
Abstract
Significance:
This article develops a holistic view on production of reactive oxygen species (ROS) by 2-oxo acid dehydrogenase complexes.
Recent Advances:
Catalytic and structural properties of the complexes and their components evolved to minimize damaging effects of side reactions, including ROS generation, simultaneously exploiting the reactions for homeostatic signaling.
Critical Issues:
Side reactions of the complexes, characterized in vitro, are analyzed in view of protein interactions and conditions in vivo. Quantitative data support prevalence of the forward 2-oxo acid oxidation over the backward NADH oxidation in feeding physiologically significant ROS production by the complexes. Special focus on interactions between the active sites within 2-oxo acid dehydrogenase complexes highlights the central relevance of the complex-bound thiyl radicals in regulation of and signaling by complex-generated ROS. The thiyl radicals arise when dihydrolipoyl residues of the complexes regenerate FADH2 from the flavin semiquinone coproduced with superoxide anion radical in 1e− oxidation of FADH2 by molecular oxygen.
Future Directions:
Interaction of 2-oxo acid dehydrogenase complexes with thioredoxins (TRXs), peroxiredoxins, and glutaredoxins mediates scavenging of the thiyl radicals and ROS generated by the complexes, underlying signaling of disproportional availability of 2-oxo acids, CoA, and NAD+ in key metabolic branch points through thiol/disulfide exchange and medically important hypoxia-inducible factor, mammalian target of rapamycin (mTOR), poly (ADP-ribose) polymerase, and sirtuins. High reactivity of the coproduced ROS and thiyl radicals to iron/sulfur clusters and nitric oxide, peroxynitrite reductase activity of peroxiredoxins and transnitrosylating function of thioredoxin, implicate the side reactions of 2-oxo acid dehydrogenase complexes in nitric oxide-dependent signaling and damage.
I. Overview
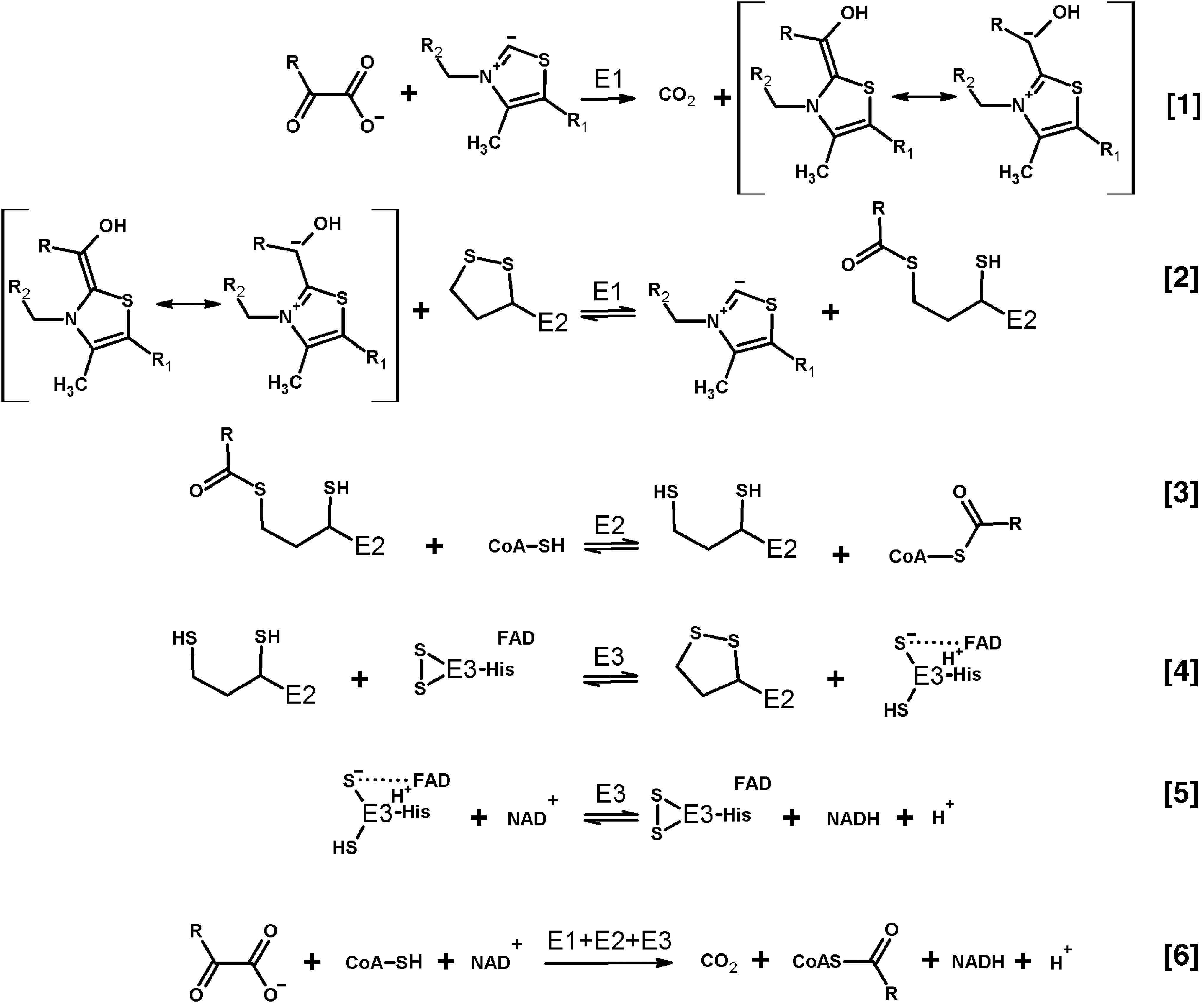
Decades after the initial discovery of generation of superoxide anion radical by dihydrolipoamide dehydrogenase in its nonphysiological reaction of NADH oxidation (130, 131), interest to the phenomenon has been renewed by the finding that the dihydrolipoamide dehydrogenase-involving multienzyme complexes of 2-oxo acid dehydrogenases are effective producers of superoxide anion radicals not only in the backward nonphysiological reaction of NADH oxidation but also in the physiological direction of 2-oxo acid oxidation. Figure 1 depicts the physiological reactions catalyzed by the three enzymatic components of these complexes (Eqs. [1–5]), along with the overall process of oxidative decarboxylation of 2-oxo acids (Eq. [6]). Using electron paramagnetic resonance (EPR) and spin-trapping, Bunik and Sievers have shown that under limitation in the terminal substrate NAD+, oxidation of pyruvate or 2-oxoglutarate by the isolated mammalian pyruvate or 2-oxoglutarate dehydrogenase complexes, respectively, leads to 1e− reduction of molecular oxygen, with the resulting superoxide anion radical released at about 1 nmol/min per milligram of protein (48). These findings on reactive oxygen species (ROS) generation by the complexes have been later confirmed by independent groups using purified and recombinant 2-oxo acid dehydrogenase complexes from bacterial and mammalian sources (5, 148, 149, 191) as well as the complexes within synaptosomes (191) and isolated coupled mitochondria (79, 160, 180). At the same time, model studies on ROS production by isolated dihydrolipoamide dehydrogenase and its mutants have added information on different factors that increase the NADH-dependent generation of ROS by this enzyme in vitro (5, 17, 78).
The accumulated data on ROS-producing side reactions of 2-oxo acid dehydrogenase complexes and their components conform to a general notion that enzymes are capable to catalyze many processes in addition to their physiological reactions. The question naturally arises to which extent and under what conditions would the side reactions affect in vivo homeostasis. The intracellular milieu is known to enhance physiological processes and limit side reactions. In contrast, perturbed homeostasis may promote the side reactions (47). Unavoidable side reactions are used in evolution to signal perturbed homeostasis, which is the first step in cellular organization of adaptive responses and homeostatic mechanisms. Examples of such mechanisms are well known in aerobic organisms, where enzymatic activities generating ROS and/or reactive nitrogen species (RNS) affect the function of proteins sensitive to these species. However, the sulfur-centered radicals are of significance in this regard too (48, 58, 76, 98), adding yet another aspect to novel insights on the role of sulfur in regulation of biological processes (189). Although the sulfur-based radicals have not attracted such attention as ROS or RNS in biological studies, the ability of thiyl radicals to promote not only oxygen reduction but also nitrosylation and transnitrosylation reactions (98) certainly warrants more attention to production of these radicals in biological systems and their potential involvement in homeostatic mechanisms. This is supported by recent findings on production of thiyl radicals by 2-oxo acid dehydrogenase complexes (48), nitrosylation of lipoyl-bearing components of the complexes (90), transnitrosylating function of thioredoxins (TRXs) (208), and specific interaction of mitochondrial thioredoxin with 2-oxo acid dehydrogenase complexes, shown for both animal (33) and plant (16, 211) proteins.
Special focus of this review on the mechanistic details of ROS generation by the complexes highlights the coproduction of ROS with the complex-bound thiyl radicals, related to thioredoxin use as a thiyl radical scavenger (115) and an acceptor of electrons from dihydrolipoyl residues, alternative to NAD+ (29, 34, 91). The alternative electron flow from the complex-bound dihydrolipoyl residues has also been shown for glutaredoxin (71). Within supramolecular structures that may be formed by 2-oxo acid dehydrogenase complexes with thioredoxins, thiol-based peroxidases (peroxiredoxins), and glutaredoxins, the side reactions catalyzed by the complexes are linked to a number of reactions of signaling and/or damaging significance. They include the superoxide-dependent mobilization of iron from proteins with iron/sulfur clusters, such as mitochondrial glutaredoxins and aconitase. Furthermore, the supramolecular structure supports the peroxynitrite reductase and nitrosylation reactions participating in the protection and damage, correspondingly, under nitrosative stress (26, 55, 90, 122, 208). Providing for the balance between the physiological and side reactions of 2-oxo acid dehydrogenase complexes, the complex-bound thiyl radicals and associated ROS may thus be concertedly involved in signaling by ROS and RNS. As a result, the coproduction of ROS and thiyl radicals, mostly overlooked in the studies on the ROS generation by the complexes, increases the biological significance of the ROS produced by the complexes. The review therefore highlights the generation and reactivities of the thiyl radicals of the complex-bound dihydrolipoyl residues.
Understanding both the damaging and signaling roles of enzymatic side reactions should take into account the enzyme function in vivo. In living organisms, not only concentrations/activities of reactants significantly differ from those in vitro but also the enzymes themselves are not isolated, but integral parts of a system evolved to stabilize homeostasis. This review scrutinizes in vitro data regarding their potential significance for in vivo conditions. To achieve the goal, available quantifications of the side reactions, which may be stimulated or inhibited in the supramolecular structure of 2-oxo acid dehydrogenase multienzyme complexes, are presented and analyzed.
II. Physiological Reactions Catalyzed by 2-Oxo Acid Dehydrogenase Complexes
A. Reaction steps and enzymatic components of 2-oxo acid dehydrogenase complexes
Independent of oligomeric organization, which may vary for the complexes of different substrate specificities and/or isolated from different sources, as reviewed by Bunik (28), the complexes catalyze the sequence of physiological reactions, presented in Figure 1 by Equations [1–5]. The sequence results in the substrate-specific oxidative decarboxylation of pyruvate, 2-oxoglutarate, 2-oxoadipate, or branched chain 2-oxo acids. The overall reaction yields CO2, acyl-CoAs, may be used for biosynthesis of high-energy phosphate bonds, and NADH (Fig. 1, Equation [6]). Well-characterized members of the family of substrate-specific 2-oxo acid dehydrogenases (E1) include pyruvate dehydrogenase (E1p), 2-oxoglutarate dehydrogenase (E1o), and dehydrogenase of branched chain 2-oxo acids (E1b). These enzymes catalyze the first and rate-limiting step of the overall process, the thiamine diphosphate (ThDP)-dependent decarboxylation of their cognate 2-oxo acid (Fig. 1, Equation [1]). The decarboxylation is followed by transfer of the reactive intermediate from ThDP to the second substrate of E1, the lipoyl-bearing domain of the second catalytic component, dihydrolipoamide acyltransferase (E2). The transfer (Fig. 1, Equation [2]) is coupled to oxidation of the “active aldehyde” intermediate to the corresponding acyl residue, concomitant with reduction of lipoyl disulfide to the acylated dithiol group. Transacylation between the acyldihydrolipoyl residue and CoA is catalyzed by E2 components of the complexes (Fig. 1, Equation [3]), including E2p (dihydrolipoamide acetyltransferase), E2o (dihydrolipoamide succinyl transferase), and E2b (dihydrolipoamide branched-chain acyltransferase), for the transfer of acetyl, succinyl/glutaryl, or branched chain acyl residues, respectively. Released after the transacylation, the dihydrolipoyl residues of E2 are oxidized by the E3 component, dihydrolipoamide dehydrogenase (the third component enzyme of the 2-oxo acid dehydrogenase complexes), which in mammals is common to the complexes of different substrate specificities.
The redox activity of E3 depends on the protein dithiol/disulfide group interacting with the tightly bound flavin adenine dinucleotide (FAD) (Fig. 1, Equations [4] and [5]). During the E3-catalyzed physiological reaction, the enzyme disulfide group accepts two electrons from the E2-bound dihydrolipoyl residue, sharing the reducing equivalents with the E3-bound FAD. Previously, spectral properties of the 2e− reduced E3 were interpreted as interaction between the FAD semiquinone radical and thiyl radical of the E3 cysteine residue (128, 129). However, EPR at low temperature did not reveal paramagnetic signal (170). Owing to this, the electronic structure of the 2e− reduced E3 under equilibrium conditions is best described as a charge transfer complex between thiolate anion and oxidized FAD (133, 187). However, a highly dynamic protein surrounding the flavin coenzyme supports multiple subtypes of the major electronic structure of the 2e− reduced E3, not excluding the FAD semiquinone radical among them (63, 64). Reduction of the two redox centers of E3, that is, catalytically active disulfide and FAD, may occur if reducing equivalents are in excess (Fig. 2). The 4e− reduced E3 is called over-reduced enzyme. It is formed less efficiently by the physiological reductant dihydrolipoamide (forward reaction according to Equation [4] of Fig. 1), compared to NADH (backward reaction according to Equation [5] of Fig. 1). The over-reduced E3 is inactive in its physiological reaction, but may catalyze a number of side reactions, including ROS production.
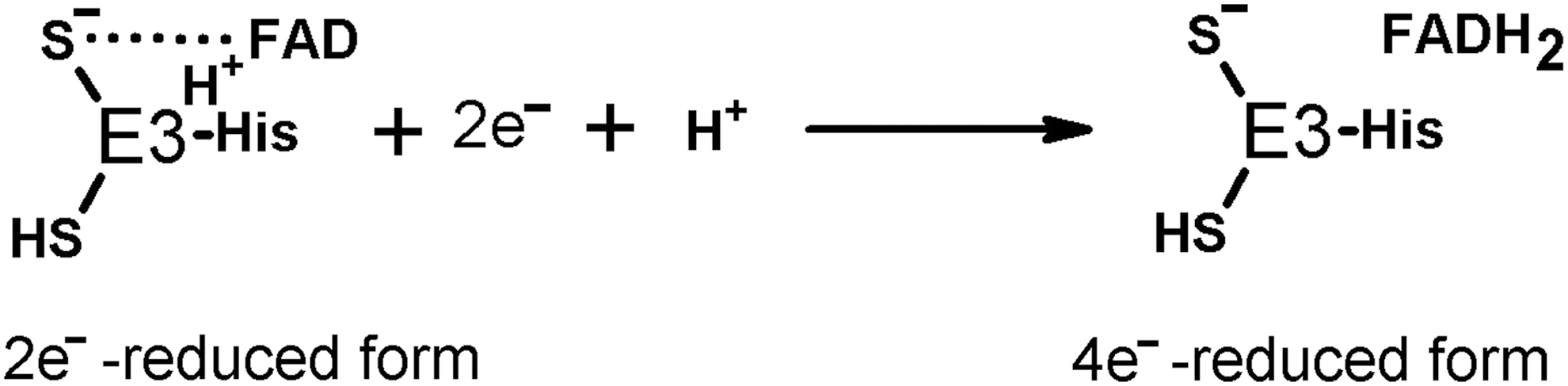
B. Catalysis by 2-oxo acid dehydrogenase complexes is tightly linked to major components of cellular redox state
Several features of the catalytic process performed by 2-oxo acid dehydrogenase complexes deserve attention regarding intracellular balance of ROS and related signaling in biosystems. First of all, each component of the complex undergoes redox reactions involving biological thiols/disulfides. They may be either protein bound, such as the lipoyl residues of E2 and the redox-active disulfide of E3, or of low molecular mass, such as CoA. In particular, CoA and dihydrolipoyl/lipoyl residues (Fig. 1, Equations [2–6]) form mixed disulfides with other cellular disulfides or thiols, respectively, of both low and high (proteins) molecular mass. Dependent on the thiol/disulfide redox potentials of the reaction participants, or their steady-state concentrations in a cellular microcompartment, this feature may underlie the inactivation of the complexes (27, 120, 152), ability of 2-oxo acid dehydrogenase complexes to reduce cellular disulfides (29, 34, 71, 105, 175), or protein “CoAlation” (197). In particular, one should take into account that availability of the thiol group of CoA for the complex catalyzed reactions (Fig. 1, Equation [3]) may be changed under oxidative stress due to formation of mixed disulfides between CoA and other participants of thiol/disulfide exchange reactions. The participants include a great number of proteins whose thiols exist as mixed disulfides with CoA, as has recently been revealed by high-throughput proteomic studies where such proteins have been named “CoAlated proteins” (197). It is obvious that massive protein CoAlation under oxidative stress conditions would simultaneously deplete CoA available for the physiological and side reactions of 2-oxo acid dehydrogenase complexes. Worth noting in this regard is an observation that CoA enters the E2-active site from the inside of the complex cores, with the intracore residence of CoA shown to stabilize the oligomeric structure of the pyruvate dehydrogenase complex from Azotobacter vinelandii (20, 23, 132). The finding implies a certain degree of CoA compartmentalization by the oligomeric structure of the complexes, potentially securing some of CoA pool for the overall reaction (Fig. 1) also under conditions of oxidative stress.
The tightly bound coenzyme of the dihydrolipoamide dehydrogenase component of the complexes, FAD, may undergo reduction in the course of the forward physiological reaction, catalyzed by the 2-oxo acid dehydrogenase complexes, or in the backward reaction of NADH oxidation. Both the free (15, 131, 207) and enzyme-bound states of reduced flavins (78, 130, 131) are known to produce ROS on oxidation with molecular oxygen. Importance of the NADH/NAD+ ratio for E3-catalyzed ROS production links the ROS generated by the 2-oxo acid dehydrogenase complexes at a high NADH to the NADH-dependent production of NADPH by nicotinamide nucleotide transhydrogenase, a protein of the inner mitochondrial membrane, which is crucial for redox homeostasis (137, 163, 171). Obviously, in vivo utilization of NADH in the transhydrogenase reaction would not only increase the disulfide reductant NADPH but also decrease NADH available for the backward side reaction of E3 yielding ROS. On the contrary, the physiological flux through pyruvate and 2-oxoglutarate dehydrogenases supports NADPH sources alternative to the transhydrogenase, such as the mitochondrial NADP+-dependent reactions catalyzed by malic enzyme, isocitrate dehydrogenase 2, and glutamate dehydrogenase (163).
Thus, not only cellular ROS levels but also the physiological function of 2-oxo acid dehydrogenase complexes is strongly coupled to the major components essential for mitochondrial redox status and overall function (102). They comprise cellular thiols/disulfides, including glutathione redox buffer, and the ratios of FADH2/FAD and NAD(P)H/NAD(P)+. In this regard, the relative catalytic rates of 2-oxo acid dehydrogenase complexes in the physiological (Fig. 1, Equation [6]) and ROS-generating reactions integrate availability of specific substrates of the complexes with overall cellular redox state. As a result, the complexes translate multiple biochemical parameters of the medium into the rates of irreversible reactions they catalyze.
C. Multienzyme structure of 2-oxo acid dehydrogenase complexes regulates catalytic properties of the component enzymes
Well-characterized 2-oxo acid dehydrogenase multienzyme complexes comprise multiple copies of the three catalytic components, organized around trimeric or oligomeric (24, 42, or 60 subunits) cores (28). Remarkably, under saturation with substrates and coenzymes in vitro, smaller 2-oxo acid dehydrogenase complexes with lower oligomerization degrees have been shown to catalyze the overall reaction with an efficiency, similar to that of the highly oligomerized structures (126). Thus, a high degree of core oligomerization may be of regulatory significance rather than a prerequisite for efficient catalysis. According to the results of long-standing studies, the whole network of lipoyl groups of the E2-formed core is coupled to each of the peripheral subunits E1 and E3, resulting in the multiple random coupling mechanism supported by the E2 oligomer (82, 86, 87). The mechanism implies that any of peripheral subunits of E1 may reductively acylate all catalytically competent lipoyl residues of the E2 core, which, in their turn, may be oxidized by any of the E3 subunits of the complexes. These experimental findings on the multiple random coupling mechanism have provided a sound basis for current models of function of complexes (145, 159).
E1 components do not readily react with free lipoate or lipoamide, and require a second substrate, the lipoyl residue (Fig. 1, Equation [2]), in its specific protein-bound form (28). Owing to this, E1 catalysis in vivo requires formation of the multienzyme complex. Integration into oligomeric structures of complexes is also associated with stabilization of activated conformations of E1 components through heterologous subunit interactions. As a result, a higher activity of the complex-bound E1 is detected not only in its physiological reaction with the lipoyl-bearing domain (Fig. 1, Equation [2]) but also in the partial E1 reaction (Fig. 1, Equation [1]), which is observed in model systems with artificial electron acceptors (47).
