
Abstract
Significance:
Once considered to be mere by-products of metabolism, reactive oxygen, nitrogen and sulfur species are now recognized to play important roles in diverse cellular processes such as response to pathogens and regulation of cellular differentiation. It is becoming increasingly evident that redox imbalance can impact several signaling pathways. For instance, disturbances of redox regulation in the brain mediate neurodegeneration and alter normal cytoprotective responses to stress. Very often small disturbances in redox signaling processes, which are reversible, precede damage in neurodegeneration.
Recent Advances:
The identification of redox-regulated processes, such as regulation of biochemical pathways involved in the maintenance of redox homeostasis in the brain has provided deeper insights into mechanisms of neuroprotection and neurodegeneration. Recent studies have also identified several post-translational modifications involving reactive cysteine residues, such as nitrosylation and sulfhydration, which fine-tune redox regulation. Thus, the study of mechanisms via which cell death occurs in several neurodegenerative disorders, reveal several similarities and dissimilarities. Here, we review redox regulated events that are disrupted in neurodegenerative disorders and whose modulation affords therapeutic opportunities.
Critical Issues:
Although accumulating evidence suggests that redox imbalance plays a significant role in progression of several neurodegenerative diseases, precise understanding of redox regulated events is lacking. Probes and methodologies that can precisely detect and quantify in vivo levels of reactive oxygen, nitrogen and sulfur species are not available.
Future Directions:
Due to the importance of redox control in physiologic processes, organisms have evolved multiple pathways to counteract redox imbalance and maintain homeostasis. Cells and tissues address stress by harnessing an array of both endogenous and exogenous redox active substances. Targeting these pathways can help mitigate symptoms associated with neurodegeneration and may provide avenues for novel therapeutics. Antioxid. Redox Signal. 30, 1450–1499.
I. Introduction
Production of free radicals is an inevitable consequence of metabolic processes in cells. This review covers aspects of redox regulation and nodes that have been disrupted in neurodegenerative disorders. In several instances, reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive sulfur species (RSS) are integral components of signal transduction processes in cells (Fig. 1A–C). These free radicals can modify susceptible amino acid residues on proteins and other cellular components to alter their structure or function. In particular, cysteine residues are highly susceptible to redox modifications. Basal levels of ROS or RNS are always present in cells; however, when they cross a limit, antioxidant defense systems are activated. The body has evolved multiple mechanisms to cope with increased levels of these free radicals and redox-active molecules. When the antioxidant mechanisms of cells cannot handle increased oxidative stress, increased free radicals lead to antioxidant imbalance or oxidative stress. The term “oxidative stress” was classically defined as a disturbance in the prooxidant–antioxidant balance in favor of the former. However, with increasing research on redox signaling, oxidative stress may be better defined as a disruption of redox signaling and control (252). Dysregulated redox regulation has been implicated in the pathology of neurodegenerative disorders. Diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS) are associated with increased levels of free radicals and oxidative damage, which contribute to disease progression.
Although the brain constitutes only 2% of total body weight, it consumes 20% of inhaled oxygen. It is one of the most metabolically active organs in the entire body, accounting, in part, for its generating higher levels of free radicals. Paradoxically, the brain is also one of the most vulnerable tissues to oxidative stress (166). The brain is rich in lipids with high levels of polyunsaturated fatty acids (PUFAs) in neuronal membranes as well as a predominance of membrane components over cytoplasmic components. The double bonds present in PUFAs such as eicosapentanoic (C20:5) and docosahexanoic (C22:6) acids are susceptible to attack by ROS such as hydroxyl radicals (•OH) leading to a chain reaction of lipid peroxidation. Moreover, antioxidant systems in the brain are less efficient than those operating in peripheral organs such as the liver (158). The brain is composed of numerous cell types, whose relative numbers, composition, and functional interactions vary in different regions, leading to the concept of selective neuronal vulnerability (SNV). This concept may explain why specific populations of neurons degenerate selectively in different neurodegenerative diseases and may account for individual predisposition to brain disease (451). In this review, we cover some of the basic principles in redox biology as well as advanced mechanistic concepts that underpin some of the signaling events in neurodegenerative diseases.
II. Free Radicals and Their Generation
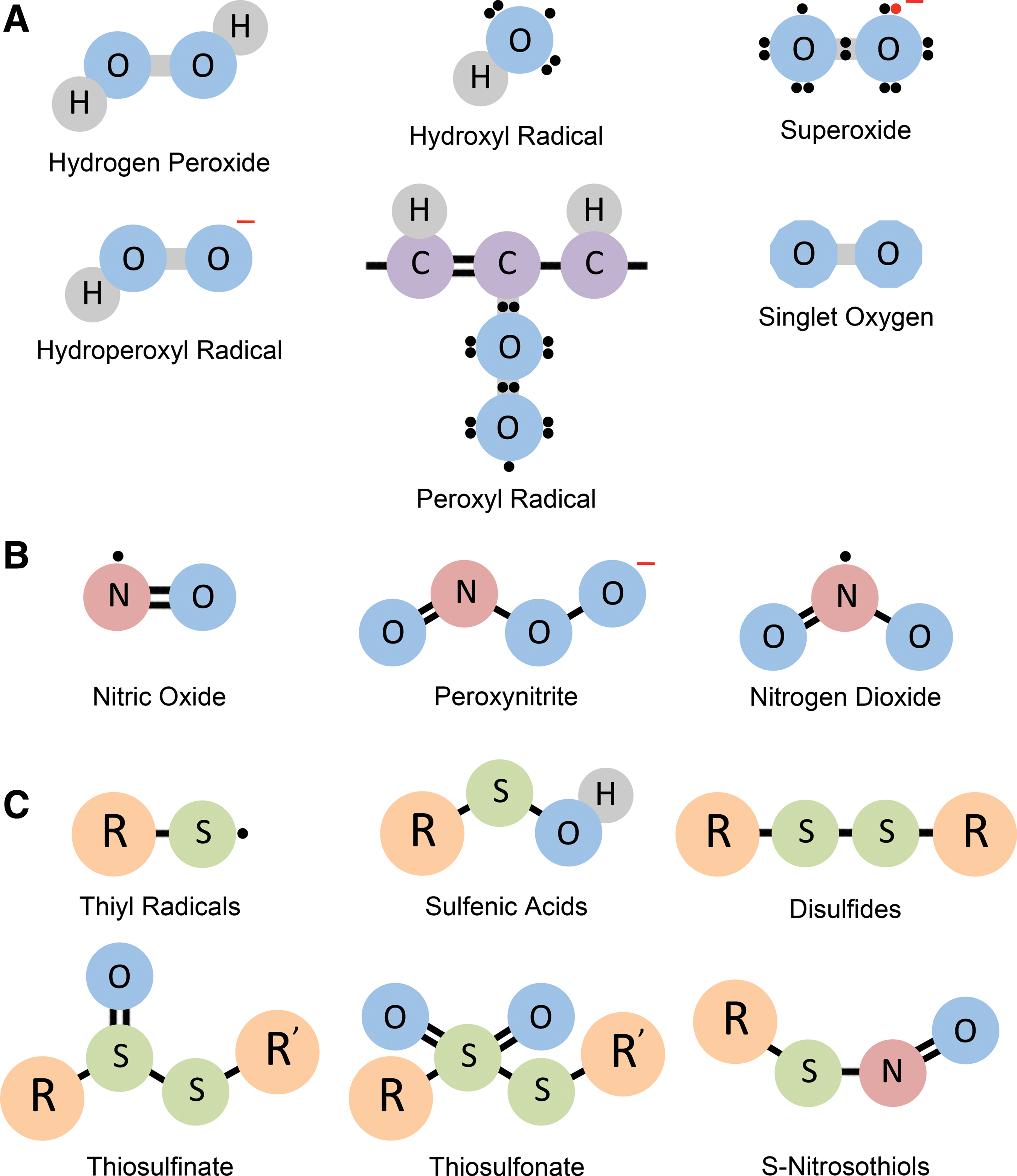
Free radicals are molecules containing one or more unpaired electrons in their molecular orbitals, capable of existing independently (104, 340, 396). In biological systems, free radicals are often termed ROS because a large proportion of biologically relevant radicals are oxygen centered. ROS in cells include hydroxyl radical (•OH), superoxide (O2 •−), singlet oxygen (1O2), and hydrogen peroxide (H2O2), although H2O2 is technically not a free radical because it does not have unpaired electrons. RNS are also free radicals that are nitrogen centered and include species such as nitric oxide (•NO), peroxynitrite (ONOO−), and nitrogen dioxide (•NO2). Radicals can form whenever there is a loss or gain of an electron. Thus, a nonradical can lose an electron or gain an electron to become a radical. Whenever electron transfer occurs in a chemical reaction, the reaction is termed a redox reaction. Removal of electrons is oxidation, whereas addition of electrons constitutes reduction. In this section, we describe the major reactive species, their sources in vivo, and their effects on cellular components.
A. Reactive oxygen species
ROS, as the name implies, involves oxygen (Fig. 1A). In addition to external agents, several enzymatic processes in cells contribute to H2O2 production (Table 1). Several distinct types of ROS participate in signaling processes.
Enzymes Generating Free Radicals
FAD, flavin adenine dinucleotide; H2O2, hydrogen peroxide; NADPH, nicotinamide adenine dinucleotide phosphate; SOD, superoxide dismutase.
1. Hydrogen peroxide
H2O2, although not a free radical, is an ROS that is deleterious to cells at higher concentrations. H2O2 is the dominant ROS influencing signaling pathways as well as inducing damage under pathophysiological conditions. H2O2 in turn can give rise to hydroxyl radical (•OH) by either the Fenton reaction or the Haber–Weiss reaction (239, 548).
2. Hydroxyl radicals
•OH are highly reactive and hence have a very short half-life of the order of 10−9 s. They are generated from H2O2 and act within a few Ao of their site of production. They can damage most cellular components, including DNAs, lipids, and carbohydrates. Lipids in cell membranes readily undergo peroxidation when exposed to •OH radicals. Transition metals present in cells can promote formation of •OH radicals. Transition metals are those elements (manganese, iron, cobalt, zinc, and molybdenum) that belong to the d series (Groups 3–12 of the periodic table), with a partially filled d subshell, or that can give rise to cations with an incomplete d subshell (100). Electrons in the d orbital can participate in bond formation (alongside the 4s electrons) such that the element can exist in different oxidation states and are redox active. For instance, iron and copper are redox active and can transfer an electron by alternating between two redox states (3+/2+ in the case of iron and 2+/1+ for copper). These ions play important roles in biological redox reactions but can also be toxic participants in free radical generating processes such as the Fenton reaction. Thus, the reaction between ferrous (Fe2+) ions and H2O2 produces •OH and HO2 to oxidize lipids, proteins, and DNAs via the reactions.
3. Superoxide anions
O2
•− are generated predominantly by the mitochondria and are by-products of aerobic metabolism as a result of one electron reduction of molecular oxygen by enzymes in the mitochondrial respiratory chain. O2
•− produced by the mitochondria are capable of reducing Fe3+ to Fe2+ by the Haber–Weiss reaction (262).
In addition, O2 •− can react with •NO to produce ONOO−. Other sources of O2 •− include enzymes in the NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX) family, xanthine oxidase, autoxidation reactions of reduced flavins, quinones, metal ions, metalloproteins, and exposure to ionizing radiation or photochemical irradiation.
4. Hydroperoxyl radical
Hydroperoxyl radical (•HO2) is the protonated form of O2 •− and is also termed as perhydroxyl radical. About 0.3% of O2 •− present in the cell cytosol exists in the protonated form (122). •HO2 can induce lipid peroxidation and also mediate tumor formation.
5. Peroxyl radical
The primary pathway of peroxyl radical, ROO•, formation in biological systems is autoxidation viz. lipid peroxidation (419, 480). Lipid peroxidation occurs when free radicals attack lipids containing carbon/carbon double bond(s), especially PUFAs. In addition, lipid peroxidation can also be caused by enzymes such as lipoxygenases, cyclooxygenases, and cytochrome P450 (19). ROO• radicals participate in a chain reaction through which PUFAs are attacked and generate more radicals. These radicals can damage cells by reacting with diverse macromolecules, such as lipids, carbohydrates, proteins, nucleic acids, and enzymes. The effects of ROO• are neutralized by vitamin E or αtocopherol, which is a major chain breaking antioxidant.
6. Singlet oxygen
1O2 is formed when photosensitizers such as chlorophyll absorb light energy and transfer it to oxygen. 1O2 can be beneficial as well as harmful to cells (371). For example, 1O2 can be bactericidal or antimicrobial (157), but it can also cause cell death by necrosis and apoptosis (373). 1O2 can damage DNA and proteins (62). The photoaging-linked mitochondrial common deletion is mediated by 1O2 (44). The common deletion refers to a 4977 bp deletion, which is considered to be a marker for mutations in the mitochondrial genome. Antioxidants, such as α-tocopherol and β-carotene, and thiols, such as ergothioneine and methionine, scavenge 1O2 (128, 139, 279).
B. Sources of ROS generation
The generation of free radicals is positively correlated with metabolic rate and is inversely proportional to life span (9). Several organelles and physiological processes contribute to the generation of ROS. A few of the major ROS-generating sources are discussed below.
1. Mitochondria
Among the organelles generating ROS, the mitochondria are the major contributors that, if not sequestered, can damage the organelle itself. Mitochondria are the sites of aerobic respiration via oxidative phosphorylation (OXPHOS), accounting for 90% of oxygen taken up by cells and provide about 80% of the energy requirements, the remaining 20% being met by glycolysis (378). Mitochondria are bilayered organelles, with outer and inner membranes. Five multiprotein complexes (designated complex I–V) constitute the mitochondrial OXPHOS system (98). Electrons are relayed from NADH, an intermediate of the Krebs cycle, to NADH coenzyme Q reductase (complex I), which passes them onto ubiquinone or coenzyme Q, which also receives electrons from succinate dehydrogenase (SDH; complex II). Coenzyme Q passes electrons to complex III (cytochrome bc1), which passes them to cytochrome C, which transfers them to complex IV (cytochrome C oxidase) that in turn uses these electrons to reduce molecular oxygen to water (Fig. 2).

O2 •− is generated predominantly by complexes I and III of the mitochondria, especially when there is an abundance of respiratory substrates derived from the diet (358, 411). Complex I is a large multisubunit membrane-bound complex, comprising at least 45 polypeptides, transferring electrons from NADH to the cofactor of complex I, flavin mononucleotide (FMN), to quinone via a series of proteins harboring iron/sulfur (Fe-S) clusters (75, 411). Although early studies provided estimates of O2 •− production, these studies were conducted in vitro on isolated mitochondria or mitochondrial fractions (50). In cells, O2 •− is acted on by superoxide reductases and dismutases to produce H2O2 (469). Thus, accurate in vivo measurement of O2 •− is difficult (358). O2 •− production by complex I can occur by different modes. Briefly, the first mode is operational during the conventional forward electron transport when FMN accepts electrons from NADH and is reduced. Here two electrons are transferred from NADH to reduce coenzyme Q. Another mode of O2 •− production occurs during reverse electron transport. Here electrons flow backward through complex I to FMN from where they can reduce NAD+ to NADH and also cause O2 •− formation (97, 358). Complex III is composed of 11 polypeptides, three hemes and an Fe-S center, and transfers electrons from coenzyme Q pool to cytochrome c and can also be a source of O2 •−. The mechanisms of O2 •− production by complex I and III have been covered by several detailed reviews (53, 97, 358).
Most intracellular ROS owe their origin to O2 •−, which is converted to H2O2 by the action of catalase or react with •NO to form ONOO−. Oxidative stress can damage mitochondrial components such as mitochondrial DNA (mtDNA) and proteins in addition to nuclear DNA, which can further inflict damage (144, 424). The mitochondrial genome is not protected by histones and is highly susceptible to damage due to its proximity to the electron transport chain (ETC). Similar levels of oxidants induce more lesions in mtDNA compared with nuclear DNA (325, 524). On an average a single mammalian cell is expected to undergo 2000–10,000 depurination (hydrolysis of the N-glycosyl linkage connecting a purine base to the deoxyribose sugar is cleaved, producing an abasic site) events per generation. In terminally differentiated cells such as neurons, ∼108 such events are expected to occur (306). Damage to mtDNA prevents optimal expression of proteins in the ETC leading to a toxic cycle of free radical generation and mitochondrial malfunction, which culminates in neuronal death (518). It is not surprising that the process of respiration has evolved to be carried out by a distinct organelle to shield the other cellular compartments such as the nucleus.
2. Xanthine oxidoreductases
In addition to external agents and the mitochondria, several cellular enzymes contribute to H2O2 production. Xanthine oxidoreductase (XOR) catalyzes the conversion of hypoxanthine to xanthine and further to uric acid during purine metabolism, generating H2O2 in the process. XOR is a homodimer, with each subunit comprising a molybdopterin cofactor, two iron/sulfur centers and a flavin adenine dinucleotide (FAD) cofactor. Expression of XOR is regulated by oxygen tension, cytokines, and glucocorticoids (45). XORs exist either as the dehydrogenase form (xanthine dehydrogenase [XDH]) or the oxidase form (XO), generated from the reversible oxidation of cysteine residues or by irreversible proteolytic cleavage (284, 368, 486). Electrons are relayed from xanthine to oxygen and NAD+, respectively, yielding O2 •−, H2O2, and NADH. In addition, XOR can generate O2 •− via NADH oxidase activity and can produce •NO via nitrate and nitrite reductase activities (45).
