypeptide. Thus, it seemed likely that contacts among the Rev-erb N-terminus, DNA-binding domain, and hinge region may impart unique biochemical properties and heme-dependent activities unobservable in experiments utilizing isolated LBDs.
To discover whether the in vitro and cellular experiments would coincide if we used the full-length Rev-erb instead of the LBD alone, we developed strategies to purify full-length Rev-erbβ (FLRev-erbβ) and an NCoR1 construct (instead of the short peptide) containing all three IDs (9). First, we found through the use of electromobility shift assays and UV-vis spectroscopy that heme and NCoR1, indeed, form a ternary complex with Rev-erbβ under equilibrium conditions, indicating the LBD is flexible in solution and can accommodate both ligands. However, heme was not required for the interaction of NCoR1 with the FLRev-erbβ•DNA complex in a purified system and heme does not promote NCoR1 binding when present at stoichiometric levels with respect to Rev-erbβ. In stark contrast to those results, heme is required for coimmunoprecipitation of endogenous NCoR1 from HEK 293 cell extracts with Rev-erbβ. Further, wild-type recombinant FLRev-erbβ co-precipitated with NCoR1, however a variant in which the His axial ligand is substituted with Phe (H568F), which exhibits a micromolar K d for heme binding, is incapable of interacting with NCoR1. Similar results were previously observed with Rev-erbα and the cognate H602F variant, indicating that heme is essential for the interaction of both Rev-erb isoforms with NCoR1 (102). Thus, we hypothesize that an unidentified cellular component present in soluble extracts promotes the heme-dependent NCoR1 recruitment to Rev-erbs. Numerous candidates are worth investigating, especially those proteins shown to modulate Rev-erbα and β stability or repressor activity, including glycogen synthase kinase 3β, which phosphorylates Rev-erbα promoting its stability (101); Tip60, an acetylase, and histone deacetylase 1 (HDAC1) that regulate Rev-erbβ repressor activity by adding, or removing an acetyl group from an RXKK motif (90); and ZNHIT-1, a zinc-finger protein that interacts with Rev-erbβ at the apo-CIII promoter, inhibiting its repressor activity (89). Heme-dependent interaction between any of these proteins and Rev-erb has not been investigated (Fig. 4).
An alternative hypothesis to explain the missing link between heme binding and NCoR1 recruitment is that diatomic gaseous signaling molecules, primarily nitric oxide (NO) and CO, could regulate Rev-erbα/β repressor activity. Both the truncated Rev-erbβ LBD and FLRev-erbβ bind CO with low nanomolar K ds, but CO appears to have no effect on repressor activity in a cellular system (9, 27, 66). NO, on the other hand, has been shown to inhibit Rev-erbα/β repressor activity in cultured cells (66), but those results have yet to be validated in vivo, or the mechanistic details revealed. For example, does NO binding cause dissociation of NCoR1? What is the affinity of Fe2+-heme•Rev-erbα/β for NO, and is that value consistent with the intracellular concentrations of NO? Does NO have secondary effects such as nitrosylation of Rev-erb zinc-finger thiolates as has been observed with other NRs (10)? In sum, additional studies are required to clarify the role, if any, of diatomic gases in Rev-erb regulation of target genes.
UV-vis spectrophotometric titrations revealed that Fe3+- and Fe2+-heme bind FLRev-erbβ with at least one order of magnitude higher affinity (low nanomolar K ds) than those early estimates using ITC. Further, based on competition experiments, dissociation of Fe3+-heme from the FLRev-erbβ•heme complex was too slow to allow rapid equilibration of Rev-erbβ with RH (10−6 s−1 = <1 day−1), negating its role as a sensor of intracellular Fe3+-heme (9). If so, does the HRM serve any regulatory function other than to bind heme and poise the receptor for interaction with NCoR1? In the next section, we discuss results suggesting that Rev-erbs may exploit the redox properties of the HRM cysteine thiolate and Fe3+-/Fe2+-heme couple to regulate heme affinity and corepressor recruitment.
