
Abstract
Aims:
The mechanisms coordinating maturation with an environment-driven metabolic shift, a critical step in determining the developmental potential of human in vitro maturation (IVM) oocytes, remain to be elucidated. Here we explored the key genes regulating human oocyte maturation using single-cell RNA sequencing and illuminated the compensatory mechanism from a metabolic perspective by analyzing gene expression.
Results:
Three key genes that encode CoA-related enzymes were screened from the RNA sequencing data. Two of them, ACAT1 and HADHA, were closely related to the regulation of substrate production in the Krebs cycle. Dysfunction of the Krebs cycle was induced by decreases in the activity of specific enzymes. Furthermore, the activator of these enzymes, the calcium concentration, was also decreased because of the failure of influx of exogenous calcium. Although release of endogenous calcium from the endoplasmic reticulum and mitochondria met the requirement for maturation, excessive release resulted in aneuploidy and developmental incompetence. High nicotinamide nucleotide transhydrogenase expression induced NADPH dehydrogenation to compensate for the NADH shortage resulting from the dysfunction of the Krebs cycle. Importantly, high NADP+ levels activated DPYD to enhance the repair of DNA double-strand breaks to maintain euploidy.
Innovation:
The present study shows for the first time that exposure to the in vitro environment can lead to the decline of energy metabolism in human oocytes during maturation but that a compensatory action maintains their developmental competence.
Conclusion:
In vitro maturation of human oocytes is mediated through a cascade of competing and compensatory actions driven by genes encoding enzymes.
Human oocytes are invaluable resources in clinical and basic research, but there is no information about the mRNA transcription changes during oocyte maturation. Recently, metabolism has been highlighted as a mediator that maintains its developmental potential during maturation. The present study shows for the first time that exposure to an in vitro environment can lead to dynamic changes in the global transcriptome in human oocytes, and it demonstrates that exposure to an in vitro environment can lead to dysfunction of the Krebs cycle in human oocytes during maturation; however, a compensatory action mediated by nicotinamide nucleotide transhydrogenase maintains their energy requirements, supporting developmental competence.
Introduction
M
The poor developmental potential of IVM oocytes has been noted in various species, particularly in humans, although >5000 human babies have been born via the IVM procedure worldwide (6), which has been attributed to an in vitro environment that can support the resumption of meiosis among immature oocytes. Failure of synchronization between the cytoplasmic and nuclear maturation leads to the poor acquisition of developmental competence by IVM oocytes (17). Factors that impair the resumption of meiosis and spindle assembly in IVM oocytes have been noted in previous studies using animal models and human materials (15, 46, 61), but the underlying molecular mechanism of cytoplasmic maturation driven by an in vitro environment is unknown, especially in human oocytes.
Metabolism plays an important role in regulating gene expression, protein translation, and protein modification. Insight into oocyte maturation has come from studies of specific metabolic pathways, including the pentose phosphate pathway, the Krebs cycle, and NADH catabolism (50). The normal function of these metabolic pathways provides necessary products for cellular physiology, such as the cell cycle, cytokinesis, and intracellular transport; however, dysfunction of these metabolic pathways results in apoptosis, autophagy, and failure of meiosis resumption (11, 57). In considering the differences between the in vitro and in vivo environments in which oocytes mature, it is necessary to investigate the dynamic changes in enzymes and their encoding genes under in vitro conditions.
Aberrant metabolism usually induces the occurrence of oxidative stress in oocytes, and it is therefore harmful to the subsequent developmental potential. In humans, oxidative stress has often been identified in the follicular fluid of aged women (34) or of women with polycystic ovary syndrome (PCOS) (36), and it is regarded as a risk factor to induce immature oocyte and decrease oocyte quality. Moreover, studies in mouse proved that antioxidant supplementation can effectively improve oocyte maturation and developmental potential in aged or PCOS models (8, 26). The molecular mechanism of the maturation failure in oocytes caused by oxidative stress has been addressed from the perspective of histone acetyltransferase (60, 66), DNA methylation (37), and chromosome segregation errors (43), but the previous studies were focused on the effects of oxidative stress on oocytes in pathological or physiological environments, and studies that explored oxidative stress during oocyte maturation in vitro remain scarce.
In 2009, Tang et al. successfully completed mouse oocyte and embryo transcriptome sequencing using only single cells (54). Furthermore, Xue et al. analyzed the transcriptome in human oocytes and embryos using single-cell transcriptome sequencing technology and described the differences in genetic programs between human and mouse (58). Yan et al. also described the characteristics of human oocytes and embryos based on the transcriptome and analyzed not only mRNA but also long noncoding RNA (59). The expression profile of the RNA transcriptome in human oocytes during maturation remains unknown.
Here we investigated the transcriptome characteristics of human oocytes matured in vitro and in vivo to gain a transcriptome-level understanding of how oocytes mature and to illuminate the differences between human IVM and in vivo (IVO) matured oocytes at the transcript level. By analyzing the key genes and pathways that regulate the maturation of human oocytes from the perspective of metabolism, we elucidated the mechanisms that maintain the developmental potential of human IVM oocytes.
Results
Single-cell transcriptome analysis in human oocytes
Single-cell transcriptome sequencing was performed using human IVM and IVO oocytes (Supplementary Fig. S1A; Supplementary Data are available online at
To avoid effects from an individual's genetic factors, we first compared sequencing data from paired IVM and IVO oocytes from each woman. In the comparison groups, there were 8290, 9168, and 11,263 differentially expressed genes (DEGs), respectively (Fig. 1A). We then found the DEGs in common across the three comparison groups, which resulted in 2230 genes that displayed common differences between human IVM and IVO oocytes (Fig. 1B). Samples from the IVM and IVO groups were further distinguished by supervised hierarchical clustering and principal component analysis (Fig. 1C, D). These genes mainly consisted of protein-coding mRNAs (1523) and long noncoding RNAs (232) (Fig. 1E and Supplementary Table S1). The top six categories of biological process, cellular component, and molecular function of these DEGs are shown in Supplementary Figure S2.
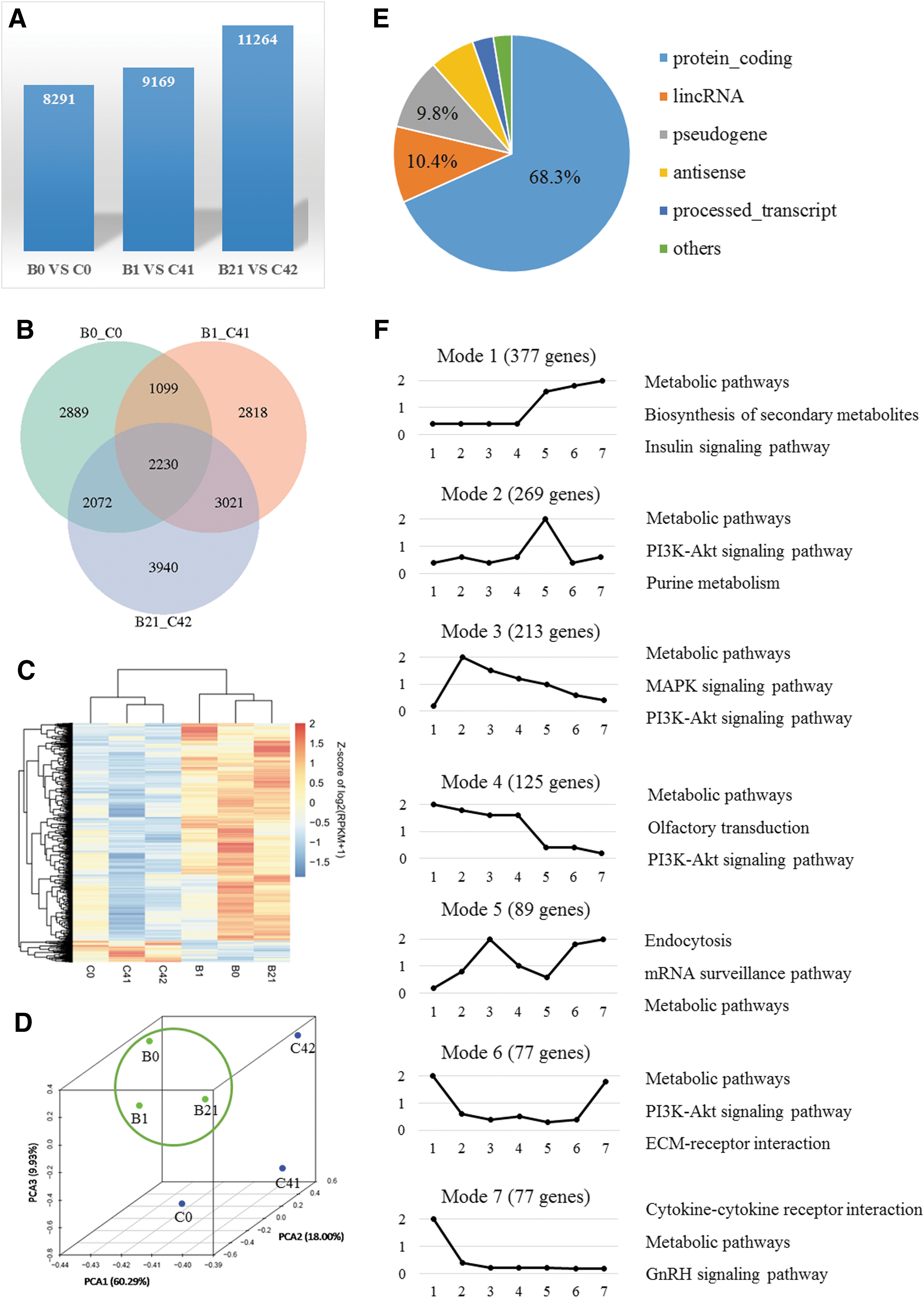
The expression profiles of the 1523 protein-coding genes were checked for all early developmental stages from unfertilized oocytes to blastocysts using published data (59). Of these, 1461 genes could be categorized into 11 expression profiles (Supplementary Table S2). The seven most prevalent profiles were selected, and the three pathways that included the genes showing the greatest differential expression are shown in Figure 1F.