3. NADPH oxidases
The NOX family of enzymes are protein complexes that utilize cytosolic NADPH to reduce oxygen to O2 •− anion (287, 321, 365). Each of these enzymes contains a core catalytic subunit (NOX) and regulatory subunits that are located in the cytosolic and membranous compartments. Neutrophils and other phagocytic cells were the first cells shown to possess NOX activity, which was utilized for defense against microorganisms. The catalytic subunit of this enzyme gp91phox or NOX2 is now recognized to be one of seven known members constituting the family namely, NOX1, NOX2, NOX3, NOX4, NOX5, dual oxidase 1 (DUOX1), and DUOX2 (539). These enzymes transfer an electron from NADPH to O2, forming O2 •−, which in turn is converted to H2O2. The physiologic functions of these enzymes include host defense, regulation of gene expression, and cell differentiation. It is becoming increasing clear that NOX activity is elevated in several neurodegenerative conditions and inhibition or genetic deletion of these enzymes has therapeutic benefits (37, 94, 95, 377, 476, 544).
Several other enzymes such as cytochrome P450 reductase also produce free radicals and are listed in Table 1.
C. Reactive nitrogen species
Although redox imbalance was selectively attributed to ROS in the past, it is being increasingly clear that other free radical species such as RNS also contribute to fluctuations in redox homeostasis (Fig. 1B).
1. Nitric oxide
•NO is the predominant RNS that plays central roles in cell signaling processes (46, 236). •NO is also the precursor of other RNS such as peroxynitrites. In mammals, •NO is produced by nitric oxide synthases (NOSs) from
2. Peroxynitrite
ONOO− is a highly reactive radical formed by the interaction of •NO and O2 •− and mediates several of the toxic effects of these radicals in cells (408, 409). The radical is short lived and causes extensive cytotoxicity. ONOO− affects mitochondrial function and triggers cell death via oxidation and nitration reactions. ONOO− formation leads to decreased bioavailability of •NO and therefore affects most processes regulated by •NO.
3. Nitrogen dioxide
Once believed to be predominantly an environmental pollutant, nitrogen dioxide has been reported to be produced endogenously by enzymes such as myeloperoxidases (318). Nitrogen dioxide is derived from •NO and oxygen, a reaction that takes place most efficiently in a hydrophobic environment. •NO2 can trigger lipid peroxidation reactions and cell death (269). More recently, it was shown that the in vivo formation of 3-nitrotyrosine depends on the availability of •NO2 radicals. Nitrogen dioxide can deplete endogenous antioxidants such as ascorbic acid, alpha-tocopherol, and bilirubin, which help scavenge this radical (207).
D. Reactive sulfur species
For many years the redox field largely focused on reactive oxygen and nitrogen species. Sulfur was considered to be only a building block of proteins and small molecules such as the reduced form of glutathione (GSH). Sulfur compounds such as sulfur dioxide (SO2) and hydrogen sulfide (H2S) were considered to be toxic environmental components. However, recent advances in the field of sulfur biology have established RSS as signaling molecules, and it is becoming increasingly evident that RSS also play significant roles in redox reactions in vivo (177, 187). Oxidation products of the thiol groups of cysteine residues, the disulfides, sulfenic acids, sulfinic acids, and sulfonic acids, were known due to studies of thiol oxidation in proteins. RSS can be generated from thiols by reaction with oxidizing agents such as H2O2, 1O2, ONOO−, and O2 •−. The effect of endogenous and dietary sulfur containing thiols such as GSH, cysteine, and ergothioneine in redox buffering and mitigation of oxidative stress was also known. The role of cysteine modifications such as glutathionylation was also gaining importance and continues to be a major field of study (173 –175). The large number of RSS owes its origin to the multivalent oxidation states of sulfur, ranging from −2, in H2S, to +6, in sulfate (SO4 2−). The concept of RSS was first introduced in 2001 and included thiyl radicals (RS•), sulfenic acids (RSOH), disulfides (RSSR), thiosulfinate [RS(O)SR], thiosulfonate [RS(O)2SR], and S-nitrosothiols (SNT) (178) (Fig. 1C). These were proposed to be formed during conditions of oxidative stress and act as oxidizing agents themselves.
The mitochondria are a rich source of RSS during sulfide oxidation. Although initial studies focused only on thiosulfate and sulfate, other sulfur species such as glutathione persulfide (GSSH) are generated during mitochondrial functions (304, 348). Sulfide oxidation in the mitochondria is catalyzed by sulfide quinone oxidoreductase, a flavoprotein, via a two-electron oxidation of H2S, forming a persulfide intermediate during the reaction (241, 304). The persulfide is transferred to a low-molecular-weight acceptor such as GSH. The released electrons are relayed to complex III of the ETC by ubiquinone. The GSSH formed during the process can be acted on by enzymes such as persulfide dioxygenase to form sulfite. Alternatively, GSSH can be utilized in a sulfurtransferase reaction by rhodanese, which transfers the sulfane sulfur to sulfite, forming thiosulfate in the process. The sulfite formed is oxidized by enzymes such as eosinophil peroxidase, prostaglandin H synthase, or myeloperoxidase to yield RSS, sulfite anion radical, peroxymonosulfate radical, and sulfate radicals, which can damage proteins in cells (416). Sulfur amino acids are major targets of redox modifications and thus the identification of an increasing number of sulfur-derived radicals adds additional layers of complexity in redox signaling processes (457).
Recently, the gasotransmitter, H2S, has been identified as a bona fide signaling molecule. H2S, derived from cysteine, has pleiotropic roles in cell function, ranging from inflammation to cardiovascular functions. H2S functions as an endothelial-derived relaxation factor and like •NO increases cyclic GMP in cells. Like •NO, H2S participates in physiological pathways in the brain. One of the first identified neural functions of H2S was the regulation of the N-methyl-
E. Effects of ROS and free radicals on cellular components
ROS affect several cellular components, including proteins, nucleic acids, lipids, and carbohydrates.
1. Proteins
ROS can cause oxidation of side chains of constituent amino acids in proteins, which can result in protein carbonylation (482). The thiol group (–SH) of cysteine residues is readily oxidized when exposed to ROS. The–SH group of cysteine residues on amino acids can be oxidized to sulfenic (-SOH), which can then be further oxidized to sulfinic (-SO2H) or sulfonic (-SO3H) acid groups or form inter- or intramolecular disulfide bonds (6). Oxidation of cysteine residues can also facilitate other post-translational modifications such as glutathionylation and sulfhydration, which play important signaling roles in cells (150). Methionine can also be oxidized to methionine sulfoxide (MetO) both during normal and stress conditions (326, 483). Recent studies show that the molecule interacting with CasL proteins, which harbor flavin-monooxygenase domain with an NADPH-dependent methionine sulfoxidase activity, oxidizes methionine residues in vivo (231, 296). In addition to these changes, free radicals such as ONOO− can elicit protein tyrosine nitration, which can participate in both physiological and pathophysiological processes (29, 408).
2. Nucleic acids
The DNA base, guanine, is prone to oxidative damage and is converted to 8-oxoguanine, which pairs with adenine instead of cytosine, leading to mutations. More complex modifications such as strand crosslinking and cyclization of nucleotides also occur when DNA is exposed to oxidants and have been extensively discussed (61, 63, 152).
3. Lipids
Lipids undergo a modification generally termed “lipid peroxidation” through free radical chain reactions (478, 555). Lipids such as cholesterol esters, phospholipids, and triglycerides are particularly vulnerable to modification by free radicals as they comprised PUFAs. Lipid peroxidation results in oxidation products such as reactive aldehydes of which 4-hydroxynonenal (4HNE), malondialdehyde (MDA), and acrolein have been extensively studied. These compounds can react with proteins and other cellular components to modulate their function. 4HNE and MDA have been recognized as markers of lipid peroxidation.
4. Carbohydrates
ROS and RNS mediate damage to carbohydrate components of the cells too, causing several undesirable effects such as oxidative degradation or depolymerization of polysaccharides via scission of carbohydrate chains (135). In rheumatoid arthritis, ROS generated by neutrophils cause fragmentation of hyaluronan, a polymer of the disaccharide [-
III. Antioxidant Systems in the Brain
As summarized above, a diverse array of free radicals and related damaging species are generated in vivo both during normal metabolism and during pathological conditions. To counteract them, a large arsenal of antioxidants and enzymes are harnessed by cells. When these countermeasures are not successful, redox balance is perturbed and oxidative stress ensues. However, a note of caution is to be mentioned. Antioxidants may not always be beneficial, especially under conditions where the production of free radicals is necessary for cell function as in the case of immune system and during certain aspects of cellular differentiation. Regardless, defense molecules can be broadly categorized into small-molecule antioxidants and protein/enzymatic antioxidants.
A. Small molecules
Several low-molecular-weight compounds such as cysteine, ascorbate (vitamin C), GSH, ergothioneine, α-tocopherol (vitamin E), carotenoids, uric acid, and bilirubin function as antioxidants (Fig. 3) (73). Several of these molecules are synthesized endogenously, whereas others are derived from the diet. Among these molecules, some are lipophilic and are effective in preventing lipid-rich components of cells. For instance, bilirubin, produced by the enzyme biliverdin reductase (BVR), and vitamin E are potent inhibitors of lipid peroxidation (359, 463, 487). Several of these small molecules form a redox couple (a pair of molecules that can be interconverted by the addition or loss of electrons are referred to as a redox couple or pair) with their oxidized forms, for instance, GSH/GSSG and CysSH/Cys-SS, where GSSG and Cys-SS are the oxidized forms of GSH and cysteine, respectively. Other redox duos in cells include the NADPH/NADP+ and NADH/NAD+ couples. In cells, there is no single or uniform redox state, as various subcellular compartments have different redox systems operating at distinct set points. Thus, the activity of any one particular redox regulating system may not be representative of the overall scenario. These redox systems are not in equilibrium with each other (264). The redox potentials (the electromotive force E h or inherent tendency to accept or donate electrons, which is expressed in millivolts) of these redox couples vary within different cellular compartments and conditions. For instance, the E h of the GSH/GSSG couple is approximately from −260 to −200 mV in cultured cells. E h for the GSH/GSSG couple is calculated using the Nernst equation: E h = E o + RT/nF ln [(GSSG)/(GSH)2], where E o is the standard potential, R is the gas constant, T is the absolute temperature, n (here 2) is the number of electrons transferred, and F is the Faraday's constant. The concentrations of GSH and GSSG can be estimated by methods such as high-performance liquid chromatography. In the plasma of mammals, this value is about −128 ± 9 mV (253). In the endoplasmic reticulum (ER), the redox environment is more oxidized than in the cytoplasm (234). The ratio of GSH to GSSG within the secretory pathway ranges from 1:1 to 3:1. The GSSG/GSH couple is also more oxidized during aging and during pathological conditions (445). The major redox couple in the extracellular milieu is the CysSH/Cys-SS pair. The redox potential of this redox duo is about −80 mV in normal healthy humans, a value that is significantly more oxidized than that of the GSSG/GSH couple, which in turn is significantly more oxidized than the NADPH/NADP+ system, which oscillates in the −300 to −400 mV range. Similar to the GSSG/GSH dynamics, the redox potential of the CysSH/Cys-SS redox couple also varies between various cellular compartments and during response to oxidative stress or pathological conditions (264). Emerging evidence suggests that measurements of redox states in cells may be more complicated than previously believed. Cells have multiple compartments and organelles with different redox changes controlled temporally and spatially. Very often, measurements of redox potential using whole cell extracts are considered cytosolic redox potentials, which is not the case. Ex vivo oxidation during sample handling can also contribute to variation in measured values of redox potential as has been reported in the case of the ER (130). Genetically encoded redox sensors that can be targeted to specific cellular compartments can provide information on the redox states of specific compartments. Interestingly, the redox potentials reported by these methods are much more negative (about −320 mV for cytosolic glutathione redox potentials) in a variety of models due to rapid removal of GSSG into the luminal compartments (119, 353).

A systematic analysis of changes in these redox couples in various disease conditions is yet to be conducted. Although the redox potential provides information on redox changes or disturbances, a living cell is complex, with several competing pathways, and kinetic pathways and rates associated with reactions, contributing to whether a particular reaction will occur or not. These aspects have been discussed in detail elsewhere (153, 402, 537).
B. Enzymatic/protein components of redox balance
Proteins involved in antioxidant defense can be classified as direct or indirect antioxidant enzymes (514). The direct antioxidant proteins neutralize or scavenge free radicals and include superoxide dismutases (SODs), catalase, glutathione peroxidases, and peroxiredoxins (73) (Table 2). The indirect antioxidant enzymes contribute to reduction of free radicals by indirect means. An example is glutamate–cysteine ligase and CSE, which are required for biosynthesis of GSH and cysteine, respectively (145, 382). The antioxidant enzymes play significant roles in maintenance of redox balance and probably constitute the most important arm of antioxidant defense mechanisms. The efficacy of small molecules such as GSH requires the action of such enzymes to exert their effects. Dysregulation of these antioxidant enzymes is frequently encountered in neurodegeneration, which contributes to pathophysiology and disease progression. A few of the major direct antioxidant enzymes are discussed below.
Redox Regulating Enzymes
ATP, adenosine triphosphate; GSH, glutathione, reduced form; GSSG, glutathione, oxidized form; GSTs, glutathione S-transferases.
1. Superoxide dismutases
SODs are metalloenzymes that scavenge O2 •− by catalyzing the dismutation of two molecules of O2 •− into one molecule each of O2 and of H2O2 (341). Three SODs have been identified in mammals so far: copper zinc superoxide dismutase (CuZnSOD/SOD1), manganese superoxide dismutase (MnSOD/SOD2), and extracellular superoxide dismutase (ECSOD/SOD3). SOD1 functions as a homodimer, with each monomer being ∼19 kDa and binding an atom each of copper and zinc (286). SOD1 is predominantly cytosolic, but has also been localized to mitochondria and endosomes (260, 443). Nascent SOD1 is inactive in its monomeric form and is converted to its active dimeric form by the copper chaperone for superoxide dismutase (CCS), which mediates the insertion of the copper ion and an intramolecular disulfide bond (76, 115, 148). The disulfide bridge is essential for the catalytic activity of SOD1. The copper reacts in a ping-pong mechanism where one O2 •− molecule is reduced to H2O2 and a second O2 •− is oxidized to O2. Interestingly, SOD1-deficient mice are viable and similar to wild-type mice at young ages but as they age, these mice suffer from a variety of maladies ranging from macular degeneration, oxidative stress, hepatocarcinogenesis, and infertility to muscle wasting, denervation of motor neurons, and behavioral changes (140, 221, 238, 418, 557). Mutations in SOD1 have also been linked to neurodegeneration as discussed later.
SOD2 is present exclusively in the inner mitochondrial space with the human enzyme existing as a tetramer of four identical 22 kDa subunits (337, 532). SOD2 is encoded by nuclear DNA, imported into the mitochondrial matrix posttranslationally, and assembled into the active enzyme, involving the incorporation of a manganese ion in the mitochondrial matrix (540). Unlike SOD1, SOD2 does not exhibit product inhibition. In addition to scavenging O2 •−, SOD2 has been reported to inhibit ONOO− formation, itself being inactivated by excess ONOO− (407, 504). SOD2-deleted mice survive up to 3 weeks of age and exhibit several abnormalities, including severe anemia, degeneration of neurons in the basal ganglia and brainstem, and progressive motor disturbances characterized by weakness, rapid fatigue, and circling behavior (295).
SOD3, the most recently discovered member of the family, is also a Cu/Zn SOD, but is secreted and exists as a homotetramer of 30 kDa subunits (165, 330). This extracellular enzyme was first detected in human plasma, lymph, ascites, and cerebrospinal fluids (331). It is also present in the extracellular matrix and on cell surfaces, anchored by interactions with the heparan sulfate proteoglycan, collagen, and fibulin-5 (330, 366, 395, 446). SOD3 is modified by N-linked glycosylation, which can be used to separate it from the other SOD enzymes (329, 509). Similar to SOD1, deletion of SOD3 is not lethal in mice but predisposes the mice to developing various pathologies such as sensitivity to hypoxia, hyperoxia, renal injury, and hypertension among others (72).
2. Catalases
Catalases are enzymes that degrade H2O2 to water and O2 with high efficiency (10, 180, 406). Human catalase is a homotetramer of 62 kDa monomers, each subunit containing a prosthetic heme group (406). Catalases have a peroxisome targeting signal and are predominantly present in peroxisomes, where tetramerization and heme incorporation occur (293, 405). Mice deleted for catalase develop normally and exhibit no gross abnormalities, but exhibited slower decomposition of extracellular H2O2 compared with wild-type mice and also higher susceptibility to mitochondrial dysfunction induced by traumatic brain injury (222).