Role of heme dissociation in redox signaling by Rev-erbβ
Early biophysical studies of Rev-erbβ described UV-vis, resonance Raman, EPR, and magnetic circular dichroism (MCD) spectra consistent with a low-spin 6-coordinate His/Cys ligated Fe3+-heme, as observed in the crystal structure shown in Figure 3A (58). However, reduction by dithionite to Fe2+-heme leads to dissociation of the C384 axial thiolate and its replacement by an exchangeable neutral ligand (Fig. 5). Since the loop containing the HRM is largely disordered and is not present in the structure of the apoprotein, we assume that dissociation of C384 from heme would result in significant conformational changes. UV-vis equilibrium titrations suggested that Rev-erbβ binds both Fe2+- and Fe3+-heme with similar low nanomolar affinity (9, 27). However, as discussed later, most equilibrium techniques vastly underestimate heme affinities and Fe2+-heme may not remain bound under the limiting heme concentrations in the cell. Several important questions require investigation to determine whether Fe3+/Fe2+-heme switching plays a role in regulating Rev-erb activity. For instance, what is the midpoint potential of the Fe3+/Fe2+-heme•Rev-erb couple, and what is the redox state of heme bound to Rev-erbs in the cell? Is the Fe3+/Fe2+-heme switch sensitive to changes in intracellular redox poise? If it is, what one-electron carrier or reductase is responsible for mediating Rev-erbα/β reduction?
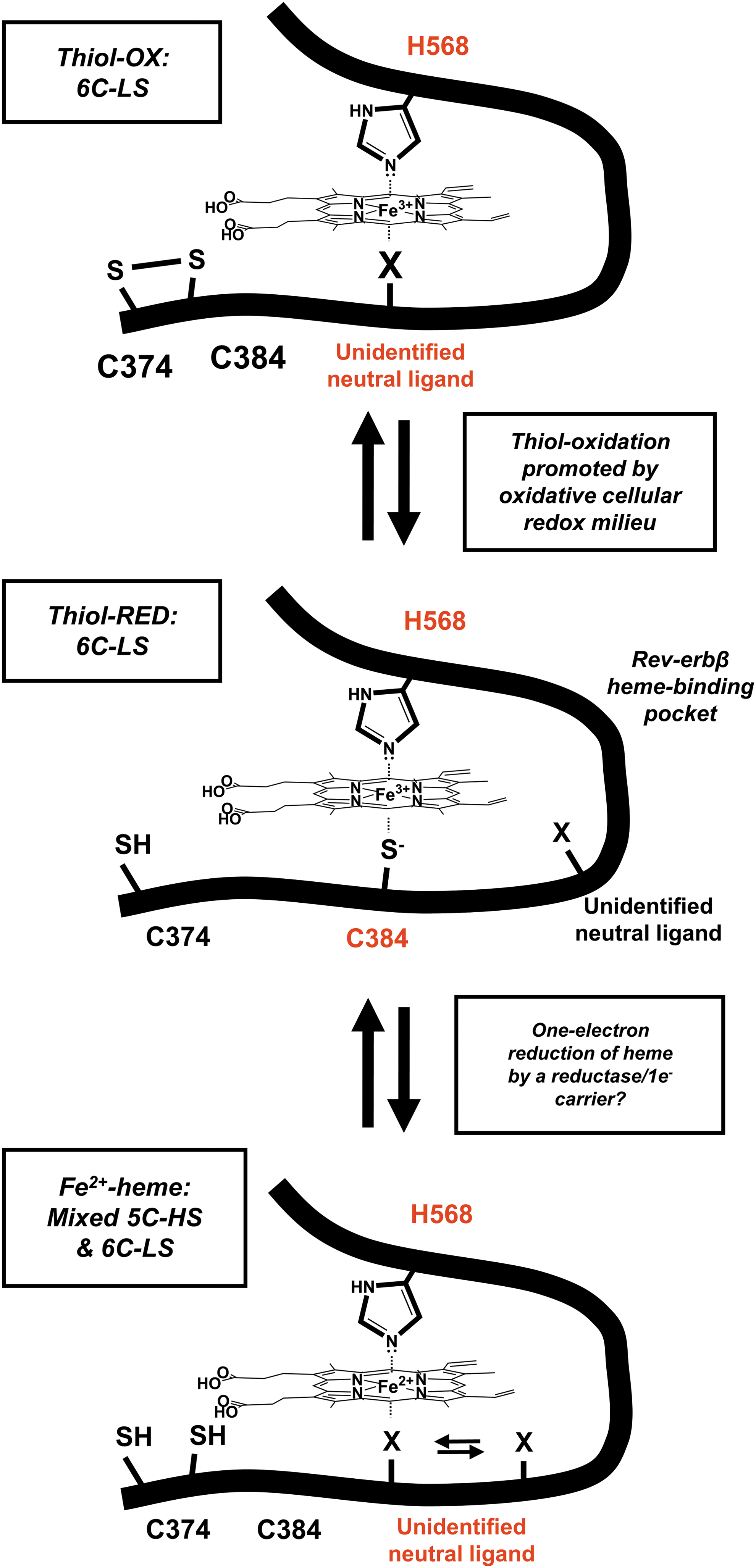
Another potential redox switch regulating Rev-erbβ repressor activity involves thiol-disulfide interconversion of the HRM cysteine. Under oxidizing conditions, C384 forms a disulfide bond with neighboring C374, causing an unidentified neutral ligand to assume the position of C384 in the Fe3+-heme complex (Fig. 5) (27). The K d of the thiol-oxidized protein for Fe3+-heme (117 nM) is approximately five-fold higher than that for the thiol-reduced form (23 nM), suggesting that thiol-disulfide interplay could regulate Rev-erbβ heme occupancy. Further, the switch occurs in Escherichia coli cells overproducing the Rev-erbβ LBD, which exhibit a whole-cell EPR spectrum consistent with that of a thiol-reduced, six-coordinate low-spin Fe3+-heme; then, treatment of cells with the thiol