
Abstract
Significance:
Stiffness of endothelial cells is closely linked to the function of the vasculature as it regulates the release of vasoactive substances such as nitric oxide (NO) and reactive oxygen species. The outer layer of endothelial cells, consisting of the glycocalyx above and the cortical zone beneath the plasma membrane, is a vulnerable compartment able to adapt its nanomechanical properties to any changes of forces exerted by the adjacent blood stream. Sustained stiffening of this layer contributes to the development of endothelial dysfunction and vascular pathologies.
Recent Advances:
The development of specific techniques to quantify the mechanical properties of cells enables the detailed investigation of the mechanistic link between structure and function of cells.
Critical Issues:
Challenging the mechanical stiffness of cells, for instance, by inflammatory mediators can lead to the development of endothelial dysfunction. Prevention of sustained stiffening of the outer layer of endothelial cells in turn improves endothelial function.
Future Directions:
The mechanical properties of cells can be used as critical marker and test system for the proper function of the vascular system. Pharmacological substances, which are able to improve endothelial nanomechanics and function, could take a new importance in the prevention and treatment of vascular diseases. Thus, detailed knowledge acquisition about the structure/function relationship of endothelial cells and the underlying signaling pathways should be promoted.
Introduction
Cellular biophysical parameters and their impact on cell and tissue function have long been underestimated. Over the last decade, however, biophysical techniques were established, which enable the quantification of mechanical properties of a variety of samples, ranging from molecules to tissues. During the last years, great efforts have been made in the development of adequate methods for a detailed examination of the nanomechanical properties of proteins, cells, and tissues. It became clear that specific mechanical properties of single molecules or higher structures such as cells determine their function, indicating a direct link between mechanics and function.
At the single-molecule level, the elasticity of a single protein, for instance, the muscle protein titin, has a direct impact on muscle function as an increased titin stiffness is linked to heart failure (72). On the larger scale, stiffness of macromolecular networks such as the extracellular matrix is sensed by cells and affects their migratory behavior (88) as well as differentiation. In several studies it could be shown that cell differentiation depends on the stiffness of their substrate (76, 79, 127). Even on the level of cell compartments, the mechanical properties influence the behavior and function of cells. Especially, endothelial mechanics is known to be linked to cell function, as several biochemical and biomechanical stimuli that affect endothelial cells are known to be linked to changes in elasticity [reviewed in Fels et al. (32)]. Here, especially two compartments of the vascular endothelium show a strong mechanics-to-function relationship, namely the endothelial cell cortex and the endothelial glycocalyx (eGC). This outer shell (i.e., the first 150–200 nm) of endothelial cells determines a variety of vascular functions in that it regulates the release of vasoactive substances and provides a dynamic scaffold or platform for the expression of particular proteins. Sustained stiffening of the outer layer of endothelial cells contributes to the development of endothelial dysfunction defined as Stiff Endothelial Cell Syndrome (SECS) (68).
Thus, the mechanical properties are strongly linked to the function of the cells. In this review, the link between endothelial nanomechanics and vascular function is discussed with special regard to endothelial dysfunction and vascular inflammation.
The Cell Cortex
The cell cortex comprises the plasma membrane and the underlying cytoskeleton (cortical cytoskeleton, CSK), as well as their associated proteins (105). Its average thickness is about a few 100 nm (32, 59, 96). As interface between intra- and extracellular space, the cortex can be described as autonomous functional compartment of cells. Biochemical signals are recognized by plasma membrane proteins and transmitted either outward or inward. The cortex regulates the uptake and the release of nutrients, ions, and signaling molecules. In addition, mechanical forces, generated during proliferation, growth, and migration, are triggered, sensed, and counteracted within the cell cortex. Hence, the cortex represents a signaling hub for all cells with a strong influence on cell physiology.