Metabolic pathways and related genes were significantly impaired in IVM oocytes
Pathways that could potentially be affected by IVM were analyzed. A total of 81 pathways were screened, and the top 10 pathways are shown in Supplementary Figure S3A. Among the pathways, 27% were related to metabolism, and 19% were signaling pathways (Supplementary Fig. S3B, Supplementary Tables S3 and S4).
To further screen the key genes regulating metabolism, we listed all of the genes in these 75 metabolic pathways, regardless of whether they were DEGs, in a sequencing database. Twenty-four genes that were involved in at least nine pathways were analyzed and were found to belong to the following six categories: the aldehyde dehydrogenase (ALDH) family, the mitogen-activated protein kinase (MAPK) family, the protein kinase (PRK) family, the phosphatidylinositol 3-kinase (PI3K) family, the adenylyl cyclase (ADCY) family, and CoA-related enzymes (Supplementary Fig. S3C). Using the STRING database, the DEGs were clearly divided into two groups, one of which included the MAPK, PRK, PI3K, and ADCY families, and the other included the ALDH family and CoA-related enzymes (Supplementary Fig. S3D). The sixteen genes in the first four families encode kinases, and eleven of them displayed significant differences in expression between IVM and IVO oocytes (Supplementary Fig. S4). Among the other eight enzymes (ALDH family members and CoA-related enzymes), we found that four genes, encoding ACAT1, HADHA, DPYD, and ALDH2, showed the most significant differences (Supplementary Fig. S5).
To narrow down the number of important genes that are relevant to oocyte maturation, we collected high-quality (HQ) and low-quality (LQ) IVO oocytes based on our previously established criteria (64) and IVM oocytes (Fig. 2A). Three genes that encode CoA-related enzymes had the same expression profile in IVM oocytes as in LQ IVO oocytes, but their expression differed notably in HQ IVO oocytes (Fig. 2B).
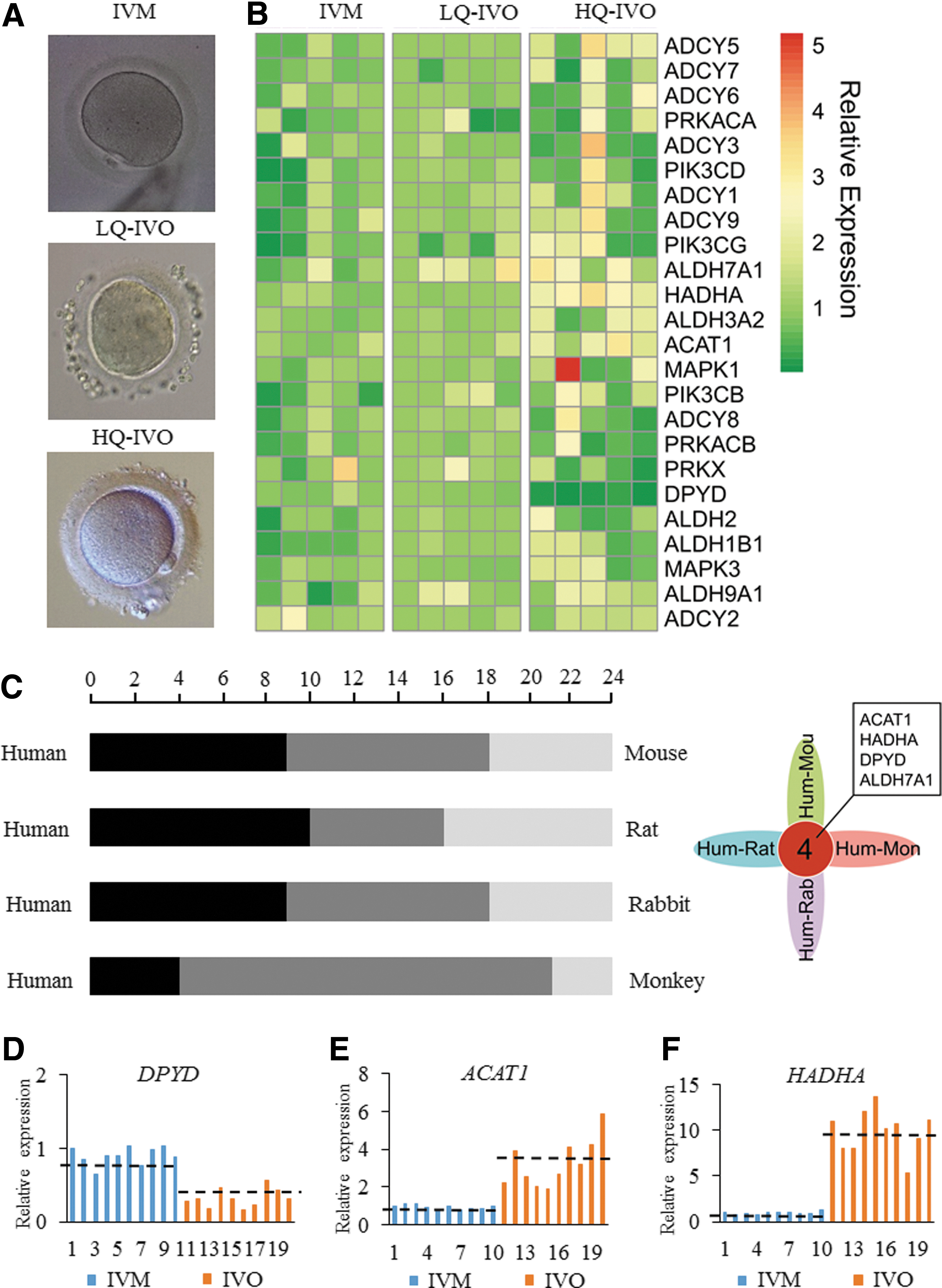
The expression levels of the 24 genes in IVM and IVO oocytes were detected from different species, including mouse, rat, rabbit, and monkey. The results showed that gene expression profiles were more similar between human and monkey oocytes. There were four genes with consistent expression profiling in each group, including three genes encoding CoA-related enzymes and ALDH7A1 (Fig. 2C and Supplementary Table S5). Using qPCR, we found that in IVM oocytes compared with IVO oocytes, expression of ACAT1 and HADHA was significantly decreased, whereas DPYD expression was significantly upregulated (Fig. 2D–F).