3. Glutathione peroxidases (enzymes)
Glutathione peroxidase (GPx) enzymes utilize GSH to reduce peroxides to produce glutathione disulfide and water (154). At least eight different isoforms of GPx exist (GPx1–8), of which GPx1 is most abundant, present in the cytosol and mitochondria (55). Among these enzymes, GPx1–4 and GPx6 in humans are selenium-containing enzymes (298). GPx5, 7, and 8 use cysteine in place of selenium (55). Gpx1 −/− mice are phenotypically normal with normal life spans, but are more susceptible to oxidants, mitochondrial toxins, and ischemia/reperfusion injury (123, 271, 558). GPx2 is enriched in the gastrointestinal tract and GPx3 in the kidneys (143, 156, 374). Similar to Gpx1 −/− mice, Gpx2 −/− and Gpx3 −/− do not have gross morphological abnormalities, but exhibit increased sensitivity to stress (155, 248). Of these isoforms, GPx4 has several unique features (105). GPx4, a monomeric enzyme, exists as three isoforms through alternative splicing and transcription initiation: cytosolic GPx4, mitochondrial GPx4 (mGPx4), and nuclear GPx4 (nGPx4). Cytosolic GPx4 is essential for embryonic development and cell survival as systemic Gpx4 caused lethality, while nGPx4 and mGPx4 play roles in male fertility and spermatogenesis (456, 551). Besides acting on hydroperoxides and lipid hydroperoxides, substrates for all glutathione peroxidases, GPx4 has the unique ability of reducing phospholipid-associated hydroperoxides in biological membranes to the corresponding alcohols. This enzyme can also utilize thiols other than GSH as a reductant. GPx4 is abundant in the brain and is present in both glial cells and neurons and protects them against oxidative damage (398, 563, 567). Among the GPx proteins, GPx7 and GPx8 are ER-localized protein disulfide isomerase (PDI) peroxidases, functioning as stress sensors and transducers (367). GPx7 binds to its target proteins such as PDI and 78 kDa glucose-regulated protein, also known as immunoglobulin heavy chain binding protein, and modulates disulfide bond formation in response to stress stimuli (89).
4. Peroxiredoxins (enzymes)
The other large class of enzymes that reduce peroxides are peroxiredoxins (Prxs), which utilize cysteine, often called the “peroxidatic” cysteine, Cp, the site of oxidation by peroxides (422, 423). Oxidation of this residue (CP-SH) generates cysteine sulfenic acid (CP-SOH), which then reacts with another cysteine, termed the resolving cysteine, CR, to form a disulfide, which can then be reduced by an electron donor. The Cp residue can be oxidized by H2O2, lipid peroxide, or ONOO− very rapidly, with rate constants 1 × 106–108 M −1·s−1, which are 5–7 orders of magnitude higher than those for small thiols (538). Depending on the location of the resolving cysteine, Prx enzymes have been classified into 2-Cys, atypical 2-Cys, and 1-Cys Prx subfamilies. Prx enzymes are homodimeric and contain two conserved (CP and CR) cysteine residues per subunit, where intersubunit disulfides are formed. Atypical 2-Cys Prx enzymes, on the contrary, form intramolecular disulfide bonds. 1-Cys Prx, as the name indicates, has only one cysteine participating in the reaction. The CP here forms a disulfide with CR-SH of other proteins or thiols (151). Mammals possess six Prx isoforms of which PrxI to PrxIV are 2-Cys (typical) Prx enzymes. PrxV is a typical 2-Cys and PrxVI a 1-Cys enzyme. The disulfide bond formed during detoxification of peroxides can be reduced by the antioxidant enzyme sulfiredoxin to restore peroxidase activity (47, 541). More recently, Prx enzymes have been reported to participate in a relay system, which leads to oxidation of target proteins (as opposed to reduction) to facilitate ROS signaling, suggesting additional roles as sensors and transmitters of H2O2 signals (488, 489).
In addition to these direct acting enzymes, several other proteins act to maintain redox homeostasis in cells some of which reverse the effects of free radicals (Table 2). One of these types of repair enzymes are the methionine sulfoxide reductases (MSRs: MsrA and B), which convert oxidized methionine residues (MetO) on proteins to their original state (356, 483). MSRs use thiol/selenothiol chemistry to reduce oxidized methionines using the reducing power generally provided by the thioredoxin system (506). Apart from these repair proteins, others regulate the redox mileu in cells by synthesizing antioxidants or by eliminating the toxic metabolites. For example, glutathione S-transferase can help eliminate 4HNE, a toxic metabolite that accumulates during lipid peroxidation (11, 24). Similarly, biosynthetic enzymes of antioxidants such as CSE, the enzyme that produces cysteine, can be upregulated by cells in response to stress (382, 384, 453, 454). Thus, cells have evolved multiple mechanisms to maintain redox signaling during different conditions and stages of development.
IV. Dysregulation of Redox Homeostasis in Neurodegeneration
Several neurodegenerative disorders are associated with elevated oxidative stress. Given the fact that the brain is one of the most metabolically active tissues, it is not surprising that the brain is also highly susceptible to oxidative damage. Some of the antioxidant enzymes such as catalase are present in lower levels in the brain (202, 204). The brain is also rich in transition metals such as iron, which can inflict damage via the generation of •OH radicals. Consistent with these findings, in the stroke-prone spontaneously hypertensive rats, levels of oxidized proteins were significantly increased in the brain, but not kidneys or serum (347). However, systematic studies of oxidative damage in various organs in comparison with the brain are still lacking. Generation of free radicals is intimately linked to metabolism so that disruption of specific physiological processes can elicit neuronal death. It has also been observed that selective vulnerability of specific regions in the brain occurs in different neurodegenerative diseases (529) (Fig. 4). A common feature of these disorders is that redox imbalance involves dysregulation at multiple levels viz, increased production of free radicals and damaging species, malfunctioning antioxidant defense mechanisms, and repair pathways. Here we discuss redox disturbances in a monogenic disorder, HD, as an example, in which several features pertaining to redox signaling are also encountered in other neurodegenerative disorders, including AD, PD, ALS, ataxias, and autism.

A. Huntington's disease
HD is an autosomal dominant neurodegenerative disease characterized by expansion of polyglutamine repeats in the protein huntingtin (1). Mutant huntingtin aggregates and causes widespread damage ranging from transcriptional dysregulation to motor and cognitive deficits. Greater than 40 repeats predispose an individual to the disease, which is characterized by involuntary chorea and cachexia during disease progression. Presently, there is no cure for the disease, although several drugs that mitigate symptoms are available. The various drugs used to treat symptoms of HD range from antidepressants to antipsychotics and those used to treat chorea (517). The food and drug administration (FDA) has approved the use of tetrabenezine for treatment of chorea. Although tetrabenezine effectively reduces chorea in HD patients, its use has been associated with side effects such as suicidal thoughts, depression, and restlessness (232, 294). HD primarily affects the corpus striatum of the brain, which regulates motor functions (Fig. 4). The medium spiny neurons in the striatum degenerate, leading to striatal shrinkage and atrophy. The cerebral cortex is also affected during the later stages of the disease. At the molecular level, several abnormalities have been reported in HD, which include elevated oxidative stress in affected regions and compromised redox signaling pathways.
1. Oxidative stress in HD
Numerous studies have reported oxidative damage in HD cells and tissues (59, 85, 282, 481). Higher levels of lipid peroxidation and low GSH content have been reported in the plasma of HD patients (270). Oxidative damage to both nuclear and mtDNA is caused by mutant huntingtin with the basal ganglia being especially vulnerable (58). Postmortem caudate tissue from HD patients displays elevated 8-hydroxydeoxyguanosine (8-OHdG), a marker for oxidative DNA damage. Increases in 8-OHdG have also been observed in serum and leukocytes of HD patients (85, 217). In both 3-nitropropionic acid-treated mice, a chemical model of HD, and the R6/2 model of HD, quantitative PCR reveals extensive mtDNA damage (7). In mouse embryonic cells derived from YAC128 HD mice, increased O2 •− production and dysregulated calcium signaling cause elevated oxidative stress compared with wild-type cells. These findings were recapitulated in YAC128 HD striatal medium spiny neurons. Similar results were observed in fibroblasts derived from HD patients and a mouse model of HD (525).
2. Compromised low-molecular-weight antioxidant metabolism in HD
HD has been associated with decreased levels of endogenous antioxidant molecules, which contribute to the oxidative stress linked to pathogenesis.
a. Reduced cysteine and GSH levels in HD
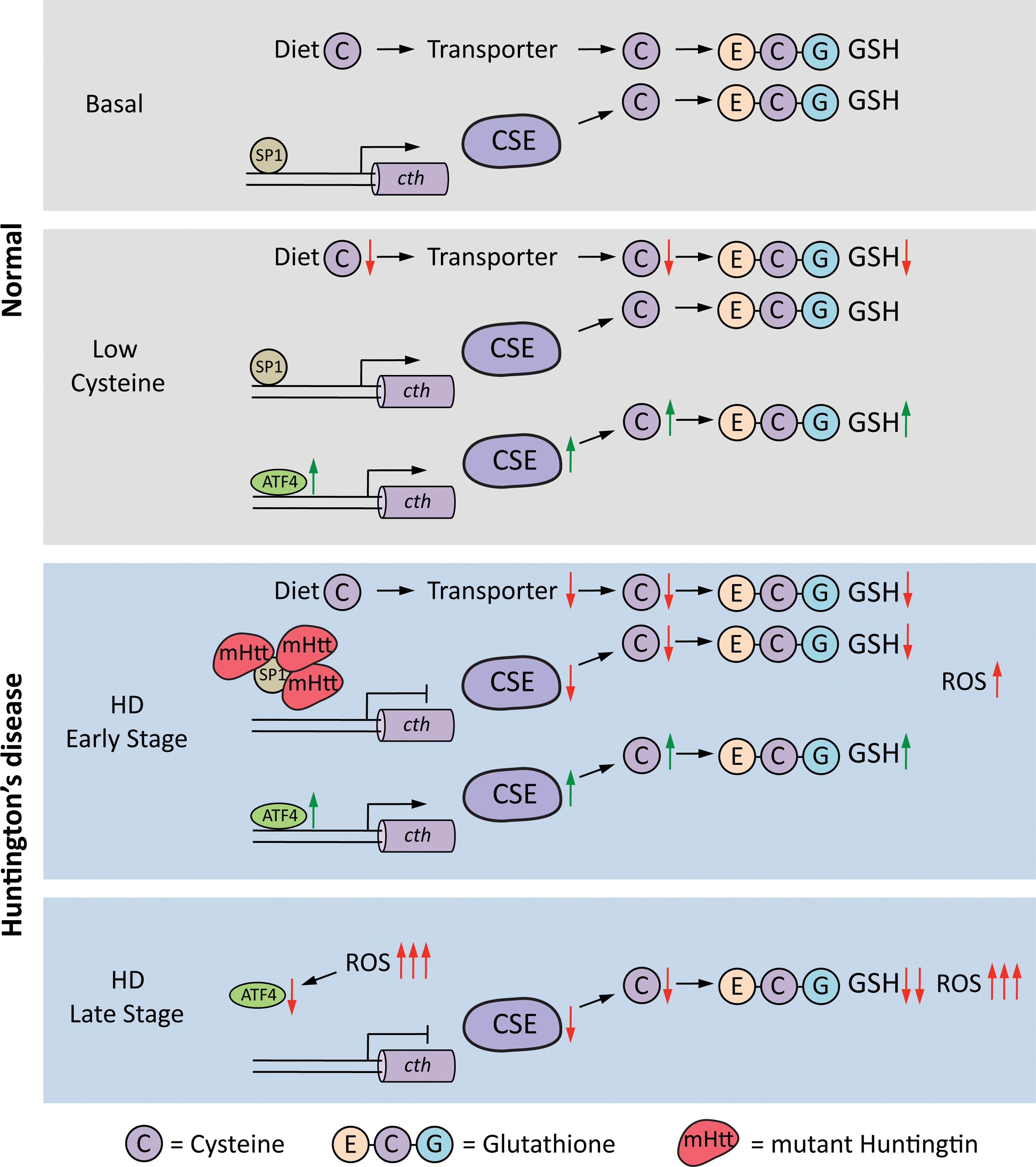
The metabolism of small-molecular-weight thiols such as cysteine and GSH is compromised in HD (381, 383, 453). We have shown previously that depletion of CSE, the biosynthetic enzyme for cysteine and the gaseous signaling molecule, hydrogen sulfide, contributes to neurotoxicity in HD. The transcription factor for basal expression of CSE, specificity protein 1 (SP1), is sequestered by mutant huntingtin, which reduces CSE expression in HD (137, 381). Low levels of CSE were observed in striatal cell culture models of HD, mouse models, and in postmortem striatal tissue from HD patients. The depletion is specific to the striatum and the degree of decrease correlates with the severity of the disease. The cortex exhibits a decline in CSE but during later stages of the disease. CSE is a key enzyme in the reverse transsulfuration pathway, in which cysteine is synthesized by transfer of a sulfur atom from homocysteine, which in turn is derived from dietary methionine (Fig. 5). The reverse transsulfuration pathway plays a central role in redox-regulated signaling nodes in cells. The decrease in CSE is accompanied by a reduction in cysteine and H2S production (381, 454). In addition to its biosynthesis, the transport of cysteine or its oxidized form of cystine is compromised in HD (161, 303). During conditions of stress, such as amino acid deprivation or ER stress, expression of CSE is dependent on the activating transcription factor 4 (ATF4). ATF4 is a stress-inducible protein that regulates the expression of amino acid biosynthetic and transport genes during conditions of amino acid deprivation, ER stress, as well as other stimuli (453, 454). The induction of ATF4 in response to cysteine deprivation is blunted in striatal progenitor cells harboring mutant huntingtin (453). Interestingly, the muted response of ATF4 in HD striatal cells occurs selectively for cysteine deprivation and not in response to deprivation of other amino acids or other forms of stress, indicating that specific properties of cysteine are compromised in HD. The abnormality stems from the chronic oxidative stress occurring in HD due to depletion of CSE leading to a vicious cycle of oxidative stress and impaired response of restorative pathways leading to further damage and ultimately cell death (453) (Fig. 6). Cysteine is a component of GSH, and the availability of cysteine is the rate-limiting step for GSH production. In addition, cysteine is a potent antioxidant on its own; therefore, its scarcity is associated with redox imbalance. Depletion of CSE and cysteine results in elevated levels of protein carbonylation, protein nitration, and lipid peroxidation in mouse models of HD (381, 453). Cysteine is also the precursor for several cytoprotective molecules such as H2S, lanthionine, taurine, and glutathione (Fig. 7). Of these, H2S plays pleiotropic roles in cell physiology, modulating signaling pathways via sulfhydration/persulfidation of reactive cysteine residues on target proteins (360, 382, 384 –386). Thus, disrupted cysteine homeostasis has multifaceted consequences. Accordingly, supplementation of cysteine in the diet and N-acetylcysteine (NAC) in the drinking water of the R6/2 mouse model of HD ameliorates symptoms and increases life span (381). These studies also showed that mice on a high cysteine diet exhibited improved motor function and decreased brain pathology. Independent studies utilizing NAC report beneficial effects on behavioral aspects of HD (542).


b. Reduced ascorbate uptake in HD
Ascorbic acid or vitamin C is an antioxidant that is highly enriched in the brain, where it can regulate neuronal metabolism (80, 477). Ascorbate defends neurons against oxidative damage and modulates neuronal metabolism during synaptic activity (110). Ascorbate mediates lactate utilization as an alternative neuronal energy substrate (79). During synaptic activity, the ascorbate released prevents neuronal glucose utilization and stimulates lactate utilization to sustain brain activity. Ascorbate, thus, acts as a switch to regulate use of other substrates such as lactate for energy production via a GLUT3 (glucose transporter 3)-dependent mechanism (38). High-affinity, sodium-dependent vitamin C transporters (SVCTs) transport vitamin C into neurons (78). Striatal cells derived from HdhQ111 mouse model of HD have impaired trafficking of SVCT2 to the membrane leading to lowered ascorbate transport and altered antioxidant and bioenergetic capacity (8). Flux of ascorbate from astrocytes to neuronal cells in brain slices of R6/2 mouse model of HD is impaired and the ascorbate metabolic switch is affected in HD. Synaptic activity generates oxidants that oxidize ascorbate to dehydroascorbate, which is released by neurons and taken up by astrocytes to regenerate ascorbate via GSH-dependent processes (109, 275). In presymptomatic stages of HD, astrocytes do not release sufficient ascorbate to be utilized by neurons. Interestingly, HD is also associated with lowered surface expression of GLUT3, which disrupts utilization of lactate as an energy substrate (167). It was proposed that the increased oxidative stress associated with HD would utilize ascorbate and minimize its availability to regulate energetics (110).
