
Abstract
Significance:
In contrast to structural elements of the extracellular matrix, matricellular proteins appear transiently during development and injury responses, but their sustained expression can contribute to chronic disease. Through interactions with other matrix components and specific cell surface receptors, matricellular proteins regulate multiple signaling pathways, including those mediated by reactive oxygen and nitrogen species and H2S. Dysregulation of matricellular proteins contributes to the pathogenesis of vascular diseases and cancer. Defining the molecular mechanisms and receptors involved is revealing new therapeutic opportunities.
Recent Advances:
Thrombospondin-1 (TSP1) regulates NO, H2S, and superoxide production and signaling in several cell types. The TSP1 receptor CD47 plays a central role in inhibition of NO signaling, but other TSP1 receptors also modulate redox signaling. The matricellular protein CCN1 engages some of the same receptors to regulate redox signaling, and ADAMTS1 regulates NO signaling in Marfan syndrome. In addition to mediating matricellular protein signaling, redox signaling is emerging as an important pathway that controls the expression of several matricellular proteins.
Critical Issues:
Redox signaling remains unexplored for many matricellular proteins. Their interactions with multiple cellular receptors remains an obstacle to defining signaling mechanisms, but improved transgenic models could overcome this barrier.
Future Directions:
Therapeutics targeting the TSP1 receptor CD47 may have beneficial effects for treating cardiovascular disease and cancer and have recently entered clinical trials. Biomarkers are needed to assess their effects on redox signaling in patients and to evaluate how these contribute to their therapeutic efficacy and potential side effects. Antioxid. Redox Signal. 27, 874–911.
Table of Contents
B. Historical evidence linking matricellular proteins and redox signaling
C. Thrombospondin-1 regulation of soluble guanylate cyclase and downstream targets
D. Physiological functions of TSP1/CD47 regulation of NO signaling
3. Regulation of other matricellular protein expression by NO
IV. Thrombospondin-1 Regulation of H2S Biosynthesis and Signaling
VIII. CD47 Regulation of Redox Homeostasis in Irradiated Cells
B. CD47-dependent regulation of redox metabolites in irradiated cells
IX. TSP1/CD47 Regulation of Stem Cell and Tissue Self-Renewal
B. TSP1 and CD47 in stem cell self-renewal and tissue regeneration
A. Dysregulation of NO signaling by TSP1/CD47 in clinical disease
1. Association of elevated TSP1 with impaired NO signaling in cardiovascular disease
I. Introduction
A. What are matricellular proteins?
M
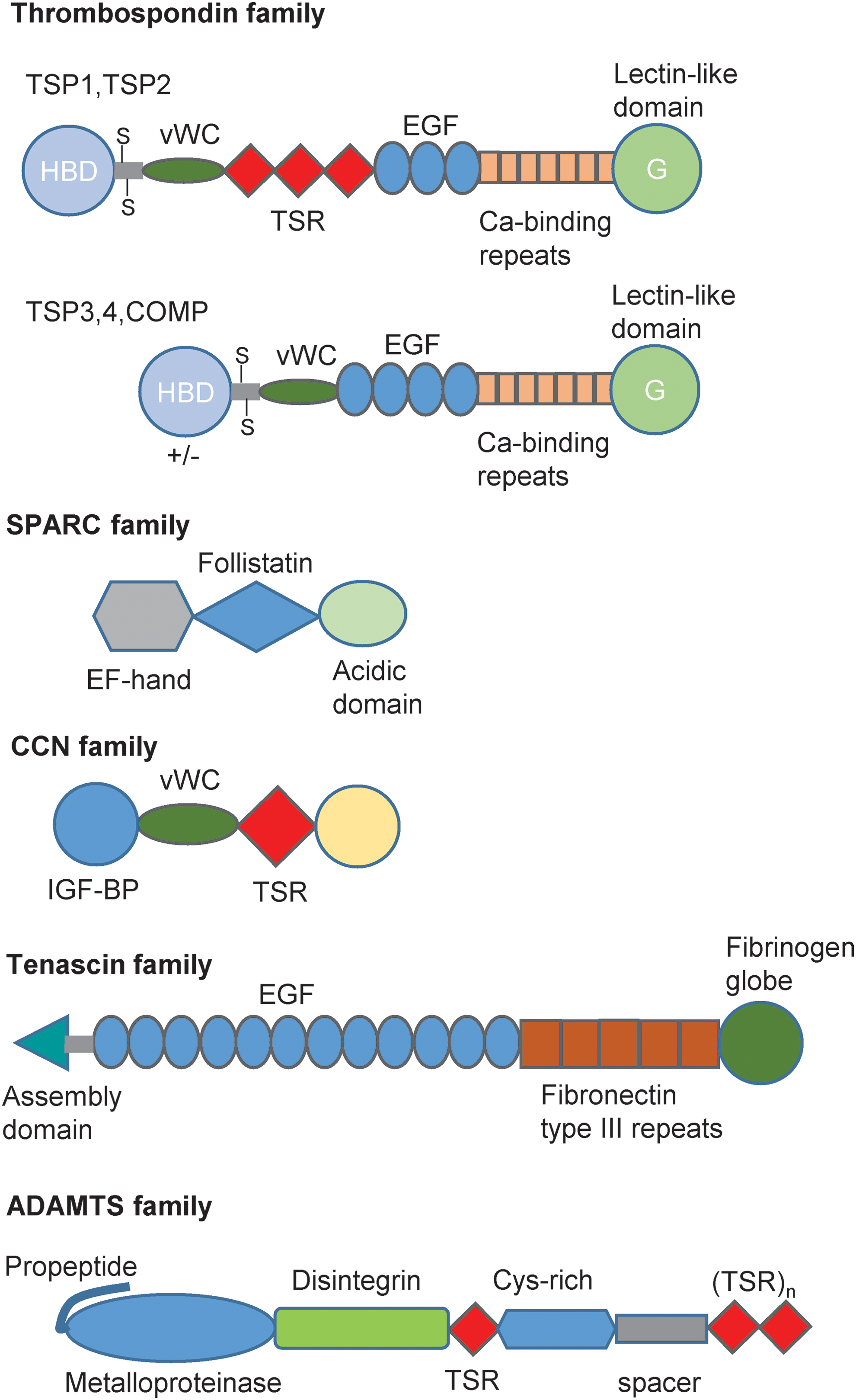
The first members of the TSP and SPARC families emerged in early metazoa and underwent gene duplications that diverged into their current family members (169). Gene deletion studies indicate relatively little overlap in functions of the five TSPs (3, 196), although some receptor interactions are conserved between family members. Integrins are the only known receptor class interacting with the primordial TSP in insects that remains functional for the modern proteins (26). Modern trimeric forms of TSP1 and TSP2 trace back at least to fish. TSP1 and TSP2 share >80% homology between their C-terminal domains, which mediates TSP1 binding to CD47, and homology decreases to 25% between their N-terminal domains. The TSP1 receptor CD47 originated in early land-dwelling vertebrates, roughly coincident with the appearance of endothelial nitric oxide synthase (eNOS/NOS3) (63).
In common with classical extracellular matrix proteins, most matricellular proteins contain several functional domains that mediate interactions with other extracellular matrix components and with specific receptors. This capacity for multifarious interactions has contributed to confusion in the literature because a given matricellular protein can elicit opposing responses in cells that express different subsets of its receptors, and responses of a given cell type can change over time as the expression of receptors and composition of the surrounding extracellular matrix microenvironment respond to external stimuli.
With a few exceptions, matricellular protein genes are generally not essential for life. Phenotypes of null mutants may only become evident when mice are subjected to a specific stress that induces matricellular protein expression or signaling. Despite these challenges, studies of matricellular protein function in transgenic mouse models and identification of genetic diseases linked to structural and regulatory mutations in specific matricellular protein genes have proven that they play important roles in physiology and pathophysiology (126, 142, 171, 243).
B. Historical evidence linking matricellular proteins and redox signaling
Earlier studies of TSP1 provided clues that altered redox signaling could mediate some cellular responses controlled by this protein. Immobilized TSP1 inhibited the ability of tumor necrosis factor-α (TNFα) to induce an oxidative burst response in polymorphonuclear cells (PMN) (179). Conversely, TSP1 enhanced superoxide production by neutrophils stimulated with the chemoattractant formyl-Met-Leu-Phe (fMLP), and fMLP upregulated the binding of TSP1 to presumed receptors on these cells (257).
In retrospect, several earlier studies suggested an association between TSP1 and nitric oxide signaling. The ability of TSP1 to disrupt focal adhesions in vascular smooth muscle cells (VSMCs) required the activity of cGMP-dependent protein kinase (170), and treatment with TSP1 acutely decreased cGMP levels in a human melanoma cell line (68). A specific role of TSP1 in regulating NO-induced synthesis of cGMP was first identified in endothelial cells (93), and subsequent studies have revealed a broad role for TSP1 signaling through its receptor CD47 to adversely regulate cytoprotective cellular redox signaling (243).
Here we review studies that have established specific mechanisms by which TSP1 and other matricellular proteins including CCN1, periostin, osteopontin, and ADAMTS1 regulate the production of nitric oxide, H2S, superoxide, and other reactive oxygen species (ROS) (Fig. 2A), and TSP1, conversely, regulates cellular redox signaling downstream of NO and H2S (Fig. 2B).
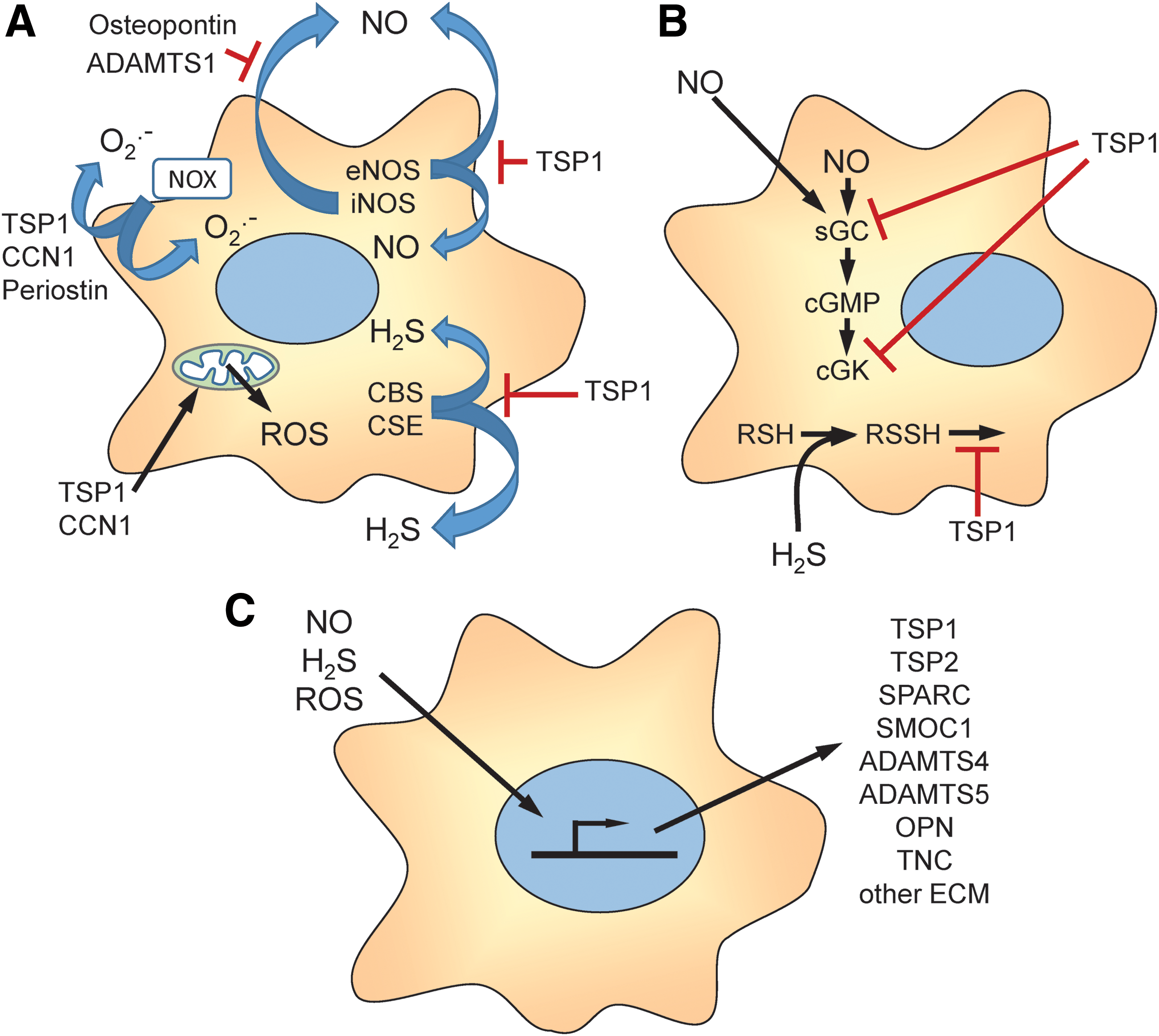
Redox and hypoxia signaling, in turn, regulate the expression, secretion, post-translational modification, and extracellular interactions of matricellular and other extracellular matrix proteins (Fig. 2C), which is discussed in more detail elsewhere in this Forum (34, 81, 119, 124, 128, 178). We review the emerging physiological functions of this regulation and the evidence that pathological dysregulation of matricellular protein expression contributes to acute and chronic disease states that are characterized by insufficient NO signaling and/or increased oxidative stress. Finally, we consider the progress made toward developing therapeutic approaches to restore NO function and other aspects of redox homeostasis by targeting the TSP1 receptor CD47.
II. Thrombospondin-1 and NO Signaling
A. Thrombospondin-1 structure and receptors
TSP1 is a trimeric protein consisting of identical subunits linked covalently through intersubunit disulfide bonds in the oligomerization domain (Fig. 3). The N-terminal domain of each subunit consists of a globular heparin-binding domain that interacts with several β1-integrins, sulfated glycoconjugates, several hyaluronan-binding proteins, and calreticulin. This is followed by a coiled-coil oligomerization domain, a von Willebrand C domain, three TSRs, three epidermal growth factor (EGF)-like repeats, seven calcium-binding repeats, and the C-terminal lectin-like G-domain (Fig. 3) (23). The TSRs mediate binding of TSP1 to the cell surface receptor CD36 and to transforming growth factor-β1 (TGFβ). Additional low-affinity integrin binding sites are present in the TSRs and EGF repeats (21), and the EGF repeats mediate binding to the gabapentin receptor α2δ1 (49). In the presence of physiological Ca2+ concentrations, the calcium-binding repeats wrap around the G domain to form the “signature domain” of TSP1 (23). The signature domain mediates binding of TSP1 to CD47 and to stromal interaction molecule 1 (STIM1) (48). The last Ca repeat also contains a RGD sequence that can be recognized by αvβ3 integrin but is cryptic in the Ca-replete protein (259).
