
Abstract
Significance:
Radiation therapy (from external beams to unsealed and sealed radionuclide sources) takes advantage of the detrimental effects of the clustered production of radicals and reactive oxygen species (ROS). Research has mainly focused on the interaction of radiation with water, which is the major constituent of living beings, and with nuclear DNA, which contains the genetic information. This led to the so-called target theory according to which cells have to be hit by ionizing particles to elicit an important biological response, including cell death. In cancer therapy, the Poisson law and linear quadratic mathematical models have been used to describe the probability of hits per cell as a function of the radiation dose.
Recent Advances:
However, in the last 20 years, many studies have shown that radiation generates “danger” signals that propagate from irradiated to nonirradiated cells, leading to bystander and other off-target effects.
Critical Issues:
Like for targeted effects, redox mechanisms play a key role also in off-target effects through transmission of ROS and reactive nitrogen species (RNS), and also of cytokines, ATP, and extracellular DNA. Particularly, nuclear factor kappa B is essential for triggering self-sustained production of ROS and RNS, thus making the bystander response similar to inflammation. In some therapeutic cases, this phenomenon is associated with recruitment of immune cells that are involved in distant irradiation effects (called “away-from-target”
Future Directions:
Determining the contribution of targeted and off-target effects in the clinic is still challenging. This has important consequences not only in radiotherapy but also possibly in diagnostic procedures and in radiation protection.
I. Introduction
F
In the 1990s, a shift in the radiation biology paradigm occurred on the basis of the observation that biological effects could be observed also after irradiation of non-nuclear cell compartments (248, 276, 335). Particularly, the existence of dynamic signaling pathways between the various subcellular compartments (nucleus, endoplasmic reticulum [ER], mitochondria, and cell membrane) needs to be taken into account when assessing radiation-induced effects. The rather naive idea that only the nucleus is sensitive to radiation and that the rest of the cell is inert was progressively replaced by a new view that considers intra- and intercellular signaling mechanisms, leading to the expansion of the cell response to radiation in time (long-lasting radiation-induced effects) and also in space (off-target effects, also called nontargeted effects, such as bystander and abscopal effects).
In this review, we first summarize the main targeted and off-target effects and the underlying molecular mechanisms. Then, we discuss the currently available models to predict the therapeutic efficacy and side effects of radiation exposure by taking into account both targeted and off-target effects.
II. Targeted Effects: Oxidative Damage to DNA, Lipids, and Proteins
The effect of ionizing radiation on cellular constituents has been extensively studied, particularly the formation of DNA lesions because the biological consequences of radiation were mostly attributed to the formation of DNA lesions. In contrast to other biomolecules that are continuously synthetized and decomposed within cells, the cell genome is replicated only once per cell cycle. Therefore, the integrity of the DNA structure is of major importance to maintain the genetic information. For this reason, cells have developed several repair systems to remove DNA lesions and restore DNA integrity. Conversely, altered RNA, protein, and lipid molecules are discarded and replaced by newly synthesized molecules.
A. DNA damage, targeted effects
Following irradiation, DNA damage can be produced
During the last four decades, a considerable amount of work has been done to understand the chemical nature, the mechanism, and the yield of radiation-induced DNA lesions in irradiated cells. Concerning the chemical nature of the DNA modifications, most of the work was performed with isolated nucleosides used as DNA model systems (41). Today, about 80 different DNA modifications (including isomers) have been identified (45). The chemical nature of these modifications is not described in this review article, but information can be found in previous publications (41, 44, 254). Only few examples to highlight the complexity of the undergoing reactions are presented, focusing on lesions that have been observed at the cellular level.
1. Direct effect
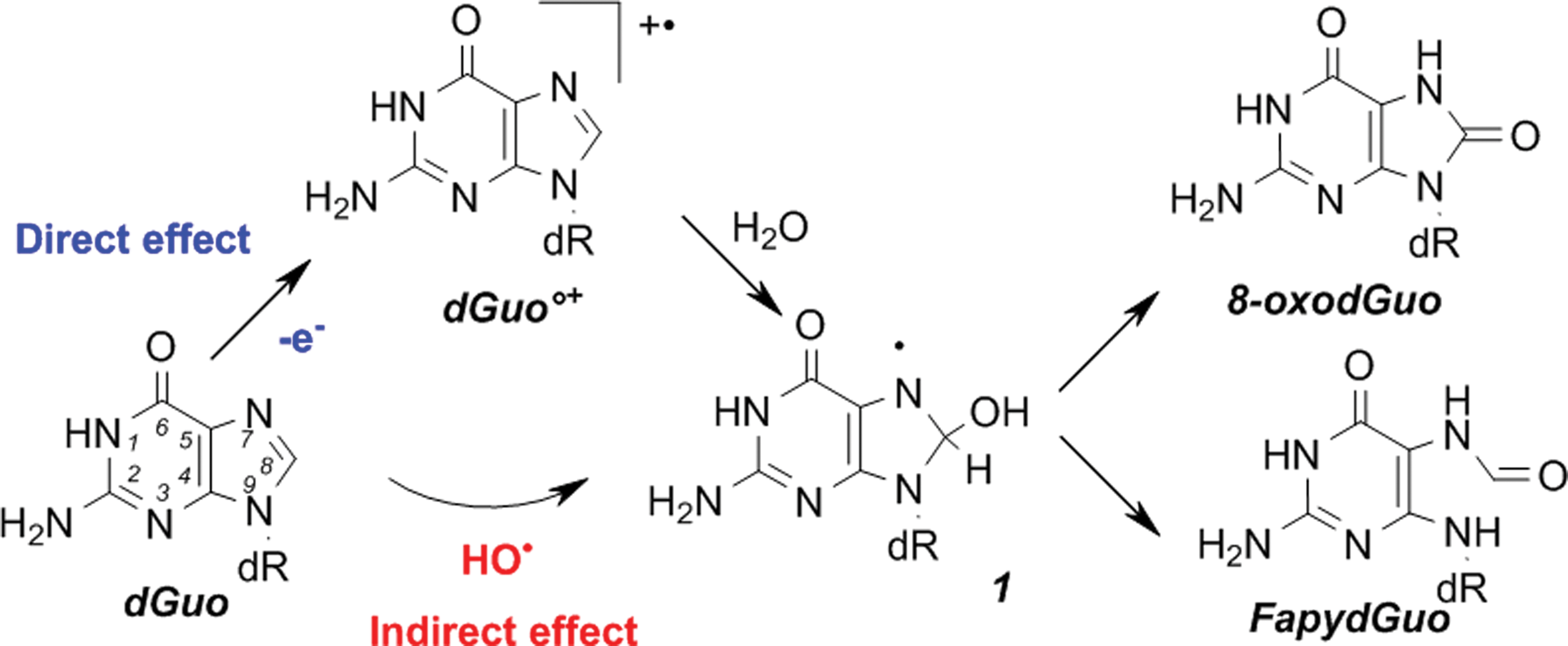
Through the direct effect, DNA molecules are directly ionized (loss of an electron), thus generating a DNA radical cation. For each nucleoside, the decomposition of the corresponding radical cation has been described in detail, but the chemistry is different in double-stranded DNA (dsDNA) (46). Indeed, among the DNA constituents, guanine has the lowest oxidation potential. Therefore, even if oxidation occurs on another base or sugar moiety, a fast electron transfer reaction occurs from guanine to the generated radical cation, thus repairing the initially produced radical and generating a guanine radical cation (G•+).
Consequently, in dsDNA and in cells, the direct effect of radiation produces mostly unstable guanine radical cations that, after decomposition, give rise typically to two guanine chemical modifications (Fig. 2): 8-oxo-7′8-dihydro-2′-deoxyguanosine (8-oxodGuo) following oxidation and the corresponding formamidopyrimidine derivative FapydGuo on reduction. Interestingly, it has been shown that in irradiated cells, FapydGuo production is two times higher than that of 8-oxodGuo, suggesting that cellular DNA is in a reducing environment.
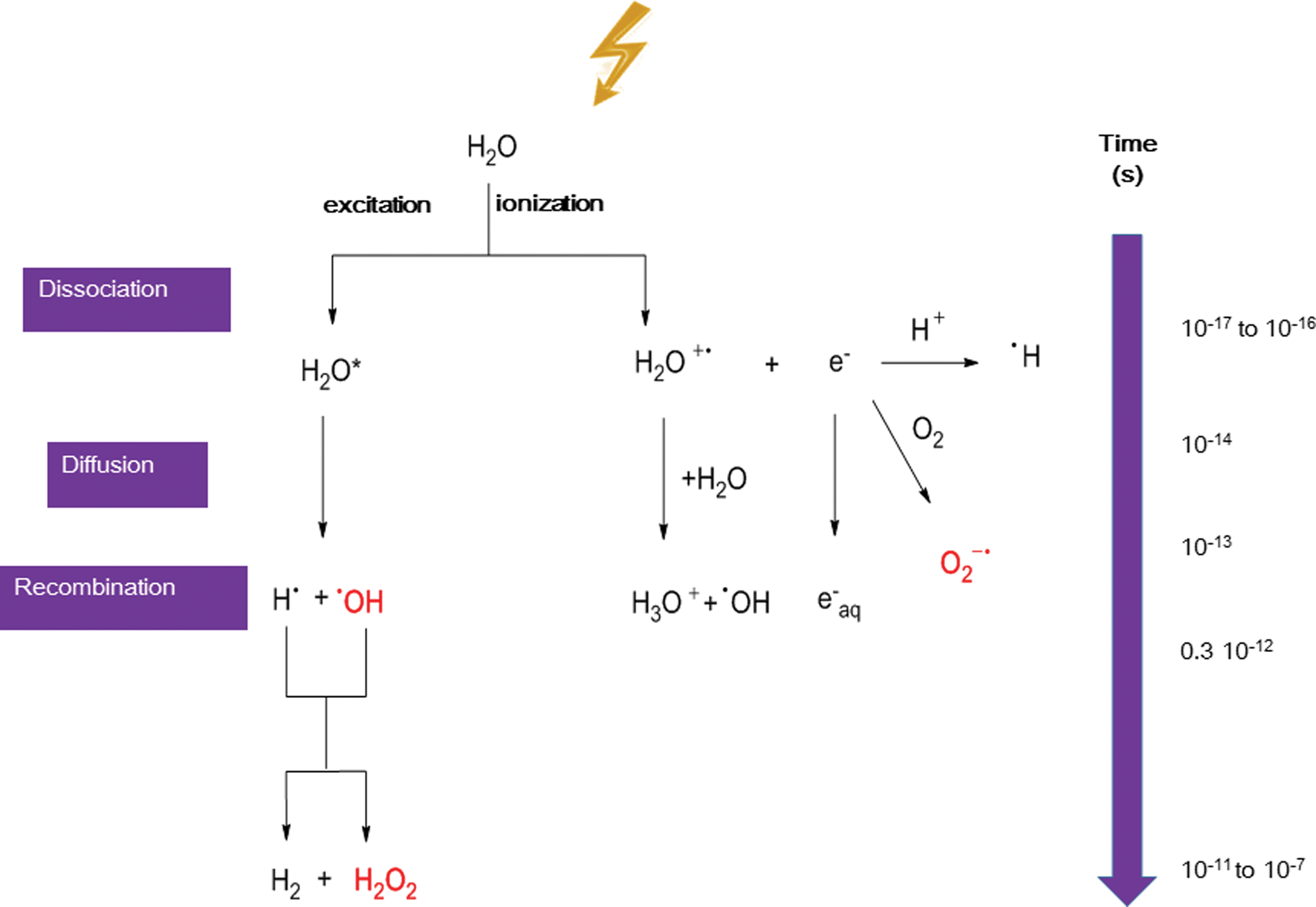
2. Indirect effect
The radiation-induced DNA lesions produced through the indirect effect are mediated by the initial formation of ROS due to water radiolysis. Exposure of water to ionizing radiation rapidly leads to the generation of HO•, ionized water (H2O+), hydrogen radicals, and hydrated electrons. Then, the reaction of the initially produced radicals generates hydrogen peroxide (H2O2) and superoxide anion (O2 −•). As these ROS are also produced endogenously and their concentration is regulated by several antioxidant defense mechanisms, the radiation-induced increase of ROS cellular level could damage cellular constituents and also induce oxidative stress (113).
Among the ROS produced upon exposure to radiation, HO• plays a predominant role because it can react, at a diffusion-controlled rate, with almost all biological constituents. Its reactivity within DNA is well documented (43), and about 30% of HO• reacts with the sugar moiety or phosphate group of DNA. Such reaction (69) produces SSBs. HO• reaction with the four different DNA bases has been studied in detail. This reaction produces a plethora of DNA lesions, but is limited by HO• diffusion. Interestingly, the DNA lesions generated by such mechanism are not different from those produced
The relative importance of the direct and indirect effects in the formation of radiation-induced DNA lesions is still a matter of debate. The general idea is that for low LET (linear energy transfer) radiation such as X- or γ-rays, the direct effect accounts for about 30% of DNA lesions (and thus 70% is attributed to the indirect effect), and that this proportion increases for higher LET radiations such as protons, carbons, and α-particles. A low mean LET of 0.2 keV/μm is observed with γ/X-rays and beta radiation, while LET increases up to 4–26 keV/μm with Auger electrons and up to 50–230 keV/μm with alpha particles. However, according to the chemical mechanism of formation of radiation-induced DNA lesions, the direct effect should mostly produce lesions on the guanine moiety (8-oxodGuo,
This suggests that the direct effect is not increased upon exposure to high LET radiation. Moreover, the strong correlation between HO• radiolytic yield and the number of produced DNA lesions strongly suggests that DNA lesions are predominantly produced
Finally, it is interesting to note that the chemical nature of radiation-induced DNA lesions is not different from that of DNA lesions produced in cells subject to endogenous oxidative stress (45). This is not surprising because ROS produced by radiation through water radiolysis are similar to those produced endogenously, for example, HO• through the Fenton reaction (204). However, the major difference concerns the lesion three-dimensional (3D) localization (212, 215, 330). Endogenous oxidative stress produces oxidative lesions that should be randomly distributed on the DNA macromolecules. These lesions, including modified DNA bases and SSBs, can be rapidly and efficiently repaired by the cell machinery, simply by removing the modification and using the complementary strand to resynthesize the original DNA sequence. After exposure to ionizing radiations, ROS are locally produced along the particle track and this could generate several DNA modifications at the same site (called “multiple damage sites,” MDS, or clustered DNA lesions) (Fig. 3). These lesions are defined as two or more modifications per helix turn (172, 219). DNA DSBs are one of the best examples of MDS. DSBs are produced in cells even after exposure to very low doses of radiations, and their formation yield increases almost linearly with the dose (102, 260, 329).
