
Abstract
Aims:
Previous studies indicate that hippocampal synaptic plasticity and spatial memory processes entail calcium release from intracellular stores mediated by ryanodine receptor (RyR) channels. In particular, RyR-mediated Ca2+ release is central for the dendritic spine remodeling induced by brain-derived neurotrophic factor (BDNF), a neurotrophin that stimulates complex signaling pathways leading to memory-associated protein synthesis and structural plasticity. To examine if upregulation of ryanodine receptor type-2 (RyR2) channels and the spine remodeling induced by BDNF entail reactive oxygen species (ROS) generation, and to test if RyR2 downregulation affects BDNF-induced spine remodeling and spatial memory.
Results:
Downregulation of RyR2 expression (short hairpin RNA [shRNA]) in primary hippocampal neurons, or inhibition of nitric oxide synthase (NOS) or NADPH oxidase, prevented agonist-mediated RyR-mediated Ca2+ release, whereas BDNF promoted cytoplasmic ROS generation. RyR2 downregulation or inhibitors of N-methyl-
Innovation:
The present novel results emphasize the key role of redox-sensitive Ca2+ release mediated by RyR2 channels for hippocampal structural plasticity and spatial memory.
Conclusion:
Based on these combined results, we propose (i) that BDNF-induced RyR2-mediated Ca2+ release and ROS generation via NOS/NOX2 are strictly required for the dendritic spine remodeling and the RyR2 upregulation induced by BDNF, and (ii) that RyR2 channel expression is crucial for spatial memory processes. Antioxid. Redox Signal. 29, 1125–1146.
Here, we show that cross talk between ryanodine receptor type-2 (RyR2)-mediated Ca2+ release and reactive oxygen species generation is a key feature of the signaling pathways that underlie RyR2 upregulation and the structural remodeling of dendritic spines induced by brain-derived neurotrophic factor (BDNF) in primary hippocampal neurons. We also present results showing that RyR2 channels play a central role in spatial memory processes, since after RyR2 downregulation, rats exhibited strikingly defective performances in a previously memorized spatial memory task. Consequently, the decrease in ryanodine receptor (RyR) levels exhibited by early-stage Alzheimer's disease patients may be a relevant factor for the initial memory defects associated with this neurodegenerative condition.
Introduction
S
Neurotrophins exert key roles as stringent regulators of synaptic plasticity (91). Earlier studies demonstrated that neurotrophin-induced hippocampal synaptic plasticity exhibits an immediate requirement for protein synthesis (50). Brain-derived neurotrophic factor (BDNF), a neurotrophin that exerts multiple actions, has a central role in the modulation of neuronal activity (18, 73, 107). Increasing hippocampal neuronal activity stimulates the production and release of BDNF (10, 27, 69). High-frequency electrical stimulation of hippocampal cultures promotes BDNF release (38), as does the application of theta-burst trains of action potentials to the same cells (62). BDNF promotes learning and memory tasks and stimulates memory persistence (10, 27, 60, 80, 96).
The central role BDNF plays in synaptic plasticity processes has been described extensively (118). By binding to specific membrane receptors, BDNF promotes changes in the expression of genes involved in synaptic function (75) and plays an important role in neuronal survival and maintenance of several neuronal systems (14). Moreover, by promoting axonal and dendritic growth of neuronal processes, BDNF controls neuronal morphology through the formation and maturation of glutamatergic and GABAergic synapses (19, 36, 44, 91). BDNF mediates pre- and postsynaptic changes related to neuronal activity, which in turn might contribute to sustained synaptic plasticity (22, 45, 64, 65). In addition, BDNF modulates synaptic efficacy by promoting presynaptic transmitter release (5, 105) and by inducing postsynaptic changes that cause long-lasting increases in synaptic plasticity (45, 67).
Neuronal dendritic spines are the primary postsynaptic loci of excitatory synapses and have long been associated with synaptic plasticity (37). Activity-induced release of BDNF from presynaptic neurons prompts the growth of postsynaptic dendritic spines and the induction and maintenance of long-term potentiation (LTP) in hippocampal area CA1 (10, 11, 27, 110).
BDNF enhances synaptic strength by increasing the open probability of N-methyl-
Binding of BDNF to TrkB triggers TrkB dimerization, promoting autophosphorylation of tyrosine residues present in the receptor intracellular domains. In turn, TrkB autophosphorylation leads to the recruitment and activation of adapter proteins involved in a range of cellular signaling pathways, including mitogen-activated kinases, phosphatidylinositol 3-kinase, and phospholipase Cγ (PLCγ) (16). Hydrolysis of phosphatidylinositol 4,5-bisphosphate by activated PLCγ generates diacylglycerol, a transient activator of protein kinase C, and inositol 1,4,5-trisphosphate (IP3), which through activation of IP3 receptors (IP3R) triggers Ca2+ release from intracellular stores (19).
Calcium release from the endoplasmic reticulum mediated by IP3R and ryanodine receptor (RyR) channels contributes to the generation of Ca2+ signals in response to neuronal activation [reviewed in Baker et al. (9), Johenning et al. (46), and Paula-Lima et al. (93)]. Both channels types, and RyR channels in particular, mediate Ca2+-induced Ca2+ release (CICR), a cellular process with a potential role as an amplification mechanism of postsynaptic Ca2+ entry signals (13). Immunohistological techniques have revealed heterogeneous IP3R and RyR expression patterns within neuronal cells of rodent brain (32). Of the three mammalian RyR isoforms, ryanodine receptor type-2 (RyR2) is the most abundant isoform expressed in rat and chicken brain (28, 31).
Earlier work showed that BDNF release induced by high-frequency stimulation of primary hippocampal cultures requires RyR-mediated CICR (10). BDNF also mobilizes Ca2+ from intracellular stores via IP3R (19, 40), which through CICR may lead to subsequent Ca2+ signal amplification by RyR channels (1, 13, 63, 93). Moreover, BDNF elicits via TrkB a nonselective cationic current, which is insensitive to tetrodotoxin and saxitoxin and requires PLC activity and IP3R activation; this pathway engages intracellular Ca2+ stores and extracellular Ca2+ entry, suggesting the involvement of TRPC channels in the signaling pathways induced by BDNF (5).
Adding to the complexity of the signaling pathways induced by BDNF, we reported that in primary hippocampal neurons, BDNF engages RyR channels to increase RyR2 and RyR3 protein contents and to induce dendritic spine remodeling (1). These last results agree with previous reports showing that treatment of primary hippocampal neurons with the RyR agonist caffeine promotes RyR-dependent dendritic spine remodeling (58, 59).
Neuronal calcium signals play key roles in memory processes (90). In particular, RyR-mediated calcium release participates in the acquisition and/or consolidation of spatial memory processes (9, 93). In addition to Ca2+ signals, reactive oxygen species (ROS), including superoxide anion, hydrogen peroxide (H2O2), and hydroxyl radical, together with reactive nitrogen species (RNS), have important roles in synaptic plasticity processes associated with learning and memory (48, 49, 76, 106). To our knowledge, however, there are no reports showing enhanced ROS generation as part of the complex signaling pathways induced by BDNF in hippocampal neurons.
Here, we report that BDNF promotes ROS generation; this ROS increase is required for BDNF-induced RyR2 upregulation and structural plasticity. Moreover, hippocampal RyR2 downregulation prevented BDNF-induced RyR2 upregulation and dendritic spine remodeling in primary hippocampal neurons, and significantly impaired the performance of rats in a memorized spatial memory task.
Results
Downregulation of the RyR2 isoform significantly inhibits RyR-mediated calcium release
To test the specific involvement of the RyR2 isoform on BDNF-induced synaptic plasticity, we decreased RyR2 expression by transfection of neuron-enriched primary hippocampal cultures (1) with a specific short hairpin RNA (shRNA) against RyR2 (shRyR2).
Immunofluorescence analysis of neuronal cells expressing a red fluorescent protein (RFP) coded in the same vector as the shRyR2 or the shRNA against the scrambled RyR2 sequence ([shScr]; Fig. 1A), indicated that ∼30% of the neuronal cells present in the cultures were transfected with shRyR2 or shScr; in addition, RyR2 immunofluorescence in the neuronal soma was 70% lower in shRyR2-RFP-positive neurons (shRyR2, RFP) when compared with shScr-RFP-positive cells (shScr, RFP), or with RFP-negative neurons transfected with shRyR2 (shRyR2) (Fig. 1B), determined by one-way analysis of variance (ANOVA; F = 12.5; p < 0.0001), followed by Tukey's post hoc test (***p < 0.001; n = 4).
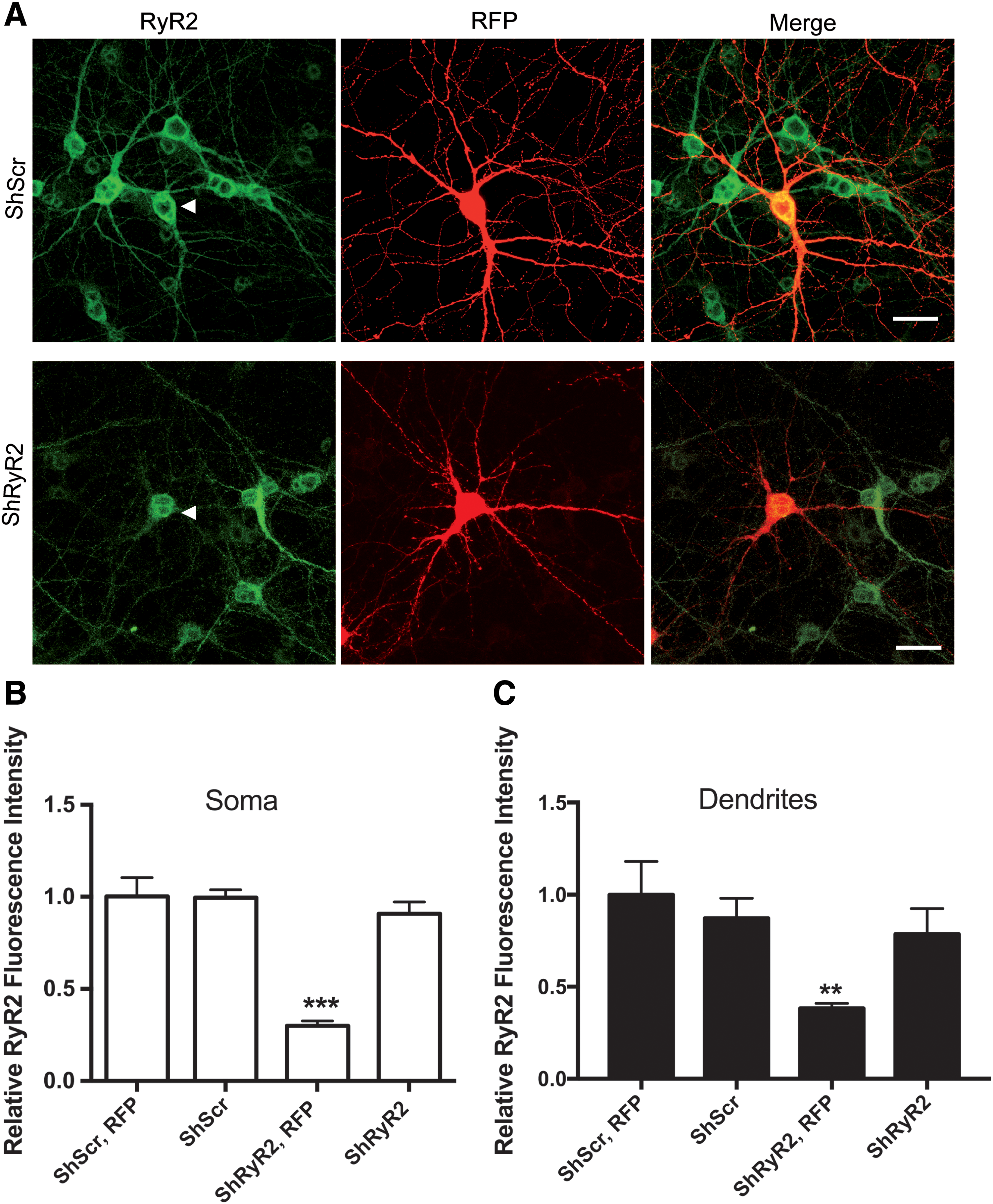
Neurons transfected with shScr displayed comparable RyR2 immunofluorescence levels as untransfected controls. Similar results were found at the level of the dendrites (Fig. 1C); statistical analysis was performed by one-way ANOVA (F = 5.0; p < 0.01), followed by Tukey's post hoc test (**p < 0.01; n = 4).
