
Abstract
Significance:
Type 2 diabetes mellitus and hyperglycemia can lead to the development of comorbidities such as atherosclerosis and microvascular/macrovascular complications. Both type 2 diabetes and its complications are related to mitochondrial dysfunction and oxidative stress. Type 2 diabetes is also a chronic inflammatory condition that leads to inflammasome activation and the release of proinflammatory mediators, including interleukins (ILs) IL-1β and IL-18. Moreover, sirtuins are energetic sensors that respond to metabolic load, which highlights their relevance in metabolic diseases, such as type 2 diabetes.
Recent Advances:
Over the past decade, great progress has been made in clarifying the signaling events regulated by mitochondria, inflammasomes, and sirtuins. Nod-like receptor family pyrin domain containing 3 (NLRP3) is the best characterized inflammasome, and the generation of oxidant species seems to be critical for its activation. NLRP3 inflammasome activation and altered sirtuin levels have been observed in type 2 diabetes.
Critical Issue:
Despite increasing evidence of the relationship between the NLRP3 inflammasome, mitochondrial dysfunction, and oxidative stress and of their participation in type 2 diabetes physiopathology, therapeutic strategies to combat type 2 diabetes that target NLRP3 inflammasome and sirtuins are yet to be consolidated.
Future Directions:
In this review article, we attempt to provide an overview of the existing literature concerning the crosstalk between mitochondrial impairment and the inflammasome, with particular attention to cellular and mitochondrial redox metabolism and the potential role of the NLRP3 inflammasome and sirtuins in the pathogenesis of type 2 diabetes. In addition, we discuss potential targets for therapeutic intervention based on these molecular interactions. Antioxid. Redox Signal. 29, 749–791.
A. Diseases related to inflammasome activation associated with mitochondrial dysfunction
B. Insulin resistance, type 2 diabetes, and the NLRP3 inflammasome
D. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic vascular complications
I. Introduction
T
Insulin resistance, one of the main characteristics of type 2 diabetes and cardiometabolic diseases, is related to clinical complications such as atherosclerosis, obesity, and alterations of androgen levels. It is important to point out that the main action of insulin is the maintenance of glucose homeostasis through stimulation of glucose uptake in peripheral tissues such as skeletal muscle and by decreasing liver gluconeogenesis.
Type 2 diabetes is a clinical condition characterized by a metabolic imbalance in whose development mitochondria are key players. Mitochondria are the main source of reactive oxygen species (ROS) and play a crucial role in redox homeostasis, metabolism, and multiple functions, including apoptosis and cell death (199, 343). Indeed, these organelles mediate cardiometabolic diseases in general and diabetes in particular. For example, serine/threonine protein kinase 25 (STK25) has recently been highlighted as a regulator of the complex interplay between lipid storage, mitochondrial energetics and insulin action in skeletal muscle, which underlines the potential role of STK25 antagonists in the treatment of type 2 diabetes (60). Mitochondria modulate homeostasis and play a key role in the metabolism by controlling adenosine triphosphate (ATP) production and energy levels through nutrient metabolism and heat generation.
ROS production is a function of mitochondria. ROS are key signaling molecules in multiple physiological pathways. However, when there is mitochondrial dysfunction or a change in the mitochondrial membrane potential (ΔΨm), ROS production is exacerbated and ATP synthesis reduced due to the energetic imbalance (36), conditions that can lead to cell death, including apoptosis. Insulin sensitivity and mitochondrial function are regulated by multiple genetic and environmental factors (diet, exercise, or stress) (260). In type 2 diabetes, insulin resistance, hyperglycemia, and mitochondrial impairment have been observed in several tissues, such as liver, skeletal muscle, spleen, adipose tissue, lung, heart, and kidney (13, 37), and in different cell types, including leukocytes (117, 269). In light of all this evidence, mitochondria are considered a key target in the treatment of type 2 diabetes.
There is an important interrelationship among mitochondria, inflammation, and metabolism, and diverse signaling pathways are involved in the regulation of this complex interplay. Specifically, mitochondria are involved in the regulation of inflammatory responses in immune cells by controlling inflammasome assembly through ROS release as the integrating signal. For this reason, mitochondria are considered key organelles, not only in innate immunity but also in pathological situations involving chronic inflammation, such as type 2 diabetes. The regulation of inflammasome activity by mitochondria and its alteration in type 2 diabetes will be analyzed in depth in this review.
In contrast, the cell metabolic status is sensed by sirtuins, a family of deacetylase enzymes involved in different processes, including energy production and cell survival. Sirtuin 1 (SIRT 1) is the best characterized sirtuin; it is activated during starvation in response to a rise in the nicotinamide adenine dinucleotide (NAD+)/nicotinamide adenine dinucleotide phosphate (NADP+) ratio and has anti-inflammatory properties. Sirtuins are reduced in specific tissues during chronic inflammation, such as in fatty tissue in obesity or in arterial walls during atherosclerosis. Besides SIRT1, other family members, such as SIRT6 and SIRT3, sense nutrient availability and changes in NAD+ production, and alterations in their activity are characteristic of type 2 diabetes. Thus, growing evidence points to sirtuins as potential therapeutic targets for metabolic diseases and particularly type 2 diabetes.
In this review, the involvement of mitochondrial dysfunction in type 2 diabetes, the regulation of inflammasome activity by mitochondria and its alteration during type 2 diabetes and the role of sirtuins in that context will be analyzed in depth. In addition, the potential of mitochondria, inflammasomes, and sirtuins as therapeutic targets in metabolic diseases will be evaluated.
II. Mitochondrial Dysfunction
A. Insulin resistance and mitochondrial dysfunction
Insulin modulates plasma glucose levels via different mechanisms that control the rate of glucose uptake, gluconeogenesis, and glucogenolysis. Insulin signaling is essential for proper cardiovascular, neural, and renal functions, among others. The crucial role of insulin supports the involvement of insulin resistance in a wide variety of comorbidities, such as diabetic foot, retinopathy, cardiovascular diseases, hypertension, neuropathy, and nephropathy (301).
Insulin resistance is a typical characteristic of type 2 diabetes and consists of a decreased capacity of cells to respond to fluctuating glucose levels. Different conditions contribute to insulin resistance, including excess weight, obesity, stress, pollution, and altered protein, lipid, and glucose metabolism (212).
Free fatty acids (FFAs) in the circulation are usually a consequence of excess energy intake and obesity. The accumulation of FFAs, diacylglycerol (DAG), and triglycerides in key tissues including skeletal muscle, liver, and adipose tissue can generate lipid peroxidation and disturb the energy balance, leading to mitochondrial dysfunction.
The dyslipidemia that is typical in type 2 diabetes and metabolic syndrome is related to oxidative stress, high levels of triglycerides, and low-density lipoprotein (LDL) cholesterol, together with reduced levels of high-density lipoprotein (HDL) cholesterol and antioxidants. This situation can lead to a decrease in insulin-stimulated glucose disposal (111). Thus, an abnormal lipid metabolism can impair insulin signaling and consequently induce insulin resistance (28). In fact, Fayyaz et al. have demonstrated that fatty acids such as palmitate are metabolized to sphingosine 1-phosphate (S1P) by hepatocytes, and S1P impairs insulin signaling by stimulating the S1P2 receptor, which attenuates insulin-dependent protein kinase B (Akt) phosphorylation (85). This evidence supports S1P2 inhibition as a new therapeutic strategy for pathologies related to insulin resistance (85).
There are various insulin signaling pathways involving different types of receptors (280). In short, the mechanism of action of insulin is triggered when it binds to its receptor, after which receptor residues are autophosphorylated and further phosphorylate insulin receptor substrates (IRS1 and IRS2). This mechanism activates phosphoinositide 3-kinase (PI3K), enhancing phosphatidylinositol 3,4,5-triphosphate and activating Akt, protein kinase C (PKC), and other kinases that modulate pleiotropic metabolic actions of insulin (Fig. 1). It is crucial that this complex pathway be correctly regulated.
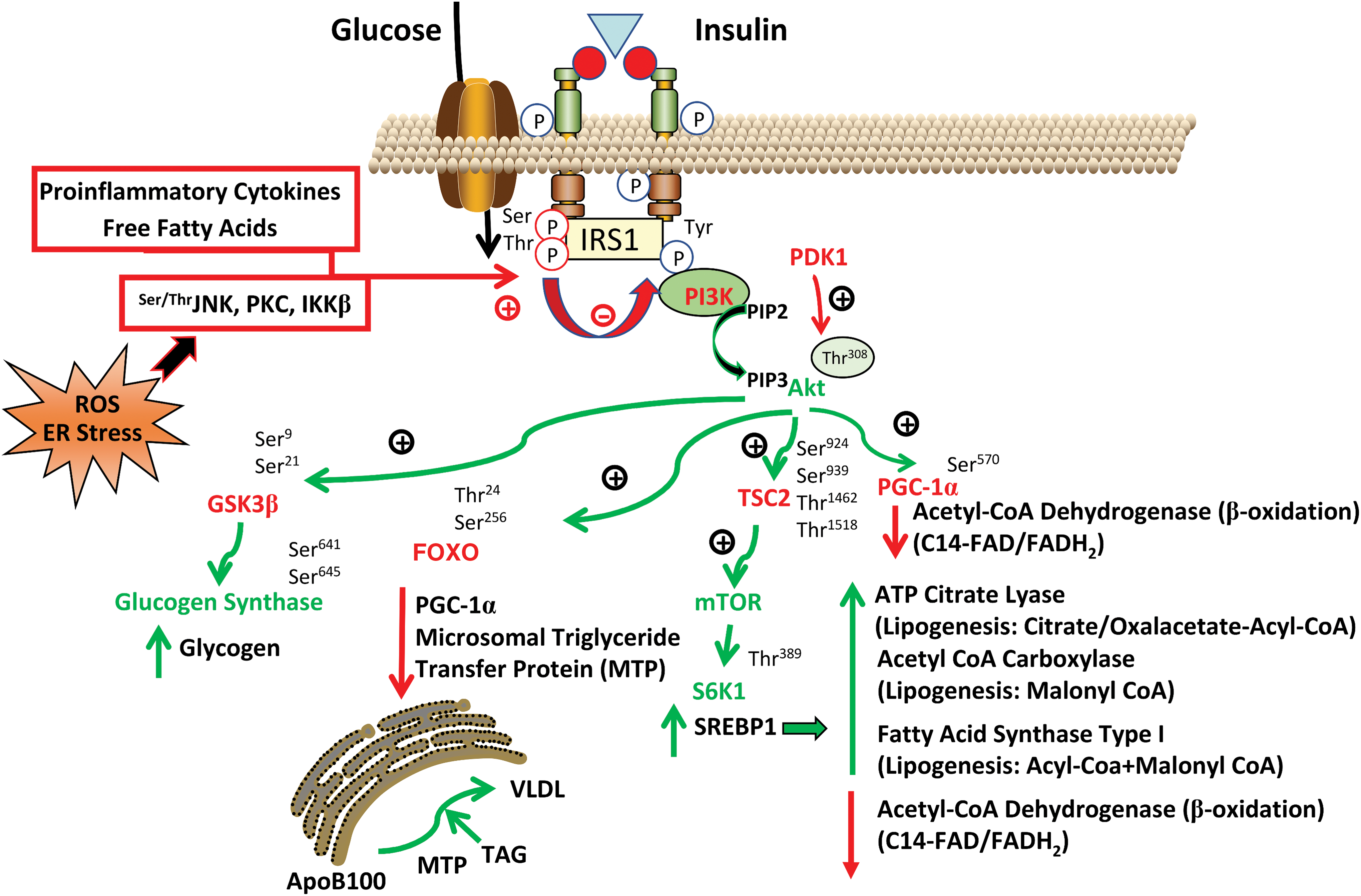
Insulin resistance can be produced by different mechanisms, such as enhanced levels of serine phosphorylation of IRS (390), excessive degradation of IRS proteins (382), decreased activation of insulin downstream signaling molecules such as PKC and Akt (302), and an increase in the activity of phosphatases (340). Furthermore, in insulin-resistant human and animal models, it has been shown that there is a decrease in IRS1 tyrosine phosphorylation (68). Consequently, it has been argued that phosphorylation of IRS at key residues plays a key role in the modulation and response to insulin levels by reducing PI3K activation (340).
Inflammation and proinflammatory cytokines are mediators of insulin resistance. For example, the activation of toll-like receptors (TLRs) by FFAs promotes proinflammatory signaling through c-Jun N-terminal kinase (JNK) or inhibitor of kappa B kinase subunit beta (IκKβ), increasing levels of several cytokines, including interleukin (IL)-1, IL-6, IL-12, IL-18, and tumor necrosis factor alpha (TNFα) (144). Both JNK and IκKβ directly inhibit insulin signaling by phosphorylating IRS (12, 296). Research points to the importance of inflammation in the development of insulin resistance (Fig. 1). In fact, it seems that the release of proinflammatory signals from adipose tissue would be, at least in part, responsible for the development of insulin resistance (122).
It should be taken into account that not only insulin signaling per se influences systemic insulin resistance, but that body fat composition also plays a crucial role. Insulin receptors in adipocytes are less capable of promoting glucose uptake than insulin receptors in muscle; thus, when fat proportion is high, hyperinsulinemia develops to ensure adequate glucose disposal. Both high fat proportion and inflammation are circumstances exacerbated by a sedentary lifestyle.
Endoplasmic reticulum (ER) stress has been described as a key mechanism of insulin resistance. ER stress can activate different kinase pathways involved in inflammatory processes, such as JNK kinases. In this sense, several compounds have been employed to reduce ER stress and insulin resistance by undermining IRS1 serine phosphorylation or JNK activity, such as taurine-conjugated ursodeoxycholic acids and 4-phenyl butyric acid, which are also capable of resolving fatty liver disease (232). Moreover, since the ER is the organelle in which lipid synthesis takes place, ER stress can induce lipid accumulation and should, therefore, be considered a target mechanism in the context of insulin resistance (93). In this sense, the ER stress X-box binding protein 1 and PKR-like ER kinase branches regulate key pathways in lipogenesis (156, 225).
One of the leading hypotheses regarding the onset of insulin resistance is that enhanced ROS production, and therefore mitochondrial impairment, can induce signals that activate the serine kinases that phosphorylate IRS proteins (211). Both mitochondria and NADPH oxidase are considered important sources of ROS that contribute to ROS-induced phosphorylation of IRS-1 and suppression of downstream insulin signaling (292, 385). In fact, inhibition of ROS production using uncouplers of mitochondrial oxidative phosphorylation or by directly inhibiting NADPH oxidase with apocynin improves glucose metabolism (146). Both extramitochondrial and mitochondrial ROS promote damage to mitochondrial complex I, the most vulnerable electron transport chain (ETC) complex. Mitochondrial ROS production increases as a result, which leads to a vicious circle in which mitochondrial dysfunction is enhanced (10). However, the participation of ROS-induced mitochondrial ROS release in the development of insulin resistance is still unknown.
ROS stimulate inflammatory signaling by activating IκKβ, which phosphorylates IRS1 (Fig. 1). The link between excessive ROS production and insulin resistance is reinforced by the fact that insulin sensitivity is improved by different types of antioxidants (263). In contrast, mitochondrial impairment could be the result of increases in DAG, long-fatty acyl-CoA, or fatty acid metabolites (129). In addition, DAG, an activator of PKCs, can increase the phosphorylation of IRS, thus promoting insulin resistance. Moreover, DAG regulates PKCθ in the induction of muscle insulin resistance in obese and type 2 diabetic subjects (314). Indeed, PKCθ knockout mice do not develop insulin resistance when subjected to hyperlipidemic conditions (147). This evidence suggests that mitochondrial dysfunction is involved in the induction of insulin resistance through the activation of PKCs.
Studies in humans have highlighted the association of insulin resistance with several mitochondrial abnormalities, such as changes in mitochondrial morphology, reduced number of mitochondria, presence of mitochondrial dysfunction, or reduction in mitochondrial oxidative enzymes (139, 239). Mitochondrial function in several organs has been closely linked to insulin resistance. For example, mitochondrial activity in skeletal muscle has been positively associated with insulin sensitivity and negatively with hepatic lipid accumulation (313). Moreover, in hepatic cells, the antioxidant alpha-lipoic acid can attenuate ER stress-induced insulin resistance by improving mitochondrial function (168).
Glucose is not sensed adequately by β-cells of insulin-resistant patients, which results in the disruption of glucose homeostasis and, eventually, a decrease in insulin production as a consequence of hyperglycemia-induced β-cell damage and development of type 2 diabetes. Insulin secretion in β-cells is tightly modulated by mitochondria, and this occurs in an ATP/adenosine diphosphate (ADP) ratio-dependent manner. When the ATP/ADP ratio is high, the ATP-dependent potassium channels (kATP) are closed, and insulin is secreted (292) (Fig. 2).
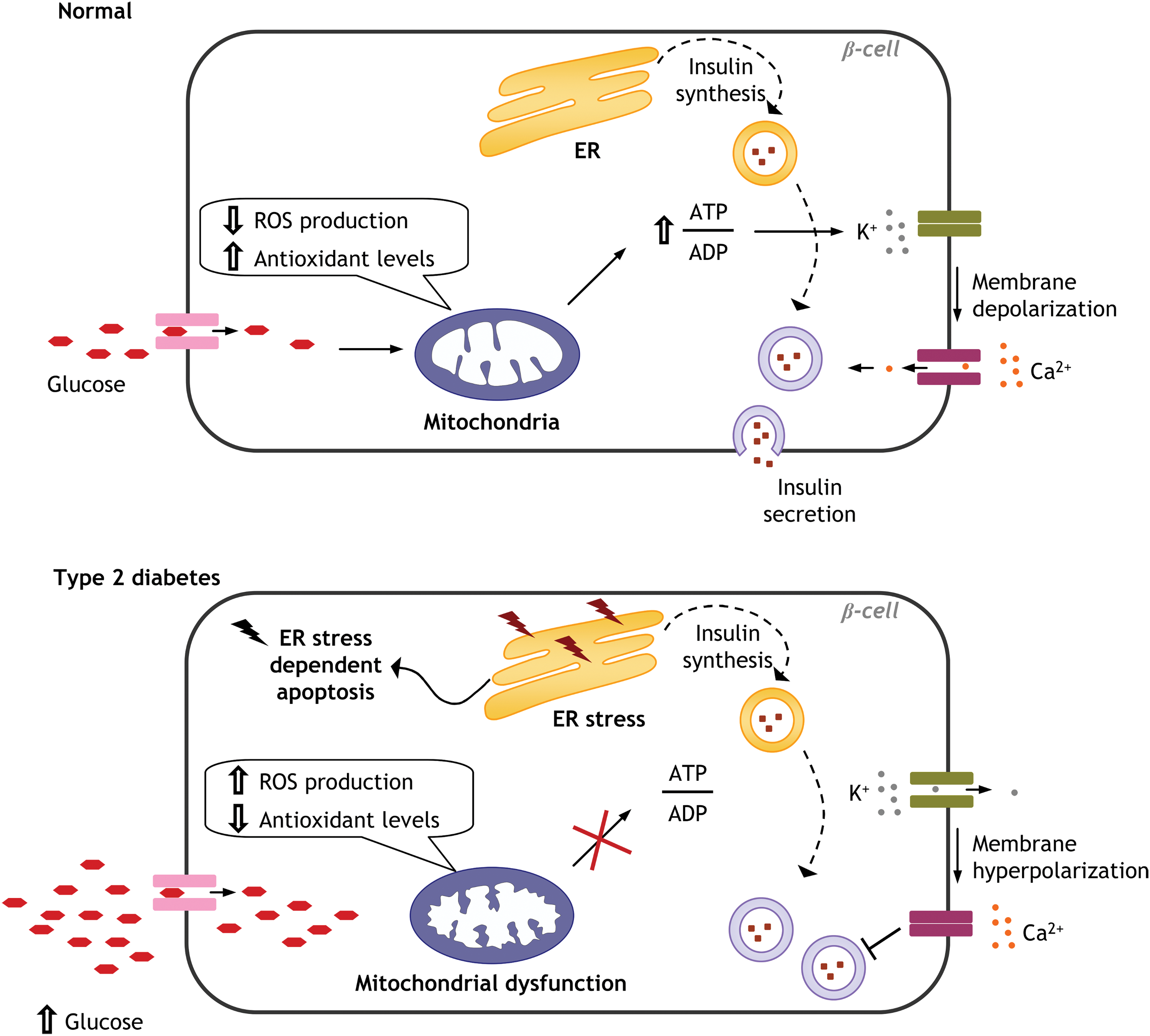
Han et al. have shown that taurine enhances the glucose sensitivity of β-cells that overexpress uncoupling protein (UCP) 2, probably by increasing mitochondrial Ca2+ influx through the mitochondrial Ca2+ uniporter, which promotes mitochondrial function and eventually raises the ATP/ADP ratio (113). Furthermore, it has been shown that insulin release is decreased in pancreatic β-cells when mitochondrial DNA (mtDNA) is depleted, suggesting that mitochondrial respiratory function is crucial in the glucose-stimulated influx of Ca2+ into pancreatic β-cells and the subsequent induction of insulin secretion. Nevertheless, if this situation is modulated by introduction of exogenous mtDNA, β-cells can recover their ability to secrete insulin (294).