Abnormal expression of ACAT1 and HADHA is a reflection of the impaired metabolic function in IVM oocytes
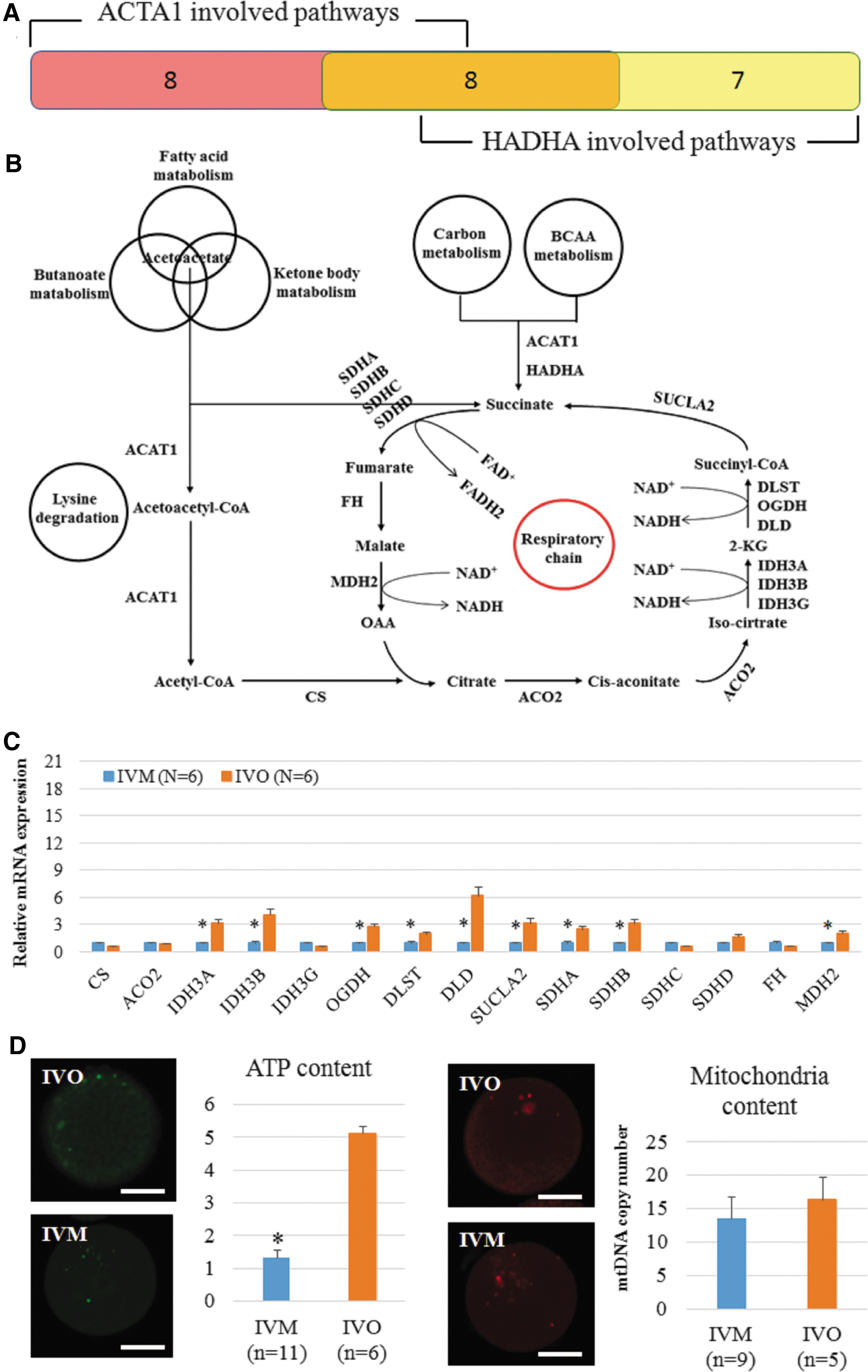
Based on a bioinformatic analysis, we found that ACAT1 is involved in 16 metabolic pathways and HADHA is involved in 15 metabolic pathways. There are eight pathways in common between the two genes (Fig. 3A and Supplementary Table S6). Based on these pathways, we propose the following metabolic scenario (Fig. 3B). Deficient expression of ACAT1 and HADHA blocked the production of acetyl-CoA and succinate, which are the key substrates for the Krebs cycle, and therefore weakened the energy metabolism. Based on qPCR results, relative expression levels of 9 of the 15 genes encoding enzymes in the Krebs cycle were lower in the IVM oocytes than in the IVO oocytes (Fig. 3C). Immunofluorescence data showed that the ATP content of IVM oocytes was significantly lower than in the IVO group, however, mitochondrial content was not significantly decreased in the IVM group compared with IVO group. (Fig. 3D).
Mouse oocytes were used to study the effects of decreased expression of ACAT1 and HADHA on maturation, fertilization, and subsequent development. First, we compared the expression levels of these 15 genes in mouse IVM and IVO oocytes (Supplementary Fig. S6A). Comparison of DEGs from human and mouse oocytes indicated that ten of these genes showed similar changes in expression (Supplementary Fig. S6B).
siRNAs for ACAT1 and HADHA were coinjected into mouse GV oocytes, which resulted in effective knockdown of ACAT1 and HADHA expression (Supplementary Fig. S7A). Enzyme-linked immunosorbent assay (ELISA) results indicated that the activity of ACAT1, HADHA, OGDH, IDH3A, and SDHA was significantly decreased in siRNA oocytes relative to controls (Supplementary Fig. S7B). The gene expression results were similar to the results from comparing mouse and human IVM oocytes with their IVO controls (Supplementary Fig. S7C). Moreover, Immunostaining results suggested that oxidative stress as represented by ROS was significantly increased in both the cytoplasm and mitochondria, but the GSH level decreased in the siRNA group relative to the controls. Furthermore, the level of oxidized FAD (FAD++) was significantly enhanced, whereas the level of reduced NADH was significantly decreased in the siRNA injection group compared with the other two control groups (Supplementary Fig. S7D–G).
The maturation ratio was not impaired in the siRNA group (Supplementary Fig. S8A). siRNA and control oocytes were then fertilized by intracytoplasmic sperm injection (ICSI). The blastocyst formation and implantation rate was not affected by the injection with ACAT1 and HADHA siRNA (Supplementary Fig. S8B, C), but the birth rate was significantly decreased (Supplementary Fig. S8D). ACAT1 and HADHA siRNA treatment led to a significant delay in the hatching of developing blastocysts, a decrease in the number of cells per blastocyst, and a higher incidence of DNA damage as indicated by the frequency of apoptotic cells (Supplementary Fig. 9A–E).