The effects of transfection with shRyR2 or shScr on RyR-mediated Ca2+ signals were evaluated in single transfected neurons (RFP positive) loaded with the fluorescent Ca2+-sensitive dye Fluo-4 AM.
As illustrated in a representative experiment, addition of the specific RyR agonist 4-chloro-m-cresol (4-CMC) at a concentration of 0.5 mM generated large fluorescence signals in shScr-transfected neurons, whereas shRyR2-transfected neurons did not respond to the agonist (Fig. 2A).
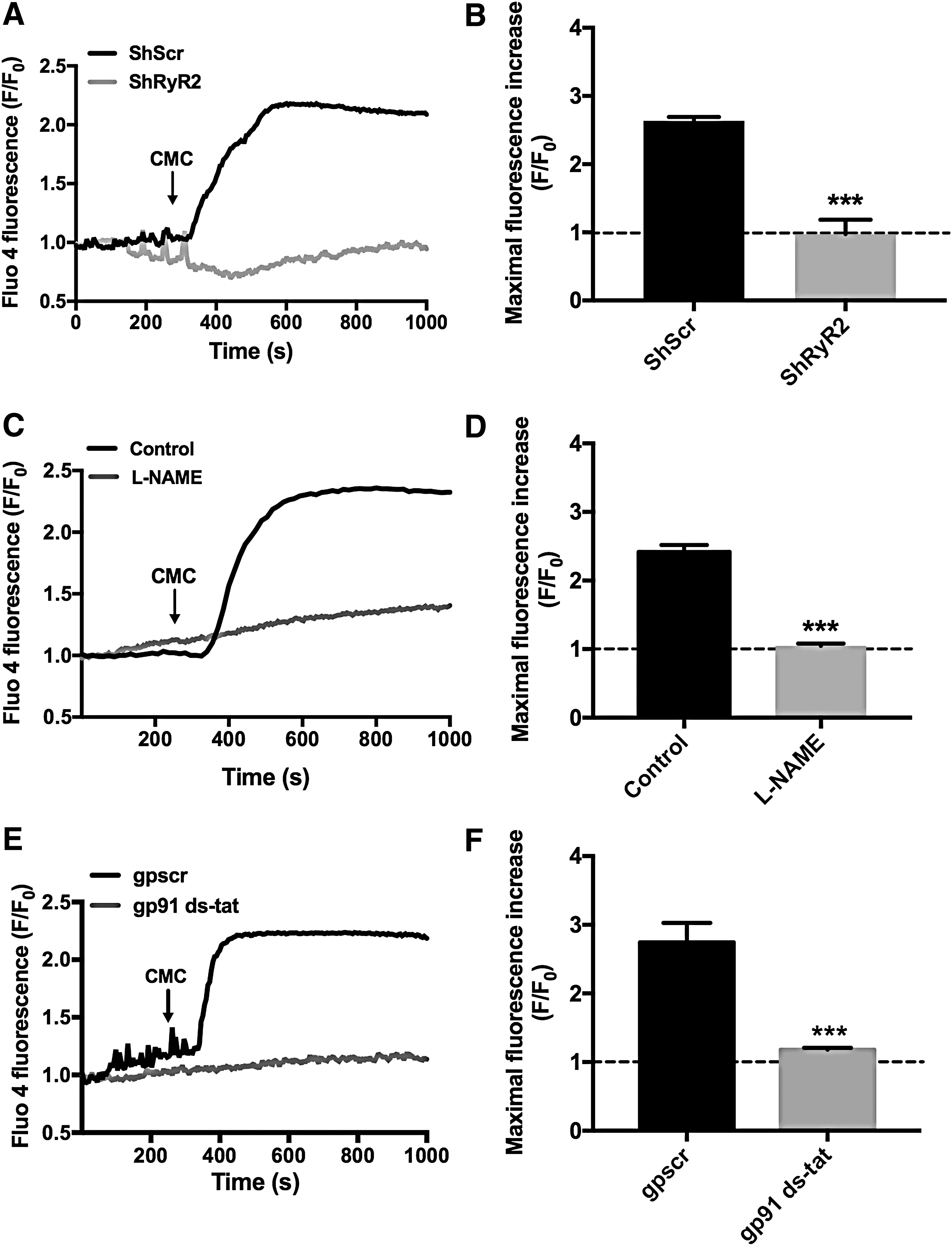
In contrast, nontransfected neurons present in the same cultures exhibited a significant fluorescence increase of 2.2 ± 0.1-fold [mean ± standard error (SE), n = 4] in response to addition of 0.5 mM 4-CMC. Addition of 4-CMC to neurons transfected with shScr caused a significant increase of maximal Fluo-4 fluorescence, normalized to the average fluorescence value recorded before agonist addition; in contrast, Fluo-4 fluorescence did not increase following 4-CMC addition to shRyR2-transfected neurons (Fig. 2B). The statistical analysis of these results was performed with unpaired Student's t-test (***p < 0.001; n = 4). Based on these combined results, we propose that the RyR2 isoform mediates 4-CMC-induced RyR-mediated Ca2+release in primary hippocampal neurons.
Inhibition of nitric oxide synthase or NADPH oxidase type-2 prevents agonist-induced RyR-mediated Ca2+ release
Several reports indicate that ROS/RNS stimulate neuronal RyR activity (34, 35, 41, 121). Accordingly, we tested whether pharmacological inhibitors of the nitric oxide synthase (NOS) or NADPH oxidase type-2 (NOX2) enzymes affected agonist-induced RyR-mediated Ca2+ release. Primary hippocampal neurons in cultures treated with the NOS inhibitor
BDNF-induced ROS generation engages RyR2 channels, NMDA receptors, and NOS and NOX2 activities
To test whether BDNF promotes ROS generation, hippocampal cultures were transfected with the HyPer-Cyto probe to monitor cytoplasmic H2O2 production (12). As shown in the representative time-lapse experiment illustrated in Supplementary Figure S1A (Supplementary Data are available online at
Representative time-lapse experiments (Supplementary Fig. S1B) show that addition of BDNF (50 ng/mL) significantly increased HyPer-Cyto fluorescence in neurons transfected with shScr, whereas it caused similar reductions of fluorescence intensity in neurons transfected with shRyR2 or treated with inhibitory ryanodine. The average results illustrated in Figure 3B show that BDNF addition (50 ng/mL) significantly increased probe fluorescence (1.59 ± 0.02) in neuronal cells transfected with the shScr sequence; in contrast, neurons transfected with shRyR2 displayed a lower level of fluorescence (0.55 ± 0.11) than that exhibited by neurons transfected with shScr or by control neurons. Statistical analysis of the results presented in Figure 3A and B was performed with unpaired Student's t-test (***p < 0.001; n = 5).
Next, we tested whether MK801, a synaptic NMDA receptor inhibitor, affected BDNF-induced ROS generation. A representative time-lapse experiment (Supplementary Fig. S1C) shows that control cells transfected with the HyPer-Cyto probe displayed increased fluorescence following BDNF addition (50 ng/mL); in contrast, neuronal cells preincubated with MK801 (10 μM, 30 min) displayed a significant fluorescence decrease following BDNF addition. Quantification of results shows that while control neurons displayed a significant increase in HyPer-Cyto fluorescence following BDNF addition, neurons preincubated with MK-801 displayed a significant reduction (Fig. 3C). Statistical analysis of the results presented in Figure 3C was performed with unpaired Student's t-test (***p < 0.001; n = 5).
Previous studies in primary cortical neurons have shown that stimulation of NMDA receptors promotes ROS generation via sequential stimulation of the NOS and NOX2 enzymes (31). Accordingly, we tested next whether NOS or NOX2 inhibitors affected BDNF-induced ROS generation. To this aim, we preincubated primary cultures transfected with the HyPer-Cyto probe for 30 min with
A previous report in neocortical neuronal cultures described that NMDA receptor activation induces a sequential signaling cascade entailing calcium-dependent NOS activation, nitric oxide generation, increased cyclic GMP production, activation of protein kinase G (PKG), and NOX2 activation (33). A subsequent report described BDNF-induced NOS activation in primary hippocampal neurons (57). Accordingly, we tested the effects of the PKG inhibitor KT-5823 (116) on BDNF-induced ROS generation. As illustrated in Figure 3F and Supplementary Figure S1F, addition to the cultures of 5 μM KT-5823 15 min before addition of BDNF (50 ng/mL) suppressed BDNF-induced ROS generation, showing that PKG activity mediates the ROS increase induced by BDNF. Statistical analysis of the results presented in Figure 3F was performed with unpaired Student's t-test (***p < 0.001; n = 3).
Based on these combined results, we propose that BDNF-induced cytoplasmic H2O2 generation requires sequential NOS and NOX2 activation in response to BDNF-induced NMDA receptor stimulation, plus Ca2+ signals initially produced by Ca2+ influx via NMDA receptors and subsequently amplified through RyR2-mediated CICR.