oxidant, diamide, yields a spectrum indicative of a mixture of thiol-reduced/thiol-oxidized species. These data support the hypothesis that the Rev-erbβ HRM is redox active and that the disulfide bond forms under oxidizing conditions. Future work should focus on whether the Rev-erbβ thiol-disulfide rheostat occurs in mammalian cells in the context of the full-length protein and whether it interfaces with cellular redox buffers, for example, the glutathione:glutathione disulfide and dehydroascorbic acid:ascorbic acid systems that are under circadian control (92). Because Rev-erbα/β protein levels are tightly regulated by the transcription/translation feedback cycle of the molecular clock, redox regulation might represent an additional mechanism of controlling Rev-erb repressor activity.
Transient kinetic experiments support the hypothesis that the HRM in Rev-erbβ is a redox sensor
A caveat of our early estimates of K d for the Fe3+/Fe2+-heme complexes with FLRev-erbβ and thiol-reduced/oxidized LBDs was the use of equilibrium titrations. Because K ds for these complexes are in the low nanomolar range, UV-vis spectrophotometric titrations performed with micromolar concentrations of heme and protein yield typical, “dog-leg” curves that sharply saturate at 1:1 stoichiometry of heme and protein. Under such conditions, the concentration of free ligand cannot be assumed to equal the total concentration since most of the ligand is protein bound below a 1:1 ratio. The quadratic equation accounts for the concentrations of total versus free heme in such titrations but is limited in that the accuracy of the K d is dependent on the sharpness of the curve; below a 10 nanomolar K d, the standard error of the fit becomes rather large. To circumvent these issues, we used a kinetic approach, analyzing individual rate constants for the association (k on) and dissociation (k off) of heme and Rev-erbβ, ultimately establishing accurate K ds using the basic expression, K d = k off/k on. As detailed in our recent manuscript (11), we utilized a unique buffer system containing caffeine, which has been shown to dissociate heme dimers and oligomers that usually complicate measurements of k on using stopped-flow techniques because of their first-order, rate-limiting dissociation.