Failure of the calcium signaling pathway accounts for the reduced activity of enzymes
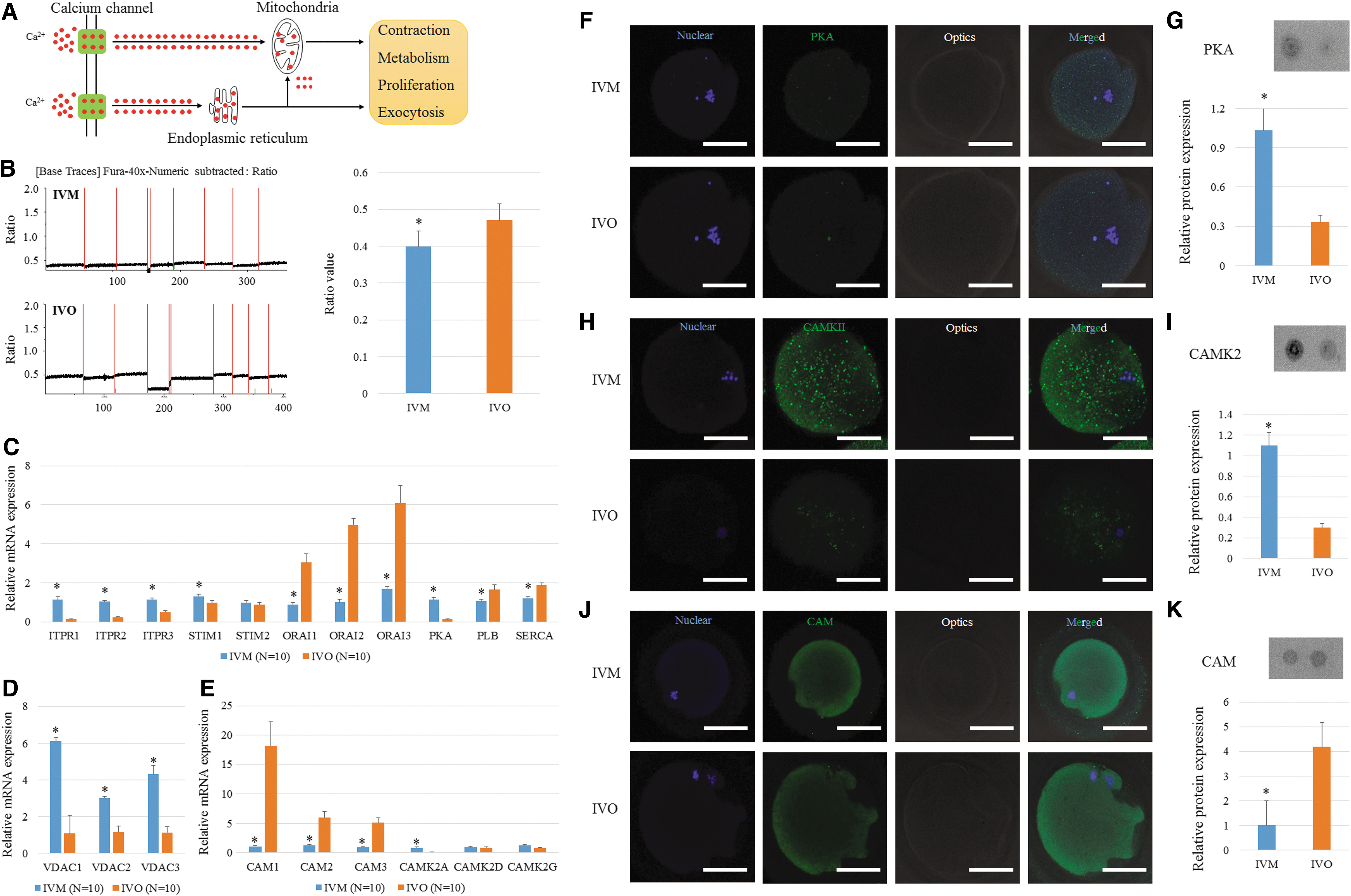
Calcium often acts as an activator for enzymes and is generally controlled via the ER or mitochondria (Fig. 4A). By analyzing the transcriptome data, we found that the calcium signaling pathway in IVM oocytes was potentially dysfunctional (Supplementary Fig. S10A). A supervised clustering result indicated that IVM and IVO oocytes can be clearly separated based on the expression of the 52 genes involved in this pathway (Supplementary Fig. S10B). Moreover, the average intensity of the calcium signal was also decreased in human IVM oocytes compared with human IVO oocytes (Fig. 4B). Genes encoding plasma membrane proteins that are related to calcium transport were all downregulated; however, genes encoding endoplasmic reticulum (ER) and mitochondrial membrane proteins were all upregulated (Fig. 4C, D). Moreover, genes encoding calmodulin (CAM) and Ca2+/calmodulin-dependent protein kinase II (CAMKII) showed different expression profiles between IVM and IVO oocytes (Fig. 4E). These differences were maintained at the protein level, as well as for PKA, based on immunostaining and dot blot analysis (Fig. 4F–K).
To confirm the effects of calcium concentration on human oocyte development, the human IVO oocytes were fertilized, and then, the fertilized embryos were cultured in a calcium-free medium. The fertilized embryos developed to the blastocyst stage with lower efficiency, and the derivation efficiency of embryonic stem (ES) cells was also decreased compared with normal controls (Supplementary Fig. S11A, B). Moreover, these embryos showed microduplications or microdeletions in multiple chromosomes in trophectoderm (TE) cells of the resulting blastocysts (Supplementary Fig. S11C, D), and gene expression results indicated that energy metabolism was impaired (Supplementary Fig. S11E, F).
Compensatory production of NADH and NADP+ provides a developmental opportunity for IVM oocytes
NADH is mainly produced by glycolysis and the Krebs cycle; however, glycolysis does not occur in oocytes, and the Krebs cycle is inactive during maturation in vitro. A potential compensatory mechanism is that NADPH transfers a hydrogen ion to NAD+ and therefore generates NADP+ and NADH (Fig. 5A). Encoding mRNA of NADPH-dependent antioxidants in our sequencing data was shown in Figure 5B, and the expression levels of GPX and PRDX family members have no notable changes. We analyzed the expression of the enzyme that catalyzes this reaction, nicotinamide nucleotide transhydrogenase (NNT). Gene and protein expression levels of NNT were both upregulated in IVM oocytes (Fig. 5C, D). Moreover, inhibition of NNT expression by siRNA in IVM oocytes resulted in developmental arrest at the eight-cell stage and significantly decreased blastocyst efficiency (Fig. 5E, F). We examined the mRNA and protein expression of NNT in multiple mouse IVM and IVO oocytes to confirm this difference (Supplementary Fig. S12A, B). When NNT expression was blocked by siRNA in mouse oocytes (Supplementary Fig. S13C), IVM oocytes were arrested at the two-cell stage more frequently than in the controls (Supplementary Fig. S12D). Although a few of the IVM embryos formed blastocysts, the quality of the blastocysts was still poor (Supplementary Fig. S12E). Immunostaining indicated that the levels of ROS in the cytoplasm and mitochondria (indicated by MitoSOX) and oxidized FAD were significantly increased, and that of GSH and NADH were significantly decreased in IVM oocytes with NNT-siRNA injection, compared with control samples (Supplementary Fig. S13A–D).
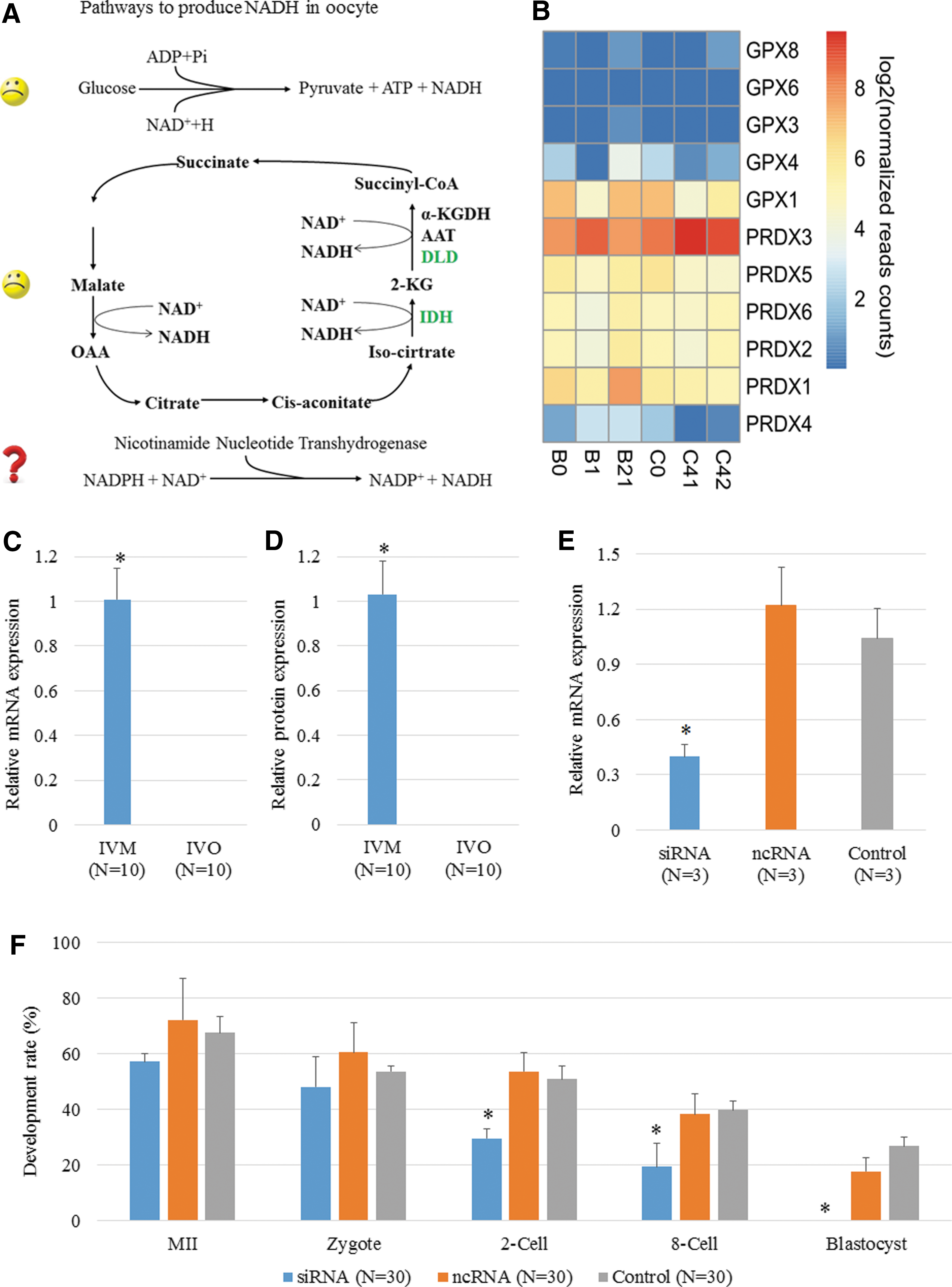
γH2AX-positive signal, which reflects DNA double-strand breaks (DSBs) in chromosomes, was higher in IVM oocytes than in IVO oocytes (Fig. 6A, B), indicating the genome instability induced by the IVM process. With higher expression of NNT in human IVM oocytes, NADP+ production was increased, which was consistent with the upregulated expression of DPYD, an NADP+-dependent enzyme that specifically repairs DSBs. In vivo, DPYD was highly expressed in the primary and secondary follicle but was not expressed in oocytes and preimplantation embryos (Fig. 6C); however, expression of DPYD increased in primary and secondary follicles incubated in vitro and was dramatically upregulated in IVM oocytes (Fig. 6D–F). Immunostaining results showed that the DPYD protein was often distributed around the cytomembrane in oocytes at the MII stage but was also localized to the GV in oocytes at the GV stage (Fig. 6G).