BDNF-induced RyR2 upregulation entails RyR2-mediated Ca2+ release plus ERK1/2, NOS, and NOX activities
RyR2-mediated Ca2+ release
Previously, we reported that BDNF-induced RyR2 upregulation requires functional RyR channels (1).
To test the specific contribution of the RyR2 isoform to BDNF-induced RyR2 upregulation, primary hippocampal neurons were transfected with shRyR2 or shScr and subsequently exposed to BDNF. Transfected cultures displayed a percent reduction of RyR2 messenger RNA (mRNA) levels of 54 ± 7 (n = 5) and a percent reduction of protein content of 38 ± 8 (n = 8) relative to cultures transfected with shScr, as illustrated in Figure 4A and B, respectively. Following incubation with BDNF (50 ng/mL) for 6 h, primary cultures transfected with shScr displayed a significant increase (1.8-fold) in RyR2 mRNA (Fig. 4A), determined by one-way ANOVA (F = 48.0; p < 0.0001), followed by Tukey's post hoc test (***p < 0.001; n = 5). Likewise, RyR2 protein levels increased significantly following BDNF addition (50 ng/mL), (Fig. 4B), determined by one-way ANOVA (F = 6.6; p < 0.005), followed by Tukey's post hoc test (*p < 0.05; **p < 0.01; n = 8). Cultures transfected with shRyR2 displayed lower RyR2 mRNA and protein levels than cultures transfected with shScr; BDNF addition (50 ng/mL) increased RyR2 mRNA and protein in these cultures only to the levels exhibited by shScr-transfected cultures not treated with BDNF (Fig. 4A, B; see also original immunoblots in Supplementary Fig. S7A). These modest increments presumably reflect the response to BDNF of untransfected neurons present in the culture.
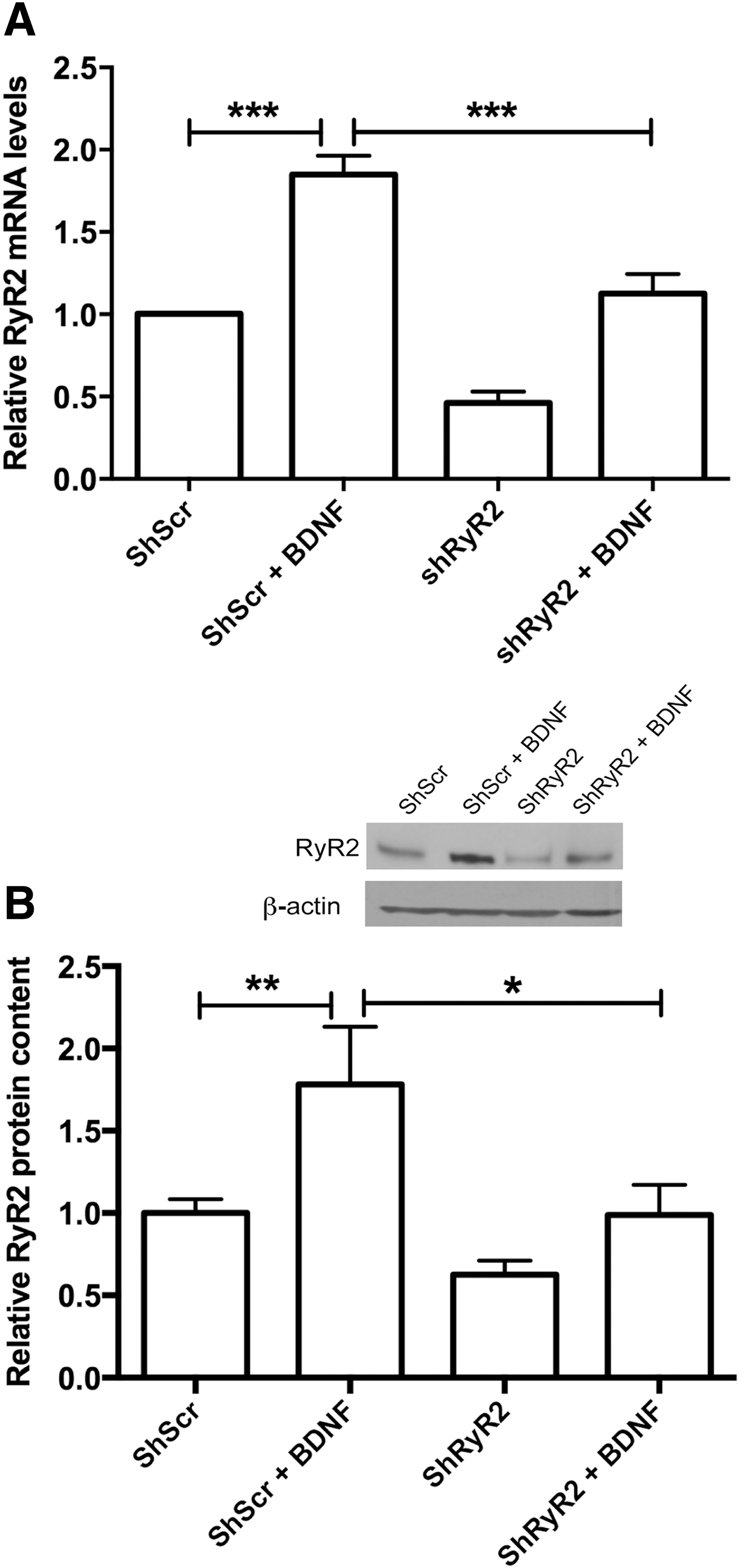
The primary hippocampal cultures used in this work displayed significant RyR2 and RyR3 mRNA levels, with average cycle threshold (Ct) values for RyR2 and RyR3 of 24 and 26, respectively. In contrast, the average Ct value for RyR1 was 35, beyond the quantitative polymerase chain reaction (qPCR) detection range; this value did not reflect the use of an inadequate primer, because this primer yielded an average Ct value of 20 for RyR1 in rat skeletal muscle homogenates. Likewise, the primary hippocampal cultures used in this work exhibited negligible RyR1 protein levels in immunoblots performed with a highly specific RyR1 antibody (manuscript in preparation).
Cultures transfected with shRyR2 showed similar RyR3 mRNA levels as cultures transfected with shScr; incubation for 6 h with BDNF (50 ng/mL) produced similar increases in RyR3 mRNA levels in Src-transfected (2.2-fold) as in shRyR2-transfected (2.0-fold) cultures (Supplementary Fig. S2A), showing that transfection with shRyR2 did not affect basal RyR3 mRNA levels or the increase in these levels produced by BDNF. In addition, transfection with shRyR2 did not modify basal RyR3 protein content and did not prevent the RyR3 protein increase induced by BDNF (Supplementary Fig. S2B).
Based on these combined results, which confirm the specificity of the shRyR2 sequence used here, and on previous reports showing that the RyR2 isoform predominates in the hippocampus (31), we propose that RyR2-mediated Ca2+ release plays a prominent role in the RyR2 upregulation induced by BDNF.
ERK1/2
To prevent downstream ERK1/2 activation in response to BDNF (4), we used U0126 to inhibit the mitogen-activated protein kinase MEK. Cultures preincubated for 30 min with U0126 (10 μM), which was maintained during the subsequent incubation with BDNF (50 ng/mL), did not exhibit increased RyR2 mRNA and protein levels following incubation with BDNF for 6 h; in contrast, a significant increase in RyR2 mRNA and protein content occurred in control neurons treated with BDNF (Fig. 5A, B; see also original immunoblot in Supplementary Fig. S7B). Statistical analysis of these results was performed with one-way ANOVA (F = 11.5; p = 0.0002 for Fig. 5A; F = 8.7; p = 0.0003 for Fig. 5B), followed by Tukey's post hoc test (**p < 0.01; ***p < 0.001; n = 5).