The essential role of the protein-bound lipoyl group as the second substrate of E1-catalyzed physiological reaction may contribute to inability of exogenous lipoate to compensate for the impairment in 2-oxo acid dehydrogenase reactions due to defective lipoylation of E2 or oxidative modification of the E2-bound lipoyl group. However, the primary concern regarding the pharmacologically introduced lipoate is its rapid degradation and the disulfide group reduction in vivo (168). Unfortunately, this is not taken into account in many studies on the multiple pharmacological effects of lipoate, such as Ref. (173) and cited therein. In particular, one should be cautious about inhibition by the lipoate degradation products of reactions catalyzed by the 2-oxo acid dehydrogenase complexes and their components (116). In contrast to E1, the E2 and E3 components are known to use free lipoate/dihydrolipoate, their amides, and some of the degradation products as the substrates (28, 34, 116). Nevertheless, also E2 and E3 components acquire important properties on their integration into oligomeric structures.
Function of E2 within the oligomeric structure of the complexes is intimately related to regulatory mechanisms involving post-translational modifications of complexes. The lipoyl-bearing domains participate in binding the LipB component of the lipoylation system, which is copurified with the pyruvate and 2-oxoglutarate dehydrogenase complexes, on one hand, and acyl-carrier protein, strongly interacting with LipB, on the other hand (88). The insertion of the two sulfur atoms in the octanoyl residue is catalyzed by the iron/sulfur cluster protein lipoyl synthase. Remarkably, the octanoyl residue of the acyl-carrier protein for the E2 modification is synthesized de novo inside mammalian mitochondria, thus involving the mitochondria-specific fatty acid synthesis pathway (204). Dependent on the level of reduction and/or acylation, the E2-bound lipoyl-bearing domains participate in post-translational modifications of the starting E1 components of complexes. These mechanisms include the well-characterized inactivation of eukaryotic dehydrogenases of pyruvate and branched chain 2-oxo acids by phosphorylation catalyzed by the pyruvate dehydrogenase kinase bound to the lipoyl-bearing domains and sensing their state (85, 109, 195 –197). The 2-oxoglutarate dehydrogenases and bacterial pyruvate dehydrogenases, which are not regulated by phosphorylation/dephosphorylation, are inactivated under accumulation of dihydrolipoyl residues in the course of catalysis (27, 48). While the former mechanism involves the action of lipoyl domain-bound kinase, the latter mechanism, whose details are considered in the next sections, involves thiyl radicals of the E2-bound dihydrolipoyl residues. The thiyl radicals acquire additional stabilization within a highly interactive network of the lipoyl residues of the E2 core. The dihydrolipoyl-intermediate-dependent regulation of the complexes is in good agreement with experimental findings that genetic reduction of the number of tandem lipoyl domains in E2 decreases the viability of bacteria with modified E2 protein, despite no detectable effect on the catalytic rate of such complexes assayed in vitro (53, 62, 82 –84). The decrease may be related to alternative electron flows from the dihydrolipoyl intermediate to thioredoxin, glutaredoxin, and thiol peroxidases, essential for viability under specific conditions (26, 71, 122), which are considered in detail in the next sections. Thus, the oligomeric core and/or tandem lipoyl domains of 2-oxo acid dehydrogenase complexes have essential functions in regulation of complexes in vivo, which may be not readily obvious on in vitro assays of enzymatic activities under saturation with substrates and coenzymes.
An important feature of E3, acquired by enzyme incorporation into the multienzyme structure of the complex, is a reduced sensitivity of the complex-bound versus free E3 to over-reduction (Fig. 2) causing the product inhibition of the enzyme by NADH (64). That is, formation of the oligomeric structure of multienzyme complexes stabilizes the catalytically active 2e− reduced form of E3 (Fig. 1, Equation [4]) and inhibits the enzyme over-reduction leading to the 4e− reduced form (Fig. 2), which is inactive in the physiological reaction. Biological relevance of this species-specific feature of E3 is demonstrated by the correlated sensitivities to high NADH/NAD+ ratios of the whole bacteria in vivo and their dihydrolipoamide dehydrogenases in vitro (177). It is important to note that even for an enzyme that is relatively stable to the NADH inhibition, such as E3 from A. vinelandii, the true catalytic rate can be detected only in the stopped-flow experiments, because the steady-state rates are affected by the NADH formed already within the dead-time of the registration (63). Accordingly, the common E3 assay system, using the backward direction of NADH oxidation under steady-state conditions, is not appropriate to reveal the catalytic potential of E3. Side reactions of E3, such as NADH oxidase activity, are affected much less, if at all, by the enzyme over-reduction. Indeed, many of the side reactions occur when E3 is over-reduced. This potential may obfuscate conclusions regarding the ratio of the physiological and side reactions, inherent in different dihydrolipoamide dehydrogenases, especially when the activities are determined at their different pH optima and the product inhibition parameters are unknown. For example, the disproportional changes in various activities of human E3 and its mutants were interpreted as increased ROS generating ability of the mutants (6). However, data on sensitivity of E3 mutants to over-reduction and pH dependence of assayed activities of mutants have not been presented. Hence, the conclusion of the study is likely inaccurate due to underestimation of the physiological activities of E3 mutants. This criticism is supported by the lack of correlation between the activities of E3 mutants and the protein FAD content (6), which obviously contradicts the well-known requirement of FAD for both the physiological (Fig. 1) and ROS-producing (48) functions of the enzyme.
Dimensions of the complexes self-assembled from their purified or recombinant components in vitro (up to 50 nm in diameter) (92, 125, 140) have raised questions on their compatibility with the intercristae space of mitochondria where the eukaryotic complexes are located, supposedly forming supramolecular structures, so-called metabolome, with other enzymes of specific pathways (183, 184). Usual allocation of the complexes to the mitochondrial matrix, presumed to be a solution of matrix enzymes, is misleading, as such a view does not take into account that the mitochondrial matrix space is mostly filled in with cristae, representing invaginations of inner mitochondrial membrane. As a result, the size of mitochondrial metabolomes built around the 2-oxo acid dehydrogenase complexes whose dimensions already are ≈5 × 10−8 m should be compatible with the intercristae space. This space is not defined by total diameter of whole mitochondria (≈10−6 m), being rather comparable with the dimensions of the complexes. This view is further supported by experimental studies that reveal a strong interaction of the complexes with mitochondrial membrane [(31) and references therein], despite the widespread opinion that the complexes occupy mitochondrial matrix. Thus, in vivo crowding of biological molecules and membranes creates conditions different from those of oligomerization experiments in vitro. Spatial constrains may be present in vivo, which are absent in vitro, where the oligomerization is usually driven by symmetry considerations and maximization of the complex activity at substrate saturations. Owing to this, the ratio of enzymatic subunits obtained on oligomerization in vitro (108) does not necessarily mean the same fixed stoichiometry of natural oligomerization in vivo. Indeed, for the same type of 2-oxo acid dehydrogenase complex, physiological heterogeneity of both the oligomeric core structure and saturation with peripheral components is known from functional and electron microscopy studies (28). A significant amount of data on native complexes have been accumulated, pointing to the regulatory potential of the oligomerization degree and component ratio in vivo. Even if the maximal catalytic efficiency of different oligomers does not significantly differ, as shown in some studies discussed above, the variability in oligomeric state could address the tissue-specific morphology and regulation of mitochondria, associated with differences in metabolic fluxes and steady-state concentrations of substrates of complexes (50, 61, 101, 166). Recent data indicate that variations in the complex core oligomerization may depend on post-translational modifications of the E2 component (126). Existence of different splice isoforms of mammalian E2 (67) may also underlie the structural variability of complexes in vivo, in contrast to those reconstituted in vitro from a single splice form of E2. Besides, availability of peripheral components in the medium (141) and/or structural differences in the isoenzymes of these components (31, 40, 65) are known to affect saturation of the core with the 2-oxo acid dehydrogenase and dihydrolipoamide dehydrogenase components. Furthermore, the saturation may depend on specific proteins binding the peripheral components. Earlier, only the eukaryotic pyruvate dehydrogenase complex has been known to possess the E2 isoenzyme, specific for binding the E3 component (201). More recently, also the 2-oxoglutarate dehydrogenase complexes of fungi and animals have been found to include a subunit mediating the specific type of E3 binding to the E1–E2 core (89). Nevertheless, the mammalian 2-oxoglutarate dehydrogenase complex isolated from heart demonstrates direct interaction between E1o and E3 (136), and self-assembly of human 2-oxoglutarate dehydrogenase complex from its three enzymatic components occurs without specific E3-binding subunits (147, 148). Probably, the fourth E3-binding subunit of the 2-oxoglutarate dehydrogenase complex may be tissue and/or E1o-isoenzyme specific. Indeed, structural differences between the 2-oxoglutarate dehydrogenase isoenzymes are suggested to define variations in the affinity of the 2-oxoglutarate dehydrogenase (OGDH and 2-oxoglutarate dehydrogenase like [OGDHL] genes) and 2-oxoadipate dehydrogenase (dehydrogenase E1 and transketolase domain-containing protein 1 [DHTKD1] gene) complexes for their flavin-dependent component, dihydrolipoamide dehydrogenase (31, 40). Mass spectrometry-based estimation of relative abundance of the components in preparation of the 2-oxoglutarate dehydrogenase complex isolated from the heart (OGDH-encoded isoenzyme) and brain (OGDH- and OGDHL-encoded isoenzymes) indicated a lower content of E3 component in the brain complex (31).
As a result, structural differences in the E2 core oligomerization and its saturation with peripheral components E1 and E3 may be observed for the same functional type of complexes in vivo. These differences may underlie mechanisms of natural regulation of the physiological and side reactions of the 2-oxo acid dehydrogenase complexes. However, unless specific conditions and/or constraints existing in vivo are mimicked, the different oligomeric structures and/or their significance for regulation may remain undetectable in vitro.
III. Oxygen-Dependent Side Reactions of Isolated Enzymatic Components of 2-Oxo Acid Dehydrogenase Complexes
A. Reactions of the 2-oxo acid dehydrogenase components with oxygen
Holoenzyme complexes of 2-oxo acid dehydrogenases (E1) with their coenzyme ThDP may undergo slow oxygen-dependent oxidation of the coenzyme. ThDP bound at the active sites of human branched chain 2-oxo acid dehydrogenase and pyruvate dehydrogenase reacts with oxygen, which results in catalytically inactive thiamin thiazolone diphosphate, as revealed by crystallographic studies (119). Although the reaction is suggested to occur in the course of the catalytic process, that is, be “paracatalytic” (182), the presence of reaction substrates is not obligatory to induce the modification (119). Nevertheless, stimulation of oxygen reactivity in the course of catalysis cannot be excluded by published data. The oxygen reactivity of ThDP in E1 holoenzymes depends on the active site structure. When a tyrosine residue interacting with the thiazolium ring of ThDP (Tyr113 in human E1b-alfa and Tyr89 in human E1p-alfa) is mutated to phenylalanine, the oxygen resistance of the wild-type enzymes decreases, leading to oxygen-dependent mutant inactivation (119). The natural occurrence of phenylalanine instead of tyrosine in an analogous position in some 2-oxo acid dehydrogenases, such as E1p from Mycobacterium genitalium, is therefore indicative of potential differences in species-specific reactivity of the enzyme-bound ThDP to oxygen. Existence of enzymatic systems repairing the oxidized forms of ThDP coenzyme [reviewed in Bunik and Aleshin (37)] supports the biological significance of the process.
Similar to E1 holoenzymes, reactive catalytic intermediates at the dehydrogenase-active sites, which are formed after the substrate decarboxylation step (the active aldehydes of corresponding 2-oxo acids in Equation [1] of Fig. 1), may react with oxygen. First of all, such oxygen reactivity would depend on specific stabilization of multiple protonation states and radical forms of the active aldehydes bound to ThDP at the dehydrogenase-active sites. For instance, a carbanionic structure of the postdecarboxylation intermediate in human E1b is resistant to reaction with molecular oxygen (119), whereas enamine structures of the active aldehyde intermediates in other 2-oxo acid dehydrogenases may promote their reaction with oxygen (95). The oxygen resistance of the E1b-stabilized carbanion correlates with the finding that among the three members of 2-oxo acid dehydrogenase complexes, the complex of branched chain 2-oxo acid dehydrogenase shows the lowest level of ROS production (24, 160). Besides, distal carboxyl groups of 2-oxoglutarate and 2-oxoadipate contribute to stabilization of the enamine radical intermediate (147), explaining the consistent finding of EPR-detectable enamine radical species in bacterial and human 2-oxoglutarate and 2-oxoadipate dehydrogenases, but not in pyruvate dehydrogenases (73, 147, 148). Thiazolium ring with its π-electron system may stabilize the radical intermediates known to arise also in the course of physiological reactions, such as those catalyzed by the bacterial ThDP-dependent oxidases of 2-oxo acids with [4Fe-4S] clusters (162).
The EPR-detectable radical of ThDP enamine intermediate, which is observed on aerobic incubation of 2-oxoglutarate with the isolated recombinant E1o holoenzymes, is supposed to arise on 1e− oxidation of the active aldehyde intermediate (Fig. 3, Pathway 1) by molecular oxygen, leading to generation of superoxide anion radicals (147). However, there are certain disagreements between the EPR-detected enamine radical and ROS-producing activity of 2-oxoglutarate dehydrogenases. First, although the enamine radical was detected by EPR in both Escherichia coli and human 2-oxoglutarate dehydrogenases (73, 147), detection of the superoxide anion radical by the cyt c reduction assay was possible only with the human enzyme (5, 147). Second, when 2-oxoglutarate dehydrogenase was exposed to its alternative substrate, 2-oxoadipate (43, 194), a threefold lower concentration of the EPR-detectable enamine radical was observed, compared to the reaction with 2-oxoglutarate, but H2O2 was produced at a sevenfold higher rate, compared with the enzyme incubated with 2-oxoglutarate (148). Third, in the reaction with 2-oxoglutarate, the site occupancy of the EPR-detectable radical showed a threefold increase on integration of E1o into the multienzyme structure (147), but the ROS-producing activity did not increase accordingly (148). Fourth, the rates of superoxide and H2O2 production are incompatible with peroxide generation by the dismutation of superoxide anion radial (Fig. 4, Equation [8]). As quantified by Nemeria et al. (147), the superoxide anion radical is produced at a rate (2 nmol/min per milligram E1o, 0.26% of the physiological reaction rate) that is twofold lower than that of hydrogen peroxide production detected using the AmplexRed assay (4 nmol/min per milligram E1o, 0.52% of the physiological reaction rate). However, taking into account the stoichiometry of the reaction of superoxide anion radical dismutation to H2O2 (Fig. 4, Equation [8]), the peroxide should be formed at a twofold lower rate compared to that of superoxide production, because production of 1 mole of H2O2 requires 2 moles of superoxide anion radical. Finally, no pathway for regeneration of active enzyme from the EPR-detectable radical intermediate on E1, which is required for the catalytic production of superoxide anion radical, has been suggested (5, 147, 148). Overall, the data point to the mechanisms of H2O2 generation by isolated 2-oxoglutarate dehydrogenases that are alternate or additional to that through the superoxide dismutation.
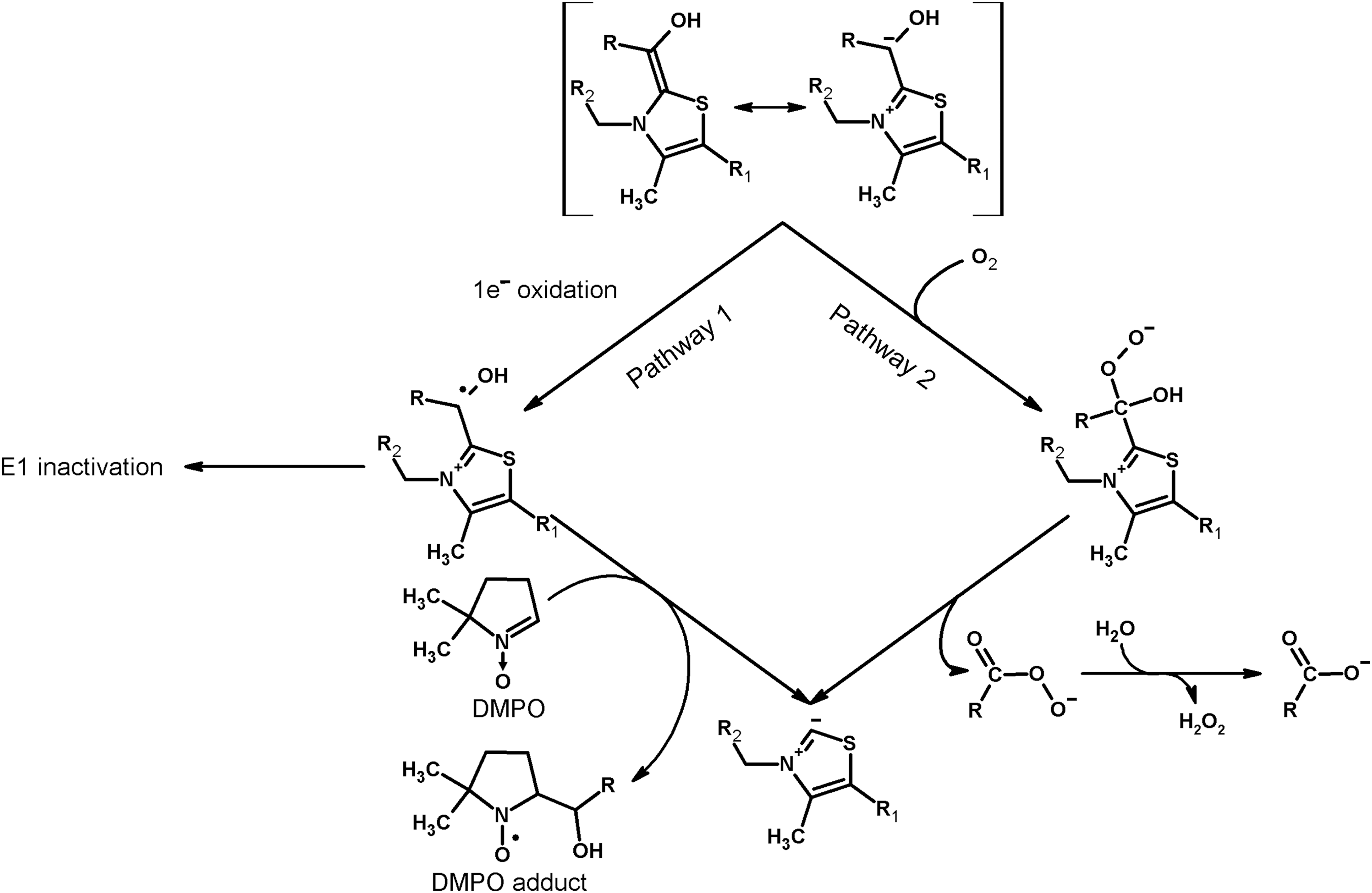
It is known that oxygen addition to the enamine intermediate may produce peracid (alternative name: peroxy acid) according to the reaction shown as Pathway 2 of Figure 3 (1, 167, 193). On aerobic incubation of muscle 2-oxoglutarate dehydrogenase with 2-oxoglutarate, the peracid has been detected by its reaction with thionitrobenzoate anion (47). Release of peracid as a reaction product may lead to AmplexRed-dependent peroxide detection, because peracids are in equilibrium with the corresponding acids and H2O2 (Fig. 3). On the contrary, one cannot exclude the direct detection of peracids using the AmplexRed system (47). As a result, oxygen addition to the ThDP enamine intermediate of isolated 2-oxoglutarate dehydrogenase with formation of peracid decomposing into succinate and H2O2 (Fig. 3, Pathway 2) may be another mechanism of ROS generation by E1o, in good agreement with independent quantifications of the reaction rates discussed above. Compared to H2O2 production through Pathway 2 (Fig. 3), the EPR-detectable radical intermediate of 2-oxoglutarate dehydrogenase arising on 1e− oxidation in Pathway 1 (Fig. 3) may represent a minor pathway, contributing to the known paracatalytic inactivation of 2-oxoglutarate dehydrogenase (43, 44, 46).
B. Dihydrolipoyl groups of the dihydrolipoamide acyltransferase components are resistant to oxidation by molecular oxygen under physiological conditions
In contrast to E1 or E3, E2 is not an oxidoreductase and therefore cannot catalyze any redox processes involving its dihydrolipoyl residues. Although dihydrolipoate and its precursor lipoate are known to have pro-oxidant effects in vivo, the mechanisms of the pro-oxidant action include nonenzymatic catalysis by dihydrolipoate of redox cycling of quinones at physiological pH (7), or E3-dependent reactions (41) considered in Section IV. The thiyl or disulfide anion radicals of dihydrolipoate, arising in these reactions, possess much higher reactivity to oxygen, compared to the corresponding thiols.
Nevertheless, thiolate anions are principally known to be directly oxidized by oxygen, forming superoxide anion radical and the oxygen reactive thiyl radicals (13). Owing to this, the dihydrolipoyl residues arising in the course of the complex catalyzed reactions (Fig. 1) are sometimes suggested to be an independent source of superoxide anion radicals produced by the complexes (4). However, such suggestions contradict available results, which clearly show that this mechanism does not take place. Although the isolated lipoylated E2 component has not been studied in this regard, the E1–E2 subcomplexes of the pyruvate and 2-oxoglutarate dehydrogenase complexes do not show increased ROS generation, compared with isolated E1, when assayed under conditions of ROS generation in the forward direction leading to the dihydrolipoyl intermediate (5). Generation of superoxide anion radical by direct reaction of E2-bound dihydrolipoyl intermediate with oxygen has also been disproved by Bunik and Sievers in experiments on chemical modification of 2-oxo acid dehydrogenase complexes with diphenyliodonium chloride (48). The compound specifically disables redox cycling of E3-bound FAD. The dihydrolipoyl residues formed on reduction of this modified complex by 2-oxo acid and CoA do not support the ROS production. Thus, at physiological and acidic pHs, used in the studies of ROS generated by the enzyme complexes, the complex-bound dihydrolipoyl residues are not highly reactive to oxygen. The finding is in agreement with chemical properties of thiols in general and dihydrolipoate in particular, as the oxidation would require deprotonation of the thiols forming the oxygen reactive thiolate anion. Below pH 8, the noncatalyzed oxidation of thiol groups by molecular oxygen cannot be comparable to the catalytic ROS production by the complexes. As a result, under physiologically relevant pH values, dihydrolipoyl residues of E2 do not contribute significantly to ROS-generating activity of the complexes, if E3 component is disabled or absent.