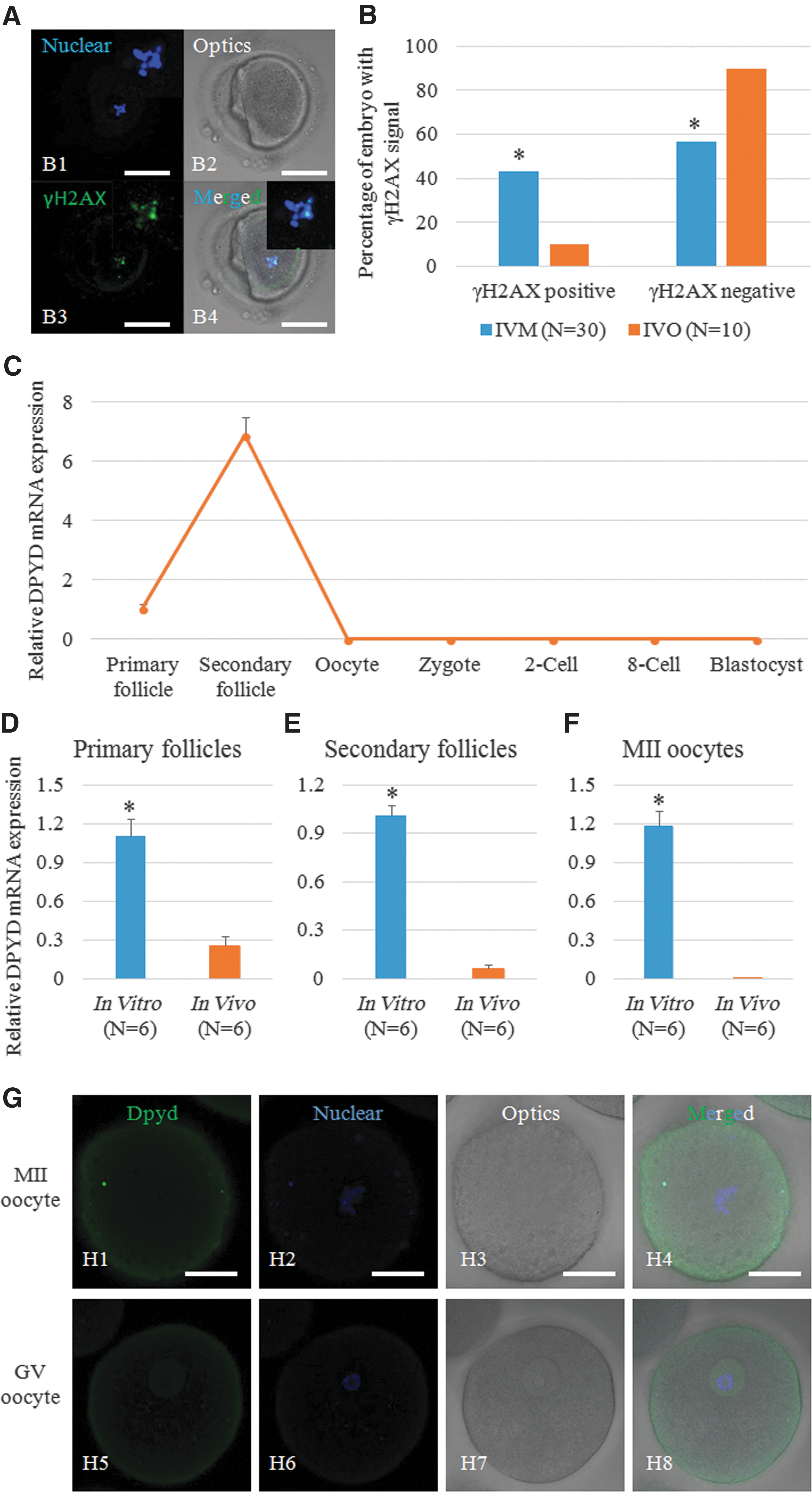
We then treated the oocytes with 100 μM H2O2 to induce DSBs and found that the proportion of embryos with a γH2AX-positive signal was significantly increased. These embryos were still able to develop to the blastocyst stage, although with lower efficiency, and DPYD expression was significantly upregulated (Supplementary Fig. S14A–C). After knocking down DPYD using DPYD-specific siRNA, the embryos derived from H2O2-treated oocytes showed a high frequency of aneuploidy, and fewer of these embryos were able to form normal blastocysts (Supplementary Fig. S14D–F). Representative images of developed embryos from the three groups are shown in Supplementary Figure S14G.
Discussion
In the present study, we analyzed the dynamic changes in global gene expression in human IVM and IVO oocytes. Single-cell sequencing with a next-generation sequencing platform has been widely applied in the fields of genetics (25), epigenetics (20), and RNA transcriptome (58, 59) analysis to establish relevant databases and explore the mechanism of development and differentiation. For human oocytes and fertilized embryos, there have been only two RNA sequencing studies because of the preciousness of human oocytes (58, 59). Xue et al. identified a sequence of transcriptional changes in multiple developmental pathways and suggested conserved key members (or hub genes) in the human and mouse networks (58). Subsequently, Yan et al. discovered 2733 novel long noncoding RNAs in human oocytes and embryos, providing a more comprehensive framework of the transcriptome landscape of human early embryos (59). The present study reveals the dynamic changes in the transcriptome during human oocyte maturation in vitro and therefore provides a series of molecular targets to improve the developmental outcome of human IVM oocytes.
Our study revealed that two groups of genes, those encoding kinases and those encoding enzymes related to CoA production, had significantly different expression levels between IVM and IVO oocytes. Kinases have an important role in activating the expression of key molecules during meiosis resumption (42), and CoA, in conjunction with enzymes, can catalyze reactions in the cytoplasm (56). Therefore, these two groups of genes represent the key regulators for nuclear and cytoplasmic maturation, which is recognized as fundamental for oocyte maturation. Although previous studies of nuclear maturation have analyzed changes in single genes or single gene families, ours is the first global analysis of these changes.
Cross talk between the nucleus and cytoplasm determines the eventual development outcome of IVM oocytes (1). Most previous studies focused on nuclear maturation and screened for the key molecules and pathways involved (48, 62, 65); however, resulting increases in the efficiency of IVM oocytes were negligible. Recently, more studies have focused on cytoplasmic maturation (24, 33, 52). Organelles in oocytes undergo changes in biogenesis, morphology, and cellular pathways in IVM oocytes that lead to cellular stress and apoptosis, and these factors eventually contribute to the failure of cytoplasmic maturation. These results were confirmed in our study in the point view of global transcriptome.
Three CoA-related enzymes encoding genes were isolated in our analysis, ACAT1, HADHA, and DPYD. ACAT1 and HADHA are mainly involved in metabolic processes and contribute to the Krebs cycle (13, 35), which is impaired in IVM oocytes. ACAT1 encodes a mitochondrial enzyme that catalyzes the reversible formation of acetoacetyl-CoA from two molecules of acetyl-CoA (27). Transcription of ACAT1 is promoted by leptin (23), angiotensin II (28), and insulin (18) in human monocytes/macrophages. As mitochondrial acetoacetyl-CoA thiolase is involved in β-oxidation, deficiency in this enzyme is marked by increased levels of cholesterol compounds (18, 23, 28). In addition, the isoleucine pathway is affected, such that proper metabolism of this amino acid is halted (29). González-Serrano et al. found that blastocysts produced in vitro and their in vivo counterparts differed significantly in the homeostasis of cholesterol and free fatty acid metabolism (19), suggesting that embryonic lipid metabolism is affected by in vitro incubation, and ACAT1 is a marker that reflects upstream lipid metabolism.