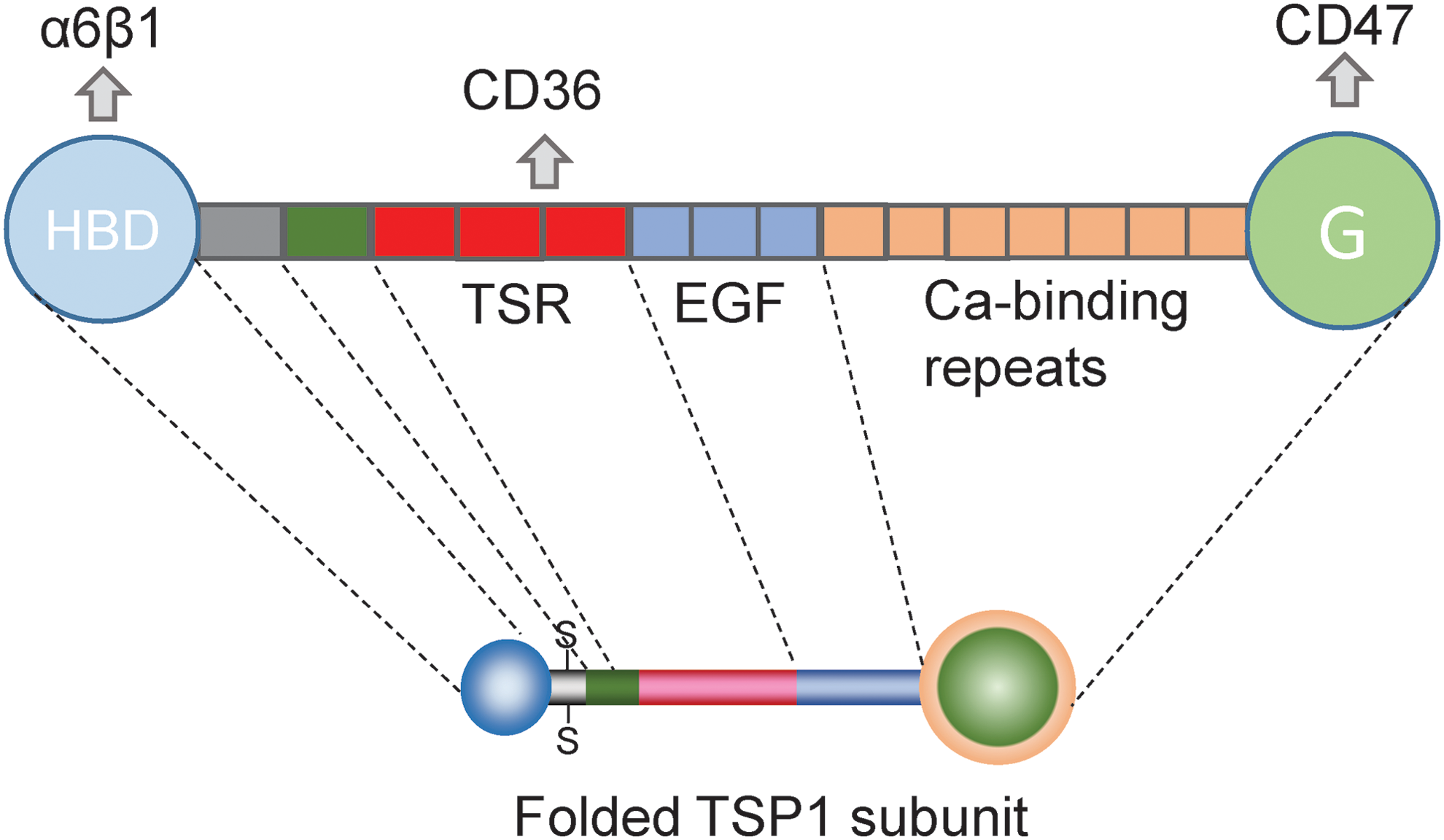
Specific peptide sequences have been identified that are involved in binding of TSP1 to several of its receptors. Although two TSP1 peptides containing a Val-Val-Met (VVM) consensus sequence were initially identified as potential CD47 binding sites and could be used to affinity purify CD47 (31, 46, 58), the crystal structure of the C-terminal domain of TSP1 indicated that the VVM sequences are not exposed to solvent (121). A potential conformation change that exposes one of the VVM sequences has been proposed based on dynamic modeling studies (54), but structure function and mutagenesis studies have not been performed to determine whether the VVM sequences are involved in binding of native TSP1 to CD47. Furthermore, both TSP1-derived CD47 binding peptides have well-documented CD47-independent activities, and they cannot be used as reliable surrogates for TSP1 to investigate CD47 signaling (9, 129, 279).
In addition to the undefined protein–protein interaction, binding of TSP1 to CD47 requires post-translational modification of CD47 with a heparan sulfate glycosaminoglycan at Ser64 (110). Because glycosylation can be cell-type specific, CD47 may vary in its ability to interact with TSP1 to control NO signaling. Such variation in functional activity to control NO signaling has been reported in a T cell line (202), but further studies are needed to define the molecular mechanism.
B. Thrombospondin-1 regulation of NO synthesis
TSP1 regulates both the production of NO in vascular cells (Fig. 4) and responses of such cells to exogenous NO mediated by soluble guanylyl cyclase (Fig. 5). Depending on the cell type, TSP1 can regulate NO production by several mechanisms. In endothelial cells, TSP1 signaling inhibits the basal and acetylcholine-stimulated conversion of arginine to NO and citrulline by eNOS, whereas Thbs1−/− and Cd47−/− murine endothelial cells display increased basal eNOS activity compared with wild type (WT) cells (10). This result was subsequently confirmed in choroidal endothelial cells (53). Choroidal capillary endothelial cells from the eyes of Thbs1−/− mice had elevated phosphorylation of eNOS relative to WT cells, and intracellular NO in the null cells assessed using 4-amino-5-methylamino-2,7-difluorofluorescein (DAF) was sixfold higher than in WT cells. One should bear in mind that DAF is primarily detecting an oxidative product of NO rather than NO itself (176).
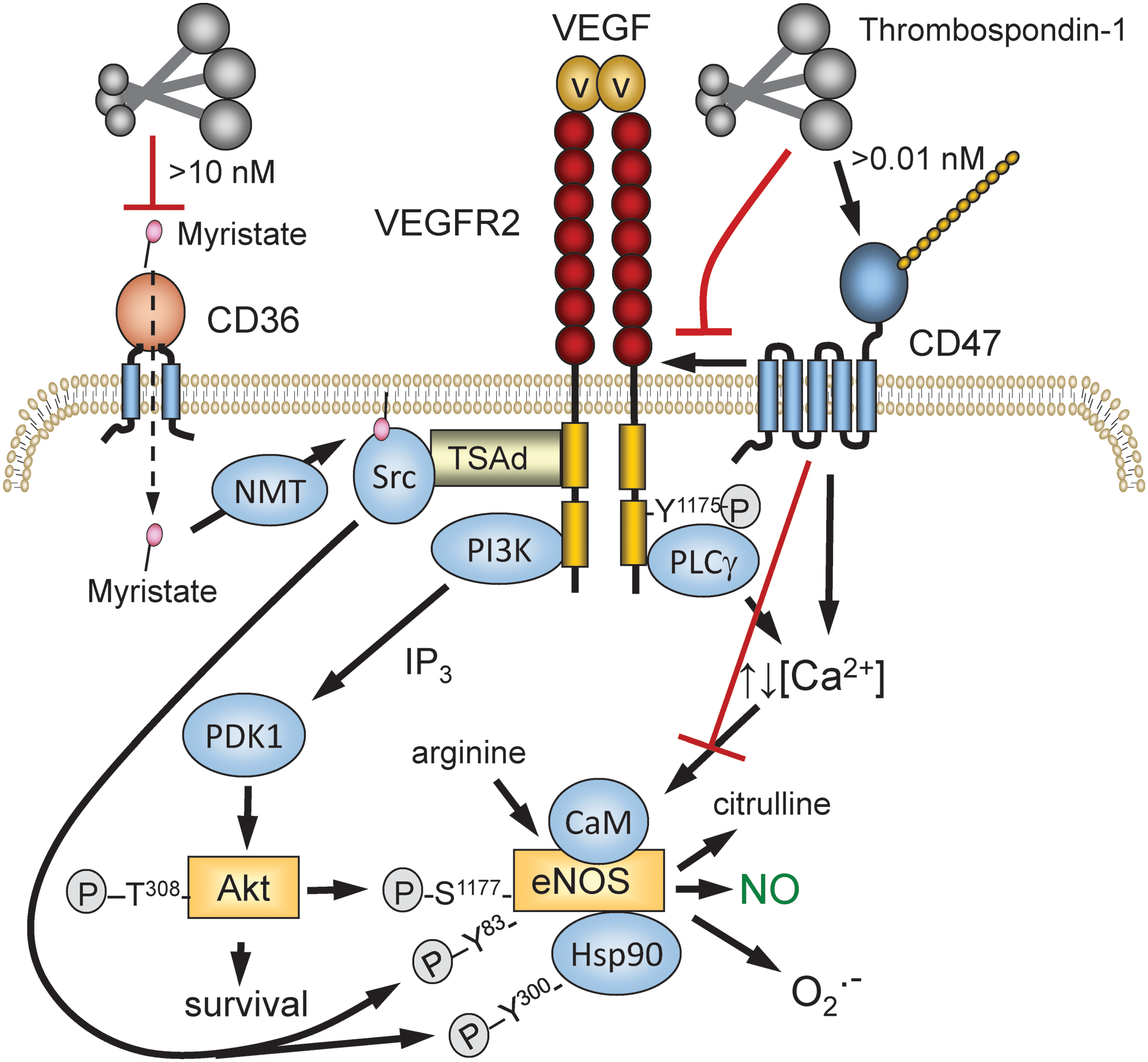
eNOS is a highly regulated enzyme (55), and CD47 controls several of the pathways known to regulate eNOS activity. CD47 constitutively associates with the tyrosine kinase vascular endothelial growth factor receptor-2 (VEGFR2) in endothelial cells and T cells (111). TSP1 binding to CD47 displaces CD47 from VEGFR2 and inhibits VEGFR2 auto-phosphorylation. Activated VEGFR2 controls several downstream pathways that control eNOS activation (Fig. 4). TSP1 inhibits vascular endothelial growth factor (VEGF)-stimulated phosphorylation of Akt at Ser473 (111). The PI-3-kinase/Akt pathway activates eNOS by phosphorylation of Ser1177 on eNOS (55), and TSP1 inhibits this phosphorylation (10). TSP1 inhibits VEGFR2-mediated phosphorylation of Src kinase at Tyr416 in a CD47-dependent manner (108). Src phosphorylates Tyr residues on eNOS and the associated Hsp90 that regulate eNOS activity, and TSP1 inhibits acetylcholine-mediated coassociation of eNOS and Hsp90 (10).
VEGFR2-mediated activation of phospholipase Cγ controls cytoplasmic calcium levels, which activate eNOS by inducing calmodulin binding (55). TSP1 signaling via CD47 has been reported to regulate calcium signaling in endothelial cells and T cells, but in opposing directions (10, 202). Therefore, the coupling between CD47 ligation by TSP1 and cytoplasmic calcium levels may be cell-type specific and may depend on whether ligand binding induces clustering of CD47 (155). Acylation by the fatty acid myristic acid is also important for maximal eNOS function (14, 143). In endothelial cells, TSP1, TSP1-derived peptides, and a CD36-binding TSP1 mimetic further limit eNOS activity by restricting myristic acid uptake through the fatty acid translocase and TSP1 receptor CD36 (87, 97) (Fig. 4), although the in vivo relevance of this remains to be determined.
Notably, Fei et al. also reported that inducible nitric oxide synthase (iNOS) expression was elevated in Thbs1−/− choroidal endothelial cells relative to WT cells, suggesting that negative regulation of NO synthesis by TSP1 is not limited to eNOS (53), although it was not determined whether these findings in null cells were reversed by exogenous TSP1. Phosphorylation of STAT3 was markedly elevated in the Thbs1−/− cells, which was suggested to mediate the upregulation of iNOS, but additional studies are needed to confirm this mechanism. Negative regulation of STAT3 phosphorylation by TSP1 was also reported in colon tissues (69). Phosphorylation of STAT3 at Ser727 was increased twofold in Thbs1−/− colon tissue, and this was inhibited by a TSP1 mimetic designed to engage the TSP1 receptor CD36 (ABT898). Functional inhibition of STAT3 by TSP1 was further indicated by increased plasma interleukin (IL)-6 levels in the Thbs1−/− mice.
C. Thrombospondin-1 regulation of soluble guanylate cyclase and downstream targets
1. Soluble guanylate cyclase
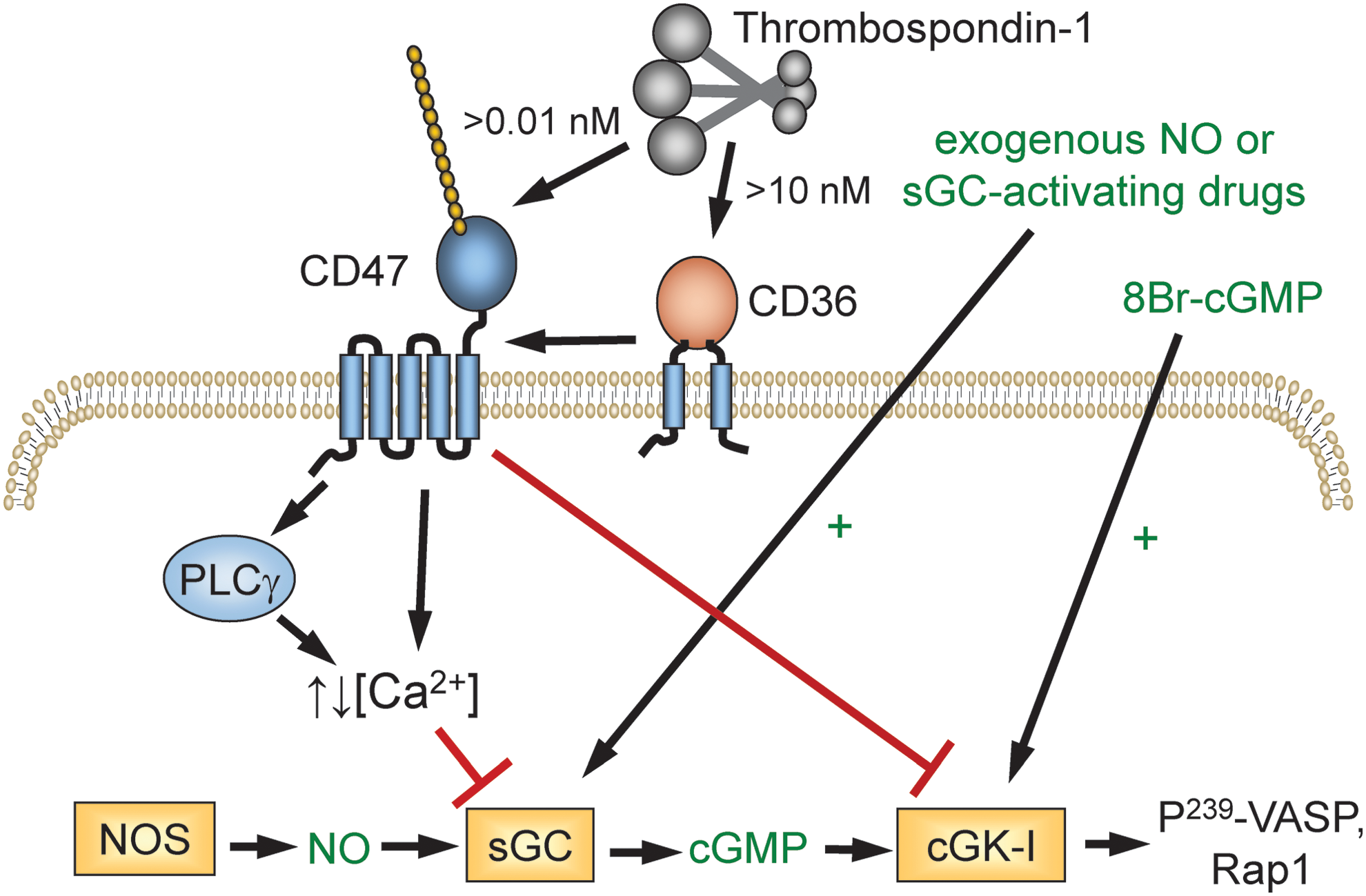
TSP1/CD47 signaling also controls signaling downstream of NO by limiting the activation of the primary NO sensor/receptor soluble guanylate cyclase (sGC) (Fig. 5) (93). This opens the potential for controlling signaling by NO produced by all three nitric oxide synthase (NOS) isoforms and for regulating paracellular signaling wherein NO produced by one cell diffuses into an adjacent cell.