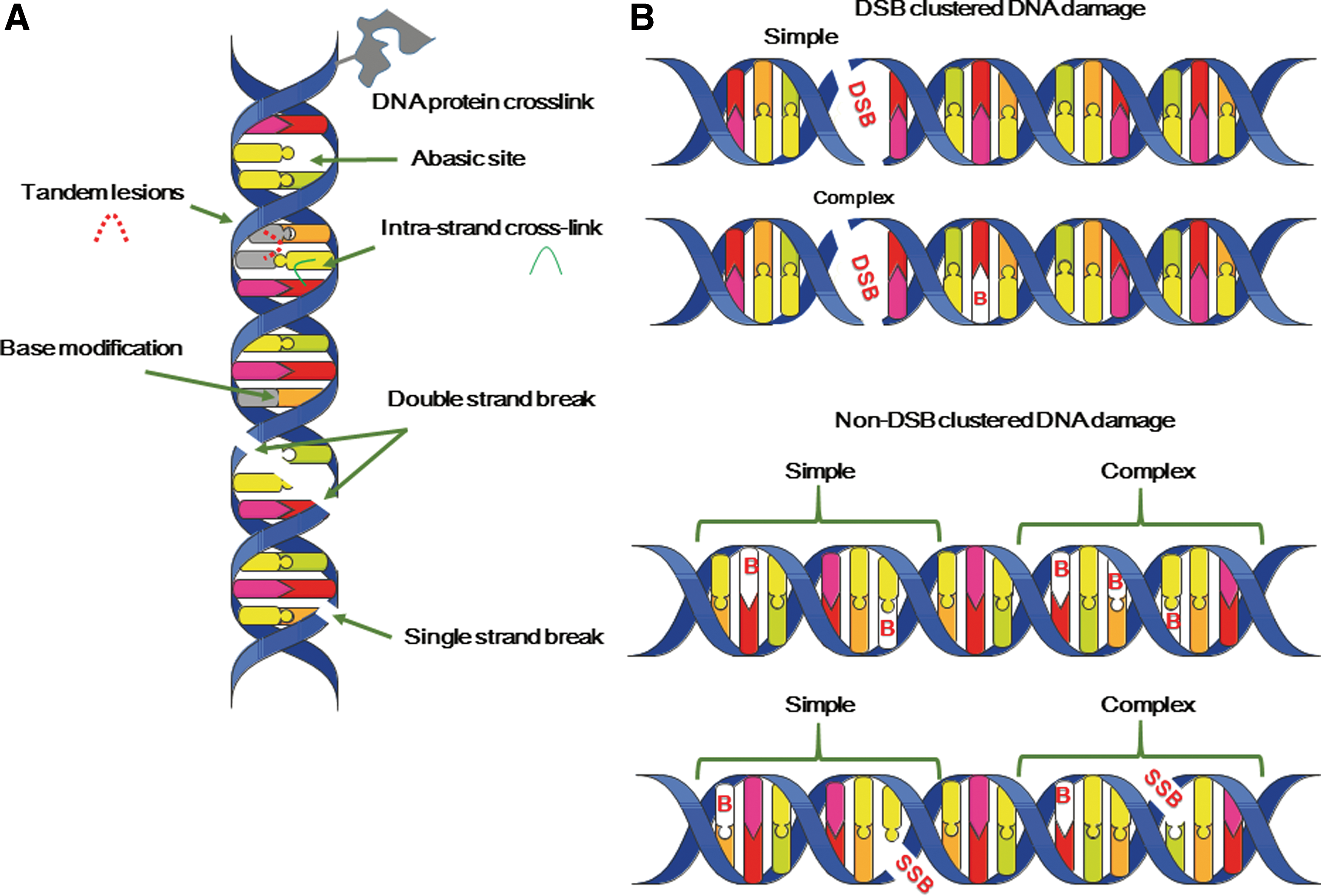
In addition, high-LET radiations, which induce more dense ionizations, produce, relatively to SSBs, more DSBs compared with low-LET radiations. In fact, when the LET of the radiation increases, the number of DNA lesions per unit dose (Gy) decreases (242) (due to an increased probability of radical recombination), but at the same time, MDS complexity increases (
However, recent works have highlighted the fact that a single oxidation event also can produce several modifications (called tandem lesions), composed of two adjacent modifications. For example, as illustrated in Figure 4 for a dG-dC sequence, in the absence of oxygen, crosslinks between two adjacent DNA bases could be produced, generating intrastrand crosslinks (G[8-5]C adduct) (128). Within the same sequence, in the presence of oxygen, the reaction of the initially produced radical with O2 produces a hydroperoxide radical that can add to the C8 position of an adjacent guanine (or adenine) base. The resulting unstable endoperoxide undergoes decomposition, producing tandem DNA lesions that involve two adjacent DNA modifications (253), such as 8-oxodGuo and a formylamine residue (8-oxodGuo-dF, Fig. 4) (29). The latter observation, although obtained using isolated DNA, indicates that a radical produced on a DNA base can react with surrounding molecules and particularly with adjacent bases, giving rise to possibly two adjacent modifications.
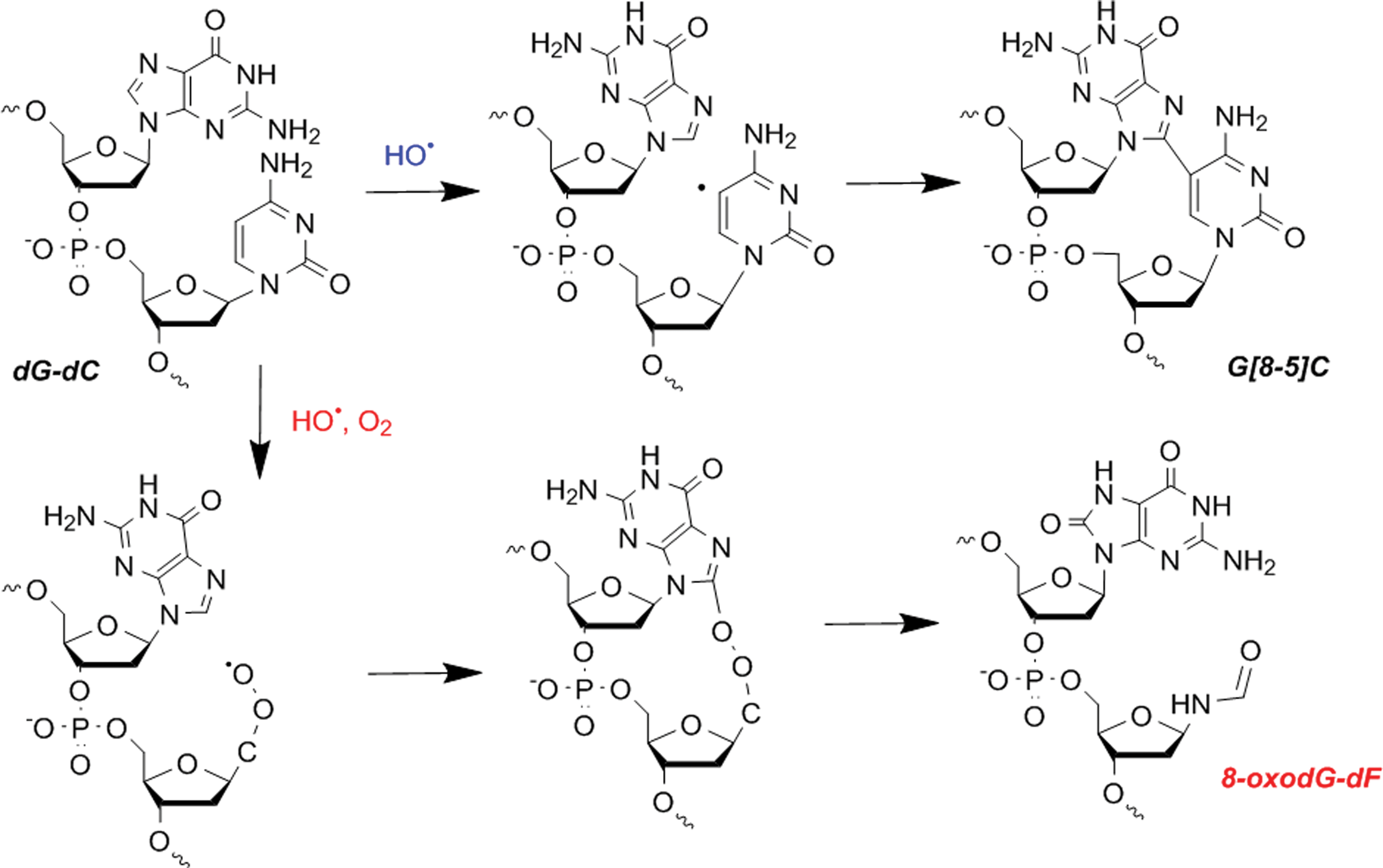
Moreover, it has been shown that the repair of 8-oxodGuo involved in tandem lesions is significantly reduced compared with the repair of a single lesion (19). At the cellular level, the decomposition reactions of an initially produced radical will strongly depend on its environment. The fact that carbon-centered radicals can react efficiently with molecular oxygen provides a partial explanation for the well-known oxygen enhancement effect, observed in radiation biology. As illustrated in Figure 4, the DNA lesions generated in the absence of oxygen could be different from those created in oxic conditions. Low oxygen concentration could indirectly increase the lifetime of the initially produced radicals, thus favoring the possibility of radical recombination and ultimately decreasing the amount of radiation-induced DNA lesions (250). In addition, the increased lifetime of radicals could also promote repair, for example, by electron donation from mild reducing amino acids (193) or antioxidant molecules that could be used as radioprotectors (192).
This could provide a possible explanation for the reduced level of lesions produced under hypoxic or anoxic conditions (154). Nevertheless, increasing the lifetime of a radical could also favor its reaction with other cellular constituents, leading to the generation of more complex DNA lesions that could compromise DNA repair (154). Indeed, the presence of polyamines, which are highly concentrated in the nucleus of eukaryotic cells, or of a nucleophilic amino acid could lead, respectively, to the formation of DNA adducts (285) and DNA-protein crosslinks (206). These DNA damage types are repaired less efficiently than single lesions (19). These examples illustrate the complexity of the chemical reactions that can occur in cellular DNA following exposure of cells to ionizing radiation (47) and that can vary in oxic and hypoxic conditions.
Although the literature on damage to mitochondrial DNA (mtDNA) is less abundant, mtDNA also is affected by irradiation (358), and additional work is needed to better understand the consequences of this damage (143). This is also true for the nucleotide pool (110) and RNA that are more sensitive to oxidative stress (126) than DNA, which is compacted and protected within the nucleus.
B. Effects of irradiation on lipids
The lipid layer of cell membranes is also a radiation target (59, 296). Damage to lipids is mediated by ROS reaction with polyunsaturated fatty acids (PUFA) (209). The initial step involves the abstraction of the bisallylic position of PUFA (Fig. 5A) either by HO• or mediated by a thiyl radical (RS•). The produced carbon-centered radical is converted into a peroxyl radical after reaction with molecular oxygen at a diffusion-controlled rate. Then, the peroxyl radical can abstract a hydrogen atom from another PUFA, inducing the so-called peroxidation reactions (271). Decomposition of the hydroperoxide generates breakdown molecules that contain reactive carbonyl groups, such as malondialdehyde (MDA), acrolein, and 4-hydroxy-2-nonenal (4-HNE). These decomposition products could be used as markers of lipid peroxidation reactions. Moreover, if they are not trapped by the cellular defenses (particularly glutathione [GSH]), they could generate secondary decomposition products because they can react with cellular biomolecules (
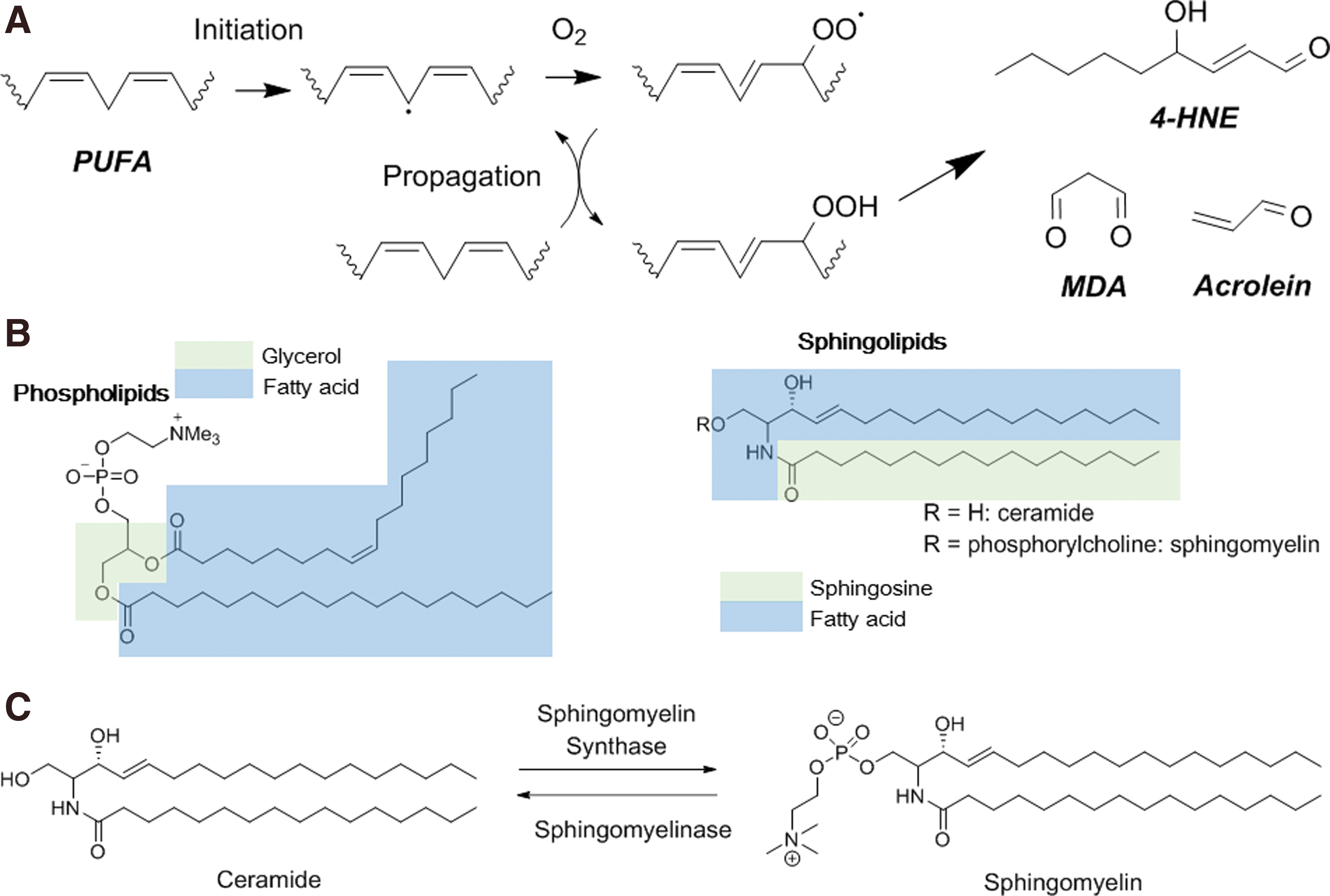
Lipid peroxidation can cause various biological effects: increase in membrane permeability, disruption of ion gradients and transmembrane processes, as well as alteration of the activity of membrane-associated proteins. Besides peroxidation reactions, irradiation could also induce
It has been clearly shown that the sphingolipid metabolism is altered following exposure to ionizing radiations. Sphingolipids are one of the four major lipid constituents of the cell membrane, in addition to phospholipids, sterols, and glycolipids. Irradiation induces rapid formation of ceramide through the hydrolysis of sphingolipid hydrolases and the concomitant decrease of sphingomyelin at the plasma membrane. Ceramide could be considered a second messenger that not only has an effect on the plasma membrane but also on intracellular signaling molecules (59).