Heme association to Rev-erbβ was complex and proceeded through several different kinetic steps. However, the initial binding phase, which made up >50% of total amplitude change, was hyperbolic with respect to protein concentration, indicative of a two-step binding mechanism (81). Similar hyperbolic dependence of k obs on protein concentration has been observed in other heme:hemoprotein systems, suggesting that they have similar mechanisms of heme association (20, 54, 64). Interestingly, the k on values for Fe3+-heme binding to wild-type protein, a C384A variant lacking the HRM cysteine thiolate, and the thiol-oxidized form were between 3.6 × 105 M −1 s−1 and 4.1 × 105 M −1 s−1, indicating that loss of the axial thiolate has a negligible influence on heme binding. Fe2+-heme association, however, proceeded with a second-order rate constant of k on = 2.7 × 106 M −1 s−1, demonstrating that the heme redox state is an important determinant of k on.
The measurement of k on was relatively straightforward, whereas heme dissociation was biphasic, that is, affording two k off values. For these experiments, one follows heme dissociation from Rev-erbβ by monitoring formation of the complex between heme and the H64Y/V68F myoglobin variant, which exhibits a unique UV-vis spectrum. For wild-type Rev-erbβ, off-rates were between 10−4–10−5 s−1, with a K d range of 104–639 pM; the slow kinetic phase predominated and made up >80% of the total amplitude, indicating that most of the bound heme dissociates with a K d of ∼100 pM. In contrast, the Fe3+-heme off-rates from thiol-oxidized and C384A proteins were between 10−2 and 10−3 s−1, giving K d ranges of 14–50 nM and 4–32 nM, respectively. Further, the k off values of Fe2+-heme from wild-type protein were 1.4 s−1–6.5 × 10−2 s−1, yielding a K d range of 24–519 nM. Thus, dissociation of the axial thiolate via either redox process (oxidation of the HRM thiolate to a disulfide or heme reduction) significantly lowers Rev-erbβ heme affinity. In other words, Rev-erbβ exhibits features of a redox sensor rather than a direct heme sensor. This hypothesis gains momentum given coimmunoprecipitation results demonstrating that the C384A variant of FLRev-erbβ loses >70% NCoR1 binding capacity with respect to wild-type protein, corroborating the importance of the HRM cysteine in stabilizing both heme and NCoR1 interactions.
These redox sensor-related results take on an even greater impact in the context with recent work by Reddi and colleagues describing the accurate determination of intracellular RH levels in Saccharomyces cerevisiae (30). In that study, fluorescent heme sensors were targeted to different subcellular organelles and RH levels were determined by using elegant normalization techniques that defined fluorescence emission ratios (with respect to an internal fluorescence standard) of the sensors in heme-free and heme-replete conditions. RH in the cytosol was found to be 20–40 nM, whereas heme levels are kept at a minimum (<2.5 nM) in the mitochondria and nucleus. RH in the nucleus and mitochondria is likely maintained at these extremely low levels to limit the amount of free heme available to participate in Fenton reactions, leading to damaged DNA or respiratory chain elements. A complicating factor is that the heme sensor employed is insensitive to Fe3+-heme at the low concentrations present in the cell; thus, the aforementioned values are reflective of the Fe2+-heme RH pool, although Fe3+-heme RH is also likely to exist due to varying ligand conformations with glutathione or protein side chains in the cellular milieu. In any case, those values provided in the aforementioned study lay the groundwork for discussions on the likelihood of heme:protein interactions with an RH pool in the low nanomolar range. Since the majority of the thiol-reduced, wild-type Rev-erbβ population binds Fe3+-heme with a K d of 0.1 nM, it would predominantly exist as holoprotein with <2.5 nM RH in the nucleus. However, thiol oxidation or heme reduction drives the K d above 14 nM. Because these values lie on the upper end of the RH estimation, Rev-erbβ may exist as an apoprotein under those conditions. Another hypothesis is that Rev-erbs might bind Fe2+-heme after being translated in the cytosol, with heme then oxidizing to Fe3+-heme to “lock in” a particular conformation of the protein. Regardless of this, the initial estimates of Rev-erbα/β affinity of 0.35–3.52 μM with ITC are problematic in that they suggest the protein would exist within the cell in the apo form exclusively (59, 70). Our results underscore the importance of using highly sensitive spectroscopic and kinetic studies to investigate the molecular mechanism of heme-dependent NCoR1 recruitment, and the possibility of Rev-erbs acting as redox sensors either through the Fe3+/Fe2+-heme or the thiol-disulfide couple (11).