The CSK has been recognized as a key component of the cell cortex that determines its mechanical stiffness. It is built by the dynamic actomyosin web directly beneath the plasma membrane and its huge variety of associated proteins. Network-like actin filaments span the whole cortex and influence cell morphology via their membrane anchors such as the ERM proteins (29, 110). The actin filaments are organized in bundles spanning the cortex as well as a fine meshwork of single filaments (27, 52, 62, 98). Crosslinkers either connect actin filaments among themselves or to other cellular compartments. Via binding to integrins the CSK generates forces, facilitating movement during migration and force transition to intracellular compartments (18, 135). Myosin motor proteins generate lateral tension within this network, facilitating movement and mechanical integrity (39, 54, 116). F-actin nucleating Arp2/3 and formins, as well as filament stabilizing or severing factors such as gelsolin or cofilin, are responsible for a steady and dynamic turnover of cytoskeletal elements (103). Together with the crosslinkers and the motor proteins, this leads to a mechanical elastic and also rigid integrity of the cell cortex. The cortex is able to rapidly change its mechanical properties to react to functional challenges and physiological adaptations. It is important to mention that the mechanical flexibility of the endothelial cortex depends on the polymerization state of actin in that the shift from (depolymerized) G- to (polymerized) F-actin stiffens the cortical region, which is, among others, under the control of small GTPases (49, 105).
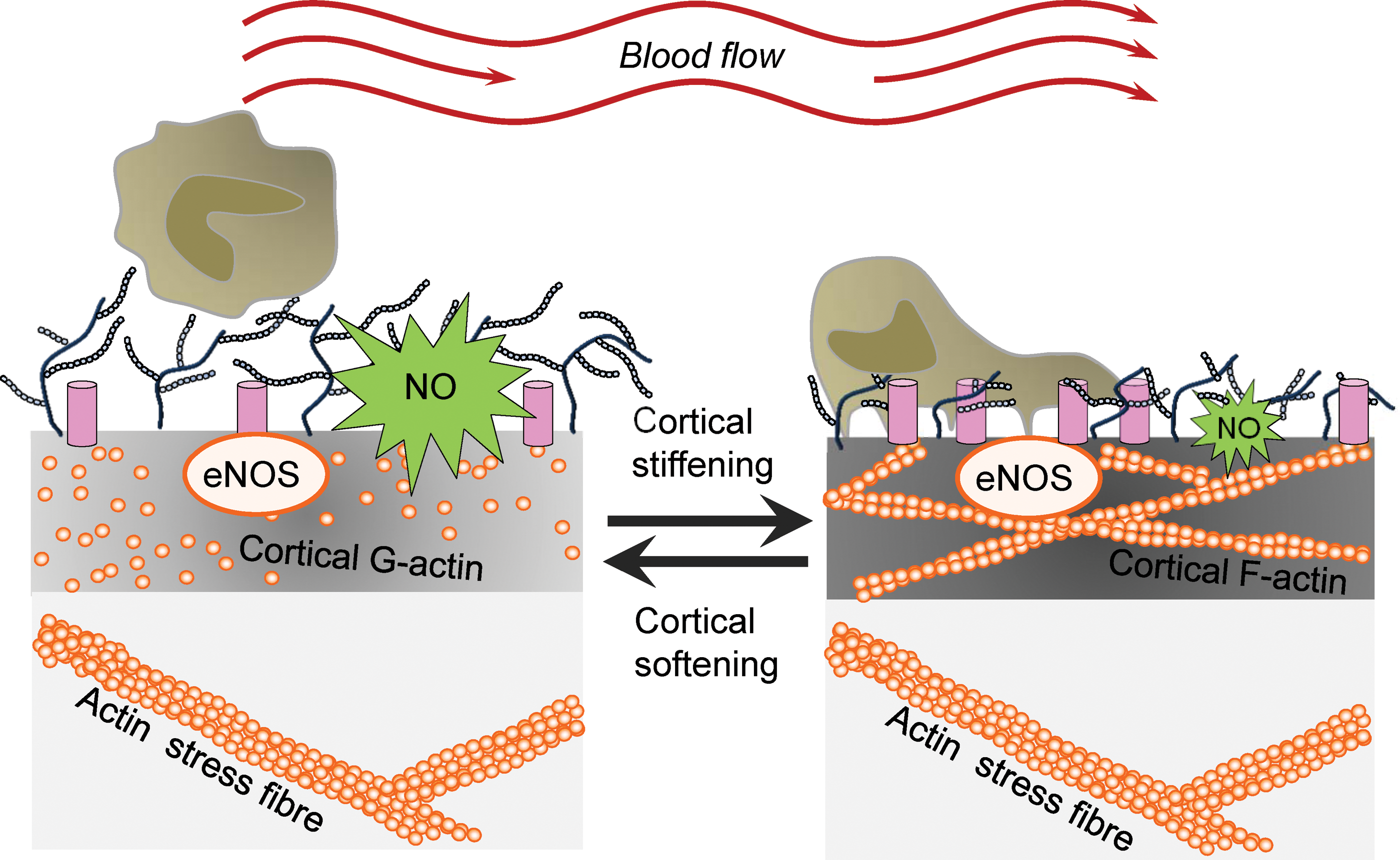
Ion channels are known to function as mechanosensors in that they are activated by mechanical stimuli and forces, which are converted into biochemical signals and transmitted into the interior of the cell. During the last years, a plethora of mechanosensitive ion channels have been identified, for example, transient receptor potential (TRP) channels, Piezo ion channels, DEG/ENaC/ASIC channels, and mechanosensitive potassium channels [for review, see Ranade et al. (107)]. However, in addition to their ability to sense forces acting on cell membranes, ion channels are recognized as mediators of the mechanical properties of the outer layer of cells. Recently, it could be shown that the presence of the endothelial ENaC (EnNaC) in the plasma membrane of endothelial cells stiffens the cortical region, which is crucial for the functional plasticity of the cell [for review, see Kusche et al. (66) and Warnock et al. (137)]. A mechanism is postulated, in which, on a specific stimulus, ENaC molecules are inserted into the plasma membrane. Physical and/or functional interaction of the channel with components of the CSK induces a shift from G- to F-actin leading to an increased rigidity of the endothelial cortex. In such a situation, the release of nitric oxide (NO) is decreased (see Cortical Stiffness and Endothelial Function section) (Fig. 1). Importantly, for epithelial ENaC, it was demonstrated that laminar shear stress increases the activity of the channel (2). The fact that ion channels are (i) regulated by mechanical forces and (ii) are able to sense and transduce mechanical forces indicates a regulatory feedback loop, which is poorly understood up to now.
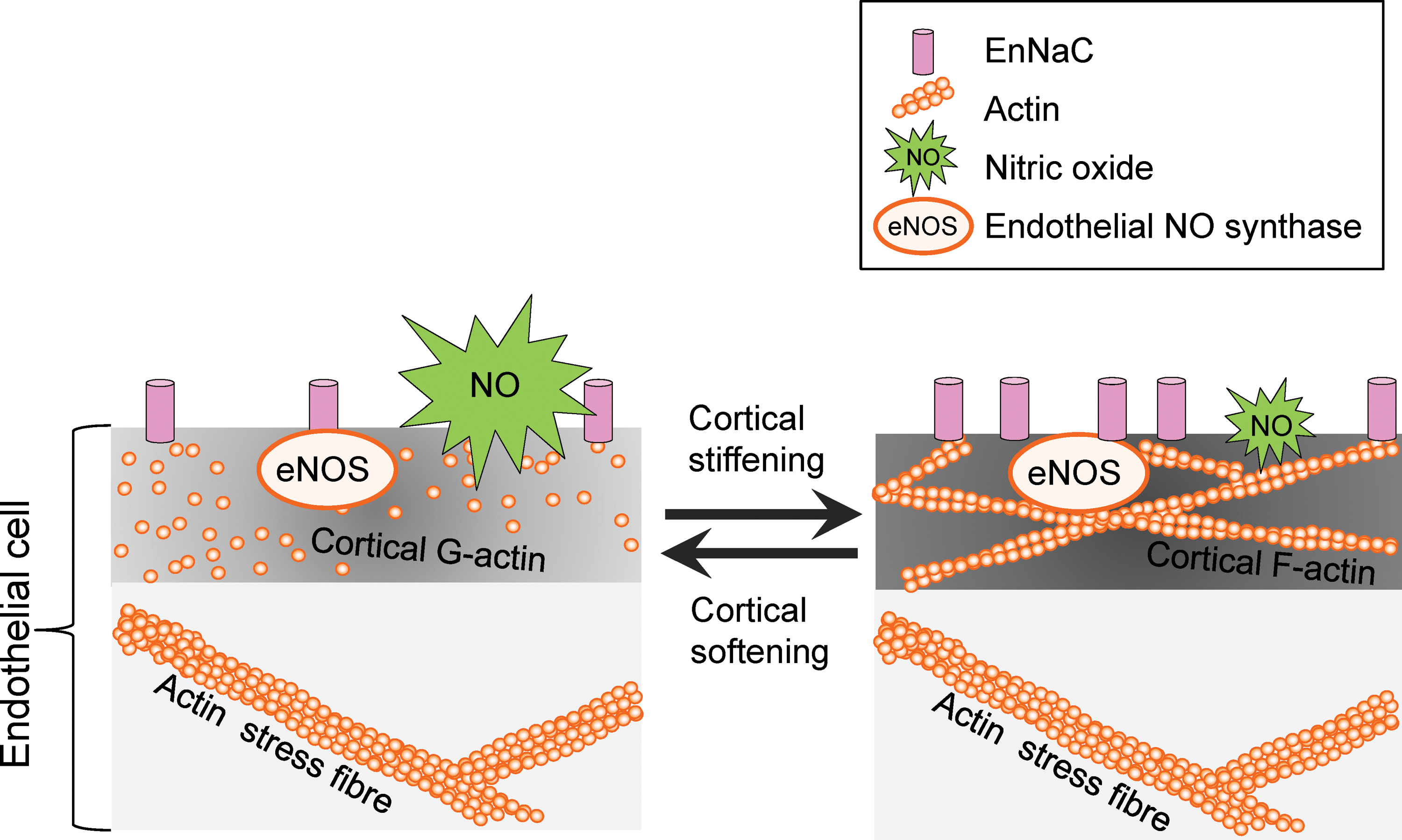
Cortical Stiffness and Endothelial Function
Endothelial function is defined by antithrombotic and anti-inflammatory activity, barrier function, and blood pressure regulation. All these characteristics are influenced by the mechanical properties, that is, the stiffness of the endothelium. Hereby, NO is the key endothelium-derived relaxing factor (also known as EDRF). Synthesized by the endothelial nitric oxide synthase (eNOS, NOS3), NO diffuses to the adjacent smooth muscle cells and activates the soluble guanylyl cyclase. The increased cytosolic level of cGMP induces a decrease in intracellular calcium within the smooth muscle cells, which results in vasorelaxation (47). In addition, NO directly acts within the endothelium as it is linked to vasoprotection via s-nitrosylation. It turned out that stiffness of the apical endothelial cortex presents a parameter, which directly correlates to eNOS function. Generally, a decrease in apical cortical stiffness (hereinafter referred to as cortical stiffness) is associated with an elevated NO release, whereas cortical stiffening diminishes eNOS activity. Thus, a shift from G- to F-actin relates to the degree of cortical stiffness and NO release (30, 31), intriguingly demonstrating the link between nanomechanics and function (Fig. 1).
