
Abstract
Significance:
The complex etiology of type 1 diabetes (T1D) is the outcome of failures in regulating immunity in combination with beta cell perturbations. Mitochondrial dysfunction in beta cells and immune cells may be involved in T1D pathogenesis. Mitochondrial energy production is essential for the major task of beta cells (the secretion of insulin in response to glucose). Mitochondria are a major site of reactive oxygen species (ROS) production. Under immune attack, mitochondrial ROS (mtROS) participate in beta cell damage. Similarly, T cell fate during immune responses is tightly regulated by mitochondrial physiology, morphology, and metabolism. Production of mtROS is essential for signaling in antigen-specific T cell activation. Mitochondrial dysfunction in T cells has been noted as a feature of some human autoimmune diseases.
Recent Advances:
Preclinical and clinical studies indicate that mitochondrial dysfunction in beta cells sensitizes these cells to immune-mediated destruction via direct or indirect mechanisms. Sensitivity of beta cells to mtROS is associated with genetic T1D risk loci in human and the T1D-prone nonobese diabetic (NOD) mouse. Mitochondrial dysfunction and altered metabolism have also been observed in immune cells of NOD mice and patients with T1D. This immune cell mitochondrial dysfunction has been linked to deleterious functional changes.
Critical Issues:
It remains unclear how mitochondria control T cell receptor signaling and downstream events, including calcium flux and activation of transcription factors during autoimmunity.
Future Directions:
Mechanistic studies are needed to investigate the mitochondrial pathways involved in autoimmunity, including T1D. These studies should seek to identify the role of mitochondria in regulating innate and adaptive immune cell activity and beta cell failure.
I
Mitochondria have long been viewed as a participant in many of the normal and pathogenic features of diabetes. These essential organelles produce energy, reducing equivalents and reactive oxygen species (ROS). In addition, they are critical metabolic centers important in regulating cellular homeostasis and signal transduction. The production of ROS and other oxidants by mitochondria has implicated these organelles in the pathogenesis of various diseases, including T1D. Oxidants are considered to be a pathogenic contributor in most forms of diabetes, but the cellular sources and types of oxidants remain controversial. In this review, we discuss the specific role(s) of mitochondria in pancreatic beta and immune cells during the pathogenesis of autoimmune T1D.
Mitochondrial Function in Beta Cells and T1D
In the postprandial state, beta cells respond to absorbed nutrients by releasing insulin into the blood stream. Secretion of insulin can be induced by sugars and amino acids with potentiation by fatty acids. With the exception of positively charged amino acids (which induce secretion through an electrogenic mechanism), nutrients require mitochondria for the induction of insulin release. Therefore, the role of mitochondria in pancreatic beta cell physiological function has long been recognized and widely studied.
Under normal conditions, beta cells sense glucose levels and transport glucose via glucose transporters. The transported glucose molecules are subsequently phosphorylated and converted to glucose-6-phosphate by glucokinase (the beta cell glucose sensor). Glucose-6-phosphate is metabolized via glycolysis to produce pyruvate and then acetyl coenzyme A (acetyl-CoA). Acetyl-CoA enters the mitochondrial tricarboxylic acid (TCA) cycle to facilitate adenosine triphosphate (ATP) generation by oxidative phosphorylation (OXPHOS). The production of ATP by mitochondria as a result of rising circulating nutrient concentrations is a key and essential physiological function of these organelles in beta cells.
ATP exchange for cytoplasmic adenosine diphosphate (ADP) by adenine nucleotide translocases increases the cytoplasmic ATP/ADP ratio allowing for ATP to displace ADP bound to the Kir6.2 subunit of the ATP-sensitive K+ channel, an inward-rectifier potassium ion channel (62). ATP binding inhibits this channel, triggering plasma membrane depolarization, opening of L-type voltage-dependent calcium channels, and the influx of calcium. Increased intracellular calcium directly triggers fusion of insulin granules and insulin exocytosis. The increase in cytosolic calcium also enhances both mitochondrial metabolism and mitochondrial ATP production. As such, secretion of insulin is tightly regulated by mitochondrial function, specifically through ATP production and regulation of intracellular calcium concentrations (54, 94, 95, 97). The strong requirement for mitochondria during beta cell function has also been extended to roles during cell survival and death (14, 30, 31). This concept will be discussed in detail later.
During the pathogenesis of T1D, pancreatic beta cells are targeted and destroyed by an autoimmune attack by islet infiltrating beta cell antigen-specific autoreactive T cells (59, 67). Preclinical models including the nonobese diabetic (NOD) mouse and biobreeding–diabetes-prone (BB-DP) rat [reviewed by Pearson et al. (68)] have provided significant information on the kinetics of cellular infiltration during the progression of insulitis. Although it was initially observed that macrophages and/or dendritic cells were the first immune cell types to infiltrate the islets, the more recent thought is that these are tissue-resident macrophages (11, 26). These tissue-resident antigen-presenting cells (APCs) take on inflammatory characteristics early (3–4 weeks of age in NOD females) and produce chemokines that recruit lymphocytes (CD4+ and CD8+ T cells as well as B cells) into the islets (3). The signals that induce the islet APCs to mature and become inflammatory remain unknown; however, long-term depletion of these cells results in protection from T1D, highlighting the essential nature of macrophages in T1D pathogenesis (8, 43, 85). T lymphocytes (both CD4+ and CD8+ cells) are also required for T1D initiation (70). The cellular components of insulitis that are essential for T1D onset have provided clues as to the effector mechanisms that induce beta cell death. However, the experimental models used to identify these mechanisms remain controversial. Early knowledge of cellular component and patterns of insulitis has been from animal models, including NOD mice (43, 102, 103), BB rats (37), and transgenic animals (1, 80). With the increased availability of human pancreas samples for research in recent years, it is evidenced that animal models do not represent human insulitis (9, 10, 40, 41).