In addition, when β cells are exposed to hyperglycemia, intracellular Ca2+ increases, which results in enhanced consumption of ADP and higher membrane potential that, together with high levels of reducing equivalents, contribute to elevated ROS production (292). In this context, adequate intramitochondrial Ca2+ concentration is critical to ensure glucose-stimulated insulin secretion, which is regulated by the Ca2+ uniporter, embedded on the inner mitochondrial membrane (IMM) (113). Indeed, both blocking mitochondrial Ca2+ efflux and improving mitochondrial Ca2+ sequestration promote glucose-stimulated insulin secretion (113).
Not only Ca2+ transport has an impact on β-cell homeostasis and insulin secretion. During type 2 diabetes, heart mitochondria are damaged and their dysfunction leads to altered Ca2+ transport resulting in enhanced mitochondrial permeability transition and consequently apoptosis (292).
This proves that mitochondrial Ca2+ homeostasis is highly important in type 2 diabetes in general, and essential for the proper functioning of β-cells in particular, and implicates disruption of mitochondrial homeostasis in the onset of the disease through its modulation of insulin secretion and/or action (Fig. 2). All of this highlights the potential role of mitochondria as a target for the prevention and treatment of insulin resistance-related diseases.
Therefore, accumulating evidence suggests that mitochondria are key targets for the treatment of insulin resistance. Obesity is associated with increased levels of circulating lipids, which accumulate in different tissues, but especially in adipose tissue, and consequently alter glucose metabolism, and induce systemic insulin resistance. Furthermore, it has been demonstrated that lipodystrophy induces type 2 diabetes and insulin resistance in human subjects (17). Adipocytes release adipokines, including leptin, adiponectin, resistin, and TNFα, thereby regulating different metabolic pathways (366). Insulin resistance is related to a decreased number of mitochondria in adipocytes, decreased mitochondrial gene expression, and ATP synthesis, together with changes in mitochondrial morphology. Reduced mitochondrial biogenesis, along with decreased expression of mitochondrial biogenesis genes, has been reported in different types of cells, including adipocytes from type 2 diabetic patients, both overweight and obese (30). Recently, it has been shown that human white adipocytes develop mitochondrial dysfunction and insulin resistance in a peroxisome proliferator-activated receptor alpha (PPARα)-dependent manner when they are deficient in lipase, pointing to the modulation of lipases in adipose tissue as a promising target for the treatment of insulin resistance in obesity and type 2 diabetes (134).
B. Regulation of mitochondrial function by caloric load
Mitochondria metabolize multiple nutrients, and it has been demonstrated that caloric and/or nutrient restriction can activate AMP-activated protein kinase (AMPK), a major cellular energetic sensor that detects increases in the AMP/ATP ratio. Once activated, AMPK triggers several mechanisms to restore ATP levels and, therefore, cellular survival. Caloric restriction and nutrient deprivation can increase NAD+ levels (264), subsequently activating a group of proteins denominated sirtuins. The deacetylase activity of sirtuins requires NAD+ as a cofactor. Although we mention some characteristics of sirtuins in this section, these proteins are explained in more detail later on in this review.
The sirtuin family is constituted by seven proteins in mammals: SIRT3, SIRT4, and SIRT5 are located within the mitochondria; SIRT1, SIRT6, and SIRT7 are nuclear proteins; and SIRT2 is mainly cytoplasmic (272).
Sirtuin proteins are lysine deacetylases, with the exception of SIRT5, which is involved in other relevant activities, including demalonylation and desuccinylation of lysines (319).
During caloric restriction, SIRT1 and AMPK reciprocally regulate each other, thus constituting one of the main functions of sirtuins (270). In fact, AMPK is activated through phosphorylation by serine–threonine liver kinase B1 (LKB1), which in turn, is activated by SIRT1. In contrast, AMPK can activate SIRT1 by enhancing NAD+ levels (45). SIRT1 and AMPK have common targets, including peroxisome proliferator-activated receptor coactivator-1α (PGC1-α), an important regulator of mitochondrial gene expression. AMPK and SIRT1 can also regulate mitochondrial quality control signaling; for example, the former is known to activate mitophagy (349). SIRT1 interacts with and deacetylates mediators of autophagy such as the autophagy proteins (Atg) 5, 7, and 8 (349) and consequently modulates cellular remodelling.
Therefore, caloric restriction can improve mitochondrial function by regulating mitochondrial turnover and mitochondrial biogenesis and, in turn, redox signaling and energy balance. In addition, caloric restriction activates SIRT3 by increasing the levels of mitochondrial NAD+ (367).
SIRT3 is a vital protein, as its deacetylase activities can improve mitochondrial function. In fact, it is capable of deacetylating the mitochondrial complex I protein NADH ubiquinone oxidoreductase subunit A9 (NDUFA9) (4), superoxide dismutase 2 (SOD2) (322), and other proteins involved in fatty acid oxidation, such as long-chain acyl-CoA dehydrogenase (118), or in cell death, such as cyclophilin D (54). One study has demonstrated that activation of SIRT1/3 improves vascular hyporeactivity in a model of severe hemorrhagic shock by enhancing mitochondrial function (173). In summary, SIRT3 modulates mitochondrial function by protecting against ROS and increasing ATP levels.
AMPK is inhibited under a hypercaloric diet or chronic excess of nutrients, as levels of ATP are enhanced and NAD+ levels are decreased in the said conditions (375). In addition, PGC1-α is reduced by hyperacetylation, thus undermining mitochondrial biogenesis (66), whereas nutrient overload can reduce SIRT3 levels, therefore, triggering mitochondrial dysfunction (119).
C. Mitochondrial dysfunction and cardiovascular diseases
Cardiovascular diseases, such as coronary artery disease, myocardial ischemia, heart failure, and stroke, are usually characterized by oxidative stress, endothelial and mitochondrial dysfunction, and insulin resistance (155). Moreover, hypertension is generally related to hyperglycemia and is present in >60% of diabetic patients (87). Cardiac efficiency is reduced in insulin-resistant animal models, such as ob/ob mice, in which high levels of FFAs reduce mitochondrial oxidative capacity and limit ATP synthesis (34). Furthermore, intramyocardial lipid accumulation in different animal models of obesity alters mitochondrial function and promotes lipotoxic injury and cardiac dysfunction (99).
Glucose intolerance and intramyocardial lipid accumulation, typical for both type 2 diabetes and cardiovascular diseases, are related to endothelium damage and a subsequent impairment of endothelium-dependent vasodilation (200). Specifically, hyperglycemia induces increased superoxide production, from mitochondria and from other sources, such as NADPH oxidase. Superoxide anion may quench endothelium-derived vasodilator nitric oxide (NO), thus undermining endothelium-dependent vasodilation. In addition, superoxide can also interact with NO to form peroxynitrite, a reactive oxidant that exerts cytotoxic effects in endothelial cells.
The heart has high caloric requirements and undergoes considerable FFA oxidation; however, it contains low levels of endogenous antioxidants, especially glutathione, which can result in oxidative stress and mitochondrial impairment (354). In fact, an association between cardiac dysfunction and mitochondrial morphology/function alterations, together with oxidative stress, has been demonstrated in insulin-resistant Zucker obese rats (137). Different insulin-resistant rat models have shown high levels of morphologically abnormal mitochondria in myocardial tissue, revealed by transmission electron microscopy (226). It has also been reported that the mtDNA copy number decreases in hypertrophied rat hearts under oxidative stress conditions (307), probably due to enhanced ROS damage. Therefore, pathological hypertrophy involves a reduction in mtDNA content and in the number of mitochondria (90).
All this evidence demonstrates that mitochondria are essential for cardiac function and that mitochondrial dysfunction in the heart can lead to numerous cardiovascular diseases, including myocardial ischemia, heart failure, and cardiomyopathy (Fig. 3).
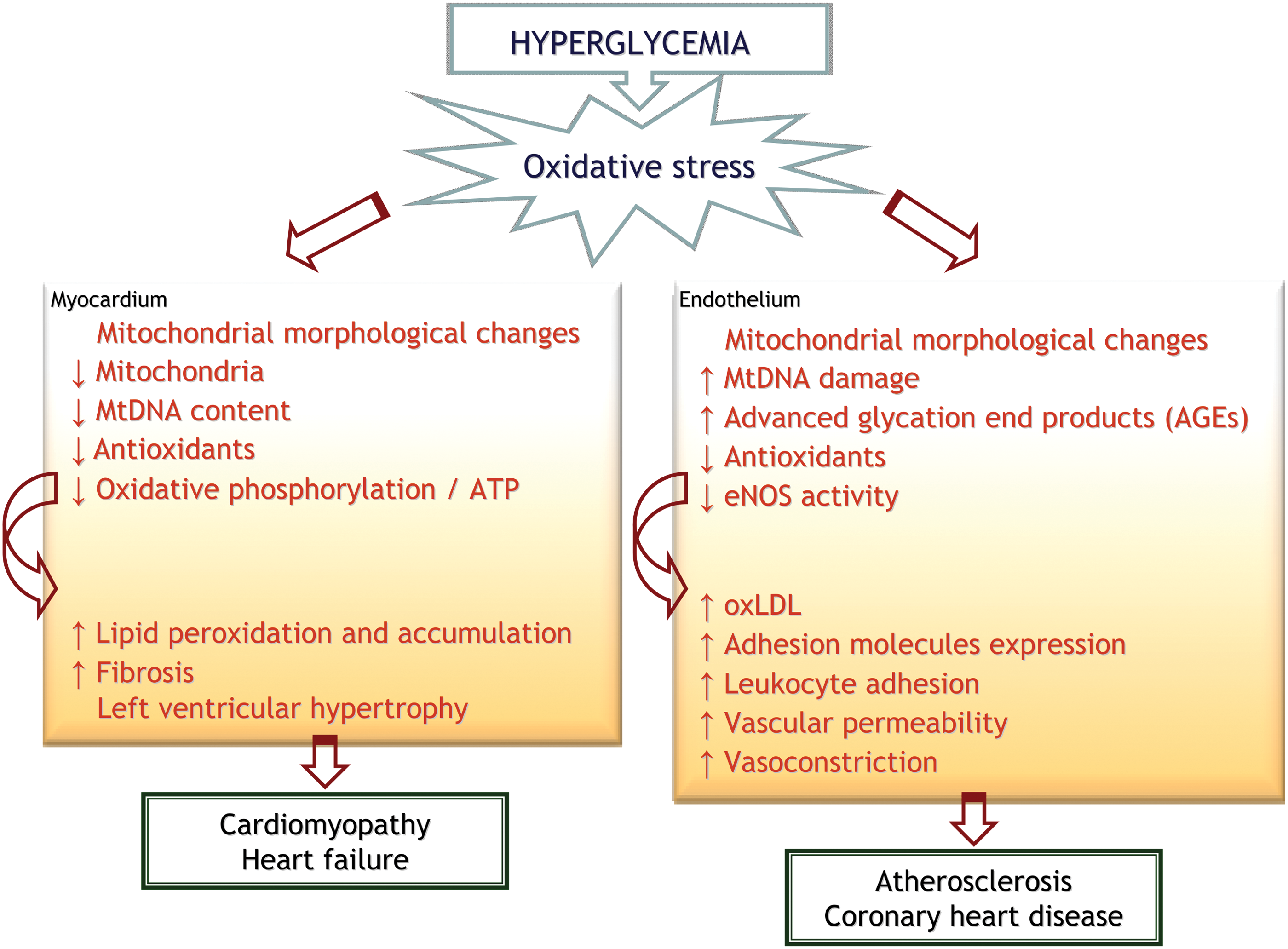
The relationship between endothelial impairment and insulin resistance is widely recognized (146); however, the underlying mechanism involved in this relationship remains to be elucidated. Endothelial cells are glycolytic, but mitochondria have been shown to play a key function as sensors of local O2 concentration and as regulators of intracellular [Ca2+] in the endothelium (70). Furthermore, mitochondrial dysfunction and high oxidant species levels are related to vascular damage (Fig. 3). In this sense, different studies have shown that endothelial dysfunction associated with hyperglycemia can be prevented by using antioxidants to block high mitochondrial ROS production, which highlights the potential of antioxidants as an emerging therapy for cardiovascular diseases, particularly mitochondria-targeted antioxidants (263).
The control of vasorrelaxation is orchestrated by vasodilatory factors released by the endothelium to regulate vascular tone. Among said factors, NO is the most important endogenous vasodilator, although prostacyclin and thromboxane A2 are also important. NO is formed by the endothelial nitric oxide synthase (eNOS) enzyme, which also participates in insulin-stimulated NO production and mitochondrial biogenesis (210). In this sense, it has been observed that eNOS knockout mice can develop insulin resistance, inflammation, dyslipidemia, atherosclerosis, oxidative stress, mitochondrial impairment, hypertension, and cardiovascular disease (79). Vasodilatory-stimulated phosphoprotein has been shown to act as a mediator of the NO/cGMP pathway, providing potential protection for the development of insulin resistance and vascular damage.
Therefore, insulin resistance can disrupt NO homeostasis, and the mitochondrial damage that subsequently occurs may alter cardiac functions and eventually lead to cardiometabolic diseases (188). Viswambharan et al. have pointed out that enhancing insulin sensitivity specifically in endothelial cells leads to a paradoxical decline in endothelial function, mediated by increased phosphorylation of inhibitory tyrosine residues of eNOS and excess NADPH oxidase 2 (Nox2)-derived superoxide (342).
D. Mitochondrial dysfunction and immune activation
Mitochondria are related with inflammation and immune function. For example, T cell polarity is modulated by the metabolic state of immune cells. Depending on the metabolic state—whether glycolytic or oxidative—T cells divide or differentiate, respectively (97). Mononuclear cells, such as macrophages and monocytes, change their metabolism depending on the inflammatory program that they activate, including the nod-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome (371).
Furthermore, it has been described that activation of TLRs on myeloid cells exacerbates glycolysis instead of oxidative phosphorylation. Another study demonstrated that proinflammatory macrophages (M1) enhance the pentose phosphate pathway and glycolysis to obtain NADPH and ATP (331). In this context, NADPH is used for the synthesis of fatty acids as precursors of prostaglandins and other proinflammatory molecules, and ATP is required to fulfill the functional demands of M1 macrophages. Therefore, glycolysis is key for immune function; in fact, it has been described that its inhibition avoids the inflammatory response in macrophages treated with lipopolysaccharide (LPS) (321). In addition, the enzyme NADPH oxidase, located in the plasma membrane, releases high amounts of ROS, especially superoxide, to eradicate infections.
Finally, mitochondria release ROS from complexes I and III of the ETC (219). ETC in M1 macrophages and leukocytes can be inhibited by treatment with LPS, with ROS production increasing particularly at complex I due to a rise in the NADH/NAD ratio (324). Furthermore, it has been demonstrated that mitochondrial dynamics can regulate immune function by forming oxidative phosphorylation supercomplexes through mitochondrial fusion, which favor a more efficient oxidative phosphorylation—thus preventing the shift to glycolysis—or, on the contrary, disrupt supercomplex assembly by fission, which is accompanied by increased glycolysis (Fig. 4).
ROS production and proinflammatory cytokine release (TNFα and IL-6) are related, since ROS signaling prevents the dephosphorylation of different kinases involved in proinflammatory signaling pathways, including mitogen-activated protein kinases, p38, extracellular signal-regulated kinase or JNK (38). Furthermore, a high number of phosphatases are sensitive targets of oxidative stress and may be inactivated under oxidizing conditions, which would amplify the effect of redox-linked activation of key protein kinases (358).
Given that the balance between phosphatases and protein kinases determines the overall phosphorylation state of cellular phosphoproteins, aberrant protein phosphorylation may exacerbate the pathophysiology of a high number of human diseases. Therefore, targeting them with different compounds would ameliorate clinical symptoms (190). In the case of Fanconi anemia, for example, abnormal phosphorylation/dephosphorylation of signaling molecules has been linked to clinical complications, suggesting that dysfunction of kinases/phosphatases is implicated in the pathophysiology of human diseases (172)
The tricarboxylic acid (TCA) cycle is another metabolic pathway that modulates immune function. This has been shown specifically for succinate dehydrogenase and NADP+-dependent isocitrate dehydrogenase (227). Isocitrate dehydrogenase reduces citrate to α-ketoglutarate. M1 macrophages undergo a break in the Krebs cycle at isocitrate dehydrogenase, which raises citrate levels. When citrate levels are high, this metabolite is used to synthesize fatty acids and proinflammatory molecules, which contribute to the inflammatory response. Moreover, succinate, whose levels depend on glycolysis, increases upon LPS stimulation, leading to enhanced interleukin-1β (IL-1β) production (321) and promoting an inflammatory response. In this case, the source of succinate is not the TCA cycle, but glutamine metabolism.
Recently, it has been described that IL-10 can inhibit glycolysis and LPS-induced glucose uptake, and can even activate oxidative phosphorylation, therefore, controling the metabolism of immune cells. In addition, IL-10 can activate the mammalian target of rapamycin inhibitor DDIT4, thus inducing mitophagy and eliminating dysfunctional mitochondria characterized by high ROS production and low membrane potential. In a study that included both patients with inflammatory bowel disease and an animal model of colitis, Ip et al. demonstrated that macrophages accumulate damage when levels of IL-10 are decreased or depleted, and that this impairs activation of the NLRP3 inflammasome and IL-1β release (128).
Another interesting article has recently shown that mitophagy regulated by BCL2/adenovirus E1B 19-kDa-interacting protein 3-like is crucial for mitochondrial clearance during macrophage polarization toward the proinflammatory and glycolytic M1 phenotype. In contrast, differentiation to the M2 macrophage relies primarily on oxidative phosphorylation (84). The authors concluded that mitophagy is essential for promoting a metabolic switch toward glycolysis, which, in turn, contributes to cellular differentiation and cellular homeostasis.
The NLRP3 inflammasome is regulated by mitochondria, not only through metabolic remodelling but also by mitochondrial integrity. This effect occurs especially under insulin resistance conditions such as diabetes and/or obesity (233). In this context, the mitochondrion itself can activate the NLRP3 inflammasome by acting as a damage-associated molecular pattern (DAMP), releasing mitochondrial content to the cytosol (289) and to the blood (384). Thus, the mitochondrial membrane lipid cardiolipin and mtDNA contribute to the inflammatory response by interacting with NLRP3 (130, 222) (Fig. 5). The NLRP3 inflammasome will be expanded on in a later section of this review article.