HADHA encodes the mitochondrial protein trifunctional enzyme subunit alpha. HADHA catalyzes the last three steps of mitochondrial β-oxidation of long-chain fatty acids (67). While NAD alone is present, HADHA can convert medium- and long-chain 2-enoyl-CoA compounds into 3-ketoacyl-CoA, whereas it results in the production of acetyl-CoA when NAD and CoA-SH are present (4).
A common characteristic of ACAT1 and HADHA is that both are mitochondrial and both are involved in mitochondrial β-oxidation. In mouse, bovine, and porcine oocytes, β-oxidation is involved in the regulation of meiosis resumption (41). In those studies, however, the cumulus–oocyte complex was often used, and it was therefore difficult to illuminate the importance of β-oxidation in the ooplasm rather than in the oocyte itself during maturation (41). Dunning et al. found that β-oxidation can be detected in denuded mouse oocytes, which indicates a potential role for β-oxidation in the ooplasm (10). In our study, we first indicated a potential relationship between β-oxidation and human oocyte maturation by finding two related genes, ACAT1 and HADHA.
In mouse, the mechanism by which impaired β-oxidation blocks maturation was mainly attributed to the effect on energy metabolism via the Krebs cycle and the electron transport chain. In our study, we also observed abnormal expression of genes involved in the Krebs cycle. Nine of a total of fifteen genes encoding ten enzymes or enzyme complexes in TCA showed abnormal expression profiling, including almost all enzymes that control NADH and FADH2 production.
There are three IDH isozymes in humans. IDH3 catalyzes the third step of the Krebs cycle while converting NAD+ to NADH in the mitochondria, whereas IDH1 and IDH2 catalyze the same reaction beyond the context of the Krebs cycle and use NADP+ as a cofactor instead of NAD+ (21). Sutton-McDowall et al. observed that IDH is a potential substrate for BMP15 and FSH, both of which can improve oocyte maturation (51). SDH is an enzyme complex that is bound to the inner mitochondrial membrane of mammalian mitochondria and to many bacterial cells. It is the only enzyme that participates in both the Krebs cycle and the electron transport chain (40). Boerjan et al. suggested that SDH activity is correlated with the location of mitochondria in preovulatory mouse oocytes, and its mitochondrial distribution is closely related to chromatin organization. Therefore, there is a potential correlation between SDH activity and chromatin morphology (2). The SDH complex has four isoforms, SDHA, SDHB, SDHC, and SDHD. In our results, we found that the expression of these four genes was dynamic and changed stochastically, with more than two isoforms being affected in human IVM oocytes. Regardless of which isoform is affected, when one isoform is mutated, the overall SDH complex becomes unstable and therefore loses its function, leading to reduced function of the Krebs cycle and electron respiration chain (31). That is, the overall dysfunction of enzymes resulted in reduced maturation and development of human IVM oocytes.
We then noticed that calcium is a common activator of these enzymes (5). From the beginning of fertilization, dynamic changes in the calcium concentration have a key role in promoting development (47). Impaired calcium homeostasis leads to the failure of oocyte maturation, fertilization, and development (22). Calcium in the cytoplasm is derived from two sources: one is intake from the extracellular environment, and the other is release from intracellular stores in the ER and mitochondria. Our study indicated that almost all genes encoding membrane proteins in the calcium metabolic pathway were downregulated, which indicates a potential intake barrier; however, intracellular calcium can still be detected in IVM oocytes, which may be attributable to an excessive release of calcium from the ER and mitochondria to complement this insufficient intake. Excessive calcium release would result in ER stress and therefore induce apoptosis and autophagy (30). In our study, genes involved in ER stress pathways were not stably expressed (data not shown), which suggests that the ER is involved in both releasing calcium and maintaining the calcium concentration in the cytoplasm, thereby inducing apoptosis and blocking development because of stress. Thus, inhibiting the ER stress response would improve the maturation and development of human IVM oocytes. In our study, we demonstrated that treating oocytes and embryos in in vitro culture with a lower calcium concentration would decrease the potential for embryonic development and increase the incidence of aneuploidy. The results again show the importance of calcium for regulating oocyte maturation and embryonic development.
Dysfunction of the Krebs cycle would impair NADH and ATP production, thereby compromising intracellular metabolism and mRNA transcription (3, 7). NADH, the reduced form of NAD+ that is generated during cellular metabolism, is used to transfer the reductive potential captured from catabolic reactions into NADH ubiquinone oxidoreductase (complex I), which generates the membrane potential for ATP regeneration (44). In human IVM oocytes, we found that the two main pathways to produce NADH were weakened (deficient ability to carry out glycolysis (49) and an impaired Krebs cycle), but the activity of mitochondrial NNT was enhanced, which should facilitate the transfer of electrons between NADPH and NADH. Nickel et al. suggested that transformation from NADH to NADPH is a forward mode, but transformation from NADPH to NADH is a reverse mode and often induces a pathological outcome (39). In our study, we found that the NADPH concentration was decreased, which indicated a potential hydrogen loss and transformation from NADPH to NAD+. Human IVM oocytes also showed a decrease in developmental competence when NNT activity was inhibited. These findings agree with the increasing ROS level in human IVM oocytes.
Interestingly, the third key gene we found in sequencing results, DPYD, is a NADP+-dependent enzyme encoded by DPYD (53). It has a key role in the repair of DNA DSBs (45). Its expression in human oocytes is described here, and higher expression of DPYD was found in secondary follicles in which DNA DSBs and homologous recombination are both occurring. Importantly, we found that the genes and proteins of DPYD were activated excessively when primary follicles, secondary follicles, and MII oocytes were cultured in vitro, which suggested that the risk of DNA DSBs or chromosome segregation was increased in the in vitro environment. Considering that embryos from human IVM oocytes had many more tiny chromosomal defects than those from human IVO oocytes, increasing the expression of DPYD may facilitate the repair process and overcome the risk of aneuploidy. Such overexpression may explain why previous studies found that although euploidy was decreased in human IVM oocytes, 60% of them still displayed euploid chromosomes (32).
In conclusion, here we revealed a mechanism to explain why IVM oocytes can be matured in vitro but often display reduced developmental potential after fertilization (Supplementary Fig. S15). The in vitro environment can induce errors in the expression of abundant genes; however, a compensatory mechanism can help oocytes avoid failure of maturation or development. Unfortunately, the continuous increase in the demand for cellular fuel during development would probably induce organelle dysfunction and thus disrupt the balance among energy production, cell cycle progression, and nutrient metabolism. Targeting the production of a sufficient energy supply in the mitochondria is therefore a promising approach to ameliorate the poor outcome of human IVM oocytes.
Methods
Collection of human oocytes
The present study was approved by the Institutional Review Board of Peking University Third Hospital and the Third Affiliated Hospital of Guangzhou Medical University. All of the oocyte donors signed informed consent voluntarily after they were clearly informed of the content and details of this study. Two groups of women were involved in the present study. One group consisted of women who had immature oocytes after oocyte retrieval and removal of cumulus cells. These women were all undergoing ICSI cycles because of male fertility factors. In general, such immature oocytes are discarded as medical waste. Here we collected them for the analysis of specific genes. The other group consisted of women who donated their oocytes voluntarily, with no financial benefit except for the necessary nutritional and transportation fees. Three women in this group were selected for single-cell transcriptome sequencing because they had at least one IVM oocyte and one IVO oocyte. No differences were found among three donors for the main physiological indexes (Supplementary Table S7).
IVM of human oocytes
Immature human oocytes were cultured in the IVM basal medium supplemented with 0.075 IU/mL follicle-stimulating hormone, 0.075 IU/mL luteinizing hormone, 10 ng/mL epidermal growth factor, 10 ng/mL brain-derived neurotrophic factor, and 10 ng/mL insulin-like growth factor-1. After 24–28 h, oocytes that expelled the first polar body were collected, transferred into Gm culture medium (LGGG-050; LifeGlobal Group, LLC), and prepared for molecular analysis.