3. Elevated production of free radicals in HD
Several factors contribute to the elevated levels of free radicals in HD. A few of these pathways are given below.
a. Activation of NOX enzymes
Increased activity of NOX has been observed in HD and may contribute to neurotoxicity. NOX catalyzes the formation of O2 •− from oxygen, which damages cellular components as described earlier. Synaptosome fractions derived from the cortex and striatum of HD (140Q/140Q) mice display elevated NOX activity with the striatum exhibiting the greatest increase. Treatment with NOX inhibitors such as diphenyleneiodonium, apocynin, and VAS2870 attenuates cell death and toxicity (516).
b. Redox metals and HD
Dysregulated iron metabolism is a common feature of several neurodegenerative disorders (263, 438). Excess iron deposition has been reported in the basal ganglia of the brain in HD (30). Furthermore, neonatal iron supplementation in the YAC128 mouse model of HD accelerates disease progression (43). Copper has also been linked to disease progression in HD. Elevated copper deposition increases oxidative stress and aggregation of mutant huntingtin, which can be reversed by copper chelators (160, 546).
c. Defective DNA repair in response to oxidative stress
Nuclear DNA damage occurs in HD. The ataxia-telangiectasia mutated (ATM) DNA repair cascade is affected, and base excision repair regulated by the ATM protein in response to oxidative stress is defective (316). Huntingtin acts as a scaffolding protein in the ATM repair complex (323). Oxidative stress mediates CAG expansion in HD fibroblasts. The expansion arises during the process of removal of oxidized DNA bases (8-oxo-G lesions), and is dependent on the DNA glycosylase, OGG1 (278). A “toxic oxidation cycle” model was proposed in which somatic mutations mediate onset and progression of the disease. As oxidative lesions in the brain accumulate with age, a DNA damage response involving OGG1 repair through a single-stranded break mechanism is mounted. The repair is error prone and thus leads to expansion of the CAG repeats, which further exacerbates the cycle.
d. Excessive NMDAR stimulation
Overactivation of the NMDA type of glutamate receptors is a feature of HD. Excessive stimulation of NMDARs causes elevated oxidative and nitrosative stress (136, 362). Neurons from the subthalamic nuclei in the BACHD model of HD exhibit elevated oxidative stress, which is decreased by treatment with NMDAR antagonists (16).
e. Impaired antioxidant response pathways in HD
The nuclear factor erythroid 2-related factor 2 (Nrf2) is a master transcriptional regulator of the cellular antioxidant stress response and detoxification pathways. It is a basic leucine zipper protein that cooperates with other proteins and activates genes with an antioxidant response element/electrophile response element or ARE/EpRE (213, 224, 440, 497). Under basal conditions, Nrf2 is sequestered in the cytoplasm by kelch-like ECH-associated protein 1–Cul3 E3 ligase complex, which ubiquitinates it, targeting it for degradation by the proteasome system (114, 240, 273, 562). The Nrf2 pathway is dysregulated in HD, which could increase free radical levels and cause mitochondrial dysfunction (249). Homologous to the E6-AP carboxyl terminus domain and ankyrin repeat containing E3 ubiquitin–protein ligase 1, an activator of Nrf2, is downregulated in HD leading to suboptimal responses to oxidative stress (435).
f. Mitochondrial dysfunction in HD
The involvement of mitochondrial dysfunction in HD was first suggested by nuclear magnetic resonance spectroscopy, which revealed elevated lactate levels in the cortex and striatum of HD patients, which correlated with the CAG repeat length (247, 276). Biochemical studies also confirmed the reduced activity of mitochondrial complex II, III, and IV in postmortem HD caudate putamen and cerebral cortex (54, 58, 189). Lymphocytes derived from HD patients also exhibit defective mitochondrial bioenergetics, which are dependent on the CAG repeat length (467). In addition, 3-nitropropionic acid, a mitochondrial complex II inhibitor, induces HD-like symptoms in mice (57). In HD, two major components of mitochondrial complex II, the 30-kDa iron/sulfur (Ip) subunit and the 70-kDa FAD (Fp) subunit, are preferentially decreased in the striatum of HD (40). Decreases in SDH activity have deleterious consequences, as it is a component of both the ETC and the Krebs cycle and would affect both energy-producing pathways. Deficits in complex II play pivotal roles in mediating neurodegeneration, and overexpression of complex II ameliorates disease progression (118). Mitochondrial deficits in HD are also caused by dysregulation of transcription factors and their coactivators that modulate mitochondrial function. Peroxisome proliferator-activated receptor gamma (PPARγ) regulates several processes such as fatty acid oxidation, mitochondrial biogenesis, turnover, energy homeostasis, immune responses, and antioxidant defense. The peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) expression is reduced in HD leading to mitochondrial dysfunction. PGC1α plays important roles in mitochondrial biogenesis, respiration, and oxidative stress response (251). Mutant huntingtin represses transcription of PGC1α by cAMP response element-binding protein (CREB)/transcription initiation factor TFIID subunit 4, the transcription factors regulating PGC1α. Deleting PGC1α in HD knockin mice by crossing these mice with PGC1α knockout mice exacerbates degeneration of medium spiny neurons of the striatum leading to motor and behavioral deficits. Thus, restoring PGC1α expression in the striatum by lentiviral injection mitigates symptoms (113). Imbalanced redox in mitochondria is a component of another vicious cycle in which free radical-induced damage compromises basic mitochondrial functions such as respiration, leading to further oxidative stress. Thus, removal of mitochondrial damage by mitochondrial fission or mitophagy becomes necessary to curtail excessive free radical generation and damage. In HD, aberrant mitophagy has been observed in which mutant huntingtin binds to valosin-containing protein (VCP), an ATPase and component of the ubiquitin proteasome system, and causes excess mitophagy and thus neurotoxicity (194). VCP associates with its cofactor UBX-domain containing protein 1 to cause degradation of the outer mitochondrial membrane protein myeloid cell leukemia sequence 1. Mutant huntingtin overactivates this process and compromises mitochondrial integrity (193).
4. Huntingtin as a sensor of oxidative stress
Although most studies have focused globally on oxidative stress mediating cytotoxicity in HD, the role of wild-type huntingtin itself as a redox sensor has been less explored. The N17 domain, the first 17 amino acids at the amino terminus of huntingtin, determines its subcellular localization. N17 is an amphipathic alpha helix (428) and can direct nuclear export (346) and anchoring to ER membranes to target huntingtin to the cytoplasm under normal conditions (18). Post-translational modifications of the N17 domain, such as phosphorylation, acetylation, and SUMOylation, regulate huntingtin localization (190, 485, 508). Stress-dependent phosphorylation of two serine residues Ser13 and Ser16 promotes nuclear localization by preventing interaction of huntingtin with chromosome region maintenance protein 1 (CRM1) thereby preventing nuclear export (17). During oxidative stress conditions, the oxidation of a methionine residue at Met8 on the N17 domain can stimulate phosphorylation of the domain and thus nuclear localization. The response of mutant huntingtin to oxidative stress could be a slower process becoming more pronounced in later stages of disease progression where the oxidative burden is higher. While oxidation of Met8 on mutant huntingtin also triggers its nuclear translocation, export from the nucleus is hampered due to the aberrant interaction with CRM1 resulting in nuclear accumulation and pathology. An unanswered question is why huntingtin translocates to the nucleus in response to oxidative stress. Wild-type huntingtin has been shown to have roles in the nucleus, such as stimulation of transcription of brain-derived neurotrophic factor (573). Thus, it is possible that huntingtin could be involved in transcriptional regulation of stress response genes.
B. Alzheimer's disease
AD is the most prevalent neurodegenerative disease reported till date. Symptoms of AD include dementia, impaired spatial memory, and other cognitive deficits. The disease has multifactorial origins unlike HD, which is caused by a single mutation in a single gene and therefore monogenic. AD can arise due to sporadic or familial causes. The region of the brain first affected in AD is the entorhinal cortex. The disease predominantly affects the cerebral cortex and the hippocampus (Fig. 4). Aggregation of β-amyloid (Aβ) and tau proteins is the hallmark of the disease, causing deposition of amyloid plaques and neurofibrillary tangles, respectively. The amyloid precursor protein (APP) is the precursor of Aβ peptides. APP can undergo cleavage at different sites by two major pathways: the nonamyloidogenic pathway and the amyloidogenic pathway (Fig. 8). Tau aggregation, on the contrary, is linked to its hyperphosphorylation (336). Elevated oxidative stress has also been associated with AD, similar to several neurodegenerative diseases (60, 491). Increased levels of lipid peroxidation, protein carbonylation, and protein nitration have been reported in AD. As in HD, elevated DNA damage has also been associated with AD (52).

1. Amyloid beta, tau, and oxidative stress
Aβ(1 –42), generated by cleavage of APP, by enzymes such as β-secretase 1 (BACE1), induces oxidative stress (Fig. 8). Aβ has been shown to reduce metal ions such as Fe3+ and Cu2+ to Fe2+ and Cu1+, which can generate •OH radicals by Fenton chemistry (229, 230). The levels of oxidative damage have been positively correlated to expression of BACE1 (49). BACE1, a transmembrane aspartyl-protease, is the major β-secretase, which cleaves APP to generate the toxic Aβ(1 –42) fragment (521). Deletion of BACE1 prevents APP processing to generate Aβ both in mice and cell culture (64, 319, 426). Formation of Aβ leads to activation of the Jun N-terminal kinase (JNK) pathway, which has been implicated in upregulation of BACE1 (191, 552). Thus, Aβ and BACE1 are components of a toxic feedforward cycle where increased oxidative stress promotes BACE1 production, which further increases Aβ production leading to oxidative stress and further BACE1 activity (Fig. 8). Increased levels of activated JNK have been reported in postmortem AD samples (570). Thus, JNK signaling constitutes a therapeutic target for AD (553). Soluble Aβ can impair cysteine and GSH disposition in cells by inhibiting the excitatory amino acid transporter 3 (EAAT3), the neuronal cysteine transporter (223). EAAT3 plays a critical role in regulating redox balance in neurons, and its depletion can promote elevated oxidative stress and age-dependent neurodegeneration (15). This component of redox imbalance can feed into the toxic cycle described earlier.
Similarly, accumulation of hyperphosphorylated tau has been reported to cause oxidative stress, and ROS have been shown to stimulate tau hyperphosphorylation (490, 569). Tau is the major constituent of neurofibrillary tangles and higher order structures generated by formation of disulfide bridges via cysteine residues (441). Interplay between tau and amyloid beta peptides has been reported. Amyloid beta peptides cause aggregation of tau to promote neuronal dysfunction. A variety of Aβ peptides are generated by cleavage of APP, which include Aβ40 and Aβ42. It was shown recently that Aβ*56, a 56-kDa oligomer that accumulates before early symptoms of AD manifest, alters neuronal signaling by activating CamKII, a kinase that phosphorylates tau (13). In addition to these changes, the metal content of AD brains is also higher with the concentration of iron in amyloid plaques almost twice as that of neighboring tissues, while copper and zinc content are threefold higher, which mediate oxidative stress (412).
2. Mitochondrial dysfunction
Similar to HD, mitochondrial dysfunction has been reported in AD. Sporadic mtDNA deletions up to 9 kb long with a commonly occurring specific 5-kb deletion have been attributed to oxidative damage (554). The mtDNA of cortical neurons in AD patients <75 years of age has 15 times more of the 5 kb mtDNA deletion mutations than age-matched controls (107).
3. Transcriptional dysregulation
In AD, the repressor element 1-silencing transcription factor (REST), also known as neuron-restrictive silencer factor, is depleted causing oxidative stress and suboptimal stress responses (315). REST is a repressor with functions during embryonic development of neuronal genes and becomes downregulated once terminal neuronal differentiation has occurred. REST is also induced during aging and regulates genes that mediate cell death and stress resistance. Some of the cell death proteins regulated by REST include the p38 MAP kinase (MAPK11), BAX, BID, and PUMA (315). Nuclear REST levels are diminished in neurons of affected regions in AD, including prefrontal cortical neurons as well as the hippocampus. Neurons lacking nuclear REST are vulnerable to oxidative stress and Aβ toxicity and display elevated levels of apoptosis inducing genes. In cell culture, knockdown of REST results in elevated oxidative stress, which is rescued by REST overexpression or by treatment with the antioxidant NAC (315).
4. Aberrant nitrosylation
Nitrosylation is a post-translational modification elicited by the gaseous signaling molecule •NO on reactive cysteine residues (484). Nitrosylation modulates several physiologic processes in cells and influences protein activity, protein/protein interactions, and localization. Low levels of •NO are usually beneficial to cells, but higher concentrations can elicit cytotoxicity. In the brain, neuronal nitric oxide synthase (nNOS) is the predominant enzyme that generates •NO, although it is also formed by inducible nitric oxide synthase (iNOS) under conditions of stress. Excess production of •NO can cause protein misfolding. For example, PDI is nitrosylated in brains of AD and PD patients, which causes improper folding of toxic proteins (512). PDI is a chaperone enzyme present in the ER that modulates formation of disulfide bonds during their synthesis and maturation (162, 186). In neurodegenerative disorders and during ischemia, accumulation of denatured proteins causes ER dysfunction. Under such conditions, PDI induction occurs as an adaptive response, which is compromised by nitrosylation (226, 502). Cell death induced by the ER stressors, thapsigargin and tunicamycin, in the cell line SH-SY5Y was largely prevented by wild-type PDI, but this protection was abrogated in the presence of an •NO donor. •NO-mediated nitrosylation of PDI inhibits its catalytic activity, causing accumulation of polyubiquitinated proteins and activating the unfolded protein response (512). In accordance with these findings, in the brains of AD and PD patients, accumulation of nitrosylated PDI was observed (512). Nitrosylation also disrupts metabolic processes such as glycolysis and antioxidant defense in cells. In addition, ONOO− generated from •NO mediates protein nitration. A well-characterized mode of •NO action in the brain is the activation of NMDARs. The neurotransmitter glutamate stimulates NMDARs and causes Ca2+ influx, which activates nNOS to generate •NO (501). The outcome depends on whether the NMDAR is synaptic or extrasynaptic. Under normal conditions, activation of synaptic NMDARs results in production of physiological levels of •NO, required to promote neuronal differentiation and survival as well as normal synaptic plasticity (317). •NO activates the CREB pathway to long-term potentiation. In contrast, excess stimulation of “extrasynaptic” NMDARs results in elevated levels of •NO and free radicals, which contribute to synaptic dysfunction by nitrosylating dynamin-related protein 1 (Drp1) and cyclin-dependent kinase 5 (Cdk5) to mediate neurodegeneration (350). Dynamin-1-like protein is a GTPase that regulates mitochondrial fission. The S-nitrosylation of Drp1 causes dimer formation and increased GTPase activity, thus accelerating the process of mitochondrial fragmentation and contributing to neuronal synaptic damage or cell death (93). Cdk5 is a cyclin-dependent, neuron-specific kinase, which has roles in cell survival, axon guidance, neuronal migration, and regulation of synaptic spine density. S-nitrosylation of Cdk5 enhances its kinase activity leading to hyperphosphorylation of its substrate tau, in AD (32). In addition, SNO-Cdk5 can transfer the NO group by transnitrosylation to Drp1, its substrate (forming SNO-Drp1), in this manner possibly mediating the synaptic damage. Soluble Aβ extensively demonstrated to preferentially activate extrasynaptic GluN2B (NR2B)-containing NMDARs (265, 302, 498). Thus, targeting the extrasynaptic receptors has therapeutic benefits. The FDA-approved drug, memantine, preferentially blocks extrasynaptic oversynaptic NMDARs (545) and delays symptoms in AD.
C. Parkinson's disease
PD was first described over 200 years ago as a shaking palsy and was considered to be a disease, which affects the substantia nigra pars compacta (SNpc) and striatum causing motor deficits (Fig. 4) (399). It is the second most prevalent neurodegenerative disease and affects 2–3% of aging populations older than 65. The incidence of the disease is 5 to >35 cases per 100,000 individuals (511). PD is associated with loss of dopaminergic neurons in the SNpc, leading to depletion of the neurotransmitter dopamine (DA). With advancements in PD research, it is now clear that the disease affects not only the motor functions but other physiological processes such as cognition, sleep, and smell as well. Similar to AD, PD can also arise due to genetic causes as well as occurring sporadically, with the sporadic forms comprising the vast majority of cases reported. Familial or heritable forms of PD constitute only 15% of the cases, of which 5–10% of PD patients have monogenic forms of the disease, exhibiting a classical Mendelian type of inheritance (127, 399). Several genes have been linked to developing PD. SNCA, encoding α-synuclein, was the first gene definitely associated with familial PD (400, 401). In addition, mutations in leucine-rich repeat kinase 2 (LRRK2), parkin (PARK2), PTEN-induced putative kinase 1 (PINK1), and DJ-1 (PARK7) are linked to familial PD in various populations. Mutations in LRRK2 are most commonly associated with sporadic and familial late-onset PD (117). LRRK2 is a large multidomain protein that regulates several cellular processes, including neurite outgrowth and synaptic morphogenesis, membrane trafficking, autophagy, and protein synthesis, and has been considered a therapeutic target for PD. Mutations in PARK2, PINK1, and DJ-1 are associated with autosomal recessive forms of PD and tend to have an earlier age of onset (458). Other genes linked to autosomal dominant forms of PD include SNCA, LRRK2, vacuolar protein sorting 35 (VPS35), DNAJC13 (encodes a chaperone protein named receptor-mediated endocytosis 8), and coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2), among several others.