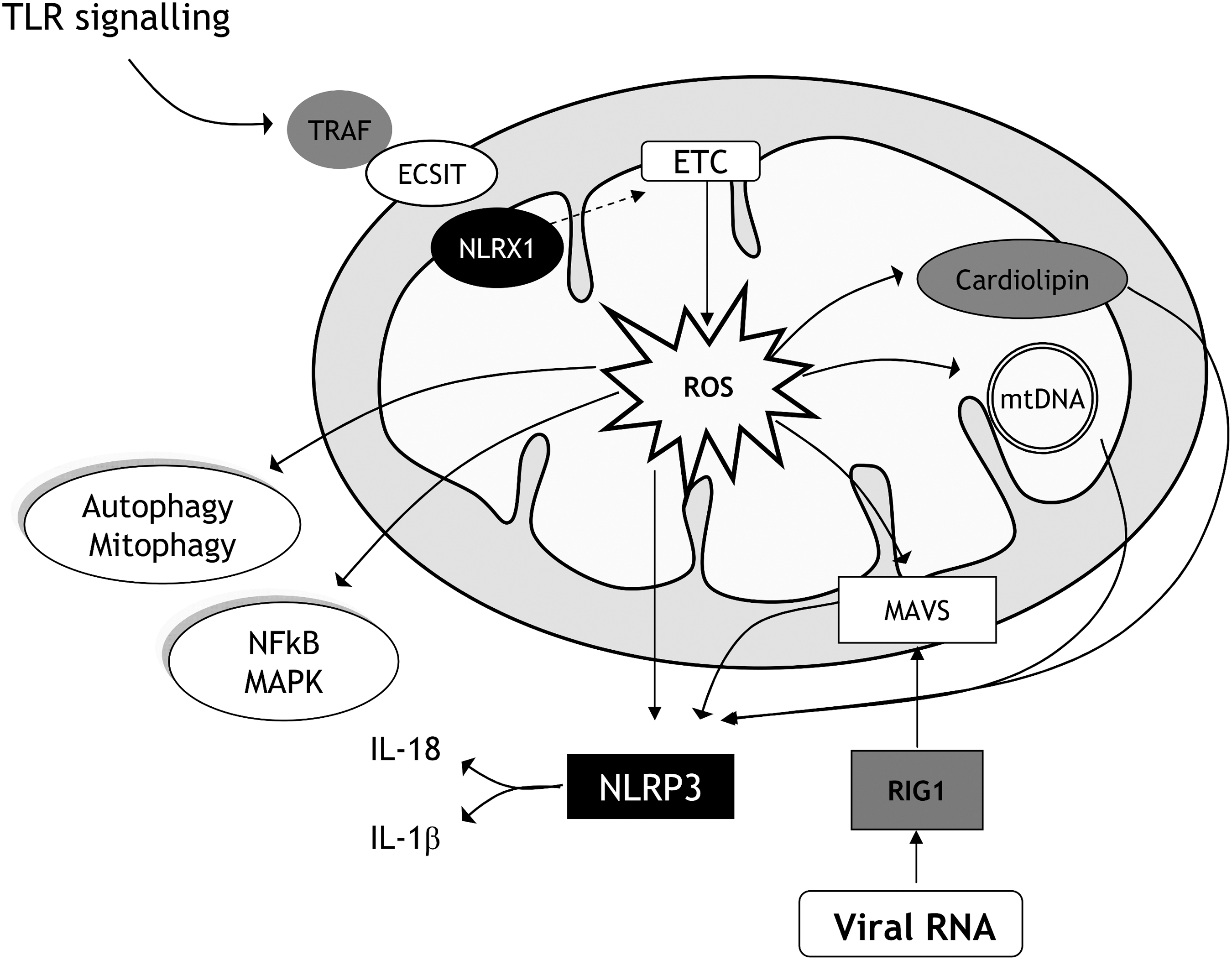
E. Mitochondria as a therapeutic target
Mitochondria perform multiple functions, including ATP and ROS production, control of Ca2+ homeostasis, and regulation of cellular survival or cell death, among others. As we have previously mentioned, oxidative stress and mitochondrial dysfunction are related with diseases such as cancer, aging, neurodegenerative, and cardiometabolic diseases. For all of these reasons, mitochondria are key targets for treatment, and in the past 15 years, important progress has been made in understanding their function, structure, and physiology in multiple diseases including obesity, diabetes, and heart disease. In addition, a large amount of research has explored the efficacy of lifestyle interventions (exercise and diet) and pharmacological strategies such as mitochondria-targeted therapies, discussed in detail by Camara et al. (43).
First, we describe how lifestyle interventions can improve mitochondrial function. Different reports have described that physical activity can improve mitochondrial biogenesis and insulin sensitivity in diabetic patients. Furthermore, it has been demonstrated that oxidative phosphorylation increases after a 12-week exercise intervention program (201).
One of the main beneficial effects of exercise is the activation of AMPK, which results in the phosphorylation of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) (131) and eventual induction of mitochondrial biogenesis. Other beneficial effects of regular exercise are the stimulation of mitochondrial biogenesis in skeletal muscle and the metabolism in aged humans (271).
Furthermore, regular exercise slows down the decline in neuromuscular function and loss of muscle mass by modulating muscle wasting. Whereas a sedentary life style is closely related with the appearance of metabolic diseases such as type 2 diabetes and/or obesity (25), physical exercise exerts beneficial pleiotropic actions that modulate mitochondrial function and mitochondrial dynamics in different tissues, especially in skeletal muscle (20). Physical exercise reduces energy levels, therefore, increasing the AMP/ATP ratio and activating the essential bioenergetics regulator AMPK.
Caloric restriction is a nonpharmacological intervention that can stave off the development of metabolic diseases and prolongs the life span (230). Caloric restriction can also prevent insulin resistance by reducing ROS production and increasing mitochondrial function (21). In relation to this, one study has demonstrated that caloric restriction enhances Akt2 activity and glucose sensitivity in skeletal muscle in an animal model of aging (344).
In addition to nonpharmacological approaches to mitochondrial treatment, there are different strategies for pharmacological interventions. In this sense, several drugs can modulate ROS production and, therefore, improve mitochondrial function. A study performed by Aldakkak et al. demonstrated that temporary blockade of mitochondrial complex I activity by amobarbital protected guinea pig hearts during ischemia/reperfusion injury by reducing production of superoxide and mitochondrial Ca2+ loading (5). Similarly, Xu et al. found that transient inhibition of complex I in mice hearts decreased ROS generation and reduced cardiac injury during reperfusion (361).
Mitochondrial dynamics are crucial for mitochondrial function, with mitochondrial fission representing a key target for treatment in metabolic diseases (258); for example, in the case of the mitochondrial division inhibitor-1 (mdivi-1), a compound that inhibits GTPase activity of dynamin-related protein 1 (DRP1) (48). In fact, mdivi-1 can improve mitochondrial function and decrease ROS production under high levels of palmitic acid by modulation of DRP1 activity (133). Under hyperglycemic conditions, mdivi-1 decreases atherosclerosis, inflammation, and oxidative stress in an animal model of diabetes, thus exerting beneficial actions (346). However, other studies have shown that treatment with this compound over long periods can inhibit mitochondrial function (143), suggesting that it is effective only when administered for short periods.
Another drug that affords benefits by modulating mitochondrial dynamics is dynasore, which disrupts the GTPase activity of dynamin and inhibits mitochondrial fission (187). For example, dynasore decreases cardiomyocyte mitochondrial fission and mitochondrial ROS production in a model of ischemia/reperfusion (95). It has also demonstrated beneficial effects in type 2 diabetes (268), and can inhibit apoptosis in cardiomyocytes under oxidative stress condition (94). Another potentially promising compound is P110, which decreases DRP1 activity, therefore, improving mitochondrial function and morphology in neurons (251).
15-Oxospiramilactone (S3), a compound that inhibits Wnt/β-catenin signaling, is an anticancer drug. (178). S3 can target key mitochondria enzymes such as deubiquitinase USP30, which modulates mitochondrial morphology by deubiquitination of fusion proteins MFN1 and MFN2. This action increases MFN1/2 activity and, in turn, mitochondrial fusion (380).
In summary, all the aforementioned compounds have demonstrated beneficial effects, although long-term administration can be deleterious due to total inhibition of mitochondrial fission. Thus, alternative therapies involving partial inhibition of mitochondrial fission may be of more use in the treatment of mitochondrial dysfunction and its related diseases.
1. Mitochondria-targeted drugs
Mitochondria are key organelles for therapy in multiple pathological conditions. Targeting mitochondria is a complex process, as they depend on specific transporters. In fact, diffusion through the inner mitochondria is challenging, and compounds usually need to be encapsulated inside or attached to a carrier to preserve the pharmacological activity of their molecules.
Several molecules have been targeted to mitochondria by enclosing them inside liposomes (281) or by using lipophilic cations (218) or small cell-permeable peptides (317). Different molecules have been used in these targeting strategies, such as antioxidants, vitamin B1, succinate, proteins, and substrates of the ETC (10, 281).
a. Nanotechnology
Nanotechnology has been developed in recent years as a therapeutic tool for mitochondrial diseases. In fact, drug delivery systems based on nanoparticles have several advantages: specificity of drug concentration at a target site, better pharmacodynamic and pharmacokinetic properties, and enhanced internalization for target organ delivery. All of these conditions have drawn attention to the use of nanoparticles as antioxidant molecules and/or carriers of antioxidant compounds (105). For example, targeted delivery of geranylgeranylacetone to mitochondria by triphenilphosphonium (TPP) nanoparticles prevents aminoglycoside-induced hearing loss (348).
b. MitoQ
The most used mitochondria-targeted antioxidant is MitoQ, which is composed of an ubiquinone moiety linked to a TPP cation by a chain of 10 carbons (218). MitoQ is incorporated into the matrix-facing surface of IMM and is recycled by complex II into ubiquinol (14). This antioxidant is capable of decreasing peroxynitrite formation, thereby preventing mitochondrial impairment and lipid peroxidation (140). In addition, when MitoQ is administered, it is rapidly taken up by cells from the blood (248).
Furthermore, ubiquinone can eliminate ROS directly, and has demonstrated beneficial effects under oxidative stress conditions. For example, we have recently demonstrated that MitoQ prevents ROS formation and leukocyte–endothelium interactions in leukocytes from type 2 diabetic patients (82). Other studies have reported that MitoQ is beneficial in different pathologies, including sepsis and cardiometabolic diseases (193). Since MitoQ has positive charges and is located at the IMM, it can generate a pseudo- ΔΨm via proton displacement with exogenous positive charges, which eventually promotes autophagy in HepG2 cells (309).
Multiple studies have confirmed the antioxidant activity of MitoQ in several cellular processes, such as mitochondrial dynamics, mitophagy and cell death. For example, MitoQ inhibits mitochondrial fission in cell lines exposed to ETC inhibitors (244) and can also reduce mitochondrial fission in the 6-hydroxydopamine cell model of Parkinson's disease (295).
Mitochondria-targeted antioxidants have demonstrated beneficial effects in β-cells under hyperglycemia (177) and in human and animal models of tolerance to nitroglycerine in cardiometabolic diseases, in which MitoQ treatment improves mitochondrial function and aldehyde dehydrogenase 2 activity (83, 96). MitoQ has also shown beneficial actions in cardiometabolic diseases; for example, Chacko et al. reported that MitoQ improves kidney function in an animal model of type 1 diabetes (50).
In animal models of atherosclerosis and metabolic syndrome, MitoQ has been shown to reduce hyperglycemia and DNA damage and to prevent hypercholesterolemia (204). Other similar studies have demonstrated that MitoQ decreases lipid peroxidation in obese rats and improves metabolic profile, restores ischemia-induced coronary collateral growth, and prolongs animal survival (250).
MitoQ has also been employed as an anticancer compound as it can activate apoptosis and autophagy, thus exerting cytotoxic effects in cancer cells (257). One possible explanation for this is that cancer cells possess a high ΔΨm and that this capacity allows the accumulation of MitoQ (391). In addition, cancer cells produce high amounts of ROS and alter the redox balance. Recently, it has been demonstrated that MitoQ can decrease mtDNA integrity by modulating ROS production and membrane potential in cancer cell lines, such as MDA-MB-231 and H23 (247). The disruption of mtDNA integrity may contribute to oxidative phosphorylation dysregulation and selectively deplete ATP levels in tumor cells.
Another example of mitochondria-targeted antioxidant is MitoC (mitochondria-targeted derivative of ascorbate), which reacts with different ROS and is rapidly recycled back—within the mitochondria—to the active ascorbate moiety by thioredoxin (TXN) and the glutathione systems (91). This antioxidant has demonstrated beneficial effects against mitochondrial lipid peroxidation (91).
c. SkQ molecules and Szeto-Schiller peptides
Another group of mitochondria-targeted antioxidants is composed of the 10-(6′-plastoquinonyl)decyltriphenyl-phosphonium (SkQ) molecules. The most used is SkQ1, which accumulates 108-fold inside the IMM. SkQ1 cation can accumulate even more, because it joins to cardiolipin and is the first site for antioxidant activity (9). Like MitoQ, SkQ1 is a rechargeable antioxidant, as it is reduced by complex III. SkQ1 has shown beneficial effects by protecting against lipid peroxidation in erythrocytes and avoiding hemolysis (229) and by promoting kidney function in a model of ischemia/reperfusion (245).
SkQR1, a fluorescent SkQ derivative, has been considered a nephroprotective and neuroprotective agent in an animal model of ischemia (246). Furthermore, it can prolong the life span of different animal species (293), and delays the development of glaucoma and dry eye (293).
In a recent article, Shabalina et al. (282) have demonstrated that SkQ1 improves health span and life span in mtDNA mutator mice. In addition, SkQ1 has been used in neurodegenerative disorders such as Alzheimer's disease, in which it has been shown to reduce alterations in behavior and memory deficit by decreasing the amount of Aβ and AβPP (299). Recently, Kolosova et al. (154) have demonstrated that SkQ1 can alleviate the signs of Alzheimer's disease in old rat models of the disease by improving mitochondrial function. SkQ1 can be administered at very low doses, thus avoiding possible secondary effects; in contrast, at high doses, antioxidants can convert to pro-oxidants, as demonstrated for MitoQ, SkQ1, and SkQ3 (308).
Szeto-Schiller (SS)-peptides (SS-19, SS-02, SS-31, and SS20) are small-sized, permeable antioxidants that are rapidly taken up by different types of cells, including endothelial, renal, and embryonic cells. SS-peptides accumulate inside mitochondria, especially in the IMM (315). It is important to mention that these compounds do not require changes in ΔΨm to accumulate (315), which confers an advantage in the treatment of mitochondria-related diseases. SS-peptides are resistant to protein degradation and are able to reach mitochondria (386). SS-peptides inhibit lipid peroxidation due to a dimethyl tyrosine residue with antioxidant capacity. In this context, SS-31 seems to be the main antioxidant compound, as it has been demonstrated in animal models of mitochondrial dysfunction (356, 387).
SS-31 has displayed beneficial effects under hyperglycemic conditions; for example, it prevents ΔΨm loss and decreases cytochrome c release and ROS production in human retinal endothelial cells (171). It has also demonstrated beneficial effects in C2C12 myoblasts by decreasing autophagy and proteolysis under nutrient deprivation or exposure to rapamycin (253).
SS-31 treatment has demonstrated positive effects in human diseases and animal models in the context of myocardial infarction (57), Alzheimer's disease (189), and obesity (9). SS-31 also decreases levels of CD36, a scavenger receptor that promotes ROS production and tissue impairment under ischemia (58). SS-31 can prevent oxidative stress and autophagy in Alzheimer's disease by protecting against Aβ toxicity and increasing axonal transport of mitochondria and synaptic viability, and by reducing fission proteins (259). It has been shown to improve glomerular architecture in aged mice (312) and to protect mitochondria after acute ischemia, preventing upregulation of the proinflammatory cytokines IL-18 and IL-1 (316). In general, the aforementioned studies have highlighted the potential of SS-31 as a beneficial agent in different diseases.
Other compounds with antioxidant activity include conjugates of the plant alkaloids palmatine and berberine with the antioxidant moiety plastoquinone (SkQP and SkQB), which display antioxidant capacity in vitro (55). Berberine has also demonstrated beneficial effects by improving diabetes-associated cognitive decline through control of the inflammatory response in diabetic rats (51). Another antioxidant molecule is the derivate of thymoquine, which has antioxidant capacity in multiple diseases such as sepsis and cancer. In fact, SkQR1 can target drugs and accumulate at higher concentrations in normal cells than cancer cells (89). In summary, SkQ-based molecules are promising agents for mitochondrial treatment in multiple diseases.
d. Bioactivation of enzymes
The use of bioactivating enzymes is another strategy for modulating oxidative stress and ROS production. In fact, mitochondrial enzymes such as cytochrome P450 and monoamine oxidase are potential candidates for xenobiotic metabolism (8). One of the strategies reported is the use of mitochondrial β-oxidation enzymes, which biotransform fatty acids and xenobiotics such as hydroxydecanoic acid and tianeptine. As antioxidants, alkanoate-based drugs have been used to target at mitochondrial β-oxidation (8). For example, 5-(1-methyl-1H-imidazol-2-ylthio) alkanoates can transform into methimazole with antioxidant and cytoprotective capacity in rat cardiomyocytes in a model of hypoxia-reoxygenation (265). Such studies highlight the efectiveness of mitochondria-targeted antioxidants in different models of oxidative stress.
Other compounds with antioxidant capacity are manganese porphyrin-based cellular redox modulators (MnP), which mimic mitochondrial manganese superoxide dismutase (MnSOD). MnP concentrate inside the mitochondria, exerting beneficial effects on the mouse heart (328) by decreasing superoxide levels. A study by Ferrer-Sueta et al. demonstrated that MnTE-2-PyP5+ protects submitochondrial particles by decreasing levels of peroxynitrite (88).
e. Lipophilic cations and liposomes
Lipophilic cations use the ΔΨm and accumulate within the mitochondrial matrix (266) (Fig. 6). There are different lipophilic cations for mitochondrial accumulation of antioxidants, but the most used is TPP+. The plasma membrane potential allows the uptake of different biomolecules, which leads to accumulation in the mitochondrial matrix. In this sense, a large number of compounds with antioxidant activity have been targeted to mitochondria using TPP; namely, vitamins, ebselen, nitrones, ubiquinones, and resveratrol (26, 132, 332). However, this strategy has some disadvantages, such as sublocalization in the matrix surface of the inner membrane, potential toxicity of high doses, and the capacity for transferring only neutral and low molecular weight molecules.