Heme Oxygenase-2
HO2 is an HRM-containing protein
A variety of mechanisms are involved in the regulation of heme homeostasis to ensure that heme levels are maintained at concentrations that are high enough for cellular processes but low enough to avoid the cytotoxic effects of heme. One such mechanism involves heme degradation, a pathway in which heme is converted to biliverdin, and biliverdin is, subsequently, converted to the more readily excreted product bilirubin (55). The first step is rate limiting and is catalyzed by HO, the only enzyme in mammals known to degrade heme.
HO2, one of the two major isoforms of HO identified in mammals, contains three HRMs (Fig. 1). One HRM, centered at Cys127-Pro128 in human HO2, is located in the α-helical catalytic core region, consisting of residues 28-248 where heme binds and undergoes conversion to biliverdin. The other two HRMs, centered at Cys265-Pro266 and Cys282-Pro283, are located in an unstructured region of the protein between the catalytic core and the membrane-spanning region that tethers the protein to the endoplasmic reticulum. HO2 was among the proteins first identified as having an HRM (105), and it was immediately thereafter noted that the presence of HRMs in HO2 distinguished it from HO1, which contains no cysteines (60). HO1 is an inducible protein expressed in most tissues but with particularly high levels in the liver and spleen, whereas HO2 is constitutively expressed and has been primarily identified in the brain and testes (55). However, the two proteins convert heme to biliverdin with similar catalytic efficiencies (56) and share a high level of homology (55% identical and 76% similar for the human proteins). A comparison of the structures of HO2 and HO1 illustrates how similar the two proteins are in the catalytic core region, consisting of residues 1-230 in human HO1 (4, 78). However, the HO2 and HO1 sequences diverge significantly between the catalytic core and the membrane-spanning region where the HRMs of HO2 are located, a region for which there is little structural information. As the HRMs appear to be the major difference between the two proteins, attention has been given to the heme-binding properties and the function of the HRMs of HO2 in an effort to understand why nature has selected a new form of HO that catalyzes the same reaction with a similar catalytic efficiency as the ancient form (HO1).
Heme binding to the HRMs of HO2
To infer that any differences between HO2 and HO1 are linked to the presence of HRMs in HO2, heme binding to the HRMs needed to be confirmed. An initial study of rat HO2 (60) demonstrated that adding heme to a 10-mer peptide spanning one of the HRMs (Val-Arg-Lys-Cys264-Pro-Phe-Tyr-Ala-Ala-Gln) results in a dramatic color change, suggesting heme ligation. A more subtle color change was observed for a peptide spanning the other C-terminal HRM (Gly-Ser-Asn-Cys281-Pro-Phe-Arg-Thr-Ala-Met). Further, subtle differences in the absorbance spectra of a Cys264Ala/Cys281Ala variant of rat HO2 as compared with the wild-type led to the hypothesis that the wild-type was binding ∼2 more equivalents of heme per monomer of protein than the variant, suggesting that the HRMs were involved in binding heme independently of heme binding to the catalytic core (60). The results were slightly ambiguous but enough to pique the curiosity of others.
A significant finding in the further characterization of the HRMs was the discovery of a disulfide bond involving the cysteines in the HRMs of the soluble form of human HO2 (HO2 sol ), which spans residues 1-288, thus eliminating residues 289-316, which form the membrane-spanning region. Cys265 and Cys282 were shown to form a disulfide bond by the 5,5′-dithiobis(nitrobenzoic acid) (DTNB) assay and by monitoring alkylation of the cysteine residues using mass spectrometry (99). A later study using 13C-Cys-labeled HO2 sol and two-dimensional NMR also confirmed disulfide bond formation (86). Further, through thiol trapping experiments using the isotope-coded affinity tag technique (53), the thiols were shown to be 60–70% reduced in vivo under normal growth conditions (98). The dithiol/disulfide ratio also changed to reflect changes in the cellular redox state, with 81–87% of thiols reduced under reducing conditions and 86–89% of thiols in the disulfide state under oxidizing conditions. The midpoint potential for the thiol/disulfide conversion, measured by thiol trapping and fluorescence trapping, was calculated to be −200 mV, which is near the ambient cellular redox potential (71). Thus, the thiol/disulfide conversion appears to be physiologically relevant and to link HO2 to the cellular redox state.