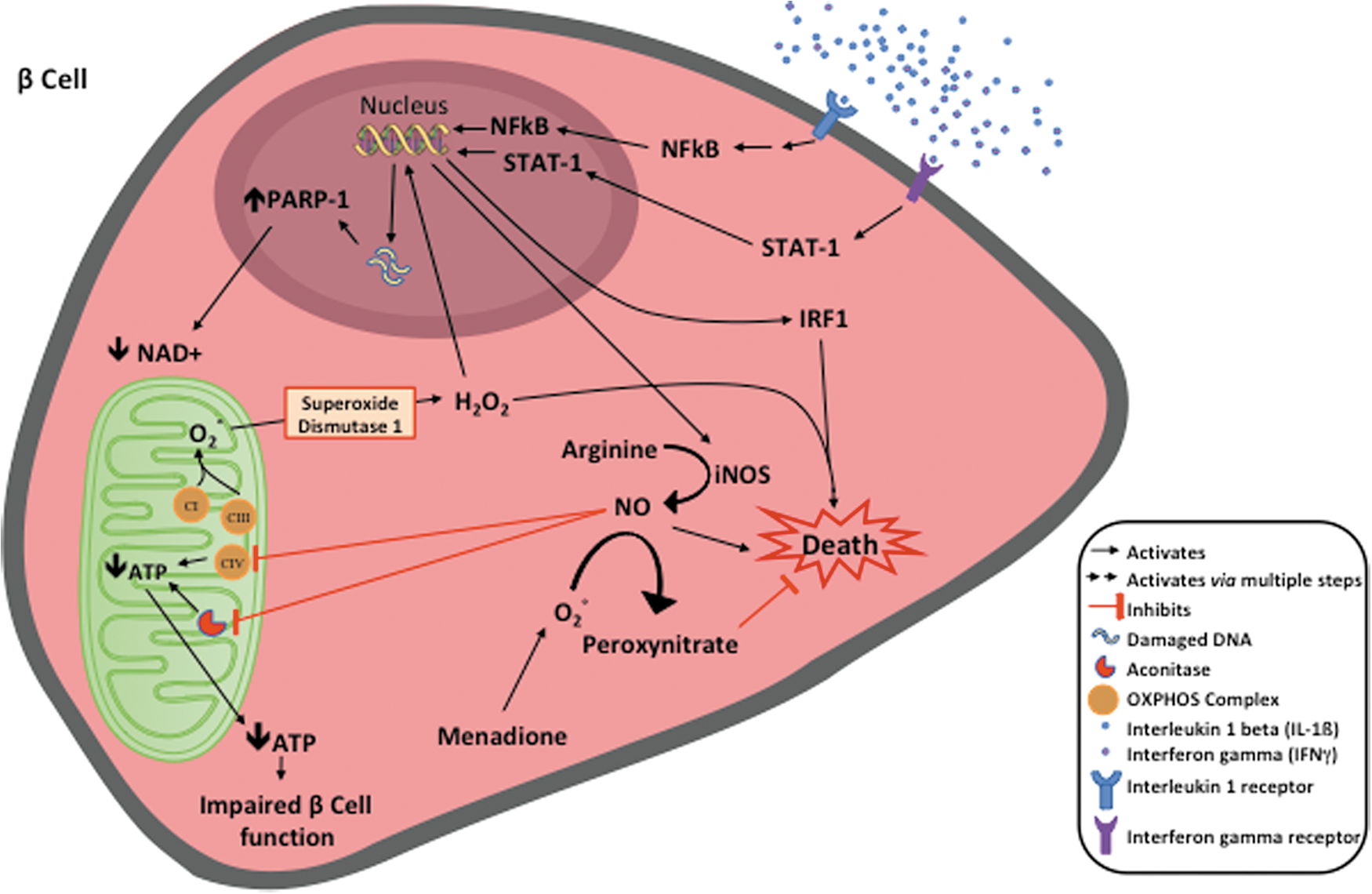
A long held notion in T1D is that macrophages within the islet produce ROS and proinflammatory cytokines, creating a beta cytotoxic environment (64). Activated proinflammatory macrophages can destroy islets in coculture systems (78). Historically, the proinflammatory cytokine combinations of interleukin 1 (IL-1), interferon gamma (IFNγ), and tumor necrosis factor alpha (TNFα) have been used in vitro to model this system. These inflammatory cytokines are produced from macrophages and CD4+ T cells and result in the activation of the inducible nitric oxide synthase (iNOS) (19, 38) through NF-κB [nuclear factor kappa-light-chain enhancer of activated B cells (24, 58)]-dependent pathways. Nitric oxide production within the beta cell results in reversible inhibition of mitochondrial OXPHOS. These inhibitions take place at cytochrome c oxidase (Complex IV) (75, 76), NADH dehydrogenase (Complex I) (72, 73), and inhibition of the TCA cycle enzyme aconitase (18). Inhibition of iNOS via pharmacological or genetic means prevents beta cell death by this cytokine combination in human and murine islets (52, 86). The major effector cytokine in this combination is IL-1; however, a number of recent studies have failed to support a role of IL-1 in the pathogenesis of human or mouse T1D (2, 29, 87). Accordingly, in human T1D, support for a pathogenic role of iNOS production and function within the beta cell has yet to materialize.