The mechanical properties of endothelial cells are recognized as important targets for specific substances. In this context, it was shown that polyphenols such as resveratrol exhibit vasculoprotective properties as it decreases cortical stiffness (100) and induces NO release. Similarly, 1-methylnicotinamide chloride leads to cortical softening and increased NO bioavailability (57). However, factors that are associated with an activated and inflamed endothelium, for example, the mineralocorticoid hormone aldosterone or the tumor necrosis factor α (TNFα), induce cortical stiffening via stimulation of actin polymerization and simultaneously inhibit NO release (33, 128). Closely linked to the aldosterone-dependent effect, it has been shown that the plasma Na+ concentration determines cortical stiffness. An acutely increased Na+ intake, resulting in a temporarily increased plasma sodium level (115, 124), elevates cortical stiffness and inhibits NO release (50, 60, 70, 92). In contrast, physiological elevations in plasma potassium concentration (86) induce a plasma membrane electrical potential-dependent decrease in cortical stiffness and increase eNOS activity (14, 90). This decrease in stiffness is again linked to cortical actin dynamics as we have shown that high extracellular potassium decreases F-actin density in the endothelial cell cortex (31).
However, the molecular basis of stiffness-mediated eNOS regulation is still not known. Three hypotheses can be taken for consideration. First of all, a stiff cell will be less deformable, which might result in a decreased susceptibility of mechanosensitive ion channels to changes in blood flow. As NO release is regulated via the Ca2+/calmodulin pathway, shear stress-induced Ca2+ entry via mechanosensitive Ca2+ channels activates eNOS (34). The reduced mechanosensitivity due to cortical stiffening would result in a decreased Ca2+ influx on mechanical stimulation. The second hypothesis is based on the fact that eNOS is able to associate with actin. Interestingly, eNOS activity is increased if bound to G-actin compared with F-actin (123). Hence, as a soft cell is likely to exhibit higher G-actin levels compared with a stiff cell, cortical softening could lead to increased NO due to eNOS/G-actin association. This hypothesis is supported by the fact that a disruption of eNOS/actin interaction leads to decreased NO release (58) even if the cortical stiffness was reduced due to high extracellular potassium levels (31). Moreover, the activating effect of eNOS/G-actin interaction seems to be independent of intracellular calcium levels as it has been shown that certain stimuli (e.g., adenosine and salbutamol) induce G-actin-dependent eNOS activation without elevations of intracellular calcium (78). The third hypothesis includes cytoskeletal control of protein expression. It has been shown by Fang et al. that eNOS messenger RNA (mRNA) synthesis is regulated by the myocardin-related transcription factor A (MRTF-A), a cofactor of the serum response factor (SRF). In the cytosol, MRTF-A can bind to G-actin, which inhibits its translocation to the nucleus. On actin polymerization, MRTF dissociates from G-actin and translocates to the nucleus where it modulates expression via the SRF pathway (97). As it has been shown that eNOS expression is downregulated on translocation of MRTF-A (28), this opens another pathway of stiffening-mediated decrease in endothelial NO release.
In summary, these findings indicate that endothelial cortical stiffness could act as a physiological readout for endothelial function. A soft cortex generally goes along with an actin depolymerization and increased NO release, whereas stiffening includes filament formation and eNOS inhibition. This hypothesis is supported by the fact that a synthetic destabilization of the cortical actin cytoskeleton, for example, by low doses of the filament destabilizing mycotoxin cytochalasin D (CyD), leads to a decrease in cortical stiffness, resulting in increased NO release (Fig. 2A, B) (30, 128).
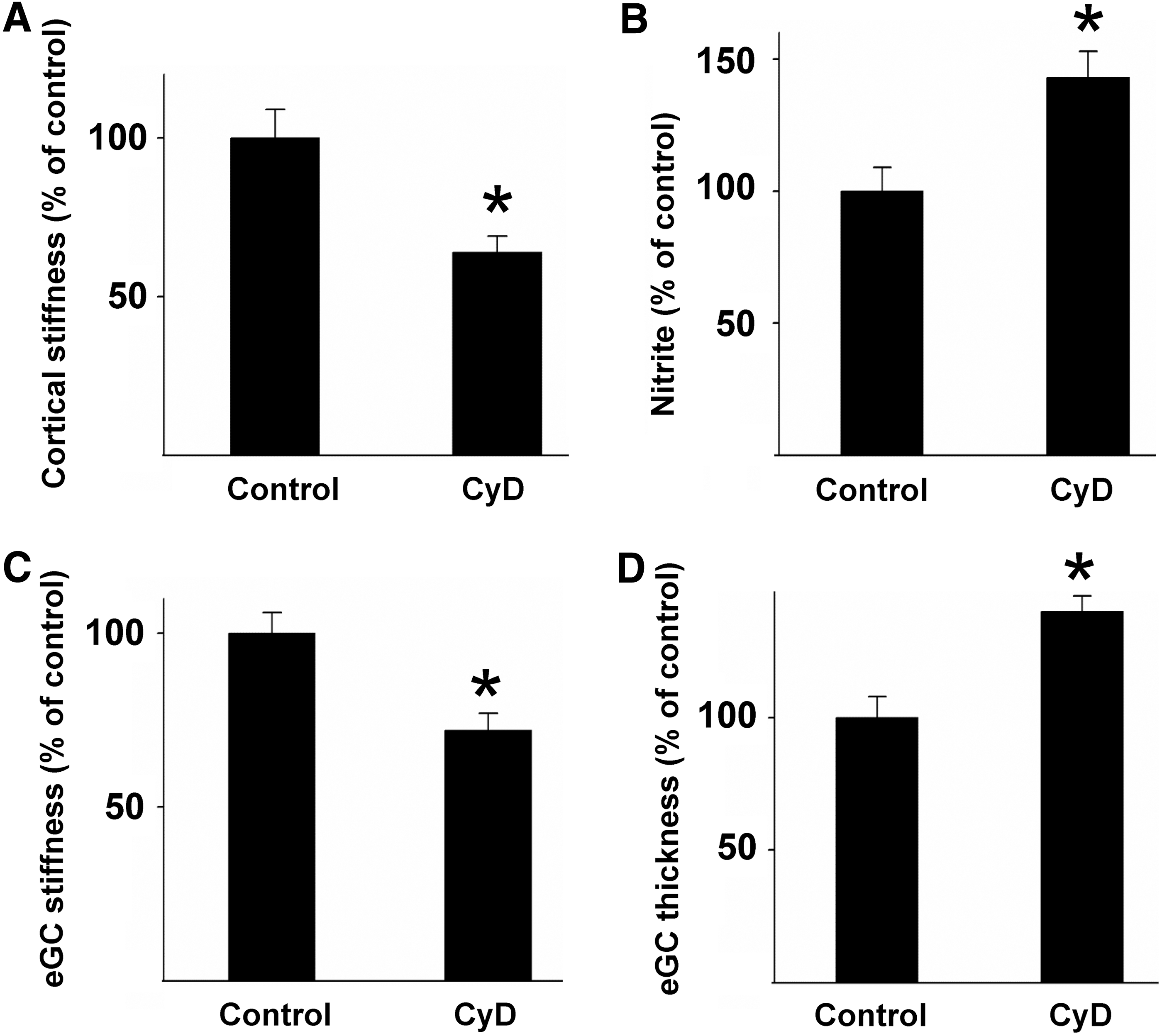
In addition to actin dynamics and the balance between filament formation and depolymerization, a number of other proteins could be identified, which play a crucial role in cortical plasticity. Myosin motor protein activity, for instance, induces stiffening of the endothelium, which is closely linked to its barrier function. Here, the Rho kinase 1 (Rock1) plays a central role in the regulation of stiffness-associated endothelial function. Rock1 increases lateral tension via actin polymerization and myosin light chain kinase (MLCK) activation (45, 143), increasing contractility. Elevated myosin activity in turn results in cortical stiffening (54, 116). The increased lateral tension leads to a barrier breakdown and an increased permeability of the endothelial monolayer (61, 74, 144). In addition to its effects on endothelial mechanics and barrier function, Rho kinase inhibits eNOS activity (125). Although stiffening and eNOS inhibition are regulated via distinct pathways, the effect of Rho kinase on endothelial mechanics correlates with its impact on endothelial blood pressure regulation.