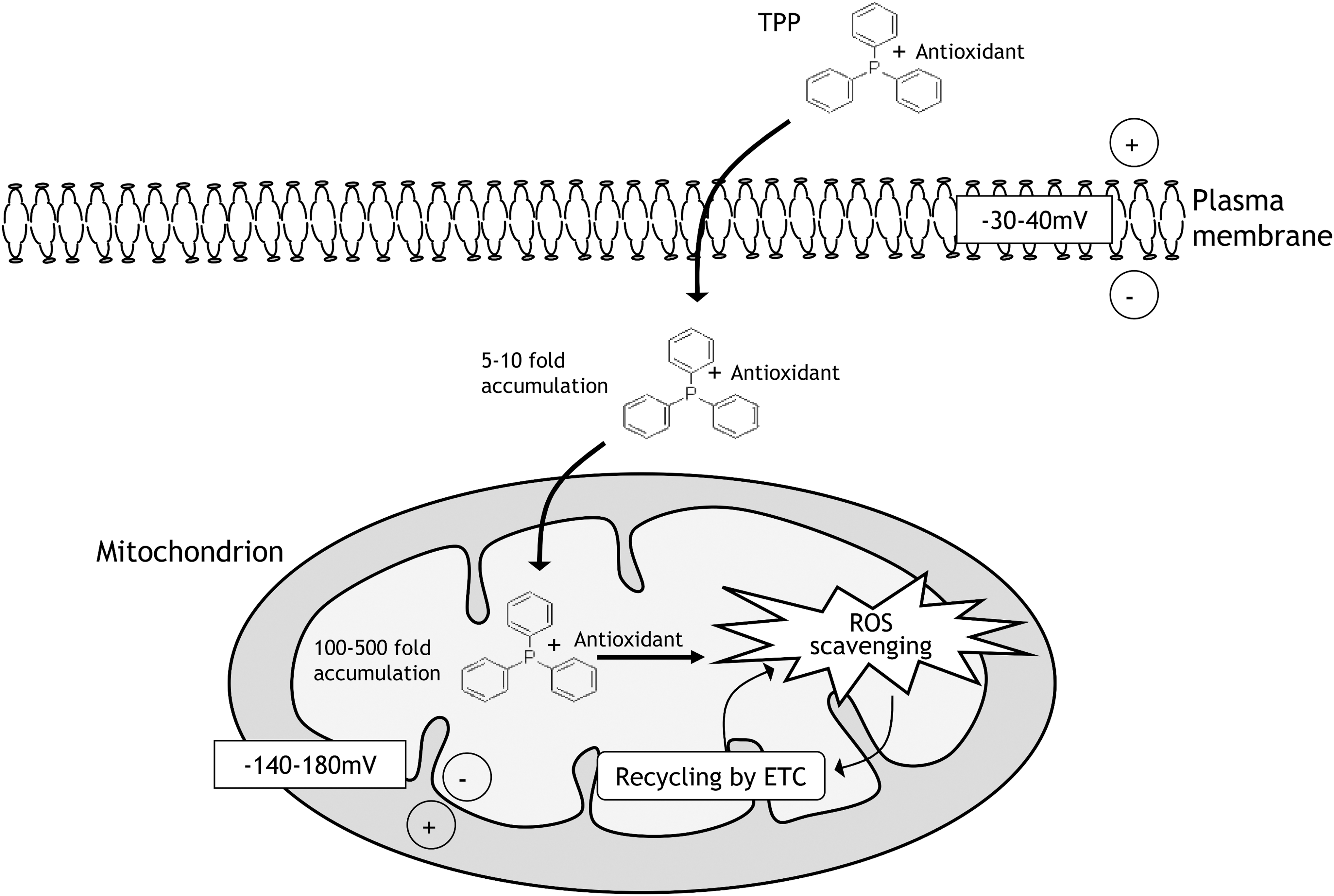
Liposomes in general are constituted by phosphatidylglycerol, cholesterol, and phosphatidylcholine. One of the main characteristics of liposomes is their capacity to carry hydrosoluble drugs or lipid compounds in their core, thus allowing them to act as nanocarrier delivery systems. One advantage is that the molecules inside the liposomes are encapsulated, thus maintaining their functionality and structure.
Liposomes can carry antioxidant enzymes, a combination of different agents or molecular antioxidants, such as encapsulated N-acetylcysteine (6), or a combination of silybin and phospholipids named Siliphos. Siliphos possesses antioxidant properties in an animal model of steatosis (71) and in nonalcoholic fatty liver disease (NAFLD) patients, improving mitochondrial function and insulin sensitivity (180). Moreover, Yue et al. (379) have demonstrated that the combination of coenzyme Q10-loaded liposomes with ultrasound-targeted microbubbles prevents kidney damage in diabetic nephropathy.
Another promising compound is MITO-Porter, a liposome that delivers cargo into the mitochondria through a membrane fusion mechanism, enabling the transport of nucleic acids, proteins, and small molecules (362). Biocompatible molecules can be transported in different ways, including delivery of the compound from the extracellular space to the cytosol, targeting of the mitochondria, and mitochondrial delivery by membrane fusion. Yamada et al. (364) have shown the MITO-Porter system to be an excellent carrier for mitochondrial delivery of a cytotoxic agent, which represents a possible therapeutic strategy for treating drug-resistant cancers.
One of the limitations of nanocarrier systems is their loss due to endocytosis, since they can be trapped within endosomes and marked for enzymatic degradation in lysosomes. To circumvent this limitation, nanocarriers are designed to induce the degradation of the endosome and facilitate their release to the cytosol. If they are to be of therapeutic value, nanocarriers should be efficient in their cytoplasmic delivery of the carried compound and in their targeting of mitochondria. For example, MITO-Porter can deliver DNAse I into mitochondria (363) and antitumor drugs can be released from liposomes into the mitochondria (27).
f. Biodegradable polymers
Another strategy for mitochondrial-targeted antioxidants is the use of biodegradable polymers (194). This method consists of the design of polymeric nanoparticles targeted to mitochondria to decrease oxidative stress and ROS. For example, the polymer PLGA-b-PEG-TPP NP has demonstrated positive effects in obesity by improving mitochondrial function. Wang et al. (347) have demonstrated that hyaluronic acid-coated chitosan nanoparticles induce apoptosis and enhance antitumor efficiency by increasing ROS production due to targeted drug delivery via CD44.
One interesting compound is ergothioneine (Egt), a molecule with antioxidant activity that accumulates inside mitochondria (105) due to an organic cation transporter. This antioxidant decreases the levels of peroxynitrite and hydroxyl radicals (105), and has demonstrated beneficial effects by protecting against DNA damage in PC12 cells in a model of cytotoxicity (64). Furthermore, Egt increases the immunomodulatory function of TLR agonists by its effects on macrophages (374).
g. Mitochondrial uncoupling
Another strategy for modulating ROS production and oxidative stress is to diminish ROS production by mitochondria; in fact, a rise in ROS production is related to an increase of ΔΨm. In this case, mitochondrial uncoupling allows protons to bypass ATP synthase, decreasing ATP synthesis due to the proton leak. This process is carried out by UCPs located in the IMM.
There are some drugs, such as 2,4-dinitrophenol, that can induce mitochondrial uncoupling. Dissipation of ΔΨm by means of mild uncoupling can decrease levels of ROS and consequently avoid oxidative damage to proteins, lipids, and nucleic acids. This is the main concept of the “uncoupling to survive theory,” which holds that the life span can be prolonged through mitochondrial uncoupling (169).
Different studies have highlighted the function of UCPs; for example, it has been shown that overexpression of hUCP2 has beneficial effects in an animal model of Parkinson disease (65). Different chemical uncouplers have been tested, including FCCP, CCCP, and 2,4-dinitrophenol, and in general have been shown to prolong life span in animals (105); however, it should be taken into account that their therapeutic range is very narrow and their use, therefore, limited. For example, 2,4-dinitrophenol was reported to produce death in patients with varying symptoms, including hyperthermia, tachycardia, and cardiovascular events (106).
The uncouplers 2,4-dinitrophenol and butylated hydroxytoluene have been conjugated with the TPP cation to generate Mito-DNP (24) and Mito-BHT (182). Interestingly, Mito-BHT increased proton leak in mitochondria of rat thymocytes and showed a wide dynamic range, in other words, the ratio of concentrations leading to maximum and minimum observable uncoupling (182).
In this context, efforts are mounting to design uncoupler drugs that work safely, with future research needing to focus on increasing the dynamic range of uncouplers to avoid side effects.
III. NLRP3 Inflammasome
Inflammasome activation is an innate immune response to pathogen invasions, but also to noninfectious stimuli, such as lysosomal destabilization, mitochondrial ROS formation, and the release of mtDNA. Microbial and endogenous stress promote the release of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), respectively (261). PAMPs and DAMPs are recognized by nod-like receptor (NLR) or absence in melanoma 2 (AIM2)-like receptors (ALR), which represent a family of pattern-recognition receptors (PRRs), and these cytoplasmic sensors can oligomerize and assemble an inflammasome.
After an activating stimulus, the NLR/ALR sensor recruits the adaptor apoptosis-associated speck-like protein, which contains C-terminal caspase-recruitment domain (CARD) namely ASC and caspase-1, a protease synthesized as procaspase-1 that becomes active after dimerization in the inflammasome complex (192, 303). Active caspase-1 cleaves the proinflammatory cytokines pro-IL-1β and pro-IL-18 into their active forms IL-1β and IL-18, and is also involved in pyroptosis, an inflammatory form of cell death (277).
Currently, four types of inflammasomes have been characterized, and are named according to the PRRs that form them: AIM2, NLRP1, NLRP3, and NLR family CARD domain containing 4 (NLRC4) inflammasomes. Whereas some sensors, such as NLRP1 and AIM2, can directly bind to stimulating molecules, others, such as the NLR family member NLRP3, require a priming step to become activated; for example, bacterial LPS-induced pro-IL-1β transcription (121). Therefore, the priming step involves the recognition of specific ligands by TLRs and the subsequent nuclear factor kappa B (NF-κB)-dependent transcription of the inflammasome components NLRP3 or pro-IL-1β.
NLRP3 is the best characterized inflammasome, and is related to the development of different metabolic diseases, including type 2 diabetes and obesity, and the early steps of the atherosclerotic process (109, 303). Interestingly, activation of the NLRP3 inflammasome is closely related to mitochondrial dysfunction and ROS production; thus, diseases that involve mitochondrial alterations may also be related to NLRP3 inflammasome activation.
The NLRP3 inflammasome is a crucial mediator of innate immune responses and is composed of the NLRP3 protein, the adaptor protein ASC, and the cysteine protease procaspase-1. The NLRP3 protein consists of a central nucleotide binding and oligomerization domain (NBD), a C-terminal leucine-rich repeat (LRR) domain and an N-terminal pyrin domain (PYD) (Fig. 7A). After an activating stimulus, NLRP3 senses the ligand via its LRR domain. The PYD and NBD domains are then exposed, thus allowing for NLRP3 oligomerization by homotypic NBD domain interaction. This allows the recruitment of the ASC adaptor through PYD–PYD interaction. ASC, in turn, recruits caspase-1 through its CARD domain, facilitating its activation via autocleavage, and triggering IL-1β and IL-18 processing and secretion (Fig. 7B) (2, 305).
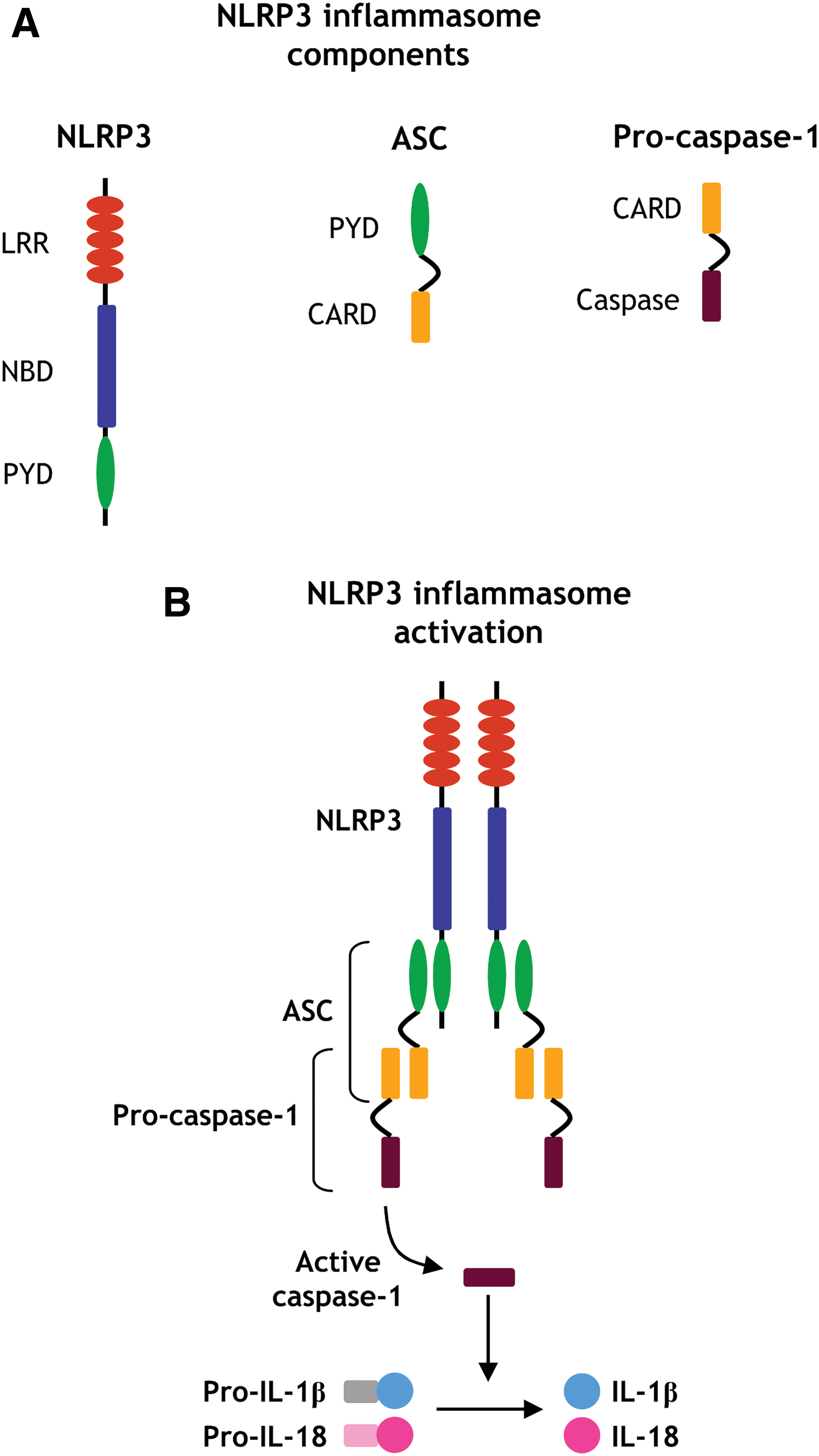
It is important to note that PYD and CARD domains are important regulation sites of inflammasome activity. PYD-only proteins (POPs) and CARD-only proteins (COPs) are endogenous negative modulators of inflammasomes that interfere with PYD- or CARD-driven interactions, respectively, and prevent caspase-1 activation (75, 298). The function of these proteins is important during inflammation as a mechanism to fine-tune inflammasome responses, thereby limiting inflammation.
The NLRP3 inflammasome can be triggered by both exogenous and endogenous stress—microorganisms or extracellular ATP, respectively—and responds to a wide array of stimuli that are usually indicative of cellular danger, including generation of mitochondrial ROS, cellular K+ efflux, release of mtDNA or cardiolipin, increases in intracellular Ca2+, and release of cathepsins from lysosomes into the cytosol, although the involvement of the latter stimulus is the subject of some controversy (108, 277). This broad spectrum of stimuli may converge in common signals that directly activate the NLRP3 inflammasome.
Two major proximal signals for NLRP3 activation have been described. First, generation of ROS seems to be critical for NLRP3 activation, and numerous studies support this hypothesis. As examples, Cruz et al. demonstrated that blockade of ROS production through inhibition of NADPH oxidase activity impairs inflammasome activation (67), whereas Dostert et al. found that IL-1β release is inhibited when cells are stimulated with inflammasome activators and treated with the ROS inhibitors N-acetyl-
Although the precise mode of activation of the NLRP3 inflammasome by high ROS production is not completely understood, the involvement of specific redox proteins, such as TXN and thioredoxin-interacting protein (TXNIP), has been well characterized. In fact, it has been demonstrated that elevated ROS production promotes dissociation of TXNIP from TXN, and this event prompts the binding of TXNIP to NLRP3, leading to its activation (Fig. 8) (389).
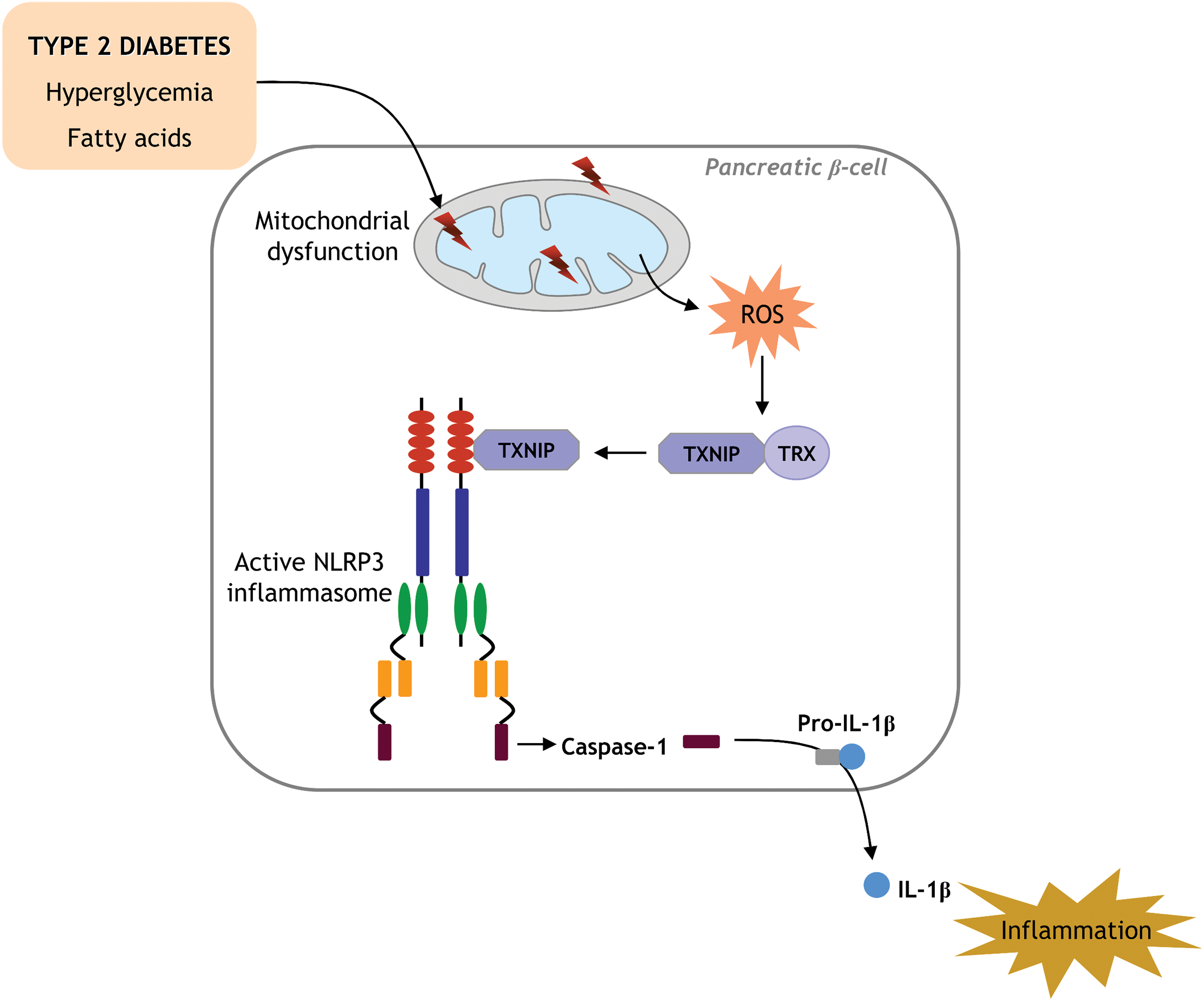
The second hypothesis proposes that a drop in intracellular K+ concentration is the main mediator of NLRP3 inflammasome activation (240). In fact, it has been reported that Candida albicans-dependent IL-1β production by the NLRP3 inflammasome is inhibited by blocking K+ efflux, suggesting that intracellular K+ decrease is critical for NLRP3 activation (104). However, ROS generation is often associated with K+ efflux, which raises the question of which is the most important of the aforementioned signals. Whether or not these mechanisms act in conjunction also requires further investigation.