The mechanism by which CD47 signaling inactivates sGC remains to be fully characterized, but some details are known. Inhibition of sGC in immortalized T cells by a recombinant C-terminal fragment of TSP1 (E3CaG1) is mediated by increased cytoplasmic Ca2+, and elevation of Ca2+ after treatment with angiotensin-II resulted in a similar inhibition (202). The NO-insensitive state of sGC persisted in cell-free lysates and was prevented by the protein kinase inhibitor staurosporine, which suggests that TSP1/CD47 signaling induces an inhibitory phosphorylation of sGC. Inhibition of sGC by TSP1 was first reported in endothelial cells, but subsequent studies have confirmed this result in VSMCs (96), platelets (95, 162), T cells (202), and macrophages (206) of human and nonhuman origin, broadly implicating TSP1 as limiter of sGC activity.
The initial studies in endothelial cells suggested that TSP1 regulates sGC in a CD36-dependent manner (93). A recombinant type 1 repeats construct from TSP1 inhibited NO-induced chemotaxis of human umbilical vein endothelial cells and cGMP accumulation in the cells, whereas a recombinant N-terminal region of TSP1 that engages integrins, calreticulin/low-density lipoprotein receptor-related protein-1 (LRP1), and heparan sulfate proteoglycans did not. Furthermore, the CD36 antibody SMΦ, which is a reported agonist of CD36 signaling (37), significantly inhibited NO-stimulated proliferation of human umbilical vein endothelial cells and dermal microvascular endothelial cells and inhibited NO-induced cGMP accumulation. Similar inhibition of NO responses by the CD36-binding domain of TSP1 and the CD36 antibody was reported in VSMCs (96).
These studies established that engaging CD36 is sufficient to inhibit NO-stimulated activation of sGC. However, additional studies challenged the hypothesis that CD36 directly mediates this inhibitory activity of TSP1 (92). NO-stimulated vascular outgrowth was inhibited by TSP1 to the same extent in skeletal muscle explants from WT and Cd36−/− mice. TSP1 also inhibited NO-stimulated adhesion of VSMCs from Cd36−/− mice, and NO-stimulated accumulation of cGMP in these cells. Therefore, engaging CD36 is sufficient but not necessary to inhibit sGC activity.
In contrast, the TSP1 receptor CD47 is both necessary and sufficient to mediate inhibition of NO signaling by TSP1 (92). NO-stimulated vascular outgrowth from muscle tissue explants from Cd47−/− mice was completely insensitive to inhibition by TSP1. Ligation of CD47 by two CD47-binding peptides derived from TSP1 or by recombinant E3CaG1, containing the CD47 binding domain of TSP1, potently inhibited cGMP accumulation and functional responses to NO signaling. In the absence of exogenous TSP1, ligation of CD47 by the CD47 antibodies C1Km1 and B6H12 also inhibited NO signaling. This inhibitory activity of B6H12 is notable because B6H12 inhibits TSP1 binding to CD47 (84), suggesting that this antibody can, under TSP1-depleted conditions, act as a mimic of TSP1 and activate CD47 signaling to inhibit NO stimulation of sGC independent of its ability to prevent TSP1 binding to CD47.
Further examination of the roles of CD36 versus CD47 revealed that CD47 is also necessary for the inhibitory activity of CD36 ligands, but CD36 is not necessary for the inhibitory activity of CD47 ligands (92). A CD47-binding peptide from TSP1 and a recombinant C-terminal domain of TSP1 inhibited NO-stimulated cGMP accumulation in VSMCs from Cd36−/− mice, but a CD36 binding peptide did not inhibit cGMP accumulation in VSMCs from Cd47−/− mice. Finally, basal and NO-stimulated cGMP levels were elevated relative to WT in Cd47−/− cells but not in Cd36−/− cells. Therefore, CD47 is the necessary and sufficient receptor for mediating the inhibitory effect of TSP1 on NO-mediated activation of sGC. CD47 is also necessary for CD36-mediated inhibition of this pathway, but the molecular mechanism of this receptor cross-talk remains to be determined.
From a physiological perspective, another important distinction between the CD36 and CD47 pathways is their TSP1 dose dependence. Inhibition through CD47 is observed at 10 pM TSP1, but inhibition through CD36 requires 10 nM TSP1. Circulating TSP1 levels in healthy individuals are 100–200 pM. Therefore, physiological TSP1 concentrations are sufficient to tonically limit sGC activation in vascular cells through CD47 but not through CD36. The higher relative affinity of TSP1 for signaling through CD47 also has pathophysiological significance, which is demonstrated by the observation that Thbs1−/− mice and Cd47−/− mice show similar resistance to ischemic injuries and vascular remodeling after ischemic injury, but Cd36−/− mice rather than exhibiting an advantage over WT mice when subjected to the same ischemic injuries tended to be more sensitive to injury (90, 94).
A major focus of the pharmaceutical industry has been to develop drugs to overcome impaired NO signaling by directly activating sGC. TSP1/CD47 signaling in platelets inhibits sGC activation by the heme-dependent sGC activators YC-1 and BAY 41-2272 and the heme-independent activator meso-porphyrin IX (162). Because TSP1 expression is now known to be elevated in many of the diseases for which sGC activators could be useful, such pathological elevation in extracellular TSP1 levels may limit the efficacy of drugs that target sGC. However, YC-1 and BAY 41-2272 could overcome sGC inhibition by E3CaG1 in Jurkat T cells, suggesting that such regulation of sGC by TSP1 may be context dependent (202). However, the ability of these drugs to overcome TSP1 inhibition of sGC in primary vascular cells and in vivo remains to be determined.
CD47-dependent inhibition of sGC activation was also observed when cells were exposed to β-amyloid (163). β-Amyloid interacts with the TSP1 receptor CD36 but not with CD47. β-Amyloid inhibition also extends to activation of sGC by BAY 41-2272. The NO inhibitory activity of β-amyloid is maintained in Thbs1−/− endothelial cells, indicating that this effect is TSP1 independent (163). Given the role of β-amyloid in certain neurodegenerative processes, these findings in vascular cells invite further inquiry.
2. cGMP-dependent protein kinase
TSP1 also inhibited endothelial cell adhesion stimulated by 8-Br-cGMP, indicating that TSP1 has a second inhibitory target downstream of sGC (93). TSP1 limited the antithrombotic activity of NO and the cell-permeable cyclic analogue 8-Br-cGMP in WT platelets (95) (Fig. 5). 8-Br-cGMP activates cGMP-dependent protein kinase, and this was identified as the downstream target of CD47 signaling based on the ability of TSP1 to inhibit phosphorylation of the cGMP-dependent protein kinase-I selective substrate Arg-Lys-Arg-Ser-Arg-Ala-Glu stimulated by NO or by 8-Br-cGMP.
TSP1 also inhibited phosphorylation of the cGMP-dependent protein kinase target protein vasodilator-stimulated phosphoprotein (VASP) at Ser239 that was induced by 8-Br-cGMP. The downstream activation of the GTPase Rap1 was also inhibited in a CD47-dependent manner. The molecular mechanism by which CD47 inhibits cGMP-dependent protein kinase remains to be determined. To summarize, TSP1 via CD47 acts redundantly on the canonical NO pathway, limiting production, receptor activation, and cGMP signaling.
D. Physiological functions of TSP1/CD47 regulation of NO signaling
1. Platelet homeostasis
NO is a physiological inhibitor of platelet activation and aggregation. NO is produced by eNOS expressed in platelets and vascular endothelium (199). The resulting tonic inhibition of platelet activation contributes to preventing thrombosis but must be suppressed for platelets to effectively limit bleeding at sites of vascular injury. Because TSP1 is a major protein in platelet α-granules and is rapidly released after their activation by thrombin, it was an obvious candidate for mediating this function.
Earlier studies reported contradictory evidence regarding the role of TSP1 in platelet activation (135, 280), and the first evaluation of platelet function in Thbs1−/− mice reported normal hemostatic function as assessed by tail bleeding and thrombin activation of washed platelets (127). Therefore, TSP1 was concluded to not be required for normal platelet function. However, the buffer used to assess activation of the washed platelets did not provide the arginine required for platelet eNOS to maintain physiological NO concentrations. When this experiment was repeated in the presence of either arginine or an exogenous NO donor, Thbs1−/− and Cd47−/− platelets showed clear defects in thrombin-mediated activation (95). Correspondingly, cGMP levels were elevated in Thbs1−/− platelets but restored to normal levels by addition of exogenous TSP1 or a CD47-binding peptide. Concentrations of TSP1 that limited platelet NO signaling were comparable with those that circulate in plasma.
Consistent with its capacity to engage in multiple interactions, other studies have revealed additional physiological functions of TSP1 in platelet hemostasis that are independent of NO signaling (13), but an NO-dependent prothrombotic activity is one clear physiological function of platelet TSP1. This function is consistent with the evolutionary record. The presumed gene duplication that gave rise to TSP1 in tetrapods (2) was followed soon thereafter in amphibians and higher land-dwelling vertebrates by the gene duplication that yielded eNOS to enable local vascular control of NO production in high pressure circulatory systems (274) and the appearance of CD47, presumably by a recombination event that joined a primordial IgV domain with a presenilin transmembrane domain (208, 210).
2. Vascular perfusion
Functional magnetic resonance imaging of WT and Thbs1−/− mice provided the first evidence that TSP1 functions as a physiological regulator of NO in the local control of tissue vascular perfusion (85). Functional imaging of blood oxygenation in the proximate hind limb muscle of mice was assessed after intestinal administration of a rapidly releasing NO donor. The resulting increase in perfusion in the Thbs1−/− mice was faster and greater than (roughly double) that observed in WT mice. This demonstrated that endogenous TSP1 in the vasculature constitutively limits responsiveness to NO-mediated vasodilation by ∼50%. cGMP levels were correspondingly elevated in Thbs1−/− tissues and VSMCs.
Mechanistic studies demonstrated that TSP1 blocked the dephosphorylation of myosin light chain-2 induced by treating these cells with NO and thereby promoted F-actin assembly and contraction of the smooth muscle cells. Notably, the elevated tissue cGMP levels found in young male Thbs1−/− mice persisted as those mice were aged to 12–16 months, but cGMP levels fell significantly with age in male WT mice (86), consistent with the reported elevations in tissue TSP1 and CD47 levels with aging (19, 105, 207, 213). Thus, TSP1 physiologically opposes the activity of NO to regulate vascular tone, but this balance becomes perturbed with increasing age. The reasons for the progressive and, in regard to NO signaling, maladaptive upregulation of TSP1 with age are not known.
3. Central blood pressure
Analysis of blood pressure using telemetry implants in young Thbs1−/− and Cd47−/− mice revealed that the physiological regulation by this pathway extends to control of central blood pressure (91). Thbs1−/− mice have decreased pulse pressure than WT mice, and during the active period of their diurnal cycle they exhibited increases in heart rate and central diastolic and mean arterial blood pressure. Cd47−/− mice have normal pulse pressure but lower resting mean arterial pressure, systolic blood pressure, and diastolic blood pressure than WT mice (10). Further studies of responses to vasoactive agents in the mice and isolated perfused vessels confirmed that endogenous TSP1 in the vessels and in circulation functions as a pressor agent supporting blood pressure.
Loss of this pressor activity in Thbs1−/− and Cd47−/− mice increases the sensitivity of these mice to pharmacological challenges that impair blood pressure homeostasis, including isoflurane anesthesia, nitrovasodilators, and chemical sympathectomy with hexamethonium chloride (91), suggesting a major role for the autonomic outflow track in stabilizing blood pressure in null animals lacking the supporting role of the TSP1/CD47 axis. Therefore, in healthy young animals, TSP1 and CD47 play relatively minor roles in blood pressure homeostasis at rest, but their function is more important for cardiovascular responses required for normal activity and to adapt to stress conditions.
E. NO regulation of TSP1 expression
In parallel with the developing understanding that TSP1 regulates NO signaling, evidence was accumulating suggesting a negative feedback loop, wherein NO controls expression of TSP1. Indirect evidence for NO inhibition of TSP1 expression was first provided by studies in endothelial cells. Endothelial cells treated with the arginine analogue
Treatment of human umbilical vein endothelial cells with exogenous NO using 0.1–1 μM diethyltriamine NONOate (DETA/NO) suppressed TSP1 mRNA and protein expression at 2 and 24 h, respectively. Inhibition could be reversed by treating cells with the sGC inhibitor 1H-[1,2,4]oxadiazole[4,3-a]quinoxalin-1-one (ODQ, IC50 = 20 nM). This is consistent with the finding that inhibition of glucose-induced TSP1 expression in mesangial cells by an NO donor was reversed by ODQ and by the inhibitor of cGMP-dependent protein kinase Rp-8-pCPT-cGMPS (291). Constitutive expression of cGMP-dependent protein kinase in mesangial cells also prevented induction of TSP1 expression in mesangial cells (293). Therefore, inhibition of TSP1 expression in mesangial and endothelial cells by nanomolar concentrations of exogenous NO is mediated by sGC.
In a rat model of mesangial proliferative glomerulonephritis, oral administration of a phosphodiesterase-5 inhibitor, to increase the half-life of cGMP, resulted in decreased renal TSP1 expression (76). The mitogen-activated protein kinase kinase (MEK) inhibitor U0126 (1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene) partially reversed low dose NO-mediated inhibition of TSP1 mRNA and protein expression in human umbilical vein endothelial cells, indicating that the MEK/extracellular-regulated protein kinase (ERK) pathway is also involved in negative regulating TSP1 expression (205). Consistent with this hypothesis, addition of
DETA/NO concentrations between 1 and 100 μM modestly increased TSP1 protein expression in human umbilical vein endothelial cells, whereas 1000 μM DETA/NO strongly suppressed TSP1 expression, suggesting a triphasic regulation of TSP1 expression by NO (205). The inhibition of TSP1 expression at 1000 μM DETA/NO coincided with increased phosphorylation of p53 at Ser15 and increased expression of MAP kinase phosphatase-1 (MKP1/DUSP1). Oxidative stress induces MKP1/DUSP1 expression in a p53-dependent manner (146), and both alterations are likely mediated by reactive nitrogen species (RNS) rather than directly by NO (272). MKP1 levels varied inversely with ERK phosphorylation across the NO dose response, suggesting that MKP1 inhibits the ERK-dependent regulation of TSP1 expression.
The inhibition of TSP1 expression by high concentrations of NO donors or the resulting RNS has been replicated in a second cell type. Treatment of rat renal mesangial cells with the NO donor and nitrosating agent S-nitroso-
Data indicate that decreased/absent TSP1 expression is associated with enhanced NO responses in vascular cells and platelets (95). Furthermore, male Thbs1−/− and Cd47−/− mice enjoy elevated NO signaling, and this is associated with loss of TSP1 induction with injury or hypoxia compared with WT. These and other findings suggest possible feedback regulatory control of TSP1 by anti-inflammatory low dose NO.
VSMC chemotaxis to a supraphysiological concentration of TSP1 (20 μg/ml) was altered by exogenous NO, with low concentrations limiting TSP1 chemotaxis and high concentrations enhancing it (227). Similarly, VSMCs treated with the NO-prodrug sodium nitroprusside were resistant to TSP1-stimulated chemotaxis. Endothelial progenitor cells exposed to laminar flow, a known activator of eNOS, upregulated NO concurrent with downregulation of TSP1 protein (7). In contrast to these results in primary vascular and renal cells, treatment of human hepatoma3 B cancer cells with the NO prodrug O2-(2,4-dinitrophenyl) 1-[(4-ethoxyxarbonyl)piperazin-1-yl]diazen-1-ium 1,2-diolate (1 to 10 μM) increased TSP1 mRNA expression and cell surface expression of the TSP1 receptor CD36 (44). Conversely, treatment of NIH 3T3 cells with high concentrations of NO (1 mM DETA/NO) for 24 h suppressed TSP2 promoter activity and mRNA (149).