One factor that controls endothelial function, even under physiological conditions, is the Nox family of NADPH oxidases in that the Nox-derived intermediates scavenge NO (67) and thus determine the availability of NO. For many years, it is known that the Nox NADPH oxidases represent the major source of reactive oxygen species (ROS) in diverse systems. In particular, in the cardiovascular system, Nox-derived ROS are important modulators of stress response leading to cellular activation, hypertrophy, and inflammation but, as mentioned above, also limit NO availability [for review, see Brandes (10) and Brandes et al. (12)].
Up to now, seven isoforms of the Nox family of NADPH oxidases could be identified (Nox1–5 and Duox1/2), which produce different types of ROS. Nox1, Nox2, Nox3, and Nox5 mainly produce •O2 −, whereas H2O2 is the prominent product of Nox4 (11, 12). In vascular cells, the NADPH oxidase isoforms Nox1, Nox2, Nox4, and Nox5 are mainly expressed, which differ in their activity, response to stimuli, and as mentioned in the type of ROS released (11). Interestingly, Nox-derived signaling in the vascular system can be both detrimental and protective. Physiological amounts of H2O2 maintain endothelial integrity and stimulate anti-inflammatory systems (118a, 118b); overproduction of H2O2, however, is shown to be proinflammatory and induces oxidative stress and endothelial dysfunction (139a, 141a).
Overproduction of superoxide anions (•O2 −) provides a proinflammatory stimulus, mediates uncoupling of eNOS, and thus triggers the development of endothelial dysfunction (39a, 118a).
Mechanical forces such as shear stress stretch or pressure stimulate the cellular production of ROS. Furthermore, ROS determine cellular mechanics in that it is involved in the remodeling of the cytoskeleton. This can occur either indirectly by modification of proteins and enzymes that regulate actin dynamics or directly by oxidation of actin filaments. H2O2, for instance, decreases the rate of actin polymerization (20). ROS and reactive nitrogen species are also shown to activate Rho GTPases, including RhoA, Rac, and Cdc42 inducing stress fiber formation (1, 44). Using the atomic force microscope (AFM) as nanoindentation tool, Sun et al. demonstrated that H2O2 decreases cell stiffness and reduced RhoA expression (126). In the end, mechanical stimulation of cells, however, modulates ROS production leading either to endothelial function or dysfunction strongly depending on the quality of the applied physical forces [for review, see Brandes et al. (13)]. However, the exact relationship between cortical stiffness, the balance between NO and ROS production, and endothelial function has to be elucidated.
Cortical Stiffness and Endothelial Function During Aging
Aging of the vascular system is a major risk factor for cardiovascular disease and mostly attributed to a combination of genetic predisposition and morphological changes of the cell finally leading to organ damage (4, 5). Recently, in a clinical study, a correlation was found between endothelial stiffness and the overall arterial stiffness (69), which relates to the fact that changes in the mechanical properties of endothelial cells could be linked to disturbed endothelial function. In particular, it was found that the EnNaC abundance in the plasma membrane of endothelial cells is augmented during the process of aging, which could be prevented by low doses of the mineralocorticoid hormone receptor spironolactone (24). In parallel to an accumulation of EnNaC in the plasma membrane, an increased cortical stiffness and reduced release of NO were found (24, 94). This might be due to either changes in protein expression or structural reconstructions of proteins and cellular components. In the first case, a concept was developed in which “aging” is able to derail specific signaling pathways leading to altered metabolism and protein expression pattern. In the second case, cytoskeletal proteins such as actin, myosin, or involved linker proteins are structurally changed, which in turn directly influences the mechanical properties of the endothelial cortex (102). However, this demonstrates a time-dependent manifestation of SECS and endothelial dysfunction, which might facilitate the development of cardiovascular pathologies.
The Endothelial Glycocalyx
The eGC is a negatively charged, carbohydrate-rich gel-like mesh of membranous glycoproteins, proteoglycans, glycosaminoglycans (GAGs), and associated plasma proteins covering the luminal surface of the endothelium along the entire vascular tree (138). The GAGs are composed of heparan sulfate (HS, 50–90%), chondroitin sulfate (CS), and hyaluronic acid (HA) (109, 112, 131). Transmembrane syndecans and the membrane-bound glypicans are the major protein core families found on the endothelial plasma membrane. Syndecans are attached to GAG and their cytoplasmic tails associate with the cytoskeleton through linker molecules such as ezrin, tubulin, syntenin, syndesmos, dynamin, and α-actinin and are thus able to distribute force throughout the cell (138).
The functional thickness of the eGC was long in debate. For many years the studies revealed an eGC thickness less than 100 nm. During the last years, new techniques developed to more precisely determine the thickness and accordingly the height of the eGC. By stabilizing the anionic carbohydrate structures of the eGC, a thickness of up to 0.5 μm was demonstrated in rat left ventricular myocardial capillaries (132). By direct in vivo measurements of the eGC in hamster cremaster muscle, Vink and Duling provided evidence for a eGC thickness of 0.4–0.5 μm (133), which was supported by recent data derived from intravital microscopy (37) [for comprehensive review about eGC structure, see Dane et al. (21) and Fu and Tarbell (36)].
Under physiological conditions, the structure of the eGC layer is fairly stable but subject to a permanent dynamic balance between biosynthesis of new GAGs and shear-dependent removal of existing constituents. Thus, the eGC is neither an inflexible nor a homogeneous structure due to various electrostatic and molecular interactions between its constituents (140). According to its crucial role as vasculoprotective layer, the eGC is structurally degraded during inflammation. However, Potter et al. showed that a period of 5–7 days is required for the eGC to make a full recovery after deterioration with proinflammatory substances or enzymatic degradation of the eGC (104).
The process of eGC deterioration is mainly maintained by proteases, where heparanase is specific for cleaving heparan sulfate side chains or hyaluronidase for removing hyaluronan from the eGC (83) or TNFα (84). In vivo several pathophysiological situations have been associated with structural and functional derangement of the eGC, for example, postischemic organ damage, sepsis, inflammation, renal disease, and atherosclerosis [for review, see Becker et al. (7)]. However, protection of the eGC against damage after ischemic insult or TNFα was found to be mediated by antithrombin (42), hydrocortisone (16), or albumin (6). After shedding, the eGC can be completely recovered within a few hours. Recently, it was demonstrated that sphingosine 1-phosphate induced eGC recovery via the phosphoinositide 3-kinase (PI3K) pathway (146, 147).
As already described for the endothelial cortex, stiffness is a mechanical property that reflects the physiological function of a specific structure. Because of the complex composition of the eGC, it can be found in different conditions regarding mechanical stiffness and functional thickness. During the last years, efforts have been made to quantify and analyze the nanomechanical properties of the eGC. Although the eGC seems to be a fragile and delicate structure, it turned out that changes of the eGC nanomechanics have immense effects on the vascular function in that a soft and upright eGC is a prerequisite for proper endothelial function and protects against inflammatory processes and bacterial invasion (38, 41, 115, 117, 140). Three different eGC conformations could be identified: (i) soft and upright, (ii), soft and flat, and (iii) stiff and flat (91, 115, 140) (Table 1 and Fig. 3). Since the structure is tightly linked to the function of the eGC, each conformation reflects a specific functional condition. A soft and upright eGC can be seen as the physiologically intact conformation where the eGC perfectly fulfils its role as vasoprotective gel-like structure and mechanosensor on top of endothelial cells. During inflammatory processes, the presence of cytokines, for example, TNF-α, damages the eGC leading to a reduced thickness and stiffness indicating a shedding of the eGC (140). Enzymatic degradation also leads to a shedded eGC. In contrast, high plasma Na+ concentrations dramatically stiffen the eGC while its functional thickness is reduced, which is likely due to a collapse of the structure. The different conformations of the eGC are depicted in Figure 3 and summarized in Table 1.