C. Oxygen-dependent reactions of the dihydrolipoamide dehydrogenase component
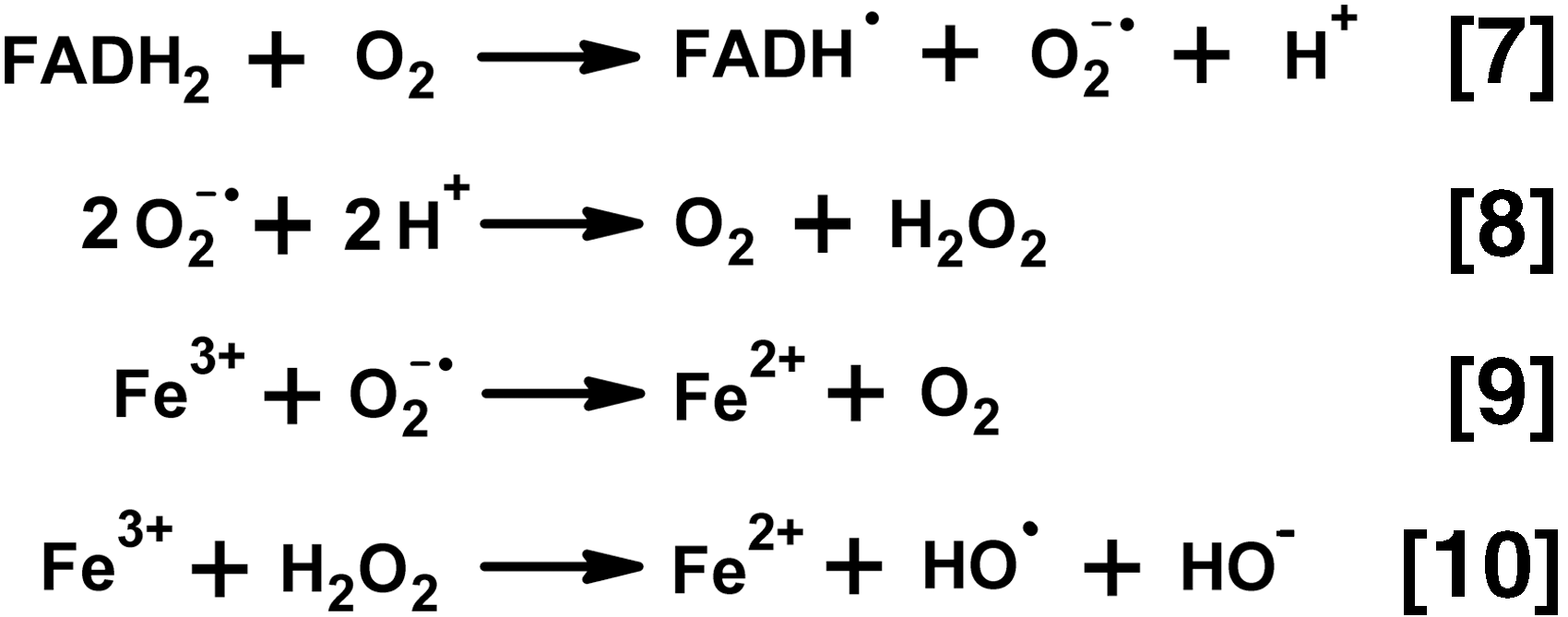
It has long been known that dihydrolipoamide dehydrogenase (the E3 component of the complexes), like other flavin-containing dehydrogenases or free flavins, catalyzes 1e− reduction of oxygen with formation of superoxide anion radical (15, 130, 131, 135). The superoxide anion radical production in the NADH oxidase reaction catalyzed by E3 purified from different sources is quantified using the cyt c assay. The reaction rates are estimated to range from 50 to 100 nmol/min per milligram of isolated E3 (0.1–0.2 mM NADH, pH 6.3–8.5) (13, 17, 99, 131). In most of the studies, the major product of the dihydrolipoamide dehydrogenase-catalyzed reaction with oxygen is H2O2, generated at a 5- to 10-fold higher rate, compared to superoxide anion radical (17, 78, 121). Reaction conditions and/or enzyme isolation/storage may affect product distribution. For example, in a study of dihydrolipoamide dehydrogenase purified from pig heart mitochondria, only a twofold higher rate of H2O2 versus superoxide anion radical generation has been observed (99). However, addition of high concentrations of ammonium stimulated the E3-catalyzed H2O2 production much more (10-fold) than the O2 •− production (2.5-fold), resulting in a 10-fold higher rate of production of H2O2 versus O2 •−. It is known that mitochondrial NADH oxidase (complex I) also produces both O2 •− and H2O2 in the reaction of its flavin site with molecular oxygen, exhibiting higher ratios of H2O2 to O2 •− with increasing NADH concentration (202). Probably, the product of E3 reaction with molecular oxygen varies for the 2e− or 4e− reduced E3 (Fig. 2), whose ratio depends on NADH excess. Limitation of the E3 redox cycle to flavin is observed on interaction of Zn2+ with the catalytic dithiol group of E3. The Zn2+-bound E3 is spectrally similar to oxidized enzyme, does not react with the (dihydro)lipoamide substrate, and may be reduced at the flavin site only. In such an enzyme, where FAD cannot share the reducing equivalents obtained from NADH, with the catalytic disulfide group, a fivefold stimulation of the NADH-dependent ROS production is observed, with H2O2 being the major reaction product (78). It may thus be suggested that H2O2 is produced when the catalytic disulfide/dithiol group of E3 does not participate in catalysis. This is observed either in the E3-Zn2+ complex or in the 4e− reduced E3 (Fig. 2, over-reduced enzyme in the right part of the equation). However, when FAD shares the reducing equivalents with the active site disulfide/dithiol group (Fig. 2, 2e − reduced enzyme in the left part of the equation), the reaction with oxygen results in the superoxide anion radical. Different protonation of the E3-active site in its varied states (2e− or 4e− reduction, native or complex with Zn2+) may affect the distribution of the product of the E3-catalyzed reaction with O2 between H2O2 or O2 •−.
According to Equation [7] in Figure 4, release of the superoxide anion radical in the enzymatic reaction implies a stabilization of the FAD semiquinone radical in the enzyme-active site. Earlier studies indicated that such stabilization is higher in FAD-dependent dehydrogenases than in FAD-dependent oxidases. The oxidases are therefore less efficient generators of superoxide anion radicals, compared to the dehydrogenases (111, 131). The different mechanisms of the FAD-dependent reactions with molecular oxygen roughly correlate with the enzyme reactivities to sulfite (130). Reduced flavin dehydrogenases readily react with sulfite, whereas reduced flavin oxidases are mostly unreactive to sulfite.
IV. Generation of ROS by Multienzyme Complexes of 2-Oxo Acid Dehydrogenases
Studies of the ROS generating activities of 2-oxo acid dehydrogenase complexes should take into account the increasing complexity of the system versus its isolated enzyme components, which may add to the primary and secondary reactions occurring in the media promoting ROS generation by the complexes. Compared to the isolated enzymes, physiological substrates are used and appropriate catalytic intermediates are stabilized in the complex-catalyzed reactions. The catalytic properties of the component enzymes are affected by formation of multienzyme complexes accordingly (Section II). Changes in the ROS-generating activity of the components of 2-oxo acid dehydrogenase complexes, which are imposed by the multienzyme structure, and biologically relevant factors affecting the process are reviewed in this section.
A. Primary and secondary reactions during ROS production by 2-oxo acid dehydrogenase complexes
Although collectively considered ROS for simplicity, discrimination between the original ROS species produced by an enzyme, and multiple secondary processes, is essential for interpreting the translational value of in vitro data. Apart from strong dependence of secondary processes on conditions, the in vitro findings may be irrelevant due to the on-site presence in vivo of the systems reacting with the primary radical species, including species-specific scavengers. Indeed, the superoxide anion radical and hydrogen peroxide produced in vivo are scavenged by different enzymes, such as superoxide dismutases (SODs) and catalases/peroxidases/peroxiredoxins, respectively. Scavengers of peroxides differ in catalytic mechanisms and substrate specificity. In mammals, the first line of defense against hydrogen peroxide is formed by the glutathione-dependent peroxidases and highly abundant thioredoxin-dependent peroxiredoxins. Different damaging and signaling potentials of superoxide anion radical and hydrogen peroxide also depend on their specific reactivities and membrane permeability. In addition to the different chemical properties (72), specific action of different radical species in biological systems strongly depends on the site of their generation (12, 142). In this regard, production of superoxide anion radical by 2-oxoglutarate dehydrogenase complex in proximity to mitochondrial aconitase containing iron/sulfur clusters may underlie mitochondrial signaling mechanisms based on the superoxide-dependent mobilization of protein-bound iron ions. These mechanisms are discussed in detail in Section VI. It is worth noting that release of the protein-bound metal ions may also affect the results of ROS generation in vitro. In particular, iron ions may be released from aconitase when superoxide anion radical production by 2-oxoglutarate dehydrogenase complex is studied using mitochondria or other aconitase-containing preparations.
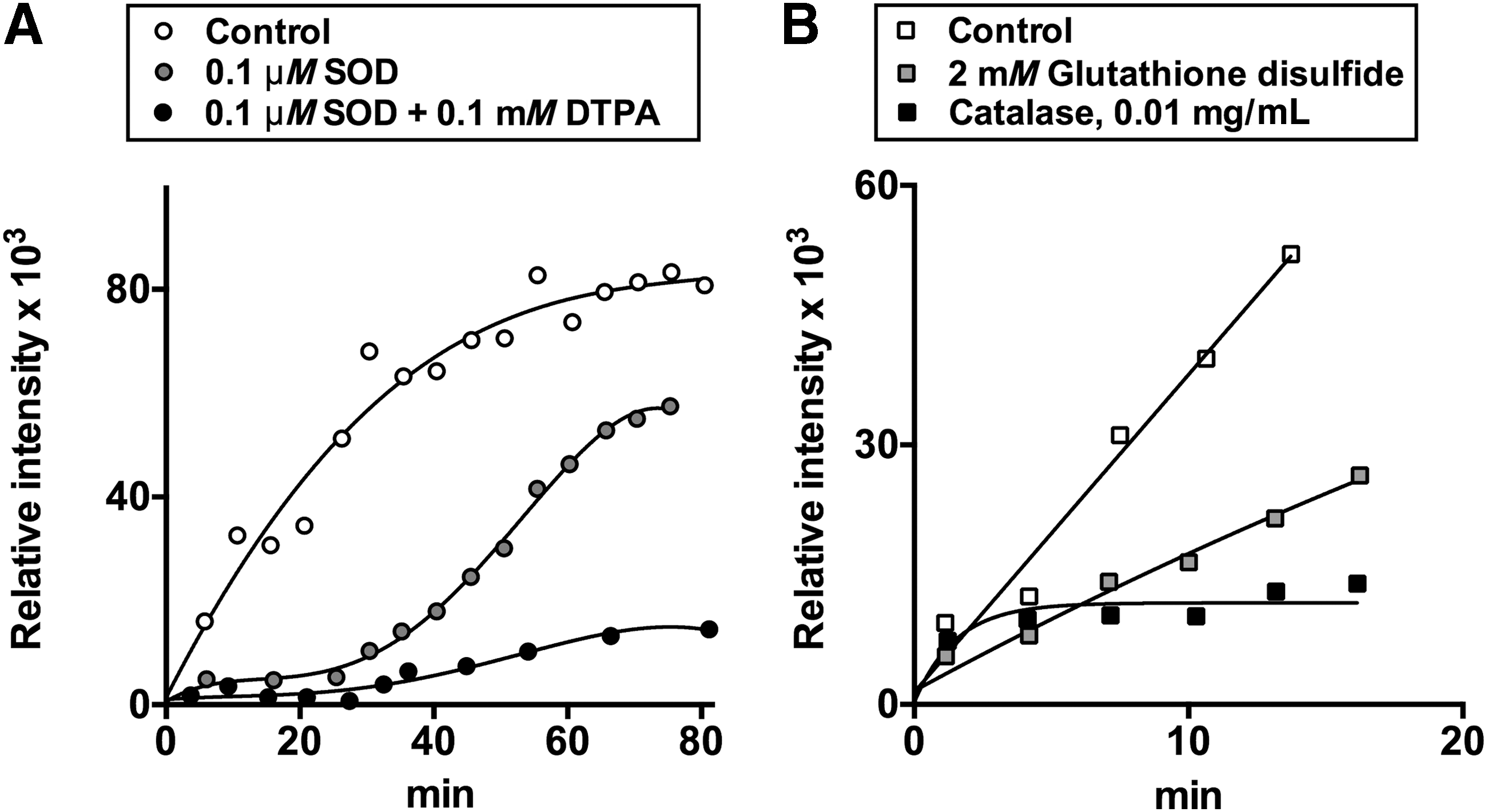
Generation of superoxide anion radical as a side product of the physiological reactions catalyzed by the pyruvate and 2-oxoglutarate dehydrogenase complexes isolated from pig heart is known from spin-trapping experiments (35, 48). Spin traps undergo addition to radical species, increasing their life time and helping discrimination of different species through characteristic hyperfine splitting constants of the adducts. Although the spin-trapped EPR adducts are detectable during catalysis in the complete reaction medium (Fig. 1, Equation [6]), the EPR signal increases when NAD+ is omitted. The condition induces accumulation of the dihydrolipoyl intermediate according to Equations [1]–[3] in Figure 1. Multiple steps in the formation of the spin-trapped adducts by the pyruvate and 2-oxoglutarate dehydrogenase complexes are seen from time courses of the reactions, as well as by effects of different radical scavengers on accumulation of the adducts (35, 41, 48). At high enzyme content in the reaction medium, accumulation of the EPR-detectable adducts is clearly biphasic, with a plateau value of EPR signal intensity separating the first and second phases (41). With lower enzyme content, the plateau may be not pronounced (control curves in Fig. 5A, B). Nevertheless, the biphasic process is effectively revealed by the action of different radical scavengers, such as SOD, catalase, and/or chelating agents. Addition of SOD eliminates the first phase of formation of the EPR-detectable adducts, leading to a lag-period in their appearance (Fig. 5A). The second phase is selectively blocked by addition of catalase (Fig. 5B). Simultaneous addition of both SOD and catalase prevents formation of EPR-detectable adducts (48). Thus, the SOD-sensitive paramagnetic adducts formed in the first phase (Fig. 5A) manifest production of superoxide anion radical by 2-oxo acid dehydrogenase complexes, whereas the catalase-eliminated second phase (Fig. 5B) is linked to accumulation of H2O2.
Production of both the superoxide anion radical and hydrogen peroxide by 2-oxo acid dehydrogenase complexes has been confirmed in a recent study using SOD and catalase in the AmplexRed system for peroxide detection (121). As discussed in previous sections, both products may be generated by the 2-oxo acid dehydrogenase and dihydrolipoamide dehydrogenase components of the complexes. In addition, H2O2 may arise from spontaneous dismutation of the superoxide anion radical according to Equation [8] (Fig. 4), which is catalyzed by SOD.
Generation of superoxide anion radical by 2-oxo acid dehydrogenase complexes has also been quantified in the cyt c assay (5, 48) and qualitatively confirmed by specific reaction of superoxide anion radical with lucigenin (48). Although lucigenin is a specific probe for superoxide anion radical (112), using the lucigenin reaction to quantify the superoxide produced by the complexes is not possible due to the potential ability of E3 to catalyze 1e− reduction of lucigenin. The reaction would be similar to the known 1e− reduction of a number of quinones (151, 200) and nitrofurans (178), catalyzed by E3. It is worth noting, however, that lucigenin fluorescence attains a plateau value, similar to the intermediary plateau observed in the formation of EPR-detectable adducts (48). The data provide further evidence that the superoxide anion radical production is limited to the first phase of the reaction, followed by formation of the secondary paramagnetic adducts.
The lag period in the formation of paramagnetic adducts, which is induced by SOD, further increases on simultaneous addition of SOD and the metal ion chelator diethylenetriaminepentaacetic acid (DTPA), with the latter chelating agent also decreasing the rate of the second phase (Fig. 5A). The additional depression of the SOD-affected kinetics by DTPA (Fig. 5A), as well as the catalase effect (Fig. 5B) point to Fenton/Haber–Weiss reactions (Fig. 4, Equations [9] and [10]) contributing to formation of the stable spin-trapped adducts in the second phase of the reaction. Besides, stability and hyperfine splitting constants of the nonprotein paramagnetic adducts formed after prolonged reaction times in the forward direction of the reactions catalyzed by the enzyme complexes are close to those of thiyl radical adducts. The finding infers potential involvement of radical-induced reactions of the substrate CoA. In the less complex medium of the backward reaction with NADH, the stability and hyperfine splitting constants of the EPR-detectable adducts pointed to the time-induced transformation of the primary adducts with superoxide anion radical to the alkyl radical adducts (48).
Secondary reactions of the radical products and/or their spin-trapped adducts, revealed by the EPR studies, add complexity to interpretation of the inhibitory action of glutathione disulfide on the EPR signal in the spin-trapping experiments (Fig. 5B). The inhibition of ROS generation by 2-oxo acid dehydrogenase complexes in the presence of glutathione disulfide has recently been observed also in an independent study using the AmplexRed assay (120). The glutathione disulfide effect may involve chelation of metal ions by glutathione disulfide, affecting the secondary Fenton/Haber–Weiss reaction according to Equations [9] and [10] in Figure 4. Besides, disulfides are known to inhibit the activity of 2-oxo acid dehydrogenase complexes due to modification of the dihydrolipoyl residues, decreasing not only the physiological (Fig. 1, Equation [6] but also ROS-generating activity of the complexes (9, 27, 48). In fact, S-glutathionylation of the 2-oxoglutarate dehydrogenase complex has long been known to mediate mitochondrial impairment on oxidative stress induced by H2O2. The peroxide oxidizes mitochondrial glutathione to the disulfide which, in turn, modifies reactive thiols of proteins, including those of the 2-oxoglutarate dehydrogenase complex (9, 152). On the other hand, both disulfides and thiols may alleviate catalysis-induced inactivation of 2-oxo acid dehydrogenase complexes in the reaction medium with limiting NAD+ concentrations (27, 30, 33), discussed in detail in the next sections. In addition, the complex-bound E3 is known to catalyze thiol/disulfide exchange reactions between the dihydrolipoyl residues of the complexes and disulfides of the medium (34, 69, 105, 175). This activity enables the complexes to transform the free disulfides into the corresponding thiols. Thus, apart from metal-chelating properties of glutathione disulfide, multiple condition-dependent thiol/disulfide exchange reactions involving dihydrolipoyl residues of 2-oxo acid dehydrogenase complexes may contribute to the effect of glutathione disulfide on ROS, seen in the EPR studies (Fig. 5B) and AmplexRed assay (120). Multiple actions may also be observed with reduced glutathione, although in most cases its effects oppose those of the disulfide. In particular, glutathione disulfide may form mixed disulfides not only with the protein but also with the substrate CoA. These thiol/disulfide exchange reactions would inhibit the complex-catalyzed reactions, including ROS production in the forward direction. In contrast, glutathione may reduce partly oxidized CoA or lipoyl groups, activating the catalysis. It is not excluded that catalytic activation of CoA in the active site of E2 and/or thiyl radicals of dihydrolipoyl residues of the complex may be involved in the formation of mixed disulfides with the protein and CoA thiols. All these factors may contribute to the phenomena, observed in recent studies on the ROS-generating activities of the 2-oxoglutarate and pyruvate dehydrogenase complexes in the presence of thiols and disulfides (120, 153), questioning the conclusions of the authors on the dependence of these phenomena on glutathionylation of the complexes only. Overall, the data obtained in such a complex system do not exclude different mechanisms/contributors to H2O2 generation over all the experimental settings used. Reversible glutathionylation may be added by nonreducible irreversible glutathionylation, involving Michael addition of a thiol group to a dehydroalanine residue of a protein (57, 152, 190). Different transformations of not only the enzyme complexes and/or their dihydrolipoyl residues but also of the substrate CoA and potential regulators (glutathione and its disulfide) are possible in the system used. Additional experiments discriminating contribution of all these factors to H2O2 generation would thus be required to distinguish among the conclusions reported.
Thus, in accordance with the results obtained with the isolated E1 and E3 components (Section III), superoxide anion radicals and hydrogen peroxide are produced also on aerobic catalysis by pyruvate and 2-oxoglutarate dehydrogenase complexes (Fig. 4). With the prolonged reaction time, multiple condition-dependent secondary reactions involving ROS, thiols, or disulfides in the reaction medium may be observed. The secondary processes include Fenton/Haber–Weiss reaction catalyzed by adventitious metal ions. Such ions could also be of enzymatic origin—for instance, when studying ROS generation in the presence of high SOD concentration, or in preparations including aconitase, which is unavoidable in mitochondrial studies. Present in the reaction medium of chemically reactive compounds, such as spin traps, thiols (including CoA, which is one of the substrates for the complex-catalyzed ROS production) and disulfides (which may react not only with the complex-bound dihydrolipoyl residues and E3 but also with the CoA substrate) may strongly affect the mechanisms of production of the reactive species and the secondary reactions involving the species.