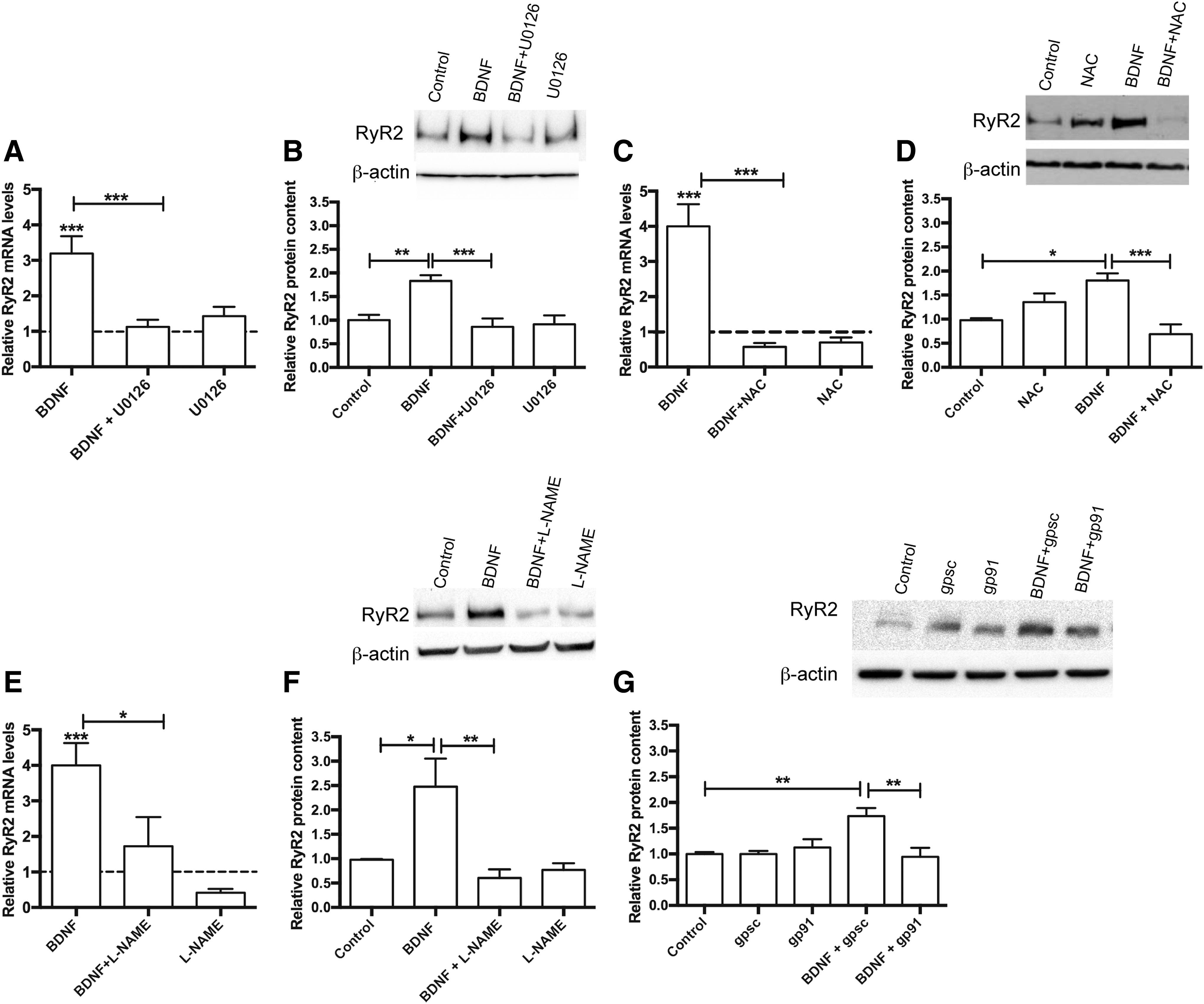
Reactive oxygen species
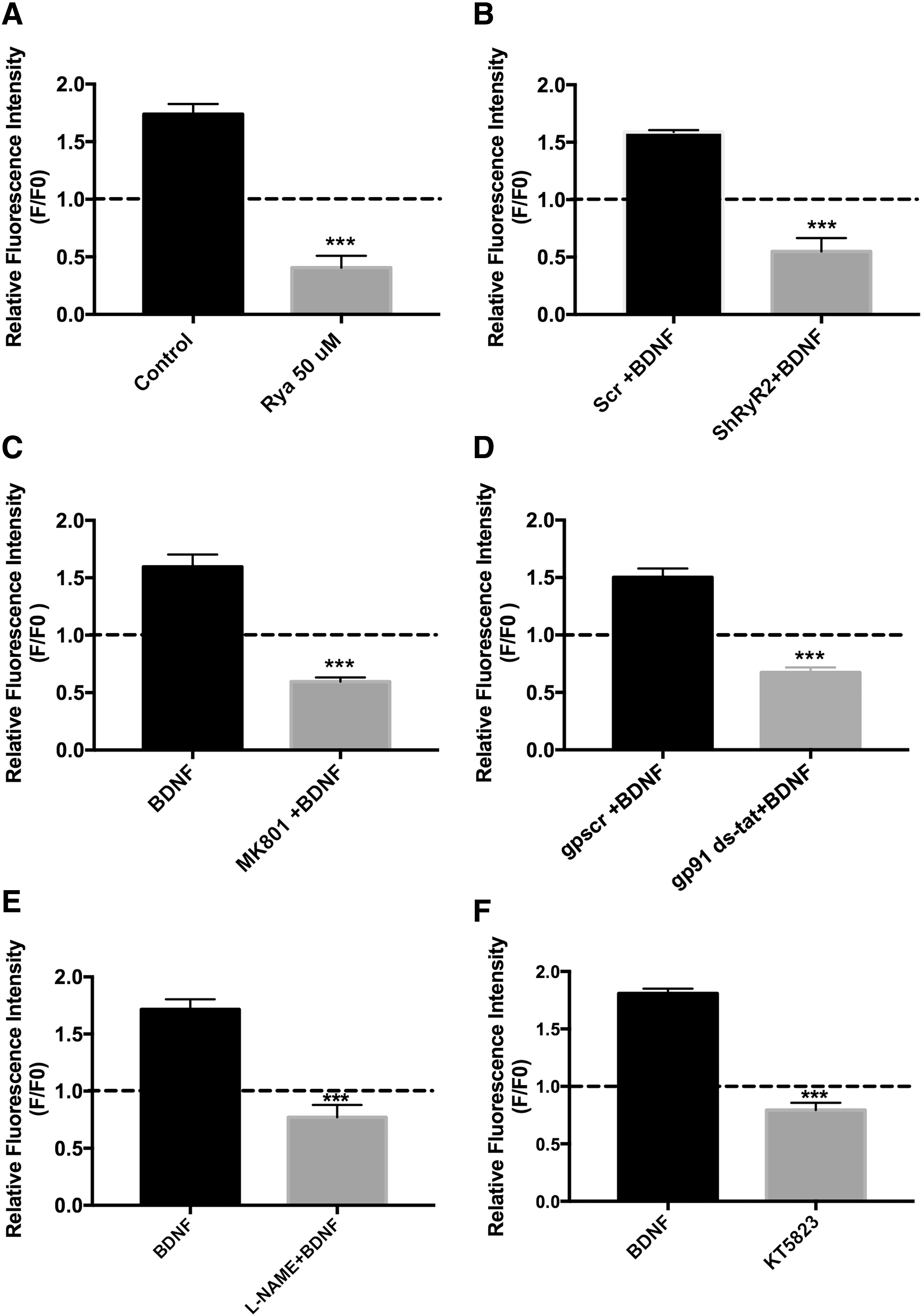
To elucidate whether BDNF-induced H2O2 generation contributed to BDNF-induced RyR2 upregulation, cultures were preincubated for 30 min with the universal ROS scavenger N-acetylcysteine (NAC, 10 mM), which was kept during the subsequent 6-h incubation period with BDNF (50 ng/mL). Control cultures incubated with BDNF displayed increased levels of both RyR2 mRNA and protein, with values five- and twofold higher, respectively, over the controls, while NAC treatment prevented these increments (Fig. 5C, D; see also original immunoblot in Supplementary Fig. S7C). Statistical analysis of these results was performed with one-way ANOVA (F = 16.3; p < 0.0001 for Fig. 5C; F = 10.0; p = 0.0007 for Fig. 5D), followed by Tukey's post hoc test (*p < 0.05; ***p < 0.001; n = 5).
NOS and NOX2
Cultures preincubated for 30 min with
Based on these combined results, we suggest that BDNF-induced RyR2 upregulation entails a complex set of Ca2+-dependent signaling cascades, including RyR2-mediated Ca2+release and ERK1/2, NOS, and NOX2 activation, leading to H2O2 generation.
RyR2-mediated Ca2+ release, ROS, NOS, and NOX2 mediate BDNF-induced dendritic spine remodeling
Changes in dendritic spine morphology induced by caffeine (58) or BDNF (1) require functional RyR channels. Here, we investigated if RyR2 downregulation affected the short-term and long-term changes in spine morphology induced by BDNF or caffeine. Addition of BDNF (50 ng/mL) to neurons transfected with shScr (detected by their green fluorescence) promoted spine elongation, as illustrated by a representative experiment (Fig. 6A). These changes, which on average took place as early as 15 min after BDNF addition (Fig. 6B), did not occur in neurons transfected with shRyR2, identified by their green fluorescence (Fig. 6A, B). Statistical analysis of the results presented in Figure 6B was performed with two-way ANOVA [interaction: F (8,87) = 4.5; p = 0.0001; row factor: F (4,87) = 0.7; p = 0.57; column factor: F (2,87) = 49.8; p = 0.0001], followed by Bonferroni's post hoc test (n = 5). The corresponding p-values are given in Figure 6B legend.
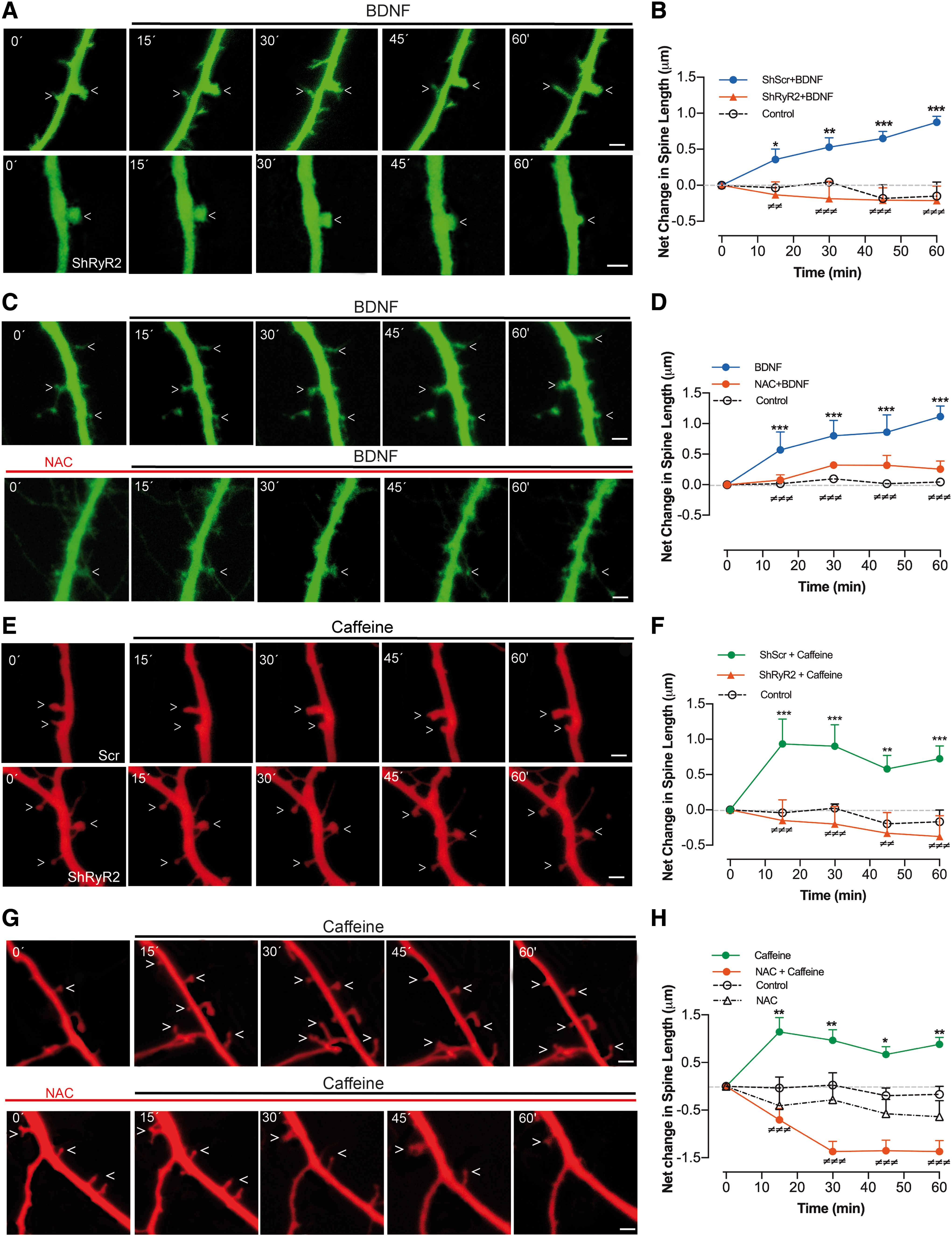
To test if BDNF-induced ROS generation was required for BDNF-induced spine remodeling, we preincubated cultures for 30 min with 10 mM NAC. This treatment produced a significant reduction in the spine lengthening induced by BDNF (50 ng/mL) (Fig. 6C, D). Statistical analysis of the results presented in Figure 6D was performed with two-way ANOVA [interaction: F (8,105) = 7.4; p = 0.0001; row factor: F (4,105) = 18.0; p = 0.0001; column factor: F (2,105) = 88.9; p = 0.0001], followed by Dunnett's post hoc test (n = 8). The corresponding p-values are given in Figure 6D legend.