The added significance of the thiol/disulfide conversion in HO2 is that a disulfide bond necessarily limits the ability of Cys265 and Cys282 to coordinate Fe3+-heme. Indeed, no thiol coordination of Fe3+-heme was observed with fully oxidized (disulfide state) as-purified HO2
sol
(99), and its spectral features were identical to those of a truncated form of HO2 (HO2
core
), which spans residues 1-248 and lacks the C-terminal HRMs (20, 99). Titration of oxidized HO2
sol
(HO2
sol
O) or HO2
core
with Fe3+-heme resulted in an increase at 406 nm that saturated at 1:1 protein to Fe3+-heme and an EPR signal (signal

), and HO2
sol
R (—) in 50 mM Tris (pH 8.0) and 50 mM KCl at 20°C.  ), and HO2
tail
(C265A)R (…) in 50 mM Tris (pH 8.0) and 50 mM KCl at 20°C.
), and HO2
tail
(C265A)R (…) in 50 mM Tris (pH 8.0) and 50 mM KCl at 20°C.
Surprisingly, on reduction of the disulfide bond of HO2 and subsequent incubation with Fe3+-heme, the EPR spectrum of HO2
sol
R (reduced HO2
sol
) included both signal
Various spectroscopic techniques were used to probe for the thiol-coordinated Fe3+-heme in HO2. MCD spectra of Fe3+-heme-bound human HO2 and the Cys variants suggested that a six-coordinate, Cys265-ligated Fe3+-heme is formed as a minor species on reduction of the disulfide bond (22); Fe K-edge EXAFS and XAS data supported thiol coordination by both Cys265 and Cys282, although Cys265 appeared to bind with a somewhat higher affinity than Cys282 to the Fe3+ center (2); and 1H and 13C ENDOR experiments using reduced, Fe3+-heme-bound HO2 that had been 2H-Cys or 13C-Cys labeled conclusively demonstrated Fe3+-heme coordination at the HRMs (2). Thus, collectively these various lines of enquiry unambiguously showed that the reduced state of HO2 contains three hemes (Fig. 6D): one at the catalytic core (His45/aquo) and two additional heme-binding sites at the C-terminal HRMs: Cys265/His256 and Cys282. Regardless of this, one study proposed that the HRMs of HO2 do not stably coordinate to Fe3+-heme (86); however, in that study involving equilibrium titrations, Fe3+-heme was added to HO2 in the presence of excess tris(2-carboxyethyl)phosphine, and phosphines are reasonably strong Fe3+-heme ligands (57, 75, 83).
Kinetics of heme binding to HO2
Spectral data suggested that the three binding sites of HO2 had varying affinities for Fe3+-heme. At less than saturating concentrations of Fe3+-heme, the spectra were dominated by features associated with Fe3+-heme binding at the His45/aquo site, suggesting that the catalytic core had a higher affinity for Fe3+-heme than the HRMs. Further, Fe K-edge EXAFS and XANES data suggested that Cys265 had a higher affinity for Fe3+-heme than Cys282 (2). However, Fe3+-heme K d values were difficult to ascertain from equilibrium titrations of HO2 sol R due to the complexity of the UV-vis spectra. Thus, equilibrium titrations of the truncated forms of HO2 were performed and revealed K d values of 14 nM for HO2 core (Fe3+-heme binding to the His45/aquo site), 90 nM for HO2 tail (C282A)R (Cys265/His256 site), and 900 nM for HO2 tail (C265A)R (Cys282 site) (20), confirming that the three sites have varying affinities for Fe3+-heme.