B. Influence of multienzyme structure of 2-oxo acid dehydrogenase complexes on the ROS-producing activities of component enzymes
The multienzyme structure of 2-oxo acid dehydrogenase complexes is characterized by heterologous interactions between the active sites, which are absent in the isolated enzymes. As a result, additional side reactions and mechanisms of their regulation arise on the assembly of isolated components into the complex. At the same time, available data indicate that ROS production, inherent in the isolated E1 and E3 component enzymes, decreases on formation of 2-oxo acid dehydrogenase complexes. However, direct comparisons may be complicated by potential activation of the components through heterologous interactions in the multienzyme structure (Section II.C), and different conditions applied in different assays of the isolated components and multienzyme complexes.
1. Maximal rate of ROS generation by 2-oxoglutarate dehydrogenase is decreased in the complex-bound state
Higher rates of ROS production by isolated recombinant 2-oxoglutarate dehydrogenase components, compared to the complex-bound enzyme, are observed in the presence of allosteric activator of 2-oxoglutarate dehydrogenase, ADP (148). The effector is known to increase the enzyme affinity for 2-oxo substrate without affecting the maximal reaction rate (Vmax ) [reviewed in Bunik et al. (45)]. Nevertheless, in some studies, including the work on recombinant human 2-oxoglutarate dehydrogenase, ADP increases Vmax . Remarkably, the ADP effect on the Vmax for enzymatic ROS production is absent with the complex-bound 2-oxoglutarate dehydrogenase (148). This finding suggests that ADP stabilizes an active conformation of the isolated 2-oxoglutarate dehydrogenase, similar to the stabilization induced by heterologous interactions in the complex, observed in independent studies (47). The ADP mimicking of the activation induced by the heterologous interactions in the complex enables comparison of the isolated and complex-bound E1o regarding ROS production. Assayed under otherwise identical conditions, production of H2O2 by the ADP-activated 2-oxoglutarate dehydrogenase at the expense of either 2-oxoglutarate or 2-oxoadipate is characterized by a twofold lower rate after incorporation of the enzyme into the complex (148). The reduced rates of ROS production by E1o are indicative of the competition between the side reaction(s) with molecular oxygen (Fig. 3) and the physiological reaction with the native E1o substrate, the lipoyl-bearing domain of E2o (Fig. 1, Equation [2]). This interpretation is further favored by the finding that increased lipoylation of the 2-oxoglutarate dehydrogenase complex decreases superoxide anion radical production in the physiological direction of the reaction (5), where the contribution of E1-produced ROS (Fig. 3) is possible.
Thus, the presence of the physiological substrate (E2-bound lipoyl residues) and/or reaction product (E2-bound succinyl-dihydrolipoyl residue) protects the enamine intermediate of E1o from reaction with molecular oxygen. The protection may involve a competition of the native substrate or product with molecular oxygen and/or conformational change of the complex-bound E1, shielding the active site from side reactions. Similar shielding is known to occur for the acylated lipoyl residues (94). The side reaction is even less likely to be efficient in vivo, where intracellular concentration of O2 is significantly lower compared to that in solutions at atmospheric oxygen pressure, with mitochondrial respiration further decreasing the local oxygen concentration.
Ability of isolated 2-oxo acid dehydrogenase components to stabilize the EPR-detectable radical, presumed to be a coproduct of the superoxide anion radical generation by the E1o-bound enamine intermediate (Fig. 3, Pathway 1), is limited to 2-oxoglutarate dehydrogenase only (73, 147). However, both the pyruvate-dependent production and 2-oxoglutarate-dependent production of superoxide anion radical by the 2-oxo acid dehydrogenase complexes in the forward reaction with 2-oxo acids and CoA have been repeatedly shown (5, 48, 121). The observations point to yet another mechanism of E1-dependent ROS production within the complexes. Sensitivity of this mechanism to chemical modification of either the E2-bound dihydrolipoyl residues or E3-bound FAD indicates involvement of all components of 2-oxo acid dehydrogenase complexes in the 2-oxo acid plus CoA-dependent ROS production (48). Thus, production of ROS by 2-oxo acid dehydrogenase complexes in the forward reaction does not necessarily involve the direct oxidation of the enamine intermediate of their first components. Nevertheless, in the case of the 2-oxoglutarate dehydrogenase complex, the ROS produced in the forward direction may include independent action of E1o, shown with the isolated enzyme (73, 147, 148). The relative contribution of the two mechanisms, that is, the E1o-dependent and E1-E2-E3-dependent ones, may vary, particularly due to the assay conditions. For example, chemical modification of the E2o-bound dihydrolipoyl residues in the 2-oxoglutarate dehydrogenase complex, isolated from pig heart, prevents appearance of the spin-trapped adducts in the forward direction (48). This finding points to insignificant contribution of the oxidation of the complex-bound E1o enamine intermediate to ROS production by native complexes in the forward direction. This is supported by quantifications of ROS in the experiments with recombinant enzymes. Recombinant human 2-oxoglutarate dehydrogenase complex catalyzes the superoxide anion radical production in the forward direction at about sixfold higher rate, compared to that exhibited by the isolated E1o or its subcomplex with E2o (5). These experiments indicate that no more than 15% of total ROS produced by the complex in the forward direction could be attributed to the independent action of the complex-bound E1o. The major part of such ROS is thus produced according to the mechanism involving all components of the complex (48).
2. Dihydrolipoamide dehydrogenase-generated ROS are decreased in the complex-bound state
Assayed in the NADH (0.17 mM) oxidation reaction (backward electron flow) by cyt c reduction, the isolated recombinant E3 from E. coli exhibits a higher rate of superoxide anion radical production (73 nmol/min per milligram of E3), compared with that of the E3 bound to the pyruvate dehydrogenase (60 nmol/min per milligram of E3) or 2-oxoglutarate dehydrogenase (50 nmol/min per milligram of E3) complexes from E. coli. Compared to the bacterial enzyme, a higher ROS-generating activity is inherent in the isolated recombinant human E3 (100–115 nmol/min per milligram of E3). The activity does not change on binding to the human pyruvate dehydrogenase complex, but reduces to 40 nmol/min per milligram of E3 on binding to the human 2-oxoglutarate dehydrogenase complex (5). Thus, with both the E. coli and human E3, incorporation of the dihydrolipoamide dehydrogenase component into the multienzyme complexes results in a greater decrease in its ROS-generating activity in the backward reaction within the 2-oxoglutarate dehydrogenase complex, than in the pyruvate dehydrogenase complex. These findings agree with independent observations on the complex-specific regulation of the side reactions of E3, such as enzyme over-reduction (Fig. 2).
No data are available to estimate the influence of E3 incorporation into the complex on its ROS generation in the forward, that is, physiological, direction, as only the backward reaction is usually used for the assays of isolated E3. Nevertheless, in vitro data on purified enzymes allow one to compare the reaction rates of ROS production by the complexes in the direction, catalyzed by E1, E2, and E3 (forward electron flow), and that catalyzed by E3 only (backward electron flow). Estimating these reactions under nonphysiological pH (6.3–6.6), which is optimal for the backward reaction, but minimizes the rate of physiological reaction, Ambrus et al. (5) claimed that the major pathway of the ROS production by the complexes is the NADH oxidation. In fact, at these nonphysiological pHs, the in vitro rates are of the same order of magnitude. The backward reaction of ROS production from NADH is about twofold or threefold more rapid for the pyruvate dehydrogenase complex or 2-oxoglutarate dehydrogenase complex, respectively, than the forward reaction. However, for native 2-oxoglutarate dehydrogenase complex purified from pig heart and catalyzing the reaction at physiological pH (7.0), ROS generation is more efficient in the forward than backward reaction. Under otherwise equal conditions, a fourfold higher EPR signal of the spin-trapped superoxide anion radical has been detected on incubation with 2-oxoglutarate and CoA (2–4 mM each), compared to the backward reaction with NADH (2.5–5 mM) (41). Only when NADH in the experiments is increased up to a nonphysiological concentration of 10−2 M, the rates of superoxide anion radical production by 2-oxoglutarate dehydrogenase complex in the backward and forward (2-oxoglutarate and CoA, 2 mM each) reactions become comparable (41). Similar rates of the superoxide anion radical generation (about 1 nmol/min per milligram of protein) quantified by cyt c assay in the forward and backward reactions under these conditions confirm the conclusions from semiquantitative EPR studies (41, 48). As a result, catalysis of ROS production by the native complex-bound E3 in the backward reaction attains the rates comparable with the ROS generation in the forward reaction, at NADH concentrations that are an order of magnitude higher (10−2 M) than the total physiological pool of NAD++NADH in mitochondria, estimated to be up to 10−3 M (66). Thus, compared to the forward reaction catalyzed by all the components of the complexes, only a small contribution of the E3-catalyzed NADH oxidation may be expected under physiological conditions, which agrees with the studies of ROS production by native coupled mitochondria, considered in the next section.
Generation of ROS at the expense of NADH (the backward reaction), catalyzed by the complex-bound E3, depends on the ratio of subunits used for the complex assembly and lipoylation level of the complex (5). These findings provide additional evidence that incorporation of E3 into the multienzyme complex changes its ROS-producing activity. The changes may affect both kinetic parameters and reaction mechanism, such as O2 •− or H2O2 production by the 2e− or 4e− reduced E3. Natural variability in the component stoichiometry and a number of lipoyl residues in different complexes (Section II) may thus contribute to the observed variations in relative rates of ROS generation by E3 within the complexes both in vitro and in vivo. For example, with recombinant human complexes, which are assembled in vitro, the pyruvate dehydrogenase complex exhibits a twofold higher ROS production in the backward reaction, compared with the 2-oxoglutarate dehydrogenase complex (5). With the native pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase complexes purified from pig heart, the same reaction is characterized by an opposite relationship, that is, E3 within the 2-oxoglutarate dehydrogenase complex catalyzes NADH-dependent ROS production more efficiently than E3 within the pyruvate dehydrogenase complex (121).
C. Dihydrolipoamide dehydrogenase component is essential for both the forward and backward reactions of ROS production by the complexes, but the forward direction prevails in vivo
As discussed in Section IV.A, in the presence of CoA, which releases dihydrolipoyl groups in the E2-catalyzed transacylase reaction (Fig. 1, Equation [3]), both E1p and E1o components within the 2-oxo acid dehydrogenase complexes are involved in generation of the 2-oxo-acid-dependent superoxide anion radicals. The process requires all the components of the complexes and occurs more efficiently when the terminal substrate NAD+ is limiting. These features distinguish this mechanism of ROS generation by the multienzyme complexes from the ROS production specific for the 2-oxoglutarate dehydrogenase component. In contrast to the forward reaction that depends on 2-oxo acid and is disabled by the chemical modification of either the complex-bound dihydrolipoyl groups or the E3-bound FAD, the backward reaction of the NADH-dependent ROS generation by the complex-bound E3 is prevented by the modification of FAD only (48). This is due to the different possibilities of FAD reduction (Fig. 1). In the complex-bound state, FAD may be reduced by NADH, dihydrolipoamide, or 2-oxo acid with CoA. In the isolated state, or in the 2-oxo acid dehydrogenase complexes with the electron flow from 2-oxo acids blocked by modification of the dihydrolipoyl residues, the E3-bound FAD may be reduced only by the enzyme substrates, dihydrolipoamide or NADH, but not by the combination of 2-oxo acid and CoA. Thus, the coupled action of all the enzymatic components of the complexes is required for the superoxide-generating activity at the expense of 2-oxo acids and CoA (so-called forward electron transport [FET]), but is not obligatory for the backward direction of the E3-catalyzed 1e− oxidation of NADH by oxygen (so-called reverse electron transport [RET]).
Similar to the above estimations based on in vitro EPR studies, the physiological (FET) direction, dependent on 2-oxo acid and CoA, prevails over the nonphysiological (RET) process of NADH oxidation in studies of isolated coupled mitochondria (24, 79, 160). Before the contribution of 2-oxo acid dehydrogenase complexes to mitochondrial ROS production has been widely recognized, the mitochondrial ROS produced at high NADH were mainly attributed to complex I of electron transport chain. Similar to the flavin-dependent E3 component of 2-oxo acid dehydrogenase complexes, the NADH-reduced flavin site of complex I may react with oxygen when its native electron transfer is impaired (24, 100, 160, 202). Later, specific settings to discriminate maximal capacities of ROS production by complex I and different 2-oxo acid dehydrogenase complexes have been elaborated in situ using preparations of native mitochondria. Comparison of the enzymes of the NADH/NAD+ isopotential group, that is, enzymes that operate at the redox potential of the NADH/NAD+ pool, indicates that the physiological direction of the 2-oxoglutarate oxidation by 2-oxoglutarate dehydrogenase complex has the greatest impact on mitochondrial ROS production at a high NADH/NAD+ ratio. Maximal contribution to mitochondrial ROS of other 2-oxo acids oxidized by their cognate complexes, including the newly characterized 2-oxoadipate dehydrogenase complex (79, 194), is less than that of the 2-oxoglutarate-dehydrogenase-dependent reaction. It is important to note that these quantifications correspond to upper limits of the ROS-producing activities of specific 2-oxo acid dehydrogenase complexes in the physiological direction of the reactions inside mitochondria. The estimated values are thus similar to the Vmax parameter in enzyme kinetics, and do not mean that this maximal potential is realized under all conditions. Real contributions of the different reactions and hence their physiological relevance are supposed to vary, dependent on the tissue- or cell-specific expression of enzymes, their regulation mechanisms, or substrate fluxes. For instance, different functional states of mitochondria may affect the relative contribution of different 2-oxo acid dehydrogenase complexes to mitochondrial ROS production, because the complexes strongly vary in dependence of their ROS-producing capacities on ADP and ATP (∼10−3 M) (79). Neither ADP nor ATP affects the ROS-producing capacity of intramitochondrial pyruvate dehydrogenase complex. ATP does not affect ROS production by intramitochondrial 2-oxoglutarate dehydrogenase complex either, but inhibits the mitochondrial ROS production by the branched chain 2-oxo acid dehydrogenase complex and 2-oxoadipate dehydrogenase complex (79). ADP addition to mitochondria also affects these complexes in a different way. Mitochondrial ROS production by 2-oxoglutarate dehydrogenase complex is activated by an order of magnitude, whereas that by branched chain 2-oxo acid dehydrogenase complex is inhibited twofold. Some inhibition by ADP is also observed for intramitochondrial 2-oxoadipate dehydrogenase complex, although it does not reach statistical significance under used conditions. The ADP stimulation of the mitochondrial ROS production by 2-oxoglutarate dehydrogenase complex relies not only on the ADP activation of the first (E1o) component of the complex as considered above but also on intramitochondrial ADP phosphorylation in the succinyl-CoA-dependent reaction. The reaction removes the inhibitor of 2-oxoglutarate dehydrogenase complex succinyl-CoA, simultaneously regenerating an ROS-producing substrate CoA. Molecular mechanisms of the ADP and ATP effects on the mitochondrial ROS production by other 2-oxo acid dehydrogenase complexes have not been deciphered. One may suggest that the ATP-induced inhibition of ROS production by intramitochondrial branched chain 2-oxo acid dehydrogenase complex may involve regulatory phosphorylation of this complex.
Overall, quantitative mitochondrial studies indicate that the contribution of the backward E3-catalyzed reaction to ROS generation in the native coupled mitochondria is much less than that of complex I (160). Only in permeabilized mitochondria, the E3 contribution becomes equal to that of complex I (100). This difference may be due to different levels of saturation of E3 with the reaction substrate NADH, which may be increased much more easily in the permeabilized than native mitochondria. The observation agrees with a strong dependence of the E3-produced ROS on NADH in the nonphysiological interval of NADH concentrations (up to 10−2 M) (41), as discussed above. It is worth noting in this regard that comparison of different studies of the NADH saturation of the ROS-generating activity of E3 is complicated due to the significantly varied assay conditions and E3 preparations used. The factors that may affect the dependence of E3-catalyzed reactions on NADH include, but are not limited to, the presence of other enzymes binding NADH, different productions of the assayed species (superoxide anion radical or H2O2) by the 2e− and 4e− reduced E3 (Fig. 2), and different levels of inhibition of the studied reactions by E3 over-reduction.
Based on the kinetic constants determined in the study on purified E3 in vitro, Gazaryan et al. estimate production of H2O2 by intramitochondrial E3 at 0.1 mM NADH and saturating oxygen at 2 nmol/min per milligram of mitochondrial protein (78). Mitochondrial studies in situ point to a 40-fold lower rate of H2O2 production with a fully reduced NADH pool of the mitochondria, that is, 0.05 nmol/min per milligram of mitochondrial protein (pH 7.0) (160). Taking into account that this value is close to the ROS produced by complex I, one may conclude that production of the E3-dependent ROS in situ is more than an order of magnitude less efficient, compared to the reaction observed in vitro.
Protein binding of intramitochondrial NADH, limiting the free NADH available for E3, may be among the factors, preventing E3 from realizing its in vitro potential for NADH-dependent ROS production in situ (e.g., in native mitochondria or cells) and in vivo. In vivo mechanisms diminishing the interaction of E3 with its product NADH may include formation of supramolecular structures to organize the fluxes in the physiological direction (3, 184). In eukaryotic complexes, such mechanisms may be heightened by the regulation of E3 binding to the mammalian pyruvate dehydrogenase complex and 2-oxoglutarate dehydrogenase complex through the specific E3-binding isoform of E2 (201) and MRPS36 (Ymr31, Kgd4) adaptor protein (89), correspondingly.
At saturating nonphysiological concentration of Zn2+, that is, 2 μM free ion at 40 μM total added, the ROS production by E3 is activated up to fivefold, resulting in a rate of 10 nmol/min per milligram of protein. The sensitivity of E3-catalyzed ROS production to Zn2+ or acidosis has been considered to be of (patho)physiological significance. However, the suggestions do not take into account that even if Zn2+ stimulates the NADH-dependent production of ROS by isolated E3 in vitro (78), irreversible loss of the physiological activity of E3 on Zn2+ addition to mitochondria also decreases mitochondrial NADH levels (77). Similarly, increased rates of the backward reaction of NADH oxidation by E3 at pH 6.3–6.4 in vitro do not necessarily activate the reaction in vivo, where inhibition of oxidative decarboxylation of 2-oxo acids in acidosis would decrease NADH, which is the substrate of E3-dependent ROS production. According to the same reasoning, it is also unlikely that relatively moderate increases in the ROS-generating activity on pathogenic mutations in E3, revealed in vitro, could greatly affect the enzyme contribution to mitochondrial ROS production through E3-catalyzed oxidation of NADH in vivo, as suggested by Ambrus and Adam-Vizi (4). Such speculations certainly need their validation in vivo before they are published and discussed. In cultured neurons exposed to glutamate, increasing the 2-oxoglutarate dehydrogenase inhibition causes a biphasic response in cellular ROS (217). First, ROS decrease exposing the contribution of the forward electron flow in the 2-oxoglutarate dehydrogenase complex to glutamate-elevated neuronal ROS production. The decrease contributes to preservation of mitochondrial function under conditions of glutamate excitotoxicity (42, 97). However, increasing the inhibition of the complex is not accompanied by further decrease in mitochondrial ROS, which increase instead. This finding points out that general metabolic impairment on a strong 2-oxoglutarate dehydrogenase inhibition overweighs contribution of the complex to mitochondrial ROS, with other ROS sources activated as a result. The nature of these sources remains to be established.
An increase in NADH is observed in some studies on inhibition of 2-oxo acid dehydrogenase complexes, supposedly manifesting importance of specific metabolic fluxes for appropriate oxidation of NADH by mitochondria (3, 210). Remarkably, the ROS production does not increase, rather it decreases under these conditions, elevating NADH (210). This finding agrees with a decreased contribution of impaired 2-oxoglutarate dehydrogenase to ROS produced in the physiological direction. At the same time, there is no significant increase of the NADH-dependent ROS, which include ROS produced by E3, despite NADH elevation.
D. Heterologous interactions in the multienzyme complexes enable formation of thiyl radicals of E2-bound dihydrolipoyl residues, regulating ROS production by the complexes
Increasing complexity of the multienzyme structure, compared with the isolated enzymes, noted at the beginning of Section IV, includes heterologous interactions between active sites of the enzyme complex, which are absent in its isolated components. This feature causes essential changes in ROS-generating side reactions of the complexes, often overlooked when the complexes are viewed only as a sum of their components. Most important in this regard is an exposure of the E2-bound dihydrolipoyl residues to the active sites of the E1 and E3 components, not occurring with isolated E1, E2, and E3. In contrast to E2, E1 and E3 are oxidoreductases. Hence, the oxidoreductase reactions of dihydrolipoyl residues, which are not possible in the isolated E2, become possible within the multienzyme complex, where these residues serve as substrates of the two oxidoreductases. As shown below, such exposure enables catalytic formation of thiyl radicals of the complex-bound dihydrolipoyl residues. Under aerobic conditions, this thiyl radical species arises concomitantly with ROS production by multienzyme complexes.
1. Detection of thiyl radicals of the E2-bound dihydrolipoyl residues under anaerobic conditions
In contrast to aerobic conditions, anaerobiosis excludes oxygen-dependent reactions of the redox centers and reduces associated secondary reactions. Under these conditions, a better resolution of different radical species in the catalytic sites of 2-oxo acid dehydrogenase complexes is possible. As seen from Equations [1] to [5] (Fig. 1), the overall physiological process of 2-oxo acid oxidation (Fig. 1, Equation [6]) involves a sequence of redox steps occurring in interacting active sites of the complexes. Although the charge-transfer complex between the catalytic disulfide and FAD is the major species under the redox equilibrium, minor occupancy of the E3-active site with the FAD semiquinone radical is not excluded. The FAD semiquinone radical is observed not only in the reactions with molecular oxygen (Fig. 4, Equation [7]) but also in the free FAD- or E3-dependent reactions involving quinones (7, 10, 151, 169, 200) and lipoic acid radical (54, 68). Some of the reactions with lipoic acid radicals are exemplified by the backward reactions described by Equations [11] and [12] of Figure 6. Due to the multiple random coupling mechanism supported by the interaction of all active sites in the oligomeric structure of 2-oxo acid dehydrogenase complexes (Section II), minor occupation of the redox sites with radical species carrying an uncoupled electron, such as depicted by generic Equation [11] (Fig. 6), implying both the lipoate and protein disulfides, may be suggested to occur on partial reduction of the complex redox sites. Indeed, addition of a small excess of the reducing substrates (2-oxo acid and CoA in the forward reaction or NADH in the backward reaction) over the redox-active sites of the complexes under anaerobic conditions enables spin-trapping of the complex-bound thiyl radical species (48). The finding agrees with partial occupation of the E3-active sites by the FAD semiquinone radical, equilibrating with the redox-active disulfides/dithiols of the complexes according to Equation [11] of Figure 6.