Beyond cell culture, limiting NO for 28 days in male rats by daily feeding of L-NAME in the drinking water was associated with increased TSP1 protein expression in distal mesenteric arteries and elevated blood pressure (17). In a small clinical study, 4 weeks of aerobic training in healthy young men was associated with increased eNOS protein expression in vastus lateralis muscle samples. Although TSP1 protein levels were not significantly changed, TSP1 levels trended lower in postexercise samples (77).
The mechanism by which NO limits TSP1 expression at the pre- and post-transcriptional levels remains to be determined. Furthermore, it will be interesting to consider the effects of NO and NO surrogate drugs on changes in TSP1 and TSP1 receptor expression as a possible index of clinical effect. Some data suggest a role for ROS to increase TSP1 expression (infra vide Section VI). If confirmed, the chemical quenching of superoxide by NO may represent yet another mechanism by which the biogas limits TSP1 expression.
III. NO Signaling and Other Matricellular Proteins
A. NO signaling and other thrombospondins
1. Regulation of NO signaling
The C-terminal domain of TSP1 that interacts with CD47 shares 53–82% sequence homology with the other four members of the TSP family, the highest being with TSP2 (23). This suggested that TSP2 and other TSPs may also inhibit NO signaling via interaction with CD47. A comparison of recombinant signature domains of TSP1, TSP2, and TSP4 showed moderate competition between the signature domains of TSP2 and TSP1 for binding to cells expressing CD47 but no significant inhibition by the signature domain of TSP4 (84). Consistent with these data, the three paralogs showed decreasing potency for inhibiting NO-stimulated cGMP accumulation in VSMCs in the order TSP1>TSP2>TSP4.
One study has also implicated CD47 as a receptor for cartilage oligomeric matrix protein (COMP) (211). Attachment of ligament cells to COMP was inhibited by a peptide derived from the globular C-terminal domain of COMP (SFYVVMWK) that resembles the known CD47-binding peptide 4N1 from TSP1 (RFYVVMWK). A CD47 blocking antibody also inhibited adhesion, but the study did not establish that CD47 directly binds to COMP or exclude the possibility that the ligands including TSP1, by engaging CD47 on the ligament cells, indirectly inhibit adhesion by inactivating αvβ3 integrin, which also serves as a COMP receptor on these cells.
To assess whether TSP2 contributes significantly to regulating physiological NO signaling, primary lung endothelial cells were prepared from Thbs2−/− mice (84). No difference was seen in basal cGMP levels, but surprisingly NO-stimulated cGMP was moderately but consistently lower in the Thbs2−/− cells. No difference in NO-stimulated proliferation was observed. These results demonstrate that endogenous TSP2 does not contribute significantly to regulating basal NO signaling in vascular endothelial cells, but it should be noted that expression of TSP2 in these cells was significantly lower than that of TSP1, which may mask potential inhibitory signaling from TSP2. Thbs2−/− mice also exhibited no significant advantage over WT mice when analyzed for recovery from an ischemic injury (84).
Thus, to date no physiological or pathophysiological regulation of NO signaling by TSP2 has been demonstrated, but conditions may be found in the future wherein increased TSP2 expression is sufficient to inhibit NO signaling. However, given the homology in structure and domains, it is predicted that TSP2 will act via CD47 to limit NO in such instances.
2. Regulation of expression by NO
Studies in endothelial cells lacking the monooxygenase cytochrome P450 1B1 first implicated regulation of TSP2 expression by eNOS-derived NO (266). Genetic evidence for this regulation was later provided using Nos3−/− mice, which exhibited elevated TSP2 expression in ischemic and wound tissues (149). Studies using Akt1−/− mice demonstrated that Akt1 signaling, which phosphorylates eNOS, also negatively regulates TSP2 expression at sites of injury (8). However, a recent study reported elevated TSP2 levels produced by Thbs1−/− choroidal endothelial cells despite having elevated eNOS phosphorylation and NO production as assessed by 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate fluorescence (53).
Additional RNS may be involved in the observed induction of TSP2 because iNOS was also strongly induced in the Thbs1−/− cells, and 4-amino-5-methylamino-2′,7′-difluorofluorescein is currently believed to directly detect NO2 • rather than NO (176). Further investigation is needed to identify the RNS that proximally regulates TSP1.
B. Regulation of iNOS-derived NO production and signaling
1. ADAMTS1
A recent study has extended matricellular protein regulation of NO signaling to the TSR family. Marfan syndrome is a genetic disorder caused by inherited or sporadic mutations in fibrillin-1 (223). Fibrillin-1 provides a scaffold for deposition of elastin, and in Marfan syndrome patients the resulting defect in elastic tissue integrity is associated with the formation of thoracic aortic aneurysms (Fig. 6).
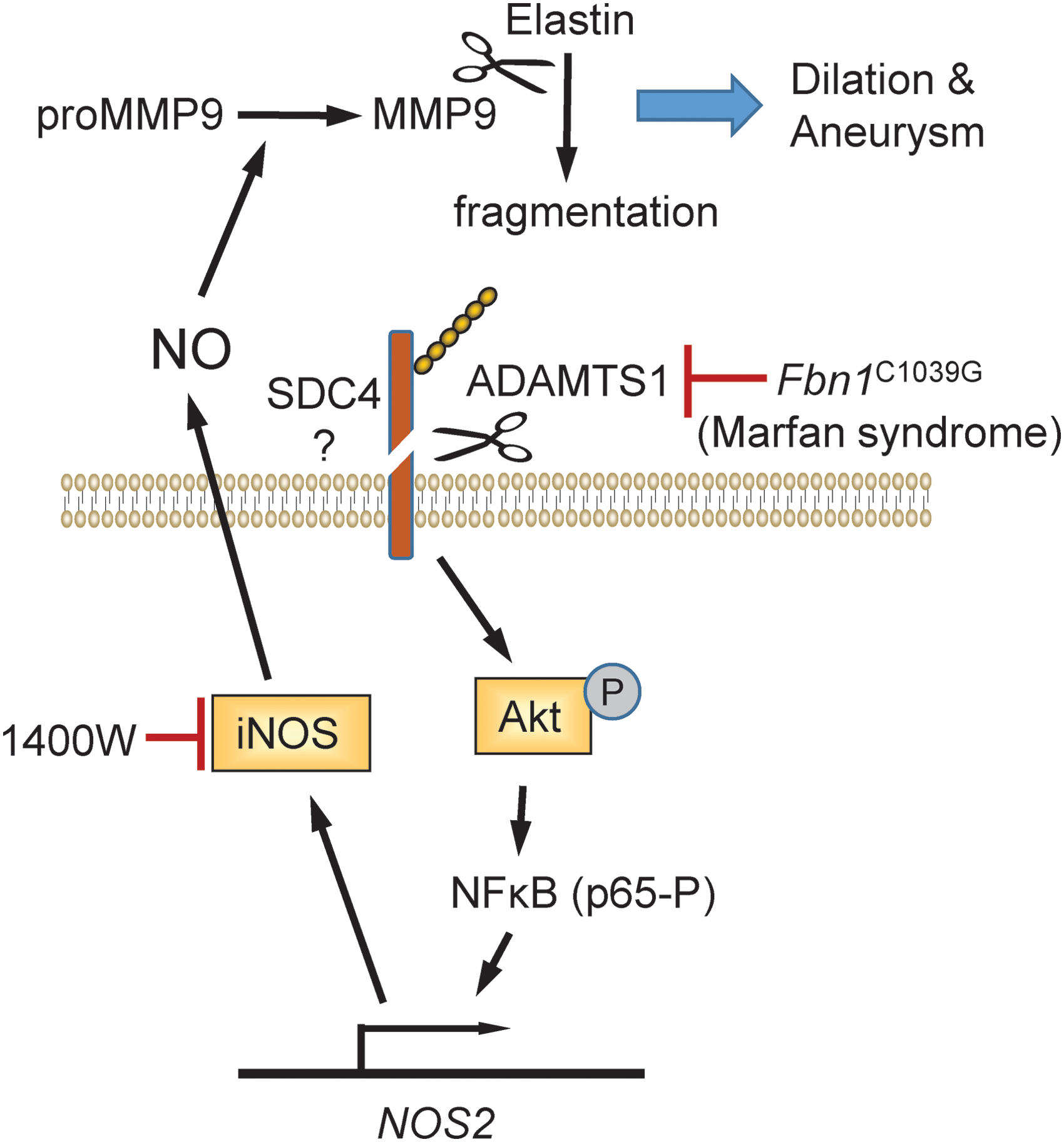
Expression of the matricellular protease ADAMTS1 was found to be lower in the medial layer of aortic sections from patients with Marfan syndrome, whereas iNOS (NOS2) expression was generally elevated at the same location (186). A role of altered ADAMTS1 expression in Marfan syndrome was suggested by similarities in the aortic pathology of Adamts1 +/− and Fbn1 C1039G/+ mice. Aortic sections from Fbn1 C1039G/+ mice had reduced levels of ADAMTS1, comparable with that seen in Marfan syndrome patients, whereas aortic NO levels and NOS2 expression were elevated. Aortic NO levels and NOS2 expression were similarly elevated in the Adamts1 +/− mice. Lentiviral siRNA knockdown of ADAMTS1 in WT mice rapidly induced aortic dilation, hypotension, and medial degeneration, which could be prevented by treating the mice with the pan-NOS inhibitor L-NAME.
In contrast, siRNA knockdown of ADAMTS1 in Nos2 −/− mice exhibited no aortic pathology, and aortic NO levels remained normal. Specific inhibition of NOS2 using the selective inhibitor 1400 W also prevented aortic dilation when used to treat both young and aged Adamts1 +/− or Fbn1 C1039G/+ mice. The elevated levels of NO produced by NOS2 were proposed to increase MMP9-mediated fragmentation of elastin because MMP9 activation and elastin fragmentation in the aorta of Adamts1-deficient mice were sensitive to NOS inhibition.
Others have reported that NO (as S-nitrosoglutathione, GSNO) enhances the expression of MMP9, its inhibitor TIMP1, and elastin in VSMCs (240). In addition, the resulting active MMP9 can lead to fragmentation of other matricellular proteins in the vessel wall, including osteopontin and the TSP1 receptor CD36 (38, 141). These studies suggest that a selective therapeutic NOS2 inhibitor could be protective for patients with Marfan syndrome. However, such a therapeutic strategy would need to be balanced against compromising the role of NOS2 in host defense.
The effects of ADAMTS1 knockdown on NOS2 expression in VSMCs were mediated by increased Akt phosphorylation and nuclear factor kappa B (NF-κB) activation (186). Inhibition of Akt signaling in Adamts1
+/− mice using the mammalian target of rapamycin (mTOR) inhibitor AZD8055 decreased aortic dilation, inhibited NO production in the aortic wall, and reduced NOS2 levels. It is interesting to consider whether mTOR blockade could provide therapeutic benefit to Marfan syndrome patients. However, as mTOR is a proximate promoter of VSMC growth, long-term mTOR blockade could lead to vascular hypoplasia, although this may be of benefit in conditions such as pulmonary hypertension (PH) wherein pulmonary vascular smooth muscle growth is accentuated (see
2. Osteopontin and NOS2 signaling
Earlier characterization of the matricellular protein osteopontin revealed that it inhibits NO synthesis induced by interferon-γ and lipopolysaccharide (LPS) in primary mouse kidney proximal tubule epithelial cells (82). Purified osteopontin inhibited NOS2 mRNA and protein expression in these cells. Similar inhibition of NOS2 induction by inflammatory mediators was reported in RAW264.7 macrophages (218) and rat thoracic aortas (226). Conversely, LPS in the presence of NO induced transcription of the Opn gene, suggesting a negative function of osteopontin to limit inflammatory production of NO (67). A similar negative feedback role was found for limiting NO production in rat pancreatic islets and RINm5F β cells (6).
Several correlative studies support a protective role of osteopontin in animal injury models and patients, but the strongest evidence for an NO-dependent protective role comes from a liver ischemia–reperfusion model. After 45 min of warm hepatic ischemia, Opn−/− mice exhibited elevated release of liver enzymes, increased necrosis, and increased expression of NOS2 and inflammatory cytokines (189).
Two potential mechanisms were identified: (i) loss of osteopontin cell autonomously impairs hepatocyte resistance to stress and (ii) osteopontin limits the production of toxic iNOS-derived NO by macrophages. The relative contributions of these mechanisms to the protective function of osteopontin remain to be determined.
In contrast, osteopontin was proposed to enhance lung injury secondary to intestinal ischemia–reperfusion, and an osteopontin blocking antibody protected mice from this injury (73). Comparing renal ischemia–reperfusion in WT versus Opn−/− mice found no differences in functional impairment in the first 7 days after reperfusion, but collagens I and IV expression was decreased and macrophage infiltration was significantly diminished in the nulls consistent with decreased inflammation and fibrosis (191). Another study found decreased natural killer (NK) cell recruitment in reperfused kidneys of Opn−/− mice, and osteopontin activated NK cells to kill tubular epithelial cells in vitro (310). Therefore, consistent evidence is currently lacking to conclude that osteopontin regulation of NO production has a major role in ischemic injuries or that osteopontin is tissue protective.
Characterization of bone cells from mice lacking osteopontin revealed a deficit in NO production, assessed as nitrite using the Griess reagent, when the cells were subjected to pulsatile flow (40). However, it is unclear which NOS isoform is regulated by osteopontin in these cells.
3. Regulation of other matricellular protein expression by NO
Induction of NOS2 in rat mesangial cells by treatment with IL-1β and TNFα resulted in a cAMP-dependent decrease in SPARC mRNA and protein levels (287). SPARC expression was also decreased in cells treated with NO donors, and downregulation of SPARC in kidneys of rats treated with endotoxin was reversed by the NOS2-specific inhibitor L-N6-l-(iminoethyl)-lysine dihydrochloride.
A subsequent study identified that the SPARC/BM-40 family member secreted modular calcium-binding protein 1 (SMOC1) as another matricellular protein downregulated by NO signaling in rat mesangial cells (47). The same NOS2 inhibitor prevented downregulation of SMOC1 in kidneys of nephritic rats. Expression of TSP2 in ischemic tissues is regulated by NOS3-dependent NO signaling (149).
Treatment of human meniscal cells obtained from nondegenerative hip meniscus with the NOS inhibitor L-NAME inhibited the mRNA expression of two ADAMTS family members that function as aggrecanases (ADAMTS-4 and −5) (231). This was identified as an upregulated catabolic pathway associated with induction of autophagy by the NOS inhibitor. An earlier study using bovine cartilage explants reported no effect of inhibiting NOS using L-NG-monomethyl arginine citrate on ADAMTS-4 and ADAMTS-5 mRNA expression, but found that their enzymatic activity induced by TNFα was significantly inhibited (255). The mechanism by which NO post-transcriptionally regulates ADAMTS-4 and ADAMTS-5 remains to be defined.
IV. Thrombospondin-1 Regulation of H2S Biosynthesis and Signaling
A. Role of H2S in T cell activation
H2S is a toxic gas at high concentrations, but it is also synthesized in mammalian tissues by three enzymes that yield physiological H2S concentrations of 0.1–2 μM (233, 234). Several functions of endogenous H2S signaling have been identified using mice lacking specific genes required for H2S biosynthesis. H2S can act as an intracellular and paracrine signaling molecule through sulfhydrylation of thiols on specific target proteins (172, 173) and by formation of persulfides and polysulfides (66, 83). H2S plays both pro- and anti-inflammatory roles in immune cells. This apparently contradictory statement can be rationalized by the bimodal dose dependence of H2S responses. Higher concentrations of H2S generally induce proinflammatory responses, whereas concentrations <10 μM are anti-inflammatory. H2S inhibits IL-2 production by and proliferation of T cell lymphocytes (283). Similar effects were also reported for polymorphonuclear cells, wherein 1 mM H2S induced PMN apoptosis (153).
Duration of H2S exposure is also important in defining its effects on cells. For example, the slow-releasing H2S donor GYY4137 inhibited an LPS-induced proinflammatory response and increased expression of the anti-inflammatory chemokine IL-10, whereas rapid exposure to H2S produced from NaHS induced the opposite responses on the same type of cells (298). In addition, H2S effects can be species- and organ-specific, depending on differences in the activities of different H2S biosynthetic and catabolic pathways (290).