Taking into account all of this evidence, the following sections deal with the function and importance of the NLRP3 inflammasome and its derived cytokines in instigating several human diseases, with a special focus on insulin resistance and type 2 diabetes.
A. Diseases related to inflammasome activation associated with mitochondrial dysfunction
Different types of stimulus-mediated induction of the NLRP3 inflammasome and the subsequent enhanced activation of caspase-1 and release of IL-1β and IL-18 cytokines have been related to several human conditions, such as cardiometabolic diseases and their associated complications (203). These diseases have classically been associated with mitochondrial impairment, which prompts one to consider whether inflammasome–mitochondria interplay is the key underlying pathological process.
Mitochondrial function is closely related to the activity of the NLRP3 inflammasome. The release of mitochondrial ROS as a consequence of mitochondrial damage is one of the main activation signals of the NLRP3 inflammasome (69). In this sense, both mitochondrial and extramitochondrial ROS are capable of activating the NLRP3 inflammasome, as suggested by studies in which the specific inhibition of mitochondrial ROS production (222) or NADPH oxidase (67) prevented NLRP3 inflammasome activation.
Besides the direct inflammasome activation by ROS, mitochondrial damage could lead to the release of several mitochondrial components, such as mtDNA or cardiolipin (108), which act as DAMPs and induce NLRP3 inflammasome activation (130, 289). Usually, mitochondrial damage originates from excessive ROS production, once again highlighting the primary role of ROS in the complex interplay between mitochondria and inflammasome (69).
Another factor linking mitochondria and inflammasome activation is cellular Ca2+ influx, induced by ATP or K+ efflux, which, in turn, promotes mitochondrial Ca2+ overload and, as a consequence, mitochondrial dysfunction (378). Murakami et al. demonstrated that blocking Ca2+ mobilization by different Ca2+ signaling inhibitors, such as thapsigargin or 2-aminoethoxydiphenyl borate, inhibits the assembly and activation of the NLRP3 inflammasome complex. These authors pointed to Ca2+ mobilization as a common, proximal step in activation of the NLRP3 inflammasome (217). However, Ca2+ mobilization may not be sufficient for NLRP3 inflammasome activation, since the Ca2+ ionophore ionomycin is unable to induce IL-1β production (217). Thus, Ca2+ may act together with other signals, such as mitocondrial ROS production, to activate the NLRP3 inflammasome.
An important mechanism of clearance of damaged mitochondria is mitophagy, a specific form of autophagy. It has been demonstrated that the absence of autophagic machinery in mice promotes the accumulation of damaged mitochondria, increasing mitochondrial ROS, and enhancing NLRP3 inflammasome activation (222). Furthermore, it has recently been reported that inducing mitophagy in macrophages by sestrin 2, a stress-inducible protein, leads to the suppression of NLRP3 inflammasome activation (148). Therefore, mitophagy represents a preventive mechanism against NLRP3 inflammasome activation (Fig. 9).
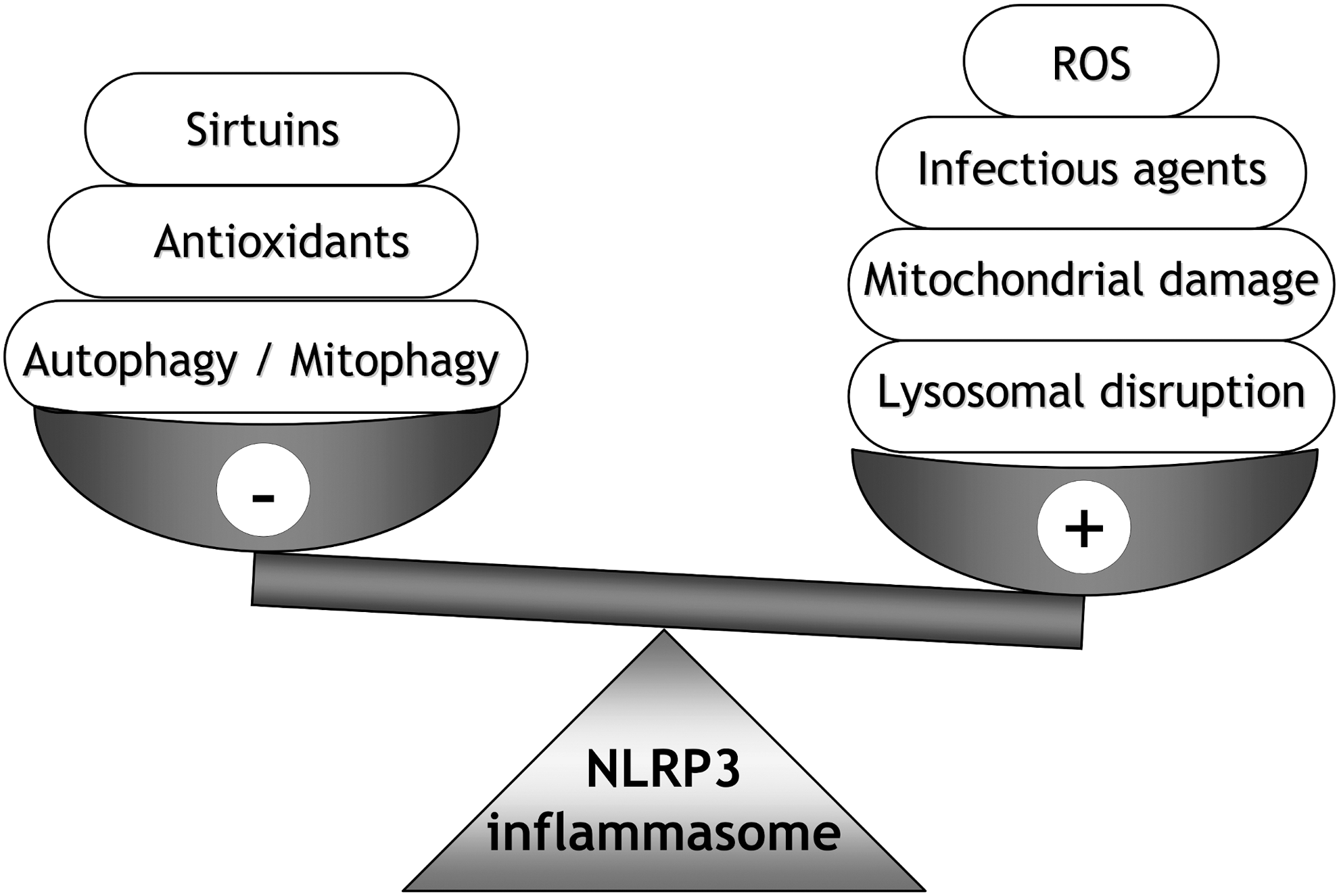
Hyperactivation of the NLRP3 inflammasome, together with altered mitochondrial function and enhanced ROS production, has been related to several human autoinflammatory diseases, such as gout or multiple sclerosis, and to metabolic disorders, such as obesity or type 2 diabetes (7, 124, 164).
Gout is characterized by elevated blood uric acid levels that induce the deposition of monosodium urate (MSU) crystals in joints, causing acute inflammation as a result. The deleterious effects of MSU crystals have been attributed to oxidative stress, mitochondrial pathway-dependent apoptosis, and NLRP3 inflammasome activation (149, 381). In relation to this, Martinon et al. showed that crystal-mediated peritonitis in mice is characterized by NLRP3 inflammasome activation and induces recruitment of neutrophils to the inflammatory focus (196). However, other authors have suggested that additional signals other than MSU are needed to activate NLRP3 inflammasome in patients with gout, such as FFA, which may act synergistically with MSU through TLR-2 signaling to drive inflammasome-induced IL-1β release (135).
Numerous studies endorse a key role of mitochondrial dysfunction in the physiopathology of multiple sclerosis (163, 224, 306), a neurodegenerative disease characterized by neuroinflammation and demyelination in the central nervous system. The immune-mediated axon injury that takes place in experimental autoimmune encephalomyelitis (EAE), a mice model of multiple sclerosis, involves alterations in mitochondrial morphology and membrane potential (224). In addition, increased caspase-1 expression has been observed in brain samples of patients with multiple sclerosis (207). EAE induction in mice lacking NLRP3 (Nlrp3 −/−) results in delayed onset and reduced severity of the disease, pointing to a crucial role for NLRP3 in multiple sclerosis (103).
The NLRP3 inflammasome has also been shown to be activated as a consequence of amyloid β (Aβ) deposition in the brain, the main feature of Alzheimer's disease (115), a condition classically associated with the mitochondrial dysfunction that occurs early on in its pathological course (39). The involvement of NLRP3 inflammasome activation in Alzheimer's disease has been the subject of numerous studies in the past decade (115, 274). In fact, caspase1(−/−) and Nlrp3(−/−) mice that also carry mutations associated with familial Alzheimer's disease display decreased activation of caspase-1 and IL-1β in the brain and enhanced Aβ clearance and are largely protected against Alzheimer's disease (115).
It is important to stress that the role of inflammation in Alzheimer's disease is ambiguous, since it is not completely understood whether inflammatory signals promote the disease-associated neurodegeneration or are induced with the aim of slowing down the accumulation of Aβ plaques in the brain, since monocytes and macrophages cross the blood–brain barrier to trigger the clearance of Aβ deposition (274).
Besides neuroinflammatory disorders, it has been demonstrated that NLRP3 inflammasome activation and the subsequent IL-1β secretion are key events in the early stages of atherosclerosis (78). Specifically, cholesterol crystals act as DAMPs and induce lysosomal damage, which activates the NLRP3 inflammasome and promotes acute inflammation in murine macrophages, constituting an early step in the atherosclerotic process, rather than a consequence of inflammation (Fig. 10) (78). Similarly, human macrophages respond to cholesterol crystals by secreting IL-1β, a response that is abolished when the NLRP3 receptor is silenced (254).
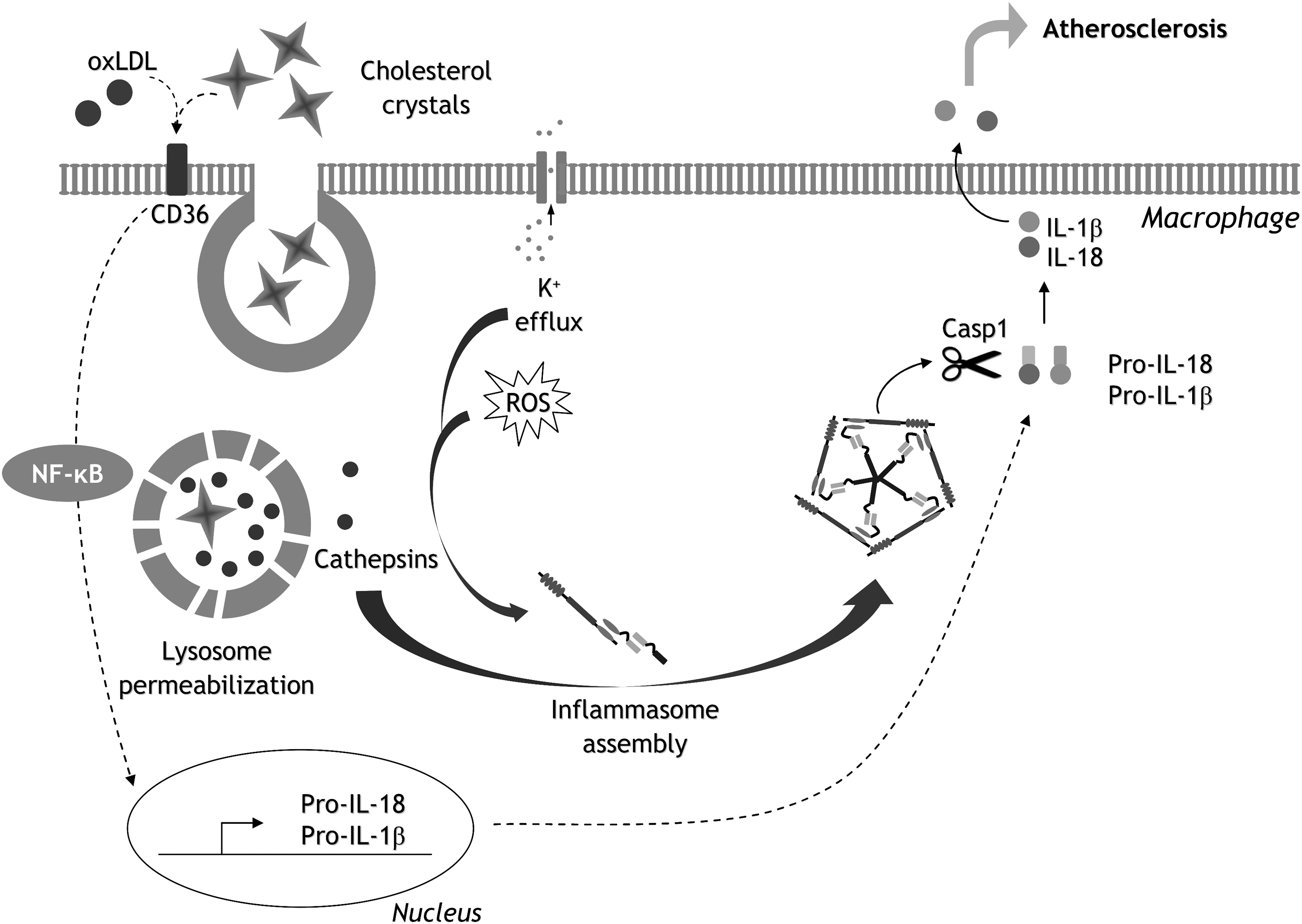
Interestingly, it has recently been reported that inhibition of inflammasome activity in the context of atherosclerosis is exerted by HDL, suggesting a novel anti-inflammatory property of this particle (325). Such studies suggest that inflammasome activation is a hallmark of atherosclerosis. Indeed, a positive correlation has been detected between NLRP3 expression levels in different cell types, such as monocytes or adipocytes, and the severity of coronary atherosclerosis, supporting a potential prognostic value of NLRP3 in the context of cardiovascular diseases (3, 16). In addition, studies in animal models endorse the targeting of NLRP3 as a promising therapeutic strategy for cardiovascular diseases (186, 327, 333). For example, van Hout et al. demonstrated that treating pigs with an NLRP3 inhibitor after the induction of myocardial infarction reduces neutrophil influx and infarct size and preserves cardiac function with respect to nontreated animals (333).
A role of NLRP3 inflammasome activation in the physiopathology of chronic kidney disease (CKD) has also been demonstrated (338). NLRP3 protein expression rises in parallel to CKD progression in mice, and genetic ablation of Nlrp3 in this murine model of CKD results in the attenuation of tubular injury, reduced leukocyte infiltration, and renal fibrosis (338).
The aforementioned associations between NLRP3 inflammasome activation and diverse human diseases highlight the urgent need for therapeutic approaches to prevent the detrimental consequences of inflammasome overactivation.
B. Insulin resistance, type 2 diabetes, and the NLRP3 inflammasome
In recent years, intense research has highlighted the role of the NLRP3 inflammasome in the development of insulin resistance and type 2 diabetes. Type 2 diabetes is characterized by chronic inflammation, which is manifested by enhanced circulating levels of proinflammatory cytokines such as TNFα, IL-6, IL-2, or IL-1β in diabetic patients (122).
For example, IL-1β is related to type 2 diabetes, as it can activate JNK and, therefore, the serine phosphorylation of IRS1, consequently interfering with the signaling pathway induced by insulin-PI3K-Akt and generating insulin resistance in several cells and insulin-targeted tissues such as skeletal muscle, liver, and adipose tissue. IL-1β can also induce oxidative stress and ER stress (336). In addition, IL-1β can enhance other inflammatory mediators through the IL-1R-amplifying cytokine network (11). Therefore, IL-1β is a key target for the control and modulation of type 2 diabetes (162). In fact, several clinical trials have evaluated the therapeutic potential of anti-IL-1β antibodies in type 2 diabetes, finding that gevokizumab and canakinumab effectively reduce glycemia and inflammation in diabetic patients (Table 1) (49, 116).
ASC, apoptosis-associated speck-like protein that contains a CARD; ROS, reactive oxygen species; TNFα, tumor necrosis factor alpha.
There is evidence that metabolic DAMPs can activate the NLRP3 inflammasome; for example, a fasting/refeeding study demonstrated decreased NLRP3 inflammasome activation in the fasting versus refed condition (330). Furthermore, the authors found that depletion of the mitochondrial-enriched deacetylase Sirtuin 3 enhanced NLRP3 inflammasome activation due to the increased mitochondrial ROS production in macrophage cell lines. Traba et al. (330) demonstrated that genetic and pharmacologic SIRT3 activation undermined NLRP3 activation in parallel with increased mitochondrial function in leukocytes from healthy and refed subjects, a trend that was not observed during fasting.
These findings indicate that nutrient levels can modulate the NLRP3 inflammasome and that this modulation occurs, at least in part, as a response to SIRT3-mediated mitochondrial homeostatic control. Supporting the importance of nutritional status for NLRP3 inflammasome regulation, Youm et al. reported that the ketone body β-hydroxybutyrate, which is enhanced under fasting conditions to serve as an alternative energy source, has anti-inflammatory properties and inhibits NLRP3 inflammasome activation by preventing K+ efflux (377). However, the potential beneficial effect of β-hydroxybutyrate or ketogenic diets on inflammasome activation during metabolic diseases such as diabetes has not yet been determined.
Studies have demonstrated that islet amyloid polypeptide induces an amyloid structure in the islet interstitium and the activation of the NLRP3 inflammasome in mouse macrophages (197). In this case, Meier et al. reported that the formation of islet amyloid in β-cells is necessary for the induction of islet inflammation in a high-caloric diet mice model, pointing to an important role of islet amyloid formation in β-cell dysfunction in type 2 diabetes (202). Furthermore, it has been described that activation of the NLRP3 inflammasome after amyloid accumulation is mediated by the PRR CD36 (286), which puts the spotlight on CD36 as a therapeutic target in pathological processes involving amyloid formation, such as type 2 diabetes. Indeed, apolipoprotein AI-mimetic peptide 5A, a novel drug that antagonizes CD36, has been reported to reduce the expression of Nlrp3 and Il-1β in the kidney and to slow the progression of the disease in a CKD mouse model (297).
Insulin resistance encompasses a variety of metabolic disturbances, promoting a vicious circle in which hyperglycemia induces glucotoxicity, which, in turn, triggers β-cell damage and alters insulin release, again favoring altered glucose metabolism. In addition, hyperglycemia promotes the release of IL-1β in β-cells (Fig. 11), which may not be merely a collateral effect, but rather a mediator of hyperglycemia-induced insulin resistance. The mechanism through which excess glucose promotes this inflammatory response and the role of inflammasomes in insulin resistance has been extensively explored. Furthermore, high levels of Ca2+ have been shown to contribute to NLRP3-inflammasome activation by instigating ER stress and the release of mitochondrial ROS or mtDNA (352, 353).