Transcriptome sequencing using single oocytes
Single-cell transcriptome sequencing was performed by Annoroad Gene Technology Co. Ltd (Beijing, China) using six human oocytes, including three IVM and three IVO oocytes. Oocytes were rinsed in Ca2+/Mg2+-free PBS solution and individually placed in 0.2-mL PCR tubes containing a 5-μL mixture of lysis buffer and RNase inhibitor. RNA from a single oocyte was amplified using the SMARTer Ultra Low RNA kit for Illumina sequencing (634936; Clontech Laboratories, Inc.). In brief, reverse transcriptase, buffer, oligo(dT) primers, and template switch oligo primers were added to obtain the first-strand cDNA. Then, PCR amplification solution and in situ PCR primers were added for second-strand synthesis (∼1–2 kb in length). The resulting cDNA was purified and recycled using AMPure XP Beads (A63882; Beckman Coulter, Inc.) and dissolved in elution buffer (EB) solution. The cDNA concentration was determined using a Qubit® 2.0 Fluorometer, and the cDNA fragment length distribution was quantified using the Agilent 2100 High Sensitivity DNA Assay Kit (5067-4626; Agilent Technologies). The cDNA library was generated as follows. The quantified cDNA (20 ng) was fragmented into 200-bp lengths by ultrasonic waves using a Bioruptor® sonication system. The cDNA fragments then underwent end repair, adaptor addition, and purification using AMPure XP Beads. The products with adaptors were amplified by PCR. The different samples were marked with different index tags. PCR amplification products were electrophoresed on 2% agarose gels, and 200–300-bp DNA fragments were recovered using the CWBIO Gel Extraction Kit (CW2302A; Cwbiotech, Inc., China) and dissolved in EB. cDNA library quality was tested using an Agilent 2100 Bioanalyzer, and the effective concentration was quantified by quantitative real-time PCR. For sequencing, the effective concentration was required to be >2 nM. The cluster was produced in c-Bot using a TruSeq PE Cluster Kit v4-cBot-HS (PE-401-4001; Illumina, Inc.), and then, 100-bp dual-sequence reads were obtained using a HiSeq 2500 machine.
Read mapping, expression quantification, and clustering analysis
All reads were mapped to the human genome (version hg19) using TopHat (version 2.0.6) with default settings. Cufflinks (version 2.0.2) was used to estimate fragments per kilobase of exon per million fragments mapped (FPKM) values (55), and genes with an FPKM value of ≥3 were considered to be expressed. For subsequent analysis, the biotype classifications of genes and transcripts in the ENSEMBL annotation were used to identify noncoding genes. Hierarchical clustering was carried out using Cluster 3.0 and visualized using Java Treeview. For differential expression analysis, we aligned reads against the RefSeq human transcriptome using Bowtie software (v. 2.0.2). Expression levels were then estimated using eXpress (version 1.3.0), with gene-level effective counts and RPKM values derived from the sum of the corresponding values for all isoforms of a gene. The effective count values were then used as input for DESeq to assess differential expression, and the genes with log2-fold change ≥2 and Q-value <0.05 were considered as differential genes. Principal component analysis was performed by a singular value decomposition of the center-scaled gene expression data matrix. All sequencing data were deposited in the Gene Expression Omnibus under accession number GSE110798. As for clustering analysis, the complete method was used for clustering, and the selection of distance and linkage methods was Euclidean.
Gene ontology annotation and STRING analysis
Gene ontology (GO) analysis of DEGs was performed using DAVID and GOstat. GO terms with a p-value of <0.05 were considered to be statistically significant terms. WebGestalt was used for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. Pathway with adjusted p-values <0.05 were determined to be statistically significant pathways. Cytoscape3.1.1 was used to build the networks to show the relationships between candidate genes and GO terms or KEGG pathways. STRING analysis was performed using the STRING database. Generally, genes were grouped based on text mining, and the edges represent known protein/protein interactions. The thickness of edges represents the tightness of the interaction.
Quantitative real-time PCR analysis
As described in our previous study (14), total RNA was extracted from single oocytes using the SMARTer Ultra Low RNA kit for Illumina sequencing. Synthetic cDNA was used for quantitative real-time PCR (qPCR) with an ABI 7500 PCR machine. Gene expression was normalized to the expression of the housekeeping gene GAPDH. Primers were synthesized by Sangon Biotech (Shanghai) Co., Ltd. For mRNA expression identification, 16 IVM and 16 IVO human oocytes were used to extract RNA from each oocyte, respectively.
Enzyme-linked immunosorbent assay
An ELISA was performed to detect enzyme activity in culture medium from mouse oocytes (∼1000 oocytes per assay). Reagents for ELISAs were purchased from Shanghai Jijin Chemistry Technology Co. Ltd (IDH3A, CK-E94849 M; OGDH, CK-E94851 M; SDHA, CK-E94855 M; ACAT1, CK-E94860 M; HADHA, CK-E94857 M). The absorbance was measured at 450 nm, and the concentration of the indicated enzyme was calculated. The intra- and interassay coefficients of variation were <15% and 15%, respectively, with a sensitivity of 0.38 ng/mL. A standard curve was plotted using software capable of generating a four-parameter logistic curve fit, and enzyme levels were calculated with the software Curve Expert 1.3 (Curve Expert).
Measurement of ATP content
The oocyte samples were washed in PBS three times and then treated using 1 μM BODIPY FL adenosine 5′-triphosphate (A12410; Thermo Fisher Scientific, Inc.) for 1 h at room temperature in a cassette. After that, the oocyte samples were rinsed again using PBS three times. Samples were assessed under a Zeiss 710 confocal fluorescence microscope. The average fluorescence intensity per oocyte was calculated using NIH ImageJ analysis software. A total of 17 human oocytes, including 11 IVM and 6 IVO oocytes, were used here.
Detection of mitochondrial content
The oocyte samples were washed three times with PBS and then incubated with MitoTracker@ Red CMXRos dye (M7512; Thermo Fisher Scientific, Inc.). After a 45-min incubation, the oocyte samples were washed three times again using PBS and assessed under a Zeiss 710 confocal fluorescence microscope. The average fluorescence intensity per oocyte was calculated using NIH ImageJ analysis software. A total of 14 human oocytes, including 9 IVM and 5 IVO oocytes, were used here.
Measurement of intracellular glutathione
To detect intracellular glutathione (GSH), mature oocytes were incubated with 50 μM monochlorobimane (mBCI) (M1381MP; Thermo Fisher Scientific, Inc.) at 37°C for 40–60 min, until a steady state was reached, before acquisition of images. mBCI fluorescence was excited at 360 nm and measured at 470 nm. To compare across experiments, GSH fluorescence was expressed as a percentage of the fluorescence emitted from untreated mature oocytes imaged simultaneously. A total of 60 mouse oocytes, including 30 IVM and 30 IVO oocytes, were used here.
Detection of intracellular reactive oxygen species
Intracellular reactive oxygen species (ROS) in living oocytes were stained using the general oxidative stress indicator (CM-H2DCFDA, C6827; Thermo Fisher Scientific, Inc.). Specifically, mature oocytes were incubated with 1 μM CM-H2DCFDA for 30 min at 37°C. After extensive washing, intracellular ROS production was assessed with a Zeiss 710 confocal fluorescence microscope at excitation and emission wavelengths of 488 and 515 nm, respectively. The average fluorescence intensity per oocyte was calculated using NIH ImageJ analysis software. A total of 60 mouse oocytes, including 30 IVM and 30 IVO oocytes, were used here.
Measurement of mitochondrial superoxide levels
Oocyte superoxide production was quantified using MitoSOX™ red mitochondrial superoxide indicator (M36008; Thermo Fisher Scientific, Inc.), following the manufacturer's instructions. Samples were assessed under a Zeiss 710 confocal fluorescence microscope at excitation and emission wavelengths of 510 and 580 nm, respectively. The average fluorescence intensity per oocyte was calculated using NIH ImageJ analysis software. A total of 60 mouse oocytes, including 30 IVM and 30 IVO oocytes, were used here.
Measurement of NADH and oxidized mitochondrial flavoprotein (FAD++) autofluorescence
Autofluorescence intensities of NADH and FAD ++ in oocytes and zygotes were used to estimate mitochondrial redox potential, as described previously (9). Reduced forms of NADH were excited by ultraviolet light at 351 nm, and emitted fluorescence was collected using a 435–485 nm bandpass filter. The oxidized form (NAD+) is nonfluorescent. In contrast to NADH, the reduced form of the flavoprotein FADH2 is nonfluorescent. Fluorescence of the oxidized form of the flavoprotein (FAD++) was excited using the 458 nm line of an argon laser, and emitted fluorescence was collected through the 505–550 nm bandpass filter. Fluorescence signals were normalized to 100 on maximal oxidation of FAD++ and maximal reduction of NADH and to 0 on maximal reduction of FADH2 and NAD+. The relative redox state of each tested oocyte was evaluated. A total of 60 mouse oocytes, including 30 IVM and 30 IVO oocytes, were used here.