Higher pharmacological concentrations of H2S inhibit T cell functions by inhibiting mitochondrial function. Exposure of Jurkat T cells to 1–5 mM NaHS at pH 6.0–8.0 induced blebbing and apoptosis by activation of the Rho-Rock pathway and cleavage of caspase-3 and poly-ADP ribose polymerase (PARP) (107). In contrast, H2S at physiological concentrations (<10 μM) is a positive physiological enhancer of T cell function (166) (Fig. 7). In primary mouse CD3+ T cells, OT-II CD4+ T cells, and the human Jurkat T cell line, physiological levels of H2S potentiated T cell receptor-induced activation. H2S at 50–500 nM enhanced T cell activation assessed by CD69 expression, IL-2 expression, and CD25 levels. H2S dose-dependently enhanced T cell receptor-stimulated proliferation. Furthermore, activation increased the capacity of T cells to make H2S by increasing expression of cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS). Disrupting this response by using siRNA to target these enzymes impaired T cell activation and proliferation, which could be rescued by the addition of 300 nM H2S. At physiological concentrations, H2S also induced formation of the immunological synapse by altering cytoskeletal actin dynamics in the microtubule organizing center during T cell activation (166).
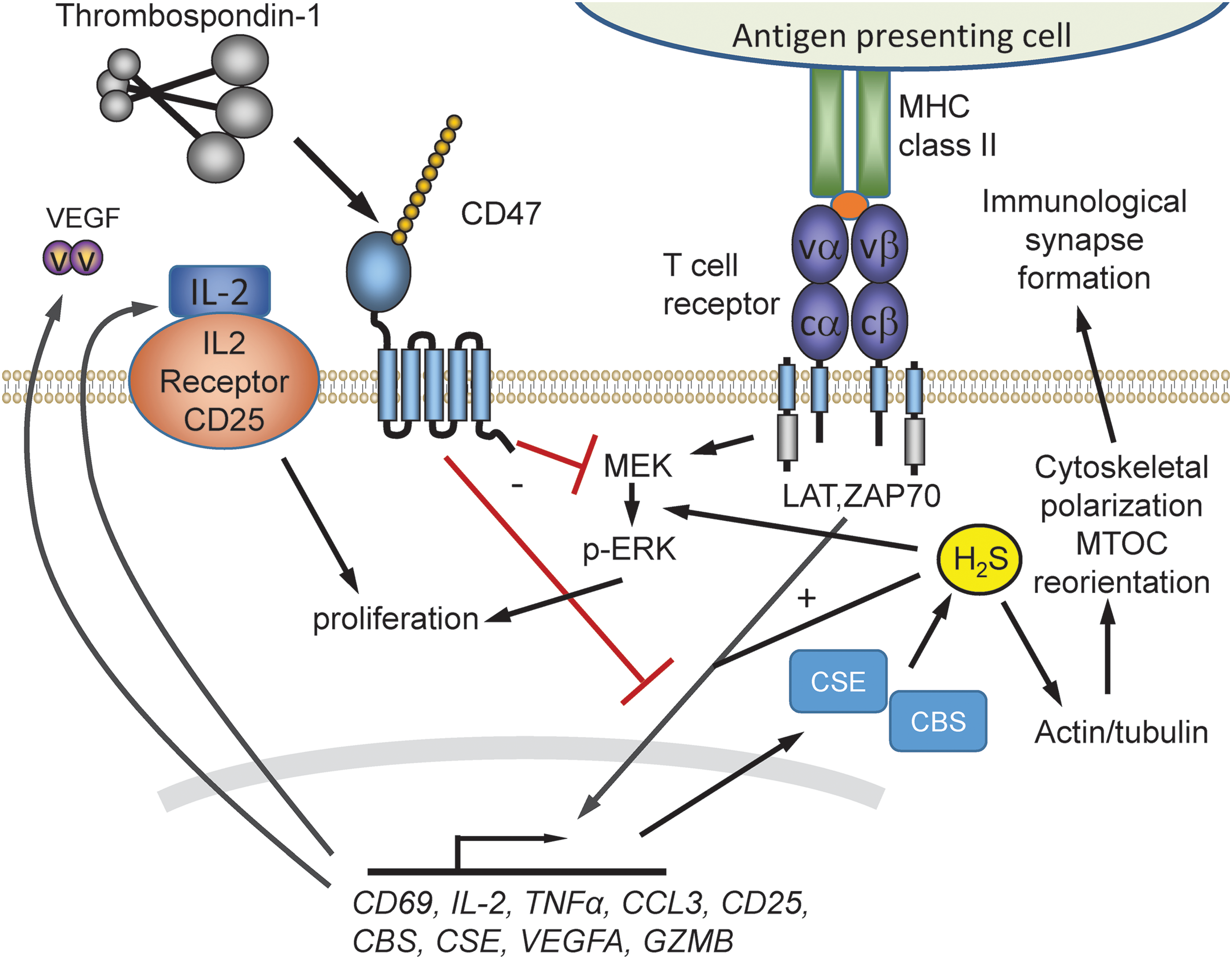
B. TSP1 inhibition of H2S biosynthesis in T cells
Analogous to its inhibition of NO signaling, TSP1 signaling through CD47 inhibits H2S signaling in T cells. Murine Thbs1−/− T cells are more sensitive to activating signals in the presence of H2S, resulting in elevated levels of IL-2 and CD69 mRNA and cell proliferation than WT-activated murine T cells (164). The increased IL-2 and CD69 expression is reversed by physiological concentrations (0.2–2 nM) of exogenous TSP1. Resting and activated Thbs1− /− murine T cells have upregulated CBS and CSE mRNA expression. This suggested that TSP1 endogenously regulates H2S signaling by limiting the expression of Cbs and Cse genes, which, in turn, limits T cell activation.
A CD47-binding peptide derived from TSP1 (7N3, FIRVVMYEGKK) recapitulated the inhibitory activity of TSP1, whereas Cd47− /− murine T cells were resistant to inhibition by TSP1 (164). This indicates that TSP1 signaling through CD47 has a homeostatic inhibitory role that limits the autocrine role of H2S in T cell activation and, therefore, may play a role in inflammatory diseases that are associated with induction of H2S.
C. Potential role of TSP1 in other H2S signaling
H2S signaling also plays important roles in angiogenesis, including activation of VEGFR2 (267). CBS expression is necessary for optimal VEGF signaling (222). H2S is also a physiological regulator of VSMCs (104). Extrapolating from its regulation of H2S biosynthesis and signaling in T cells, TSP1 could be a global antagonist of H2S biosynthesis and signaling. If so, TSP1 would be an even more comprehensive regulator of angiogenesis and blood flow than indicated by its highly redundant inhibition of VEGF/NO signaling pathways (88). The emerging role of H2S signaling in cancer also merits future studies to investigate whether TSP1 and CD47 regulate tumor cells in a H2S-dependent manner (261).
V. H2S Regulation of Matricellular Protein Expression
A. Osteopontin
Osteopontin has emerged as an important target of H2S signaling in several cell types, but the nature of this regulation appears to be cell-type specific. Osteopontin plays a major role in regulation of vascular calcification and mineral depositions in humans (61). During vascular calcification of arteries, smooth muscles cells, pericytes, fibroblasts, and macrophages transform into an osteoblast-like phenotype, which is characterized by upregulation of osteopontin. During calcification induced with vitamin D3 plus nicotine in rats, aortic H2S content and CSE mRNA were decreased significantly but osteopontin mRNA was increased (302). Daily treatment of vitamin D+nicotine exposed rats with NaHS at 2.8 or 14 μmol/kg downregulated osteopontin mRNA to control levels. This suggests that H2S inhibits mRNA expression of osteopontin. Similarly, osteopontin mRNA was upregulated in hyperhomocysteinemia, and H2S/CSE expression was downregulated (294). This led to enhanced proliferation of VSMCs from rat thoracic aortas. Treatment with 100 μM NaHS significantly decreased proliferation of VSMCs via an ERK1/ERK2-dependent pathway and partially inhibited the induction of osteopontin by homocysteine.
In contrast, endogenous H2S produced by CSE induces differentiation and maturation of primary osteoblasts, resulting in induction of osteopontin and BMP2 expression (315). This is mediated by sulfhydration of the transcription factor RUNX2 at Cys123 and Cys132 and results in elevated BMP2, osteopontin, and osteocalcin expression (Fig. 8A). The H2S donor GYY4137 induced the same sulfhydration-dependent induction, whereas mutation of these Cys residues on RUNX2 or treatment with the CSE inhibitor
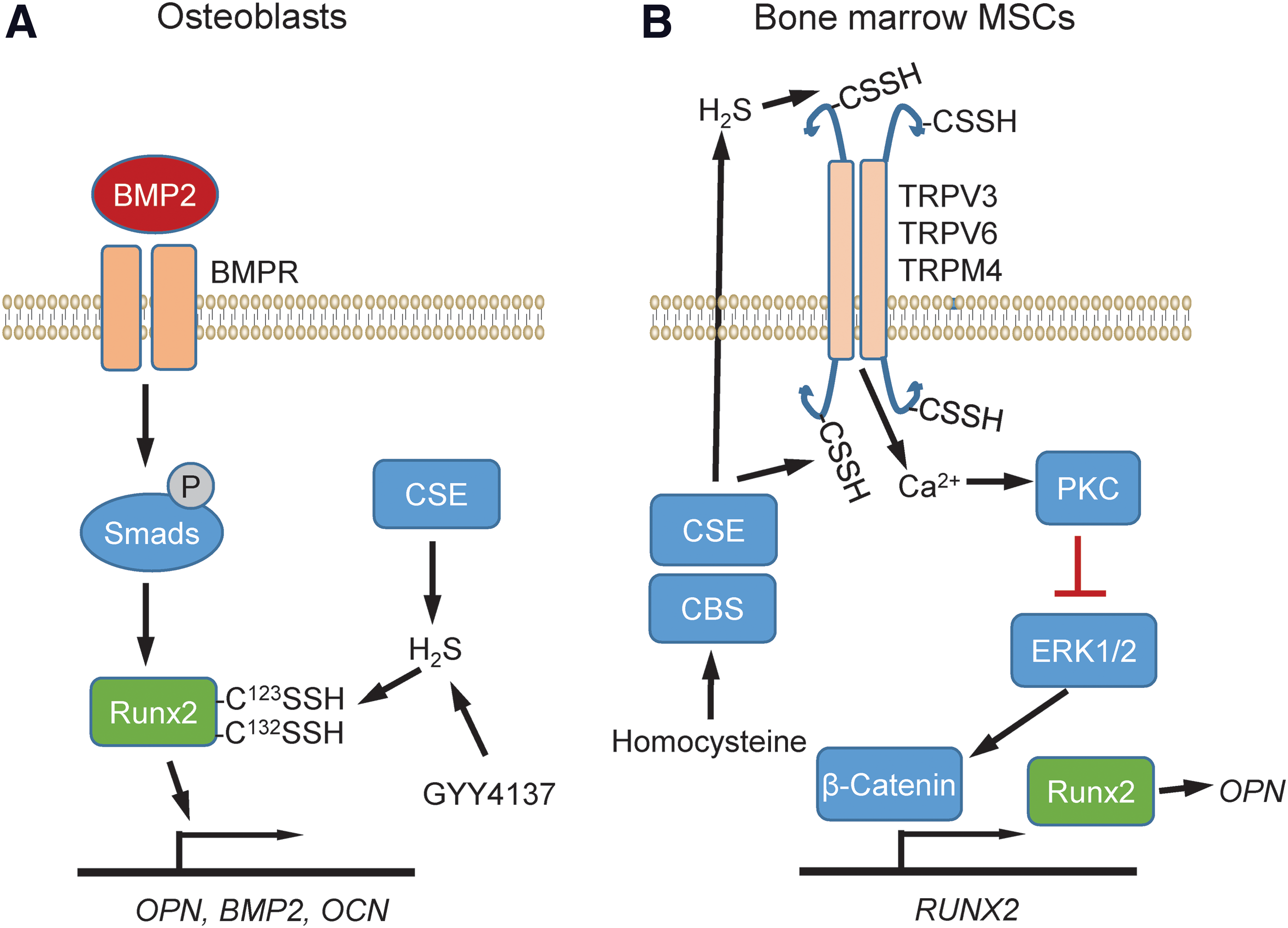
Studies using bone marrow mesenchymal stem cells (MSCs) identified another sulfhydration target that controls the expression of osteopontin (145) (Fig. 8B). H2S deficiency has been linked with a perturbed balance between osteoblast and osteoclast cells that leads to osteopenia and bone marrow MSC impairment in mice. Injection of the H2S donor GYY4317 rescued abnormal osteoclast-mediated bone resorption and bone mineral density in Cbs+/− mice. The H2S deficiency perturbed Ca2+ influx mediated by TRPV6, TRPV3, and TRPM4 channels, which are regulated via sulfhydration. Mass spectrometry identified sulfhydration of TRPV6 at Cys172 and Cys329. Overexpression of the double mutant TRPV6C172+C329mu reduced H2S donor-induced protein kinase C phosphorylation and downstream ERK-dependent activation of β-catenin, which induces RUNX2 and in vitro osteogenic differentiation. These data suggest that H2S is essential for maintaining a homeostatic function of osteopontin for differentiation of MSCs during osteogenesis.
B. Tenascin C
Complementary to regulation of H2S production and signaling by TSP1 in the extracellular matrix, several recent studies implicate H2S as an important regulator of extracellular matrix remodeling that may involve additional matricellular proteins. H2S treatment of spontaneous hypertensive rats and isolated VSMCs inhibited collagen levels and hydroxyproline secretion (312). H2S treatment also protected lung tissues from ventilator-induced lung injury (249). Gene enrichment analysis of microarray data showed significant regulation of extracellular matrix remodeling pathways, including altered expression of the matricellular protein tenascin C.
Chronic lung fibrotic diseases increase deposition of extracellular matrix, including collagens, proteoglycans, and adhesive glycoproteins. These structural alternations in lung tissues resemble those found in hibernating animals during the transition from torpor to arousal state. The golden Syrian hamster is an accepted model for lung remodeling during hibernation (265). Cooling of hamster cells increased endogenous H2S production by CBS. During the torpor phase, H2S suppressed immune responses and elevated collagen and CBS expression, which became normalized during euthermia or arousal state. Treating golden Syrian hamsters with exogenous H2S inhibited gelatinase activity, potentially by quenching of Zn2+ and increased expression of CBS (264). This suggests a central role for lung extracellular matrix remodeling in the protective function of H2S during hibernation.
C. Laminin-γ1
H2S inhibited glucose-induced laminin γ1 synthesis in mouse podocytes and kidney proximal tubular epithelial cells via the mammalian target of rapamycin complex 1 (mTORC1)-AMP kinase pathway (131). CBS and CSE expression was significantly reduced in mice with type 1 or type 2 diabetes. The resulting deficiency in H2S led to increased glucose-dependent inhibition of AMP kinase and activation of mTORC1. This dysregulation, in turn, resulted in increased synthesis of extracellular matrix proteins and renal cell hypertrophy. Similar results were reported for mouse glomerular endothelial cells cultured with low and high glucose, wherein H2S treatment downregulated matrix protein expression induced by high glucose (118). H2S treatment increased phosphorylation of LKB1-AMPK and induction of several autophagy genes. The latter result suggests a mechanistic link between the ability of TSP1/CD47 signaling to inhibit both H2S biosynthesis (164) and autophagy responses (244).
NO and cGMP signaling also regulates the expression of laminin γ1 and other extracellular matrix proteins in an AMP kinase-dependent manner (130). In some contexts, H2S enhances NO signaling (262). Further studies are needed to determine how simultaneous regulation of NO and H2S signaling by TSP1/CD47 influences the cross-talk between these pathways. In addition, the proangiogenic effects of H2S, by increasing VEGF expression and VEGFR2 phosphorylation in endothelial cells and decreasing levels of the antiangiogenic soluble fms-like tyrosine kinase 1 (sFlt1), may be relevant to this model (79, 289).