1. Redox stress in PD
Dysregulation of redox homeostasis occurs at multiple levels in PD (Fig. 9). A strong link between oxidative stress and cell death of dopaminergic neurons has been established. DA is synthesized from tyrosine by the action of tyrosine hydroxylase, which requires iron as a cofactor, and aromatic amino acid decarboxylase (471). Loss of DA leads to symptoms such as resting tremor, rigidity, bradykinesia, sleep disorder, cognitive deficits, and depression (429). PD has been associated with decreased levels of GSH and other thiols, which are vital for the maintenance of redox balance (387, 391, 392, 523). The generation and sources of ROS in PD include the metabolism of DA, mitochondrial dysfunction, iron deposition, inflammation, aberrant calcium handling, and aging. PD-associated gene products, including DJ-1, PINK1, parkin, alpha-synuclein, and LRRK2, also impact mitochondrial function leading to augmented ROS generation and susceptibility to oxidative stress. In addition, cellular homeostatic processes, including the ubiquitin–proteasome system and mitophagy, are impacted by oxidative stress. On the contrary, increased uptake of DA itself can cause oxidative stress (335). It has been observed that injection of DA into the rat striatum resulted in loss of dopaminergic cells, which could be rescued by antioxidant coinjection (212). Preventing DA degradation caused accumulation of cytosolic DA and caused neurotoxicity, while blocking the conversion of

Modified adducts of DA derived from docosahexaenoic acid (C22:6/omega-3) and arachidonic acid (C18:4/omega-6), the major PUFAs in the brain, can mediate neurotoxicity. Of these, hexanoyl dopamine (HED), an arachidonic acid-derived adduct, is extremely toxic to human dopaminergic neuroblastoma SH-SY5Y cells. Generation of ROS and mitochondrial dysfunction has been linked to HED-induced cell death (309).
2. Aggregation of α-synuclein
A hallmark of PD is the deposition of α-synuclein in the cytoplasm of certain neurons in several regions of the brain (51). Aggregated α-synuclein is a constituent of Lewy bodies (181, 479), which initially accumulate in cholinergic and monoaminergic brainstem neurons and also in the neurons of the olfactory region. As the disease progresses, Lewy bodies are also found in limbic and neocortical brain regions. Mutations in the α-synuclein gene, SNCA, confer an increased risk for PD (401). The A53T mutant of α-synuclein accelerates its aggregation and disease progression (106). Inflammation also contributes to aggregation of α-synuclein, including the wild-type protein (528). The function of α-synuclein, a 140 amino acid protein, is not well understood, but has been linked to modulation of mitochondrial morphology and function, protein chaperone function, and intracellular trafficking. Similar to most neurodegenerative diseases involving protein aggregation, soluble oligomeric forms of α-synuclein are thought to be the toxic species (522, 536). Both soluble and fibrillar synuclein bind to metal ions to induce oxidative stress (124). α-Synuclein can also bind to proteins and alter their activity or conformation. Aggregated α-synuclein binds to SOD1 and increases its aggregation (215). The aggregated α-synuclein also binds to tyrosine hydroxylase and inhibits its activity (388). Complex I activity of mitochondria is affected by soluble prefibrillar α-synuclein via Ca2+-mediated mitochondrial swelling and depolarization and cytochrome c release. (320). These findings are in agreement with recent findings that administration of fibrillated α-synuclein to primary ventral midbrain neuron cultures decreased synaptic protein levels, caused alterations in axonal transport-related proteins, and DNA damage. Mitochondrial impairment (including modulation of mitochondrial dynamics-associated protein content) enhanced oxidative stress, and an inflammatory response was also observed in these studies (505). Activating stress-responsive pathways regulated by the master regulator of antioxidant response genes, Nrf2, rescues neurotoxicity by reducing levels of α-synuclein by promoting its degradation (474).
3. Mitochondrial dysfunction in PD
Evidence for the involvement of mitochondrial dysfunction in PD first came from symptoms developed by users of drugs, which were identified as 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) with trace amounts of 1-methyl-4-phenyl-4-propionoxy-piperidine that appeared to damage neurons of the substantia nigra (289). Within hours of publication of this discovery, MPTP, which was a promising and valuable tool to study PD, was sold out, as declared by its manufacturer Sigma (288). MPTP is a by-product during the chemical synthesis of the opiate meperidine (571). Later it was shown that MPTP is converted to the toxic metabolite, 1-methyl-4-phenylpyridinium or MPP+, which selectively damages the dopaminergic regions of the brain, destroying neurons in the substantia nigra (290, 328). Autoradiography using [3H]MPP+ in slices of rat brain shows high densities in the caudate-putamen and nucleus accumbens (245). It was later shown that MAO converts MPTP into MPP+ (92). Later MAO B, but not MAO A, was identified as the specific enzyme involved in this conversion (214). Once formed, MPP+ is enriched in mitochondria, where it inhibits the mitochondrial complex I of the ETC (112, 413 –415). Thus, MPP+ causes decline in adenosine triphosphate (ATP) levels and also induces oxidative stress by redistribution of DA (313, 314).
Complex I defects have been widely observed in PD. Analysis of postmortem samples from PD patients revealed a decrease in complex I in the substantia nigra and prefrontal cortex (379, 455). The components of complex I also exhibited increased oxidative damage as assessed by increased protein carbonylation, a marker of oxidative damage (261). Reduction in complex I activity in PD has also been reported in several other studies (170, 261). Exposure to the pesticide, rotenone, which inhibits complex I has been linked to an increased risk of developing PD (258). Dopaminergic neurons are highly vulnerable to complex I inhibitors whose toxicity is partly mediated by generation of ROS (192, 334, 430). Studies have revealed that inhibition of complex I is not sufficient to induce cell death of dopaminergic neurons (96). In addition to generation of oxidative stress, failure of mitochondrial bioenergetics to generate ATP has been observed in PD. Several studies have reported that cell death caused by inhibition of complex I in PD cannot be fully ascribed to ROS or prevented by antioxidants as complex I inhibitors have other effects such as destabilization of the cytoskeletal network in addition to other effects such as activation of inflammatory pathways (27, 70, 142).
In addition to suboptimal electron transport in PD, abnormal mitochondrial homeostasis has also been observed in PD (397). Healthy neurons remove damaged mitochondria by a quality control process termed mitophagy involving the PD-linked proteins PINK1 and parkin. The loss of mitochondrial membrane potential causes recruitment of PINK1 accumulation to the outer mitochondrial membrane. PINK1 recruits the E3 ubiquitin ligase, parkin, to the mitochondria leading to the ubiquitination of mitochondrial membranes, and removal of the defective mitochondria (364). Loss of parkin or PINK1 compromises the ability to remove damaged mitochondria leading to an accumulation of these dysfunctional organelles leading to early-onset PD.
4. Iron accumulation in PD
Iron accumulation in PD was observed as early as 1924 (301). Initial studies utilized Prussian blue staining to localize iron deposits (138). Nuclear magnetic resonance studies have revealed that iron deposition and striatal atrophy are associated with disease progression in PD (225). Iron deposition is not unique to PD, as elevated iron content is also observed in AD, HD, and ALS prompting an additional categorization of diseases involving iron: the neurodegeneration with brain iron accumulation syndromes. In PD, iron deposits are especially high in the substantia nigra. The accumulation of iron is directly correlated with disease severity and motor deficits (333). Elevated iron levels can mediate aggregation of α-synuclein (515). Proteins involved in iron metabolism are dysregulated in PD. Expression of ferritin, the iron binding protein, is diminished in PD patients. On the contrary, the transporter of iron, the divalent metal ion transporter, is upregulated (444). In addition, ceruloplasmin, an iron export ferroxidase, is decreased by more than 80% in the substantia nigra of idiopathic PD brains. Ceruloplasmin knockout mice have elevated iron levels and develop some symptoms of PD, which can be rescued by iron chelation. Similarly, in the MPTP chemical model of PD, infusion of ceruloplasmin decreases neurodegeneration (20). One of the upstream events and contributing factors to iron accumulation is nitrosative stress (403). The inhibitor of NO synthase, 7-nitroindazole, prevents MPTP-induced toxicity in mice (461). The expression of β-APP, which promotes neuronal iron export, is decreased in dopaminergic neurons of the substantia nigra of PD brains. In agreement with these findings, APP −/− mice develop iron-dependent nigral cell loss (20). Recently, a new form of cell death involving iron, ferroptosis, was identified (131). Initiation of ferroptosis is specific to intracellular iron, but not other metals, and has been shown to be genetically, morphologically, and biochemically distinct from apoptosis, necrosis, and autophagy (131, 550). Although ferroptosis has several features similar to oxytosis, another form of cell death mediated by glutamate toxicity, there are several differences (550). In oxytosis, elevated calcium ions activate a number of serine proteases, calpains, and phospholipases, leading to mitochondrial damage and nuclear translocation of AIF (apoptosis-inducing factor). These changes are necessary for executing oxytosis, whereas they are dispensable for ferroptosis. Thus, it has been suggested that oxytosis may be a combination of the more general ferroptosis and a specialized type of damage induced by glutamate. In ferroptosis, depletion of cysteine or GSH can initiate the process due to elevation of iron. Lipid peroxidation is a consequence of ferroptosis induction. Induction of ferroptosis due to cysteine depletion causes degradation of ferritin (i.e., ferritinophagy), the iron binding protein, which releases iron via the nuclear receptor coactivator 4-mediated autophagy pathway (292). Ferroptosis was characterized in Lund human mesencephalic cells and confirmed in organotypic slice cultures ex vivo and in the MPTP mouse model. The cell death pathway in this instance differs from the classical ferroptosis reported previously, in that activation of protein kinase Cα (PKCα) can initiate ferroptosis. Ferroptosis inhibitors, such as ferrostatin-1, are beneficial in PD models (132). Targeting iron disposition in PD therefore is being explored to manage disease symptoms (112, 129).
5. Dysregulated calcium signaling in PD
Several studies show that disturbed Ca2+ contribute to neurotoxicity in PD (496, 560). Because of its involvement in cellular processes ranging from the regulation of enzyme activity to programmed cell death, calcium is under very tight homeostatic control.
6. Inflammation in PD
PD is also characterized by microglial activation, accumulation of proinflammatory cytokines, and chronic inflammation (179, 351). Analysis of postmortem brains of PD patients revealed the presence of macrophages and microglia in the SN (220, 342). Early studies revealed that administration of lipopolysaccharides (LPSs) into the brain of mice and rats selectively induces cell death of dopaminergic neurons (77, 216). Mice overexpressing α-synuclein developed persistent neuroinflammation after LPS challenge, with prolonged microglial activation, expressing iNOS and NOX enzymes that generate •NO and O2 •−, respectively (168). Thus, activation of microglia contributes to elevation of free radicals as demonstrated by these and other studies (564). In these studies, inhibition of iNOS and NOX enzymes blocked pathology and neurodegeneration. The contribution of α-synuclein to inflammatory processes has also been demonstrated in microglial cell lines, where exposure to extracellular α-synuclein led to increased secretion of the cytokines IL1α, IL1β, TNFα, and IL6 (12). A possible mechanism underlying activation of microglia by α-synuclein involves Nurr1, an orphan nuclear receptor belonging to (NR)4 family of orphan nuclear receptors. Nurr1 has been reported to inhibit expression of proinflammatory neurotoxic mediators in both microglia and astrocytes (442). Levels of Nurr1 have been shown to be decreased in human PD, and mutations resulting in reduced expression of Nurr1 are associated with late-onset familial PD (179, 352).
7. Dysregulated gasotransmitter signaling in PD
Disrupted gasotransmitter signaling involving H2S and •NO has been associated with PD. The decrease of endogenous H2S production in the substantia nigra of 6-OHDA-treated rats reveals a link between H2S and PD (227). Accordingly, administration of the H2S donor, NaHS, reverses motor deficits, reduces loss of tyrosine hydroxylase-positive neurons in the substantia nigra and diminishes markers of oxidative stress such as the elevated MDA levels in 6-OHDA-treated rats. H2S also attenuates 6-OHDA-induced NOX activation. In PD, altered nitrosylation and sulfhydration of parkin, an E3-ubiquitin ligase, mediated by •NO and H2S, have been observed. The activity of parkin is inhibited, by nitrosylation, leading to aggregation of its substrate α-synuclein and neurotoxicity (99) (Fig. 10). In contrast, sulfhydration activates parkin to degrade misfolded proteins and promote neuroprotection (519). In postmortem PD brains, sulfhydration of parkin is diminished and nitrosylation increased (519). Overexpressing cystathionine β-synthase (CBS), one of the biosynthetic enzymes of H2S, increases parkin sulfhydration and E3 ubiquitin ligase activity. Administration of H2S donors has proved beneficial in PD. Inhalation of H2S by MPTP mice ameliorates symptoms and reduces the death of dopaminergic neurons (266). Treating the 6-OHDA-induced PD mouse model with an H2S releasing
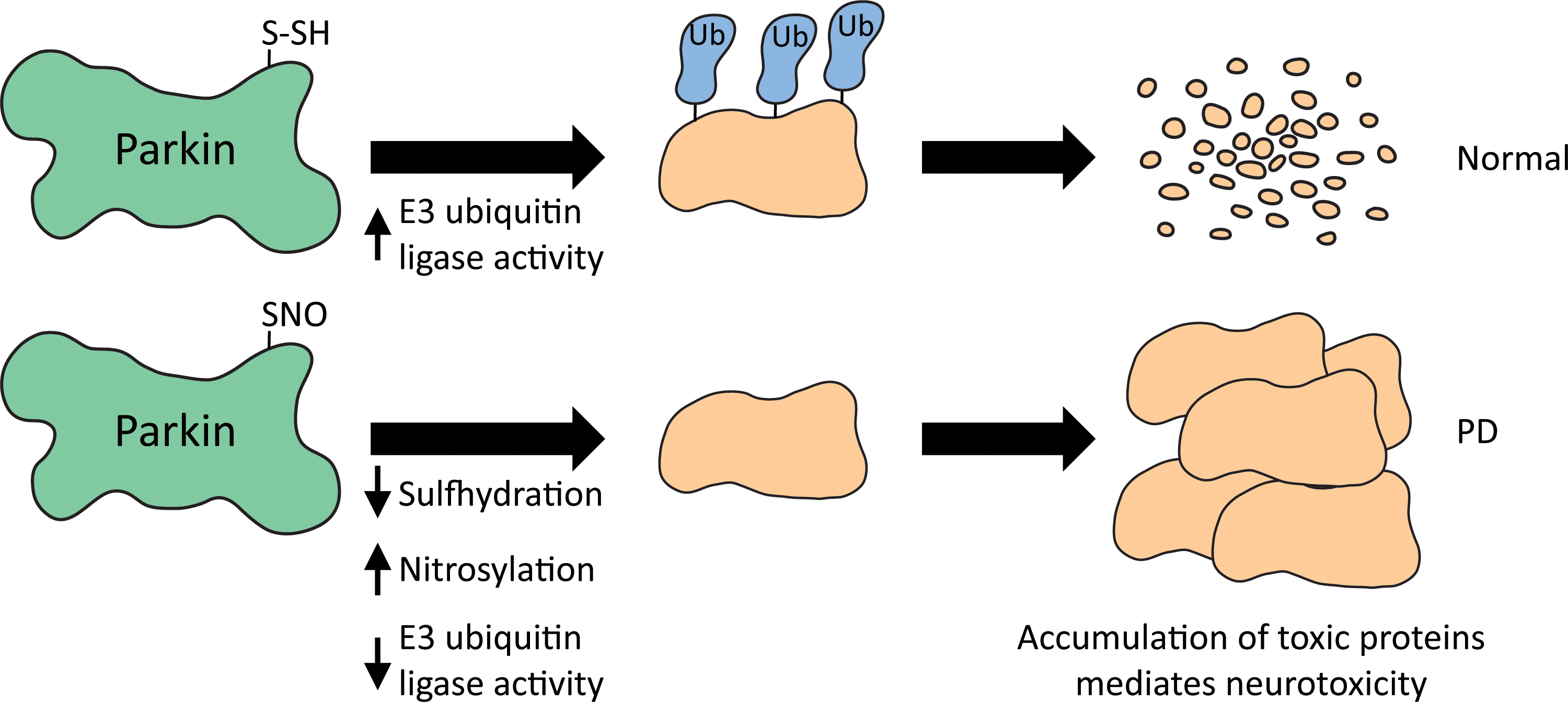
Nitrosylation of parkin also affects other pathways. As parkin is a suppressor of Drp1 expression, which is involved in mitochondrial fission, its nitrosylation abrogates this function. As a result, upregulation of Drp1 occurs with associated abnormal mitochondrial dynamics (568). Similarly, S-nitrosylation of Prx2, a peroxidase that plays important roles in regulation of oxidative stress, inhibits its enzymatic activity and antioxidant defense. Increases of nitrosylated Prx2 have been detected in mouse models of PD as well as human PD (494). Ubiquitin C-terminal hydrolase-1/UCHL1, a deubiquitinating enzyme and a constitutent of Lewy bodies, has been shown to be nitrosylated in vitro. Specific nitrosylation of Cys90, Cys152, and Cys220 occurs, altering the enzymatic activity and structural stability. Nitrosylation destabilizes its structure leading to the formation of an amorphous aggregate, which acts as a nucleating site for native α-synuclein and accelerated aggregation (283). Thus, protein S-nitrosylation can be targeted in neurodegenerative diseases to improve survival and outcome (363). For instance, thioredoxin-mimetic peptides, which catalyze the reduction of SNO, protect cells from nitrosative stress (280). Another example is the use of nitromemantine, which not only blocks overactivated NMDARs but also acts as an NO donor to nitrosylate the receptor and further inhibit its activity (500, 501).