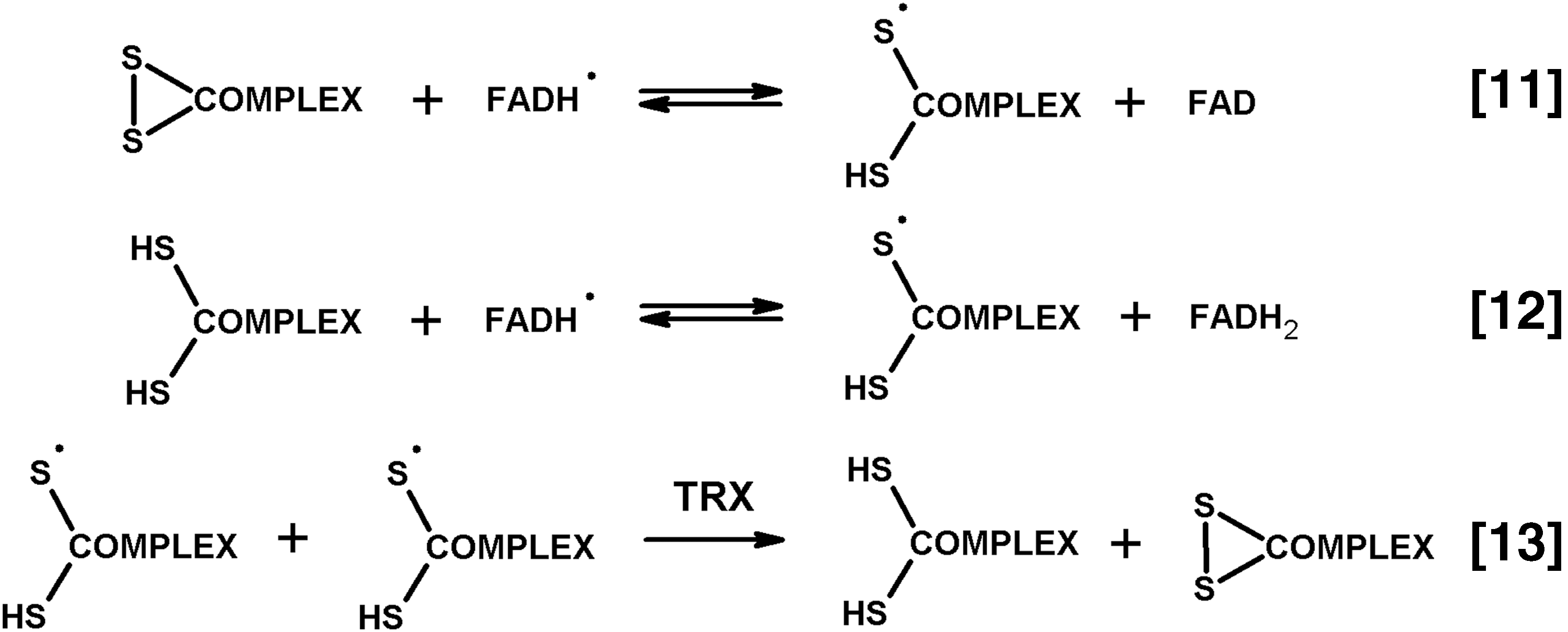
Compared to monothiols, the thiyl radical of dihydrolipoate (Fig. 7) is stabilized by formation of the three-electronic structure of the disulfide radical (13). Within the 2-oxo acid dehydrogenase complexes whose oligomeric cores comprise clusters of lipoyl residues in vicinity of each other, the thiyl radical may acquire further stabilization due to transfer of the uncoupled electron through the network of interacting lipoyl groups of the cores. Remarkably, this additional mechanism of stabilization of the thiyl radicals would not be pronounced in the minicomplexes, such as those formed around the E2 trimers (Section II), as much as in the complexes with high oligomerization degrees. The stabilization may also be perturbed by a decreased number of lipoyl residues in the second components comprising tandem lipoyl domains. In this regard, the importance of tandem organization of lipoyl domains in E2 for organism viability, but not for catalysis per se (53, 62, 82 –84), supports biological significance of the thiyl radicals and their stabilization in the structure of 2-oxo acid dehydrogenase complexes.
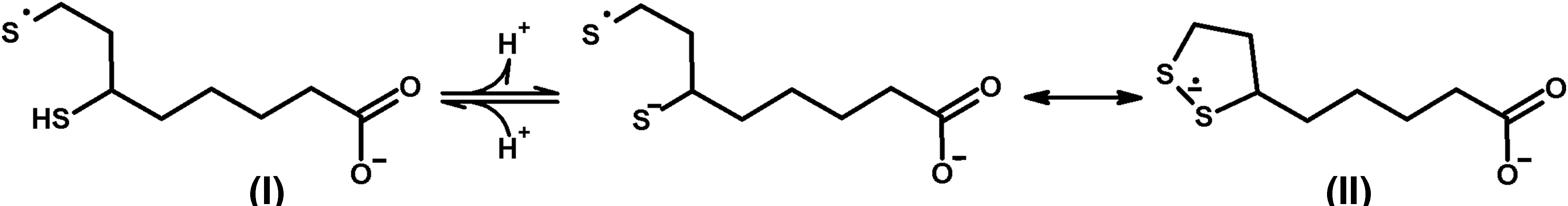
2. Thiyl radical of the dihydrolipoyl substrate of E1 is accompanied by the carbon-centered radical product of E1
Anaerobic incubation of 2-oxoglutarate dehydrogenase complex with 2-oxoglutarate and CoA in the presence of spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO) allows one to detect the DMPO-trapped carboxy radical presumably originating from the catalytic action of E1 (48). Detection under anaerobic conditions of the two types of radical species, that is, the carboxy radical related to catalysis by E1 and thiyl radicals of the complex-bound dihydrolipoyl residues, favors participation of the thiyl radical in anaerobic 1e− oxidation of the catalytic intermediate of E1. This reaction may represent an “off-pathway” of the physiological reaction of the 2e− oxidation of the same enamine intermediate by its second substrate, the lipoyl-bearing domain (Fig. 1, Equation [2]). Pathway 1 of Figure 3 depicts the reaction mechanism in the DMPO-containing medium. 1e− Oxidation of the E1 enamine intermediate formed according to Equation [1] in Figure 1 may occur during interaction of E1 with the closest analog of its substrate acceptor, the oxidizing thiyl radical of the E2-bound dihydrolipoyl residues. As a result, the radical intermediate in the E1-active site and dihydrolipoyl residue are produced. The E1-bound radical intermediate may be released as the spin-trapped adduct after its transfer to DMPO (Fig. 3). The latter reaction may be catalyzed by 2-oxoglutarate dehydrogenase, as the enzyme is known to transfer its reactive intermediate to nonphysiological acceptors, including the diazo and nitroso compounds whose chemical properties have certain similarity to those of DMPO (47). In the absence of DMPO, the E1-bound radical intermediate should have other acceptors to complete the catalytic cycle. Occurrence of such “off-pathways” generating “wrong” reactive intermediates is usually associated with so-called paracatalytic inactivation, which has indeed been observed for 2-oxo acid dehydrogenases and their complexes under a variety of conditions [reviewed in Bunik et al. (47)]. The next section considers the inactivation under conditions used in the EPR studies (summarized above), where the complex-bound thiyl and E1-related carboxy radical species are observed (48).
3. Thiyl radical-associated inactivation of 2-oxo acid dehydrogenase complexes and alleviation by thiyl radical scavengers
In the anaerobic reaction medium where the thiyl- and carbon-centered radicals are detected, 2-oxoglutarate dehydrogenase complex is inactivated (48). The finding provides evidence for the thiyl radical-dependent oxidation of the enamine intermediate as an off-pathway (Fig. 3, Pathway 1) causing the inactivating modification of E1. Studies on the dependence of inactivation on the concentration of 2-oxoglutarate and CoA, which induce the inactivation, revealed that the inactivation is less pronounced when the substrates are in excess over the redox centers of the complex (48). The inactivation kinetics is presented in the upper part of Figure 8A. It is obvious that under anaerobiosis, a higher activity is attained in the medium where concentrations of 2-oxoglutarate and CoA exceed that of the redox-active sites. In contrast, incomplete reduction of the complex due to substrate limitation promotes inactivation. Because the incomplete reduction enables exchange of redox equivalents between the simultaneously present reduced and oxidized species, also the 1e− exchange may occur as an “off-pathway,” leading to the complex-bound thiyl radical detected by EPR. In contrast, when all the redox-active sites of the multienzyme complex are reduced, neither the redox exchange nor the anaerobiosis-blocked reactions with oxygen are possible. Besides, rapid reduction of the complex with the substrate excess shortens the transient phase when the redox exchange is possible. As a result, the thiyl radical-induced inactivation is less pronounced under the substrate excess over the redox-active sites, compared with the inactivation under the low substrate concentrations, which are insufficient for complete reduction of the complex (Fig. 8A, anaerobic conditions). As seen from Figure 8B, the time course of the anaerobic accumulation of the spin-trapped thiyl radicals confirms their transient formation. Thus, irreversible inactivation of the 2-oxo acid dehydrogenase complexes on anaerobic incubation with the substrates of partial reaction (Fig. 1, Equations [1]–[3]), 2-oxo acid and CoA, may be a functional indicator of the E2-bound thiyl and E1-dependent carboxy radical species, which are spin-trapped under anaerobic conditions with N-tert-butyl nitrone (PBN) and DMPO, respectively.
Aerobic catalysis also leads to the inactivation of complexes in the presence of 2-oxoglutarate and CoA, pointing to the formation of complex-bound thiyl radicals under aerobic conditions. However, as seen from Figure 8A, the substrate concentration dependence is different in the aerobic and anaerobic systems (48). When the level of reduction of redox sites of the complexes increases, the inactivation in the aerobic system is promoted, in contrast to the alleviation of the anaerobic inactivation. This observation reciprocates the transient nature of anaerobic PBN adducts with thiyl radicals versus time-dependent increase of the aerobic PBN radical adducts (Fig. 8B). Thus, oxygen changes not only the concentration dependence of the inactivation (Fig. 8A) but also the time courses of the inactivating radical species generated in the systems (Fig. 8B). Under anaerobiosis, the radical species are transient. In the aerobic system, they are generated concomitantly with the catalytic production of superoxide anion radical, which is increased with increasing the substrates.
Formation of multiple radical species and spin-trapped adducts in the aerobic system interferes with identification of thiyl radicals under these conditions. Therefore, evidence for the thiyl radical-dependent inactivation mechanism has been obtained in functional studies (48). The inactivation is not observed in the presence of NADH or dihydrolipoamide, which are the substrates for E3-dependent ROS production. This finding excludes the ROS-dependent impairment of the complexes, in agreement with no effect of SOD (48) or anaerobiosis (Fig. 8A) on inactivation. However, the combinations of either NADH with 2-oxoglutarate or dihydrolipoamide with succinyl-CoA lead to measurable inactivation. Thus, not only the dihydrolipoyl residues but also a catalytic intermediate of E1, which is produced either in the presence of 2-oxoglutarate or on the dihydrolipoyl-dependent transfer of succinyl group from CoA to ThDP (74), is involved in the substrate-dependent inactivation of E1. Moreover, the inactivation rate coincides for the overall reaction (Fig. 1, Equation [6]) and E1 partial reaction (Fig. 1, Equations [1] and [2] in the presence of artificial electron acceptors instead of the lipoyl domain of the complex). The finding indicates that the inactivation of E1 component is responsible for the impairment of overall reaction.
The 2-oxo acid plus CoA-dependent inactivation in the absence of NAD+ (Fig. 8A) may also be observed at a low saturation of complexes with NAD+. When following the NADH accumulation in the complex catalyzed reaction under these conditions (Fig. 9A), one observes a rapid decrease in the reaction rate (Fig. 9A, control). Addition of thioredoxin, which is a scavenger of thiyl radicals (115), strongly increases the reaction rate at a delayed reaction time, that is, when the catalysis-associated inactivation becomes pronounced (Fig. 9A). Thus, thioredoxin protects 2-oxo acid dehydrogenase complexes from the inactivation under a low NAD+ saturation, when thiyl radicals of the complex-bound dihydrolipoyl groups may arise. The protective effect is in good accordance with the thioredoxin-dependent dismutation of the complex-bound thiyl radicals of dihydrolipoyl residues according to Equation [13] of Figure 6. This mechanism is further supported by the findings that mutation and/or modification of the catalytic cysteine residues of thioredoxin impair the protective effect (33), although the redox state of these residues does not change the protective ability of thioredoxin (27).
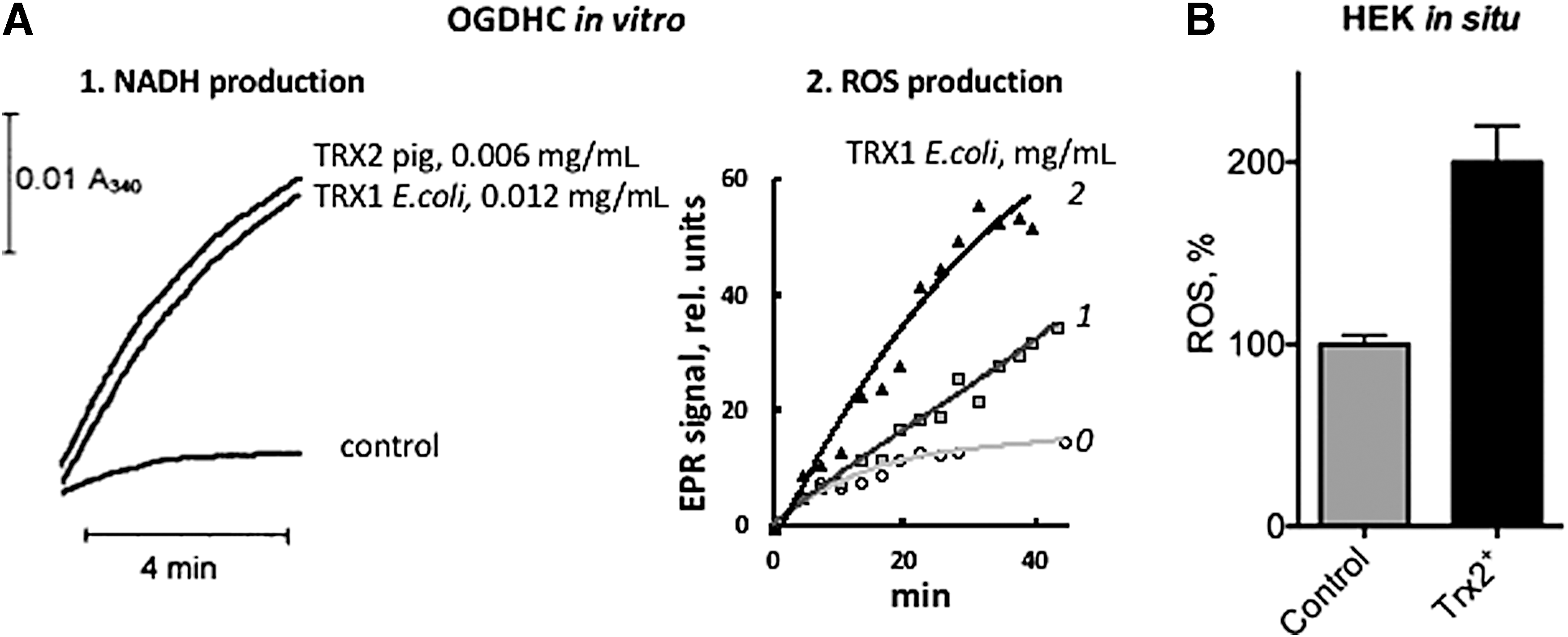
The inactivation in the course of catalysis at a low NAD+ saturation is known for bacterial and mammalian pyruvate and 2-oxoglutarate dehydrogenase complexes, and in all the cases it is alleviated by thioredoxin. As noted above, the thioredoxin protection is seen as an activation of the complex-catalyzed NADH production at a delayed reaction time (Fig. 9A). Figure 10 shows that such an activation is caused by both the oxidized and reduced forms of thioredoxin and a number of nonprotein thiols or disulfides. However, effective concentrations of the nonprotein thiols or disulfides are at least an order of magnitude higher than those of thioredoxin. These high concentrations may result in additional effects, which are not seen with the catalytic quantities of thioredoxin. For instance, as discussed above, inhibition of the complexes by formation of mixed disulfides with catalytic dihydrolipoyl residues may be observed simultaneously with the protective effect causing activation (9, 27). Superposition of the different influences will determine the dependence of apparent effect on redox potential of system components. In particular, under conditions of experiment presented in Figure 10, inhibition by cysteine disulfide is obvious. Regarding the disulfide of glutathione, its lower activation of the complex (150%), compared to the same concentration of glutathione (250%) (Fig. 10), suggests that in the presence of glutathione disulfide both the activating protection and inactivating modification of the complex occur.
In summary, the substrate dependence, no protection by the scavenger of superoxide anion radical (SOD) (48) or anaerobiosis (Fig. 8), and the protective action of thioredoxin and other thiols/disulfides confirm the dependence of aerobic inactivation in the presence of 2-oxoglutarate and CoA on generation of complex-bound thiyl radicals. Taking into account the thiyl radical scavenging properties of thioredoxin and importance of its catalytic cysteine residues, but not its redox state, for protection, a mechanism for the thioredoxin-catalyzed dismutation of the complex-bound thiyl radicals may be suggested, as shown in Figure 11.
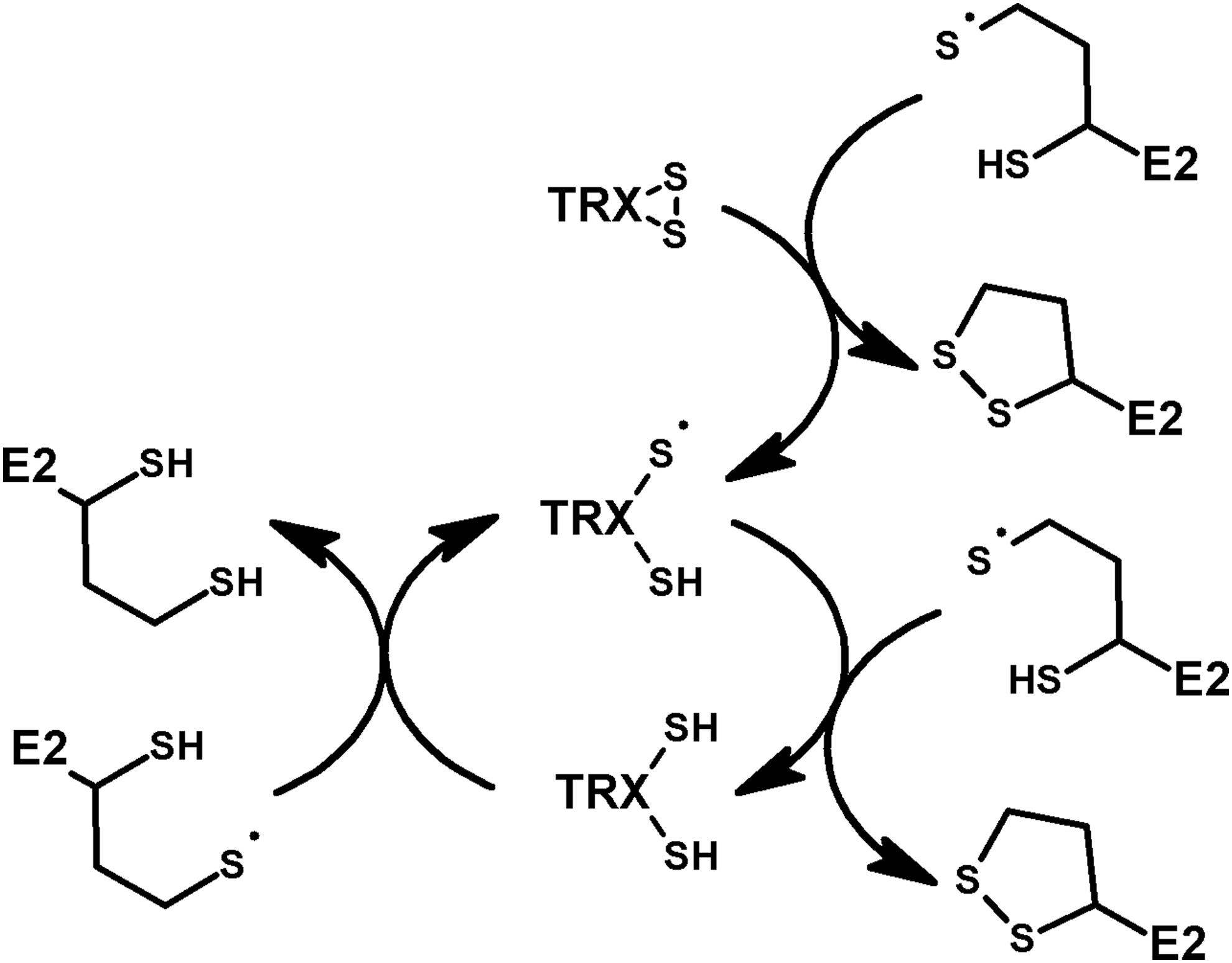
Both the E1-dependent DMPO-trapped radical species observed by Bunik and Sievers (48) and the EPR-detectable enamine radical specific for 2-oxoglutarate dehydrogenase (73, 147) may be formed by the first step of Pathway 1 (Fig. 3), representing 1e− oxidation of the enamine intermediate without specification of the oxidizing agent. In case of the EPR-detectable radical intermediate, specific for the E1o-catalyzed oxidative decarboxylation of 2-oxoglutarate and 2-oxoadipate, neither the mechanism of the active site regeneration nor potential paracatalytic inactivation caused by such a radical intermediate has been studied or suggested (147, 148). A low stability of the enamine radical intermediate in the pyruvate dehydrogenase reaction, which presumably interferes with its EPR detection, is in line with the observation of a less pronounced inactivation of the pyruvate versus 2-oxoglutarate dehydrogenase complex in the presence of cognate 2-oxo acids and CoA. However, qualitative similarity of the thioredoxin-sensitive paracatalytic inactivation of the pyruvate and 2-oxoglutarate dehydrogenases (27, 30, 33) points to a common inactivation mechanism involving the thiyl radical-dependent 1e− oxidation of the active aldehyde intermediate in the active sites of the 2-oxo acid dehydrogenases.