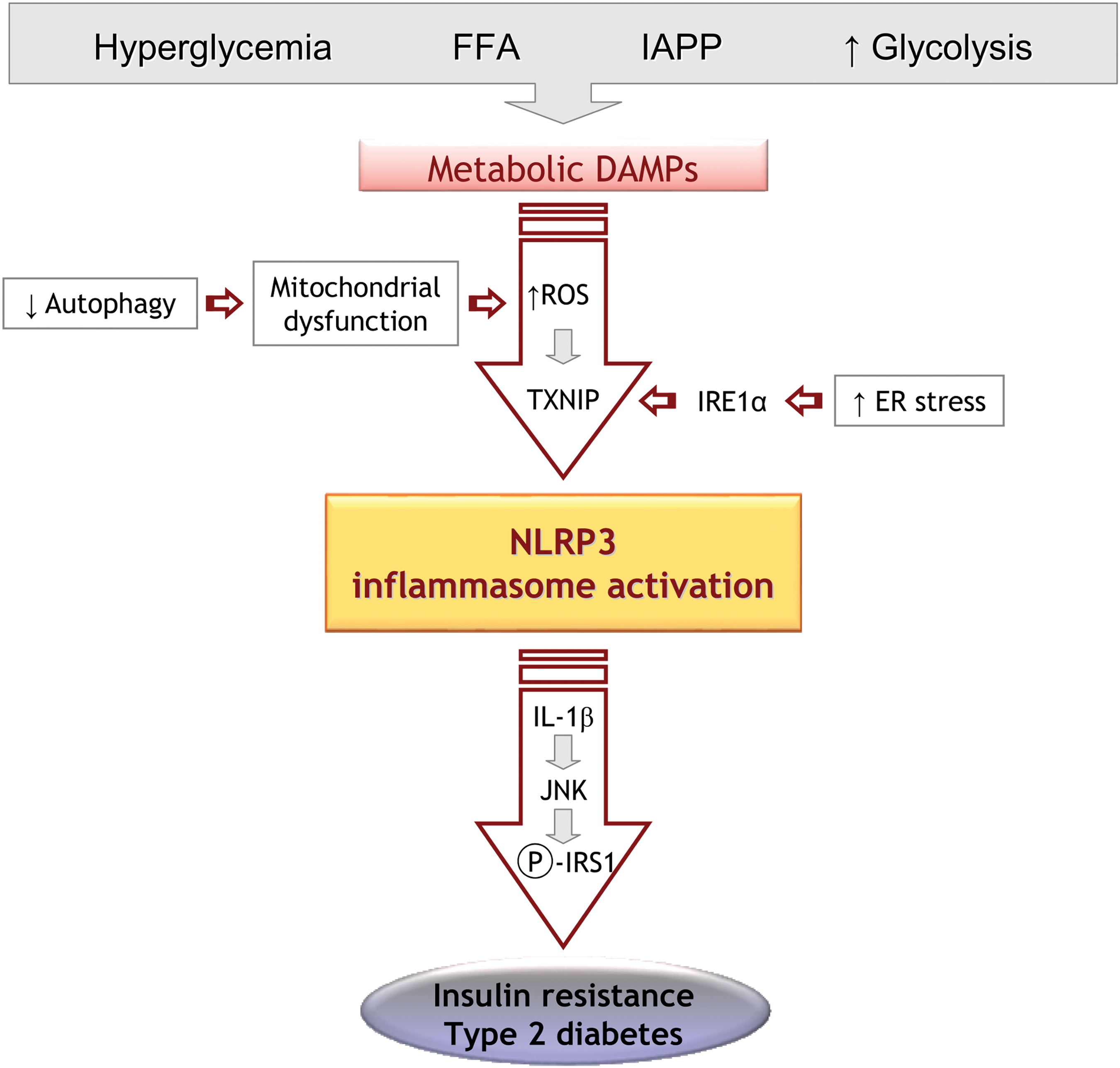
In this sense, metformin, the first-choice hypoglycemic drug for type 2 diabetes treatment, has demonstrated beneficial effects in monocyte-derived macrophages from diabetic patients, in which it decreases IL-1β levels through activation of AMPK (164), highlighting the relationship between the NLRP3 inflammasome and the regulation of glucose metabolism. This relationship has also been demonstrated in animal models of gene deletion (300, 334). Such studies have shown that deficiency of Nlrp3 or other related genes, such as Casp1 or Asc/Pycard, improves insulin sensitivity and glucose tolerance in mice models of obesity (300, 334).
Overall, these studies highlight the relationship between insulin resistance, inflammation, and the NLRP3 inflammasome. In fact, Chiazza et al. have demonstrated that BAY 11-7082, an NLRP3 inflammasome inhibitor, can prevent the activation of NLRP3 inflammasome induced by diet, thus leading to decreased levels of caspase-1, IL-1β, and IL-18 in different tissues, including the liver and kidney. Furthermore, BAY 11-7082 inhibited NF-κB nuclear translocation, demonstrating that the metabolic abnormalities attributable to chronic exposure to a high-fat high-fructose diet are attenuated when the NLRP3 inflammasome is selectively modulated (56).
Similarly, other drugs (summarized in Table 1), such as MCC950, isoliquiritigenin, or γ-tocotrienol (γT3), inhibit NLRP3 inflammasome activation and have the potential to prevent the progression of type 2 diabetes (63, 120, 150); γT3 treatment in particular, when tested in mice with diet-induced obesity, was shown to improve insulin sensitivity and preserve pancreatic β-cells in parallel to a decrease in circulating IL-18 levels (150). Therefore, NLRP3 inflammasome activation downstream from hyperglycemia or excess of FFA supply may represent an important pathway for type 2 diabetes treatment.
Nutrient excess triggers enhanced ROS production (350), which plays an important role in the control and activation of NLRP3 by enhancing interactions between NLRP3 and TXNIP (389), and which regulates NLRP3 at the transcriptional level (22). In fact, a drop in ROS levels has recently been reported to abolish hyperglycemia-induced inflammasome activation and the disruption of tight junctions in mouse vascular endothelial cells (53). In this way, ROS-dependent activation of NLRP3 inflammasomes by high glucose levels in the endothelium appears to be a critical phase in the induction of endothelial impairment.
Several pharmacological approaches that block inflammasome activation and at the same time reduce ROS production have been discovered, such as auranofin, a gold-derived compound used in rheumatoid arthritis patients (15), or minocycline (283) and mangiferin (235), both of which also exhibit this dual effect (Table 1). Thus, modulating the activity of inflammasomes concomitantly to lowering oxidative stress may be effective in metabolic and inflammatory conditions in which ROS damage to tissues is a major factor, such as type 2 diabetic vascular complications.
Numerous studies have demonstrated that ER stress activates the NLRP3 inflammasome and supports a relationship between inflammasome signaling and ER stress as a primary pathway of inflammation and β-cell death (170, 231). In this regard, Oslowski et al. identified TXNIP as a fundamental mediator of ER stress-induced inflammasome activation, since suppressing TXNIP expression in β-cells attenuated IL-1β upregulation induced by thapsigargin treatment (an ER stress activator) (231). It has recently been described that inositol-requiring enzyme 1 alpha, an ER stress mediator, also appears to mediate NLRP3 inflammasome activation, since inhibiting this protein decreased IL-1β secretion induced by saturated fatty acids or thapsigargin (Fig. 11) (89). Furthermore, ER stress increases cytosolic Ca2+ levels and activates NLRP3 inflammasome in a Ca2+-dependent manner (217).
Other potential mechanisms related to insulin resistance and inflammasomes are mitochondrial function and autophagy. Autophagy is impaired in type 2 diabetes, evident in the downregulation of autophagy markers such as microtubule-associated protein light chain 3 (LC3) and Beclin-1 in the livers of high-fat diet (HFD) animal models and genetic models (ob/ob) (368). It has been demonstrated that deletion of autophagic genes, such as Atg1611 or Atg7, enhances NLRP3, possibly as a result of mitochondrial impairment (222, 388). In general, these studies highlight the idea that autophagy can negatively regulate NLRP3 inflammasome activation by engulfing damaged mitochondria, therefore, reducing ROS.
AMPK may also be an upstream modulator of autophagy through phosphorylation of ATG1 (80). Furthermore, Han et al. have recently demonstrated that AXL receptor tyrosine kinase induces autophagy, leading to inhibition of NLRP3 inflammasome activation and resulting in the amelioration of acute liver injury in an animal model (112). Arglabin, a natural compound isolated from Artemisia glabella, has displayed inhibitory effects on NLRP3 inflammasome activation, reducing the production of IL-1β in a hyperlipidemic mice model. In addition, it increased autophagy by raising levels of Beclin-1 and LC3-II proteins (1). As a consequence, arglabin treatment leads to reductions in plasma glucose and insulin levels and in β-cell apoptosis in an ApoE2Ki mouse model of atherosclerosis under a Western-type HFD (1), again providing evidence of the relationship between autophagy and the NLRP3 inflammasome.
In summary, impairment of autophagy appears to be related to insulin resistance and may induce an inflammasome activation-enhancing inflammatory response in insulin-targeted tissues.
Another mechanism of regulation of IL-1β production is the enhanced ratio between glycolysis and oxidative phosphorylation. When this ratio increases, IL-1β transcription is enhanced through hypoxia-inducible factor-1α (HIF-1α) (321). Furthermore, after macrophage stimulation with LPS, there is an increase in glycolysis, decreased mitochondrial O2 consumption, and an accumulation of TCA metabolites such as succinate, which can induce HIF-1α and IL-1β transcription. As type 2 diabetes is characterized by high levels of blood glucose, it is possible that the macrophages of patients possess an activated inflammasome and enhanced IL-1β transcription. In fact, Xie et al. have shown that pyruvate kinase M2-dependent glycolysis triggers the activation of different inflammasomes, including AIM2 and NLRP3 (359).
It has been speculated that NLRP3 polymorphisms play a crucial role in the development of diabetic macrovascular complications, especially myocardial infarction (153). In the study in question, polymorphic NLRP3 rs35829419 allele was related to an enhanced risk of macrovascular comorbidities in type 2 diabetes. Therefore, NLRP3 polymorphisms may be a useful molecular marker to identify patients in which pharmacological treatment could be used to modulate the proinflammatory state (153). Moreover, the NLRP3 inflammasome and downstream signaling pathways are potential novel targets for the pharmacological prevention of comorbidities associated with type 2 diabetes (100).
C. FFA, NLRP3, and diabetes
Type 2 diabetes and obesity are characterized by elevated plasma levels of saturated fatty acids, of which palmitate is the most abundant (29). Several studies have demonstrated that palmitate and ceramide trigger NLRP3 inflammasome activation (262, 352). In a lipidomic study in humans, Kien et al. demonstrated that lowering dietary palmitate to an oleate ratio diminishes cytokines in leukocytes and reduces redox-sensitive gene expression in muscle (142). Palmitate can activate the AMPK-mitochondrial ROS cascade and, eventually, the NLRP3 inflammasome, thus promoting insulin resistance (Fig. 11) (352).
Interestingly, whereas saturated fatty acid HFD activates NLRP3 inflammasome in mice and contributes to insulin resistance, monounsaturated fatty acid HFD attenuates IL-1β-mediated insulin resistance by preserving AMPK activity (92). The role of dietary polyunsaturated fatty acids as inhibitors of inflammasome activity has been supported by several studies (157, 195), thus highlighting that dietary fatty acid composition is sensed by the NLRP3 inflammasomes that modulate their response depending on fat type.
Different cell types respond to FFA and assemble inflammasome components, thereby inducing IL-1β release (243, 345). In monocytes, for example, palmitate activates a pyroptotic program through caspase-4/5 and prompts the release of IL-1β and IL-18 (243). In endothelial cells, palmitate-induced NLRP3 inflammasome complex formation disrupts endothelial tight junctions, leading to the onset of endothelial injury during obesity (345). Intestinal epithelial cells respond to a high-cholesterol diet by activating caspase-1 after IL-1β-dependent accumulation of myeloid cells in the intestine (42).
Ceramide has been shown to promote NLRP3 inflammasome activation in cultured macrophages and in adipose tissue explants of diet-induced obese mice, which, upon exposure to this lipid, promote NLRP3-dependent caspase-1 activation (334).
Increased levels of circulating FFA, together with other factors related to insulin resistance, such as adipokines and cytokines, are key contributors to the development of liver diseases, with NAFLD being the most frequent among type 2 diabetic patients (42). Wree et al. reported enhanced expression of the NLRP3 inflammasome components in the livers of patients with NAFLD, which increased further as the disease progressed to nonalcoholic steatohepatitis (357). The same study illustrated the crucial role of NLRP3 in protecting against diet-induced steatohepatitis in mice with loss of NLRP3 inflammasome function (357). In contrast, Henao-Mejia et al. demonstrated more severe NAFLD in inflammasome-deficient mice and showed that NLRP3 signaling influenced NAFLD progression via modulation of the gut microbiota (114).
Thus, the specific action of inflammasome activity on NAFLD development and progression represents an interesting field to explore novel targets for therapy.
D. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic vascular complications
Recent evidence suggests that additional cellular processes other than those already described are involved in the cardiovascular problems associated with diabetes (Fig. 12). In this regard, inflammasome activation arises as an underlying pathological mechanism in diabetic heart disease. In fact, NLRP3 gene silencing in rats with diabetic cardiomyopathy ameliorates cardiac inflammation, pyroptosis, and fibrosis and restores cardiac function (186). Furthermore, inhibiting the IL-1β axis has been shown to inhibit cardiac arrhythmia induced by diabetes (209).
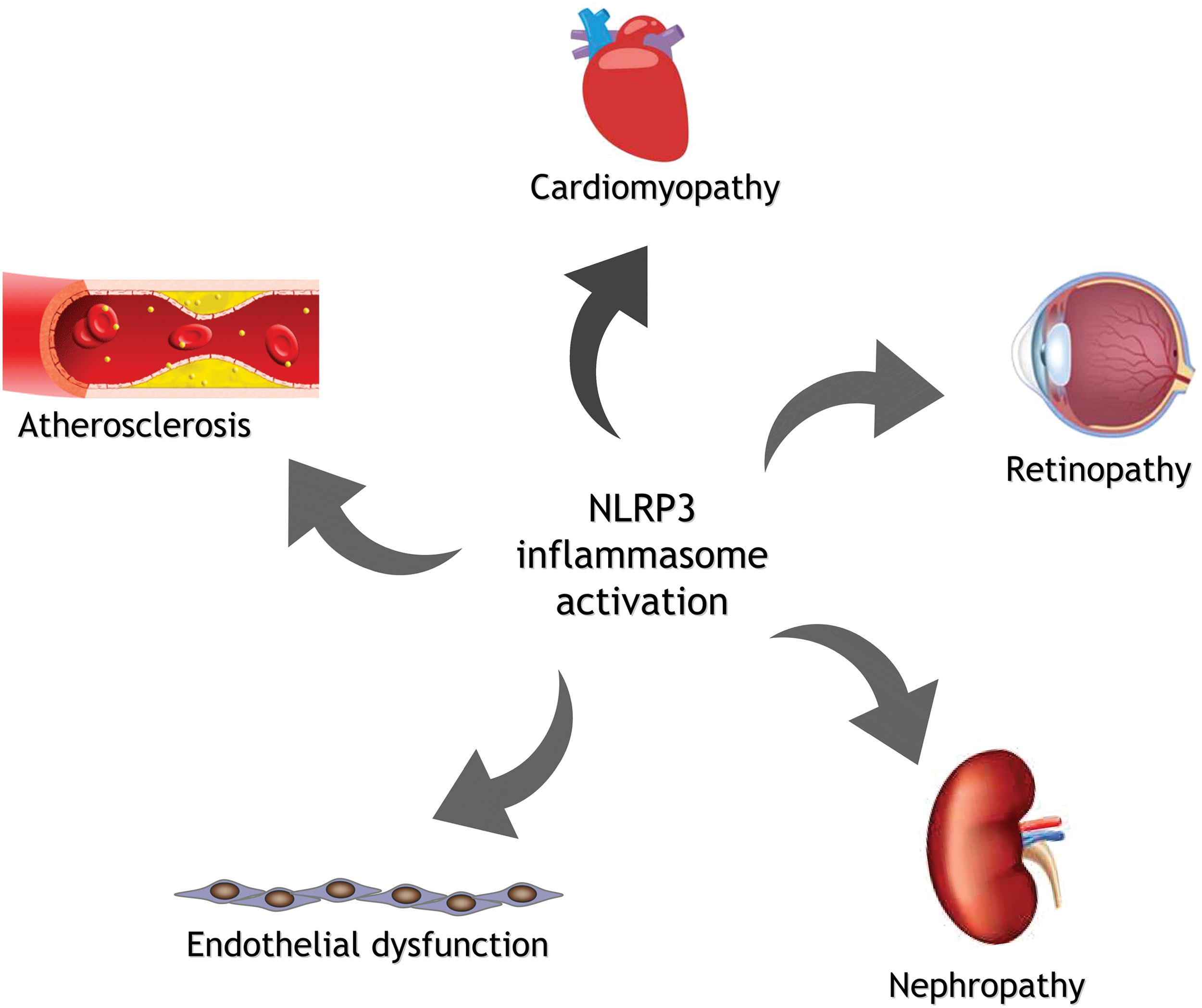
Endothelial dysfunction is a hallmark of diabetes, and prompts the onset of vascular problems. It is acknowledged that hyperglycemia induces the endothelial dysfunction that encompasses enhanced inflammation and ROS production, and some recent studies have focused on the role of inflammasomes in this context.
Li et al. demonstrated that the aortas of diabetic pigs display endothelial dysfunction associated with increased expression of Nlrp3, Asc, and IL-1β (176). Indeed, it has been demonstrated that Nlrp3 ablation in diabetic mice prevents endothelial hyperpermeability, one of the main consequences of endothelial dysfunction during type 2 diabetes (53). Endothelial barrier disruption by excess of circulating FFA in the obese seems to be mediated by inflammasome activation (345). Vascular calcification is also a general feature of patients with atherosclerosis and type 2 diabetes, and again has been associated with NLRP3 inflammasome activation (351).
Diabetes affects not only large blood vessels but also small vessels, causing microvascular complications such as retinopathy or nephropathy, in whose development and progression inflammasomes seem to participate. In fact, in vitro-cultured retinal pigment epithelial cells respond to hyperglycemia by enhancing components of the NLRP3 inflammasome, a response increased by inhibition of autophagy (287). In addition, it has recently been reported that vitreous samples of patients with diabetic retinopathy display enhanced caspase-1 and IL-18 levels, together with an increase in NLRP3 protein levels, which rise in parallel with the progression of retinopathy (183).
Diabetic nephropathy is a highly prevalent condition in long-term diabetic subjects. It is characterized by progressive abnormalities in the kidney, such as renal fibrosis and abnormal high albumin excretion in urine, together with a decrease in the estimated glomerular filtration rate (267). Immune cell infiltration occurs in renal tissues and drives the inflammatory response, which is crucial to the pathogenesis of diabetic nephropathy (223). In addition, increased ROS production from both sources, namely NADPH oxidase and mitochondrial ETC, damages renal cells and promotes glomerular sclerosis (311).
Interestingly, inflammasomes have been implicated in this complex pathological situation, since Nlrp3-deficient mice are protected against diabetic nephropathy (284). The study in question revealed that the rise in Nlrp3 and IL-1β levels was prevented when the mitochondria-targeted antioxidant MitoTempo was added to glucose-stressed podocytes, indicating that mitochondrial ROS are essential for glomerular inflammasome activation (284). Another report demonstrated that inhibition of TXNIP, the connecting signal between ROS and the NLRP3 inflammasome, decreases NLRP3 activation in parallel to a reduction in tubulointerstitial fibrosis in diabetic rats with nephropathy (318), thus highlighting the important roles of ROS production and NLRP3 inflammasome activation in the context of diabetic nephropathy.
IV. Sirtuins, Metabolism, and Inflammation
The association of histones with chromosomal DNA is fundamental in gene regulation that links epigenetics to human disease. Human histone deacetylases (HDACs) can be categorized into three classes: (I) (HDACs 1–3 and 8), (II) (HDACs 4–7 and 9–10), and (III), which contain seven evolutionarily conserved mammalian sirtuins (Sirtuins 1–7). Sirtuins, members of the Sir2 (silent information regulator 2) family, have a highly conserved NAD+-binding catalytic core domain protein with deacetylase activity, except for SIRT4, a mono-ADP-ribosyl transferase with no deacetylase activity on its histone substrates (205). The divergent N- and C-terminal of sirtuins differ according to their binding partners, substrates, and subcellular localization (110).