D. Amyotrophic lateral sclerosis
1. ALS and redox imbalance
ALS is a neurodegenerative disease manifesting selective degeneration of upper and lower motor neurons (507). In the United States, ALS is referred to as Lou Gehrig's disease after the famous baseball player who succumbed to the disease. About 90% of ALS cases are sporadic, whereas the remaining (10%) are familial (439). The familial causes include mutations in the genes Cu-Zn superoxide dismutase 1 (SOD1), Tar DNA binding protein (TARDP), fused in sarcoma (FUS), ubiquilin 2 (UBQLN2), VCP, and optineurin (OPTN) among others. Other proteins mutated in ALS include senataxin, ataxin2, HNRNPA2/B1, ELP3, HNRNPA1, alsin, FIG4, VABP, and CHMP2B (327). Similar to most other neurodegenerative disorders, several factors, including oxidative stress, have been observed in ALS (Fig. 11). GSH content in the motor cortex of ALS patients is decreased, which can promote oxidative stress (531). In addition, mitochondrial function is affected in ALS. Mitochondrial dysfunction in ALS was first identified as morphological abnormalities in the skeletal muscle, liver, spinal cord neurons, and motor cortex during postmortem analysis of ALS patients and cell culture models (219). The morphological changes are accompanied by decreased activities of complexes II and IV of the mitochondrial ETC and oxidative damage to mitochondrial protein and lipid components (338, 345).
2. ALS and mutations in SOD1
Among the familial cases of ALS, ∼20% are caused by dominantly inherited mutations in the SOD1 gene. More than a hundred mutations have been identified in the SOD1 gene, but not all of the mutants have been characterized. The most commonly studied genetic mutation in SOD1 is the G93A mutation, which is a gain-of-function mutant that causes degeneration of motor neurons (197, 433). The mutation causes the protein to aggregate and mediate toxicity. Transgenic mice expressing human SOD1 harboring the G93A mutation become paralyzed in one or more limbs, which arises due to motor neuron loss from the spinal cord, and die within 5–6 months of age (197). Human neuroblastoma SH-SY5Y cells expressing G93A SOD1 display an increase in both cytosolic and mitochondrial ROS production, which can be mitigated by NAC, which also serves as a precursor for GSH biosynthesis (42). Oxidative stress has been associated with disease progression in ALS caused by SOD1 mutations, but the precise mechanisms by which motor neurons degenerate have not been well characterized. Cysteine residues have been implicated in the aggregation of SOD1 (111). Human SOD1 has four cysteine residues, of which two, Cys57 and Cys146, form an intramolecular disulfide bond (499). Cys57 and Cys146 are highly conserved in yeast, plants, flies, fishes, and mammals. The other two cysteine residues Cys6 and Cys111, which are not conserved, do not form a bridge. SOD1 proteins aggregate when they are in the metal-free form and, depending on the mutation, rates of oligomerization vary. These oligomers are formed through oxidation of Cys6 and Cys111 and are stabilized by hydrogen bonds, between beta strands, forming amyloid-like structures. Cys111 is present in human and ape but not mouse SOD1. Cys111 is surface exposed, reactive, and prone to modifications. Cys111 has a relatively low pK a, leading to its ionization at physiological pH, which facilitates its modification by a wide variety of reactive cysteine interacting compounds (56, 68). Cys111 was shown by mass spectrometry to be modified by a persulfide group (121). Modified SOD1 is more resistant to oxidation-induced aggregation caused by copper and H2O2. Crystallographic analysis confirms that Cys111 is modified by a covalent polyheptane sulfane sulfur (559). Sulfane sulfur is elemental sulfur that has six valence electrons with no charge, which can bond together to form hydropersulfides (R-S-SH) and polysulfides (-S-Sn-S-). Sulfane sulfur can be derived from H2S and act as a signaling molecule in vivo (5, 268, 470). The interaction of SOD1 with H2S has been reported to increase its activity (462). Cys111 also plays a role in the mitochondrial localization of SOD1 (147). SOD1 enters the mitochondria as the metal-free form. In order for SOD1 to be targeted to mitochondria, interaction with the protein, CCS, which is also redox sensitive, is required (259). Oligomers formed by mutant SOD1 proteins associated with the mitochondria cause a shift in the redox state of the organelle and impairment of respiratory complexes in the motor neuron cell line NSC34. Association of mutSOD1 with mitochondria decreases the reduced/oxidized glutathione ratio (GSH/GSSG). However, NSC-34 cells expressing Cys6F/Cys111S mutSOD1 do not exhibit alteration of the GSH/GSSG ratio in the mitochondria and impairment of the respiratory chain, indicating that a link exists between mitochondrial localization and toxicity of mutSOD1. In another study, generation of the H46R SOD1-expressing mouse model of ALS where Cys111 is mutated delayed disease progression (361).
In addition to these changes, elevated H2S production has been reported in ALS (120). Increased levels of H2S are detected in the tissues of the G93A mouse model of ALS and in media of spinal cord cultures of these mice. Elevated H2S was also detected in cerebrospinal fluid of ALS patients, indicative of aberrant gasotransmitter signaling in ALS. Decreased cysteine levels have been reported in the plasma of G93A mice (25). Supplementation of the diet with a cysteine-rich whey isolate rescues GSH content in tissues and delays disease onset in the G93A mice (434). The involvement of redox imbalance in ALS is further supported by studies involving the G93A mice on a gclc deleted background. GCLC (glutamate cysteine ligase light chain) participates in the biosynthesis of GSH; its deletion leads to decreased antioxidant capacity, mitochondrial dysfunction, and accelerated disease progression of ALS (520).
3. The C9ORF72 model of ALS
One of the leading causes of ALS as well as frontotemporal dementia (FTD) is a mutation in the noncoding region of the chromosome 9 open reading frame 72 (C9ORF72) locus, where an expansion of the hexanucleotide GGGGCC occurs (125, 201, 324, 421). The repeat expansion was found in 11.7% of familial FTD and 23.5% of familial ALS (125). The wild-type C9ORF72 allele typically has less than 20–25 copies of these hexanucleotide repeats, whereas the C9ORF72 ALS/FTD patients can have repeats up to thousands (125, 176, 421). One of the first models of ALS with an expanded hexanucleotide repeat (generated by transgene delivery mediated by somatic transduction of adeno-associated virus [AAV] carrying the G4C2-repeat DNA to the central nervous system) resulted in mice that had deposition of the dipeptide repeats in the brain. In addition, these mice displayed cortical neuron and cerebellar Purkinje cell loss, astrogliosis, and lower body weight in addition to behavioral abnormalities, including hyperactivity, anxiety, antisocial behavior, as well as motor deficits (90). Bacterial artificial chromosome DNA clones harboring either partial- or full-length C9ORF72 gene with the hexanucleotide repeats were also used to generate transgenic mice, of which only the full-length transgene elicited abnormalities such as nucleolar stress and downregulation of immunomodulatory and extracellular matrix pathways (370, 394). The C9ORF72 protein has roles in membrane trafficking and autophagy (299, 464, 530). Three mechanisms have been proposed to mediate neurotoxicity: diminished C9ORF72 protein levels, generation of toxic RNA species, and noncanonical translation leading to accumulation of dipeptide repeat proteins (DPRs). Possible reasons for decreased expression of C9ORF72 protein include epigenetic silencing and transcriptional instability (39, 200). The expansion causes aberrant translation in all three frames, which leads to production of DPRs such as poly GA, poly GR, and poly GP (Fig. 12). The dipeptide proteins are produced by repeat-associated non-ATG-dependent translation or RAN translation (101). Expression of poly GR in human neurons causes mitochondrial dysfunction by binding to mitochondrial ribosomal proteins, which leads to oxidative stress. Increased DNA damage and oxidative stress have been observed in induced pluripotent stem cell (iPSC)-derived C9ORF72 motor neurons in an age-dependent manner (312). The DNA damage increases as a function of age. Transfection of a poly GR construct into wild-type cells induces oxidative stress and DNA damage, and increased toxicity. Mitochondria-generated ROS are also increased in these cells. More importantly, pharmacological or genetic reduction of oxidative stress partially suppresses these detrimental effects in human motor neurons and flies (312).
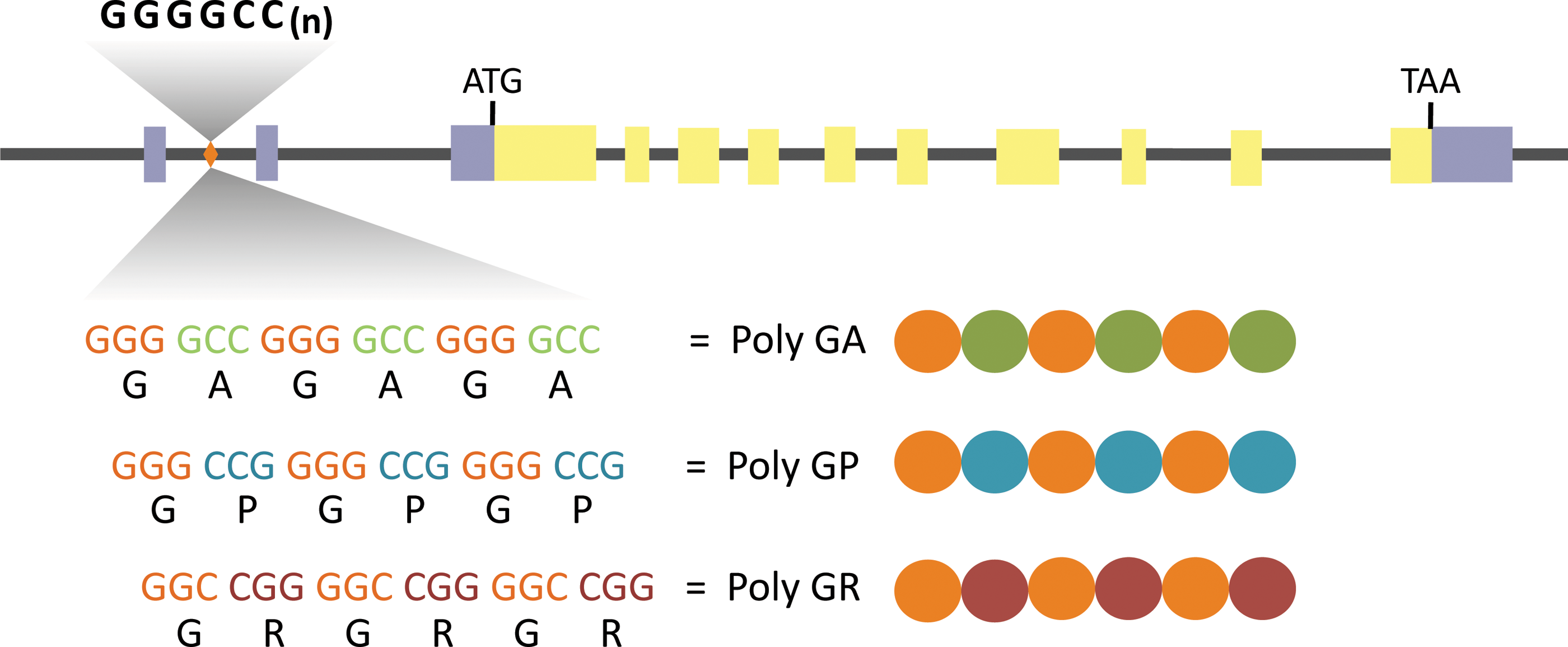
E. Autism spectrum disorders
Autism spectrum disorders (ASD) are a group of neurodevelopmental disorders characterized by impaired social interactions, communication, repetitive stereotypic behaviors, and abnormalities in language and perception. Other disorders such as Asperger's syndrome, childhood disintegrative disorder, and pervasive developmental disorders, not otherwise specified, which share certain behavioral features with autism, have been collectively designated as ASD. The exact etiology of the disorders has been elusive, but probably involves genetic components as well as environmental contributions (393). Genetic factors have been challenging to elucidate as they are highly heterogeneous, exhibiting varying gene/gene and gene/environment interactions with different degrees of penetrance. Thus far, no reliable markers of ASD have been identified. The prevalence of autism is about 7.6 in 1000 (33). ASD patients frequently report comorbidities such as sleep disorders, digestive tract problems, and impairment in motor functions. Regardless of the cause of ASD, oxidative stress and suboptimal antioxidant defense have been linked to autism in several studies (83, 188, 242, 285).
1. Mitochondrial dysfunction and oxidative stress in ASD
The first indication that mitochondrial function may be affected in ASD came from the observation that the levels of lactate are higher in the plasma of autistic subjects, suggesting a deficit in OXPHOS (103). Later it was demonstrated that in addition to high lactate levels, there is an accumulation of Krebs cycle metabolites, impaired glucose utilization, and decreased ATP production (311). Reduced expression of all five electron transport complexes has been observed in autistic individuals, with pronounced defects in complex I (84). In addition, marked increases in lipid peroxidation occur in the cerebellum and frontal cortex of autistic children, which may be linked to abnormal energy homeostasis and mitochondrial dysfunction. Mitochondrial copy number is usually proportional to the cellular DNA content (468). A cell typically harbors 2–10 mitochondria under normal conditions (427). This number can change based on the energy requirements of the cell in response to physiologic stimuli, but may not necessarily correlate with increased OXPHOS. Mitochondrial copy number variation occurs in response to stress and has been observed in leukocytes exposed to oxidative stress caused by alteration of plasma antioxidants/prooxidants and oxidative damage to DNA (307). Mitochondrial abnormalities have also been reported in cell lines derived from autistic patients (432). Higher expression of the mitochondrial fission proteins, Fis1 and Drp1, and lower levels of the fusion proteins, Mfn1, Mfn2, and Opa1, have been observed in ASD patients (503). Other studies report decreased levels of mitochondrial SOD2 and elevated oxidative DNA damage. Literature review and meta-analysis revealed decreased blood levels of GSH, glutathione peroxidase, methionine, cysteine, and an increase in oxidized glutathione, GSSG, in autistic patients relative to controls (163). Lymphoblastoid cell lines from children with ASD display higher oxidative stress and decreased GSH levels (431). Increases in cystathionine levels and decreased cysteine and methionine levels have also been observed in the plasma of autistic patients, which could contribute to oxidative stress, as the cysteine/cystine redox couple plays a central role in maintenance of redox balance (23). In addition to low GSH levels, low levels of vitamin B12 have been reported in autism and other neuropsychiatric disorders (566). It remains to be clarified whether altered redox control in patients contributes to ASD. More recently, deficits in the molybdenum cofactor sulfurase (MOCOS), an enzyme involved in purine metabolism, have been observed in autism (146). MOCOS sulfurates the molybdenum cofactor, which activates XDH and aldehyde oxidase 1 (AOX1) (235). Both XDH and AOX1 contribute to redox homeostasis in cells and hence depletion of MOCOS results in elevated oxidative stress.
2. Abnormal transmethylation, reverse transsulfuration, and vitamin B metabolism in ASD
In addition to elevated oxidative stress, abnormal transmethylation has been linked to the pathophysiology of autism. In a study published in 2004, fasting levels of plasma transmethylation and reverse transsulfuration metabolites were found to be abnormal in autistic children (242). The transmethylation pathway and the reverse transsulfuration pathway are linked by the metabolite homocysteine, which is a metabolic decision point, where either cysteine and GSH biosynthesis or S-adenosylmethionine (SAM) and related metabolites can be generated (Fig. 13). Autistic subjects had significantly diminished levels of methionine. Interestingly, a study showed that parents of autistic children have deficits in the methylation and reverse transsulfuration pathway and associated redox imbalance (244). The ratio of S-adenosylmethionine to S-adenosylhomocysteine (SAM/SAH) is decreased indicating altered methylation capacity. These changes can lead to altered epigenetic patterns. Several unbiased genome-wide analyses have implicated genes involved in chromatin remodeling events such as histone demethylation and the recognition of DNA methylation (291). Methylation of the oxytocin receptor (OXTR) promoter is increased in several cases of ASD and is frequently associated with preterm birth, a risk factor for autism (36). OXTR is involved in the regulation response to stress and anxiety, social memory and recognition, and maternal behavior and could underlie some of the behavioral abnormalities observed in autistic patients.