VI. TSP1 Regulation of ROS
A. TSP1 and ROS signaling in inflammatory cells
1. Historical context
Studies have identified roles for several matricellular proteins in regulating intracellular or extracellular ROS production, although the chemical identity of that ROS often remains unclear (Table 1). TSP1 enhanced the production of extracellular superoxide by neutrophils stimulated by the bacterial chemoattractant peptide fMLP (257). The identity of superoxide was confirmed by superoxide dismutase (SOD) inhibition of the detected reduction of cytochrome C. A similar enhancement was found when TSP1-treated monocytes were exposed to TSP1 antibodies (224). A subsequent study using fMLP-stimulated PMN from a patient with leukocyte adhesion deficiency suggested that the ability of PMN to spread on a substrate coated with TSP1 regulated superoxide production (256).
ROS indicates general detection of oxidants. Although authors often presume detection of specific oxidant species, most fluorescent dyes or other assays are not specific for detecting superoxide. Intracellular DCFDA fluorescence produced by intracellular oxidation of 5/6-chloromethyl-2,7-dichlorodihydrofluorescein diacetate and related derivatives is not specific for superoxide.
Extracellular superoxide confirmed by loss of signal after SOD treatment.
Dihydroethidium and Mitosox are selectively oxidized by superoxide to yield red fluorescent 2-OH-ethidium+. However, in the absence of supporting chemical characterization of the fluorescent product, other oxidants including OH• and HOCl can generate similar fluorescent products and can indicate activation of myeloperoxidases rather than NADPH oxidases (150).
DCFDA, 2,7-dichlorofluorescein diacetate; ROS, reactive oxygen species; SOD, superoxide dismutase; VSMCs, vascular smooth muscle cells; WT, wild type.
Leukocyte adhesion deficiency is caused by defective β2 integrin function, suggesting that TSP1 may regulate PMN superoxide production through integrin signaling. However, another study localized the adhesion activity of TSP1 for PMNs to its 140 kDa C-terminal region and provided evidence that this was not mediated by a β2 integrin or the known RGD integrin binding site in this region of TSP1 (258). Although these earlier studies established that TSP1 has a supportive role to stimulate production of ROS by PMN, a direct role for TSP1 in this process remained unclear.
2. Roles of TSP1 receptors
Several studies have examined a potential role of the TSP1 receptor CD36 in regulating ROS production by monocytes. Antibody cross-linking of CD36 on human mononuclear cells induced a strong oxidative burst, characterized by measurement of SOD-inhibitable cytochrome C reduction (277) and by chemiluminescence (225). Treatment of these cells with TSP1 alone did not induce an oxidative response, but cross-linking TSP1 bound to the surface, or two treatments with TSP1 (225), enhanced oxidative burst in a CD36-independent manner (225).
More recent studies suggest indirect mechanisms by which TSP1 signaling through CD36 can regulate oxidative inflammatory responses in macrophages. One mechanism involves TSP1 induction of toll-like receptor 4 expression on macrophages (137). TLR4-deficient macrophages did not respond to TSP1, and activation was partially inhibited by a peptide or antibody that blocks binding of TSP1 to CD36. In another study, a recombinant TSR domain of TSP1 that contains its CD36 binding site enhanced the inflammasome-dependent maturation of IL-1β in human THP-1 monocyte-derived macrophages (254). ROS production is an effector pathway induced by inflammasome activation (1), suggesting that TSP1 signaling through CD36 could increase ROS production by this mechanism.
In contrast to these proinflammatory effects of TSP1 signaling through CD36, the resolution of LPS-induced lung injury was found to be delayed in Thbs1−/− mice, which was associated with defective macrophage production of IL-10 (314). Administration of recombinant IL-10 to the Thbs1−/− mice corrected this defect in vivo. IL-10 production by Thbs1−/− macrophages in vitro could be restored by a recombinant TSP1 TSR construct, and siRNA knockdown of CD36 in macrophages inhibited IL-10 production, confirming an inhibitory role for TSP1-CD36 signaling in macrophage inflammatory responses. TSP1 signaling via CD47 may also contribute to IL-10 regulation in macrophages because circulating IL-10 levels were elevated in Cd47−/− mice infected with Candida albicans (180), and TSP1 inhibited IL-10 expression in irradiated ANA-1 macrophages (204). TSP1 at 2.2 nM also inhibited IL-10 mRNA expression in Jurkat T cells but not in a CD47-deficient mutant, demonstrating that TSP1 signaling through CD47 can also regulate expression of this anti-inflammatory cytokine (204).
Further insights into TSP1 regulation of macrophage ROS signaling were obtained by investigating how TSP1 regulates macrophage killing of tumor cells (154). Treatment of interferon-γ-differentiated U937 cells, monocytes, and murine macrophages with TSP1 (∼10 nM) for 40–130 min (depending on cell type) augmented phorbol ester-mediated superoxide production as characterized by luminol-enhanced chemiluminescence, whereas treatment with exogenous SOD quenched the ROS signal (154).
A recombinant N-terminal domain of TSP1, but not the TSR or C-terminal domains, partially recapitulated the effect of TSP1 to increase phorbol ester-stimulated superoxide production. The N-terminal domain contains binding sites for several β1 integrins. A function-blocking antibody to integrin α6β1 and an α6β1-binding peptide derived from this domain of TSP1 partially suppressed the TSP1-induced production of superoxide by differentiated U937 cells. The ability of TSP1 to enhance ROS depended on intracellular Ca2+ and was inhibited in the presence of extracellular ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid. The cytotoxic activity of activated macrophages for tumor cells in these experiments depended on extracellular release of superoxide. TSP1 consequently enhanced the killing of target tumor cells by human and murine macrophages.
3. Role of CD47 counter-receptor SIRPα
Studies of signal regulatory protein α (SIRPα) signaling in macrophages indicate that this counter-receptor for CD47 can regulate superoxide production in a cell autonomous manner, wherein CD47 serves as the ligand that activates SIRPα via cell–cell interactions (5, 136). One mechanism involves reverse CD47-SIRPα signaling (5). Binding of the IgV domain of CD47 to SIRPα on macrophages recruits Janus kinase (JAK) and induces its Tyr phosphorylation (Fig. 9). This results in JAK/signal transducer and activator of transcription (STAT)-mediated induction of NOS2 expression, and the resulting released NO can react with superoxide to form the nitrosating species N2O3. In parallel, SIRPα ligation induces phosphatidylinositol 3-kinase-dependent Rac1 recruitment to Nox1, increasing superoxide production. A subsequent study reported that the phosphatase SHP2 stimulates ROS production, measured by enhanced luminol chemiluminescence ± diphenyleneiodonium chloride, by dephosphorylating SIRPα and activating ERK (136), which can activate Nox1 by phosphorylation of p47phox (152) (Fig. 9).
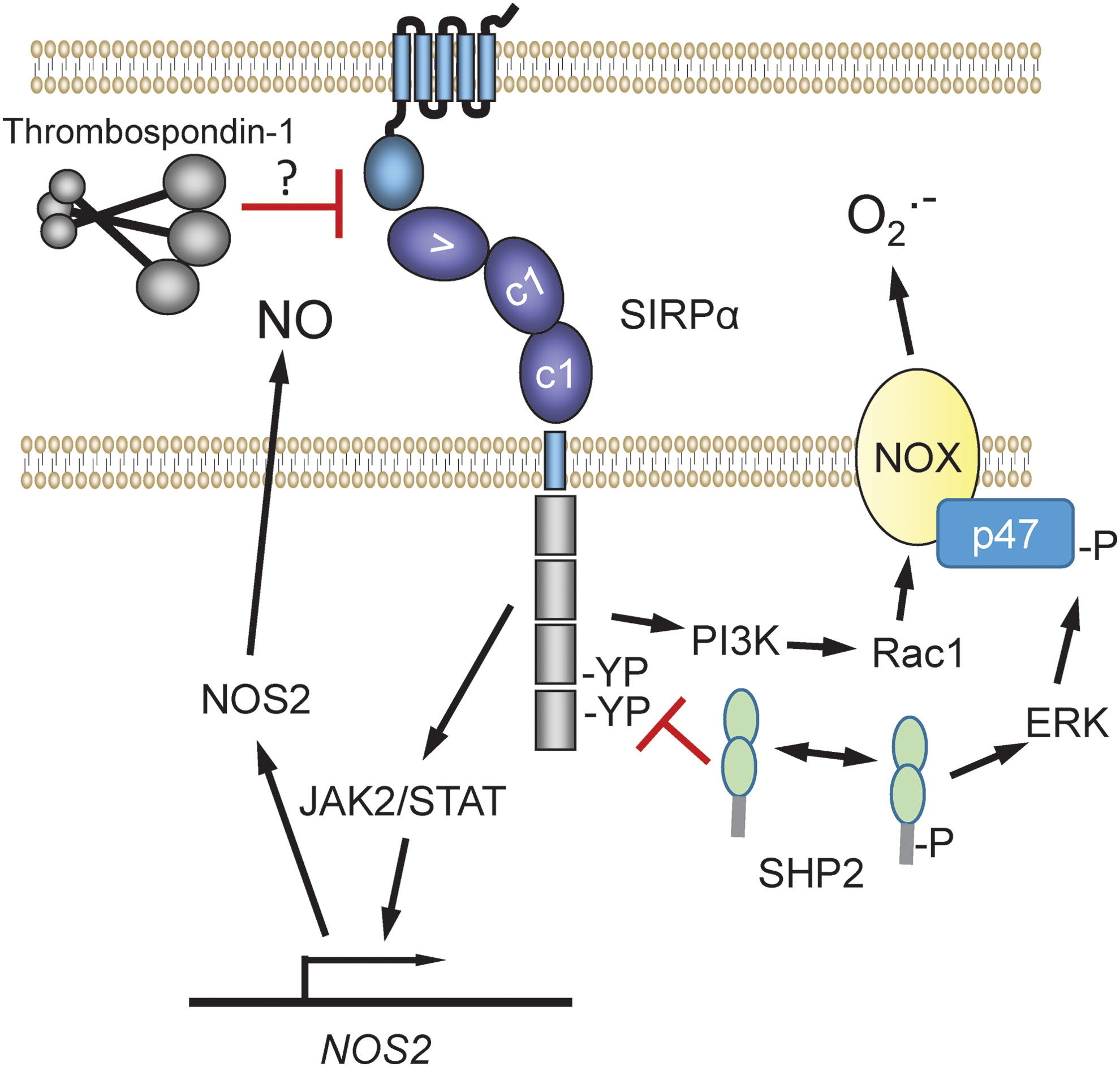
Further studies are needed to determine whether this mechanism is also relevant in nonmyeloid cell types. In addition, given that TSP1 can inhibit binding of recombinant SIRPα to cell surface CD47 (84), the possibility should be considered that TSP1 can inhibit this signaling in cells that express SIRPα by preventing CD47 counter-receptor binding (Fig. 9).
B. TSP1 and ROS signaling in vascular and renal cells
Distinct from the innate immune function of extracellular ROS production by inflammatory cells, intracellular ROS production plays important roles in signal transduction through reversible oxidation of reactive thiols on signaling proteins, including tyrosine phosphatases (282). TSP1 expression and intracellular superoxide levels were increased in primary human pulmonary arterial endothelial cells exposed to short-term hypoxia (1% FiO2, 12 h) (11). Conversely, hypoxia-mediated increases in superoxide were abrogated by treating cells with a CD47 antibody (clone B6H12, 1 μg/ml) or with the NOS inhibitor L-NAME, suggesting that eNOS was the source of ROS under these conditions. Interestingly, siRNA suppression of caveolin-1 (Cav-1) restored hypoxia-mediated ROS levels in endothelial cells treated with a CD47 antibody, suggesting that TSP1-CD47 dysregulation of Cav-1 participated in ROS production by these cells (Fig. 10).
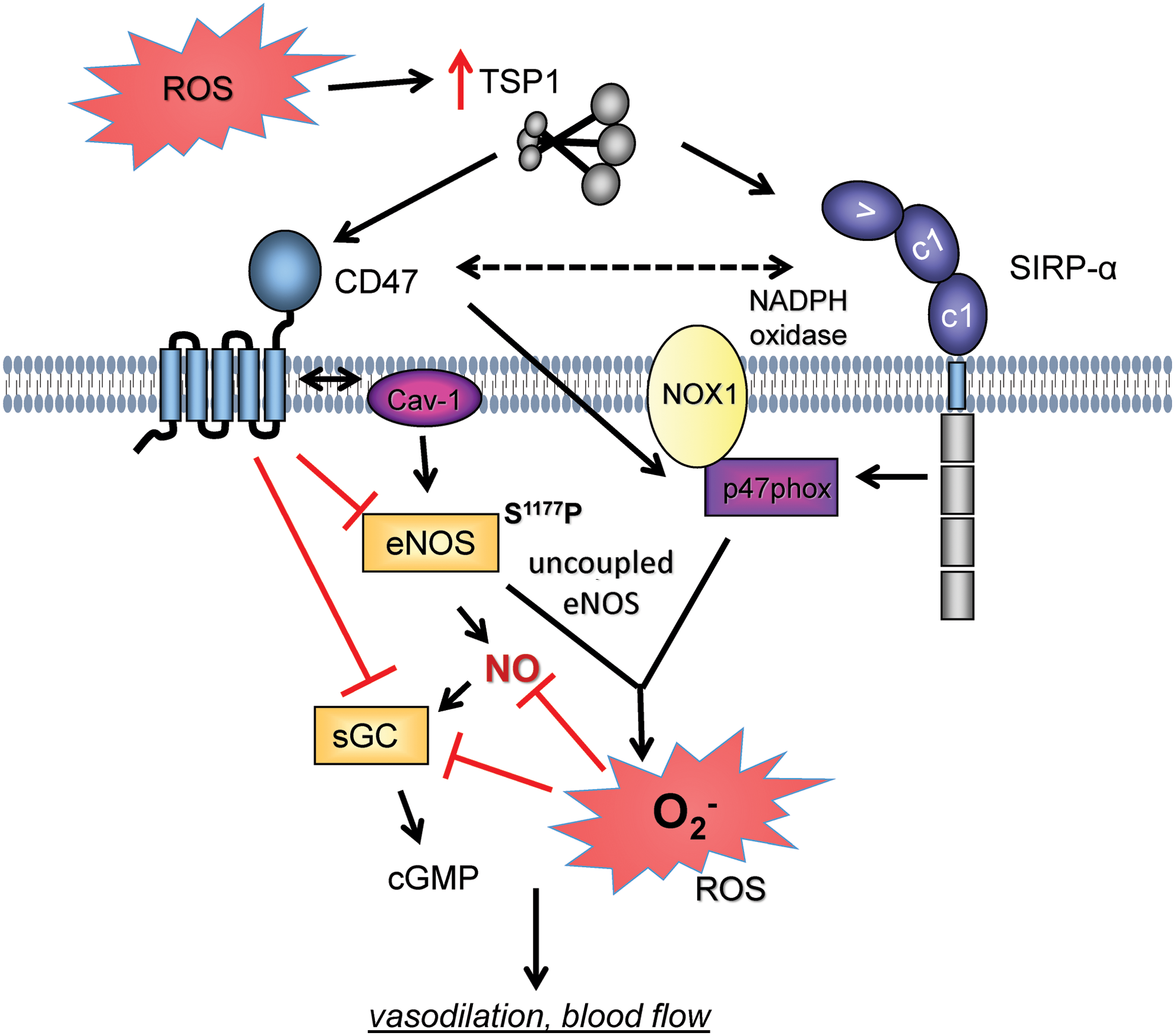
Direct evidence that TSP1 treatment stimulates superoxide production was provided in human aortic VSMCs. Treating cells with 2.2 nM TSP1, which selectively engages CD47 but does not result in significant occupancy of other known TSP1 receptors, for 60 min resulted in a significant increase in superoxide (36) (Fig. 10). Interestingly, this effect was lost at TSP1 concentrations >11 nM, which are sufficient to activate TSP1 receptors other than CD47. Treatment with a CD47-suppressing morpholino oligonucleotide or a CD47 antibody (clone B6H12) blocked the TSP1-mediated increase in superoxide.