However, as discussed earlier for Rev-erbβ, equilibrium titrations may not give the most accurate K d values. Instead, individual rate constants for the association (k on) and dissociation (k off) of Fe3+-heme provided a more detailed view of binding. Further, this kinetic approach permitted the observation of Fe3+-heme binding to some of the sites in HO2 sol R. The rate constants were very similar for the sites in the truncated proteins as the comparable site in HO2 sol R, indicating that the truncated proteins are good models for the full-length protein. Calculation of K d values using the simple equation K d = k off/k on revealed K d values of 0.08–0.2 nM for Fe3+-heme binding to the His45/aquo site and 0.3–0.4 nM for Fe3+-heme binding to the Cys265/His256 site (20). The k on value could not be determined for Fe3+-heme binding to the Cys282 site, but estimates would suggest a value of ∼1 nM. Values obtained by kinetic measurements follow the same trend as equilibrium titrations, that is, the His45/aquo site has a higher affinity than Cys265/His256, which, in turn, has a higher affinity than Cys282. However, the lower K d values are on par with RH levels in the cell, in contrast to the values obtained by equilibrium titrations that would have suggested that the HRMs only bind Fe3+-heme under conditions in which heme levels were elevated. Therefore, the redox state of the cell, by influencing the thiol/disfulfide conversion of Cys265 and Cys282, appears to be more relevant to Fe3+-heme binding to the HRMs of HO2 than previously thought, suggesting that the HRMs function as redox sensors.
Functional significance of HRMs in HO2
HRMs appear to place HO2 in a unique position at which the redox state of the cell and regulation of heme and iron homeostasis intersect. However, the exact nature of their function remains unclear. Repeated experiments have demonstrated that the steady-state activity of HO2 is not affected by substitution of any of the Cys to Ala or by exclusion of the cysteine residues by truncation of the protein (16, 20, 60, 99). A more detailed investigation of heme hydroxylation, the initial steps in heme degradation, by HO2 using a combination of radiolytic cryoreduction/annealing with UV–vis and EPR spectroscopy demonstrates that there is no involvement of the HRMs in the catalytic cycle of HO2 (16). In addition, the HRMs can bind but not oxidize heme as no steady-state activity was observed with HO2 tail R (16). HO2 and HO1 also appear to bind cytochrome P450 reductase, which provides electrons for the HO reaction, with the same corresponding residues (80), further suggesting that the degradation of heme proceeds in the catalytic core independently of the HRMs. HRMs are also not involved in the specific activation of HO2 by menadione analogs or its specific inhibition by clemizole analogs (87) nor do they participate in binding of the heme mimic Zn(II) PPIX, an HO inhibitor (36). Thus, further investigation is needed to detail the functional significance of heme binding to HRMs in a redox-regulated manner.
Conclusions and Future Directions
Here, we have highlighted HO2 and Rev-erbβ to illustrate the similarities and differences between two hemoproteins that both contain HRMs. HRMs of both proteins appear to act as redox sensors in that the participation of the HRM cysteine(s) in a thiol/disulfide switch alters affinity for heme. Therefore, future studies of HRM-containing proteins should include experiments that probe for disulfide bonds and/or differences in heme binding under oxidizing and reducing conditions. HRMs acting as redox sensors may be applicable to other HRM-containing proteins as many contain multiple HRMs and/or other cysteine residues. Further, the affinity for heme at the HRMs, based on the kinetic approach used for HO2 and Rev-erbβ, may be lower than previously proposed, suggesting that the functional significance of the HRMs may be greater than expected. The survey of HRM-containing proteins (see the Introduction section and Fig. 1) indicates that HRMs play a role in regulating protein stability in a significant number of proteins, many of which are involved in regulating circadian rhythm and metabolic pathways (Rev-erbs, p53, and Per2), suggesting that it may be advisable to probe more HRMs for this function. Thus, the study of HRMs, in general, is a growing field with many interesting questions yet to be answered.
Footnotes
Acknowledgments
This work has been funded by NIH (R21HL089837, R01-GM123513, and R01-HL102662A) to SWR, F32HL114150 to ELC and by the University of Michigan Biomedical Research Council (to SWR).