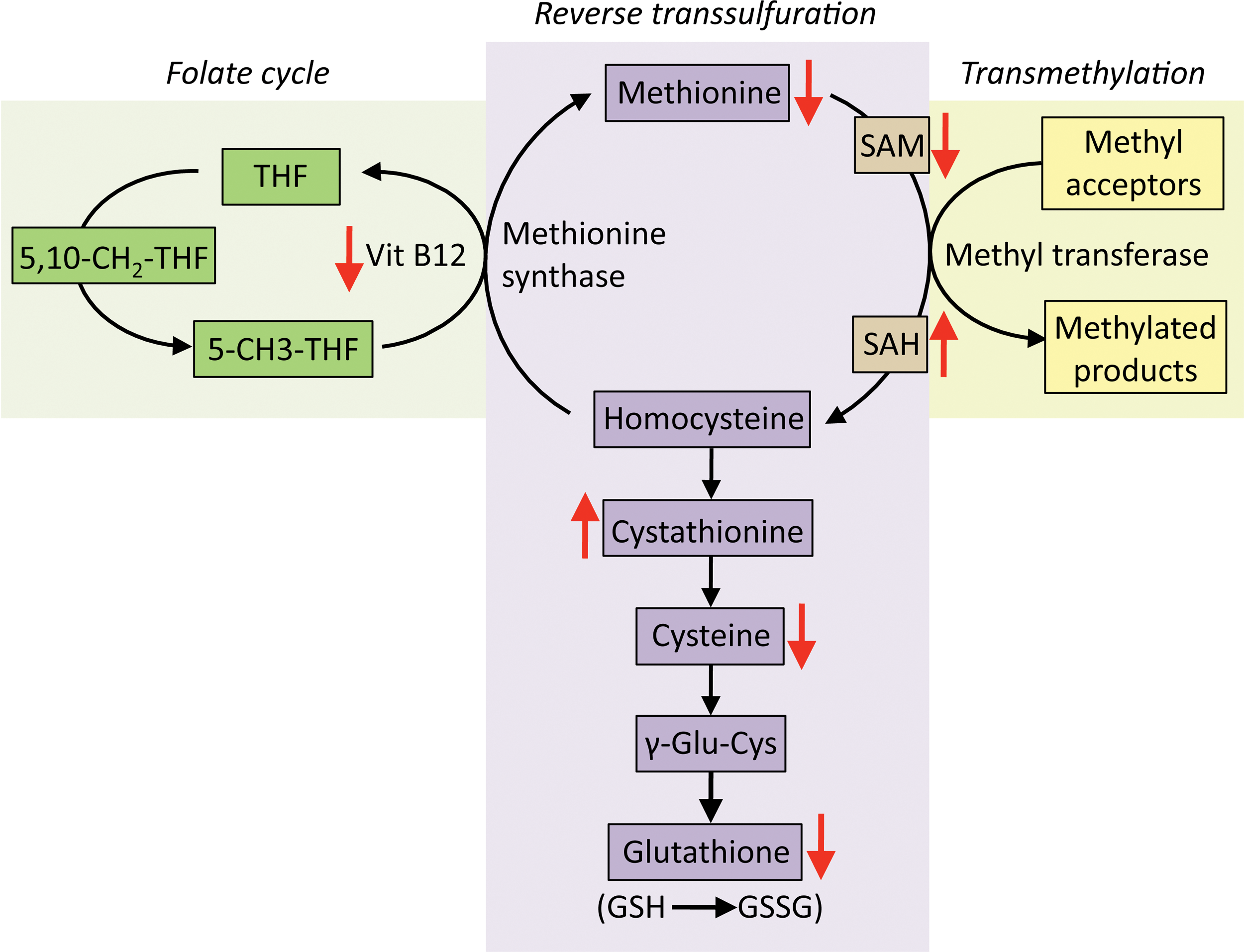
The reverse transsulfuration pathway is also affected in ASD. Total glutathione levels are decreased, with elevations in oxidized glutathione, GSSG, leading to an approximately threefold reduction in the ratio of GSH/GSSG. Cysteine, one of the components of GSH, is diminished, a contributing factor for the low GSH levels and oxidative stress associated with ASD, as the availability of cysteine is the rate-limiting step for GSH biosynthesis. In several studies, elevated homocysteine levels have been observed in autistic children along with reduced Mg2+ levels in hair samples (242, 256, 380). Mg2+ is an essential cofactor for several enzymes, for utilization of vitamin B6, and for certain ATP-dependent reactions. Mg2+ is also involved in the disposition and metabolism of neurotransmitters, which can affect mood and behaviors. Thus, a decrease in Mg2+ can explain decreased ATP synthesis and the suboptimal transmethylation and transsulfuration activities, which can lead to decreased transcription and synaptic plasticity, a phenomenon termed “Magnesium deficiency hypothesis” (256). Thus, increased homocysteine and decreased Mg2+ levels have been proposed to be diagnostic markers in combination with other symptoms. In addition to these aberrations, deficits in vitamin B12 (cobalamin), a cofactor for enzymes such as methionine synthase and methylmalonylmutase, have been observed in the postmortem frontal cortex of autistic patients, a feature also observed in neuropsychiatric conditions such as schizophrenia and aging (566).
3. Treatments for ASD
Based on the observations linking mitochondrial dysfunction and oxidative stress to ASD, treatments include reducing oxidative stress and improving mitochondrial function (164). These strategies include increasing complex I activity by fatty acid and folate supplementation (126). Trials involving antioxidants have shown promise and include treatments with ascorbate (133) and NAC (172, 210). Treatment with methylcobalamin and folinic acid increases cysteine and GSH content in children with autism (243). Other supplements evaluated include
F. The ataxias
The word “ataxia” is derived from the Greek words “a” without and “taxis” meaning order. Clinically, ataxia refers to lack of muscle coordination when voluntary movement is attempted. Ataxia results from the damage or degeneration of neurons that control movement typically in the cerebellum (Fig. 4). Most ataxias have a genetic cause and thus far more than 50 ataxias have been identified. As the topic is vast and heterogeneous with respect to underlying causes and mechanisms, only a few ataxias are described (Fig. 14). Ataxias can be those that are hereditary and those that are not. The classification of the various ataxias has been widely debated, however, the first system, introduced by Harding, broadly classifies ataxias based on clinical and genetic criteria as those with a known metabolic disorder and those with unknown etiology (211). Later, as the causes of the ataxias have become clearer, additional classification criteria have been included and categorized into ataxias that are hereditary, nonhereditary, and acquired (272). The hereditary ataxias are further classified into autosomal dominant, autosomal recessive, and X-linked ataxias.
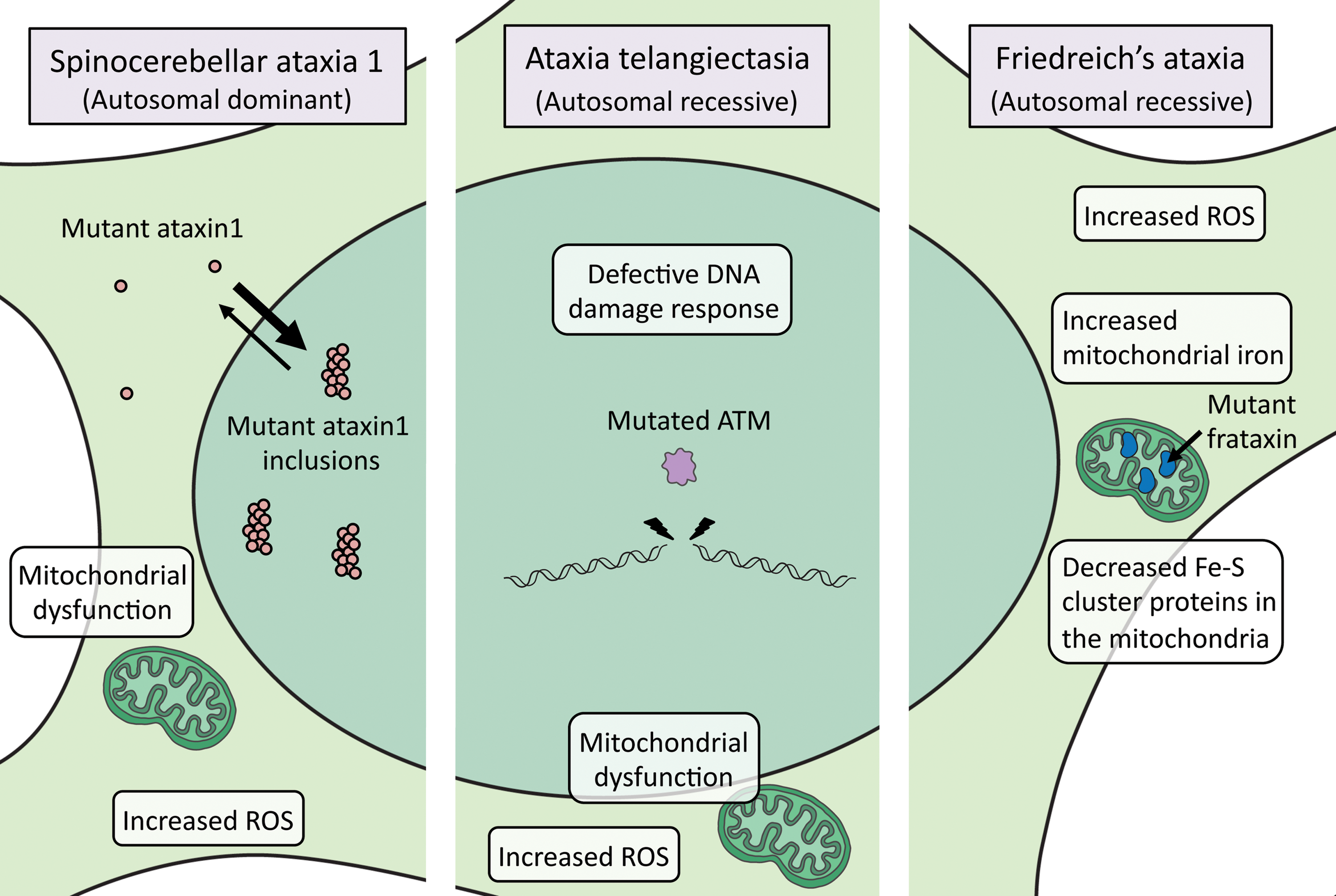
1. Autosomal dominant ataxias: spinocerebellar ataxia
Spinocerebellar ataxia 1 (SCA1) is the first ataxia with an identified genetic etiology (375). The disease is caused by expansion of CAG repeats in the coding region of ataxin-1 on the short arm of chromosome 6, which varies from 6 to 39 repeats in normal individuals and from 40 to 81 repeats in ataxia. The disease is autosomal dominant and results in degeneration of the cerebellum, spinal cord, and brainstem. Symptoms usually begin in the third or fourth decade of life with progressive deterioration in motor function and death 1–20 years after onset. In SCA1, degeneration of cerebellar Purkinje cells as well as certain brainstem nuclei occurs, leading to uncoordinated voluntary movement, impaired balance, and difficulty speaking and swallowing. The CAG repeats are unstable and prone to expansion, leading to the phenomenon of anticipation, where each successive generation has an earlier age of onset. Thus, there is a direct correlation between the size of the polyglutamine tract and the age of onset with juvenile cases of SCA1-bearing larger polyglutamine tracts in ataxin 1. Subsequently, other genetically transmitted ataxias have been identified, caused by mutations in other genes and named according to the order of discovery. CAG expansions have been identified in SCA2, MJD (Machado Joseph disease)/SCA3, SCA6, SCA7, and SCA17 in coding regions. The ataxias, SCA8, SCA10, SCA12, and SCA31, have repeat expansions in noncoding regions. Nonrepeat mutations also occur in ataxias such as mutations in beta-III spectrin (SPTBN2) in SCA5, tau tubulin kinase 2 in SCA11, a potassium channel in SCA13, PKCγ in SCA14, inositol 1,4,5-triphosphate receptor type 1 in SCA15/16, fibroblast growth factor 14 in SCA27, and ATPase family gene 3-like 2 in SCA28. All these mutations are inherited in an autosomal dominant manner.
2. Autosomal recessive ataxias: ataxia telangiectasia
Ataxia telangiectasia is characterized by an unsteady gait (ataxia) and dilation of blood vessels (telangiectases) in the conjunctiva of the eyes or facial skin (449). Positional cloning has localized the mutation to the ATM gene on chromosome 11, which encodes a polypeptide with a PI-3 kinase domain. The disease begins in childhood and is characterized by the degeneration or absence of the thymus and cerebellum and deficits in immune response leading to frequent pulmonary infections in patients. Other symptoms include marked sensitivity to ionizing radiation, premature aging, and chromosomal instability, as well as a predisposition to cancer and death within the second or third decade of life. At the molecular level, cells of telangiectasia patients undergo early senescence and have defects in DNA replication and cell cycle progression with extreme sensitivity to radiation. These cells exhibit cytoskeletal abnormalities and have a greater requirement for serum growth factors. Subsequent characterization of the gene product has revealed that ATM has sequence similarities to proteins in yeast, Drosophila, and mammals, which are involved in the detection of DNA damage and the control of cell cycle progression (450). ATM is a serine/threonine kinase that has several substrates and functions in addition to its role in DNA damage signaling (343). In response to DNA damage, induced by ionizing radiation, the multimeric inactive ATM undergoes autophosphorylation on Ser1981 to dissociate into an active monomeric form (22). ATM can also be activated in response to oxidative stress by forming dimers via a disulfide linkage between cysteine residues of monomers (195). Cells lacking ATM are hypersensitive to oxidative stress, and mice lacking ATM display oxidative injury in tissues with marked damage to the Purkinje cells of the cerebellum (28, 31). ATM-deleted cells have elevated levels of NADPH oxidase 4 (NOX4), which produces ROS such as O2 •−; knocking down NOX4 reduces DNA damage, DNA double-strand breaks, and replicative senescence (533). ATM-deficient cells have constitutively activated antioxidant defense pathways and display disturbed mitochondrial function, which could contribute to increased ROS levels (14). Structural organization of mitochondria is abnormal, and the mitochondrial membrane potential is decreased in these cells along with diminished respiratory capacity. Thus, oxidative stress and mitochondrial dysfunction play significant roles in cellular dysfunction in ataxia telangiectasia. Treating the mutant cells with the antioxidant, alpha lipoic acid, restores mitochondrial respiration rates. In addition, expression of wild-type ATM rescues impaired mitochondrial function in ATM-deficient cells (14). Targeting catalase to mitochondria alleviates symptoms in a mouse model of ataxia, which has been attributed to reduction of mitochondrial ROS (116). Targeting to mitochondria was first achieved by introducing the first 25 amino acids of the ornithine transcarbamylase leader sequence, added to the amino terminus of the catalase ORF (459). Mitochondrial catalase reduces the incidence of thymic lymphoma in ATM−/− mice improving bone marrow hematopoiesis, macrophage differentiation, and partially reversing memory T cell developmental defects. Other therapies for ataxia telangiectasia include treatment with antioxidants such as NAC, which delays disease progression (420). Treating ATM mutant mice with the nitroxide antioxidant, tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), increased the life span of these mice by prolonging the latency to thymic lymphomas. Tempol treatment also decreased oxidative stress and associated damage (460).
3. Friedreich's ataxia
Similar to ataxia telangiectasia, Friedreich's ataxia (FRDA) is an autosomal recessive ataxia. The disorder affects the central and peripheral nervous systems as well as the heart. The genetic basis of FRDA includes expansion of GAA repeats or point mutations in the gene frataxin, which maps to chromosome 9 (81). A majority of mutations map to the first intron of frataxin with 8–22 expansions occurring in normal alleles. Some patients have an expansion of GAA repeats on one allele and point mutations on the other allele, whereas most have repeat expansions on both alleles. Patients with larger expansions typically have an earlier age of onset and additional complications and disease symptoms. There is a drastic reduction in the levels of frataxin in patients. (69). The onset of the disease usually occurs during adolescence, and symptoms include unsteady gait, the absence of lower limb reflexes, dysarthria, dysphagia, eye movement abnormalities, scoliosis, foot deformities, cardiomyopathy, and the presence of pyramidal signs (neural pathways controlling voluntary motor functions).