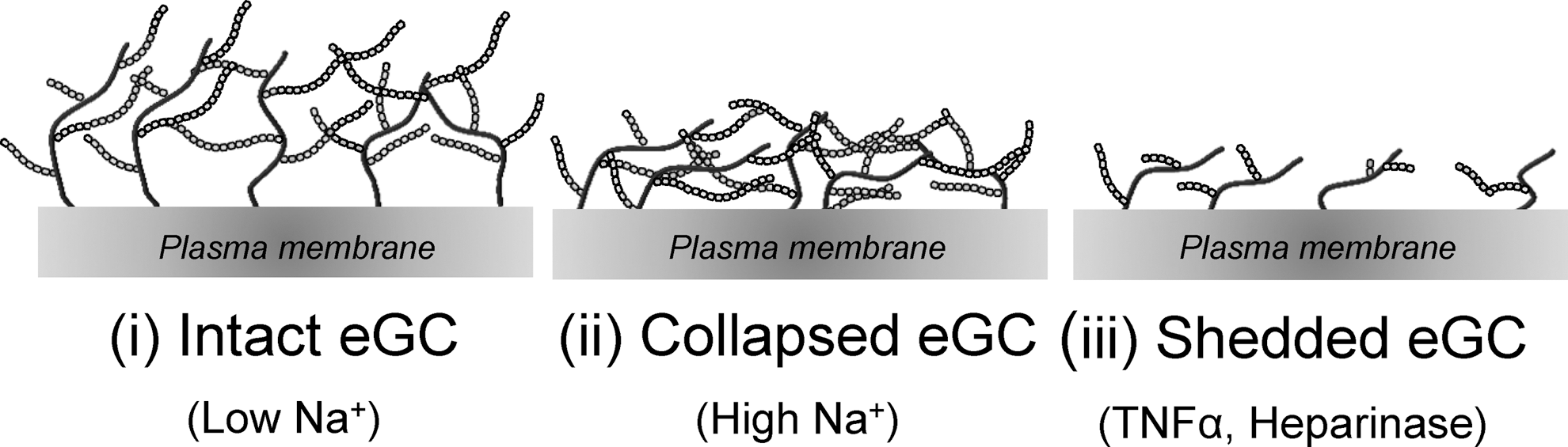
Effects of Na+, Tumor Necrosis Factor α, and Heparanase on the Shape of Endothelial Glycocalyx
Oberleithner et al. (91).
Wiesinger et al. (140).
eGC, endothelial glycocalyx; TNFα, tumor necrosis factor α.
Recently, a mechanism was described resulting in “swelling” of the eGC, which can be attributed to reorganization events (depolymerization) of the cortical actin (100).
However, despite its fundamental role in regulating vascular integrity and endothelial functions, visualization and quantification of the eGC are rather challenging. Recently, the AFM was used to measure the elastic response of bovine lung microvascular endothelial cells (87). It was found that the glycocalyx stiffness and thickness changed after removal of GAGs by specific enzymes. By using a microinterferometric technique based on reflectance interference contrast microscopy (RICM), the same group reported that enzymatic digestion of HA and nonspecific glycocalyx digestion with pronase increased the mean effective stiffness of the glycocalyx, implying that HA chains act as a cushioning layer to distribute applied forces to the glycocalyx structure (51).
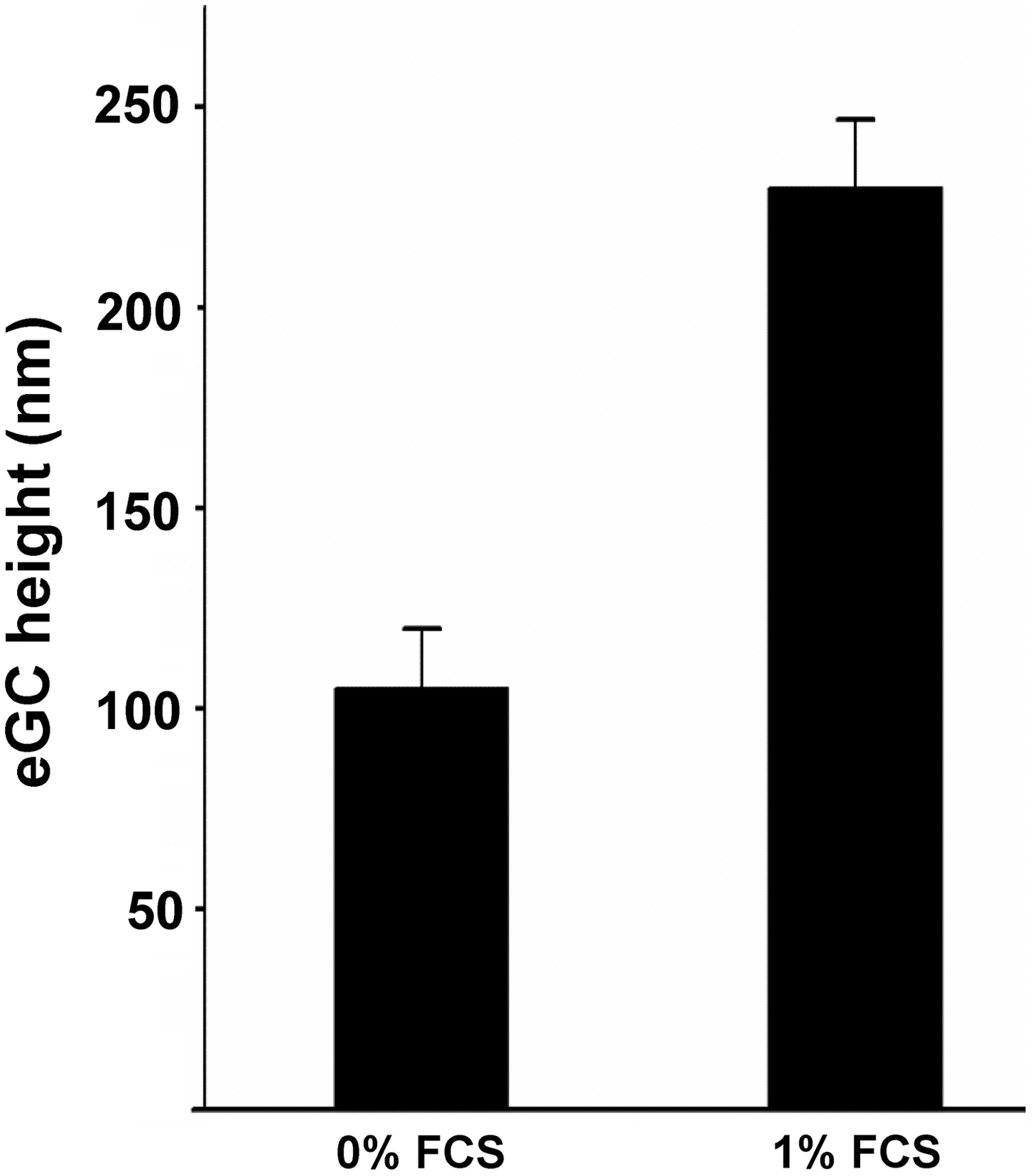
With a combination of immunofluorescence microscopy and AFM-mediated nanoindentation, it was shown that heparanase treatment leads to a reduction in eGC stiffness by 47% as well as a reduction in the HS abundance on the cell surface by 34% indicating a correlation between eGC stiffness and the amount of stained HS (99). The mechanical properties of the eGC also depend on blood proteins and specifically on the albumin concentration in the plasma. Job et al. exposed the eGC to 0.1%, 1.0%, and 4.0% albumin. The RICM images indicated that eGC thickness and its compliance increase at higher albumin concentrations (51). Recently, we could show with AFM nanoindentation measurements that a concentration of at 0.1% fetal calf serum (FCS) is required for a proper mechanical nanostructure of the eGC. The functional thickness (height) is increased with application of 0.1% FCS compared with the absence of FCS (Fig. 4).