While SIRT1, SIRT6, and SIRT7 are classified as nuclear, SIRT2 is localized predominantly in the cytoplasm, and SIRT3, SIRT4, and SIRT5 reside in the mitochondria (335). Sirtuin functions also vary; SIRT6 possesses both histone deacetylase and ADP-ribosyltransferase activities (206), whereas SIRT5 is not only a deacetylase (221) but also a demalonylase and desuccincylase (77). Sirtuins deacetylase not only histones but also many other nonhistone target proteins. As depicted in Figure 13, their activity occurs through cleavage of the nicotinamide ribosyl bond of NAD+ and transfer of the acetyl group from proteins to their cosubstrate, which means these enzymes can be considered transacetylases rather than deacetylases. This reaction renders nicotinamide, deacetylated protein, and a mixture of 2′ and 3′-O-acetyl-ADP-ribose (OAADPR) (33).
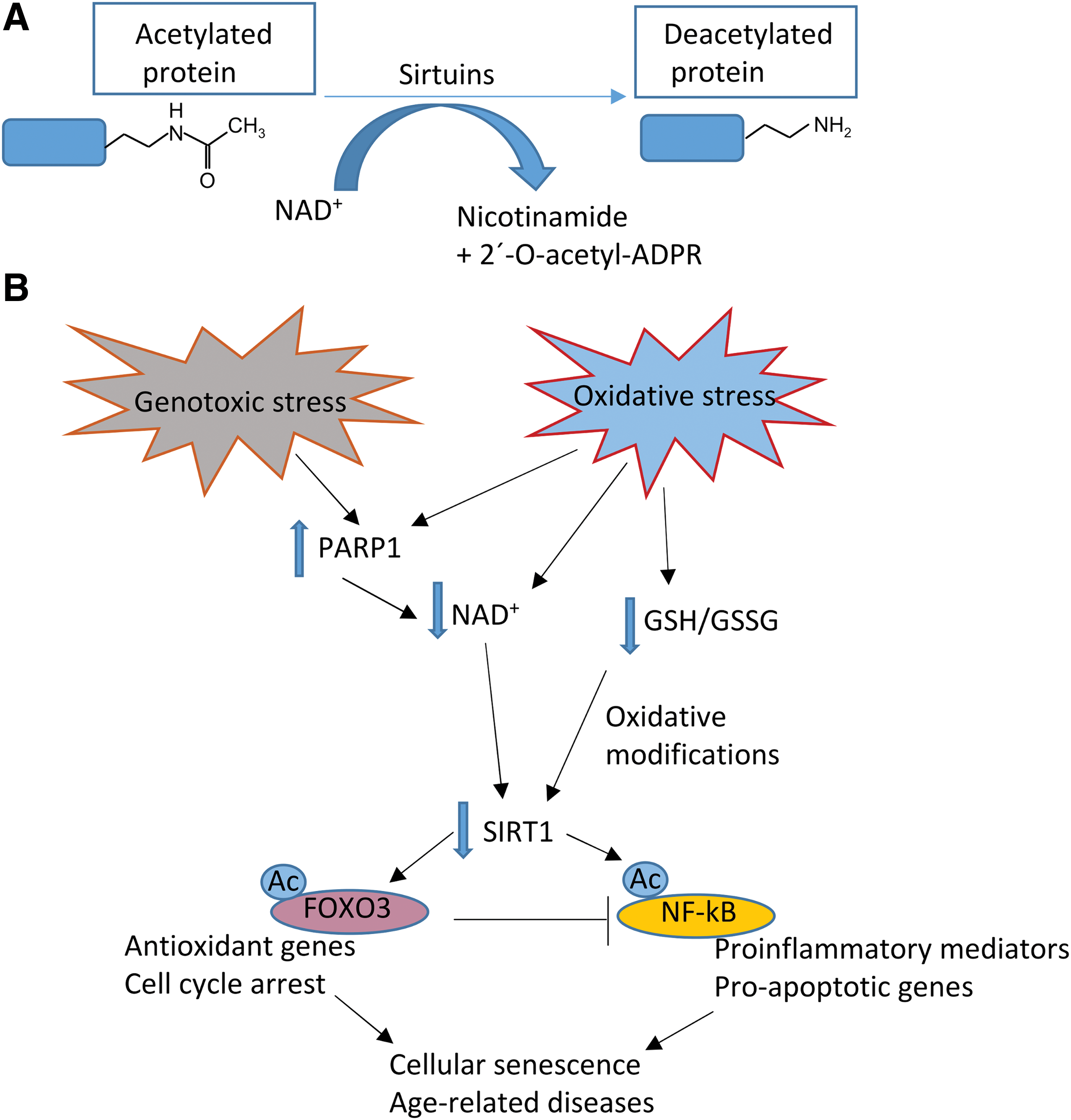
The activity of sirtuins is regulated via nicotinamide through a negative feedback loop, whereas OAADPR itself is thought to have a biological function as a signaling molecule.
The spectrum of the different sirtuins's biological activity is extremely diverse (220), and the field of research about these proteins is growing rapidly, particularly regarding their role in aging and age-associated pathologies (249). Acetylated proteins usually exhibit impaired functions, and deacetylation by sirtuins usually improves and reverses this impairment. Sirtuins can be activated by a large number of stimuli whose function is to promote cell survival, including starvation and caloric restriction, altered redox, energy, or NAD/NADH balance.
In this sense, sirtuins, heat shock proteins, and TXN are genes known as vitagenes (40, 41). In fact, hormesis, an adaptive response to continuous cellular stress, is a phenomenon by which exposure to a mild stressor confers resistance to subsequent, otherwise harmful, conditions of elevated stress. This biphasic dose–response relationship of low-dose stimulation and high-dose inhibition—as an adaptive response to detrimental lifestyle factors—determines the extent of protection against the progression to metabolic diseases such as type 2 diabetes.
Integrated responses detect and control diverse forms of stress. This is accomplished by a complex network of so-called longevity assurance processes in which several vitagenes are implicated. Furthermore, nutritional antioxidants have been demonstrated to be neuroprotective by activating hormetic pathways under the control of the vitagene protein network. Therefore, the continuous activation of sirtuins can confer protection against the progression of insulin resistance in general and type 2 diabetes in particular.
The most widely studied sirtuin is SIRT1, a mammalian homologue of the yeast Sir2. Given the well-established capacity of Sir2 to influence yeast-replicative life span and longevity in higher eukaryotes (44), research has focused on SIRT1's contribution to longevity in humans. However, yeast is aged by nucleolar fragmentation linked to the appearance of extrachromosomal rDNA circles, which are not present in mammalian aging. Although the role of SIRT1 in the life span of mammals is still a matter of debate, there is abundant evidence—obtained in animal models and human studies—that it is at the pinnacle of metabolic regulation and adaptation.
SIRT1 is abundant in the liver, brain, skeletal muscle, pancreas, and adipose tissues, where it modulates cytoplasmic targets by nucleocytoplasmic shuttling in conditions of oxidative stress. SIRT1 not only deacetylates histones but also a number of nonhistone targets, including both proteins within the nucleus (transcription factors and regulators) and cytoplasmic proteins, summarized in Table 2. This plethora of targets means that SIRT1 plays significant roles in a number of pathological conditions, including cell senescence, cardiovascular disorders, metabolic diseases, cancer, placental cell survival, and age-related diseases.
It should be noted that SIRT1 also controls the gluconeogenic/glycolytic pathway in the liver in response to fasting signals through the transcriptional coactivator PGC1α. Once SIRT1 is induced, it can interact with and deactelytase PGC1α in a NAD+-dependent manner. In this way, SIRT1 induces gluconeogenic genes and hepatic glucose output through PGC1α (264).
For example, it has been demonstrated that the NF-κB binding domain is the regulatory genetic motif associated with the aging process. In the context of HIV-1 infection, the Tat protein binds to the SIRT1 protein, a well-known longevity factor, and inhibits SIRT1-mediated deacetylation of the p65 subunit NF-κB complex. This enhances the transactivation efficiency of the NF-κB factor, leading to the activation of the immune system and, eventually, to the decline of adaptive immunity. These observations support the idea that the immune response and aging process can be enforced by the potentiation of NF-κB transactivation efficiency. SIRT1 and its activators appear to regulate the efficiency of NF-κB signaling, the major outcome of which is inflammation aging via proinflammatory responses (273).
SIRT1 activity can be enhanced by increasing NAD+ levels and by lowering its kinetic constant Km values for NAD+. SIRT1 can also be modulated at the transcriptional level in various metabolic tissues by activation through forkhead box O1 (FOXO1), cyclic AMP response-element-binding protein, and PPAR-β, or inhibition by carbohydrate response-element-binding protein (47).
Of the major cellular processes regulated by sirtuins, metabolism and inflammation are of most interest in the context of this review. Sirtuins, especially SIRT6, are known to be master regulators and corepressors of glycolysis (179). Acute proinflammatory and immune effector pathways require increased glucose uptake, pentose phosphate pathway activation, and glycolysis, which leads to lactic acid accumulation. Anabolic glucose-fueling aerobic glycolysis (the Warburg response, a major stress response to acute systemic inflammation) shifts to a fatty acid-fueling catabolic/adaptation response, generating acetyl CoA, which enters the TCA cycle inside the mitochondrion. Emerging data indicate that this “switch” from aerobic glycolysis to a fatty acid fueling/adaptation response is modulated by sirtuins.
Mounting evidence shows that NAD+ levels and SIRT transcription and/or protein levels are persistently reduced in specific tissues during chronic inflammation, such as in fatty tissue in obesity, the brain in Alzheimer's disease, and arterial walls in atherosclerosis. Sirtuins play a crucial role in inflammation, and many studies have revealed the mechanisms of activators and inhibitors of SIRT1 as pharmacological agents that prevent inflammation (86, 370).
SIRT1's effects on inflammation can be a double-edged sword, since low levels accentuate early acute inflammation-related autotoxicity by increasing NF-κB RelA/p65 activity, whereas prolonged increases in SIRT1 during late inflammation are associated with immunosuppression and increased mortality (138). It has been shown that SIRT1 interacts directly with the ReIA/p65component of the NF-κB complex (373). Deacetylation of Lys310 inhibits transcriptional activity of the ReIA/p65 subunit, therefore, suppressing the transcription of NF-κB-dependent proinflammatory genes. Furthermore, deacetylation of Lys310 in the ReIA/p65subunit makes Lys314 and Lys315 residues available for methylation, which further promotes ubiquitinylation and proteasomal degradation of NF-κB.
Another way to affect SIRT1's inflammatory signaling is through inhibition of the transcription factor AP-1 via deacetylation of its subunits c-Jun and c-Fos. This process plays a key role in regulating the functions of certain immune cells. For example, activation of SIRT1 in macrophages decreases transcription of cyclooxygenase 2, a typical proinflammatory AP-1-activated gene, whereas deacetylation of AP-1 by SIRT1 precedes proliferation in T-lymphocytes.
Sirtuins' involvement in inflammation is also related to their effect on the NLRP3 inflammasome. In fact, caloric restriction is associated with a decrease of inflammatory molecules (35), and, even more caloric restriction blunts the NLRP3 inflammasome in obese individuals with type 2 diabetes (334). In addition, caloric restriction can increase mitochondrial integrity by enhancing mitochondrial quality control and modulating mitochondrial ROS levels (158). These effects are mediated in part by activation of sirtuin enzymes, which increase mitochondrial function and integrity (107). Other studies have highlighted that fasting mimics caloric restriction in the sense that temporary nutrient deprivation enhances sirtuin-dependent increases of mitochondrial functioning (185).
Oxidative stress is involved in the relationship between NLRP3 inflammasome and sirtuins. For example, ROS can induce a reduction in intracellular NAD+ levels, which undermines the activity of SIRT2, causing acetylated α-tubulin to accumulate. Acetylated α-tubulin regulates dynein-dependent transport of mitochondria, enabling an efficient interaction between the adaptor ASC and NLRP3 (208). One study has demonstrated that prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3-mediated activation of SOD2 (329). Another study has demonstrated that SIRT1 inhibits the inflammatory response in vascular endothelial cells, partly through regulation of the NLRP3 inflammasome (175).
A. Role of sirtuins in type 2 diabetes physiopathology
There is growing evidence that shows sirtuins are linked to metabolic diseases and particularly diabetes via multiple pathways. The nuclear sirtuins SIRT1, SIRT6, and mitochondrial SIRT3 sense nutrient availability and changes in NAD+ production (or the NAD/NADH ratio) and respond by reprogramming immune, metabolic, and bioenergetic pathways, and this is probably the case with other less well-studied members of the sirtuin family.
The requirement of NAD+ as a cosubstrate for sirtuin deacetylase activity suggests that sirtuins have developed as NAD+ sensors and, therefore, energy and redox state sensors. This is supported by the fact that the estimated total intracellular NAD+ content, whose range is 0.2–0.5 mM, lies within the estimated Km values of SIRT1 for NAD+ (44), indicating that NAD+ actually limits the already mentioned rate in certain circumstances to boost SIRT1 to its maximal activity. The biological significance of NAD biosynthetic pathways for the regulation of the enzymatic activity of sirtuins, including its role in diabetes and related diseases, has been highlighted in numerous articles over the past decade.
In diabetes and its complications, the balance between NADH and NAD+ is severely altered. On one hand, NADH is overproduced due to an influx of hyperglycemia into NADH-generating pathways such as glycolysis, Krebs cycle, and the polyol pathway. On the other hand, NAD+ is depleted due to (i) overactivation of poly ADP ribose polymerase (PARP), which uses NAD+ as a substrate in the process of synthesizing ADP ribose by cleaving NAD+ into ADP ribose and nicotinamide and (ii) impairment of NAD+ regeneration enzymes such as lactate dehydrogenase in erythrocytes and complex I in mitochondria.
The consequence of NADH/NAD+ redox imbalance is, initially, reductive stress—leading to oxidative stress—and redox imbalance-triggered oxidative damage. Conversely, oxidative stress triggers NADH/NAD+ redox imbalance. NAD biosynthesis by nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in a major NAD biosynthetic pathway of nicotinamide in mammals, plays an important role in the regulation of SIRT1 activity in several cell types, including pancreatic β-cells (256).
SIRT1 is the sirtuin most widely associated with diabetes, and substantial data support that increased SIRT1 activity counters obesity, metabolic syndrome, and diabetes through actions involving enhanced insulin secretion in pancreatic β-cells, as occurs in β-cell-specific SIRT1-overexpressing transgenic mice (215). This significantly improves glucose tolerance through enhanced glucose-stimulated insulin secretion with respect to controls fed a high-fat diet (256). The enhancement of insulin secretion by SIRT1 seems to be mediated by a repression of UCP2 expression (32), as this mitochondrial inner membrane protein negatively regulates glucose-stimulated insulin secretion by uncoupling oxidative phosphorylation. However, SIRT1 also enhances insulin secretion upon artificial depolarization with KCl, indicating that SIRT1 alters insulin release by additional mechanisms downstream from depolarization and independent of UCP2 (215).
It is noteworthy that the beneficial effects of β-cell SIRT1 overexpression are restricted to young mice and are lost upon aging (256), a phenomenon possibly linked to decreased NAD+ levels in aging tissues. SIRT1 also plays an important part in protecting pancreatic β-cells from metabolic stress- and cytokine-induced β-cell death by deacetylating FOXO1 and the p65 subunit of NF-κB, respectively (152, 167), as represented in Figure 14.
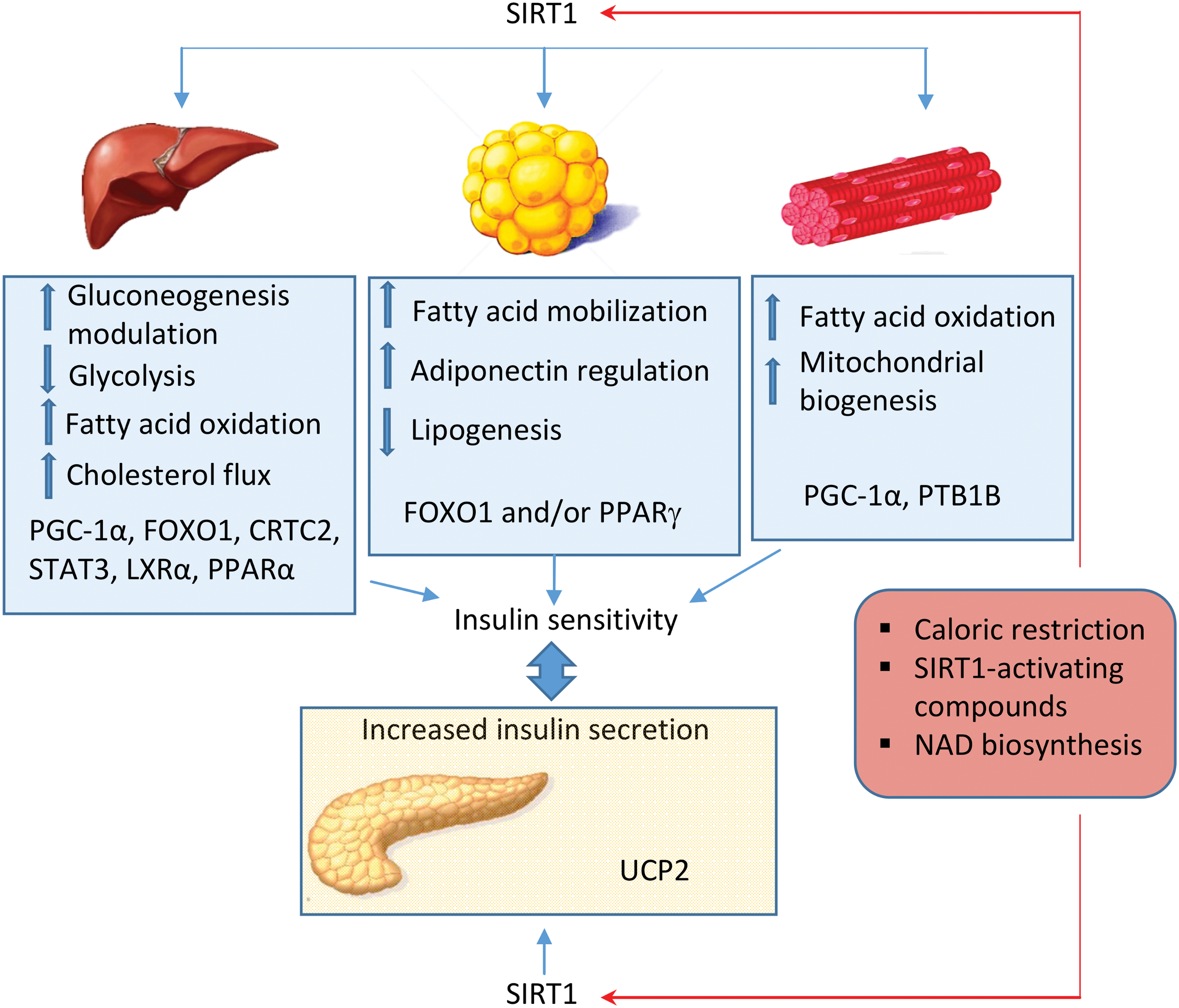
SIRT1 not only counters insulin resistance (127), increasing energy expenditure, and improving insulin sensitivity (285), but also stimulates gluconeogenesis in hepatocytes (278) and lipolysis/fatty acid oxidation in macrophages (228). Importantly, certain genetic variations of the SIRT1 gene have been associated with obesity and influence the survival of patients with type 2 diabetes in interaction with dietary niacin and smoking in Dutch populations (392).
SIRT1 is also capable of improving insulin resistance in adipose tissue, which might largely be due to its anti-inflammatory properties. Adipose tissue is an important regulator of the inflammatory status of the organism and the maintenance of glucose homeostasis, and these functions are affected by sirtuins. A chronic inflammatory response in adipose tissue has been shown to be the cause of obesity and insulin resistance, and is accompanied by reduced SIRT1 levels (102).