Frataxin is a low-molecular-weight protein of 23 kDa, which localizes to the mitochondria (277). Deletion of the frataxin homologue, YFH1, in yeast causes iron accumulation, loss of mtDNA, as well as sensitivity to H2O2 and iron (21, 159, 535). In mice, knockout of the frataxin gene causes embryonic lethality, but some studies report no change in iron accumulation, while others observe elevated iron levels (108, 404). Decrease in the iron/sulfur (Fe-S) cluster-containing subunits of mitochondrial respiratory complexes I, II, and III has been described in endomyocardial biopsies of two FRDA patients, indicating a role of frataxin in Fe-S protein homeostasis (437). In addition, activity of the Fe-S protein aconitase, which participates in iron homeostasis, is low in FRDA patients. The defects in iron metabolism and mitochondrial respiration have been attributed to elevated oxidative stress due to iron overload and its participation in the Fenton reaction to generate ROS. The role of iron in pathogenesis of FRDA has been debated, as it is not a consistent feature in FRDA cell culture and mouse models and has been considered to be a late event when Fe-S proteins are depleted (34, 108). However, more recently, Drosophila and CRISPR mouse models lacking frataxin have been shown to accumulate iron and toxicity (86, 87). Knockout of mouse frataxin in the central nervous system using AAV and CRISPR/Cas9 causes behavioral and neurological phenotypes similar to those reported earlier in a neuronal conditional knockout mouse model, including smaller body size, hunchback phenotype, impaired locomotion, and shortened life span (404). Both intracellular and extracellular accumulations of iron (both Fe2+ and Fe3+) are observed. Iron deposition has been found to trigger sphingolipid synthesis and activation of 3-phosphoinositide-dependent protein kinase-1 and myocyte enhancer factor-2 (Mef2). Mef2 then triggers transcription of downstream target genes, which mediate degeneration (87). Other studies have reported that frataxin depletion leads to elevated sensitivity to oxidative stress. Humanized transgenic mouse models of FRDA, Y87R, and Y47R harbor a human frataxin YAC with 190 + 90 GAA repeats on a mouse frataxin null background. These mice display impaired mitochondrial function and decreased GSH levels along with elevated oxidative stress, especially lipid peroxidation (3). Complex I activity is decreased, leading to increased compensatory activity of complex II, which generated higher levels of free radicals in the process. Both the mitochondrial and cytosolic compartments of cerebellar granule cells from these mice exhibit increased ROS production. Fibroblasts derived from FRDA patients show a blunted response to elevated iron levels. The O2 •− detoxifying mitochondrial enzyme MnSOD fails to respond in FRDA fibroblasts exposed to iron. (250). This abnormality, which reflects depletion of PGC1α, a protein involved in mitochondrial energetics and the coactivator for PPARγ in FRDA cells, leads to suboptimal functioning of antioxidant defense pathways (332). In concordance with these findings, the hypersensitivity of fibroblasts from FRDA patients to oxidative stress can be prevented by reducing lipid peroxidation with deuterated PUFAs (currently under clinical trials) or by activating the Nrf2 pathway (3, 4).
Thus, it is clear from the above discussion that elevated oxidative stress contributes to the pathology of the various ataxias, which profoundly affect cerebellar function. Antioxidant therapy in these ataxias has been extensively attempted with varying efficacy, and detailed studies need to be conducted to arrive at more efficient drugs for these diseases (26).
G. Aging and redox imbalance
Aging, especially when coupled to redox imbalance, is a risk factor for cognitive and motor deficits as well as neurodegeneration. Age-dependent increase in oxidative damage to mtDNA has been reported, which can lead to impaired mitochondrial energetics and elevated oxidative stress (344). The amount of 8-OHdG, a DNA damage biomarker, increases progressively with normal aging in both nuclear DNA and mtDNA, with a much greater rate of accumulation in mtDNA. A 10-fold increase in 8-OHdG content in mtDNA compared with nuclear DNA has been observed in samples tested. In addition, in a cohort older than 70 years, a 15-fold increase has been reported, further attesting to the vulnerability of mtDNA to damage. Aging has been associated with decline in the redox buffering capacity with decreases in GSH and cysteine levels (141, 417, 475). In agreement with these studies, GSH levels are maintained in aged inbred albino Louvain (LOU/C) rats where cognitive functions are preserved (357). LOU rats are considered to be a model of healthy aging due to their increased life span without obesity and due to a low incidence of common age-related diseases (35). Aging involves altered or suboptimal response to environmental or physiological stress. Aged rats treated with menadione (vitamin K), a redox cycling agent, are less responsive than younger rats. The antioxidant response is higher in younger than older rats. Supplementation of NAC is beneficial in aged rats reducing oxidative stress and improving mitochondrial function (102). There are numerous examples in literature where genes linked to maintenance of redox homeostasis are implicated in aging. In yeast, overexpression of Tsa1, an H2O2-scavenging enzyme, extends life span (208). Genetic disruption of Nrf2, the master regulator of antioxidant defense and redox balance, shortens the life span of female mice and also in males in other experimental models (556). Overexpression of glucose 6 phosphate dehydrogenase (G6PD), the rate-limiting step of the pentose phosphate pathway, increases life span in mice (369). G6PD is involved in the synthesis of nucleotide precursors and in the production of NADPH, which plays central roles in redox homeostasis in cells. G6PD-deficient mice display elevated oxidative stress and age-dependent neurodegeneration (246). Old G6PD-Tg male and female mice have decreased levels of 8-OHdG in the liver and brain with old females displaying reduced lipid peroxidation in the liver. In addition, the transgenic mice exhibit increased insulin sensitivity and decreased obesity as they age in comparison with their wild-type counterparts. Although some studies indicate that depletion or inhibition of proteins involved in antioxidant defense reduces life span, other studies report that this may not be the case. An analysis of deletion of 18 different genes participating in the antioxidant defense revealed a role for only the mice lacking Sod1, on life span (389). Interestingly, the Sod1 −/− mice displayed elevated levels of thiol antioxidants such as GSH, thioredoxin, and sulfiredoxin. In several instances, transgenic mice overexpressing antioxidant enzymes such as catalase or SOD1 do not exhibit a longer life span (228, 389). As longevity is determined by multiple factors, which range from environmental factors to diet and stress response, caution should be exercised while interpreting the contributions of individual genes or gene products to life span. Some of the knockout mice used in the studies, for instance, the peroxiredoxin 1 (Prdx1)-deleted mice, have a shorter life span due to a higher incidence of cancer, rather than elevated oxidative stress.
During aging, the redox potential of the GSSG/GSH and Cys/Cys-SS couple also shifts to a more oxidized state (254, 445). Oxidation of the Cys/Cys-SS redox couple is a risk factor for cardiovascular disease and ocular disease. Antioxidant supplements prevent oxidation of cysteine/cystine in patients with age-related macular degeneration (354). In Hutchinson–Gilford progeria syndrome (HGPS), a disease characterized by premature aging, mitochondrial dysfunction and elevated oxidative stress are key molecular features (183, 425). HGPS is caused by constitutive expression of progerin, a truncated splicing mutant of the nuclear protein lamin A. Progerin exerts its effects in a dominant manner and affects mesenchymal stem cells (MSCs), underlying several phenotypes observed at the organismal level. Early death occurs, most frequently, due to myocardial infraction or stroke. iPSC-derived MSCs from HGPS patients are impaired in their ability to respond to oxidative stress and survive under hypoxic conditions. Dysregulation of the Nrf2 signaling pathway contributes to imbalanced redox control in HGPS cells (281). Progerin binds to Nrf2 and prevents its proper localization in the nucleus, which results in suboptimal Nrf2 transcriptional function and elevated oxidative stress (308). This pathway is also frequently disrupted in neurodegenerative diseases (249, 435, 526). Thus, upregulation or stimulation of Nrf2 may have therapeutic potential.
In recent years, caloric restriction has gained appreciation as a means to increase longevity in several species, including worms, flies, yeast, and mammals. Redox signaling has been linked to dietary restriction. Caloric restriction triggers the antioxidant response program. This is not surprising, since during caloric restriction, cells undergo metabolic reprogramming to rely on OXPHOS for energy requirements more than glycolysis (310). Caloric restriction has been shown to elevate H2S, the gasotransmitter via the reverse transsulfuration pathway, plays pivotal roles in the process (218). Prior studies had shown that increased flux through the reverse transsulfuration pathway, which plays key roles in antioxidant defense, is a mediator of longevity (257, 513). Thus, upregulation of the reverse transsulfuration may have beneficial effects.
We had earlier proposed that H2S-mediated sulfhydration/persulfidation can protect proteins from irreversible oxidation on cysteine residues (382). The CySSO2H (perthiosulfinic) and CySSO3H (perthiosulfonic) oxidation products of persulfides can be recycled by the reduction of their S–S moieties, which does not occur in the case of the CySO3H oxidation product of unmodified (unsulfhydrated) cysteine residues on proteins (386). Our hypothesis of protection of SH groups of cysteine residues by persulfidation has been verified in the case of phosphatase and tensin homologue deleted on chromosome 10, PTEN, and GAPDH (169, 372). Thus, H2S production and sulfhydration could prevent oxidation of proteins and contribute to longevity, both at the level of protein half-life and overall life span. Consistent with the contribution of protein integrity to life span, in the long-living naked mole-rats, age-related accumulation of oxidation damage to thiol groups and age-associated upregulation of homeostatic proteolytic activity are significantly attenuated compared with mice (390). A point to be noted is that although young mole-rats have greater protein oxidation compared with young mice, the increase in oxidative damage over time (two decades) is minimal, indicating efficient protein homeostatic mechanisms.
V. Concluding Remarks
Fluctuations in redox state are an integral part of cellular physiology. Subtoxic levels of free radicals have roles in several signaling events. For instance, ROS are necessary for differentiation of certain cell types (448). It is only when the oxidant levels cross a “threshold” that damage to cellular components occurs, which, if not corrected by endogenous antifree radical machinery, causes irreversible damage and cell death. Thus, the concept of “oxidative eustress” has been proposed to distinguish it from “oxidative distress,” which occurs under pathological conditions and involves higher levels of oxidants (447, 472, 473). Thus, there are different metabolic set points and thresholds that are dynamic in nature. Accordingly, situations arise where elevated levels of free radicals as well as upregulated antioxidant defense can occur leading to altered metabolic set points, which could still maintain cellular functions. In normal conditions, a slight upregulation of antioxidant defense mechanisms can counter the redox imbalance created by metabolic processes. However, under pathogenic conditions or during stress, the redox balance may be tilted resulting in increased free radical generation (Fig. 15). When cells cannot counteract this stress, cell death ensues. It is also becoming increasingly clear that oxidative stress has adverse effects in neurodegeneration, while in cancers, the opposite is true. Mitigating oxidative stress in cancer may be detrimental, as cancer cells have been reprogrammed to elevate their antioxidant defenses to maximize cell growth and proliferation. In this case, inducing oxidative stress may be beneficial. In cancers, there is over proliferation and growth, whereas in neurodegeneration, there is atrophy and loss of neuronal cells. Thus, depending on the context, modulating the redox milieu of cells can have different effects.

Oxidative stress plays a significant role in disease progression and neurodegeneration. Oxidative stress, mitochondrial dysfunction, and proteostasis are intimately linked, with mitochondrial malfunction leading to elevated oxidative stress and vice versa. Thus, ameliorating oxidative stress and/or stimulating redox signaling pathways responsible for corrective responses may prevent or delay neurodegeneration. As the redox regulatory network in cells is vast, with interconnecting nodes, disruption of a major regulatory point may lead to a cascade of events that culminate in aberrant stress response. When this process remains uncorrected, as in the case of neurodegeneration, cell death ensues. Another cellular process linked to oxidative stress is autophagy, the process by which damaged proteins and organelles are cleared (41). Autophagy and oxidative stress have been increasingly interlinked in several studies, although it is still unclear which species of ROS or RNS initiates the process (88, 149, 561). Mitochondrial functions are also intimately associated with the process as they are the predominant generators of ROS in cells (297). Nevertheless, excess oxidative stress can compromise this process too, leading to buildup of toxic aggregates, which also contribute to redox imbalance. Signaling pathways operating in the lysosomes, the organelles playing central roles in autophagy have also been linked to redox regulation (565). Impaired autophagy has been observed in several different neurodegenerative disorders and promoting clearance of unfolded or aggregated proteins and dysfunctional organelles may afford neuroprotection. More recently, the Nrf2 pathway, recognized as an antioxidant pathway, has also been shown to modulate autophagic processes via p62, an autophagic receptor, indicating that a fine balance between redox signaling in mitigating excess oxidative stress while maintaining normal cellular processes is essential for optimal cell function (74, 274). Thus, it is not surprising that use of antioxidants to counter neurodegeneration can impair essential processes such as autophagy in certain instances (300).
Thus, identification of the origins of neurodegenerative processes in various diseases becomes critical. Although a plethora of studies have been conducted on neurodegenerative diseases, the differential susceptibility of specific regions of the brain in different neurodegenerative diseases is intriguing. Thus, the molecular basis of SNV is still elusive. A combination of genetic and epigenetic factors, environmental interactions, and nature of the mutation contribute to the initiation and progression of neurodegeneration. Neurodegenerative diseases such as AD, PD, and ALS can arise due to genetic causes (familial AD, PD, or ALS) or arise sporadically. HD on the contrary is the only neurodegenerative disease where all cases have mutations in the huntingtin gene. A stressor threshold model has been proposed in which the intrinsic susceptibilities of neuronal cell types to stressors and specific disease-related misfolding proteins determine neuronal toxicity. Specific combinations of genetic predispositions and environmental stressors elicit age-dependent stress and proteostasis dysfunction in vulnerable neurons (451). A common feature of diverse neurodegenerative diseases is mitochondrial dysfunction and oxidative stress, which are intimately linked (305). Whether oxidative stress elicits mitochondrial dysfunction or vice versa is a matter of debate.
Although it is becoming increasingly clear that oxidative stress is associated with several diseases, including neurodegenerative disorders, antioxidant supplementation has had limited therapeutic benefit in terms of “curing” disease, a phenomenon termed the “antioxidant paradox” (203, 206). Multiple reasons have been postulated to account for failure of antioxidants to reverse or halt disease progression (204, 205). In several instances, administration of antioxidants does not reduce oxidative stress significantly. The time and stage at which the antioxidant is to be delivered are still poorly researched. Some situations, where there is a paucity of the antioxidant or molecule under investigation, may benefit. For example, the supplementation of cysteine and GSH has proved beneficial in several age-related conditions. These interventions too depend on the bioavailability of the compound being administered. The studies on mouse models of disease do not translate to human trials due to differences in intrinsic antioxidant networks. For instance, vitamin E was reported to be beneficial for a mouse model of familial ALS, whereas this was not the case in ALS patients (196, 376). Despite these limitations, more recently, edavarone (radicava), an antioxidant that eliminates lipid peroxides and •OH, has been found to be effective in ALS patients and approved for use (436, 543). Another pitfall of utilizing certain antioxidants (e.g., polyphenols and ascorbate) is that they could interact with transition metals, such as iron and copper, and mediate pro-oxidant effects. Antioxidants have different targets in cells, both in terms of scavenging or neutralizing free radicals directly or by modulation of redox regulatory networks. Thus, an antioxidant may counter the activity of one type of ROS but leave another intact. In addition, as discussed earlier, certain thiols such as cysteine and GSH act via post-translational modifications, which can account for their activity in cells. These activities are very often confused with “antioxidant” effects. Very often, the supplementation of the depleted antioxidant molecule, by itself, does not provide a robust protective effect. Targeting the antioxidant to specific sites may also alter outcomes in clinical trials. Upregulating the biosynthetic pathway responsible for production of the molecule may be more effective. Understanding the redox state of the cell may help design better therapeutics.
In several instances, a reversible oxidized redox state precedes free radical damage and can be targeted for the treatment of neurodegenerative diseases. Priming cells with mild forms of stress may induce cytoprotective pathways that convey resistance to future insults. Thus, the concept of “hormesis” or adaptive response has been proposed (65 –67, 349). This is similar in concept to the process of vaccination, where a low or attenuated dose of the pathogen can trigger host responses that afford future protection. Hormetic mechanisms are part of the normal physiology of the brain, and their disruption may be detrimental to neuronal and glial systems. Intermittent fasting, regular exercise, and consumption of dietary phytochemicals can promote adaptive cellular stress response pathways that protect against and counteract several diseases (339, 410). Low levels of stressor molecules can induce pathways that maintain redox balance, such as the Nrf2 pathway and the PPARγ pathway. Another target is the reverse transsulfuration pathway, which is frequently disrupted in neurodegeneration. This pathway is central to the maintenance of redox balance via the synthesis of GSH and cysteine, major antioxidant cytoprotectants in cells (381, 453). In addition, this pathway is also responsible for the generation of H2S by the enzymes CSE and CBS. Recently, it was shown that H2S can also modulate Nrf2-mediated antioxidant signaling (549). The newly emerging area of gasotransmitter signaling, encompassing NO, CO, and H2S, adds several layers of complexity to the intricate network of redox signaling. The interplay of various redox active molecules is yet to be elucidated. Especially relevant is the interplay between oxygen and H2S signaling, which plays fundamental roles in cerebrovascular function. Another area of interest is the overlap of gasotransmitter signaling with the microbiota of the host, which could play important roles in redox regulation as well as modulation of the gut/brain axis.
In addition, identification of sensors of various forms of stress may reveal additional hubs and targets of redox regulation. Oxidative stress and ER stress can reprogram cells to adapt for survival. Sensor functions present on organelles such as the Golgi and lysosomes may also play significant roles in maintenance of redox homeostasis whose disruption could mediate neurodegeneration (322, 452). The Golgi is emerging as a major stress response organelle, similar to the ER. It has been proposed that changes in the structure and organization of the Golgi apparatus mediate sensing of stress stimuli well before cell death and neurodegeneration. Fragmentation of the Golgi has been observed in various neurodegenerative diseases such as AD, ALS, and HD (182, 255, 452). The spatiotemporal control of gene regulatory networks that govern the cytoprotective stress responses of an organism is especially important. Transcription factors that regulate major hubs of redox control may serve as targets of drug design and therapeutics. With the advent of high-throughput methodologies and platforms, the integration of genomics, epigenomics, proteomics, and biochemical analyses, metabolomics, and identification of signaling networks should pave the way for personalized medicine. These developments, coupled with nutritional and environmental analysis, portend a promising future for redox medicine.
Footnotes
Acknowledgment
This work was supported by USPHS grants DA000266 and MH18501 to S.H.S.