C. Protein damage
An indirect evidence that ionizing radiations do not target only DNA but also highly abundant proteins is given by the effects of irradiation directed to the cytoplasm (353). The effects observed for doses as low as 1 Gy are changes in protein expression and activity that are mostly mediated by post-transcriptional modifications (302) and also by chemical modifications, including oxidation or carbonylation (255, 295).
Like for DNA, a considerable amount of work has been done to determine the reactivity of amino acids with radicals produced by irradiation (295). Due to its fast and high reactivity, HO• can react with all amino acids with a reaction rate between 107 and 1010
In addition to oxidation, proteins could also be modified by reduction, through reductant species produced by irradiation (
D. Targeted effects: conclusions
It is now very well documented that irradiation damages cellular biomolecules, such as DNA, RNA, proteins, and lipids. Most of the decomposition reactions of biomolecules mediated by ROS produced during irradiation have been identified. However, the complexity of the cellular media and the possible reaction of transiently produced radicals with the surrounding molecules lead to a great variety of cellular processes induced by radiation that makes almost impossible their precise description/identification. This is a matter of concern for DNA lesions and it cannot be totally excluded that not yet identified lesions are produced in cells exposed to ionizing radiation. The most harmful lesions are not necessarily those produced in higher yields, such as base lesions or SSBs that can be repaired rapidly and with high fidelity. Radiation-induced lesions are not different from those produced by endogenous oxidative stress. However, due to their special distribution in clusters, which hampers their repair, they can be very harmful for the cells.
III. Nuclear-Centered View of the Cellular Response to Radiation
While SSBs and base damages are efficiently repaired by SSB repair (SSBR), base excision repair (BER), and nucleotide excision repair (NER), DNA DSBs and clustered DNA lesions in general are mainly responsible for the final cellular outcome on irradiation (269).
A. DNA DSB repair
DNA DSBs are detected by surveillance proteins of the phosphatidylinositol-3-kinase (PI3K)-like kinase family, particularly the serine/threonine protein kinase ataxia-telangiectasia mutated (ATM), which is the main sensor of DNA damage, and ATM- and Rad3-related (ATR) protein (Fig. 6) (185, 231, 282). Both are actively recruited to DNA DSB sites to monitor the DNA damage response (DDR).
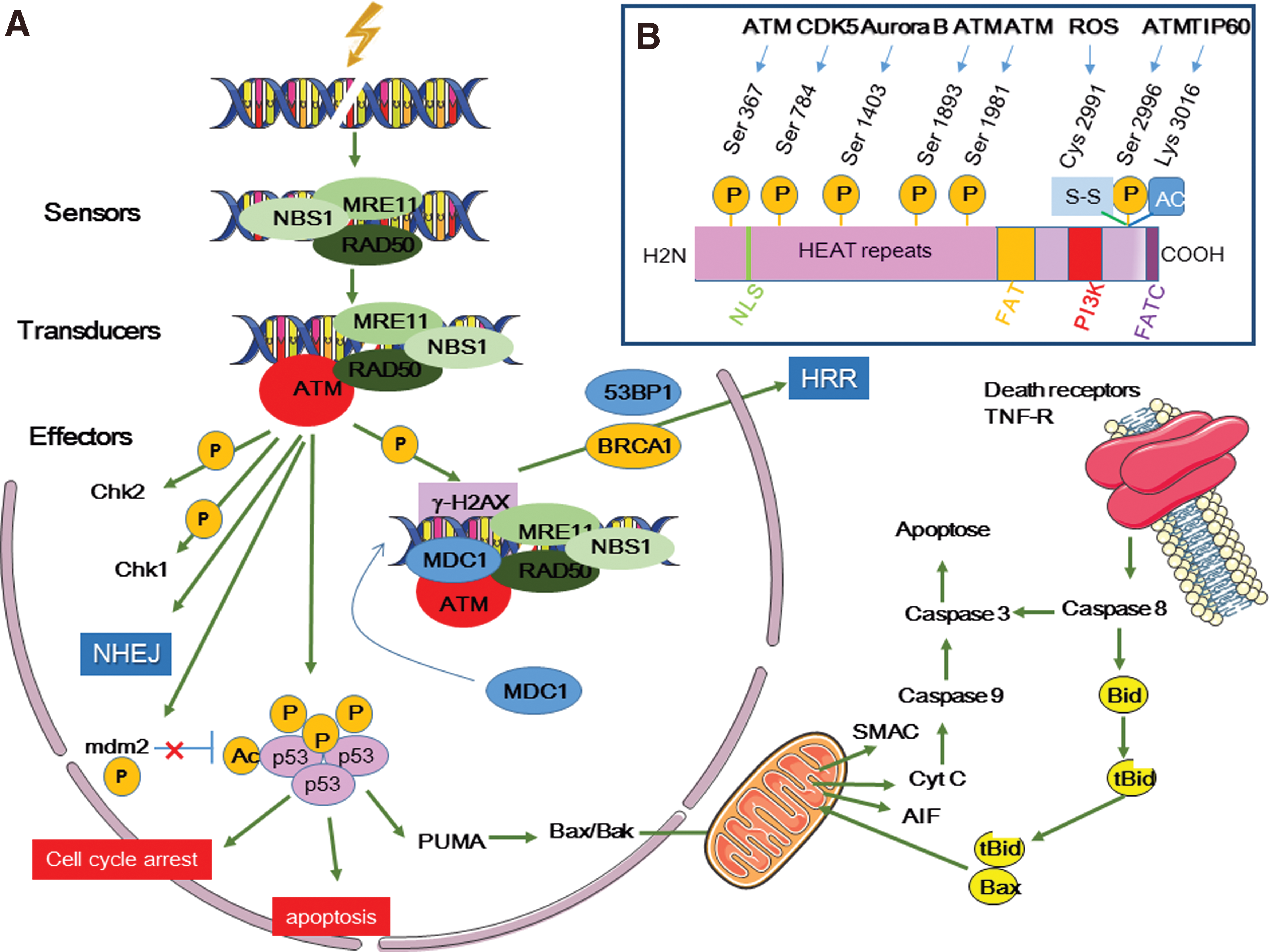
DDR is activated to avoid the transmission of erroneous genetic information to daughter cells and to protect cells from deregulated metabolism. It includes mechanisms involved in DNA damage detection, signaling, and repair, when possible (135), or in the initiation of programmed cell death or senescence.
ATM activation is mediated
However, ATM full activation requires the recruitment of the MRE11–RAD50–NBS1 (MRN) complex at DNA DSB sites before induction of the DDR systems (nonhomologous end-joining [NHEJ], and homology-directed repair, HDR). The MRN complex also is phosphorylated by ATM. This complex forms a bridge between DNA ends. Then, its nuclease activity, mediated by the nuclease MRE11, resects DNA DSB ends, a crucial step for homologous recombination repair (HRR) (282). Moreover, interaction between NBS1 and ATM is essential for maintaining ATM at DSB sites. Breast cancer type 1 (BRCA1) and p53-binding protein 1 (53BP1) are involved in this interaction. ATM also interacts with a mediator of DNA damage checkpoint protein 1 (MDC1) (170, 185, 231, 282). MDC1 is a DNA damage sensor protein located at DSB sites where it binds to phosphorylated histone H2AX (γH2AX) and ATM. Additional H2AX phosphorylation by ATM allows the recruitment of more MDC1 molecules that bind to ATM and γH2AX in a positive feedback loop, leading to ATM and γH2AX spread over large domains (>500 kb) around DNA breaks and amplification of DNA DSB signaling (136, 170, 185, 231, 282).
Other ATM substrates are proteins involved in G1-S (p21), intra-S (Fanconi anemia group D2 protein [FANCD2], BRCA1, and structural maintenance of chromosomes protein 1 [SMC1]), and G2-M cell cycle arrest (checkpoint kinase 2 [CHK2] or DNA repair [poly(ADP-ribose) polymerase 1 (PARP1), the nuclease Artemis, C-terminal-binding protein 1-interacting protein (CtIP), DNA-dependent protein kinase (DNA-PK) cs84, polynucleotide kinase 3′-phosphatase, DNA-PK, and AKT]. These factors allow DNA repair according to the NHEJ pathway before replication and mitosis (147, 163). ATM is also activated and dissociated into monomers by ROS
ATR recognizes DSBs, but can also be activated by many other DNA damage types. ATR is mainly involved in DNA DSB repair
According to this school of thought, DNA DSBs are the central lesions on irradiation and their formation, repair, or nonrepair influences the cellular outcome through complex interplays between cell death and survival signals.
B. Nuclear-centered view of the cellular response to radiation: conclusion
DDR includes a complex network of proteins activated by DNA lesions, mainly DSBs, produced in irradiated cells. ATM/ATR and MRN play a major role in DDR by recruiting proteins involved in DNA lesion detection and repair, in cell cycle progression, and in cell metabolism (
IV. Off-Target Effects: An Integrated Cell Response to Radiation
According to the United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) 2006 report, “bystander effect” is “the ability of irradiated cells to convey manifestations of damage to neighboring cells not directly irradiated,” and “abscopal effect” is “a significant response in a tissue that is physically separate from the region of the body exposed to radiation” (314). Similarly, the International Commission on Radiological Protection describes the bystander effect of radiation as the transmission of signals from irradiated to nonirradiated cells in a cell population, leading to biological changes in the recipient cells (133a, 307) (Fig. 7).
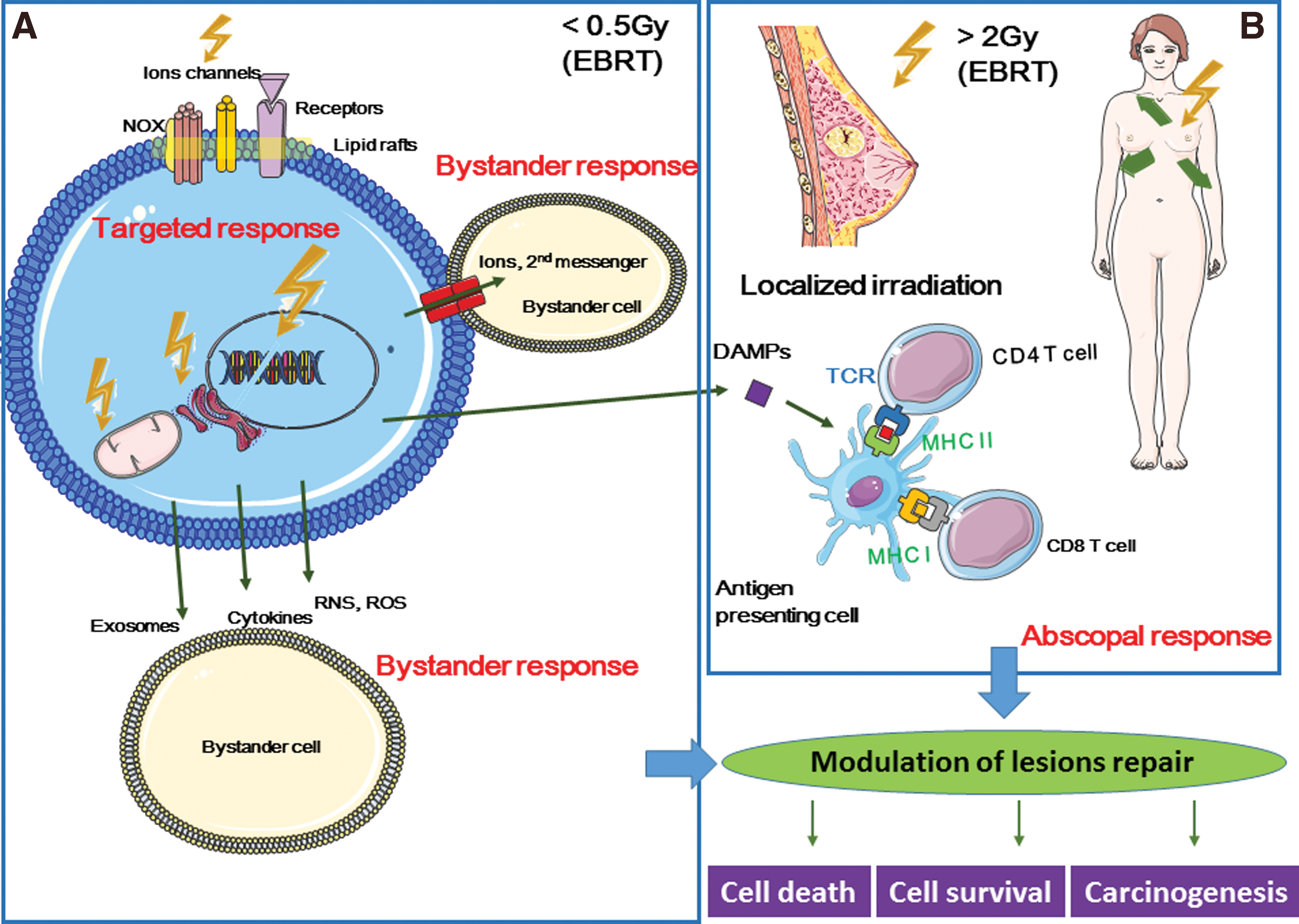
Off-target effects also include induced genomic instability. This is the delayed and stochastic appearance of genomic alterations (chromosomal aberrations, gene mutations, and reproductive cell death) in the offspring of irradiated (and also of bystander) cells, although these daughter cells were not irradiated (198, 199, 200, 201).