Addition of 10 mM caffeine to transfected cultures promoted, after 15 min (the first time point tested), significant spine elongation in neurons transfected with shScr but not in neurons transfected with shRyR2 (Fig. 6E, F). Statistical analysis of the results presented in Figure 6F was performed with two-way ANOVA [interaction: F (8,93) = 2.6; p < 0.01; row factor: F (4,93) = 1.7; p = 0.15; column factor: F (2,93) = 40.6; p = 0.0001], followed by Dunnett's post hoc test (n = 8). The corresponding p-values are given in Figure 6F legend. Of note, the effects of caffeine, which stimulates RyR activity directly, were faster than the BDNF-elicited changes. Presumably, BDNF stimulates RyR2-mediated Ca2+ release by engaging first other upstream signaling cascades.
In addition, NAC largely prevented the spine lengthening induced by caffeine (Fig. 6G, H). In fact, neurons treated with NAC (10 mM) exhibited on average a net decrease in spine length even after addition of 10 mM caffeine (Fig. 6H). Statistical analysis of the results presented in Figure 6H was performed with two-way ANOVA [interaction: F (12,165) = 5.1; p = 0.0001; row factor: F (4,165) = 2.3; p = 0.06; column factor: F (3,165) = 71.7; p = 0.0001], followed by Dunnett's post hoc test (n = 8). The corresponding p-values are given in Figure 6H legend.
Next, we evaluated at long term, spine morphology changes in neurons present in cultures incubated for 6 h with BDNF (50 ng/mL). As illustrated in Figure 7A and B, BDNF produced a marked increase in spine density in neurons transfected with shScr but not in neurons transfected with shRyR2. Statistical analysis of the results presented in Figure 7B was performed with one-way ANOVA (F = 23.0; p = 0.0001), followed by Tukey's post hoc test (***p < 0.001; n = 5).
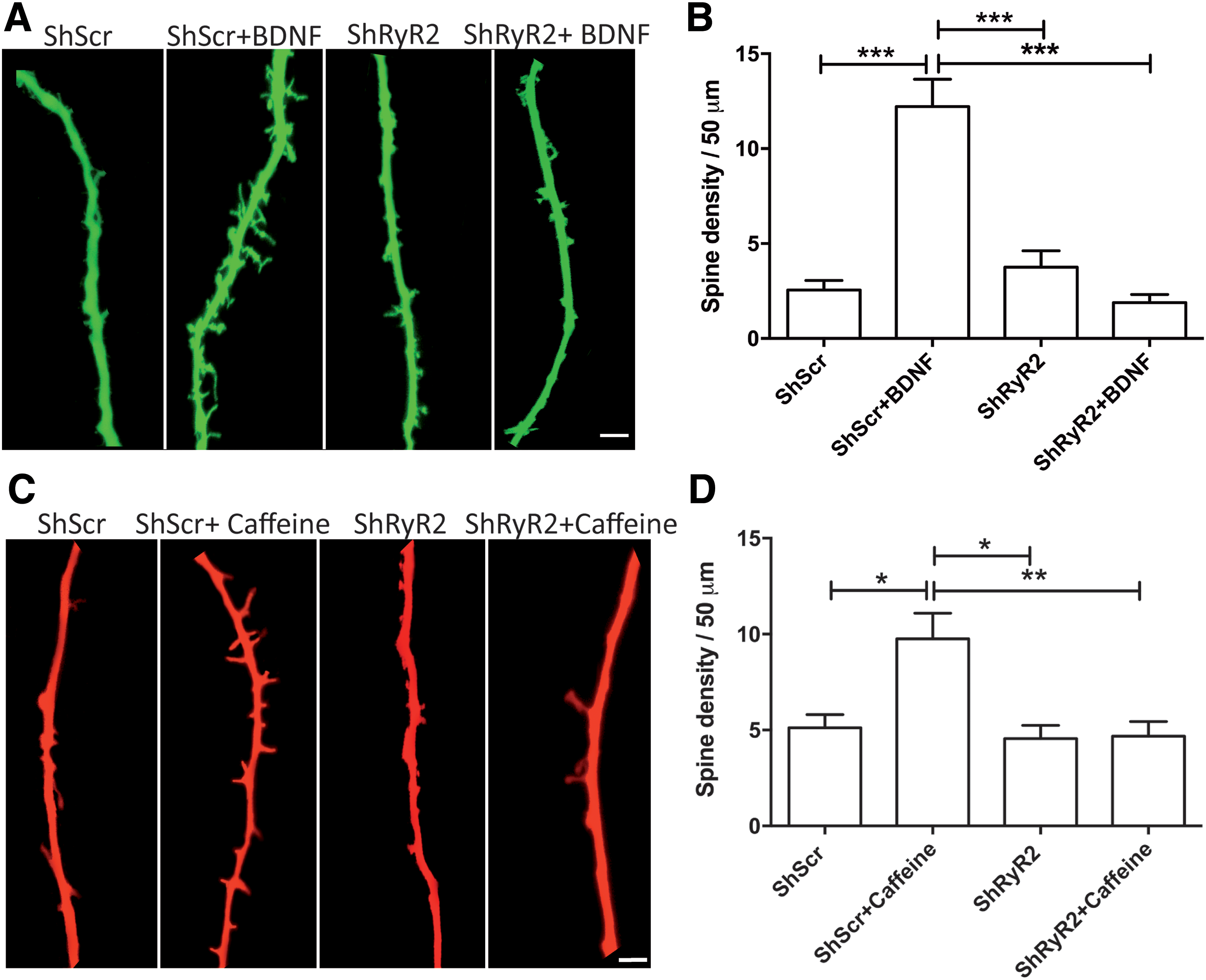
Similarly, incubation with caffeine (10 mM, 6 h) caused a substantial increase in spine density in neurons transfected with shScr, whereas shRyR2-transfected neurons did not undergo caffeine-induced spine remodeling (Fig. 7C, D). Statistical analysis of the results presented in Figure 7D was performed with one-way ANOVA (F = 6.8; p = 0.002), followed by Tukey's post hoc test (*p < 0.05; **p < 0.01; n = 5).
To examine whether the long-term structural plasticity changes induced by BDNF in hippocampal neurons required ROS, we preincubated the cultures for 30 min with NAC (10 mM) before BDNF addition. NAC-treated neurons displayed a significant decrease in dendritic spine density relative to controls and did not present the density increase displayed by control neurons incubated for 6 h with BDNF (50 ng/mL), (Fig. 8A, B). Statistical analysis of the results presented in Figure 8B was performed with one-way ANOVA (F = 38.1; p = 0.0001), followed by Tukey's post hoc test (***p < 0.001; n = 5).
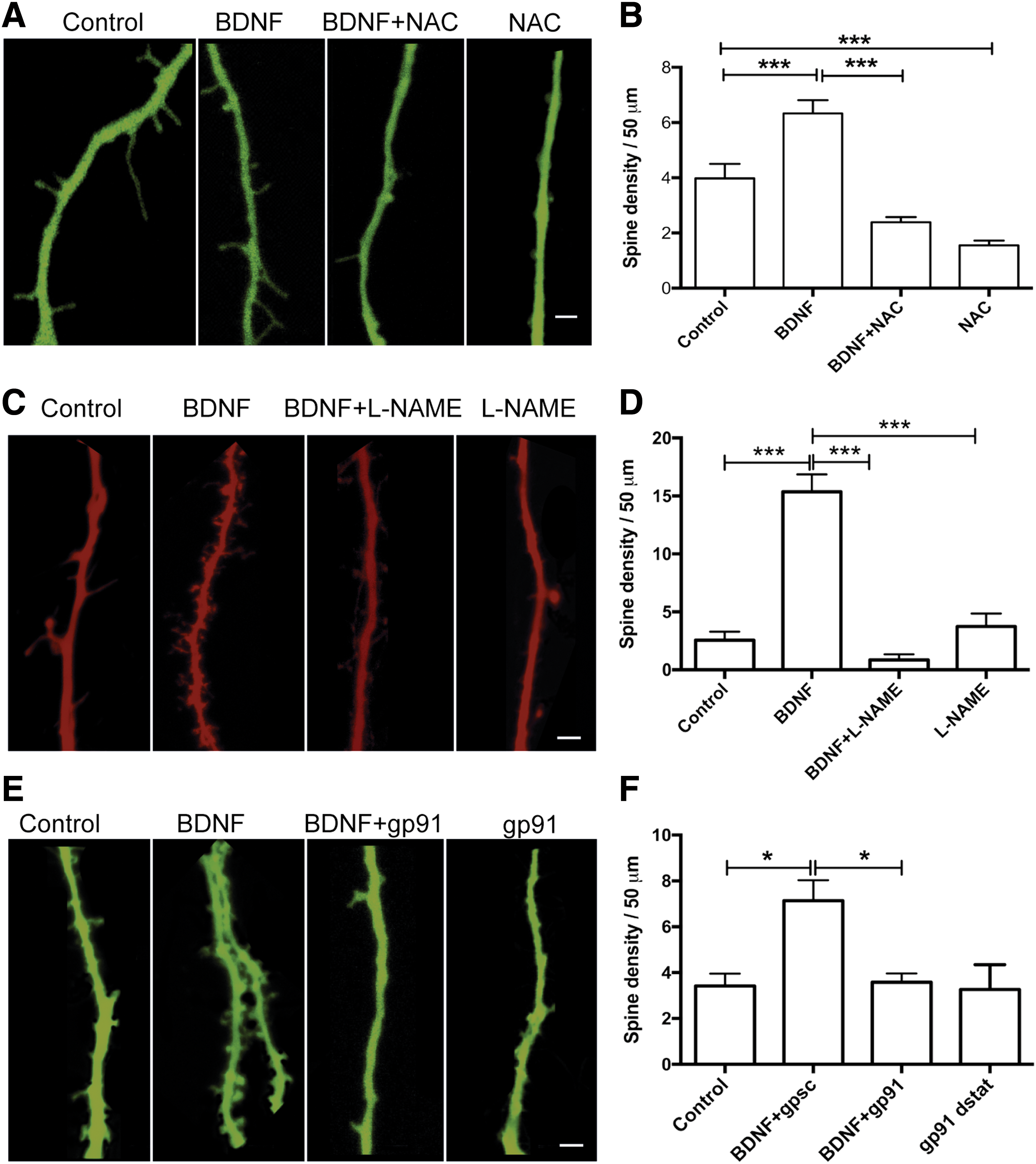
We studied next the possible participation of NOS and NOX2 as sources of ROS generation on BDNF-induced spine remodeling. Cultures preincubated for 30 min with 10 μM
We infer from these combined results that ROS generated by sequential activation of the NOS and NOX2 enzymes, together with RyR2-mediated Ca2+ release, play key roles as signaling molecules in the short- and long-term spine remodeling induced by BDNF or caffeine.