The mechanical properties of both endothelial cortex and eGC are important for proper vascular function. Apparently, the mechanical properties of the endothelial cortex determine the mechanical stiffness and functional thickness of the eGC. Application of low doses of CyD softens the endothelial cortex. In fact, CyD-induced cortical softening is accompanied by a decrease in eGC stiffness and an increase in eGC thickness (100). This is explained by the fact that the eGC is anchored to proteins of the cortical web (3). This functional and structural connection leads to a mutual interaction in that the depolymerization of cortical actin leads to changes in the anchoring of the eGC. This in turn reorganizes its structure and affects its mechanical properties. Under these conditions, the release of NO is increased, which might be explained by (i) the facilitated shear stress sensing of the thicker eGC and (ii) the better deformability of the softer cortex, which stimulates the eNOS underneath the plasma membrane (Fig. 2A–D). The functional interaction between eGC and cortex becomes relevant by detailed observation of a specific situation (e.g., high plasma Na+ concentrations or inflammation) with increased mechanical stiffness of both endothelial cortex and eGC.
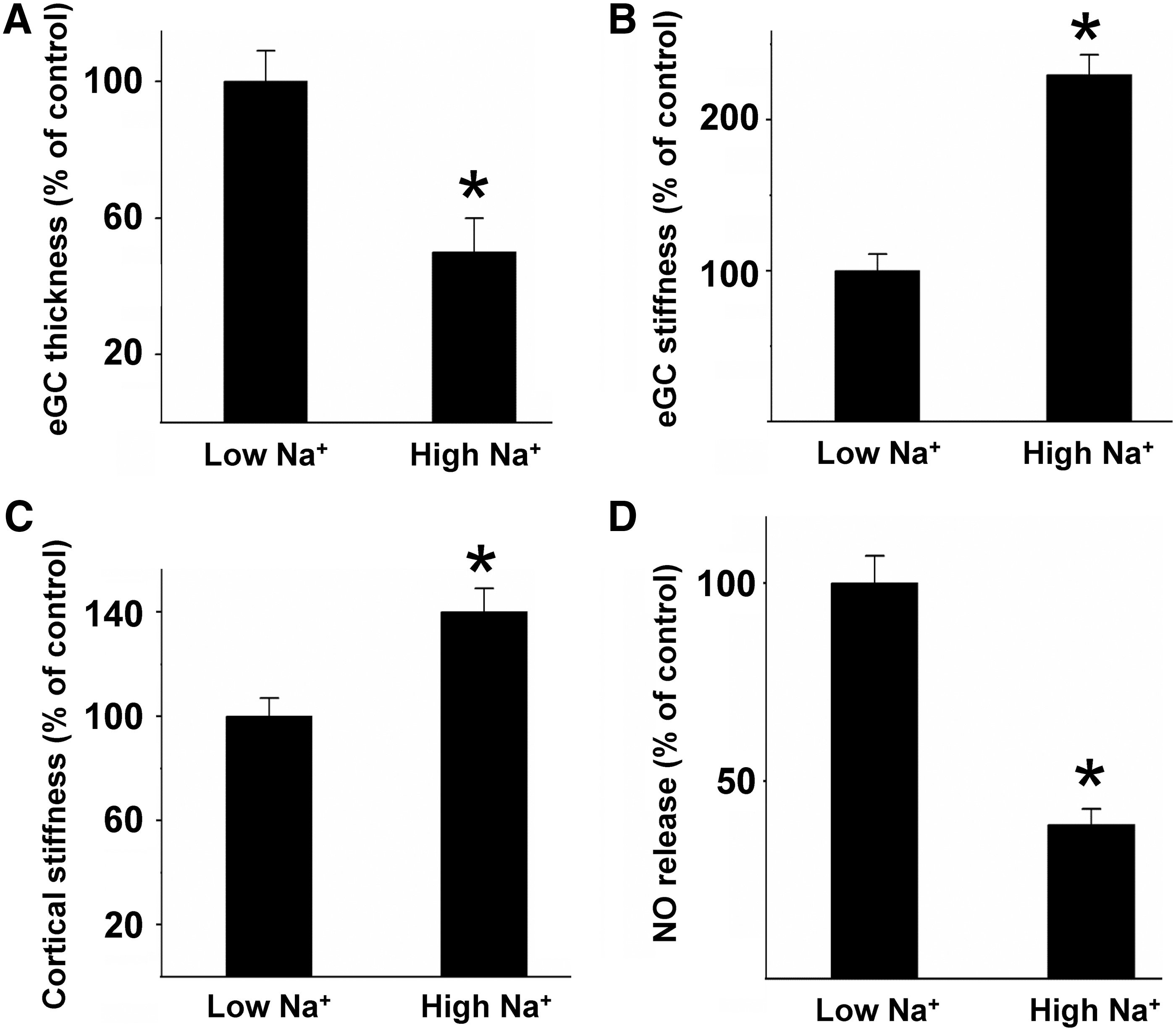
In Figure 5, an example is given for the link between endothelial nanomechanics and function. Challenge of the vascular endothelium with Na+ concentrations in the supraphysiological range (150 mM), as present after acute oral Na+ load (115, 124), damages the eGC in that the functional thickness/height is decreased by 50% (Fig. 5A), while the mechanical stiffness is increased by 130% (Fig. 5B). In other words, Na+ load makes the eGC rigid and flat, negatively influencing the barrier function of the eGC. Recent data show that under high Na+ conditions, the adhesion of monocytes is facilitated, promoting the development of vascular inflammation (115) (see paragraph below). In parallel, high Na+ load stiffens the endothelial cortex and decreases the release of NO (Fig. 5C, D). This example indicates that (i) the nanomechanical properties of the eGC and cortex sensitively react to physiological challenges and (ii) that changes of the nanomechanical properties are directly linked to vascular function. Thus, the inverse correlation between mechanical plasticity and NO secretion is physiologically relevant since a reduced release of NO due to endothelial stiffening causes impaired vasodilation of blood vessels, which promotes the development of endothelial dysfunction, arterial hypertension, and inflammatory processes (30, 71).
As mentioned above, the application of polyphenols such as resveratrol induces a softening of the endothelial cortex. AFM-based nanoindentation measurements using ex vivo endothelial cells derived from mouse aortae reveal that polyphenols also act on the eGC making it soft and thick (99). Since under these conditions the amount of HS molecules at the surface of endothelial cells was not changed, it is postulated that the nature of eGC modulation by polyphenols is a result of conformational alterations, named “swelling.” This might at least partially explain the anti-inflammatory properties of, for example, hawthorn extracts. In addition, it is possible that a softer and thicker glycocalyx results in a better detection of shear stress by the endothelium and thus higher NO synthesis and release (99). However, it can speculated that the nanomechanical properties of the eGC influence its biochemistry, for example, by regulating the number of binding sites for different blood-borne substances making it a crucial regulator and protector of vessel function.