During the last 25 years, many studies have investigated the molecular mechanisms underlying off-target effects. They showed that off-target effects are the results of a huge dynamic and integrated process initiated in irradiated cells and transmitted to neighboring cells and to some extent to the whole organism through the activation of the immune system.
A. Bystander effects
Although off-target effects were first described by Parsons in 1954 (202), Nagasawa and Little performed one of the key early studies that highlighted the need to rethink radiobiology by also considering bystander effects. They used very low mean doses of alpha particles (0.31 mGy) to demonstrate that although <1% of cell nuclei were actually hit by radiation, 30% of cells showed an increased frequency of sister chromatid exchanges (205). Since then, many
These studies showed that bystander effects depend on the radiation dose, dose rate, and LET (104). They proposed that the contribution of bystander effects to radiation-induced biological effects in conventional external beam radiotherapy (EBRT) and also in therapies using heavy ions, protons, or radionuclides (5, 23, 31, 222, 227, 228, 339) should be taken into account for future applications.
Many cellular components and functions contribute to the extranuclear response to radiation. They include ceramide production and lipid raft formation (111), tyrosine kinases such as epidermal growth factor receptor (EGFR) (270), cytoplasmic Ca2+ homeostasis mechanisms (304), protein kinase C (PKC), MAPKs, JNKs (297), phospholipase C (PLC), NF-κB-mediated cyclooxygenase-2 (COX-2) and nitric oxide (NO) synthase (NOS) activation, and cytokines.
Approaches for investigating bystander effects rely on conventional broadbeam and microbeam irradiation to selectively irradiate a single cell or its subcompartments while sparing neighboring cells, or on the transfer of conditioned culture medium from irradiated to nonirradiated cells. The main evidence for extranuclear radiation-induced effects came from cytoplasmic irradiation using microbeams (53, 276, 335). Moreover, Gaillard
ROS and reactive nitrogen species (RNS) initiate events in targeted cells and also participate in their propagation through self-sustained production, leading to similarities between bystander effects and inflammation/immune response. Indeed, the so-called damage-associated-molecular-patterns (DAMPs) released by irradiated tissue can be detected by the innate immune system, as observed for pathogen-associated molecular patterns. Released factors are then recognized by Toll-like receptors (TLRs), C-type lectin receptors, nucleotide binding oligomerization domain-like receptors, and cytosolic retinoic acid-inducible gene-like receptors (151) that are expressed at the surface of immune cells.
1. Intercellular communications between irradiated and nonirradiated cells
Bystander effects involve signaling from irradiated cells to nonirradiated cells. The nature of the communications between irradiated and nonirradiated cells depends on whether cells are in contact or not. This cross talk can be mediated by paracrine secretion in the extracellular space of soluble factors
Cxs and Panxs can also form full channels that are called gap junctions and mediate gap junction intercellular communication (GJIC). These intercellular channels show much higher selectivity and allow direct diffusion of factors between the cytoplasm of adjacent cells. The factors involved in bystander effects and transiting through intercellular gap junctions are small molecules (<1.5 kDa) and include ROS (9, 10), RNS (187), ions (Ca2+, K+, Na+), (178, 279), lipid peroxides (83), ATP (221), cyclic adenosine monophosphate (cAMP), glucose, glutamate, and GSH. As hemichannels are less selective than full gap junctions, it has been suggested that in normal conditions only GJIC plays a role. Conversely, in conditions of oxidative stress (
2. Reactive oxygen and nitrogen species initiate and propagate bystander effects
Early experiments highlighted the multiple roles of ROS and RNS in bystander effects. Specifically, they showed that radical scavengers (ascorbic acid, N-acetyl
Oxidative processes that involve ROS and RNS continuously occur in cells because at low concentrations (0.01–0.001 n
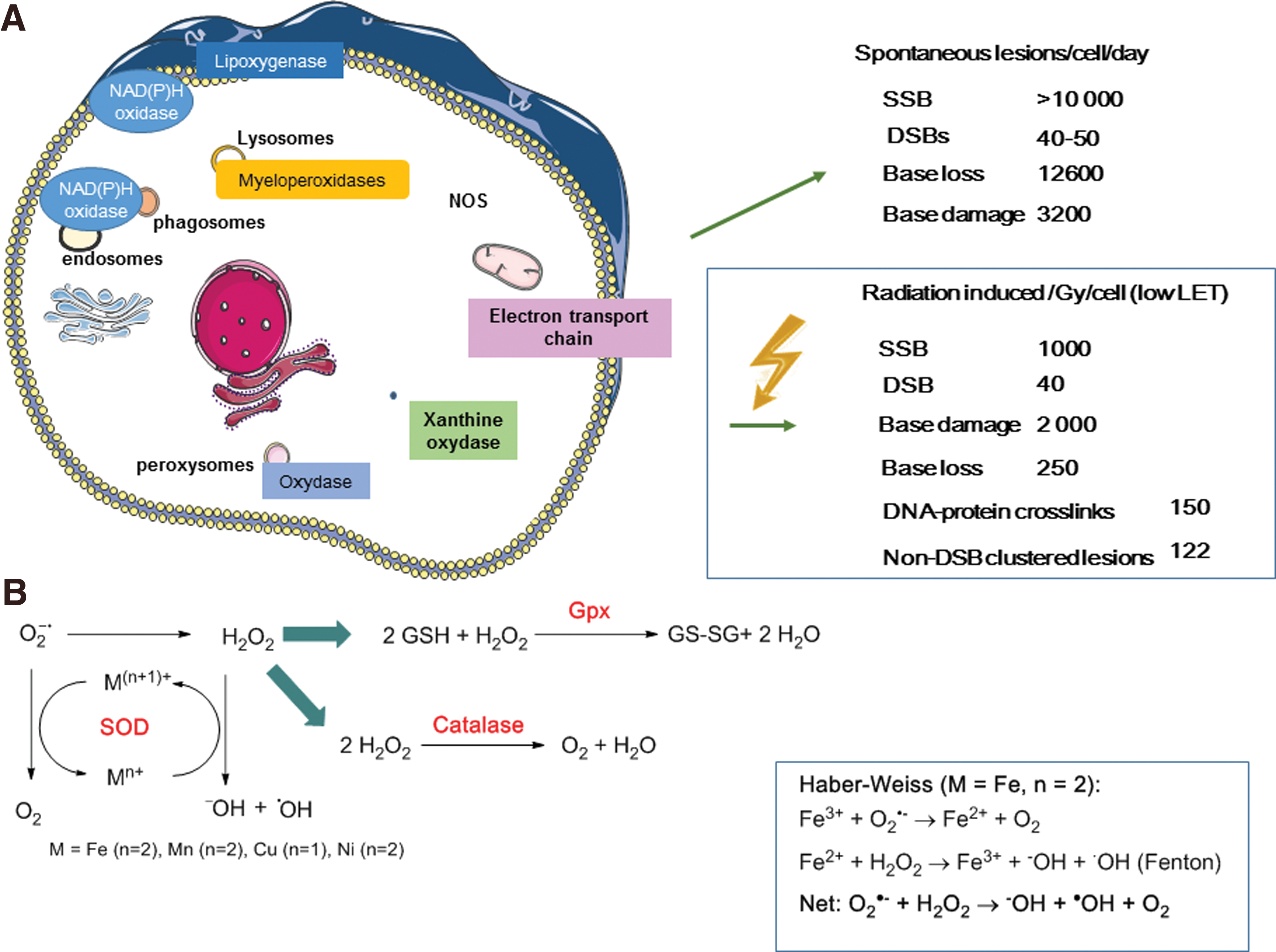
O2 −• is one of the main endogenous cellular ROS compounds (Fig. 8). It is produced by reduction of ground-state molecular oxygen (3O2) by enzymatic (nicotine adenine dinucleotide phosphate [NAD(P)H], xanthine oxidase) or nonenzymatic (semi-ubiquinone Q−, a compound of the mitochondrial electron transport chain [ETC]) systems (80).
Peroxisomes also contain enzymes, namely
O2 −•concentration is also regulated by three SODs, namely the cytoplasmic Cu–Zn-dependent superoxide dismutase (CuZnSOD or SOD1), the mitochondrial MnSOD (or SOD2), and the extracellular SOD (ECSOD or SOD3), that catalyze O2 −•dismutation into the less reactive H2O2. This reaction requires reduced transition metals, such as iron or copper ions, and leads to the formation of the precursors of the highly damaging hydroxyl radicals (HO•), according to the Fenton reaction (Fig. 8). In cells and in mitochondria, H2O2 can be decomposed into H2O and O2 by GPx, catalase, or peroxiredoxins (Fig. 8).
The initial production of ROS by water radiolysis is limited in time, space, and quantity. If one considers that 2000 ionization events are produced per cell per Gy (for low LET radiation), clinical doses of 2 Gy produce much less ROS than the amount generated during standard cell metabolism (329). For instance, a dose of 1 Gy would produce 1 × 103 DNA breaks per cell (about 1000 SSBs and 40 DSBs) (102, 241, 245), while endogenous metabolism would produce more than 10 × 103 DNA breaks per day (262, 318).
Therefore, radiotherapy efficacy in tumor cells can be partly explained by the amplification and prolongation in time (hours and weeks) of the initial radiation-induced ROS production. This would involve ROS- and RNS-mediated activation of signaling pathways and signal transmission to neighboring cells. The main sources of endogenous ROS that participate in this amplification process include mitochondria, NAD(P)H oxidases, and other oxidases, such as xanthine oxidase, lipoxygenases, and peroxisomes (80).
a. Mitochondria-dependent ROS production
Human-hamster hybrid AL cells with normal (ρ+) or depleted (ρ0) mtDNA were used to demonstrate the contribution of mitochondrial functions in the generation of bystander signals from irradiated cells (53, 127, 355).
(1) Mitochondria and ROS endogenous production
Mitochondria produce energy through the oxidative metabolism of carbohydrates, lipids, and amino acids that generate NADH
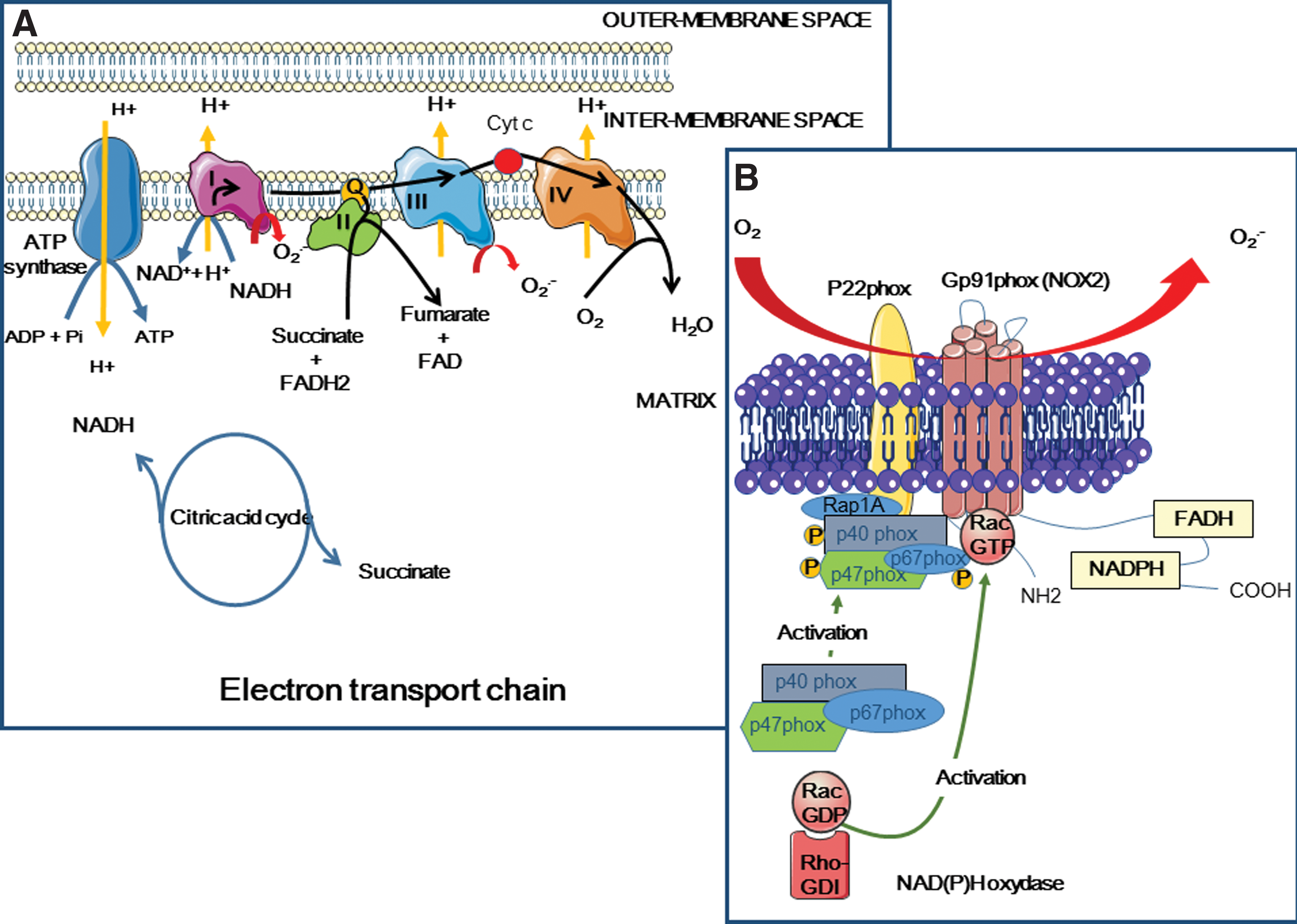
(2) Mitochondria and irradiation
Mitochondria are essential in the intrinsic apoptosis pathway (but not in the death receptor-mediated extrinsic pathway). Specifically, mitochondrial membrane permeabilization and cytochrome C release are followed by caspase activation. The intrinsic apoptosis pathway involves PUMA, a proapoptotic protein that allows BAX and/or BAK translocation to mitochondria to signal apoptosis.
The total cellular mitochondrial volume is high (4–25% of the cell volume, depending on the cell type and state), and therefore, mitochondria represent a fairly substantial target for cytoplasmic irradiation. Radiation produces many different types of mitochondrial damage, including mtDNA damage. mtDNA is a circular double-stranded genome that encodes proteins, transfer RNA, and ribosomal RNA. It lacks protective histones and shows limited repair capability. Therefore, on radiation exposure, mtDNA could be preferentially damaged or lost (217), leading to loss of mitochondrial function (149, 316). Moreover, it has been shown that microbeam irradiation with carbon ions leads to depolarization of mitochondria (323). Exposure to direct gamma radiation causes mitochondrial mass increases (218), and alpha-particle microbeam irradiation leads to mitochondrial fragmentation, involving dynamin-related protein 1 (DRP1), a member of the dynamin family involved in mitochondrial fusion and fission (348).