Downregulation of the RyR2 isoform causes significant defects in a memorized spatial memory task
To test if RyR2 downregulation affected spatial memory processes, we placed cannulas in the CA1 region. To evaluate hippocampal-dependent spatial learning and memory, cannulated animals (male rats) were trained during 9 consecutive sessions in the Oasis maze task, which represents a dry version of the Morris water maze (25, 53) and involves searching for the reward (water) in one of 21 equidistant wells on a circular arena provided with visual cues (Supplementary Fig. S3).
After the third training session, we performed bilateral injections of animals three times in a period of 25 h (see Table in Fig. 9B) with RyR2-directed antisense oligodeoxynucleotides (ODN-RyR2, 10 nmol/μL) or with the scrambled sequence (ODN-Scr, 10 nmol/μL). These bilateral injections (1 μL each) caused significant downregulation of RyR2 protein contents in the CA1, CA3, and dentate gyrus hippocampal regions, determined by immunohistochemistry analysis (Supplementary Fig. S4) of tissue sections collected 6 h after the ninth training session, but did not affect RyR3 protein levels (Supplementary Fig. S5). Quantification of the immunofluorescence images and Western blot analysis of the whole hippocampus confirmed these findings (Supplementary Fig. S6).
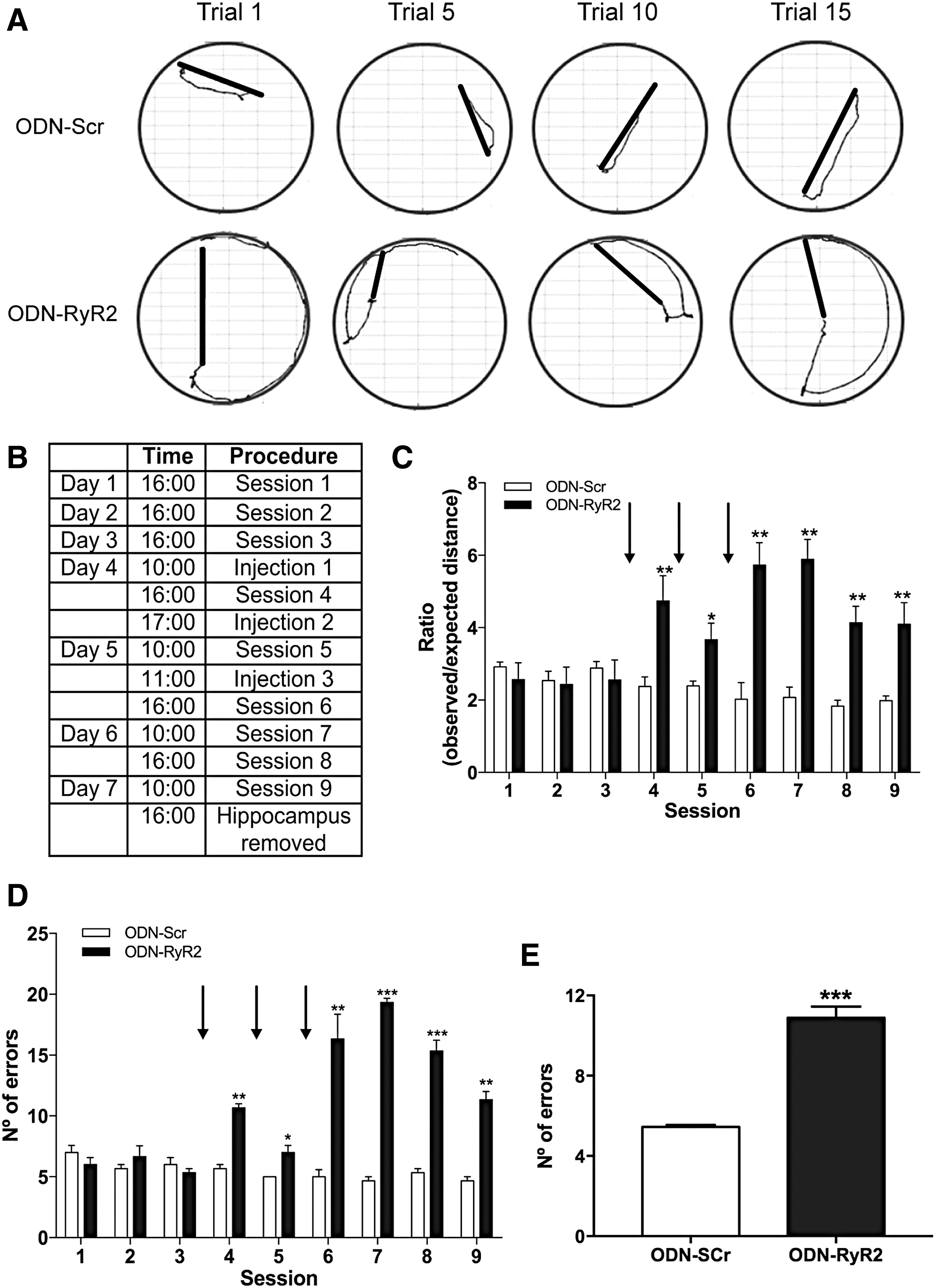
We found that the animals injected with ODN-RyR2 displayed striking defects in the previously memorized spatial task, as indicated by the increased trajectories exemplified in the traces illustrated in Figure 9A, which were collected after the ninth session (see Table in Fig. 9B).
After injection, rats displayed increased ratios of observed over expected distances (Fig. 9C). Statistical analysis of the results presented in Figure 9C was performed with two-way ANOVA [interaction: F (8,90) = 7.4; p = 0.005; row factor: F (8,90) = 3.1; p < 0.0001; column factor: F (1,90) = 67.4; p < 0.0001]. Likewise, after injection, rats displayed increased number of errors along the subsequent sessions (Fig. 9D), analyzed by two-way ANOVA [interaction: F (8,90) = 32.8; p < 0.0001; row factor: F (8,90) = 22.5; p < 0.0001; column factor: F (1,90) = 277.0; p < 0.0001], followed by Bonferroni's post hoc test. Quantification of the number of errors performed in the last session by animals injected with ODN-Scr or ODN-RyR2 revealed significant differences (Fig. 9E), determined by unpaired Student's t-test (***p < 0.001; n = 6).
Discussion
Summary of results
In this work, we report that RyR2 channels make a major contribution to agonist-induced RyR-mediated Ca2+ release in primary hippocampal neurons; NOS or NOX2 inhibitors abolished agonist-induced RyR-mediated Ca2+ release, confirming the redox sensitivity of this process. We also show that BDNF-induced RyR2 upregulation and dendritic spine remodeling required RyR2-mediated Ca2+ release, H2O2 generation, and NOS plus NOX2 activities. Moreover, inhibition of ERK1/2 activity prevented RyR2 upregulation induced by BDNF, whereas selective RyR2 downregulation drastically impaired the capacity of animals to perform a previously memorized spatial memory task.
BDNF-induced ROS generation
Previous reports showed that BDNF promotes NO generation in cortical neurons (55, 56, 104). Rat CA1 hippocampal neurons express the NOS enzyme in dendritic spines (21), while incubation of primary hippocampal neurons with BDNF induces NO generation in both soma and dendrites, in parallel with intracellular Ca2+ signals (57). In cortical neurons, BDNF increases the expression levels of the p47 NOX2 subunit, presumably implicating NOX2 as a downstream target of BDNF (54). In this work, we show that ROS generation forms part of the complex array of downstream signaling pathways induced by BDNF in hippocampal neurons. In physiological conditions, ROS act as signaling molecules critical for the induction of synaptic plasticity and memory formation (76). Accumulating evidence suggests that there is substantial cross talk between ROS and Ca2+ signaling in different cellular systems (34,117), including hippocampal neurons (41). Here, we found that inhibition of NMDA receptors or RyR2 downregulation prevented BDNF-induced cytoplasmic H2O2 generation. Accordingly, we propose that Ca2+ signals initially produced by Ca2+ influx via NMDA receptors, and subsequently amplified through RyR2-mediated CICR, are essential for BDNF-induced ROS generation, presumably by enhancing NOS activation by Ca2+.
Previous studies indicate that hippocampal neurons express the NOX2 enzyme (111), and that activation of NMDA receptors results in sequential activation of NOS and NOX2 in neocortical neuronal cultures (33). Here, we found that BDNF-induced cytoplasmic H2O2 generation required both NOS and NOX activities. Previous reports indicate that NOS-generated nitric oxide enhances RyR activity via S-nitrosylation of RyR cysteine residues (109); however, ROS-induced RyR redox modifications, and not RyR S-nitrosylation, are required for brain RyR channel stimulation promoted by increased cellular oxidative conditions (20). Our results show that PKG inhibition suppressed BDNF-induced ROS generation. Hence, we propose that BDNF triggers a signaling cascade that engages sequentially NMDA receptors, NOS, PKG, and NOX2 enzymes, and redox-sensitive RyR2-mediated CICR, as illustrated in the scheme presented in Figure 10.