The Endothelial Glycocalyx as Mechanotransducer and Mediator of NO Release
Endothelial cells are constantly subjected to the forces of streaming blood. This shear stress induces cellular responses, including morphological adaptations of the cells such as orientation in flow direction, the regulation of gene expression, and production of vasoactive substances such as NO and ROS. All these cellular responses are mediated by specific intracellular signaling pathways. However, knowledge about the cellular structures that sense and transduce shear forces is rare (130). Mechanotransduction involves both sensing of shear forces at the luminal side of endothelial cells and at the opposite site where the forces are transmitted toward the cytoskeleton. A number of transmembrane and intracellular mechanosensors have been identified over the past years, including ion channels, receptor tyrosine kinases, G protein-coupled receptors, platelet endothelial cell adhesion molecule-1 (PECAM-1) and its associated intercellular junction complex, and integrins and their basal adhesion complex [reviewed in Tarbell et al. (130)]. Especially to be emphasized is the role of eGC as mechanotransducer and mediator of NO release (129). NO is one of the most important vasoprotective molecules produced in a biphasic manner. The first rapid and acute NO release depends on blood flow (shear stress) and Ca2+, whereas the chronic NO production is independent of Ca2+ (15, 35, 64). Morphologically, the eGC is linked to proteins of the cortical web beneath the plasma membrane (139) and thus is able to transmit biochemical and biomechanical signals from the intravascular compartment toward endothelial cells making it a critical interface between the blood and vascular wall (Fig. 6). In a number of studies, it could be shown that degradation of the eGC inhibits the flow-induced NO production, suggesting that the eGC plays a key role in mechanosensing and mechanotransduction in vessels (35, 95, 145). The underlying mechanism of these observations could be that drag forces acting on the cell surface are transduced via the eGC core protein glypican to the submembranous caveolae, which triggers eNOS activation and subsequent production of NO (26).
The production of superoxide is found to increase in blood vessels under elevated shear stress, indicating that superoxide may be involved in endothelial mechanotransduction. It could be shown that HS proteoglycans and sialic acid act as important mediators for vascular oxidative stress. Removal of HS impacts the nearby superoxide dismutase (SOD), which in turn affects the superoxide balance and thus the bioavailability of NO (65).
Recently, Dragovich et al. could demonstrate that horizontal and vertical stimulation of the eGC causes reorganization of GAGs, proteoglycans, and glycoproteins, which is transduced to the cytoskeleton. The enhanced tension then may activate mechanically sensitive Ca2+ channels, such as TRP channels. The following TRP-mediated Ca2+ influx into the cell leads to vasorelaxation and stretch-induced NO release (8, 23). Unpublished data of our laboratory reveal that Na+ influx via the mechanosensitive EnNaC might also contribute to this mechanism, in that a decreased activity of EnNaC coincides with an increase in NO production. These data lead to a model where the eGC acts as an important sensor and transducer of mechanical forces at the surface of cells. As a consequence, NO is rapidly released and likely related to the action of ion channels or more specifically the influx of Ca2+ and Na+ via mechanosensitive cation channels. However, one parameter that is important for the transduction of shear forces into the cell is the mechanical stiffness of eGC.
eGC Stiffness and Inflammation
During the last years, eGC was recognized as a crucial vasculoprotective and anti-inflammatory nanobarrier on the apical surface of endothelial cells. It has both proadhesive and antiadhesive functions and thus plays a crucial role during inflammatory processes, especially during leukocyte recruitment (77, 80, 148). Due to its position on the surface of endothelial cells, the eGC serves as a “firewall” by mediating flow-induced shear stress and regulation of leukocyte/endothelium interactions. Since the eGC can reach a height of about 0.5–1 μm, leukocytes are “tip-toeing” with their cytoskeletal protrusions on the eGC and can barely reach the adhesion molecules at the endothelial surface—unless the barrier is compromised. Thus, shedding of the eGC appears to be required for leukocyte adherence to the vessel wall, because, under normal conditions, leukocytes are supposed to be shielded from contact with their adhesion molecules by eGC (19, 73, 80) (Fig. 6).
Furthermore, the eGC seems to play a role as protector of endothelial cells against damage by various mediators of oxidative stress. Under physiological conditions, the eGC contributes to its vasculoprotective effect by docking major enzymatic systems, such as the extracellular SOD bound to HS proteoglycans, contributing to a reduction in oxidative stress by quenching oxygen radicals and maintaining NO bioavailability (40, 85, 108). The eGC can also bind cytokines, which have profound effects on eGC compound synthesis and inflammatory processes by attenuating the binding of cytokines to cell surface receptors (77).
Release of inflammatory mediators induced by eGC degradation initiates the accessibility of leukocytes to adhesion molecules (17, 56). Kubes et al. demonstrated that endothelium-derived NO modulates leukocyte adhesion (63). NO seems to suppress the expression of VCAM-1, ICAM-1, and E-selectin in response to proinflammatory cytokines, indicating an anti-inflammatory role of NO.
In observational studies in critically ill patients with sepsis, it was shown that plasma levels of shed glycocalyx constituents correlate with disease severity and mortality (82, 111, 115). Both systemic and local inflammatory responses such as diabetes, atherosclerosis, surgical ischemia/reperfusion injury, and sepsis lead to a rapid loss of glycocalyx functions (6, 17, 56). As discussed in The Endothelial Glycocalyx section, the functions of the glycocalyx are dependent on an intact structure. Perturbation of the structure can range from deterioration to fundamental destruction of the glycocalyx layer (Table 1 and Fig. 3).
By applying AFM-based nanoindentation, Wiesinger et al. demonstrated eGC deterioration in septic mice, which again identifies the eGC as an important structure during inflammatory processes. Corresponding in vitro experiments reveal that nanomechanical changes, that is, decreased stiffness and reduced thickness induced by TNF-α and thrombin, respectively, are identical to those seen after lipopolysaccharide exposure in vivo, suggesting that changes of the eGC nanomechanics are a common hallmark of all inflammatory processes (140).
The stiffness of eGC determines the adhesion and transmigration of leukocytes. As mentioned before, eGC thereby acts as vasculoprotective “firewall.” In areas with increased stiffness of the extracellular matrix and thus the vascular wall, such as atherosclerotic plaques, leukocyte extravasation is enhanced [reviewed in Doring et al. (22)]. In an elegant study by Schaefer et al., endothelial α-actinin-4 could be identified as a key regulator of endothelial cell stiffness and of ICAM-1-mediated neutrophil transmigration. Since endothelial cells lining atherosclerotic plaques show elevated levels of α-actinin-4, endothelial cell stiffness was recognized as an important regulator of the adhesion and transmigration of neutrophils (113, 114). Adhesion of leukocytes was shown to occur predominantly in endothelial areas with increased stiffness, while transmigration seems to be facilitated in softer regions (113). This indicates that inflammatory processes depend on the nanomechanical properties of endothelial cells. Mechanical forces induced by age- and inflammation-mediated extracellular matrix stiffening promote endothelial stiffness, which impairs barrier function and stimulates leukocyte transmigration (48, 113).
As shown for human neutrophils, adhesion increases leukocyte stiffening which, in turn, further promotes adhesion and transmigration, indicating a positive feedback loop (136). Indeed, pharmacological inhibition of F-actin dynamics in human neutrophils, impairing leukocyte stiffening, reduces leukocyte transmigration across inflamed human umbilical vein endothelial cells (122). These events are likely to be mediated by the RhoA–ROCK–myosin pathway, which results in localized actomyosin-based contractile forces. These forces, dependent on endothelial stiffness, allow crawling and transmigrating leukocytes to retract their uropod for efficient transmigration (113, 142). Furthermore, Rho kinase-induced lateral tension correlates with an increased transendothelial migration of leukocytes (43, 118, 121).
High Na+ Induces Mechanical Stiffness and Vascular Inflammation
In addition to its function as a transmitter of signals, the eGC is known to function as an Na+ buffering system (91, 119). Importantly, the capacity of eGC to buffer Na+ is diminished after exposure to high Na+ concentrations. This phenomenon can be explained, in part, by the fact that Na+ strongly interacts with negatively charged side chains of proteins and proteoglycans within the eGC (134). Changes in the nanomechanical properties (i.e., stiffness and functional thickness) of the eGC, however, alter its function (32, 137). By studying eGC nanomechanics, it could be shown that, in the presence of aldosterone, already a small increase in Na+ concentration causes a collapse of the eGC (91). A chronic increase in extracellular Na+ concentration beyond 140 mM “neutralizes” the negative charges of the eGC leading to collapse of the structure (91). Thus, the eGC is highly sensitive to salt, and Na+-induced collapse of eGC results in breakdown of its barrier function. In addition, at high Na+ conditions, the physical interaction forces between eGC and red blood cells (RBCs) are augmented, which predisposes blood vessels to thrombotic events (89, 93).