Treating cells with TSP1 resulted in increased phosphorylation of the Nox1 organizer subunit p47 phox , whereas pharmacological inhibition of protein kinase C abrogated TSP1-stimulated increases in superoxide (36). A similar effect was found in arterial tissue. Treating endothelial-denuded murine aortic rings with 2.2 nM TSP1 for 60 min resulted in increased superoxide levels that were inhibitable by pretreating with a CD47 antibody (clone 301), as confirmed by electron paramagnetic resonance. Similarly, TSP1-mediated increases in superoxide were abated in VSMCs treated with NADPH oxidase 1 (Nox1) siRNA or in aortic rings from Nox1−/− mice but not in cells treated with control siRNA or in aortic rings from WT mice. In renal tubular epithelial cells, TSP1 also increased Nox1-derived superoxide levels and this involved interaction with SIRPα (307). Superoxide levels represent a balance between enzymatic production and catabolism by SOD. Interestingly, treating VSMCs with TSP1 (2.2 nM) for 1 and 2 h did not change SOD expression or activity (36). It remains to be determined whether TSP1-mediated increases in superoxide via SIRPα are independent of, or through, CD47 cross-talk.
TSP1 signaling via CD47 may also increase superoxide production by mitochondria. Aortic VSMCs from young WT mice displayed increased ROS levels compared with cells from arteries from Cd47−/− mice, as characterized by Mitosox fluorescence (56). Additional chromatographic analysis of the fluorescent product would be required to confirm that CD47 regulates mitochondrial superoxide production rather than other mitochondrial ROS (42). Despite a higher mitochondrial density in skeletal muscle, young cd47−/− mice consumed less oxygen and produced less heat than WT mice, suggesting that mitochondrial coupling is more efficient in the absence of CD47, which is consistent with the decreased ROS production. It is not known whether TSP1 signaling through CD47 similarly controls mitochondrial-derived ROS production in other cell types.
Ex vivo myography experiments in isolated systemic arterial rings indicate a role for TSP1 to inhibit vasodilation in conditions of oxidative stress. TSP1 (2.2 nM) decreased NO-mediated vasodilation of aortic rings from WT and Thbs1−/− mice but not Cd47−/− mice (11). Treatment of WT murine vessels with the membrane-permeable superoxide scavenger 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (Tempol) partially reversed the inhibitory actions of TSP1 (307). Tempol can inhibit the Fenton reaction, a source of hydroxyl radical that limits arterial vasodilation (263). This may be clinically relevant in conditions of dysregulated iron metabolism such as sickle cell disease wherein Fenton chemistry and circulating TSP1 are enhanced (184, 203).
In ex vivo myography studies of the microcirculation, TSP1 limited coronary arteriole vasodilation, and ROS scavenging again partially reversed the inhibitory effect of TSP1 (182). Unexpectedly, the inhibitory activity of TSP1 noted in arterioles from aged rats, while trending toward, did not reach significance in arterioles from young rats. In these experiments, effects were reported only in vessels from female animals (182). TSP1 also inhibited NO-mediated vasodilation of pulmonary arterial rings from mice as well as rats (214), indicating that TSP1 limits vasodilation of the systemic and pulmonary vasculature. It remains to be seen whether the pulmonary arterial effects of TSP1 are secondary to increased ROS.
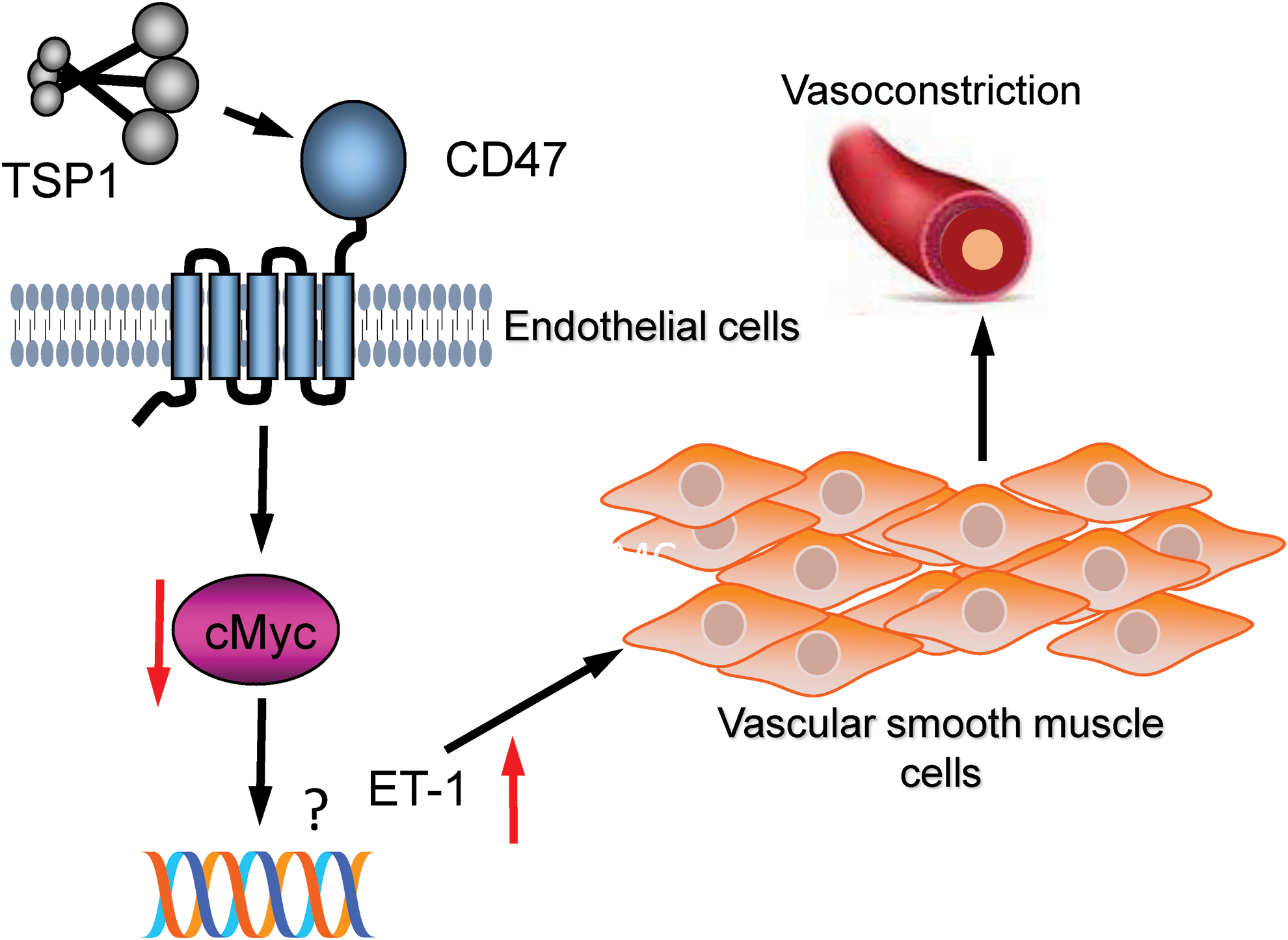
TSP1 may also function as an enhancer of and/or a direct vasoconstrictor as murine aortic and pulmonary arterial segments from WT mice treated with TSP1 and phenylephrine (PE) (10) or endothelin-1 (ET-1) (214) demonstrated increased contraction (Fig. 11). In the case of ET-1, TSP1 treatment was associated with increased vessel constriction at baseline (214). PE-stimulated vasoconstriction of aortic rings exposed in vivo to angiotensin II (Ang II) was dampened by a panel of ROS mitigators, including catalase, apocynin (a nonspecific Nox inhibitor), ML-171 (a specific Nox1 inhibitor), and Mito-TEMPO (a mitochondrial superoxide scavenger) (72). In mesenteric arteries exposed to ischemia–reperfusion in vivo, ET-1-mediated vasoconstriction, as assessed with ex vivo myography, was inhibited by superoxide scavenging with SOD (156). Whether TSP1 alteration in basal tone of ET-1-treated arteries is via increased Nox signaling or some other mechanism requires further research. Interestingly, PE-mediated vasoconstriction was increased in aortic rings from WT mice compared with that from Thsb1−/− and Cd47−/− mice, and this deficit was corrected after treating null vessels with L-NAME (10).
C. TSP1 and ROS signaling in vivo
In vivo, studies in Thbs1−/− and Cd47−/− mice indicate that the TSP1–CD47 axis limits blood flow and tissue survival under conditions of fixed ischemia (85, 100a) as well as ischemia–reperfusion (36, 271, 307), situations characterized by significant oxidative stress. Conversely, suppressing CD47 expression using an adenoviral siRNA vector protected myocardial tissue from ischemia–reperfusion injury in rats while elevating eNOS phosphorylation and NO production, decreasing gp91phox and superoxide, decreasing tissue malondialdehyde, and increasing SOD activity (288). Tissue malondialdehyde levels, produced by lipid oxidation, were correspondingly lower in a rat soft tissue flap ischemia–reperfusion model after postreperfusion treatment with a CD47 antibody (clone OX101) (159). Tissue survival was also improved in this ischemia–reperfusion model. Furthermore, exogenous TSP1 (60 μg/kg body weight) administered via bolus intravenous injection decreased hind limb reperfusion in rats in a Nox1-dependent manner, whereas pretreatment with a CD47 antibody (clone OX101, 0.4 μg/g body weight i.p.) restored the reperfusion deficit induced by exogenous TSP1 (36). Ischemia–reperfusion injury stimulates substantial oxidative stress and contributes to organ failure in clinical transplantation. Mice (217) and rats (140) treated with CD47 antibodies sustained less injury after syngeneic renal transplantation, and in rats this was associated with increased blood flow 24 h post-transplantation. In healthy and steatotic rat liver transplantation ischemia–reperfusion models, pre-treatment with a CD47 antibody (mAb400) was associated with improved animal survival, less organ injury, and lower superoxide levels, as characterized by dihydroethidium fluorescence of liver tissue sections (303, 304).
TSP1 is a stress response gene and often upregulated under conditions of increased inflammation. For example, multiple studies have reported TSP1 induction under hypoxia (11, 123, 185, 187) and in response to ischemia (52, 219, 241, 260) or ischemia–reperfusion injuries (89, 125, 138, 215, 217, 228, 271), situations well known to upregulate ROS. This raises the intriguing hypothesis that ROS signaling may increase TSP1 expression. Arterial smooth muscle cells from mice overexpressing a nonfunctional form of Nox4 showed decreased TSP1 mRNA and protein expression (275), suggesting that Nox4 is a proximate driver of TSP1 expression. Fibroblasts treated with doxorubicin displayed increased ROS levels and TSP1 protein expression (269).
Conversely, in astrocytes, CoCl2-mediated increases in ROS were associated with decreased TSP1 mRNA and protein levels, and these changes were blocked by pretreating cells with with N-acetylcysteine but not resveratrol (29). Curiously, treatment of astrocytes with resveratrol alone significantly increased TSP1 mRNA levels (29), whereas others have found that β-amyloid inhibits TSP1 release from astrocytes (200). These findings may be relevant to the accumulating evidence linking the neurodegenerative condition, Alzheimer's disease and TSP1 (19, 200, 232). Further studies employing exogenous ROS and gene silencing of ROS enzymatic sources would be useful to evaluate possible ROS-stimulated feed-forward induction of TSP1.
VII. ROS Regulation by Other Matricellular Proteins
A. CCN1
CCN1 (CYR61), a member of the CCN family of matricellular proteins, plays an essential role in TNFα-induced apoptosis. This activity is mediated by binding to its cell surface integrin receptors αvβ5 and α6β1 and the heparan sulfate proteoglycan syndecan-4 to induce the accumulation of a high level of ROS through 5-lipoxygenase and mitochondria, accompanied by cytochrome c release (28) (Fig. 12). Within 10 min, CCN1 activates neutral sphingomyelinase-1, which is a major source of CCN1-induced superoxide and hydrogen peroxide (detection methods given in Table 1), driven by a synergism between CCN1 and Fas ligand (101). CCN2 (CTGF) also synergizes with Fas ligand to induce apoptosis by elevating cellular ROS levels (101). Binding to LRP1 is required as a coreceptor for CCN1-induced ROS generation (102). CCN1 inhibited EGF receptor-dependent hepatocyte proliferation through α6β1 integrin-mediated accumulation of ROS, and this was attenuated by treatment with apocynin or the Rac guanine nucleotide exchange factor inhibitor NSC23766 (27). Loss of this signal in Ccn1dm/dm mice, in which CCN1 lacks α6β1 integrin binding, decreased hepatocyte proliferation through an ROS-mediated increase in p53 and increased hepatic carcinogenesis.
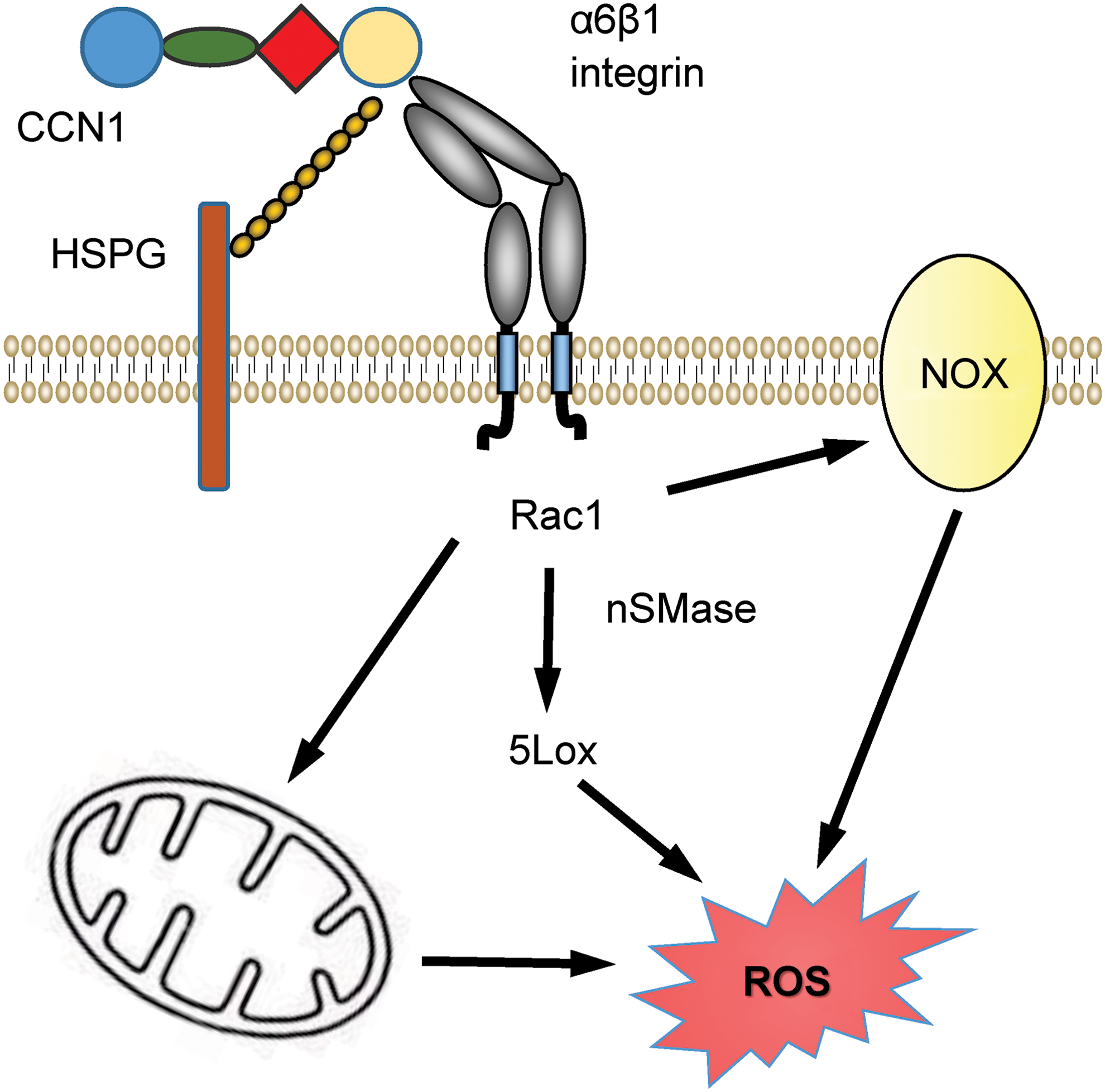
Three of the CCN1 receptors identified in these studies (LRP1, heparan sulfate proteoglycan, and α6β1 integrin) also function as TSP1 receptors, and as discussed previously, α6β1 integrin is required for TSP1 to augment phorbol ester-stimulated superoxide production by macrophages (154). Therefore, different matricellular proteins may utilize convergent receptor signaling pathways to regulate ROS production.