Cytoplasmic irradiation also has been associated with altered protein synthesis and irregular mitochondrial oxidative phosphorylation (66, 217, 348), leading to persistent oxidative stress (162, 345). Yoshida
Chen
Zhou
Finally, mitochondria are affected also in nonirradiated cells grown in culture medium from irradiated cells. Specifically, changes of mitochondrial distribution, loss of mitochondrial membrane potential, and mitochondrial mass (218), increased ROS and RNS production, and increased apoptosis rates have been observed in such cells. These effects can be blocked by antioxidant treatments (180).
b. NAD(P)H oxidase-dependent ROS production
The plasma membrane-bound NAD(P)H oxidase (NOX) is another endogenous source of ROS. It is found in phagocytic cells, such as macrophages and neutrophils (innate immunity), where it was first identified (14, 259). NOX-1 participates in pathogen killing
NOX includes several subunits, such as membrane proteins (gp91phox or NOX-2 and p22phox that constitute flavocytochrome b558, and the small G Rap1A protein) and cytosolic proteins (p40phox, p47phox, p67phox, and the G Rac2 protein). Flavocytochrome b558 contains heme subunits with low oxydo-reduction potential (−225 and −265 mV at pH 7.0) and is mainly involved,
In irradiated fibroblasts, NOX activation does not require direct nuclear or cellular “hits” by alpha particles and leads to O2
−• and H2O2 generation, and also to persistent ROS production in bystander cells (207). NOX is located in ceramide-enriched lipid raft domains, the disruption of which leads to NOX activity inhibition (347). For instance, transforming growth factor (TGF) β1 secreted by irradiated cells activates NOX-1 and the subsequent O2
−• production in bystander cells (277). Moreover, TGFβ1-mediated activation of DUOX proteins and the release of the peroxidase domain by metalloproteases (1) are required for the HOCl signaling pathways. O2
−• production by NOX might participate in the activation of ASMase, leading to NOX activation in ceramide-enriched domains (337). It was shown that irradiation induces NOX/DUOX-1-dependent H2O2 production for several days (6). This process involves p38 MAPK-mediated activation of NOX
c. ROS and RNS as second messengers
Most ROS, such as HO•, are very reactive and have a very short life, and thus react within a few nm range from their site of production. They cannot be transmitted to neighboring cells. However, H2O2 and NO can diffuse through the cell membrane. H2O2 can cross membranes through aquaporin channels (21) and can diffuse through the cytoplasm and plasma membranes of neighboring cells along several cell diameters (13, 340). NO, one of the main RNS, can damage DNA (211), is mutagenic, and is involved in proapoptotic signal transduction (211). NO also plays a role in blood compartment functions, including smooth muscle tone and blood pressure regulation, platelet activation, and vascular cell signaling. Thanks to its lipophilic properties and relative stability, NO can activate signaling processes in adjacent cells (161).
NO is upregulated on oxidative stress and therefore can compete for substrates with SOD by reacting with O2
−• to form the diffusible peroxynitrite (ONOO−) (13, 340). In agreement, incubation with L-NAME (an NOS inhibitor), but not with rotenone (an inhibitor of electron entry into complex I of the mitochondrial ETC), leads to an increase of oxidative stress. This suggests that constitutive levels of NO production contribute to the regulation of mitochondrion-derived intracellular oxidant generation (99). When NO concentration increases to the level of SOD, ONOO− can rearrange into biologically inert nitrite (NO2
−) or react with GSH to form the NO donor GSNO (161). However, it can also spontaneously and rapidly decompose to nitrogen dioxide (NO2) and HO•, thereby oxidizing lipids, thiols, amino acid residues, DNA bases, and low-molecular-weight antioxidants, as done by ROS cellular constituents (13). NO can also modify proteins, leading to S-nitrosylation (80, 164) or nitration, mainly tyrosine nitration, a marker of tissue inflammation. This posttranslational modification participates in the regulation of cellular functions. For example, NO affects the DNA repair mechanisms by downregulating the expression of BRCA1, involved in HRR and cell cycle checkpoint control, while promoting the error-prone NHEJ mechanisms. This regulation seems to be mediated
NO production from
Pretreatment with the NO scavenger c-PTIO of cells exposed to microbeam with alpha particles abolishes MN formation in bystander cells (280). Moreover, incubation of a mouse leukemic monocyte macrophage cell line (RAW 264.7) with lipopolysaccharide (LPS) induces iNOS activity and NO generation, thereby increasing DNA damage in bystander EL-4 lymphoma cells (97).
3. Cell membrane response to radiation
Compared with DNA-centered approaches, relatively few radiobiological studies have investigated the role of radiation targeted at the cell membrane that, for long time, was considered to be just an inactive phospholipid bilayer. However, in the 1990s, several studies showed the production of ceramide, a cellular second messenger of apoptosis involved in the sphingomyelin signaling pathway, during membrane irradiation (111). Using lymphoblasts from patients with Niemann–Pick disease (deficiency in ASMase activity), it was demonstrated that this enzyme is required for radiation-induced production of ceramide and apoptosis (263). Many studies now strongly support the ceramide-mediated cell membrane role in the biological effects of radiation, including cell death. Moreover, radiation resistance in Burkitt's lymphoma cells has been associated with defective ceramide signaling (189). Finally, structure-modulating agents (
a. Cell membrane and lipid rafts
Cell membrane contains lipids (sphingolipids and glycerophospholipids), proteins, and sterols (Fig. 5B).
Lipids, such as PUFA, are susceptible to free radical-initiated oxidation (49), leading to lipid hydroperoxide formation that can then be reduced by peroxidases. If not efficiently reduced, lipid hydroperoxides can be degraded into hydroxyalkenals (such as 4-HNE) that show great reactivity toward DNA, proteins, and lipids (49).
Cholesterol and sphingolipids, among which sphingomyelin is the prevalent, mainly localize in the outer leaflet of the cell membrane. They play a crucial role in signal transduction and participate in cell growth, senescence, differentiation, and apoptosis (122). Sphingolipids contain a long-chain sphingoid base (such as sphingosine) linked
The concept of the cell membrane as a fluid mosaic (289) is based on the finding that most physiological phospholipids exhibit low melting temperatures and, therefore, most likely exist in a liquid disordered phase. For instance, in
However, the notion of fluid mosaic has been reconsidered because mammalian membranes contain very small domains that are in a liquid ordered phase (35, 287). Indeed, sphingolipids, which have a much higher melting temperature than other phospholipids in the cell membrane, interact with each other
Lipid rafts can be converted into larger membrane platforms by ASMase activity that hydrolyzes sphingomyelin to ceramide in rafts (Fig. 5B). Ceramide molecules then spontaneously associate to form ceramide-enriched microdomains that fuse into large ceramide-enriched membrane platforms, thus altering the biophysical properties of these membrane domains (167) (Fig. 10). ASMase is activated by multiple stimuli, including CD95, CD40, DR5/TNF-related apoptosis-inducing ligand (TRAIL), CD20, FcgRII, CD5, LFA-1, CD28, cytokines, and chemotherapeutic drugs (doxorubicin, cisplatin), and also by ionizing radiation. Ceramide is considered the second messenger of many factors. For instance, ceramide acts as a messenger of inflammatory cytokines by binding and exerting a dual effect on the cytosolic phospholipase A2 (cPLA2) involved in eicosanoid formation (132). Ceramide is also involved in apoptosis. Its binding to the endosomal acidic aspartate protease cathepsin D results in the autocatalysis of the 52 kDa prepro-cathepsin D into the enzymatically active 48/32 kDa cathepsin D isoforms that mediate oxidative stress-induced apoptosis (120).
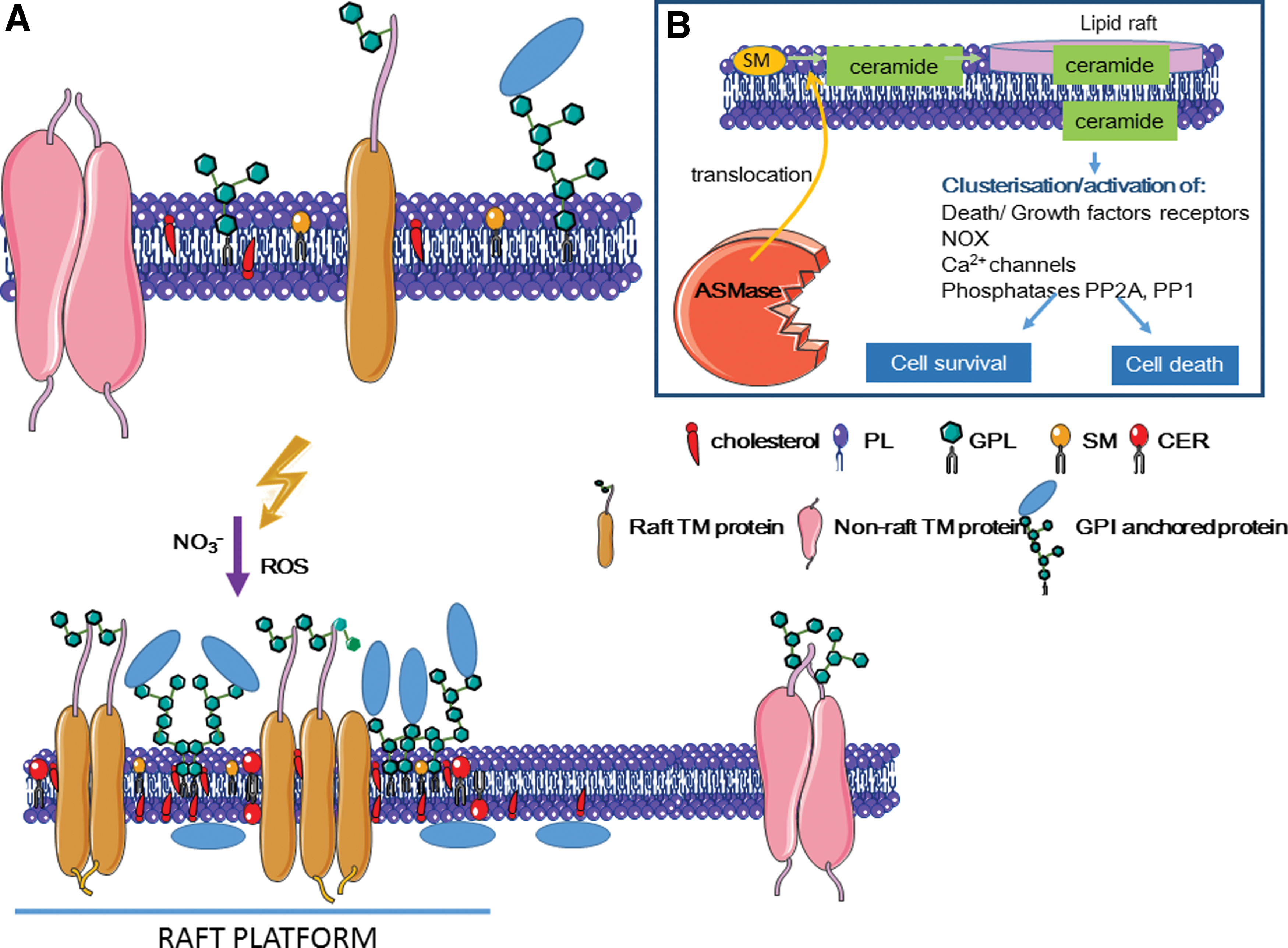
It has been proposed that oxidation of cysteine 629 in ASMase C-terminus by HO• could be responsible for a possible mechanism leading to increased ASMase enzymatic activity (251). Moreover, the RNS ONOO− specifically activates ASMase (48).
Ceramide is generated by ASMase in the outer leaflet of the cell membrane or within intracellular vesicles. It might also flip to the cytoplasmic leaflet and then interact with intracellular molecules. Although the mechanisms of ceramide translocation from one membrane leaflet to another are unknown, ceramide can interact and activate serine/threonine phosphatases, namely PP2A and PP1 (50, 58) (Fig. 10). Once activated, these phosphatases act on different signaling proteins, including MAPKs (AKT, c-JUN), PKC isoforms (PKCα and ζ), kinase suppressor of Ras (KSR), pRB, and BCL-2 (28, 349).
b. Lipid rafts in mitochondria-ER-associated domains
Several recent studies indicate that ceramide is also present in mitochondria (122). Mitochondrial ceramide could be generated
c. Caveolae, a subgroup of membrane lipid rafts
Caveolae are a subset of membrane lipid rafts that make small invaginations in the plasma membrane, ER, and Golgi apparatus. They contain caveolins that act as organizing centers for cellular signal transduction (233). Caveolin-1 contains a tyrosine 14 phosphorylation site and its phosphorylation leads to its accumulation at focal adhesion sites (
d. Ion channels and lipid rafts
Ceramide-enriched membrane platforms are involved in the regulation of potassium (27) and calcium (56) channels. Calcium ion cytoplasmic level is critical for many cellular functions
Intracellular Ca2+ can activate NOX and iNOS, thereby promoting ROS and RNS formation, respectively. It can also modulate the activity of the transcription factors NF-κB and AP1 that are involved in COX-2 and iNOS transactivation and the subsequent release of ROS, RNS, and cytokines. Moreover, it activates kinases, including PI3K/AKT and MAPK.