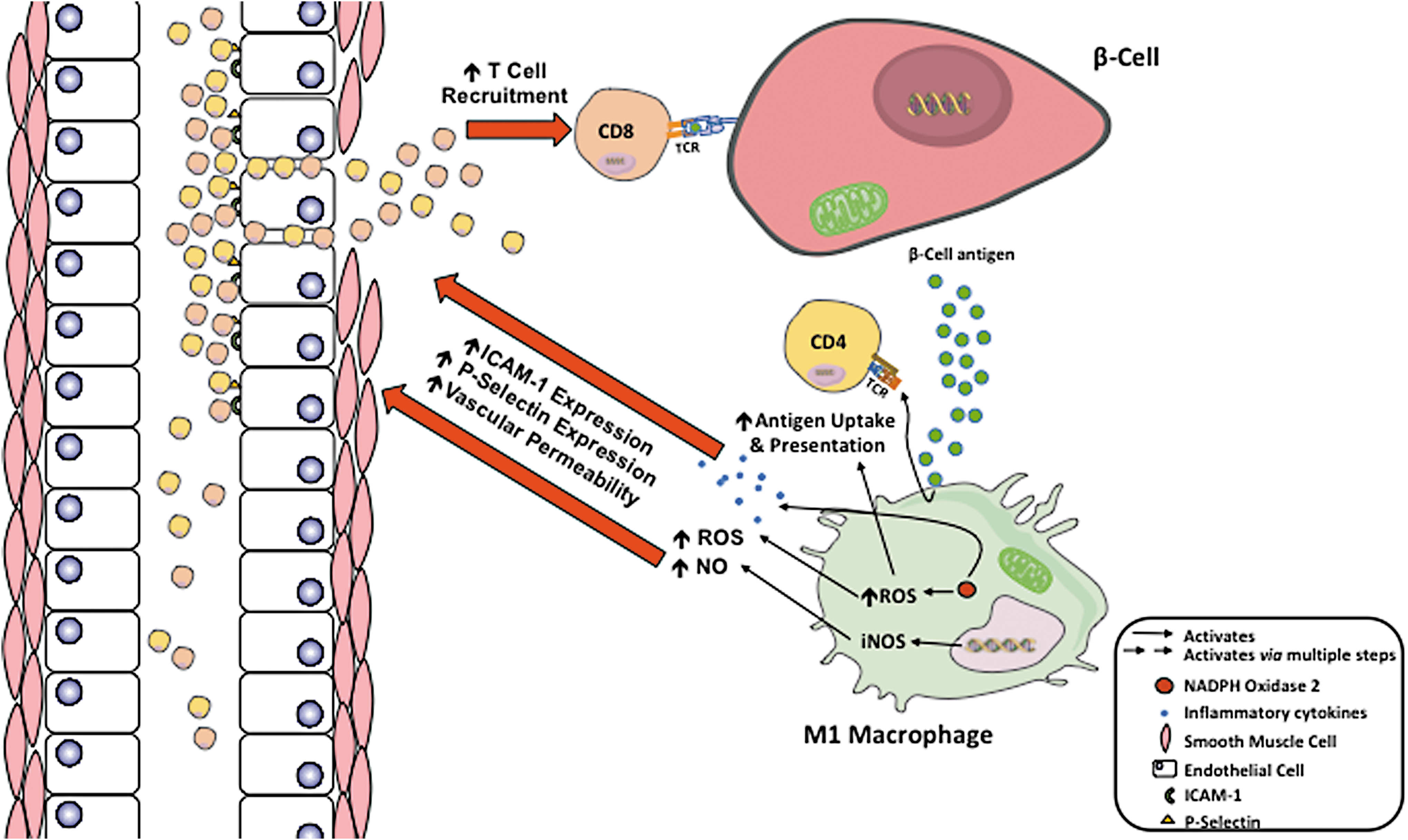
Other cells within the islet could be a source of ROS and reactive nitrogen species that damage beta cells during T1D pathogenesis. Indeed, activated proinflammatory macrophages can destroy islets in coculture systems (78). M1 macrophages produce ROS (64), which plays an important role in the recruitment of CD4+ and progression to T1D (Fig. 1). However, ROS are also critical for the regulation of M1 phenotypic macrophages (84). A lack of ROS production by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) in mice causes an increase in macrophage polarization toward an M2 phenotype (65). In addition, adoptive transfer systems have demonstrated that macrophage production of ROS via NOX2 is essential for T1D onset (85). Thus, a decrease in M1 macrophages highlights the essential role that ROS have in the islet microenvironment and is key to T1D pathogenesis
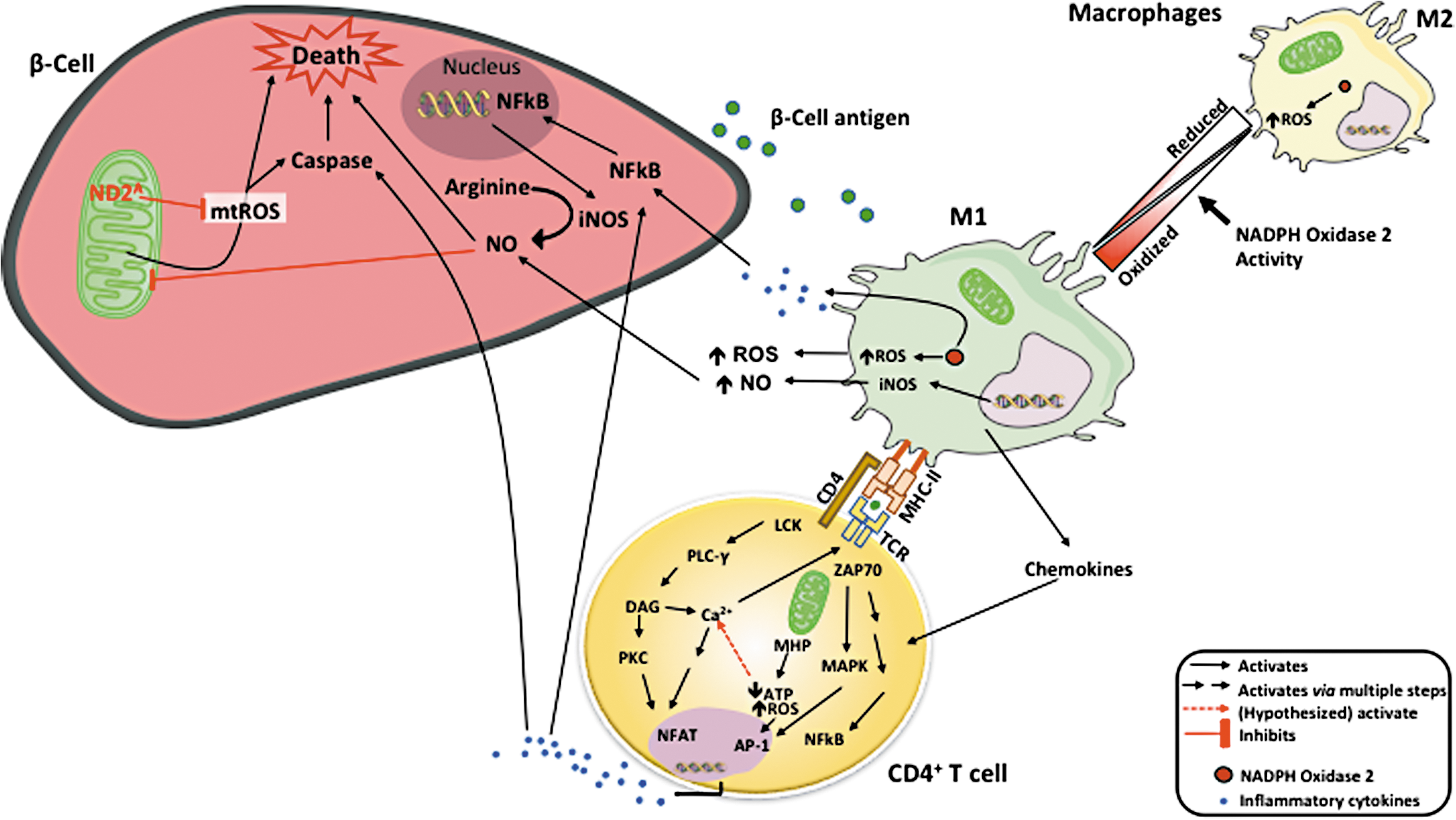
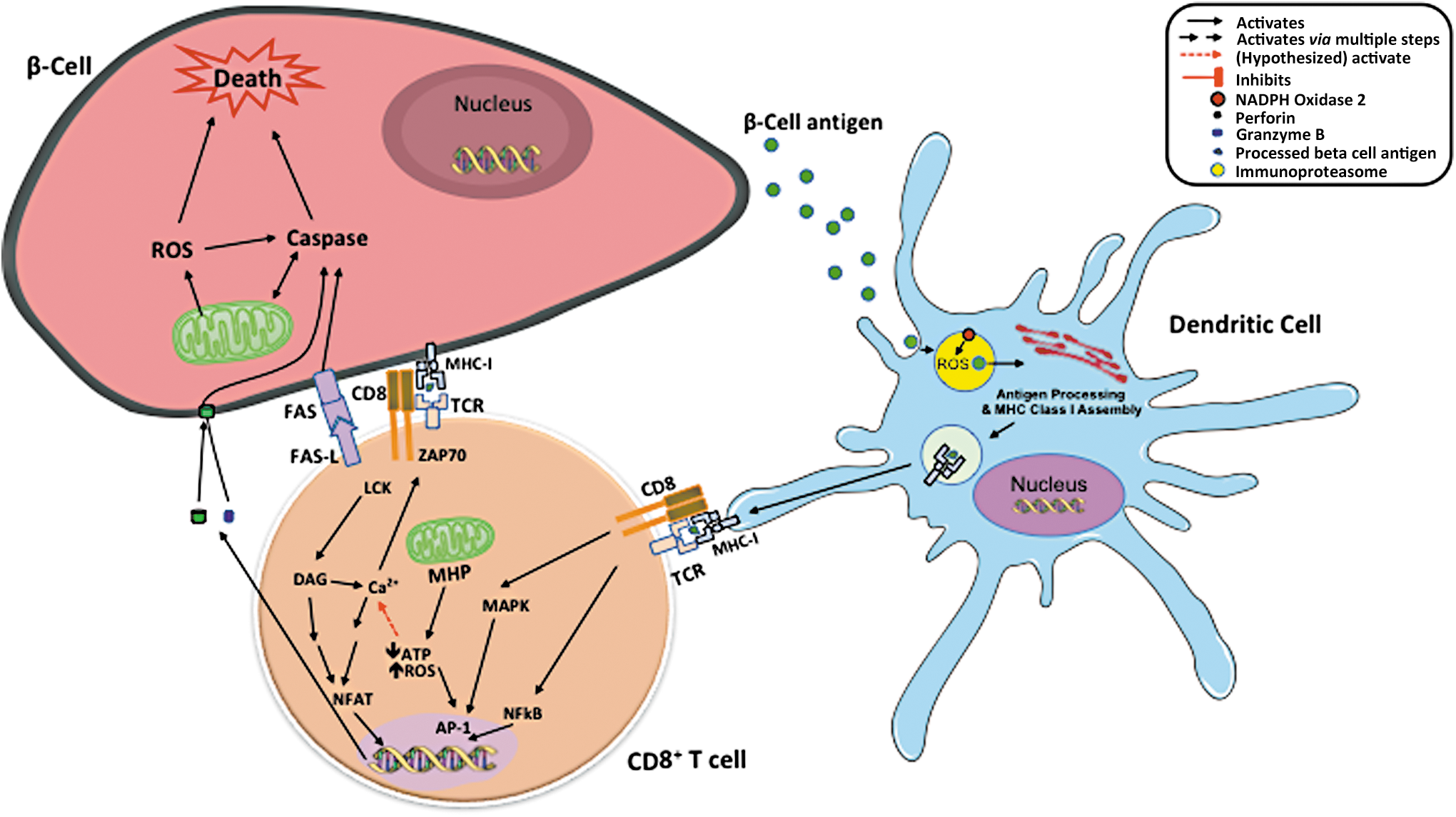
T cell induction of beta cell death has been linked to intrinsic apoptosis with involvement of mitochondria. CD8+ T cells recognize beta cell antigens in the context of major histocompatibility complex (MHC) class I and directly kill beta cells by the release of perforin and granzymes (23). Perforin and granzymes mediate cleavage of BH3 Interacting Domain Death Agonist (BID), resulting in mitochondrial permeability transition, release of cytochrome C, and eventually beta cell death (25). FAS-FAS ligand has been linked to mechanisms by which both CD4+ and CD8+ T cells mediate beta cell killing (20, 23, 91). These death receptors signal activation of caspases and mitochondrial apoptotic pathway. Therefore, mitochondrial function, particularly ROS production, plays an important role in regulating beta cell death under autoimmune attack [Reviewed by Padgett et al. (64)]. In addition, ROS produced by NOX2 within dendritic cells are essential for maintenance of a neutral pH within lysosomes where autoantigens are processed and assembled for cross-presenting to CD8+ T cells (Fig. 2).