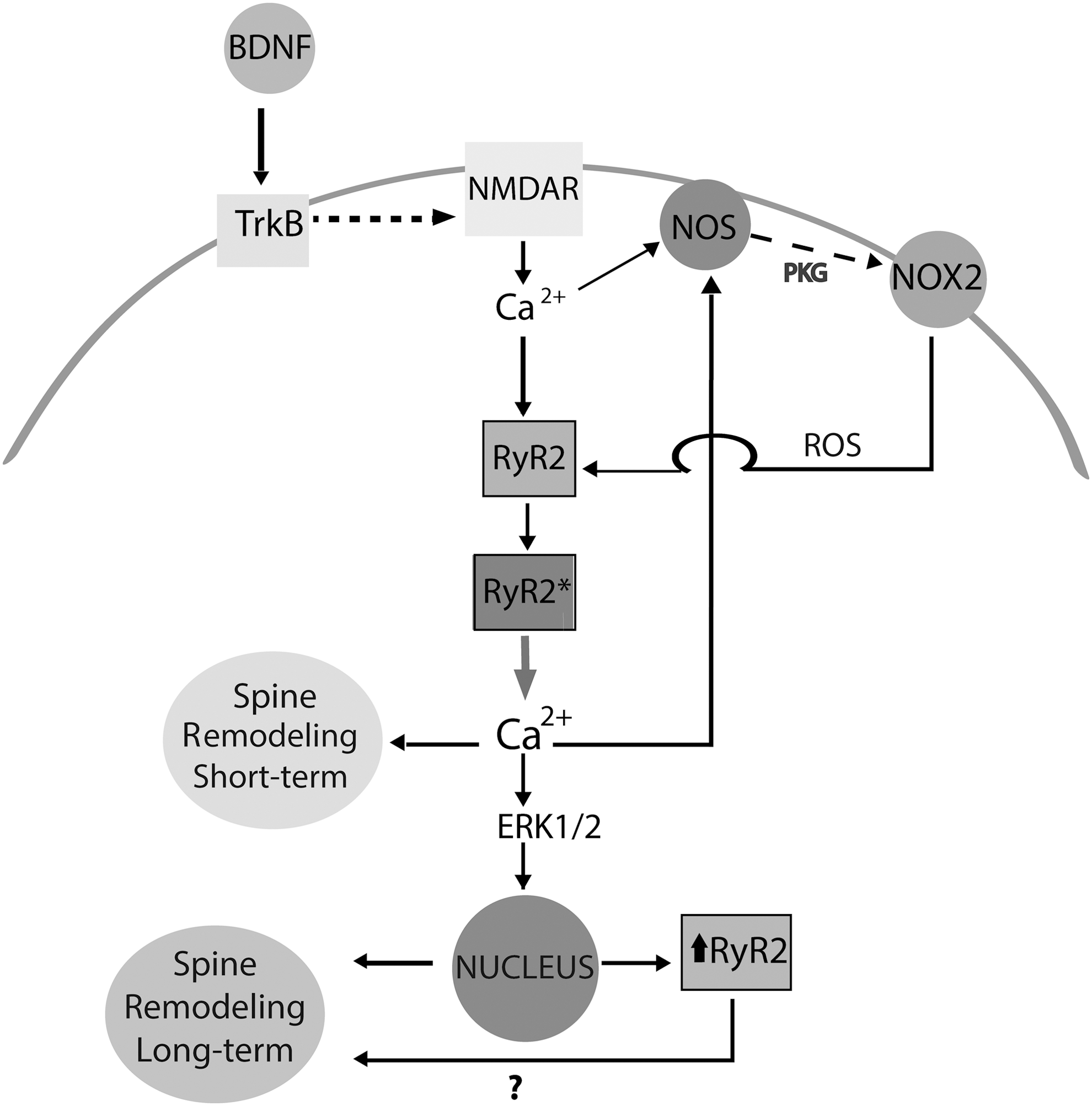
BDNF-induced RyR2 upregulation
Here, we show that RyR2 downregulation prevented Ca2+ release induced by 4-CMC, a strong indication that the RyR2 isoform is mostly responsible for this response. RyR2 downregulation hindered BDNF-induced RyR2 upregulation as well, suggesting that RyR2-mediated Ca2+ release is a key factor for its own expression. In addition, we found that the enhanced RyR2 expression induced by BDNF required ERK1/2 activity, which has a key role in the synaptic plasticity processes elicited by BDNF (4). Moreover, ERK1/2 activity is essential for the activation of the transcription factor cAMP response element-binding (CREB), which is central for long-term hippocampal synaptic plasticity and spatial memory processes (7). Nicotine administration to mice upregulates RyR2 levels in a number of brain areas associated with cognition and addiction, such as the cortex and ventral midbrain; RyR2 upregulation requires CREB activation via a mechanism dependent on CICR (122).
Future studies should address if BDNF-induced RyR2 upregulation involves CREB activation in hippocampal neurons. In addition, we found that the antioxidant NAC, or NOS or NOX2 inhibition, prevented BDNF-induced RyR2 upregulation. We suggest that cross talk between RyR2-mediated Ca2+ and ROS signaling plays a key role in RyR2 upregulation induced by BDNF.
BDNF-induced dendritic spine remodeling
Dendritic spines are miniature protrusions that compose most excitatory synapses in the mammalian brain [reviewed in Lippman and Dunaevsky (71) and Nimchinsky et al. (86)]. These structures receive, integrate, and compartmentalize the immense majority of excitatory inputs (37, 86). The shape and numbers of spines are a direct consequence of changes in neuronal activity (15, 112), which engages different Ca2+ signaling pathways (39, 42, 103). Dendritic spine remodeling, including the formation/elongation and the pruning/retraction of spines, requires changes in intracellular Ca2+ concentration (87), which are necessary for the actin-mediated spine enlargement associated with electrical stimulation (30, 87, 114). Previous work demonstrated that BDNF has an essential role in the structural remodeling of excitatory synapses (3, 4, 113). In brain slices, the gradual spine enlargement induced specifically by synaptic stimulation strongly depends on protein synthesis and BDNF signaling (110). Dendritic spine size positively correlates with an increase in AMPA-receptor-mediated currents at stimulated synapses and depends on NMDA receptors, calmodulin and actin polymerization (77, 78). In young hippocampal synapses, increased or decreased spine sizes, respectively, occur during persistent LTP or long-term depression (78, 82, 89, 120). Spontaneously released glutamate is sufficient to activate nearby spines, which can then lead to the growth of new postsynaptic processes connecting to a presynaptic site (100). Similarly, in cultured hippocampal neurons, short pulses of glutamate applied to dendrites induce spine elongation, whereas long pulses cause shrinking (59).
One of the main findings of our work is the strong dependence of BDNF-induced dendritic spine remodeling on RyR2-mediated Ca2+ release and ROS generation via NOS/NOX2. These results, which add to previous work (1, 58), show for the first time that BDNF-induced ROS generation is important for the structural remodeling of dendritic spines induced by BDNF. In addition, RyR2-mediated Ca2+ signals also mediated BDNF-induced dendritic spine remodeling, highlighting that the cross talk between Ca2+ and ROS signaling is a central process in activity-dependent neuronal function. In fact, ROS signaling and RyR-mediated Ca2+ release are key activities for neuronal polarization (115).
RyR2 downregulation impairs spatial memory
The involvement of RyR channels in the generation of postsynaptic Ca2+ signals, synaptic plasticity, and memory processes has acquired increasing relevance in the last two decades (8, 28, 29, 31, 74, 81, 97, 99, 101, 119). In chickens trained in a passive avoidance discrimination task, treatment with dantrolene to inhibit RyR channel activity causes loss of memory retention (28), while administration of the RyR agonist 4-CMC enhances memory consolidation (8). In mice conditioned in an inhibitory avoidance task (31), or trained in a radial arm-maze task (88), RyR channel inhibition with dantrolene or with high concentrations of ryanodine alters memory retention. In contrast, intrahippocampal injection of stimulatory ryanodine enhances both learning and memory consolidation in rats trained in the Morris water maze (1). Selective knockdown by intracerebrovascular injection of antisense ODNs of the RyR2 and the RyR3 isoforms in mice, but not of the RyR1 isoform, impairs memory retention in a passive avoidance test (31).
Here, we report that injection in the hippocampal CA1 region of a specific ODN anti-RyR2 caused severe defects in a spatial task previously memorized by rats. Remarkably, rats exhibited utter disorientation after downregulation of RyR2 expression, while injection of the scrambled ODN did not affect the memorized behavior. These results highlight the relevance of RyR2 expression for successful performance of hippocampal-dependent spatial tasks. Furthermore, rodent exposure to different hippocampal-dependent memory tasks, including the Morris water maze (1, 119) and novel object recognition (6), results in significant RyR2 upregulation and in less prominent RyR3 upregulation as well. Based on these combined findings, it seems reasonable to propose that hippocampal-dependent memory formation and consolidation not only require RyR2 channel expression but also entail RyR2 > RyR3 upregulation to ensure the generation of the Ca2+ signals required for memory processes.
It is worth mentioning that sublethal concentrations of soluble amyloid β-peptide oligomers (AβOs), increasingly recognized as causative agents of Alzheimer's disease, decrease RyR2 protein content and prevent BDNF- or caffeine-induced spine remodeling (92). The significant RyR2 protein downregulation induced by AβOs may contribute to the RyR decrease reported in Alzheimer's patients at early stages of the disease (51). Hence, RyR2 downregulation may represent a significant factor in the impaired synaptic plasticity and memory defects characteristic of this neurodegenerative disorder.
Materials and Methods
Materials
BDNF and 4-CMC were from Merck Millipore (Darmstadt, Germany). Fluo4-AM, calcein-AM, Alexa Fluor® 488, TRIzol reagent Hoechst 33258, GlutaMAX™, and Lipofectamine 2000 transfection reagent were from Invitrogen (Carlsbad, CA). DAKO mounting medium and Brilliant III Ultra-Fast SYBR® GREEN qPCR Master Mix were from Agilent Technologies (Santa Clara, CA). Ryanodine was from Tocris Bioscience (Bristol, United Kingdom). N-acetyl-
Mouse anti-RyR2 (MA3-916) was from Thermo Fisher. Rabbit anti-RyR3 (AB9082) was from Merck Millipore, and monoclonal anti-β actin (A5316) from Sigma-Aldrich (St. Louis, MO). A highly selective RyR1 antibody was kindly provided by Dr. Vincenzo Sorrentino. DNAase digestion (DNA-free™ Kit) was obtained from Ambion (Austin, TX), and ImProm-II Reverse Transcriptase kit was from Promega (Madison, WI). PVDF membranes (Trans-Blot® Turbo™ Mini PVDF) were from Bio-Rad Laboratories (Hercules, CA). The amplification system MX3000P was from Stratagene (La Jolla, CA). The pRFP-C-RS and pRFP-C-RS (TR30014) vectors and plasmid containing a noneffective 29-mer shScr cassette (TR30013 and TR30015) were from Origene (Rockville, MA). The HyPer™-Cyto plasmid was from Evrogen (Moscow, Russia).
Primary rat hippocampal cultures
Primary cultures were prepared from the hippocampus dissected from Sprague-Dawley rats at embryonic day 18 (1, 92). Cells were plated in minimum essential medium plus 10% horse serum for 40 min to allow adhesion of neurons and minimize glial cell adhesion. Cultures were maintained subsequently at 37°C under 5% CO2 in serum-free Neurobasal medium supplemented with B27 serum free and 2 mM GlutaMAX. Hippocampal cultures were used at 14 days in vitro (DIV) for all experiments.
All experimental protocols used in this work complied with the “Guiding Principles for Research Involving Animals and Human Beings” of the American Physiological Society and were approved by the Bioethics Committee on Animal Research, Faculty of Medicine, Universidad de Chile.
Transfection with knockdown plasmid vectors
Cells in primary culture were transiently transfected with Lipofectamine 2000 according to the manufacturer's specifications. Neurons were transfected with 1 μL Lipofectamine-2000 in Neurobasal medium containing 2 μg of shRNA expression vector specific to silence RyR2 expression. This shRNA expression vector was obtained by cloning a shRNA cassette directed against rat RyR2 mRNA (target sequence 5′-AGGAAGCAATGGTGGACAAGTTGGCTGAA-3′) in the BamHI/HindIII cloning sites of pGFP-V-RS vector or pRFP-C-RS vector. A plasmid containing a noneffective 29-mer shScr cassette (TR30013 and TR30015) was used as control. One day post-transfection, cultures were stimulated with BDNF (50 ng/mL) as described in the text to perform experiment of RyR2 expression or spine remodeling.