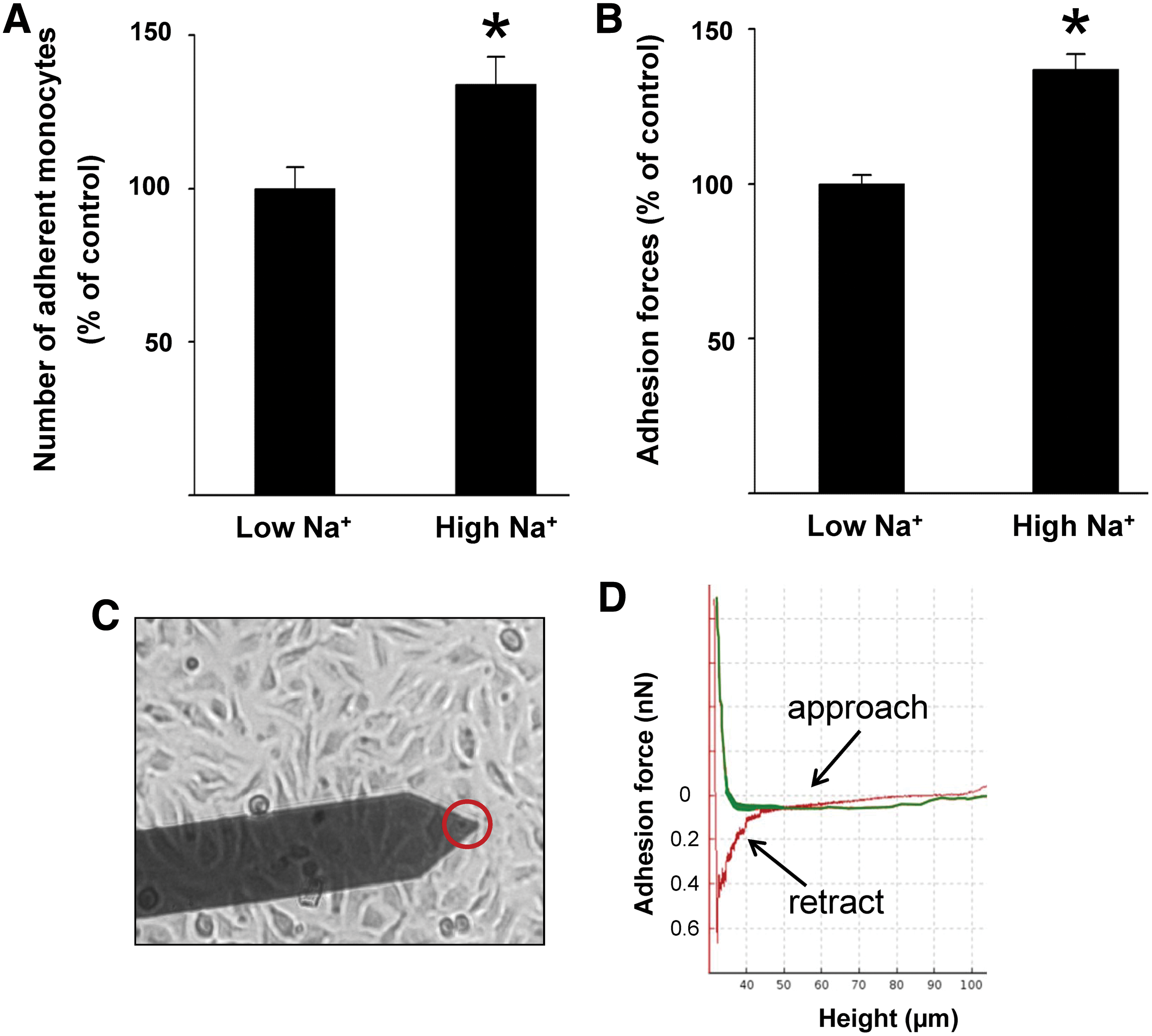
Recently, high extracellular Na+ concentrations were considered to be a risk factor for inflammatory pathologies and for the development of autoimmune diseases (9, 46, 53, 75). Wild et al. showed that the number of adherent human monocytes on endothelial cells is augmented under high Na+ conditions (141). In particular, eGC seems to play a crucial role during Na+-dependent inflammation. It could be demonstrated that high extracellular Na+ concentrations activate endothelial cells thus creating proinflammatory conditions. This leads to (i) stiffening of the endothelial cortex and reduced NO release (92), (ii) endothelial-derived production of proinflammatory cytokines (115), and (iii) damage of the eGC (91). In such a scenario, the adhesion of leukocytes is increased, which is most likely due to dramatic changes of the nanomechanical properties of eGC and thus facilitated accessibility to adhesion molecules in the plasma membranes of inflamed endothelial cells. As shown in Figure 7A, incubation of endothelial cells in media with high extracellular Na+ concentrations increases the number of adherent monocytes on endothelial cells, which can be seen as the first step toward vascular inflammation. In addition, the adhesion forces between cells can be measured by using specific AFM techniques (25, 93, 106, 120). This nanotechnical approach allows the quantification of adhesion forces between monocyte and the endothelial surface and thus the measurement of Na+-dependent cell/cell interaction forces. As shown in Figure 7B, a significantly larger force (+37%) is necessary to detach a human monocyte from the endothelial cell surface under high Na+ conditions compared with low Na+ conditions (115) (Fig. 7C, D).
By comparing the effects of TNFα and heparanase, both known to induce a shedding of eGC, it is concluded that high Na+ leads to conformational changes and collapse of the eGC (Table 1 and Fig. 3). Since the effect of Na+ on eGC contradicts that induced by TNFα and heparanase, the question is raised whether it is driven by different molecular mechanisms. High Na+ conditions per se, besides contributing to the development of endothelial dysfunction, activate vascular endothelial cells and thus create proinflammatory conditions. In this context, it can be stated that endothelial nanomechanics significantly contributes to proper endothelial function. Hence, Na+- induced changes in the nanomechanical properties of eGC and cortex contribute to the development of inflammatory processes, making them both a predictor and therapeutic target for vascular pathologies.
Conclusions
Endothelial cells are exposed to a number of different forces generated by the streaming blood, membrane tension, and traction forces acting on the vascular wall. The outer layer of the cells (cortex and eGC) senses the mechanical forces and adapts its mechanical properties in response to changes in its environment. This controls a biochemical and biophysical signaling cascade, including the release of vasoactive substances (e.g., NO and ROS). Thus, the interplay between structure and function of cortex and eGC suggests that the mechanical properties can be seen as a direct marker and readout for the integrity of the vasculature. Recently, in a clinical study, a correlation between the cortical stiffness of endothelial cells ex vivo derived from patient's arteries and the overall arterial stiffness in vivo has been shown (69). This strongly suggests that endothelium-dependent dysregulation of the vascular tone could be of relevance for the development of vascular-related diseases.
Footnotes
Acknowledgments
The authors thank Prof. Hans Oberleithner (Institute of Physiology II, University of Münster) for assistance in critically reading the article. They are grateful to Dr. Alexander Lukasz (Department of Nephrology, University of Münster) for providing unpublished experimental data used in ![]() . This work was supported by grants from the Deutsche Forschungsgemeinschaft (KU 1496/7-1 and KU 1496/7-3) and by the Centre of Excellence (Cells in Motion; CIM), University of Münster. The authors thank COST Action BM1301 for supporting networking activities.
. This work was supported by grants from the Deutsche Forschungsgemeinschaft (KU 1496/7-1 and KU 1496/7-3) and by the Centre of Excellence (Cells in Motion; CIM), University of Münster. The authors thank COST Action BM1301 for supporting networking activities.