In response to irradiation, Ca2+ levels increase (112, 319, 352). These changes can be described as oscillations or single transient changes within minutes to days after irradiation. The concentration of free Ca2+ in the cytoplasm is low (∼100 n
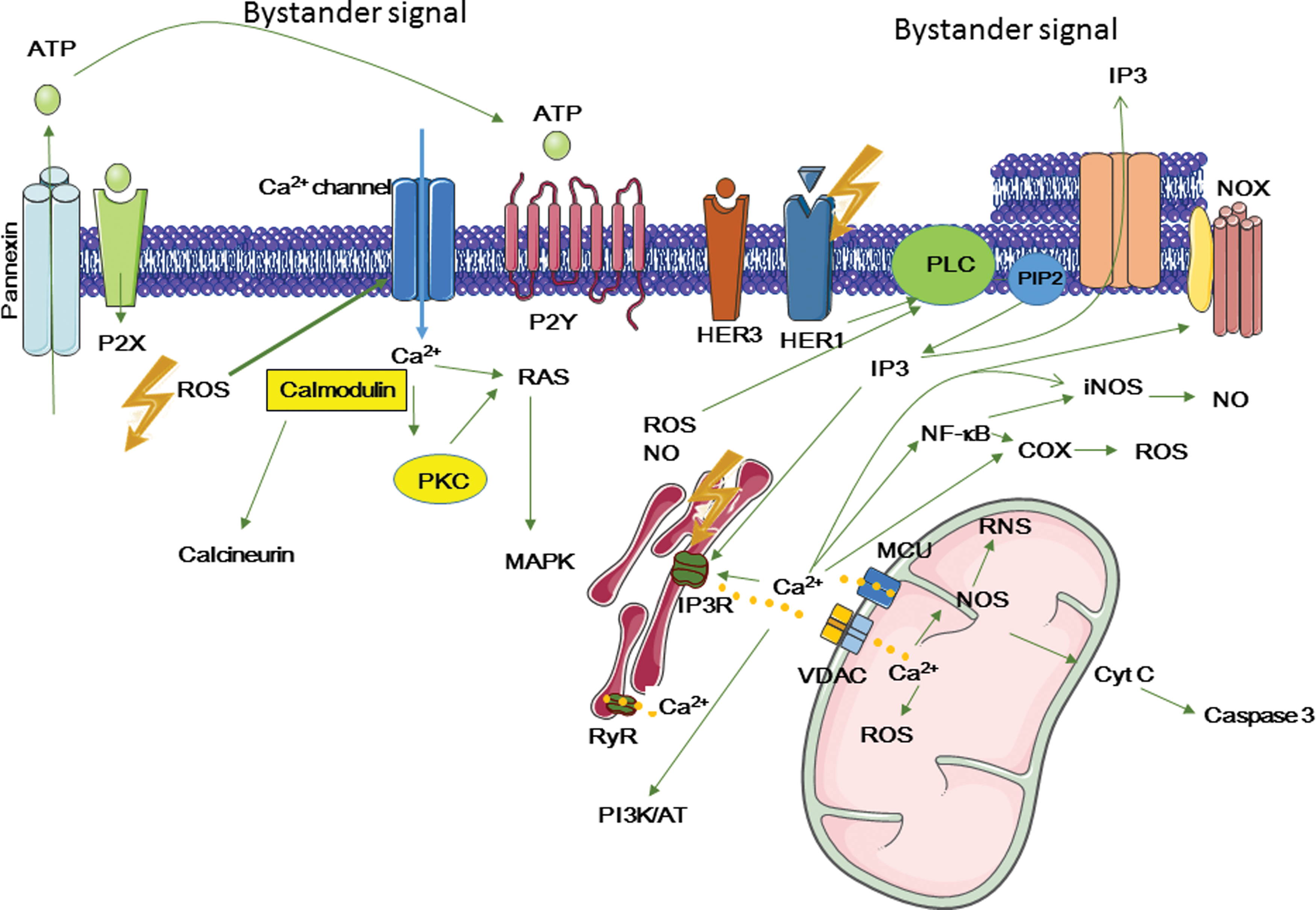
e. The role of Ca2+ ions in bystander effects
Ca2+ ion role as second messenger has been highlighted by several studies using calcium chelators or blockage of voltage-gated
Ca2+ released from ER can be transferred to mitochondria through voltage-dependent anion channels (VDACs) and mitochondrial Ca2+ uniporter (79, 257). Accumulated Ca2+ can then activate mitochondrial metabolic functions and stimulate energy production
Studies in which human keratinocytes were incubated with conditioned medium from gamma particle-irradiated cells in the presence of EGTA, verapamil, nifedipine, or thapsigargin (known to act on calcium homeostasis) showed the involvement of calcium and the activation of multiple MAPK pathways, such as the ERK, JNK, and p38, in the production of radiation-induced bystander effects (178). Similar results were obtained during
f. ROS/RNS and growth factor receptor activation
Lipid raft-mediated membrane reorganization can lead to activation of receptor and nonreceptor tyrosine kinases, such as ERBB1, that can homodimerize or heterodimerize with other members of the ERBB receptor family (ERBB2, ERBB3, and ERBB4) (91), insulin-like growth factor-1 (IGF-1) receptors, and death receptors (Fig. 12). Binding of growth factors to their tyrosine kinase receptors leads to activation of MAPK and PI3K/AKT signaling pathways. These processes are mediated by similar signaling pathways. Indeed, activation of ERBB1, 2, and 3 leads to activation of RAS family members (K-RAS, N-RAs, H-RAS) and further downstream signaling pathways that involve MAP3K (MEKK2/3, RAF-1/B-RAF), MAP2K (MEK1/2; MEK5), and MAPK (ERK1/2) and in turn activate transcription factors, such as AP1, NF-κB, and CREB (315). Similarly, death receptors activate two other MAP3K/MAP2K/MAPK pathways resulting in the activation of p38 (α−γ) kinases and JNKs (1/2). Finally, IGF-1 receptor and its downstream kinases PI3K, PDK1, and AKT participate in mTOR activation and GSK-3 regulation. Ultimately, all these different pathways result in the activation of transcription factors involved in cell death or survival.
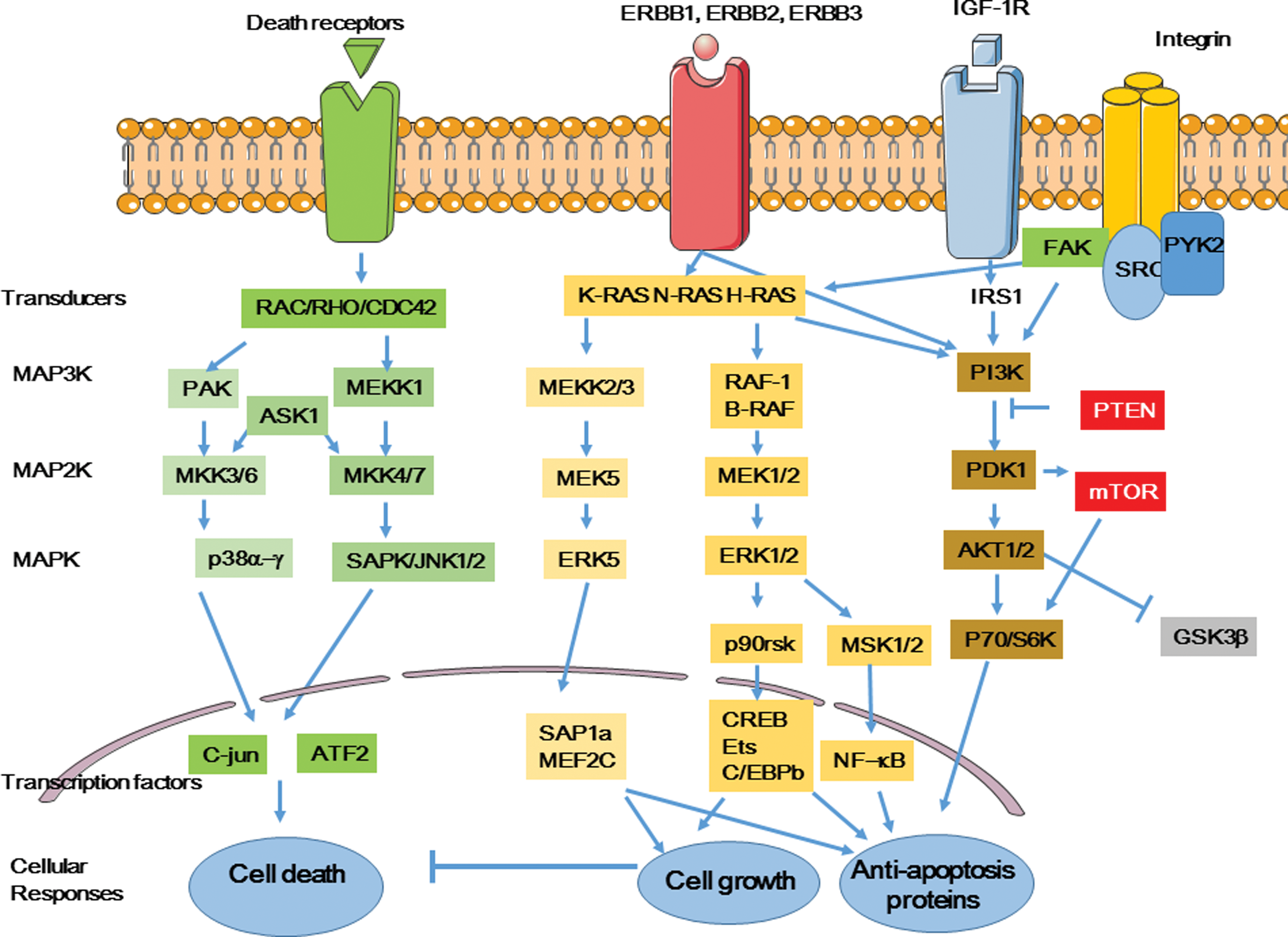
Radiation can modulate the production and expression of cytokine and growth factor receptors (54, 270, 305). For example, a 2 Gy dose can modulate the expression of cell surface receptors, such as ERBB1 (HER1) (305). As described above, NO and ROS produced by radiation can lead to defective mitochondrial function due to leakage of mitochondrial membranes, allowing the amplification of the initial signal through massive release into the cytosol,
The PI3K pathway, which plays a role in the long-term effects of cell survival, can also be activated on irradiation (315).
It was shown that activation of growth receptors is modulated by radiation according to waves. For example, Dent
g. MAPKs and the bystander response
The four major MAPK signaling pathways in mammalian cells (ERK1/2, ERK5, JNK1/2, and p38) can regulate either survival (ERK1/2 and ERK5) or apoptosis (JNK1/2 and p38). Inhibitors of the ERK pathway (PD98059 and U0126), JNK pathway (SP600125), and p38 pathway (SB203580, SB202190) helped demonstrating the role of these signaling pathways in bystander cell death induced by gamma/X-ray (55, 178), alpha particle (89), or carbon ion (77) exposure. Similar results were obtained after radionuclide therapy with Auger electron (228) or alpha particle-emitters (unpublished data). ROS and RNS have a role in MAPK direct and indirect activation. Incubation of confluent cultures of human diploid fibroblasts with CuZnSOD or catalase after irradiation with 0.003–0.03 Gy of alpha particles inhibits p21 (WAF1) upregulation by MAPK and the induction of MN formation in bystander cells (10). Moreover, p21 knockout results in inactivation of MAPK signal pathway kinases (55), and cNOS inhibition leads to reduction of ERK1/2 kinase activity (cytoprotective effect) (161), suggesting that diffusible NO could mediate MAPK activation in bystander cells.
4. Central role of NF-κB in the nuclear and extranuclear responses to radiation
Experiments showing that ATM activates NF-κB, which is involved in iNOS and COX activity, demonstrated the link between inflammation and DDR (336). Therefore, as the bystander and inflammatory responses share homologies, studying the role of NF-κB in bystander effects can be of major relevance.
a. Nuclear factor kappa B
NF-κB is a redox-sensitive transcription factor made of five different subunits that are organized in homo- or heterodimers: p50 (
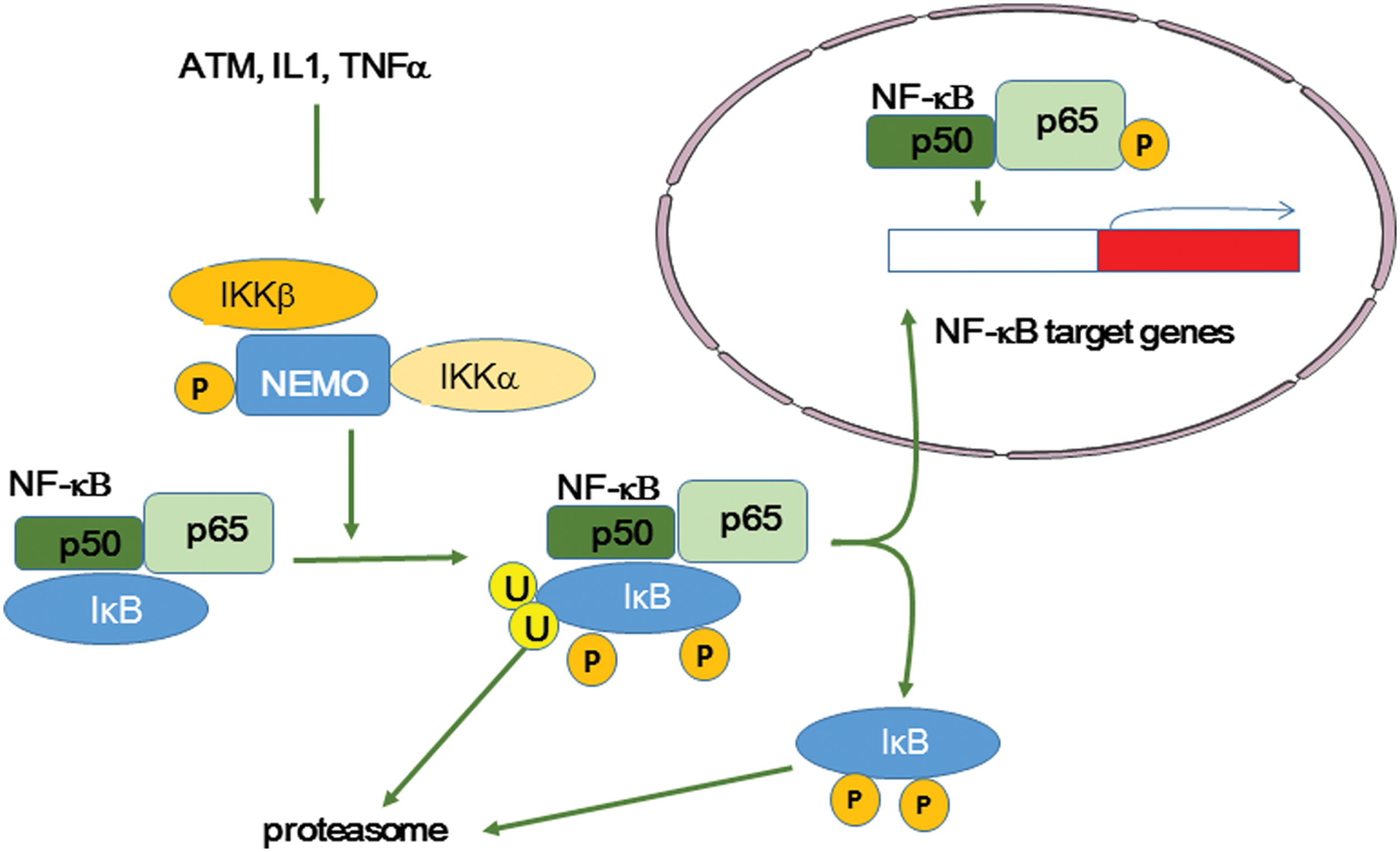
Specifically, TNFα binding to its receptor (TNFR) leads to a conformational change of TNFR that is accompanied by binding of TNFR type 1-associated DEATH domain protein (TRADD) to TNFR death domain. Two proteins, TRAF2 and receptor-interacting protein (RIP), are then recruited and in turn recruit the IκB kinase (IKK) complex, which is made of IKK-α (IKK1), IKK-β (IKK2), and the regulatory subunit IKK-γ/NF-κB essential modulator (NEMO) (Fig. 13). IKK is then activated and phosphorylates IκB before ubiquitination and proteosomal degradation to release NF-κB (121, 124). Among the plethora of targets with NF-κB responsive element sequences, COX-2, iNOS, and also cytokines (TNFα, IL1, IL6, IL33), chemokines (IL8, MCP-1), VEGF, ICAM1, and VCAM1 have been identified as major factors that can modify the microenvironment and trigger inflammation (95). These molecules may also be involved in oncogenesis (246).
b. NF-κB and irradiation
NF-κB can be activated by doses as low as 0.1 Gy of X-rays, but participates in the RIAR (84) also at higher doses (2–50 Gy) (32). RIAR was demonstrated by the observation that a priming dose (generally >0.005 Gy) reduces the detrimental effects of the challenging dose administered a few hours later (186, 333). This can be a beneficial effect for low-dose radiation, but might induce radioresistance during radiotherapy. Indeed, RIAR has been associated with reduced chromosome aberrations, MN formation, and mutation induction (223, 344, 356). RIAR shares similarities with inflammation. It was shown that low irradiation doses lead to ATM phosphorylation that contributes to NF-κB activation and to cell survival, in a process involving also ERK (but not p38/JNK) (4, 151). ATM participates in NF-κB activation by phosphorylating IκB and NEMO (165) that mediate NF-κB inhibition. NF-κB activation by low-dose irradiation leads to the expression of MnSOD 15 min postirradiation, and small interfering RNAs (siRNAs) against MnSOD reduce RIAR (84).