Overall, the data reveal the central role of thiyl radicals of the complex-bound dihydrolipoyl residues in the 2-oxo acid and CoA-dependent inactivation of the starting components of the complexes in both the aerobic and anaerobic systems. In the anaerobic system, these radical species are transiently generated during incomplete reduction of the complex redox sites (Fig. 8B, anaerobiosis) according to Equations [11] and [12] in Figure 6. In the aerobic system, the thiyl radicals are generated along with the coproduction of O2 •− and the complex-bound flavin semiquinone radical, FADH• (Fig. 4, Equation [7]). The semiquinone is reduced to FADH2 by catalytic dithiol of E3, which mediates the transfer of reducing equivalents from dihydrolipoyl residues of E2 in either the side reaction (Fig. 6, Equation [12]) or physiological process (Fig. 1, Equations [4] and [5]).
Remarkably, the ROS production by E3 is decreased on binding to the complexes (Section IV.3), but increases with increased level of the complex lipoylation (5). Although not explained by the authors, this observation provides independent evidence for the reaction depicted by Equation [12] in Figure 6. Obviously, the regeneration of FADH2 is needed for the catalytic production of superoxide anion radical (Fig. 4, Equation [7]), and the E3 reaction with the native substrate, the dihydrolipoyl residues of the complex, would do it in the most efficient way. The semiquinone-dependent formation of the highly reactive complex-bound thiyl radicals (Fig. 6, Equations [11] and [12]) demonstrates a particular case of the well-known damaging potential of quinone radicals. To escape the damage, formation of semiquinone radicals in biosystems is supposed to be prevented by NAD(P)H:quinone acceptor oxidoreductases, which have evolved to promote the 2e− transfer reactions of the quinones (199). As shown above, in case of the flavin semiquinone-dependent formation of thiyl radicals of the complex-bound dihydrolipoyl residues, the thiyl radicals are dismutated (Equation [13] of Fig. 6) by their scavenger TRX according to the mechanism suggested in Figure 11. The dismutation protects from the thiyl radical-induced inactivation of E1 components, resulting in similar changes in the time courses of reactions producing NADH and ROS (Fig. 9A).
Thus, in aerobic system, the thiyl radicals of the complex-bound dihydrolipoyl residues arise as a result of the E3-catalyzed generation of superoxide anion radical and FAD semiquinone radical (Fig. 4, Equation [7]). The dihydrolipoyl residues of the E2 core regenerate FADH2 for its next catalytic cycle of oxygen reduction (Fig. 6, Equation [12]). Under anaerobic conditions, the radicals may arise on 1e− exchange between incompletely reduced redox sites of the complexes (Fig. 6, Equation [11]). Migration of the uncoupled electron through the network of (dihydro)lipoyl residues of the complex core provides for additional stabilization of the thiyl radicals. Existence of the network also accelerates the thioredoxin-catalyzed dismutation (Fig. 6, Equation [13]), which regenerates the oxidized and reduced lipoate residues (Fig. 11).
4. Contribution of the complex-bound thiyl radicals to ROS production on addition of dithiols to the reaction medium
Compared with thiyl radicals of monothiols, the thiyl radicals of dithiols are significantly more stabilized through their interconversion with the disulfide anion radical shown in Figure 7 (13). The disulfide anion radical exhibits reducing properties (E0 = −1.6 V), while the thiyl radical is a strongly oxidizing species with E0 from +0.75 to +1.33 V. The oxidizing potential depends on the tendency of the radical to be protonated (115). The protonation prevents the thiyl radicals of dithiols to close into the 3e•− structure of the moderate reductant, the disulfide anion radical (Fig. 7). The complex-bound dihydrolipoyl residues, generated in the sequence of reactions catalyzed by E1 and E2 (Fig. 1, Equations [1]–[3]), have a distinct pro-oxidant action. The pro-oxidant action is caused by coupling of the FADH2-dependent reaction of E3 with oxygen (Fig. 4, Equation [7]) to formation of the complex-bound thiyl radicals inactivating 2-oxo acid dehydrogenases (Fig. 6, Equation [12]). The data suggest stabilization of the protonated state of the complex-bound thiyl radicals. Unlike the complex-bound dihydrolipoyl residues, free dithiols, such as dithiothreitol (DTT), seem to be more prone to lose the proton after they undergo electron transfer exchange with the complex-bound thiyl radical (Fig. 12, Equations [14] and [15]). Formation of the reducing disulfide anion radical enables a rapid transfer of 1e− to molecular oxygen (Fig. 12, Equation [15]). This mechanism is supported by the data of Mailloux et al. who have shown an increase in the O2 •− generation in the forward direction of the complex-catalyzed reaction in the presence of DTT (121). It is worth noting in this regard that a chemically incorrect mechanism was suggested by the authors, who did not take into account formation of the thiyl radicals of the complex-bound dihydrolipoyl residues as a coproduct of the catalytic generation of O2 •− by complexes. The abstraction of a single electron from reduced lipoate by reduced DTT, as depicted in the article, contradicts redox chemistry of thiols. However, taking into account the complex-bound thiyl radicals as coproducts of E3-catalyzed O2 •− generation by the complexes (48), the DTT-dependent increase in superoxide anion production may occur according to the mechanism, presented in Figure 12, discussed above. At a high concentration of DTT, compared to that of the complex-bound lipoyl residues, the oxygen reduction (Fig. 12, Equation [16]) may be amplified owing to accumulation of the disulfide anion radical of DTT in the medium due to 1e− redox cycling of the complex-bound dihydrolipoyl residues with DTT. Besides, stimulation of this redox cycling by high concentrations of DTT prevents the complex-bound thiyl radical from interaction with the E1 catalytic intermediate. As a result, DTT also increases catalytic production of superoxide anion radical by the complexes, similar to the increase observed in the presence of thioredoxin (Fig. 9).
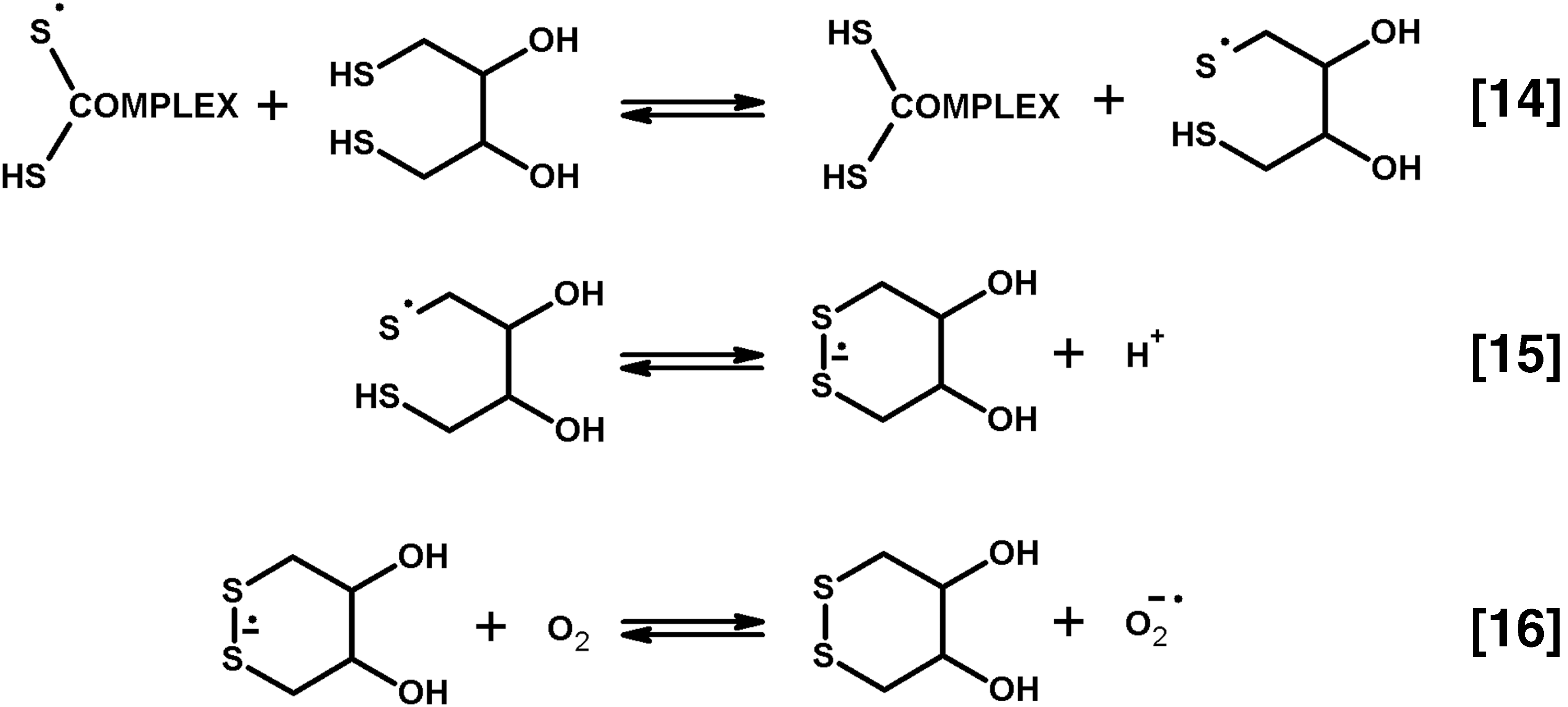
Thus, one should not overlook the coproduction of ROS and thiyl radicals by the complexes under aerobic conditions. The significance of this finding is heightened by specific functional interactions between 2-oxo acid dehydrogenase complexes with thioredoxins, glutaredoxins, and peroxiredoxins, considered in the next section.
V. Dihydrolipoyl Residues Couple ROS Generation by 2-Oxo Acid Dehydrogenase Complexes to Thioredoxins, Glutaredoxins, and Thiol-Based Peroxidases
A. Physiological significance of the protection of catalytic functions of 2-oxo acid dehydrogenase complexes by thioredoxin
As discussed above and shown in Figure 9A, the product accumulation in either the physiological or ROS-producing reactions in the presence of thioredoxin does not show the strong decay inherent in these reactions due to the catalysis-associated inactivation in the absence of thioredoxin (27, 30, 41). When 2-oxoglutarate dehydrogenase complex functions at limiting NAD+ in mitochondria in situ, linear kinetics of ROS accumulation is observed (160), similar to that observed in vitro in the presence of thioredoxin (41, 48) (Fig. 9A, NADH production). Data indicate that the catalysis by intramitochondrial 2-oxoglutarate dehydrogenase complex under limiting NAD+ is protected by endogenous mitochondrial thioredoxin. An important consequence of such protection of 2-oxo acid dehydrogenases by thioredoxin is their effective transformations of 2-oxo acids into acyl-CoAs at low saturation with NAD+. The low NAD+ saturation in vitro simulates competition between many NAD+-dependent dehydrogenases for the same substrate NAD+ in vivo. Function of the intramitochondrial complexes under subsaturating NAD+ is suggested by mitochondrial impairment under decreased saturation of the complexes with E3 (103). In particular, significance of succinyl-CoA production in the 2-oxoglutarate dehydrogenase reaction is demonstrated by a decreased level of mitochondrial substrate phosphorylation, that is, ADP phosphorylation at the expense of succinyl-CoA, on 2-oxoglutarate dehydrogenase complex deficiency due to reduced E3 content (103). Because E3 possesses orders of magnitude higher catalytic activity compared with the reaction rate-limiting E1 components, the impairment suggests function of the E3-deficient complex under a low NAD+ saturation. These conditions may increase the thiyl radical-dependent inactivation of the complex, negatively affecting the succinyl CoA production.
Physiological significance of the thioredoxin protection of the catalysis by 2-oxo acid dehydrogenase complexes is supported by data obtained in cells with manipulated thioredoxin levels. As summarized in Figure 9, increased ROS production by the complexes in the presence of thioredoxin is observed not only in vitro (41, 48) but also in cells overexpressing mitochondrial, but not cytosolic, thioredoxin (214). This pro-oxidant action of thioredoxin is unexpected in view of the well-known antioxidant function of the thioredoxin system. On the contrary, it is in good accordance with the thioredoxin protection of the activity of 2-oxo acid dehydrogenase complexes at low NAD+ (Figs. 9 and 10). Added by significant contribution of the complexes to mitochondrial ROS production in situ (24, 160, 217), the data in cells provide a confirmation for the role of mitochondrial thioredoxin in protection of catalytic function of 2-oxo acid dehydrogenase complexes, including their ROS generation, in vivo. Remarkably, not only the overexpression (214) but also downregulation of mitochondrial thioredoxin increases cellular ROS (186). The role of mitochondrial thioredoxin in the protection of 2-oxo acid dehydrogenase complexes from the thiyl radical-induced inactivation reconciles also this contradiction. Presumably, self-inactivation of 2-oxo acid dehydrogenase complexes at low NAD+ saturation is increased in the cells lacking thioredoxin. The ensuing impairment of oxidative metabolism is known to stimulate alternative sources of mitochondrial ROS production (49). Re-establishment of the thioredoxin expression in such cells should protect the complexes from the thiyl radical-induced inactivation, which normalizes homeostasis. Indeed, the antiapoptotic effect and decreased ROS are observed in cells with the re-established expression of mitochondrial thioredoxin (186).
Species-specific protein interactions determine efficiency of the thioredoxin protection of 2-oxo acid dehydrogenase complexes from the thiyl radical-induced inactivation, supporting biological significance of the phenomenon (33). Although certain cross-reactivity between the mitochondrial complexes and other thioredoxins may occur at increased concentrations, the cognate mitochondrial thioredoxin exerts the most efficient protection, obvious at its concentrations down to 10−7 M, followed by its closest homologue, thioredoxin from E. coli. Studies of structural determinants for the high protection efficiency of mitochondrial thioredoxin have revealed a correlation between the protective effect and specific electrostatic properties of mitochondrial thioredoxin (33, 161). Compared with other thioredoxins, which are less efficient protectors, the thioredoxin from mammalian mitochondria (TRX2) is characterized by the most unequal distribution of the charged residues on the molecule surface, resulting in the highest dipole moment among the thioredoxins. This feature is indicative of the role of long-range electrostatic interactions in biological function, where the dipole-mediated preorientation of the interacting proteins increases the probability of productive complex formation (93). Importance of the long-range interactions for the biological function of mitochondrial thioredoxin, first revealed on thioredoxin interaction with 2-oxo acid dehydrogenase complexes (33), has been confirmed and extended by an independent study of both thioredoxins and glutaredoxins (21).
The interaction of mitochondrial 2-oxo acid dehydrogenase complexes with mitochondrial thioredoxin has been also shown in several high-throughput proteomic studies in plants (16, 211). Remarkably, similar proteomic studies also revealed other interactions between lipoyl-bearing proteins and thioredoxins. For instance, thioredoxin h3 interacts with the enzymes of glycine decarboxylase system. They include the lipoylated protein H analogous to E2 components of 2-oxo acid dehydrogenase complexes, and dihydrolipoamide dehydrogenase (123, 124). At the same time, no other components of 2-oxo acid dehydrogenase complexes have been detected as targets of thioredoxin h3 in these studies, in good accordance with the absence of efficient interaction between mammalian 2-oxo acid dehydrogenase complexes and thioredoxin h (33). In a proteomic study of several mammalian thioredoxin-like proteins, beta-subunit of pyruvate dehydrogenase is detected among targets of the thioredoxin-like protein Rdx-12, which is anchored to the plasma membrane of cancer cells (146). In accordance with different cellular compartmentation, mitochondrial complexes are not found among nitrosylated protein targets of cytosolic thioredoxin, although components of 2-oxo acid dehydrogenase complexes are known to be nitrosylated (90, 185). Thus, the functional in vitro studies and high-throughput proteomics provide complementary data on the evolutionarily conserved and specific interaction between mitochondrial thioredoxin and 2-oxo acid dehydrogenase complexes, pointing to the biological significance of interaction.
B. Complex-bound dihydrolipoyl residues reduce thioredoxin
Specific involvement of dihydrolipoate into the thioredoxin-dependent reactions is known from in vitro studies where dihydrolipoate displays the highest efficiency among other dithiols in reducing thioredoxin (91). The complex-bound dihydrolipoyl residues also participate in the thioredoxin reduction (29, 34). The mechanism of reaction may involve the two-step 1e− reduction of oxidized thioredoxin by the thiyl radicals of dihydrolipoate, as shown in the right part of Figure 11. Existence of an electron flow from 2-oxo acids to thioredoxin, which is alternative to NAD+ reduction catalyzed by the complexes (Fig. 13), is favored by certain findings in mammalian cells. For instance, a lower ATP level is inherent in cells overexpressing mitochondrial TRX2, but not cytosolic TRX1 (215). Because mitochondrial oxidative phosphorylation uses oxidation of NADH produced by 2-oxo acid dehydrogenase complexes, increased electron flow from the complex-bound dihydrolipoyl residues to overexpressed TRX2 (Figs. 11 and 13) may reduce the NADH supply to respiratory chain. The corresponding decrease in oxidative phosphorylation is thus in good accord with the finding of decreased ATP levels in cells overexpressing TRX2 (215).
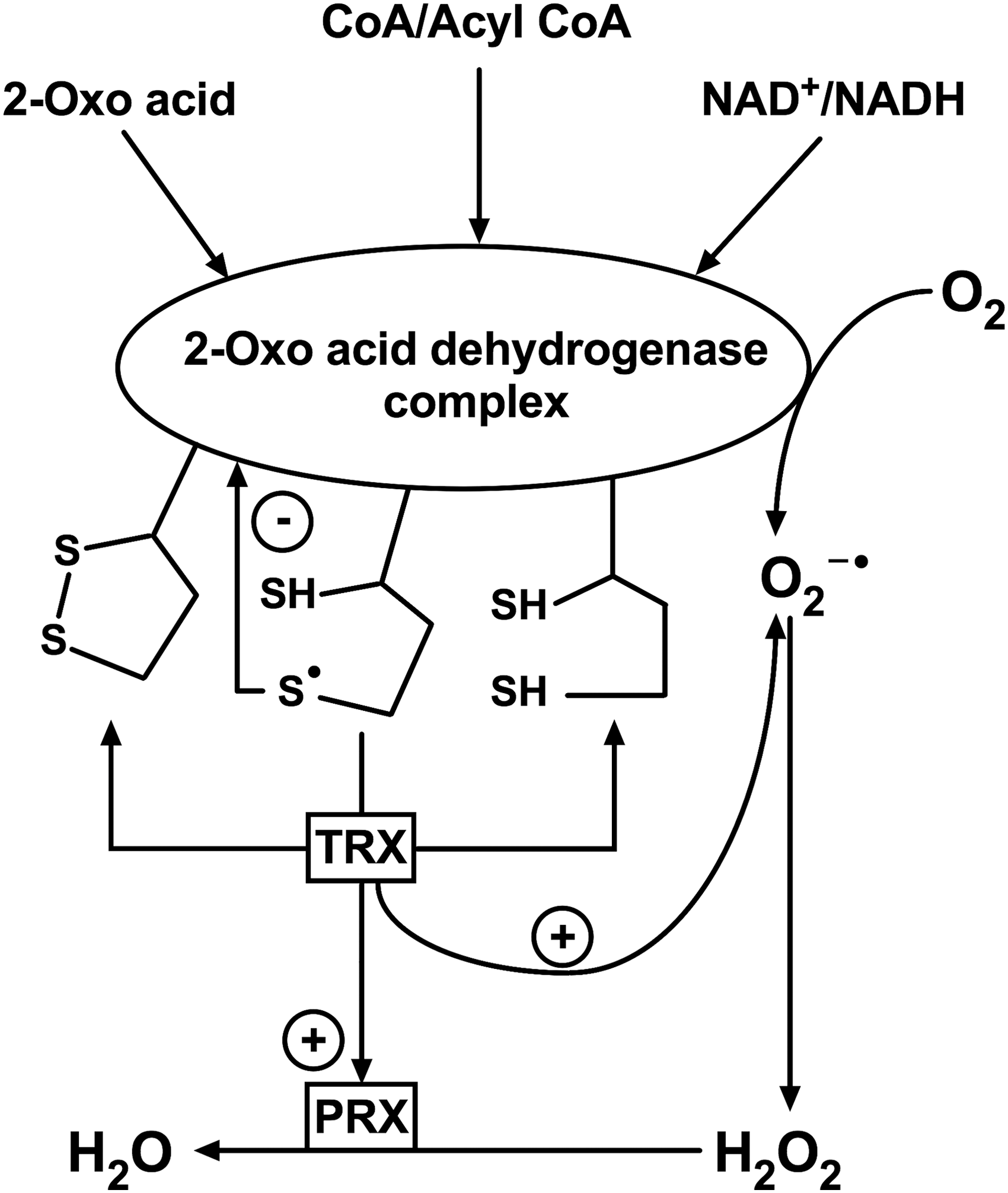
Furthermore, it is known that in yeasts, mitochondrial thioredoxin (yTRX3) remains reduced when mitochondrial thioredoxin reductase (TRR2) is deleted, in contrast to oxidation of cytosolic thioredoxins on the deletion of cytosolic thioredoxin reductase (192). In mammalian cells, inhibition of TRR2 did not change the reduction level of TRX2 in the resting state either (179). Pointing to alternative sources of the mitochondrial, but not cytosolic, pathways of reduction of the corresponding thioredoxins, these findings corroborate the reduction of TRX2 by the dihydrolipoyl residues of mitochondrial 2-oxo acid dehydrogenase complexes, discussed above. When both TRR2 and glutathione reductase (GLR1) are deleted in yeasts, the redox state of yTRX3 is shifted to a more oxidized state, resulting in slow growth and a sixfold increase in apoptosis (81). Because glutathione disulfide inactivates 2-oxo acid dehydrogenase complexes (27, 152), the glutathione dependence of the mitochondrial thioredoxin reduction may be mediated by the dihydrolipoyl residues, which become redox inactive in the complexes inactivated by the glutathione disulfide. Moreover, apoptosis induced on partial oxidation of yTRX3 in the double mutant with deletion of yeast TRR2 and GLR1 is alleviated in the triple mutant with additional deletion of yTRX3, suggesting a gain of function rather than the impaired redox cycling of yTRX3 to promote apoptosis (81). From the viewpoint of competing electron flows from the dihydrolipoyl intermediate to thioredoxin or NADH (Figs. 11 and 13), the gained function may represent the reduction of yTRX3 by the dihydrolipoyl intermediate at the expense of NADH production. In the double mutant of yeast TRR2 and GLR1, formation of this sole source for the mitochondrial thioredoxin reduction is impaired by glutathione disulfide. The ensuing decrease in the physiological NADH producing reaction of the 2-oxo acid dehydrogenase complexes may thus become critical under these conditions. As impairment of the physiological reactions catalyzed by 2-oxo acid dehydrogenase complexes is known to induce apoptosis (56), the dysregulated electron flows from the dihydrolipoate intermediate may underlie apoptosis induction in the yeast double mutant, not observed in the triple mutant where the yTRX3 cannot impair NADH production by the dihydrolipoyl intermediate (81).