Free radicals generated within the mitochondria are also players in the process of immune-mediated beta cell destruction (32). Beta cells are particularly sensitive to ROS/oxidant damage due to lower antioxidant enzyme expression in islets (49, 56, 58). In vitro studies have shown improvements in mitochondrial antioxidant defenses of beta cell lines or primary islets with the overexpression of antioxidant enzymes protecting these cells against inflammatory cytokines or oxidative stress (4, 16, 31, 53, 54). Beta cell susceptibility to cytokine-induced mitochondrial dysfunction pathways has been linked to T1D-genetic risk loci. Gli-similar 3 (GLIS3), a candidate gene for both T1D and type 2 diabetes (T2D) susceptibility, is reported to sensitize beta cells to cytokine-induced death through mitochondrial apoptosis pathway (63). Another T1D-associated candidate gene, CLEC16A (35), affects beta cell survival by changing mitochondrial morphology, function, and mitophagy (81). Downregulation of C-type lectin domain containing 16A (CLEC16A) leads to reduction of Nrdp1 (neuregulin receptor degradation protein-1), an E3 ubiqutin ligase (81). Nrdp1 regulates proteasomal degradation of Parkin (104), a key regulator of mitophagy (42). Mitochondrial ROS have been shown to initiate parkin RBR E3 ubiquitin protein ligase (PARK2)/Parkin-dependent mitophagy (93).
Studies using the T1D-prone NOD mouse, and a closely related diabetes-resistant strain, alloxan resistant (ALR), mapped a single nucleotide polymorphism (SNP), C4738A, to mitochondrial DNA (mtDNA) (57). This SNP is within the NADH dehydrogenase subunit 2 (mt-Nd2) gene and is associated with protection against both spontaneous T1D and chemically induced diabetes (16, 55, 57). To study this SNP, conplastic NOD.mtALR mice were created by crossing female ALR mice with males of the NOD strain, followed by backcrossing with male NOD for 10 generations. This process resulted in mice harboring mt-Nd2a on the NOD genetic background (34). Reciprocal ALR.mtNOD mice were also created. The protective allotype (mt-Nd2a ) has been demonstrated to be associated with lower ROS production within isolated mitochondria (33, 34). We have generated beta cell lines harboring either mt-Nd2a or mt-Nd2c on the NOD genetic background (14). Beta cells with the T1D-resistant mt-Nd2a allele display resistance to alloxan, inflammatory cytokines, and autoreactive CD8+ cytotoxic T cell-mediated lysis (14). The protection has been associated with lower mitochondrial ROS production under inflammatory conditions. We have also investigated the mechanism of protection from mt-Nd2a in vivo by comparing spontaneous T1D incidence between NOD and NOD.mtALR mice. Not surprisingly, as a complex disease under the influence of multiple genetic and environmental factors, the incidence of spontaneous T1D was unaltered by this single mitochondrial SNP. Yet, mice with mt-Nd2a were resistant to induction of diabetes after the transfer of highly pathogenic beta cell antigen responsive T cells. Mice were completely protected from transfer of T1D by the diabetogenic CD4+ T cell clone BDC2.5 (14). Induction of T1D by BDC2.5 CD4+ T cells requires macrophages to mediate beta cell death. After adoptive transfer to recipients, the CD4+ T cells recruit macrophages (8) and activate the latter to release ROS and cytokines (8, 85). BDC2.5-mediated destruction of beta cells is also FAS-FAS ligand dependent (20, 91). Since BDC2.5 T cells attack beta cells through a cytokine-mediated ROS-dependent pathway (64, 69), the protection provided by mt-Nd2a is through enhancing the ability of these cells against ROS-mediated damage. In contrast, mice harboring mt-Nd2a are also protected to a certain degree against monoclonal diabetogenic CD8+ T cell-induced diabetes (14). As already mentioned, mitochondria participate in CD8+ killing of beta cells. Decreased mitochondrial ROS from mt-Nd2a beta cells are, therefore, considered to lower sensitivity of these beta cells to CD8+ T cell-induced activation of caspases and contribute to the partial protection.
A corresponding SNP exists in the mtDNA of humans within NADH dehydrogenase subunit 2 gene, mt-ND2. This SNP, C5178A, results in an identical amino acid substitution when compared with mouse (leucine to methionine). The protective mt-ND2 A of the mtDNA haplogroup D is associated with lower prevalence of both T1D and T2D in certain populations (51, 88), as well as a series of diseases and conditions related with oxidative stresses, including cardiovascular disease, plasma lipid levels, and longevity (39, 46, 47, 83, 100). Using a human beta cell line BetaLox5 (21, 36), we were able remove the mitochondrial genome and repopulate mtDNA from platelets of donors harboring mtDNA haplotypes with either mt-ND2 A or mt-ND2 C. These cytoplasmic hybrid, or cybrid, cells were used to understand the influence of mt-ND2 alleles on the interaction of beta cells with autoimmune mechanisms. These cybrid cells have identical nuclear genetic backgrounds and only differ in the mitochondrial genome. When compared with cells with the mt-ND2C allele, human beta cells encoding an mtDNA haplotype with mt-ND2A exhibited resistance against inflammatory cytokine-induced damage, which is associated with lower mitochondrial ROS production. Inflammatory cytokines damage beta cells and impair their function through induction of nitric oxide (NO) and hydrogen peroxide (H2O2) production (Fig. 3). These cells are also resistant to diabetogenic autoreactive CD8+ T cell attack in vitro [(50) and our unpublished observation] possibly via a reduction in NO and H2O2 production. These data suggest that protective variants of this mitochondrial OXPHOS subunit exert protection at the beta cell level and potentially impact both T1D and T2D by reducing stress when beta cells encounter inflammatory conditions.