Sirtuins also regulate signaling in adipocyte/adipogenic differentiation. For example, SIRT1 suppresses a key adipogenic transcription factor, PPARγ, and promotes fat mobilization in white adipocytes (242). SIRT1 knockdown in 3T3 L1 adipocytes also inhibits insulin-stimulated glucose uptake and GLUT4 translocation mediated by a decrease in tyrosine phosphorylation of IRS1, and serine phosphorylation of Akt (with a subsequent increase in JNK phosphorylation as well as serine phosphorylation of IRS1) (376). In adipocytes, hypoacetylated FOXO1 mimics the effect of SIRT1 overexpression, suggesting a role for FOXO1 as a primary target of SIRT1 in balancing metabolic homeostasis (18).
Another key SIRT1 target tissue involved in insulin resistance is skeletal muscle, which harbors triglyceride accumulation and reduces lipid oxidation capacity in type 2 diabetes. IFN-γ, a proinflammatory cytokine that is an integral part of the metabolic inflammation circuit and contributes significantly to metabolic dysfunction, downregulates SIRT1 transcription and impairs energy expenditure in skeletal muscle cells by inducing the transcriptional modulators class II transactivator (CIITA) and hypermethylated in cancer 1 (HIC1) (174). In C2C12 myotubes, increased expression of SIRT1 has been found to downregulate both protein and mRNA levels of protein tyrosine phosphatase 1B, which acts as a negative regulator of insulin signaling, mainly via inhibition of tyrosine phosphorylation of IR or IRS1 (310).
The role of SIRT1 in glucose and lipid metabolism has been confirmed in gain-of-function mouse models. The first one possessed several features of calorie-restricted mice: animals were leaner, metabolically more active, and displayed increased glucose tolerance (31). In the other two models of SIRT1 overexpression, mild overexpression protected against the development of hyperglycemia, metabolic disease, and fatty liver (18, 241). It is noteworthy that although SIRT1 transgenic mice were protected against the onset of age-related diseases such as cancer and metabolic diseases, they did not live longer.
Although the capacity of whole-body activation of SIRT1 to exert beneficial effects on lipid and glucose metabolism is clear, controversial results have been reported regarding the tissue-specific actions of SIRT1, in particular those related to insulin sensitivity. HFD-fed transgenic mice with overexpression of SIRT1 specifically in skeletal muscle do not manifest improved insulin sensitivity (337, 355). This points to the fact that SIRT1 activation or overexpression in other metabolic tissues, such as the liver or white and brown adipose tissue, may be necessary to ameliorate whole-body insulin sensitivity.
The post-transcriptional regulation of SIRT1 is mediated by two classes of molecules: RNA-binding proteins and noncoding small RNAs. miRNAs bind to the 3′-untranslated region of target mRNAs and inhibit their expression by causing mRNA cleavage or impeding their translation. It is believed that approximately 30% of all human genes are regulated by miRNAs. The effect of SIRT1 on insulin resistance has also been described in terms of miRNA-mediated modulation.
Insulin resistance is associated with upregulation of miR-128-1 and miR-34a, the latter of which is among the best studied diabetes-related miRNA and strongly affects glucose and insulin homeostasis through SIRT1. MiR-34a, whose levels are consistently elevated in the livers of diet-induced and genetically obese mice (166), directly targets and undermines SIRT1 expression. In this model, the nuclear bile acid receptor farnesoid X receptor inhibits miR-34a in the liver, which results in a positive regulation of SIRT1 levels. SIRT1 activity is reduced further by miR-34a targeting of the rate-limiting enzyme in the salvage pathway of NAD+ biosynthesis, NAMPT (59). In the pancreas, miR-34a overexpression inhibits insulin secretion (184).
There is an interesting feedback loop between SIRT1, p53, and miRNA-34a. p53, a deacetylation target of SIRT1 itself, negatively regulates SIRT1 expression and also induces miRNA-34a, providing an additional mechanism to ensure SIRT1 repression upon p53 activation (365). Another miRNA that modulates SIRT1 levels in pancreatic β-cells is miR-9 (255).
Other miRNAs reported to affect SIRT1 expression in different tissues include miRNA-132, which downregulates SIRT1 expression in adipose tissue, prompting inflammatory responses (304), and miR-22, a senescence-related miRNA whose direct target is SIRT1. Some miRNAs are not linked directly to SIRT1, but are involved in the pathogenesis of diabetes.
For example, miR-451 is upregulated in type 2 diabetes, which counters AMPK-activating phosphorylation of LKB1 by reducing levels of the LKB1 scaffold protein calcium-binding protein 39 (159). LKB1 is a serine-threonine protein kinase that phosphorylates and consequently activates 13 downstream kinases, one of which is AMPK, a key enzyme that regulates cellular energy state, growth, inflammation, and mitochondrial function. Of special note, SIRT1-mediated deacetylation is necessary for the proper functioning of the AMPK-activating kinase, LKB1. This is because LKB1, when not associated with other proteins, is located predominantly in the nucleus, whereas SIRT1-deacetylated LKB1 moves to the cytoplasm, where it is activated after it complexes with STRAD (STE-related adapter) and MO25 (mouse protein 25). Depletion of NAD+ undermines SIRT1's enzymatic activity and this leads to diminished AMPK activity.
It is important to mention that SIRT3 can deacetylate a high number of targets in mitochondria, and for this reason has been broadly studied. Several studies have demonstrated that SIRT3 levels increase in liver, skeletal muscle, and adipose tissue during fasting and caloric restriction (118, 234, 279) and decrease under insulin-resistant conditions (372) or in response to a HFD (19, 141). Acetyl-CoA synthetase 2 has been identified as one of mitochondrial SIRT3's substrates, confering free acetate to the active metabolite acetyl-CoA for energy production in the Krebs cycle (291). Furthermore, SIRT3 activates and deacetylates 3-hydroxy-3-methylglutaryl-CoA synthase 2, a mitochondrial enzyme that, under fasting conditions, converts acetyl-CoA in the liver into β-hydroxybutyrate, acetoacetate, and acetone, a source of energy in different tissues including the brain (290).
Different studies have shown that one of the most important functions of SIRT3 is to maintain energy homeostasis by regulating the mitochondrial ETC (181). In fact, SIRT3-deficient mice display reduced basal ATP levels in the kidney, liver, and heart (4). Furthermore, SIRT3 interacts with the mitochondrial complex I component NDUFA9, and SIRT3 deficiency is associated with inhibition of complex I activity and increased complex I acetylation (19). In addition, SIRT3 can regulate complexes II (61), III, IV (141), and V (19), in this way regulating mitochondrial oxidative phosphorylation.
SIRT3 is also involved in the regulation of mitochondrial β-oxidation; for example, SIRT3-deficient animals show impaired β-oxidation and high levels of long-chain fatty acids upon fasting (279). Another study has demonstrated that SIRT3-deficient hepatocytes are more susceptible to fatty acid-induced cell death due to mitochondrial impairment and increased levels of ROS (19). Furthermore, SIRT3 enhances β-oxidation in muscle cells and decreases lipid accumulation in hepatic cells through its activation of AMPK (288). Recently, it has been demonstrated that SIRT3 is required for osteogenic differentiation through maintenance of PGC-1α-SOD2-mediated mitochondrial function (72).
There are emerging roles in diabetes for other sirtuins, particularly SIRT6. Dysregulation of SIRT6 activity has been associated with the onset of many pathologies related to its function in the context of inflammation, glucose, and lipid metabolism, including cardiovascular diseases, cancer, neurodegenerative diseases, and diabetes (214). There is a strong connection between SIRT6 and glucose metabolism, as SIRT6 promotes glucose-stimulated insulin secretion and ATP production in pancreatic β cells, and SIRT6-deficient pancreatic β cells manifest mitochondrial damage and a lower rate of oxygen consumption (360).
Moreover, activation of hepatic SIRT6 induces acetylation of PGC-1α, a key mediator of gluconeogenic gene transcription, via activation of the acetyltransferase GCN5, thus suppressing hepatic glucose production (74). Interestingly, obese/diabetic mice [Lepr(db/db)] exhibit reduced SIRT6 levels and ectopic re-expression of SIRT6-suppressed gluconeogenic gene expression (74).
SIRT6 also regulates lipid metabolism, and one mechanism by which this occurs is the deacetylation of histone H3 at lysines 9 and 56 in the promoter of genes that regulate triglyceride synthesis and fat metabolism, which represses their expression (323). For example, cholesterol metabolism involves the repression of lipogenic transcription factors SREBP1 and SREBP2 and their target genes, but also the inhibition of SREBP1/SREBP2 cleavage into their active forms and the activation of AMPK, which phosphorylates and inhibits SREBP1 (81). SIRT6-hepatic disruption in mice has been demonstrated to enhance TG synthesis and accumulation, leading to fatty liver disease (145), whereas transgenic mice overexpressing SIRT6 and fed a HFD show reduced expression of genes regulated by PPARγ (136).
B. Therapeutic potential of sirtuins
Overnutrition, such as that related to diabetes, usually produces excess NADH together with diminished NAD+ content, which often leads to attenuation of sirtuin protein content and the inactivation of SIRT enzymatic activity. Therefore, enhancing sirtuin expression in diabetic tissues that could restore or improve the NADH/NAD+ redox balance has been suggested as a therapeutic approach to treating diabetes and its complications (126, 151).
Conversely, there are concerns regarding a possible worsening of the redox imbalance due to elevated levels of sirtuins, which consume more NAD+. However, it is likely that higher levels of sirtuins modify the acetylated/deacetylated proteome, rendering more efficient metabolic pathways, which leads to more NADH utilization and, thus, more NAD+ regeneration (238). It has also been reported that sirtuin-mediated deacetylation enhances NADPH production, which may contribute to restoring the cellular redox balance (62).
One strategy to directly activate sirtuins is to increase the intracellular concentration of their cofactor NAD+. Indeed, in animal models, treatment with NAD+ precursors, such as nicotinamide mononucleotide or nicotinamide ribose, increases intracellular NAD+ levels and confers metabolic benefits, which can improve diabetes (46, 375). However, this is not a sirtuin-restricted action, as it has been reported that NAD+ also directly activates AMPK with a Km that is comparable with that of SIRT1 (252).
Numerous therapeutic and nutritional interventions have been highlighted as effective or potentially effective treatments for diabetes, and many of them involve modulation of sirtuins. One of these strategies is caloric restriction, defined as restricting the diet of an organism to fewer calories (20–50%) than ad libitum feeding without altering the levels of essential nutrients such as vitamins, minerals, and amino acids. Caloric restriction has been shown to promote higher intracellular concentrations of NAD+, which act as a natural sirtuin activator. In this sense, different mechanisms have been proposed to mediate dietary restriction longevity, including activation of NAD+-dependent deacylases such as sirtuins and increases in NAD+ derived from higher levels of respiration.
Moroz et al. have demonstrated that the nicotinamidase PNC-1, a key NAD+ salvage pathway component, is vital for dietary restriction to increase life span (213). Independently of PNC-1, dietary restriction increases the proportion of respiration that is coupled to ATP production but, surprisingly, reduces overall O2 consumption. The authors conclude that NAD+-dependent processes may be supported by a dietary restriction-induced shift toward oxidative metabolism rather than an increase in total respiration.
Caloric restriction-activated sirtuins regulate crucial metabolic signaling pathways through deacetylation of targets such as LKB1 (161), MnSOD (322), and eNOS (198). NO-regulated effects of caloric restriction are believed to be mediated via SIRT1, as caloric restriction-activated SIRT1 upregulates the activity of eNOS via deacetylation of the eNOS on lysines 496 and 506 (198).
Conceptually, there are two approaches to the pharmacological activation of sirtuins: direct and allosteric. An example of direct activation involves the nicotineamide analogue isonicotinamide (INAM), which binds directly to the catalytic domain of SIRT1, where it competes with nicotinamide, a potent endogenous inhibitor of SIRT1 (275). INAM is thought to prevent the reversal of the first step of deacetylation by inhibiting the binding of endogenous nicotinamide to the enzyme intermediate. In the allosteric model, a protein region flanking the catalytic domain of SIRT1 seems to bind to it and inhibit catalysis. This protein–protein interaction may be impeded by the binding of a small molecule called SIRT1-activating compound (STAC).
In light of the available evidence, it is clear that SIRT1 and SIRT6 are promising molecular targets for pharmaceutical and nutriceutical interventions against diabetes and its related comorbidities. Many natural antioxidant and anti-inflammatory molecules have been shown to activate SIRT1, including resveratrol, terpenylated coumarins, and several Chinese medicines. Among the hypoglycemic compounds used in the treatment of type 2 diabetes, thiazolidinediones enhance SIRT6 expression in the rat liver (369). Similarly, icariin, an important active component in Herba epimedii, has shown potential in treating cardiovascular diseases (CVD), and also acts as an activator of SIRT6 and an inhibitor of NF-κB (52).
The SIRT6/NF-κB axis is also affected by ginsenoside Rg1 (320) and by Egt, a dietary antioxidant that protects against many diseases, including CVD. Egt has been shown to exert endothelial cytoprotective effects against high-glucose cytotoxicity by reducing oxidative stress and cell senescence and, importantly, by upregulating SIRT1 and SIRT6 protein expression and downregulating their cellular targets p66shc and NF-κB (73).
In mammals, Egt is obtained only through the diet, but its widespread presence in food makes this bioactive agent a rich and free source of reducing equivalents, which ensures additional protection during oxidative stress. This occurs through a redox mechanism different from that of alkylthiols such as cysteine and glutathione and one that is specifically based on the irreversible transformation of intermediate oxidation products. Furthermore, the absence of toxicity in a millimolar range of intracellular concentration and its ability to interfere with multiple pathways that modulate SIRT1 and SIRT6 confer Egt a potentially starring role in the treatment of diabetes.
The therapeutic potential of endogenous molecules in type 2 diabetes and similar metabolic diseases has also been described to occur in an SIRT1-related manner. One of these molecules is vitamin D, whose deficiency has been related to insulin resistance and type 2 diabetes. In this vein, vitamin D has recently been shown to prevent oxidative stress and upregulate glucose uptake via the SIRT1/AMPK/IRS1/GLUT4 cascade in high-glucose-treated adipocytes and in adipose tissue of HFD diabetic mice (191).
One of the most widely studied plant derivatives with antidiabetic properties is resveratrol, a polyphenolic phytoalexin (3,5,4′-trihydroxy-trans-stilbene) present in white and red wines (216). Cis–trans isomerization of resveratrol occurs upon the action of ultraviolet light. Cis-resveratrol plays a role in protecting the organism from different pathological processes, including cardiovascular and oncological diseases. Although trans-resveratrol is known to be an antioxidant and anti-inflammatory agent, much less is known about cis-resveratrol's role in these activities.
A number of reports have been published regarding different activities of resveratrol and point to multiple uses of this versatile molecule: as a plant analogue of hormones, a regulator of epigenetic processes (326), an immunomodulator, a factor that decreases neurodegenerative processes in aging, and an apoptosis sensor, among others. Owing to its potent protective activity in metabolic diseases, resveratrol is widely considered to be a caloric restriction mimic. Abundant evidence obtained in vitro, in vivo, and in patients supports the beneficial effect of resveratrol on diabetes; it increases mitochondrial biogenesis, metabolic rate, insulin sensitivity, and glucose tolerance, and protects against premature death due to a HFD in mice (23, 98, 160, 285, 310). Moreover, these actions occur, at least partially, through SIRT1, and so resveratrol was the first STAC to be reported.
Although the capacity of resveratrol as a direct SIRT1 activator has been questioned (276), it is known to enhance SIRT1 activity (123). This appears to occur by allosteric activation of SIRT1 through a single amino acid at its terminal N-domain (125), and also indirectly, by inhibiting cAMP phosphodiesterases. This leads to an increase in cAMP levels, indirectly enhancing NAD+ (236). Resveratrol has also been shown to be protective in an SIRT1-independent manner in mouse models of diabetes (101).
Other polyphenolic plant-derived compounds, such as butein, quercetin, oligonol, piceatannol, bisdemethoxycurcumin, and myrcetin, have also been shown to activate SIRT1. As resveratrol was found not to be Sirt1-specific, a group of synthetic STACs structurally unrelated to resveratrol was subsequently developed.
Several such selective SIRT1 activators, all analogues of resveratrol, have been commercially developed by Sirtris Pharmaceuticals for the treatment of type 2 diabetes, and include SRT1720, SRT2183, SRT501, SRT2104, SRT2379, SRT1460, resVida, and Longevinex. The most potent of them, SRT1720, improves glucose homeostasis and insulin sensitivity in three different animal models of type 2 diabetes and obesity (339). However, there is much controversy regarding the role of SIRT1 in some of the protective effects of SRT1720, and there are several reports of the SIRT1-independent activity of this STAC, such as that observed recently in a diet-induced mouse model of obesity in which Sirt1-independent activation of AMPK was required for SRT1720 to improve glucose homeostasis (237).
V. Concluding Remarks
Owing to lifestyle changes and increased longevity, metabolic diseases such as type 2 diabetes, atherosclerosis, and obesity-related conditions are becoming an epidemic. Type 2 diabetes is one of the leading causes of retinopathy, blindness, nephropathy, and neuropathy, and nontraumatic lower limb amputation. The growing prevalence of type 2 diabetes calls for the need to develop novel therapeutic and preventative treatments that take into account genetic, immunological, and dietary aspects. Effective approaches will require changes in diet and exercise, as well as novel therapeutics that ameliorate the associated chronic inflammation and improve mitochondrial function. In this regard, pharmacologic and nonpharmacologic strategies have been developed to modulate mitochondrial metabolism, biogenesis, dynamics, or recycling.
Preventing oxidative damage using mitochondria-targeted antioxidants has also been extensively proved as an effective therapy for diabetes and its related comorbidities.
Furthermore, as type 2 diabetes is related to NLRP3 inflammasome activation, its products IL-1β and IL-18 are leading candidates for therapeutic targets, since they are generated in response to a diverse array of DAMPs associated with the metabolic imbalance characteristic of the disease. Several specific inhibitors of these ILs are being investigated as promising therapeutic agents for numerous diseases, including type 2 diabetes. Finally, the crucial role of sirtuins as energy sensors has conferred them a central position as potential targets for intervention in metabolic diseases.
A complex interplay exists between mitochondria, inflammation, and metabolism, which is highlighted by the key role of mitochondria in regulating immune cell metabolism, proinflammatory signaling, and inflammasome activation. Furthermore, the crosstalk between sirtuins and mitochondria represents a key calorie-sensing system whose proper regulation is key. Dysregulation of these metabolic signaling pathways occurs in type 2 diabetes. By modulating mitochondrial function itself, novel therapies for diabetes seek to exploit the potential of antioxidant/anti-inflammatory tools and to regulate inflammasome activation and/or sirtuin activity, two mechanisms closely related to mitochondrial function, all of which reinforce the mitocentric nature of metabolic diseases.
Footnotes
Acknowledgments
The authors thank Brian Normanly (University of Valencia-CIBERehd) for his editorial assistance. This study was supported by grants PI16/1083, PI16/0301, and CIBERehd CB06/04/0071 from Carlos III Health Institute and the European Regional Development Fund (ERDF “A way to build Europe”); UGP15-193 and UGP15-220 by FISABIO and PROMETEOII2014/035; and GV/2016/169 from the Department of Education of the Valencian Regional Government. V.M.V. and M.R. are recipients of contracts from the Ministry of Health of the Valencian Regional Government and Carlos III Health Institute (CES10/030 and CPII16/00037, respectively). S.R-L. is recipient of a Juan de la Cierva-Formación contract from the Spanish Ministry of Economy and Competitiveness (FJCI-2015-25040). C.B is recipient of a Sara Borrell contract from Instituto de Salud Carlos III (CD14/00043). We also thank the Instituto de Salud Carlos III (PI13/00021), Consejería de Economía, Innovación, Ciencia y Empleo (CTS-6264), Consejería de Salud.