The competitive electron flows from the dihydrolipoate intermediate to thioredoxin, O2, and NADH provide plausible explanations to a number of results originating from detailed studies on the thioredoxin and glutathione system of mammalian mitochondria. In particular, some of the studies have indicated that the effect of mitochondrial thioredoxin reductase inhibition or glutathione depletion on the reduction level of mitochondrial thioredoxin and hydrogen peroxide emission depends on the respiratory state and species-specific features of mitochondria (8, 179). Moreover, modeling studies indicate that the ROS emission during the FET in the functionally active mitochondria under oxidative phosphorylation conditions (state 3) is low because of the highest ROS scavenging under these conditions. Correction for the ROS scavenging results in an order of magnitude higher ROS production in the FET under oxidative phosphorylation conditions (8). Production of ROS by mitochondrial electron transport chain is unlikely under these conditions (203, 206). In contrast, significant emission of ROS as side products of the ADP-activated 2-oxo acid dehydrogenase complexes together with scavenging of such ROS through the mitochondrial thioredoxin/peroxiredoxin system (Figs. 11 and 13) is in good accordance with the high production and efficient utilization of ROS during oxidative phosphorylation in state 3. Moreover, inhibition of mitochondrial thioredoxin reductase in mammalian cells does not significantly affect the level of reduction of mitochondrial thioredoxin in state 3, when 2-oxo acid dehydrogenase complexes are highly active. In contrast, in state 4, where the complexes are not ADP activated, the contribution of TRR2 to the reduction of TRX2 is increased (8, 179).
Thus, in vitro data on the redox exchange between mitochondrial thioredoxin and the complex-bound dihydrolipoyl residues are supported by the consequences of genetic or pharmacological manipulations affecting cellular redox state and viability. As shown in the next sections, the interaction of the dihydrolipoyl residues with thioredoxin may transmit the signal on perturbed homeostasis from the 2-oxo acid dehydrogenase complexes to thiol-based peroxidases (peroxiredoxins) and a thioredoxin-interacting protein regulating mitochondrial function in mammals (164, 212).
C. Complex-bound dihydrolipoyl residues reduce glutaredoxin
In addition to the well-known reduction of cellular disulfides at the expense of NADPH, glutathione disulfide may also be reduced at the expense of dihydrolipoamide, which is catalyzed by glutaredoxins in vitro (157). The NADH-dependent reduction of mitochondrial disulfides has been also attributed to lipoamide dehydrogenase component of 2-oxo acid dehydrogenase complexes (175). In vivo, redox reactions between glutaredoxin and dihydrolipoyl intermediate of 2-oxo acid dehydrogenase complexes have been shown on studies of nonviable triple mutant of E. coli lacking thioredoxin reductase, glutaredoxin reductase, and peroxidase AphC. Such cells become viable on additional mutation in the E3 component of 2-oxo acid dehydrogenase complexes (71). Study of the underlying molecular mechanisms has revealed that the decreased content of E3 in the complexes allowed the reducing equivalents from oxidative decarboxylation of 2-oxo acids to be transferred from the dihydrolipoyl residues of the 2-oxo acid dehydrogenase complexes to glutaredoxin. In the mutant lacking thioredoxin reductase, glutaredoxin reductase, and peroxidase AphC, this reaction enables the glutaredoxin-dependent reduction of ribonucleotide reductase, essential for cellular viability. In mammals, the glutaredoxin interaction with dihydrolipoyl intermediate may be involved in iron homeostasis and nitric oxide-dependent signaling (75, 110, 113), considered in detail in Section VI. Such interaction may be highly relevant for pathological conditions, including neurodegenerative diseases and aging (75, 110, 155).
Thus, redox reactions of the dihydrolipoate intermediate generated by 2-oxo acid dehydrogenase complexes with thioredoxins and glutaredoxins have been shown. These reactions manifest existence of alternative electron flows from 2-oxo acids to either NAD+ or the thiol/disulfide oxidoreductases in vivo. The partition of the reducing equivalents from dihydrolipoyl residues to the acceptors depends on physiological conditions, such as the functional state of mitochondria and expression of different protein components of cellular redox system. As a result, disproportional availability of the substrates of the complexes is transformed into their ROS and thiyl radical productivity, coupled to the thioredoxin and glutaredoxin systems (36, 71), the known participants of redox-dependent regulation (8, 118).
D. Interaction between the lipoylated proteins and thiol-based peroxidases
Based on the thioredoxin reduction by the complex-bound dihydrolipoyl residues (29, 34) and thioredoxin-dependent regulation of ROS generation by 2-oxo acid dehydrogenase complexes (48), participation of thioredoxin-dependent mitochondrial peroxidases (see PRX in Fig. 13) (104) in scavenging H2O2 produced in the side reactions by 2-oxo acid dehydrogenase complexes has been suggested (36). The hypothesis has been since then confirmed in multiple independent studies considered below. Peroxiredoxins defend organisms against H2O2, organic peroxides, and peroxynitrite, participating in the redox- and nitric oxide-dependent signaling (52, 156). Physiological significance of the abundant cytosolic peroxiredoxin 2 is extensively characterized, allowing for modeling of protein participation in the H2O2 scavenging and redox signaling (19). Other mammalian peroxiredoxins may be less abundant, and molecular mechanisms of their physiological actions require further studies. Several peroxiredoxins localized to mitochondria differ in their substrate specificity and catalytic action. 2-Cys peroxiredoxin 3 (alternative name SP22) and 1-Cys peroxiredoxin 6 (PRX1 in yeasts) (81) scavenge mitochondrial hydrogen peroxide. Peroxiredoxin 5 exhibits a higher activity to organic peroxides and peroxynitrite (104). Remarkably, peroxiredoxins 5 and 6 have been identified among the proteins that bind on the thiamine-modified sorbent (144). In view of the established connections between the protein-bound lipoyl residues and redoxin family of proteins (18), the finding suggests a rather strong interaction of these peroxiredoxins with the components of the thiamine-dependent 2-oxo acid dehydrogenase complexes, identified among the thiamine-binding proteins eluted from the sorbent.
A number of bacterial lipoylated proteins have been discovered to cooperate with thiol-dependent peroxidases. In Mycobacterium, a thioredoxin-like protein AphD, which provides reducing equivalents to the thiol-based peroxidase AphC, is reduced by the complex-bound dihydrolipoyl residues arising either in the backward reaction of NADH oxidation (26) or in the forward reaction of 2-oxo acid dehydrogenase complexes (122). Interaction of AphC with thioredoxin C is also shown in Mycobacterium (205). Apart from the E2 components of 2-oxo acid dehydrogenase complexes, some other lipoylated proteins, usually related to 2-oxo acid dehydrogenase complexes, are involved in the peroxiredoxin functions as well. For instance, the organic hydroperoxide resistance protein (Ohr)/OsmC family of proteins, identified in both pro- and eukaryotes, include Ohr and osmotically inducible proteins (OsmCs) with the thiol-dependent peroxidase activities (138). The peroxidase activity of the Ohr and OsmC proteins is supported by dihydrolipoyl residues of E2 components of 2-oxo acid dehydrogenase complexes, and unusual dihydrolipoamide dehydrogenase comprising the lipoylated domain (59). Indeed, the lipoyl-bearing domains may be located to components of 2-oxo acid dehydrogenase complexes other than E2, in a number of bacteria, including such pathogens as Mycobacterium tuberculosis and Neisseria meningitidis. As mentioned above, one such protein, the dihydrolipoamide dehydrogenase of Xylella fastidiosa, encoded by the LpdA gene, supports the Ohr and OsmC peroxidase activities (59). This finding highlights potential participation of such unusually located lipoyl-bearing domains or lipoyl-bearing components found outside of their 2-oxo acid dehydrogenase complexes (59, 134, 176) in the peroxidase reactions. Evolutionary relationship of such proteins to enzymatic components of 2-oxo acid dehydrogenase complexes underlines biological significance of the peroxidase function associated with the lipoyl residues of the complexes. The peroxidases may use reducing equivalents from the protein-bound dihydrolipoyl residues either directly, as observed with Ohr/OsmC peroxidases, or mediated by thioredoxin (thioredoxin 3) or a thioredoxin-like protein (AphD), as occurs with AphC peroxidase. In view of the known nitrosylation of mammalian 2-oxo acid dehydrogenase complexes (90, 185), it is remarkable that all such peroxidases, including mammalian mitochondrial peroxiredoxin 5, detoxify not only H2O2 but also peroxynitrite (2, 26, 122).
VI. Role of Side Reactions of 2-Oxo Acid Dehydrogenase Complexes in Signaling by the Reactive Oxygen and Nitrogen Species
Discrimination between the mitochondrial ROS sources in vivo remains a general challenge. As discussed in detail elsewhere (24, 39, 206), even specific targeting of a certain ROS source, either by pharmacological means or genetic manipulations, may perturb distribution of substrate fluxes in a metabolic network. In the resulting new steady state, other sources of ROS may be activated instead of the inhibited one. An example has been discussed in previous sections, regarding inhibition of neuronal 2-oxoglutarate dehydrogenase complex, which induces a primary decrease in ROS followed by their secondary increase (49). In another metabolic network, such as that of a neuroblastoma cell line, the opposite changes in the ROS production due to inhibition of 2-oxoglutarate dehydrogenase complex are observed, with the primary increase in ROS followed by decrease (32).
A rather general approach to estimate the in vivo significance of molecular mechanisms and quantifications established in vitro is exemplified below by incorporating the in vitro data in interpretation of the results obtained in vivo. When a consistent mechanistic view on interactions between relevant components of a biosystem is achieved, and corresponding quantitative parameters are determined, mathematical modeling may be used. Comparing the model behavior on variation of certain parameters with experimental observations allows one to further refine our understanding of the biosystem under consideration (8, 19).
A. Common and substrate-specific features of signaling by ROS produced by 2-oxo acid dehydrogenase complexes
Interplay of the ROS-producing activities of different 2-oxo acid dehydrogenase complexes with cellular signaling mechanisms demonstrates many common features. The commonalities are defined by the common substrates CoA and NAD+, catalytic role of dihydrolipoyl residues, and NADH production. The most studied condition stimulating ROS production by the complexes is an increase in NADH/NAD+ ratio, which is observed in vivo under pathological situations, such as reductive stress in hypoxia. However, the link between NAD+ dependence of ROS production by the complexes and the NAD+-dependent signaling reactions (107, 150), exposed in Figure 14, is not sufficiently appreciated. The latter reactions involve NAD+-dependent protein deacetylase sirtuins and the DNA damage response protein poly (ADP-ribose) polymerase (PARP) (22, 127), demonstrating a cross talk in signaling pathways (70). In view of the different properties of mitochondrial sirtuins (154), either all the complexes may be affected or the links to sirtuins may be substrate specific. The former refers to the lipoamidase activity of sirtuin 5, whereas the latter involves sirtuin-dependent deacylation of proteins modified by the substrate-specific acyl-CoA products of the complexes. That is, mitochondrial sirtuin 3 catalyzes deacetylation of proteins modified by acetyl-CoA, whereas mitochondrial sirtuin 5 counteracts the protein succinylation by succinyl-CoA. Stimulation of the 2-oxo acid-dependent ROS generation on depletion of cellular NAD+ in the PARP reaction (Fig. 14) may underlie molecular mechanisms of cellular decision on life or death following DNA damage.
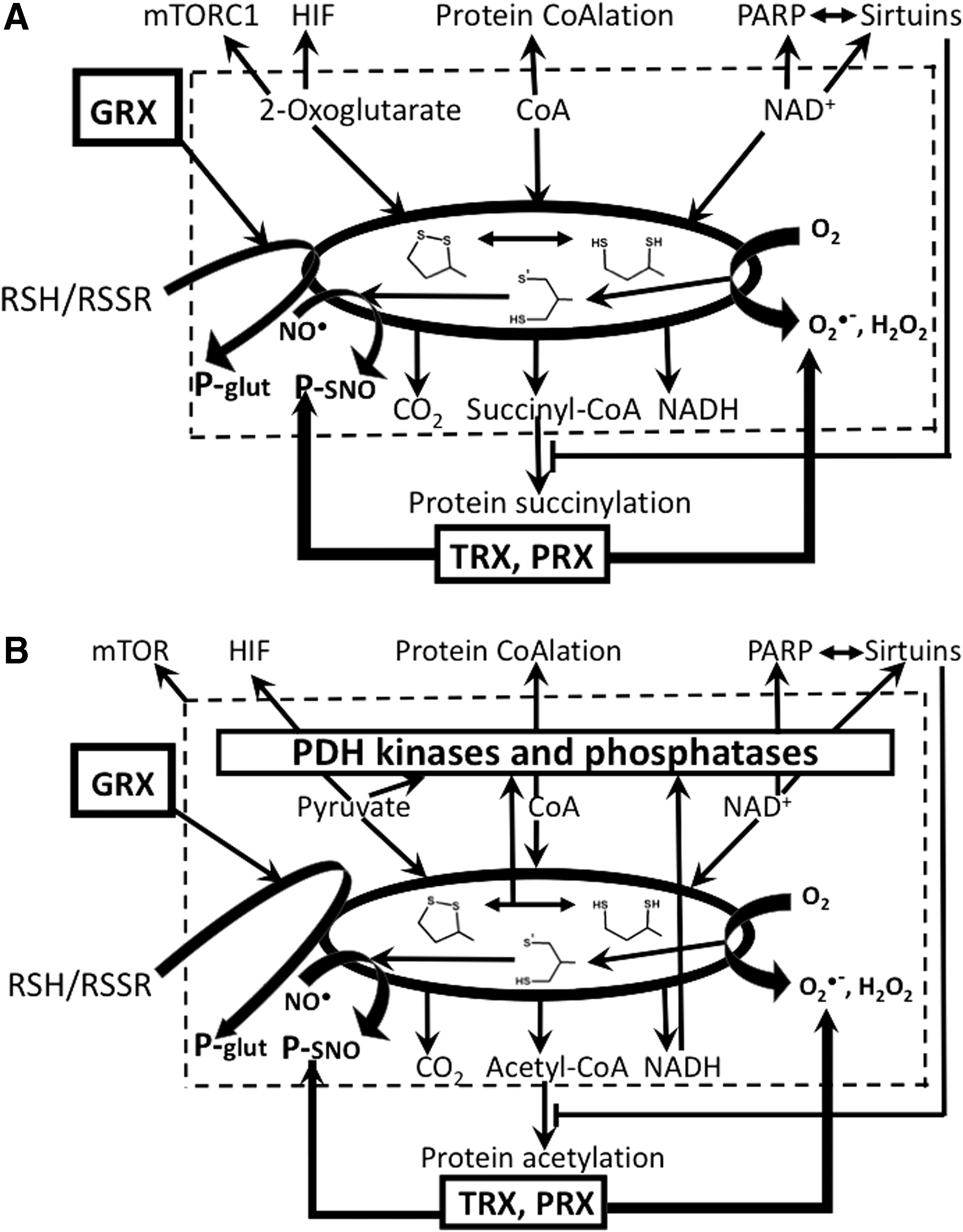
Specific features of the ROS-dependent signaling by different 2-oxo acid dehydrogenase complexes rely on the action of the 2-oxo acid substrates of the complexes in signaling pathways. It is, however, remarkable, that a common pathway is usually affected even if each of the 2-oxo acids may act through specific mechanisms. For instance, both pyruvate and 2-oxoglutarate are known players in the hypoxia-inducible factor (HIF)-dependent oxygen sensing (Fig. 14). However, 2-oxoglutarate is a cosubstrate of 2-oxoglutarate-dependent oxygenases increasing HIF decay by protein hydroxylation (158). In contrast, pyruvate participates in the feed-forward activation of HIF signaling through HIF stabilization, which cannot be counteracted by 2-oxoglutarate (117). HIF signaling is also known to be specifically linked to the mitochondrial thioredoxin system. Induction of HIF by hypoxia is increased by overexpression of cytosolic TRX1, but decreased by overexpression of mitochondrial TRX2 (215). The finding agrees with a higher flux of pyruvate into the dihydrolipoate-dependent reduction of overexpressed TRX2 (Fig. 13), compared to the cells with normal TRX2 content. As a result, the accumulation of pyruvate that would be sufficient to stabilize HIF in cells with normal level of TRX2 may be delayed in the cells overexpressing TRX2. The resulting delay in the pyruvate-induced HIF stabilization may be observed as decreased HIF induction in the experiment at a fixed time point (215).
Another signaling system that may be affected through the ROS produced by all the 2-oxo acid dehydrogenase complexes involves mammalian target of rapamycin (mTOR), whose function is known to be linked to mitochondrial ROS production and apoptosis (60, 143, 213). The interplay between the 2-oxo acid dehydrogenase complexes and mTOR has been shown in independent studies under different experimental settings (114, 210, 213). The 2-oxoglutarate-dependent regulation has been involved in mTORC1-dependent pathway (Fig. 14A). Interestingly, effects of thiamine depletion on metabolic markers in cancer cell lines were reversed by the mTOR effector rapamycin despite the fact that thiamine deficiency persisted (114). This finding suggests that mTOR participates in a homeostatic adjustment of metabolic network where 2-oxo acid dehydrogenase complexes are functionally disabled. Probably such adjustment occurs through mTOR-dependent activation of autophagy (60) and is relevant to the mTOR significance in cancer, where function of the pyruvate and 2-oxoglutarate dehydrogenases may vary in a cancer cell-specific manner (32, 38). Under thiamine depletion conditions, also the link between the branched chain 2-oxo acid dehydrogenase complex and mTORC1 signaling has been shown to depend on the cancer cell line (114).
Thus, the substrate-specific contributions of 2-oxo acid dehydrogenase complexes to mitochondrial ROS production may be linked to the signaling mechanisms supported by HIF and mTOR. Fine tuning of these signaling pathways may depend on particular metabolic positions of the 2-oxo acid dehydrogenase complexes and supported by specific molecular mechanisms of action of the 2-oxo substrates in the HIF and mTOR signaling.
B. Role of phosphorylation of the pyruvate and branched chain 2-oxo acid dehydrogenases in regulation of ROS production by 2-oxo acid dehydrogenase complexes
Regarding the in vivo role of ROS produced by the complexes at the expense of dihydrolipoyl intermediate, and the associated regulation by the catalysis-dependent inactivation, there is an important difference between the mammalian 2-oxoglutarate dehydrogenase complex, on one hand, and pyruvate and branched chain 2-oxo acid dehydrogenase complexes, on the other hand. As discussed in Section II, in eukaryotic pyruvate dehydrogenase complexes, over-reduction of the complex-bound dihydrolipoyl residues switches on the pyruvate dehydrogenase phosphorylation by pyruvate dehydrogenase-specific kinases. Branched chain 2-oxo acid dehydrogenase complex is also subjected to phosphorylation/dephosphorylation-dependent regulation, which is, however, not known for mammalian 2-oxoglutarate dehydrogenase complex. Thus, the different response of the eukaryotic complexes is expected under accumulation of dihydrolipoyl intermediate in vivo. The electron flow from pyruvate to ROS in the pyruvate dehydrogenase complex may be blocked by its kinases (Fig. 14B), whereas the 2-oxoglutarate-dependent ROS production by 2-oxoglutarate dehydrogenase complex may be disabled by the mechanism involving the complex-bound thiyl radicals (Fig. 13). The pyruvate dehydrogenase phosphorylation is counteracted by negative effectors of the kinases and the phosphatases of pyruvate dehydrogenase, while the thioredoxin protection from the thiyl radical-dependent inactivation may be a major factor regulating the dihydrolipoyl intermediate accumulation in nonphosphorylatable 2-oxo acid dehydrogenase complexes, particularly in the mammalian 2-oxoglutarate dehydrogenase complex. The different regulation may contribute to lower ROS-producing capacities of the pyruvate and branched chain 2-oxo acid dehydrogenase complexes, compared with 2-oxoglutarate dehydrogenase complex, which is observed in mitochondrial studies (160).