Mitochondrial Function in T Cells and T1D
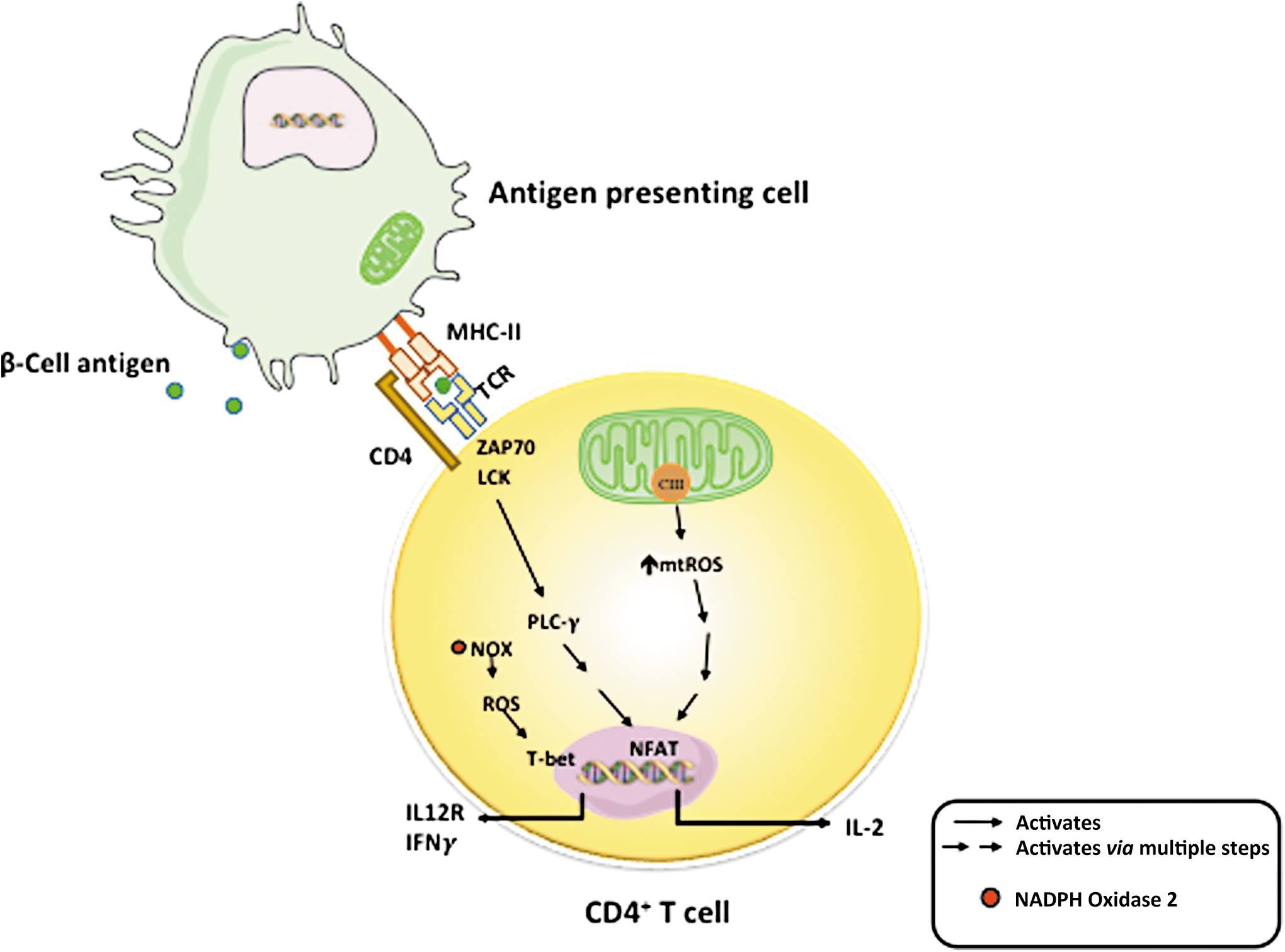
The essential role of mitochondria in T cell activity has drawn great attention in recent years (7, 89). Upon T cell activation, mitochondria are relocated to the immune synapse, where, through balanced fission and fusion processes, mitochondria regulate the local concentrations of ATP, calcium, and free radicals (5, 29, 71, 77). T cell activation and function are regulated by substrate metabolism and mitochondrial function. Naive and memory cells depend on the more efficient mitochondrial OXPHOS process for energy source. When activated, both OXPHOS and aerobic glycolysis are increased in these T cells. The increase in aerobic glycolysis is to fulfill energy demand, that is, Warburg effect (90). This increase in glycolysis allows fast ATP generation from glucose and TCA cycle intermediates to be utilized for biosynthesis of materials that are required for cell proliferation. Evidence from studies using mouse CD4+ T cells suggests that OXPHOS is needed for T cell activation, whereas activated T cells can use either OXPHOS or glycolysis for proliferation (48). T cell receptor (TCR) signaling activates ADP-dependent glucokinase (ADPGK) that catalyzes the phosphorylation of glucose to glucose-6-phosphate using ADP as the phosphate donor. ADPGK is responsible for switching between glycolysis and enhanced mitochondrial ROS generation (44). Mitochondrial ROS, specifically ROS produced at complex III, are essential for antigen-specific activation of T cells and effector function (Fig. 4) (79). In a study wherein mouse cells were differentiated and activated in vitro, it was demonstrated that mitochondrial fission and fusion activities closely control T cell development toward memory or effector phenotypes (6).