Apoptosis assay
To label fragmented 3′ DNA ends (MK500; TUNEL, CLONTECH Laboratories, Inc.) as described previously (38), blastocysts were fixed in 2% paraformaldehyde, permeabilized with 0.5% Triton X-100, and then incubated with fluorescein-labeled dUTP and terminal transferase in the dark for 1 h at 37°C. Propidium iodide was used for nuclear counterstaining. Embryos were observed with a Zeiss 710 confocal fluorescence microscope.
Immunocytochemistry
Protein localization was assessed via the immunofluorescence method following methods described in our previous study (14). Briefly, oocytes were fixed in 4% paraformaldehyde and then washed three times with 0.05 M glycine in PBS. Cells were permeabilized with 0.2% Triton X-100 for 30 min and then placed in a humid chamber and blocked in 3% BSA/PBS for 2 h. Incubation with a primary antibody was carried out overnight at 4°C. After rinsing, the embryos were incubated under the same conditions with a fluorescein isothiocyanate (FITC)-conjugated secondary antibody for 2 h. The nuclear status of embryos was evaluated by staining with 10 μg/mL propidium iodide for 10 min. Finally, the samples were mounted on glass slides and examined with a Zeiss 710 confocal microscope. All of the antibodies were purchased from Abcam, Inc. (PKA, ab75991; CAMKII, ab171095; CAM, ab2860; DPYD, ab54797; γH2AX, ab26350). A total of 120 human oocytes, including 88 IVM and 32 IVO oocytes, were used here.
siRNA injection
siRNA injection was carried out as described previously (14). In brief, siRNA powders and negative control siRNA (ncRNA) were purchased from Santa Cruz Biotechnology, Inc. [ACAT1, sc-96390 (human) and sc-108039 (mouse); HADHA, sc-75220 (human) and sc-75221 (mouse); NNT, sc-91738 (human) and sc-150013 (mouse); DPYD, SC-45326; ncRNA SC-37007]. siRNA powders were diluted in dilution buffer and then injected into the cytoplasm of mouse and human oocytes. Successful inhibition was confirmed by single-cell qPCR. Oocytes injected with negative control siRNA were used as the negative control (nc) group, and uninjected oocytes were used as the control group. After microinjection, immature oocytes were cultured in IVM medium. A total of 146 human IVM oocytes were used here.
Preparation of mouse oocytes
Collection of mouse oocytes was performed as described in our previous study. Briefly, female mice (8–10 weeks old) were superovulated by intraperitoneal injection of 10 IU of pregnant mare's serum gonadotropin (PMSG) (Ningbo Hormone Production, Ningbo, China). For IVM oocytes, the ovaries were collected and punctured repeatedly at 48 h after PMSG injection. The separated oocytes were collected and cultured in an incubator if GV could be observed under a microscope. For IVO oocytes, the mice were injected using an injection of 10 IU of human chorionic gonadotropin (Ningbo Hormone Production, Ningbo, China). The cumulus–oocyte complexes were isolated from oviduct ampulla and treated with 0.3% hyaluronidase. Oocytes with the first polar body were cultured in an incubator for further experiments.
Oocyte dot blot
Oocytes of each group were treated using 4 μL of RIPA lysis buffer (CW2333S; Cwbiotech, Inc., China), and then, 2 μL of lysate from each group was loaded onto a PVDF membrane (EMD Millipore Corporation). After drying at room temperature, the membrane was blocked for 1 h in TBST (0.2 M NaCl, 0.1% Tween-20, and 10 mM Tris [pH 7.4]) containing 5% nonfat dry milk. The blocked membranes were then incubated with a primary antibody (Abcam) in TBST overnight at 4°C. After that, membranes were incubated with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG; 1:1000; sc-2030; Santa Cruz Biotechnology) in TBST for 1 h at room temperature. After each step, the membrane was washed three times for 5 min with TBST, and bound antibody was then detected using an enhanced chemiluminescence detection system (Carestream 4000MM) according to the manufacturer's instructions. The antibodies targeted PKA, CAMKII, CAM, and NNT (ab209565). A total of 12 human oocytes, including 6 IVM and 6 IVO oocytes, were used here.
Derivation of human ES cells
The procedure for deriving human ES cells was as described in our previous studies (63). Briefly, the inner cell mass was isolated using laser equipment (ZILOS-tk; Hamilton) and was immediately transferred onto prepared feeder cells for approximately 5–7 days, and primary ESC colonies were dissociated mechanically and cultured in human ES medium consisting of 80% Dulbecco's modified Eagle's medium (DMEM) (A1286101; Thermo Fisher Scientific, Inc.), 20% serum replacement (10828028; Thermo Fisher Scientific, Inc.), 1 mM glutamine (25030081; Thermo Fisher Scientific, Inc.), 1% nonessential amino acids (11140050; Thermo Fisher Scientific, Inc.), 0.1 mM 2-mercaptoethanol, and 50 UI/mL penicillin and 50 UI/mL streptomycin mixture. ES cell colonies were mechanically dissociated every 4–5 days. All human ES cells were cultured at 37°C and 5% CO2 under a humidified atmosphere.
The remaining TE cell clusters were pinched in half with a needle. One part was transferred to lysis buffer for array CGH testing, and the other was cryopreserved at −80°C for examination of gene expression.
Array comparative genome hybridization
Genomic DNA was extracted from TE cells and amplified using a whole-genome amplification kit (150025; Qiagen, Inc., Germany). Array CGH manipulation using a BlueGnome platform was performed according to the instruction book and as reported previously (16). In brief, the amplified DNA samples were labeled with Cy3 or Cy5 and mixed with control human DNA. After dried and resuspended, the samples were loaded onto the BlueGnome 24Sure V3 arrays. The data were analyzed using BlueFuse software (BlueGnome).
Statistics
The results were compared using SPSS 17.0 software. An independent-samples t-test was performed to compare gene expression data from the two groups. A one-way ANOVA was used when comparing data from three or more groups. A chi-squared analysis was used to compare the numbers of embryos between two groups. p-Values of <0.05 were considered to indicate significant differences. Each experiment was repeated at least three times for independent biological replicates.
Study approval
Mice used in the present study were housed and bred in the Animal Center of the Medical College of Peking University according to national legislation for animal care. Human oocytes were obtained from Peking University Third Hospital and the Third Affiliated Hospital of Guangzhou Medical University, and rabbit and nonhuman primate oocytes were obtained from the Yunnan Key Laboratory of Primate Biomedical Research, Institute of Primate Translational Medicine. All the protocols in the present studies were approved by the IRB of Peking University Third Hospital, the Third Affiliated Hospital of Guangzhou Medical University, and the Institute of Primate Translational Medicine. The collection of human oocytes strictly followed the guidelines legislated and posted by the Ministry of Health of the People's Republic of China, including the “Technical Regulation for Human Assisted Reproductive Technology” and “Ethical Guiding Principles for the Research of Human ESCs.”
Authors' Contributions
H.C.Z. performed collection and IVM of human oocytes, and charged for the identification of screened genes and their encoding proteins using qPCR, dot-blot, and immunohistochemistry. Y.Z. performed bioinformatic analysis; T.J.T. performed mouse study; T.T. collected rabbit and monkey oocytes, did the qPCR experiment, and charged for bioinformatic analysis. C.Y.L. performed siRNA injection experiment; Y.L.L. performed rat oocyte collection and qPCR identification. L.C. charged for aCGH analysis; N.H. took part in critical discussion and data analysis; C.L. joined bioinformatic analysis; Y.F. and Y.Y. joined in study design, article drafting, and critical discussion and article submission. R.L. charged for the management of clinical patients, and mainly contributed to the article revision; J.Q. contributed to the conception of design and coordinated the research and article editing.
Footnotes
Acknowledgments
This work was supported, in part, by the National Key R&D Program of China (2016YFC1000601), the Ministry of Science and Technology of China Grants (2014CB943203), the National Natural Science Funds for general program (81571400, 31501201, 81771580, 81570101, and 31371521), and the Beijing Nova Program (xxjh2015011).
Author Disclosure Statement
All authors declare no competing financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.