Compared with the dehydrogenase complexes oxidizing pyruvate and 2-oxoglutarate, the branched chain 2-oxo acid dehydrogenase complex is characterized by a relatively low maximal capacity for ROS production, similar to that of a newly described mammalian dehydrogenase complex specific to 2-oxoadipate (24, 39, 206). Hence, the dehydrogenase complexes transforming branched chain 2-oxo acids or 2-oxoadipate appear to be less significant producers of ROS under physiological conditions. Their general significance as ROS producers is also limited by the tissue-specific expression and flux through these complexes. However, similarities in the molecular mechanisms of catalysis, inherent in all the complexes, do not exclude that in certain tissues and/or circumstances also these complexes of lower ROS-producing capacities could have a role in ROS-dependent signaling and/or production. For instance, reciprocal regulation of the kinases of the pyruvate and branched chain 2-oxo acid dehydrogenase complexes by pyruvate and branched chain 2-oxo acids (11, 28) may decrease ROS produced by one of the complexes, simultaneously increasing those produced by the other one. That is, high concentrations of branched chain 2-oxo acids may activate the kinase of the cognate complex, simultaneously inactivating the kinase of the pyruvate dehydrogenase complex. As a result of decreased phosphorylation of E1p, production of ROS by the pyruvate dehydrogenase complex may increase under elevated concentrations of branched chain 2-oxo acids. This reciprocal regulation may contribute to increased ROS levels in maple syrup urine disease on mutations of the branched chain 2-oxo acid complex and other pathologies associated with increased levels of branched chain 2-oxo acids or their cognate amino acids (213).
Regulation of phosphatases, which activate the corresponding complexes by dephosphorylation (28), is also important for in vivo ROS production and signaling by the complexes. In particular, emission of ROS from the pyruvate dehydrogenase complex in the forward direction of the reaction may be increased or decreased, correspondingly, by allosteric activation of the pyruvate dehydrogenase phosphatase by Ca2+ or the enzyme inhibition by the reaction product NADH.
Thus, the mechanism of production of ROS and thiyl radicals of the dihydrolipoyl intermediate is common for all the complexes, but the control of these side reactions under physiological conditions may be different due to specific regulation mechanisms of the complexes transforming different 2-oxo substrates and their tissue-specific significance (Fig. 14). The substrate-specific coupling between the side reactions and signaling systems is in good accord with particular metabolic positions of the complexes, irreversibly oxidizing important branch point metabolites of regulatory significance, such as 2-oxoglutarate, pyruvate, and branched chain 2-oxo acids.
C. Interplay between the ROS production by the complexes and cellular thiol/disulfide oxidoreductases
Accumulation of the dihydrolipoyl intermediate in 2-oxo acid dehydrogenase complexes, which induces their ROS production, may be counteracted by the dihydrolipoyl-dependent reduction of thioredoxin and thioredoxin-like proteins, participating in the peroxidase cycle of peroxiredoxins (26, 29, 34, 122). As a result, the supramolecular system of the 2-oxo acid dehydrogenase complexes, thioredoxins, and peroxiredoxins possess the scavenging functions regarding ROS and thiyl radicals produced in the side reaction of the complexes (Figs. 13 and 14). While most of the data on such supramolecular system are obtained in microorganisms, certain data suggest this mechanism to be operative also in mammals. For instance, partial inactivation of the 2-oxoglutarate dehydrogenase complex, which occurs in the rat brain cortex after acute hypoxia (11, 80), mimics the thiyl radical-dependent inactivation of isolated 2-oxoglutarate dehydrogenase complex, observed under anaerobic conditions in the presence of 2-oxoglutarate and CoA (Fig. 8) (48). The hypoxic conditions also increase the expression of mitochondrial thioredoxin in rat brain (165, 181). This finding suggests a compensatory response that may protect 2-oxo acid dehydrogenase complexes from the thiyl radical-induced inactivation at high NADH/NAD+ induced by hypoxia. Under hypoxic conditions, the dihydrolipoyl-dependent reduction of thioredoxin instead of the lacking NAD+ may simultaneously contribute to improvement of the thiol/disulfide status. As depicted in Figures 11, 13, and 14, the complex-bound thiyl radicals, which are produced by the complexes at physiological pH, may catalyze the thiol/disulfide exchange reactions between the complex-bound and medium thiols and disulfides (29, 34, 105, 175), including the known processes of glutaredoxin-controlled glutathionylation (9, 27, 152, 172).
Thiyl radicals are also highly reactive to nitric oxide (98). The corresponding nitroso thiols produced in the reaction of thiyl radicals of the complex-bound dihydrolipoyl residues and nitric oxide (Fig. 15) may be involved in the nitrosylation of the complex components, known to occur in vivo (90, 185). Given the interaction of thioredoxin with 2-oxo acid dehydrogenase complexes (Figs. 11, 13, and 14), the transnitrosylating function of thioredoxin (208), depicted in Figure 15, may be used to use the nitrosylated dihydrolipoyl residues for nitrosylation of other components of the complexes and/or their interaction partners (P-SNO, Fig. 14). Thus, in vivo, the supramolecular system of the 2-oxo acid dehydrogenase complexes, thioredoxins, and thioredoxin-dependent peroxidases may interact with the nitric oxide sources. In this case, the coproduction of the complex-bound thiyl radicals and superoxide anion radicals by 2-oxo acid dehydrogenase complexes may also lead to the local peroxynitrite production in the reaction of nitric oxide and superoxide anion radicals produced by the complexes (Fig. 15). Cooperative participation of the complex-bound dihydrolipoyl residues and thiol-based peroxidases in defense against nitroxidative stress is known in Mycobacterium (96). Thus, simultaneous production of ROS and thiyl radicals by 2-oxo acid dehydrogenase complexes may couple the signals by ROS and RNS in vivo (Fig. 15).
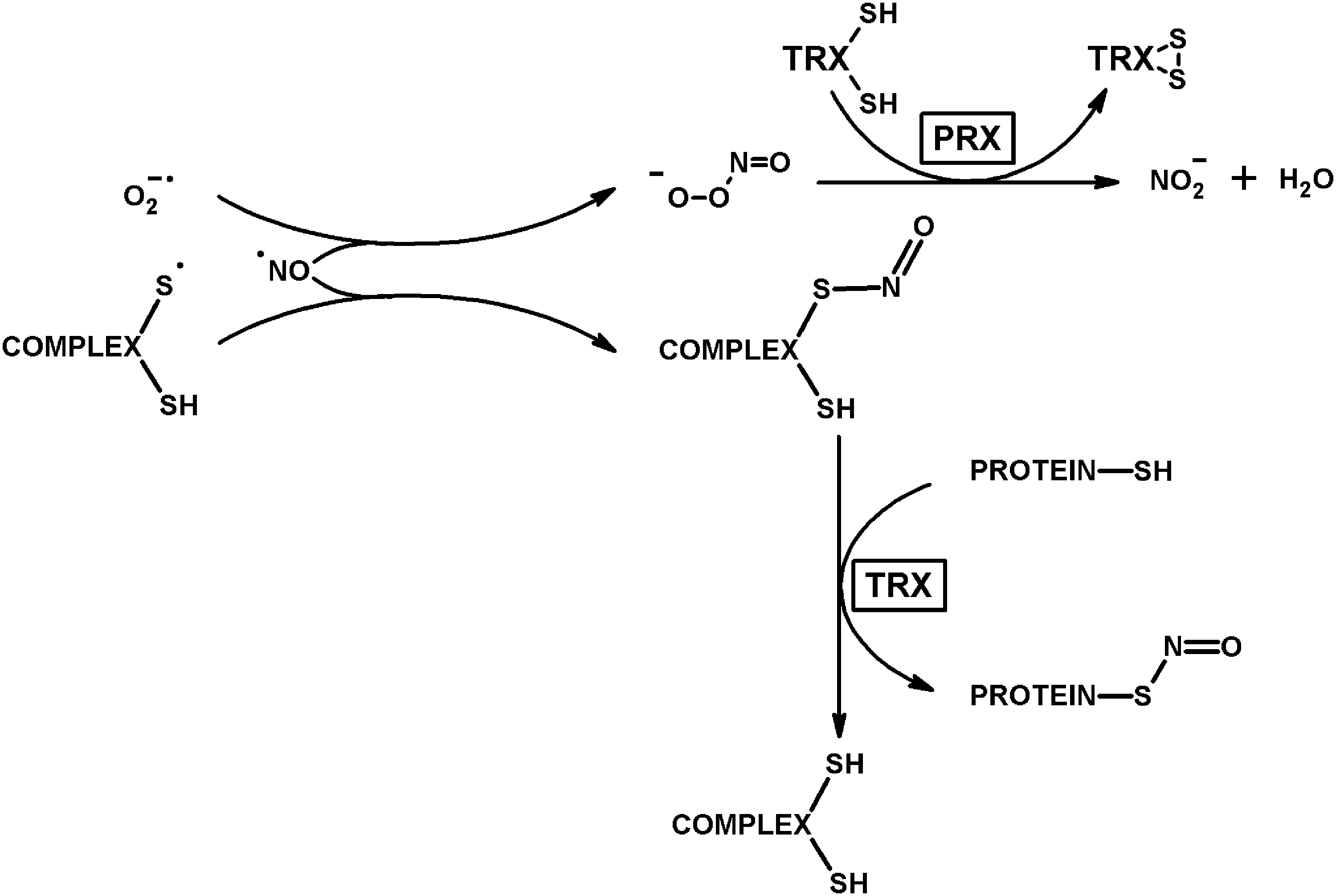
As a result, the reviewed data on ROS generating activity of 2-oxo acid dehydrogenase complexes, the regulation by intrinsic thiyl radicals, and the dihydrolipoyl-residue-mediated coupling to the thiol/disulfide-dependent antioxidant and antinitrosative functions of peroxiredoxins (Figs. 13 –15) highlight involvement of the side reactions of these heavily regulated systems in the ROS- and RNS-dependent signaling. Complexity of biological signaling, including its interactive nature and specific compartmentation, interferes with satisfactory predictions of the system behavior under so-called oxidative stress conditions. Although depletion of cellular reducing power may be easily achieved in model systems, the events occurring in such models, exposing cells to major damage, may obscure true processes in vivo, which include compensatory responses and redistribution of the substrate fluxes. Accordingly, there are many factors to be considered regarding interaction between oxidative stress and ROS generation by 2-oxo acid dehydrogenase complexes. Different activities and modifications of the complexes as well as their interactors may be involved in vivo, dependent on specific stress inductor. Effects of post-translational modifications on the catalytic properties of 2-oxo acid dehydrogenase complexes are multiple and strongly depend on conditions (45). In general, the statements on increased ROS production by the complexes under oxidative stress conditions often overlook a crucial issue that the availability of the ROS-supporting substrates CoA (in the forward direction) or NADH (in the backward direction) would be impaired under these conditions. The same is applied to the availability of molecular oxygen under hypoxia, where quinones, including flavins, may well be of damaging potential instead of ROS (Figs. 6 and 8).
D. Potential involvement of the superoxide anion radical produced by 2-oxo acid dehydrogenase complexes into iron homeostasis
Local production of superoxide anion radical by the 2-oxoglutarate dehydrogenase complex in the proximity to mitochondrial aconitase may be involved in a signaling mechanism based on iron mobilization from iron/sulfur clusters, similar to that established for cytosolic aconitase. This bifunctional protein contains iron/sulfur clusters [4Fe-4S] that not only catalyze the aconitase reaction but also regulate iron metabolism by binding to untranslated regions of specific mRNAs. Superoxide anion radical induces the aconitase transformation in the [3Fe-4S] form, accompanied by the loss of two activities. As a result, enhanced transcriptional regulation and diminished translational repression of the iron storage protein ferritin are observed, which may counterbalance the superoxide-induced effect on iron mobilization (139). Mitochondrial aconitase is smaller than the cytosolic isoenzyme, but the domain organization of both proteins is similar. Remarkably, the iron response element of mitochondrial aconitase upregulates the enzyme by increasing the iron levels through the process linked to the antioxidant response element pathway and HIF (96, 198).
The superoxide-dependent mobilization of iron from iron/sulfur proteins may also provide for metabolic adjustments involving mitochondrial 2-oxo acid dehydrogenase complexes and glutaredoxins 2 and 5 (Fig. 16). These mitochondrial glutaredoxins are known to be involved in iron metabolism and biogenesis of iron/sulfur clusters (209). In particular, mitochondrial glutaredoxin 5 is involved in protein lipoylation, acting in the pathway that provides an iron/sulfur cluster to lipoate synthase (14). Homodimerization through [2Fe-2S] clusters deprives these glutaredoxins from their glutathione transferase activity (110, 113), which appears after the loss of the [2Fe-2S] clusters. In this regard, metabolic imbalance, resulting in increased superoxide anion production from 2-oxoglutarate dehydrogenase complex at the expense of 2-oxoglutarate, is especially interesting in view of the findings that the antioxidant function of [2Fe-2S] glutaredoxins is activated by oxidative stress. On the contrary, the data obtained in mammalian systems suggest that cytosolic glutathione may be exchanged for mitochondrial 2-oxoglutarate through mitochondrial carriers of 2-oxoglutarate or dicarboxylates (51). As a result, the superoxide anion radical generated at the expense of 2-oxoglutarate may simultaneously activate antioxidant functions of mitochondrial glutaredoxins 2 and 5 and increase glutathione through 2-oxoglutarate-dependent transport (Fig. 16). Additional mechanisms of the metabolic adjustment may use regulation of the complex through its lipoylation, which requires the iron/sulfur clusters (Fig. 16).
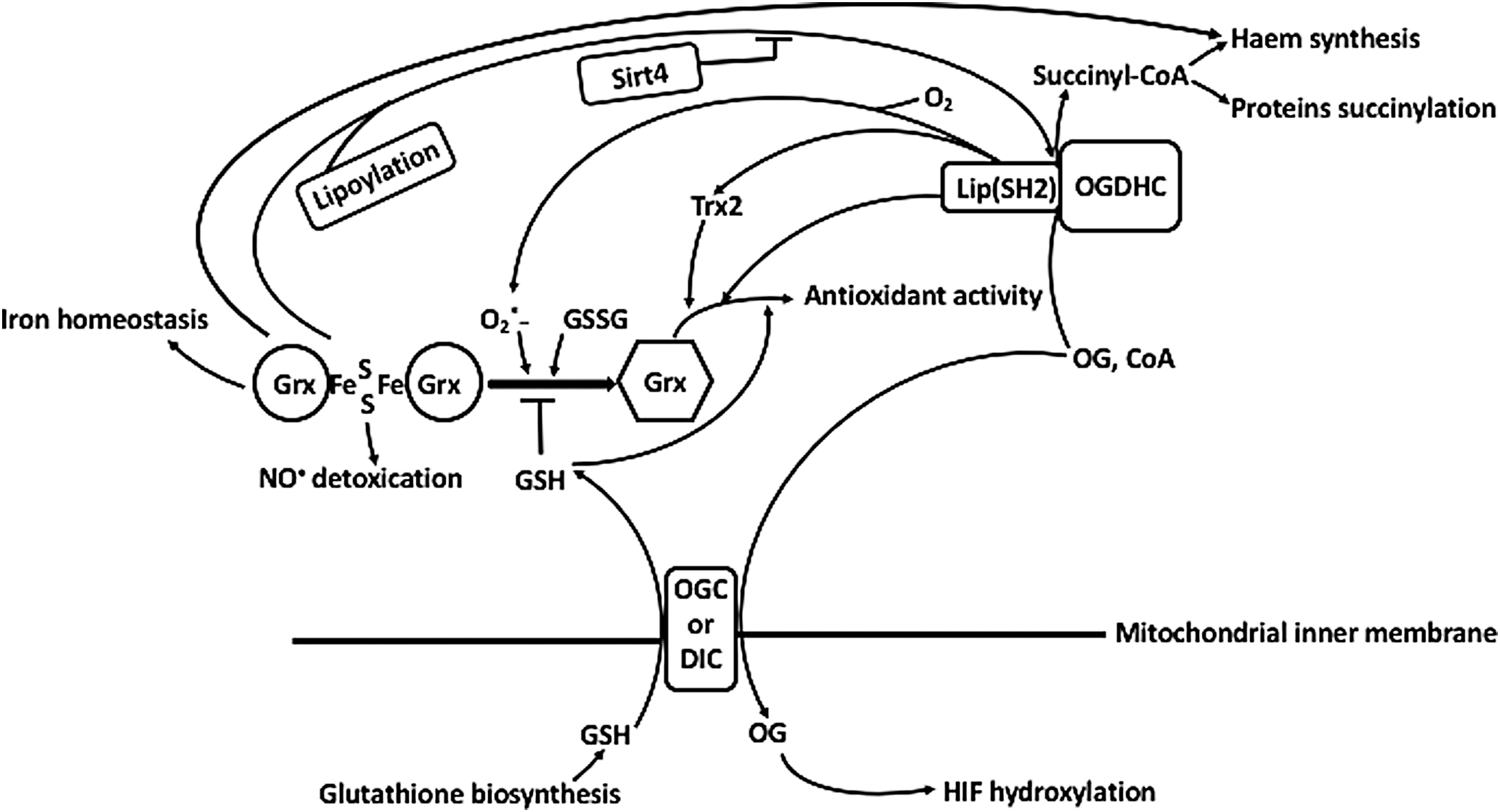
Thus, generation of superoxide anion radical by 2-oxoglutarate dehydrogenase complex may participate in signaling by mitochondrial aconitase and glutaredoxins 2 and 5 involving the iron/sulfur clusters of these proteins.
E. Concluding remarks on potential physiological roles of ROS and thiyl radicals produced by 2-oxo acid dehydrogenase complexes
Overall, the ratio between the physiological (to NAD+) and alternative (to oxygen, thioredoxins, thioredoxin-like proteins, peroxiredoxins, glutaredoxins, and low molecular mass disulfides) electron flows, catalyzed by 2-oxo acid dehydrogenase complexes (Figs. 13 –16), may be considered an indicator of homeostasis, used to switch on additional defense mechanisms when the system goes out of balance. The switches are exemplified by the systems using cooperation of peroxiredoxins and lipoyl-dependent proteins (174), or thiol-dependent regulation of gene transcription (25, 188). In these cases, when the reducing power of a cell is insufficient to compensate for the side reactions of ROS production, the protein thiols undergo post-translational modifications, concomitantly changing the protein functions. Peroxiredoxins are known to undergo oxidation-dependent transitions in their oligomerization degree, accompanied by functional changes required to generate appropriate cellular response. For instance, changed oligomerization due to hyperoxidation may manifest a transition from the thiol-based peroxidase to a chaperone under increased oxidative stress (52, 174). In critical branch points of central energy and nitrogen metabolism, which are occupied by 2-oxo acid dehydrogenase complexes, the functional switch of the thiol-based peroxidases possessing peroxynitrite reductase activity may respond to metabolic changes encoded by ROS production and nitric oxide levels. Ultimately, availability of the substrates for these essential reactions provides for a chemical signal that couples function of the branch points of metabolism with multiple defense and signaling systems. This may relate to the importance of mitochondrial ROS-induced ROS release (216) in such systemic pathologies as septic shock (106). As indicated in Figures 13 –16, the function of 2-oxo acid dehydrogenase complexes may link the actions of thioredoxins and glutaredoxins not only to the protein post-translational modifications but also to the DNA damage response, mTOR, HIF, and iron homeostasis. Further understanding of mechanistic details of this coupling will certainly help addressing a number of challenges in basic science and medicine.
VII. Conclusions
ROS production by 2-oxo acid dehydrogenase complexes is not equal to the sum of ROS generated by the isolated complex components. ROS production by the complexes operating in the physiological direction, prevailing in vivo, enables regeneration of the FAD semiquinone formed after the superoxide anion radical release, by the dihydrolipoyl intermediate undergoing oxidation to the thiyl radical. The superoxide anion radical produced in the proximity of mitochondrial aconitase and glutaredoxins with iron-sulfur clusters, may participate in iron homeostasis and related signaling through mobilization of iron from these proteins. Suppressors of 2-oxo acid dehydrogenases, the thiyl radicals may also mediate thiol-disulfide exchange, glutathionylation, and nitrosylation. Reactive products of the complex-catalysed side reactions are scavenged by thioredoxin and thiol-based peroxidases, coupling the key systems of energy production and nitrogen metabolism to cellular ROS- and RNS-dependent signaling and defense.
Footnotes
Acknowledgments
V.I.B. greatly acknowledges her cooperation with Prof. Dr. Hartmut Follmann (GhK Kassel, Germany) on the thioredoxin interaction with 2-oxo acid dehydrogenase complexes, supported by Volkswagen Foundation and DFG (Germany), and with Dr. Christian Sievers (Tubingen University, Germany) on EPR studies of radical species, supported by Alexander von Humboldt Foundation (Germany). The author thanks Dr. Garik Mkrtchyan (Department of Cellular and Molecular Medicine, University of Copenhagen, Denmark) and Mr. Artem Artiukhov (Lomonosov Moscow State University, Russia) for their qualified technical help in preparation of this article for publication. Research work of V.I.B. is currently supported by the Russian Science Foundation (Grant No. 18-14-00116).