The final barrier that prevents autoimmune T cells from invading islets is vascular walls, particularly the vascular endothelium. During the development of T1D, islet microenvironment becomes altered due to the increased production of inflammatory cytokines (IFNγ, TNFα, and IL-1β), ROS, and NO by M1 macrophages. This increased production of cytokines, ROS, and NO causes the vascular endothelium to become more permeable, allowing enhanced movement of CD4+ and CD8+ T cells to move into the extravascular space (60). In addition, ROS and NO increase the expression of intracellular adhesion molecule 1 (ICAM-1) and P-selectin on vascular endothelium (60, 82). P-selectin is responsible for the adhesion and rolling of T cells, with ICAM-1 binding to membrane proteins causing cellular arrest and allowing extravasation to occur (82). This combination increases the extravasation of CD4+ and CD8+ T cells exacerbating the inflammatory microenvironment, leading to further destruction of beta cells and T1D progression (Fig. 5).
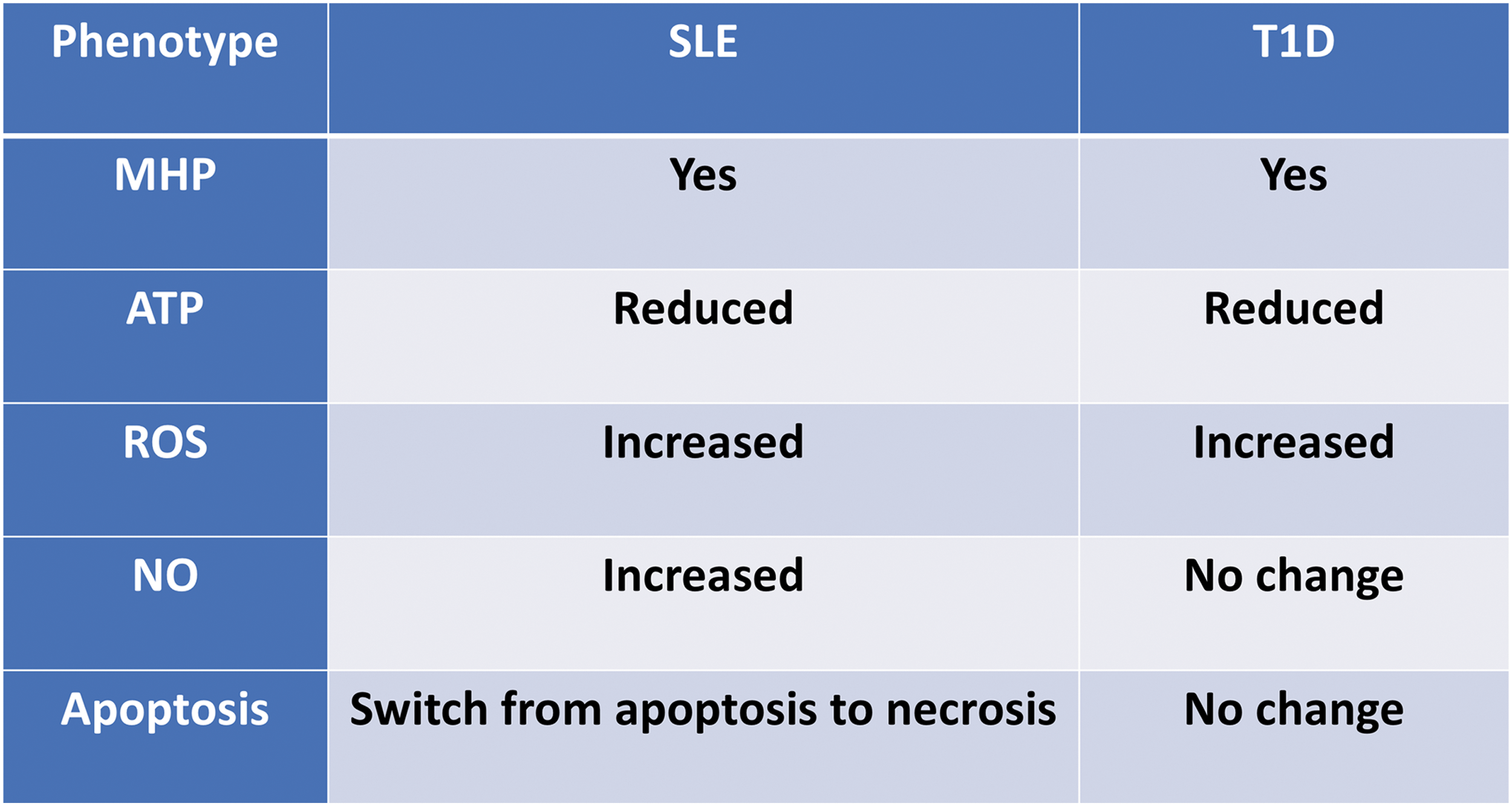
Given the important role of mitochondria in T cell function, it is not surprising that T cell mitochondrial dysfunction has been associated with autoimmune disease in humans. T cells from patients with systemic lupus erythematosus (SLE) exhibits mitochondrial hyperpolarization (28), which is thought to be the consequence of abnormal nitric oxide production from monocytes (61), leading to ATP depletion, increased ROS production, and necrotic death of T cells (28). Animal models of SLE identified that CD4+ T cells have elevated glycolysis and mitochondrial oxidative metabolism when compared with those in control mice (101). Using these mouse models, potential therapeutic approaches have been discovered linking inhibition of the mitochondrial oxidation and glycolytic rate in CD4+ T cells that may normalize their metabolism and potentially reduce the risk of SLE with targeted therapy (101). CD4+ T cells from autoimmune rheumatoid arthritis also displayed ATP depletion, elevated autophagy, and impaired reduction–oxidation status (98, 99). In autoimmune multiple sclerosis (MS), mitochondrial dysfunction in the nervous system has been long known (74, 96). CD4+ T cells from MS patients exhibit significant increases in the mitochondrial inner membrane lipid cardiolipin (92). Changes in mitochondrial enzyme activities and bioenergetic profiles are also detected. CD4+ T cells from patients with MS showed decreased respiratory control ratio, decreased mitochondrial complex I and complex IV activities, increased activities of hexokinase and phosphofructokinase, increased expression of glucose transporter 1 (GLUT1), and decreased activities of antioxidant enzymes superoxide dismutase and glutathione peroxidase (22).