RNA isolation and real time PCR
Total RNA was isolated using TRIzol reagent; DNase digestion was included to remove any contaminating genomic DNA. RNA purity was assessed by the 260/280 absorbance ratio and RNA integrity by gel electrophoresis. Complementary DNA (cDNA) was synthesized with 1 μg total RNA with the Improm TM II reverse transcriptase. One hundred nanograms of cDNA was used in 20 μL final volume for qPCR amplification. qPCR was performed in an amplification system (MX3000P; Stratagene) using the Brilliant III Ultra-Fast SYBR GREEN qPCR Master Mix. Amplification was performed after 35 cycles, each of which included 15 s at 95°C, 15 s at 60°C (RyR2 and β-actin), and 15 s at 72°C. After these cycles, a final 15-s incubation step at 95°C was added. Primers (1) were as follows (f: forward; r: reverse): RyR1 f 5′-GCCTTTGATGTGGGATTACAG-3′; r 5′-CCCCAACTCGAACCTTCTCTC-3′. RyR2 f 5′-AATCAAAGTGGCGGAATTTCTTG-3′; r 5′-TCTCCCTCAGCCTTCTCCGGTTC-3′. RyR3 f 5′-GAAGCCTGTTGGTGGACCATAC-3′; r 5′-TCCAGAGTGTTTGCATAAAGGAG-3′. β-actin f 5′-TCTACAATGAGCTGCGTGTG-3′; β-actin r 5′-TACATGGCTGGGGTGTTGAA-3′. The levels of RyR mRNA were assessed with the 2−ΔΔCT method (95) using β-actin as housekeeping gene. Dissociation curves were analyzed to verify purity of products. All samples, including controls, were run in triplicate.
Western blot analysis
Cells extracts prepared as described (52) were resolved by sodium dodecyl sulfate/polyacrylamide gel electrophoresis (4% with a base of 15% polyacrylamide gels), transferred to PDVF membranes, and incubated overnight with specific antibodies against RyR2. To correct for loading, membranes were stripped and reprobed for β-actin using a specific antibody. The image acquisition and densitometric analysis of band density were performed by means of the ChemiDoc™ MP System and the Image Lab software by Bio-Rad Laboratories, respectively.
Immunofluorescence
Hippocampal cultures transfected with shRyR2 or shScr were fixed by adding an equal volume of 4% paraformaldehyde and Neurobasal medium for 5 min. After replacing this medium with a solution containing 4% paraformaldehyde, cultures were incubated for 10 min and washed with phosphate-buffered saline (PBS) at pH 7.4. Fixed cultures were incubated for 2 h with a blocking and permeabilization solution containing 3% donkey serum and 0.25% Triton X-100 (in PBS 1 × ); next, cultures were incubated overnight at 4°C with primary antibody anti-RyR2 diluted in blocking solution. After this incubation period, cultures were washed with PBS and incubated for 2 h with the secondary antibody Alexa Fluor 488 anti-rabbit (in blocking solution) at room temperature. Cultures were washed with PBS, and coverslips were mounted in DAKO mounting medium for microscope observation. Cells were visualized in a Nikon C2+ confocal microscope (Melville, NY), with the 63 × objective lens. Images were analyzed using ImageJ software (National Institutes of Health).
Immunohistochemistry
Brain tissue fixation and immunohistochemistry were performed as described (1). Briefly, each animal was perfused transcardially and under anesthesia with saline solution (NaCl 0.9%), followed by 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4, to fix and remove their brains. Brains were postfixed for 2 h in paraformaldehyde and were cryopreserved in PBS containing 30% sucrose plus 0.03% sodium azide for subsequent slice preparation with a microtome. Coronal slices (40 μm) were incubated in the blocking/permeabilization solution for 2 h at room temperature, and were then incubated overnight at 4°C in blocking/permeabilization solution under shaking with anti-RyR2 or anti-RyR3 primary antibodies, diluted to 1:50 and 1:100, respectively. Slices were washed three times for 5 min with PBS, followed by a 5-min wash with blocking/permeabilization solution before further incubation with the secondary antibodies Alexa 488 goat anti-mouse and Alexa 488 chicken anti-rabbit, respectively. Slices were then washed three times with PBS, for 10 min at room temperature, and then incubated with Hoechst 33258 for detection of the cell nucleus for 5 min at room temperature. Slices were mounted on slides coated with mounting solution to preserve fluorescence. The ImageJ software program was used for image analysis and generation of zeta projections from stacks (1.5 mm thickness each). Total intensity of RyR2 or RyR3 fluorescence in each z-projection was measured and normalized by Hoechst fluorescence intensity.
Determination of cytoplasmic Ca2+ signals
Cells were transferred to modified Tyrode's solution (in mM: 129 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 30 glucose, and 25 HEPES, pH 7.3), preloaded for 30 min at 37°C with 2.5 μM Fluo-4 AM, and washed for 10 min in modified Tyrode's solution to allow complete dye de-esterification. Fluorescence images of intracellular Ca2+ signals in primary hippocampal neurons (14 DIV) were obtained every 5 s with an inverted confocal microscope (Axiovert 200, LSM 5 Pascal; Carl Zeiss, Oberkochen, Germany), using Plan Apochromat 63 × Oil DIC objective (excitation at 488 nm, argon laser beam). Image data were acquired from different regions of optical interest located in the cell bodies. Frame scans were averaged using the equipment data acquisition program. Fluorescence signals are shown as F/F 0 values, where F 0 corresponds to the basal fluorescence recorded before any treatment. The increase in fluorescence induced by 0.5 mM 4-CMC did not saturate the probe, as tested by addition of ionomycin at the end of the experiment (2). All experiments were done at room temperature (20–22°C).
Determination of H2O2
Hippocampal neurons were transiently transfected with the HyPer-Cyto plasmid at 14 DIV. This plasmid codes for a cytoplasmic protein, HyPer-Cyto, which has a circularly permuted yellow fluorescent protein inserted into the regulatory domain of the prokaryotic H2O2-sensing protein OxyR (12), allowing selective detection of H2O2 production in living cells. One day post-transfection, cells were washed with Tyrode's solution and were stimulated with BDNF 50 ng/mL in the same solution. Images were obtained every 5 s with an inverted confocal microscope (Axiovert 200, LSM 5 Pascal; Carl Zeiss) using the Plan Apochromat 63 × Oil DIC objective (excitation at 488 nm, argon laser beam). Changes in probe fluorescence, which reflect H2O2 levels, are presented as F/F 0 values, where F 0 corresponds to the average basal fluorescence obtained from a specific interest region in the soma.
Morphological analysis of dendritic spines
We analyzed the changes in spine morphology by confocal microscopy of hippocampal cultures (14 DIV). Confocal fluorescence images were obtained with a Zeiss LSM-5, Pascal 5 Axiovert 200 microscope, by using LSM 5 3.2 image capture and analysis software and a Plan-Apochromat 63 × /1.4 Oil DIC objective. Dendrites in 30–50 μm proximity to the soma were selected randomly independently of their spine density. We used the ImageJ software program for image deconvolution and zeta-projection reconstruction from 10 to 15 stacks (0.4 μm each) to measure spine length. Spine density in dendrites was analyzed by measuring the number of spines present in a length of 50 μm; five dendrites were analyzed per condition in five independent experiments. Short-term changes were monitored for 1 h after BDNF incubation through acquisition of dendrite images every 15 min; long-term changes were visualized after 6 h of incubation with BDNF. We used two different strategies to visualize cellular morphology; neurons were loaded for 20 min with 1 μM calcein-AM in Tyrode's solution or neurons were transfected with pGFP-V-RS or pRFP-C-RS vectors that express the green fluorescent protein (GFP) or the RFP, respectively, coupled to the expression of shRyR2. Transfection with the scrambled sequence or only with the RFP or GFP was used as control.
Spatial memory evaluation
Bilateral injections of ODN-RyR2 or ODN-Scr were performed into rat CA1 hippocampal region. Two-month-old Sprague-Dawley male rats (250–280 g) were anesthetized with isoflurane (3% in oxygen) for induction of anesthesia. Isoflurane (2% in oxygen) was also used for maintenance of anesthesia, at a constant oxygen flow (1 L/min). Two 21-gauge stainless steel guide cannulas were implanted bilaterally in the CA1 hippocampal region of each rat. Target coordinates, calculated as given in Ref. (94), were as follows relative to bregma: anteroposterior −3.30, lateral ±2.6 mm, 1.8 mm in depth, with 10° angles to target the dorsal CA1 hippocampal region. Guide cannulas were fixed to the skull with skull screws and dental cement. Following surgery, animals were individually housed with food and water ad libitum for 7 days before exposure to the Oasis maze task, a modified dry-land version of the Morris water maze (25, 53).
To evaluate hippocampal-dependent spatial learning, cannulated and water-deprived pretrained animals were trained and tested during three consecutive daily sessions in the Oasis maze task, which involved searching for the reward (water) placed in the same one of 21 equidistant wells on a circular arena provided with visual cues; each session was composed of 15 trials of 1-min duration each. After this initial training period, animals received three bilateral injections (1.0 μL each) of ODN-RyR2 (10 nmol/μL) or ODN-Scr (10 nmol/μL), with the following sequences. ODN-RyR2: 5′T*T*CGCCCGCATCAGCC*A*T-3′; ODN-Scr: 5′C*G*GCAGGAGTCTGTGC*G*C-3′, the * indicates the phosphorothioate residues.
A complete scheme of training sessions and injections is illustrated in the Table presented in Figure 9B. Animal behavior was recorded with a video camera in the zenithal position. Video recordings were analyzed using the Virtual Dub software. The position of the animal was tracked and navigation was reconstructed and analyzed with a MATLAB (MathWorks) routine. Six hours after the conclusion of session 9, animals were perfused and their brains were removed and sliced in a frozen microtome for immunohistochemistry analysis of RyR2 and RyR3 (for details, see the Immunohistochemistry section). Fluorescence images, captured in an inverted confocal microscope C2+ Spectral Nikon Eclipse TI, were analyzed with the ImageJ software.
Statistics
Results are expressed as mean ± SE from at least three independent experiments. Statistical significance was evaluated with the GraphPad Prism 7.0 Software (San Diego, CA). The particular analysis performed in each case, Student's t-test, one-way or two-way ANOVA, is detailed in the Results section and figure legends.
Footnotes
Acknowledgments
We thank Dr. Vincenzo Sorrentino, University of Siena, Italy, for his kind gift of a highly specific RyR1 antibody. This work was supported by FONDECYT (3120093, 11140580, 1140545, 1150736, and 1170053) and by BNI (P-09-015).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.