RIAR is also observed in bystander cells. Indeed, a priming dose of 0.02 Gy of γ−rays, delivered 6 h before single-cell microbeam irradiation, inhibited 50% of the bystander effects observed in control cells (no priming dose) (265).
Optimal NF-κB activation is observed at doses between 7 and 10 Gy and with high LET (90–230 keV/μm). In these conditions, NF-κB activation is mediated by ATM recruited at DNA DSB sites (336) and is an alternative pathway leading to p53-mediated signaling. p53 participates in cellular redox status by transactivating p53-induced genes (PIGs) that encode antioxidant molecules (GPx). In irradiated cells, p53 transactivates genes encoding ROS-generating enzymes, such as quinone oxidoreductase (NQO1, PIG3) and proline oxidase (POX, PIG6), BAX, PUMA, and p66SHC, thereby leading to oxidative stress and apoptosis (168, 240). However, the ATM-activated p53 signaling pathway is not required for the bystander response (95), as indicated by the finding that bystander effects can be observed in p53-null cells exposed to gamma rays (350) or to Auger-emitting radionuclides (227).
Several studies reported that common p53-regulated radiation response genes, such as cyclin-dependent kinase inhibitor 1 (
Following gamma or alpha irradiation, activation of NF-κB signaling leads to the production of cytokines and chemokines, such as IL-1α and β, IL-6, TNFα, CXCL1, CXCL2, and CXCL8 (121, 158) (Fig. 14). Activation can occur within few hours following irradiation and might be ATM dependent. A second activation wave can be observed after 24 h and this might be related to receptor binding by secreted cytokines, such as TNFα (24). Therefore, NF-κB could modulate the response of directly irradiated cells and also alert neighboring nonirradiated cells
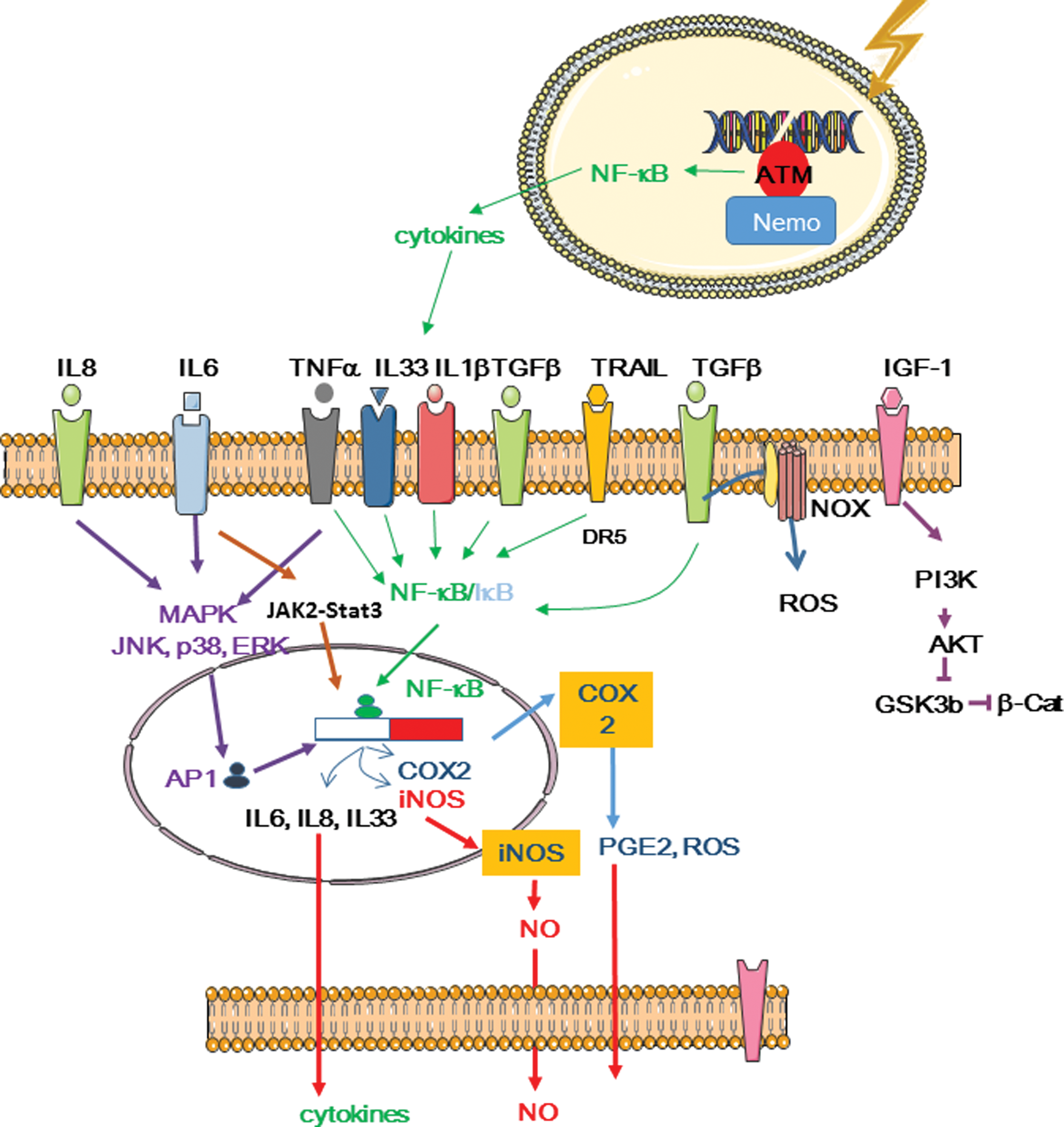
NF-κB regulates cytokine production in irradiated cells, but it is controlled by cytokine signaling in bystander cells (151). NF-κB-dependent expression of IL6
NF-κB can also interfere with the main MAPK signaling. More specifically, it suppresses the JNK cascade and ROS activity (36, 232). It must be noted that TRADD, a TNFR1-associated signal transducer, can also bind to TRAF2 that activates NF-κB and Fas-associated protein with death domain (FADD), two proapoptotic factors, whereas NF-κB is a potent antiapoptotic agent (129).
NF-κB also controls IGF-1 receptor that can activate the downstream (PI3K)-AKT survival pathway in both directly irradiated and bystander fibroblasts (94). In bystander cells, GSK3β phosphorylation by AKT is accompanied by stabilization of beta catenin that acts as a nuclear activator of transcription after GSK3b phosphorylation by WNT signaling (86).
5. The COX-2 and iNOS
The NOS and COX systems include constitutive forms (NOS1, NOS3, and COX-1), which are expressed in many cell types and are mostly involved in housekeeping tasks, and inducible forms (iNOS known as NOS2 and COX-2), which are activated in stress conditions (294). For example, activated macrophages produce clastogenic factors,
NOS1 and NOS3 are Ca2+/calmodulin dependent, while NOS2 is much less. Inducible forms lead to NO production from
COX-2, also known as prostaglandin endoperoxide synthase 2 (PTGS2), converts arachidonic acid into prostaglandin endoperoxide H2 (PGH2), a precursor of PGE2 involved in inflammation. During this reaction, singlet oxygen can also be released. COX-2 is an inducible enzyme responsible for the generation of ROS and of proinflammatory PGE2 (355).
Expression of iNOS (12) and COX-2 (133) is also controlled by NF-κB and is involved in secondary proinflammatory waves following irradiation. Conversely, cNOS can stimulate the early signaling effects of low-dose irradiation (161).
COX-2 is also upregulated in bystander normal human fibroblasts and its inhibition by NS-398 in bystander cells reduces mutagenesis and genetic instability (354). COX-2 is a downstream target of MAPK pathways, such as ERK, JNK, and p38 kinase. Zhou
Zhou
We mentioned above that post-translational modifications of p53, through phosphorylation by ATM and also by other kinases, such as CHK2 and homeodomain-interacting protein kinase 2 (HIPK2), contribute to its stabilization and subsequent induction of transcription of downstream genes involved in cell cycle arrest, programmed cell death, or cell metabolism. Accumulation of p53 also attenuates iNOS induction because p53 interacts with TATA binding protein and/or NF-κB that are essential for
B. Distant/systemic effects
1. Immune response as the mediator of radiation-induced systemic effects
As discussed above, ionizing radiation can cause a variety of effects in cells directly exposed to radiation and also in neighboring and distant nonirradiated cells (
Nikitaki
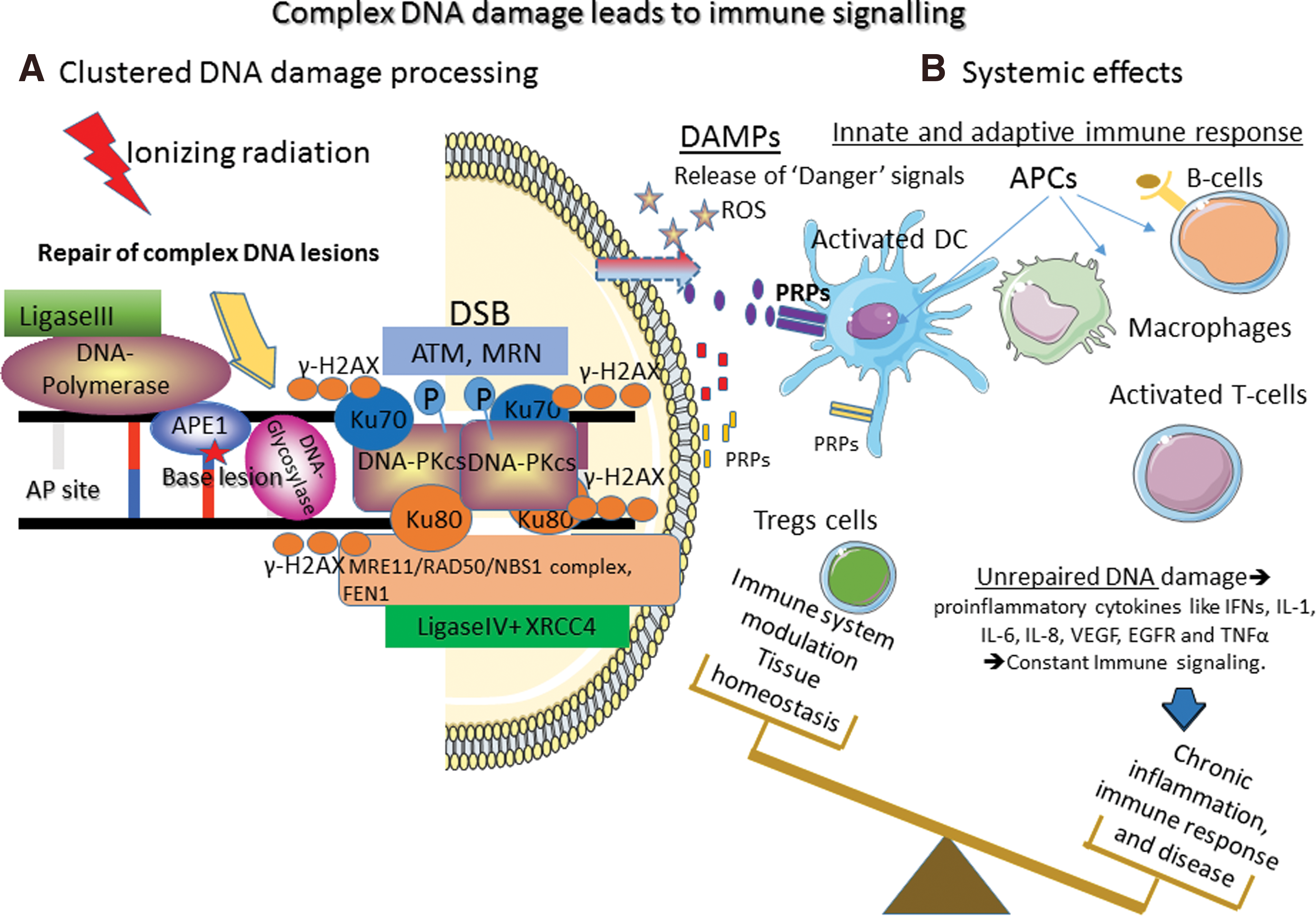
Overall, the immune response to radiation is highly related also to the radiation sensitivity (
The role of the innate immune system in coordinating these response types is a rapidly emerging research area (146). As discussed by Gasser
2. Abscopal effects: historical changes and clinical evidence
Although modern radiotherapy is getting progressively closer to its ideal target size or volume, still there are unresolved questions; particularly, it is not clear whether radiation effects are be really limited and localized. The idea of distant, abscopal effects was introduced for the first time by Mole in 1953 (196). Abscopal is made of the “
Although the idea of abscopal effects seems logical, it has been disregarded in radiotherapy for many years. It took about 50 years to show clear mechanistic evidence, at least in mice, about the immune system's role in this phenomenon (72). Specifically, mice bearing one syngeneic mammary carcinoma (mouse 67NR cells derived from mammary gland malignant neoplasms) in each flank were treated with the growth factor Flt3-Ligand (Flt3-L; daily for 10 days) to boost dendritic cell production, after local radiotherapy (or not) to only one of the two tumors (single dose of 2 or 6 Gy). Growth of the nonirradiated tumor was impaired only in mice that received unilateral radiotherapy and Flt3-L, but not in animals treated only with Flt3-L. This result indicates that immunity is involved in the abscopal effect. Radiotherapy immunostimulatory effects have created a wide interest due to the preclinical and clinical observations that tumor-localized radiotherapy can occasionally prompt anticancer immune responses that facilitate the regression of distant and nonirradiated metastatic tumors, as recently reviewed in Wennerberg
Although circumstantial evidence existed since the 1950s, abscopal effects are still considered in some cases as unexplained, obscure, or of limited clinical value due to the high specificity and dependence on the type of host organism, tumor, radiation, and so on. In the 1960s, Law and Mole (159) showed direct (
It is now quite safe to suggest that local irradiation of a tumor leads to cell death and tissue damage and the release of ROS, cytokines, and danger signals, several of which can trigger an innate immune response (144), although clinical evidence is still rare. One of the first clinical evidences was the description in 2009 of an abscopal effect in a patient with chronic lymphocytic leukemia during radiation therapy (157). One week after X-ray radiotherapy, the lymph nodes in the neck that were not irradiated and distant from the irradiated area started to regress, and after 2 weeks of radiotherapy, they showed complete regression (157). The same year, another patient was diagnosed with acute radiation pneumonitis and other signs of radiation toxicity after spine irradiation. The authors suggested that this should alert clinicians on the toxicity risk for nontarget organs that receive low-dose or zero radiation (283). For a list of the reported clinical cases concerning abscopal effects in patients with nonhematological malignancies and treated by conventional radiation (patient characteristics, treatment strategy, and outcomes), the reader can refer to a relatively recent review by Siva
Currently, the idea that abscopal effects exist and can be modulated primarily
Another early example of regression of hepatocellular carcinoma was observed after radiotherapy (total dose of 36 Gy) for bone metastasis (220). Doses of 2 and 6 Gy have been used in combination with the dendritic cell growth factor Fms-related tyrosine kinase 3 (FLT3) or with injection of dendritic cells in tumors as immune therapy (72, 150). In mice with melanoma tumors, a single fraction of 15 Gy showed similar results as 3 × 5 Gy and was accompanied by the generation of tumor antigen-specific effector cells that traffic to the tumor (175). The combination of anti-CTLA4 antibodies with fractionated irradiation (3 × 8 Gy or 5 × 6 Gy), but not single-dose irradiation (20 Gy), showed tumor growth delay outside the field of irradiation in preclinical models (300).