Early research and identification of mitochondria in T cells participating in T1D pathogenesis came from animal studies. Disruption of a mitochondrial outer membrane protein GTPase, IMAP family member 5 (GIMAP5) was determined to account for the lymphopenia that has been described as a major feature of T1D onset in the diabetes-prone BB-DP rats. The mutation in GIMAP5 regulates T cell apoptosis (66). BDC2.5 CD4+ T cell behavior was followed in fetal thymus organ culture (FTOC) of NOD and major histocompatibility complex congenic C57BL/6.NODc17-H2 g7
mice. BDC2.5 FTOC was challenged by peptide in culture and the resulting induction of genes was analyzed by hybridization of messenger RNA from sorted double-positive thymocytes to gene expression arrays (105). Data revealed that the NOD background promotes a significantly increased expression of antiapoptotic gene Bcl2 (B cell lymphoma 2) (105), which is thought to be attributed to failed central tolerance induction in NOD. In a separate study, gene array results showed that islet-specific glucose 6 phosphatase catalytic subunit-related protein (IGRP)-reactive CD8+ T cells from NOD mice failed to induce increased expression of genes associated with OXPHOS pathways, which were detected in nonautoreactive CD8+ T cells, which have otherwise the same phenotype (27). As in the case of the SLE mouse model treated with mitochondrial metabolism inhibitor 2-deoxy-
In accord with results from other autoimmune diseases, T cells from patients with T1D are characterized by mitochondrial abnormalities, specifically, mitochondrial inner membrane hyperpolarization (MHP) (13). This phenomenon is similar in some ways to the observations from T cells of autoimmune SLE patients (28), shown in Figure 6. However, key differences are present when comparing MHP T cells in T1D and SLE. T1D T cells are not necrosis prone (our observation). T cell MHP in T1D is associated with functional changes of both CD4+ and CD8+ subsets. CD4+ T cells from T1D patients that display MHP exhibit a higher IFNγ production upon activation in vitro. Activation-induced IFNγ production is further correlated with increased mitochondrial reactive oxygen species (mtROS) (12). Human CD8+ T cells from donors with MHP also showed an enhanced antigen-specific cytotoxicity when assayed using human beta cells as targets (12). We have observed a decreased mitochondrial spare respiratory capacity in enriched total T cells from patients with T1D (15). T cell mitochondrial dysfunction is not correlated with patients' hemoglobin A1c (HbA1c) level or T1D disease duration (12). Furthermore, MHP is not detected in T cells from T2D patients. Therefore, this T cell mitochondrial defect is not a consequence of abnormal metabolic control but rather intrinsic to these T cells. In addition, genetic linkages of human T1D-risk SNPs have been detected and are associated with these abnormal T cell mitochondrial metabolisms in T1D (12). These data indicate the important role of T cell mitochondrial function in the pathogenesis of human T1D.
In summary, during the development of T1D, mitochondria are involved in both sides of the autoimmune attack: in the victim (target beta cells) as well as in the attacker (effector T cells). Within the target beta cells, mitochondria are required for nutrient-induced insulin secretion. Perturbations to the mitochondria from metabolic or autoimmune stress may enhance susceptibility to autoimmunity through enhanced production of mtROS. On the effector side, mitochondria affect T cell autoreactivity through regulating T cell metabolic activities. In addition to mtROS, cytoplasmic ROS generated from NADPH oxidase also participate in macrophage phenotype switching, dendritic cell antigen cross-presentation, effector T cell response, and impaired vascular endothelium integrity. Taken together, ROS and mitochondrial function play important roles in the pathogenesis of T1D. It remains to be clarified how mitochondrial metabolism and substrate utilization affect immune balance during the development of T1D. This is possibly occurring via changes in development and function of different subsets of T cells, including, but not limited to, effector, memory, naive, and regulatory T cells. During the development of T1D, how mitochondrial function affects each T cell subset differently also remains inconclusive. Owing to their known role in intracellular calcium modulation, mitochondria are implicated in TCR signaling. Nevertheless, a major unmet need is to understand how mitochondrial dysfunction affects intracellular TCR signaling. Dysfunction of mitochondria could result from impaired calcium flux or ROS production, improper endoplasmic reticulum interactions, and/or aberrant downstream transcription factor regulation. These mechanisms of TCR and mitochondrial communication require further investigation to compile a comprehensive understanding of the intracellular pathways that lead to autoimmunity in T1D. Furthermore, studies are necessary to explore how mitochondria regulate the innate immune system and antigen presentation in the context of human T1D.
Footnotes
Acknowledgments
The authors gratefully acknowledge funding from the National Institutes of Health, R01 DK074656 (C.E.M.), P01 AI042288 (C.E.M.), and UC4 DK104194 (C.E.M.), as well as funding from the JDRF (J.C.) and American Diabetes Association (J.C.).
Author Contributions
J.C. and C.E.M. conceived the overall idea for this article, contributed to discussion, and wrote/reviewed/edited the article. G.A.F.B. and S.E.S. contributed to discussion and wrote/reviewed/edited the article.