C. Off-target effects: an integrated cell response to radiation: conclusion
It was initially thought that the harmful effects of ionizing radiation were caused by clusters of DNA lesions, involving DNA DSBs, in irradiated cells. However, several observations progressively modified this DNA-centered paradigm view of radiobiology. First, the yields of radiation-induced ROS and RNS formation and subsequently of DNA lesions were relatively low compared with the endogenous production [mainly by mitochondria, NAD(P)H oxidases]. Second, DNA lesions involving DDR activation were observed in cells that had not been traversed by radiation, but that were close to irradiated cells (bystander effects). These bystander effects are the consequence of oxidative stress signals initiated in irradiated cells and propagated to neighboring cells. In this context, not only the initial nuclear DNA breaks but also all the cell compartments (cell membrane, mitochondria, ER) and associated complex networks that involve Ca2+ release and ROS/RNS production through NF-κB, iNOS, and COX activation have to be considered. Increased oxidative stress in irradiated cells and its transmission also to neighboring cells can amplify the initial response.
Distant systemic effects of radiation, which in the clinic are often called abscopal effects, are generally accepted nowadays and probably complete the picture of radiation effects in the whole body. Similarly to bystander effects, they are initiated by local DNA damage in irradiated cells or tissue. This can be considered the triggering effect that marks the irradiated area as a “stress” area in the body. Distant radiation effects involve the release of short and long distance messengers to convey the stress signal to distant sites by the mediation of well-conserved inflammatory and immune response networks. In the clinic, the most extreme manifestation of this phenomenon is tumor shrinkage in distant sites not reached by irradiation. This knowledge could pave the way to clinical applications of this systemic effect of radiation treatment.
V. Benefit/Risk Analysis
In the clinic, irradiation is used to destroy tumor cells by triggering cell death mechanisms. Innovative technologies and procedures have been developed in conventional EBRT, and the use of radionuclides for both imaging and therapy has progressively gained interest in the last two decades. Predicting the potential consequences of off-target effects is required for both radiation effectiveness and radiation risk assessment (247).
A. Target theory
The prediction of therapeutic efficacy and of side effects of radiation exposure relies on the establishment of dose/effect relationships between radiation dose and tissue reactions. Most radiobiological studies have used EBRT, generally with gamma and X-rays (244). The biological effects induced by radiation at medium (0.5–5 Gy) to high doses (5–15 Gy) are quite well known (140). They are usually subdivided as follows: (i) nonstochastic effects (also called deterministic or tissue reaction effects) that occur above a certain threshold (>0.5 Gy) and the severity of which increases with the dose; and (ii) stochastic effects (genetic risks in offspring and cancer) for which there is no threshold, but only a probability of occurrence. The tumor tissue response to radiation (assimilated to the tumor control probability, TCP) belongs to the nonstochastic effects category. In EBRT, it is proportional to the dose according to a sigmoid curve. At the molecular level, this curve reflects the eradication of the most radioresistant clonogenic malignant cells present in the tumor. TCP can be described using mathematical models, and cell killing can be analyzed using the Poisson law as statistical model. The “
This model has been later replaced by a linear quadratic (LQ) model [S = exp(−αD − βD2) where α and β are constant parameters and D is the dose.
In this context, tumor eradication means that no clonogenic cell survives. Therefore, TCP is strictly proportional to the number of clonogenic cells, depending on the dose: TCP = exp(−
B. The clinical relevance of off-target effects might be radiotherapy dependent
On the contrary, a general rule about the benefit/risk ratio of off-target effects in radiotherapy might not exist because this response is influenced by many parameters: type of radiotherapy, dose ranges, dose rates, dose fractionation, and radiation type (photons
Radionuclide therapy is another procedure where off-target effects have to be considered. Radionuclides are used for the treatment of various diseases, such as thyroid cancer with 131I, lymphoma with radiolabeled anti-CD20 monoclonal antibodies (90Y-ibributomab tiuxetan; Zevalin™), neuroendocrine tumors with
The particularity of radionuclide therapy is that radionuclides are targeted by conjugation with peptides or antibodies and are then injected in the circulation. The dose is delivered over several hours and days, at a relatively low-dose rate. The delivered dose can be strongly heterogeneous because of tumor-site accessibility issues and also because of the particles used (beta, alpha, and Auger electrons). Heterogeneity also occurs at the whole-body scale because the whole body may be irradiated (at variable doses), although the highest dose is delivered to the tumor. Moreover, the interplay between the radiation effects and the biological effects of the vector should also be considered (243). Because of its physical features, radionuclide therapy could be, to some extent, assimilated to low-dose rate brachytherapy using sealed sources of 125I for prostate cancer treatment. Moreover, as some of the radionuclides (
C. Bystander effects and radiation protection
There is evidence of
At doses below 0.1–0.2 Gy, only stochastic effects, including cancer, are relevant for assessing the tissue response. Such doses can be encountered during environmental exposures and also during treatment in nontumor tissues located at the margin of the irradiation field, or in the presence of dose gradients, particularly in IMRT, tomotherapy, and heavy ion particle therapy. It also concerns diagnostic radiological procedures and possibly also nuclear medicine. It must be also noted that the assessment of the dose delivered to the tumor and healthy tissues is more difficult and challenging during radionuclide therapy than EBRT, making risk assessment even more complicated.
The effects of such low doses, essentially cancer induction, could be predicted by extrapolating from assessments at high dose and by correcting for factors that account for low-dose rate and low dose, according to a linear no-threshold (LNT) model. The LNT model assumes that detrimental effects decrease according to the dose rate and are proportional to the dose. According to the “target theory” that relies mainly on a DNA-centered view of radiobiology (3), detrimental effects of radiation (mutagenesis, carcinogenesis, and cell death) are only observed in irradiated cells (more precisely in their nucleus) and in their progeny. This view is now challenged by the identification of harmful effects of radiation also in nonirradiated cells (off-target effects) and their amplification. However, the subject is still a matter of debate because there are many experimental and epidemiological evidences showing that it makes sense to extrapolate the risk from high to low doses. Nevertheless, many other studies highlight some of the weaknesses of the target theory (140).
The theory behind off-target and bystander effects considers that cells can die without being crossed by particles, that is, without having absorbed any energy from radiation. The ultimate consequence is that irradiated cells cannot be considered anymore as an independent entity, the final fate of which depends solely on the delivered dose and on their intrinsic radiosensitivity, which is one of the bases of the “target theory.” Conversely, the interplays with the microenvironment and even with the whole body through systemic communication that involves the blood and lymphoid compartments have to be considered.
D. The biology of low dose and low-dose rate might differ from that of high dose and high-dose rate
Besides bystander effects, several other phenomena, such as inverse dose rate effect and low-dose hypersensitivity, challenge the LNT model and risk assessment. For example, genomic instability frequency is higher at low than at high doses (309), and bystander effects have also been observed at low EBRT doses and after one single alpha particle track. As mentioned above, the RIAR shows that a priming dose (generally >0.005 Gy) reduces the detrimental effects of the challenging dose administered a few hours later (140, 275).
Therefore, it is likely that extrapolating data from high to low dose cannot be corrected solely by considering that cells have more time to repair DNA DSBs at low-dose rate, but by considering to some extent different biological mechanisms.
E. Off-target effects and radiotherapy efficacy
The existence of off-target effects might be an advantage for tumor eradication, although they are still difficult to control. One of the challenges is now to define the best radiotherapy plan (dose, dose rate, LET) and combination for promoting these effects. There have been several approaches (247) based on gene therapy to introduce NOS2 (328) and to use radiation-iNOS promoters (334). Gene therapy with human telomerase promoters associated with targeted radiotherapy using 131I-MIBG radionuclides has also been investigated by Boyd
The parameters affected by new clinical practices are dose and fractionation (hypo- and hyperfractionation). In some way, radionuclide therapy could be assimilated to hyperfractionated therapy. It is clear that dose and dose fractionation can modify the contribution of off-target effects to the final outcome. It was shown that the bystander response resulting from fractionated irradiation is different in tumor and healthy tissues (203).
The dose influence on the bystander response was reviewed by Tomita and Maeda (307). Bystander cell death induced by doses as low as 0.01 Gy of low LET radiation (γ-rays from 60Co) is similar to the cell death caused by exposure of nonirradiated cells to conditioned medium from cells irradiated at the same dose. It seems that there is a threshold above 2–3 mGy and bystander response reaches a plateau at 0.3 Gy (169). The influence of dose and dose rate on abscopal effects was reviewed by Rodel
At doses of 0.5 and 0.7 Gy, secretion of proinflammatory cytokines (
Conversely and as previously discussed, abscopal effects have been described in preclinical models and in patients at higher doses than those causing bystander effects (2–40 Gy) (258).Their occurrence and contribution to the final antitumor efficacy depend on the tumor and its microenvironment. Moreover, they are more pronounced when using therapeutic combinations that activate the immune system, such as injection of dendritic cells (87, 261). Currently, the critical factors orchestrating the overall response of the tumor and of the organism to irradiation are not clearly defined. Nevertheless, it is accepted that there is a synergy between tumor response, radiation toxicity, and immune system.
F. Benefit/risk analysis: conclusion
The off-target effects of radiation are a major concern both for radiotherapy and radiation protection. The relative contribution of off-target effects to the final therapeutic efficacy and to the detrimental effects of radiation might depend strongly on the type of exposure (
Radiation protection systems rely on the LNT model. Although it is now clear that off-target effects (
VI. General Conclusion
In this review, we presented in an analytical way the current status of knowledge in the area of off-target and away-from-target ionizing radiation effects with special emphasis on their role on radiation therapy and clinical applications. After years of intensive
Undoubtedly, radiation harmful effects are linked to the induction of a high percentage of clustered lesions that destroy the target (
We then discussed the clinical importance of these molecular mechanisms and of the association between targeted and off-target radiation mechanisms. We described the existing evidence for the systemic nature of radiation effects (
VII. Key Points
Cell damage is mediated by the direct interaction of ionizing radiation with water and cellular constituents, such as DNA, lipids, and proteins.
Radiation generates “danger” signals that propagate from irradiated to nonirradiated cells, leading to off-target (bystander and abscopal/distant) effects.
Redox mechanisms also play a key role in both targeted and off-target radiation effects.
NF-κB is essential for triggering the self-sustained production of reactive oxygen and nitrogen species (ROS and RNS) that are involved in both targeted and off-target radiation effects.
The level of damage produced by radiation is relatively lower than that caused by endogenous oxidative stress.
The harmful effects of radiation are explained not only by the induction of closely spaced (clustered) DNA lesions but also by the amplification of the initial radiation-induced cellular response.
Immune cells can be recruited and contribute to the distant (abscopal) effects of irradiation with potential clinical implications.
Differently from targeted effects (
Although there is evidence of off-target effects existence
Off-target effects contribution is likely to depend on the irradiation physical parameters (type and level of damage) as well as on the type of tissue and organism.
Radiation-induced systemic phenomena (as opposed to localized, direct damage) represent a radiobiology paradigm shift and must be taken into account in radiation protection as well as in radiotherapy.
Footnotes
Acknowledgments
This research has been financed by Action Nu1.1 of Plan Cancer 2009–2013 (ASC 13038FSA), the French National Research Agency under the program “Investissements d'avenir” Grant agreement LabEx MabImprove: ANR-10-LABX-53, Labex PRIMES: ANR-11-LABX-0063, Ligue Nationale Contre le Cancer, Electricité de France Comité de Radioprotection, Le segment Radiobiologie du CEA, by “Research Projects for Excellence IKY/SIEMENS” and DAAD grant “DNA Damage and Repair and Their Relevance to Carcinogenesis” (No. 57339330).