A recent study demonstrated that CCN1 expression is induced by oxidative stress in human dermal fibroblasts (198). Treatment with N-acetyl-
B. Periostin
Periostin is a matricellular protein that is induced at sites of inflammation and in the tumor microenvironment (142). Several studies have implicated ROS as a target of periostin signaling and in regulating periostin expression, but the nature of this regulation may be tissue specific. Cyp1b1−/− mice exhibited decreased expression of periostin, increased lipid peroxidation, and abnormalities in their trabecular meshwork tissue (313). Trabecular meshwork cells from Cyp1b1−/− mice had increased ROS levels detected by dihydroethidium fluorescence and decreased periostin expression and increased levels of the lipid peroxidation by-product 4-hydroxyl-2-noneal on tissue section staining. Inhibition of ROS using N-acetylcysteine reversed this phenotype, but a direct role of ROS in regulating periostin was not established. In contrast, periostin was induced by oxidative stress in fibrotic hypertensive hearts, and pretreating fibroblasts with N-acetylcysteine inhibited angiotensin II-induced oxidative stress and periostin expression (301).
Conversely, periostin inhibits glucose-induced ROS production in endothelial cells (316). Growth of human umbilical vein endothelial cells in medium containing 33 mM glucose for 2 days induced periostin expression and apoptosis. However, overexpression of periostin in the endothelial cells protected them from apoptosis induced by subsequent exposure to high glucose. This protective activity of periostin was associated with upregulation of heme oxygenase-1, which produces the immunosuppressive gasotransmitter CO, and siRNA knockdown of heme oxygenase-1 restored the induction of ROS detected as 2,7-dichlorofluorescein diacetate (DCFDA) and apoptosis in high-glucose medium. Periostin was also demonstrated to protect mitochondrial function in cells exposed to high glucose as assessed by preservation of mitochondrial Δψm, increased Bcl2 expression, and decreased Bax expression.
C. Tenascin-C
The ability of antioxidants such as N-acetyl-
D. Caveats
Most studies to date of matricellular proteins and ROS relied on fluorogenic cell-permeable dyes to assess intracellular ROS production (Table 1). Although this is an acceptable starting point, methods exist that enable precise characterization of ROS. Detailed chemical and biophysical characterization of ROS has been reported for some TSP1 studies (36, 307), but this remains to be addressed for other matricellular proteins.
VIII. CD47 Regulation of Redox Homeostasis in Irradiated Cells
A. NO and radiosensitivity
Although NO under some conditions has a radioprotective activity (299), NO can also be a radiosensitizer (296), and current data do not support an NO-dependent mechanism to account for the increased radioresistance of CD47-deficient cells or WT cells in which CD47 expression was suppressed (160). Treating human umbilical vein endothelial cells with the NO donor DETA/NO or elevating its downstream effector cGMP by treating cells with 8-Br-cGMP did not confer radioprotection to the same cells that were protected by blocking CD47. Conversely, inhibiting NO production in the cells using L-NAME did not prevent the radioprotection caused by CD47 blockade. Therefore, enhancing NO/cGMP signaling is neither necessary nor sufficient to confer the radioprotection obtained by blocking CD47.
Other studies established that blocking CD47 signaling augments autophagy in irradiated cells and mice (244, 245). Inhibiting this protective autophagy response eliminates the survival advantage of irradiated CD47-deficient cells. A role for redox signaling in this radioprotective mechanism has not been identified, but redox signaling plays a well-established role in regulation of autophagy (306).
B. CD47-dependent regulation of redox metabolites in irradiated cells
Additional metabolic pathways through which CD47 blockade confers radioprotection were revealed through a global analysis of the metabolic changes that are induced after exposure to ionizing radiation in WT cells versus a CD47-deficient somatic mutant of Jurkat T cells. This analysis identified several metabolic pathways that are basally regulated by CD47 as well as a global stabilization of metabolism in irradiated CD47-deficient cells (165) (Fig. 13). Of the 342 named metabolites quantified, 27 were significantly altered at 2 h and 112 at 8 h after irradiation in WT cells. Of these, the majority progressively fell below baseline levels (81% at 2 h and 98% at 8 h, respectively). In contrast, the metabolome of irradiated CD47-deficient cells was remarkably stable, with only 6 metabolite levels significantly altered at 2 h and 29 (8%) at 8 h. Furthermore, 86% of the metabolites that were altered at 8 h in CD47-deficient cells showed increased rather than decreased abundance. Thus, CD47-deficient cells are globally resistant to the metabolic changes caused by ionizing radiation. Moreover, the preponderance of elevated metabolites at 8 h suggested that CD47-deficient cells actively compensate for radiation stress by increasing levels of key metabolites.
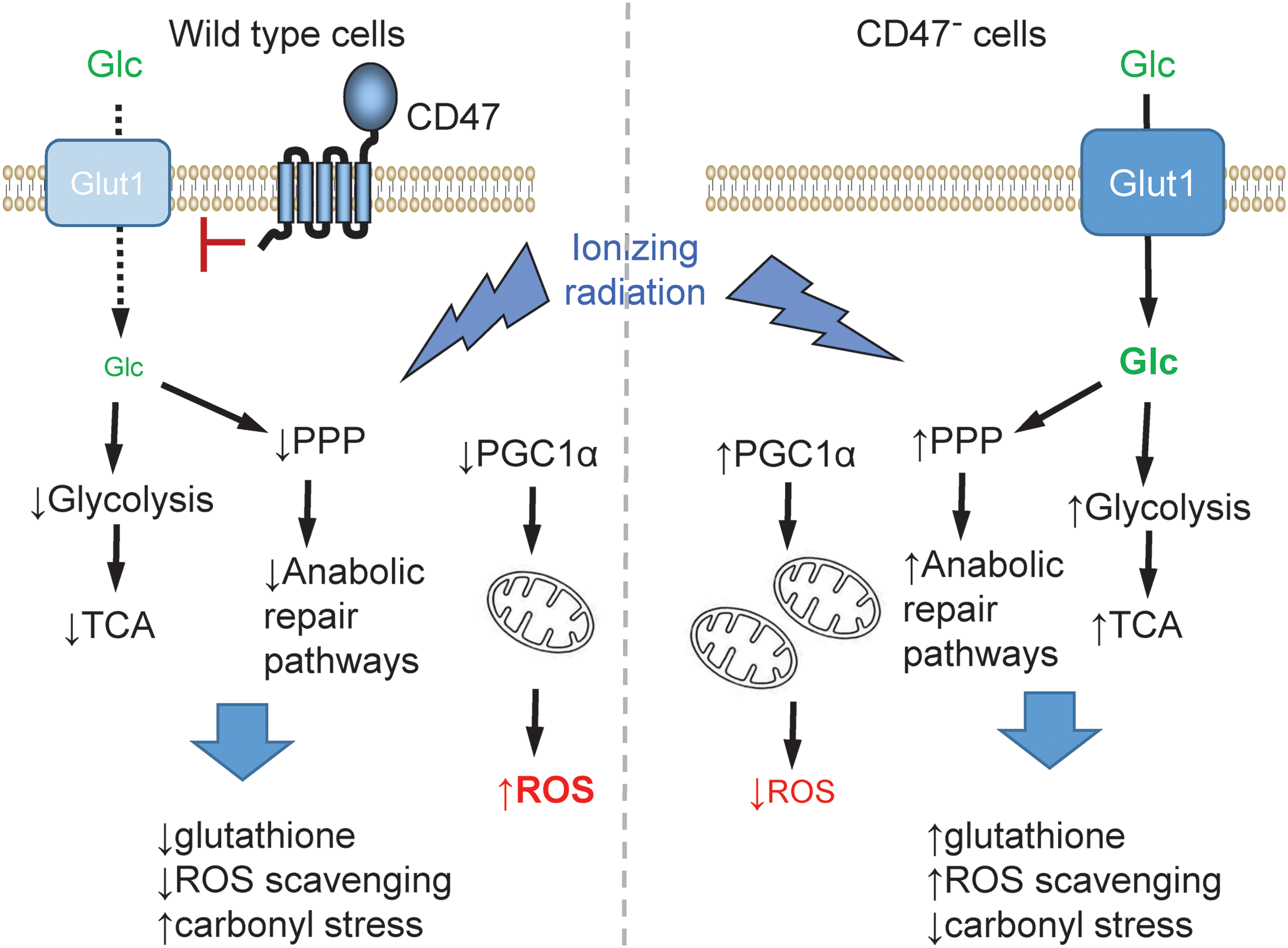
Metabolic pathways required for controlling oxidative stress and mediating DNA repair were generally enhanced in irradiated CD47-deficient cells, and the global declines in glycolytic and tricarboxylic acid cycle metabolites characteristic of normal cell and tissue responses to ionizing radiation were prevented in the absence of CD47 (165). Basal glucose uptake was higher in CD47-deficient cells, and uptake remained higher after irradiation. This was reflected by higher expression of the glucose transporter Glut1 in irradiated CD47-deficient cells. Thus, CD47 mediates signaling from the extracellular matrix that coordinately regulates basal metabolism and cytoprotective responses to radiation injury. Specific preservation of metabolites involved in de novo nucleotide biosynthesis, methyl group metabolism, glutathione homeostasis, and the glyoxalase system indicates an enhanced detoxification and repair capacity in irradiated CD47-deficient cells.
Exposure to ionizing radiation depletes cellular glutathione as it is consumed in the repair of damage caused by the resulting oxidative stress (133, 190). Levels of oxidized and reduced glutathione fell progressively as expected in irradiated WT cells but were preserved in irradiated CD47-deficient cells (165). The half-cell potential of the glutathione redox couple (EGSH) slightly decreased in irradiated WT cells but increased at 2 h before returning to basal levels in irradiated CD47-deficient cells. Consistent with the improved preservation of glutathione levels, total free thiol concentrations in cell lysates were lower basally in CD47-deficient cells and remained stable for 8 h after irradiation, whereas glutathione levels fell sharply at 2 h in irradiated WT cells. The acute loss of glutathione in the irradiated WT cells was consistent with the observed suppression of glycolysis in the same cells (139).
Several metabolic precursors of glutathione including cystathionine, glutamate, γ-glutamylcysteine, and 5-oxoproline fell progressively after irradiation in WT cells, but were preserved or elevated in irradiated CD47-deficient cells, and γ-glutamylcysteine levels were elevated above baseline at 8 h. γ-Glutamylcysteine is the immediate precursor of glutathione, and its elevated level indicates a higher capacity for biosynthetic replenishment of glutathione after irradiation of the CD47-deficient cells.
Accumulation of 2-oxoaldehydes such as methylglyoxal causes damage to cells by reacting with DNA and proteins and generating advanced glycation end products, and the glyoxalase is critical to limit this damage under conditions of oxidative stress such as caused by ionizing radiation (25, 273). Furthermore, methylglyoxal can activate Nox in certain cells to increase superoxide, leading to induction of specific matrix molecules including fibronectin (74, 120). Methylglyoxal accumulates after radiation-induced redox stress, and glutathione is required for its metabolism to lactate via S-lactoylglutathione and glyoxalase I. Ionizing radiation induces glyoxalases I and II (230). Levels of S-lactoylglutathione fell dramatically in irradiated WT cells but were preserved in irradiated CD47-deficient cells (165), which may be enabled by the preservation of glutathione homeostasis in irradiated CD47-deficient cells. Therefore, an enhanced glyoxalase pathway may enable irradiated cells more effectively limit carbonyl stress in the absence of CD47.
C. CD47 regulation of mitochondrial redox
Some of the metabolic advantages involving mitochondrial metabolism may be mediated by CD47 regulation of the transcription factor peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1α). CD47 signaling regulates expression of PGC1α in skeletal muscle (56), and its expression is induced by irradiation in CD47-deficient but not in WT Jurkat cells (165). PGC1α positively regulates several of the genes that were selectively induced in irradiated CD47-deficient cells, including glutathione peroxidase-1 (Gpx1). Cytosolic and mitochondrial Gpx1 is cytoprotective against oxidative stress by limiting hydrogen peroxide accumulation (148). Therefore, Gpx1 induction may contribute to the increased radioresistance of CD47 −/− cells. This and other transcriptional targets of PGC1α, by inhibiting the generation of mitochondrial-driven ROS (251), may account for the maintenance of a favorable redox environment in irradiated CD47-deficient cells.
As discussed hereunder, CD47 also regulates cMyc, and cMyc controls several metabolic pathways that regulate radioresistance, including glucose uptake and glutathione homeostasis (64, 168). Further studies are needed to determine whether TSP1-mediated increases in ROS separate from irradiation stress occur through suppression of counter-regulator systems such as Gpx1.
IX. TSP1/CD47 Regulation of Stem Cells and Tissue Self-Renewal
A. Redox signaling in stem cells
To date, most investigations of the functions of TSP1 in redox signaling have focused on differentiated vascular and immune cells. However, several of the redox pathways regulated by TSP1 also play important roles in stem cell self-renewal and differentiation (Fig. 14A). Stem cells can be quiescent or proliferative by a process of asymmetric division wherein one of the daughter cells is a stem cell that preserves the DNA of the parent stem cell and the other contains newly synthesized DNA and commits to a differentiation pathway determined by its tissue context.
NO has been proposed to stimulate self-renewal in several cell types (12) (Fig. 14B). The general prosurvival effects of low concentrations of NO extend to stem cells. Murine bone marrow stromal (OP9) cell apoptosis was reduced in cells treated with the NO donor S-nitroso-N-acetylpenicillamine or YC-1 (300). In embryonic stem cells, treatment with low concentrations of an NO donor delayed the process of differentiation and maintained expression of the key self-renewal transcription factors: octamer-binding transcription factor 4 (Oct4), Nanog, and sex determining region Y-box 2 (Sox2) (270). Hematopoietic stem cell sensitivity to the self-renewal effects of NO may decrease with cell age (99). In vitro, treatment with the NO donor linsidomine (3-morpholinosydnonimine, SIN-1) stimulated proliferation of muscle satellite cells (a precursor muscle cell type), and this was inhibited by concurrent treatment with L-NAME (20). Hematopoietic stem cells treated with an NO donor proliferated faster and had increased expression of the self-renewal transcription factor cMyc (183). Thus, multiple stem cell populations are sensitive to the proliferative/cell cycle stimulating effects of NO.
A link between ROS/Nox and self-renewal is emerging (Fig. 14B). siRNA suppression of Nox2 and Nox4 was associated with a decrease in murine-induced pluripotent stem cells in culture as well as a decrease in Sox2 and Oct4 expression compared with control siRNA transfected cells (106). Loss of ROS homeostasis and defense mechanisms such as transcriptional regulators that control ROS scavenging enzymes deterred the proliferative capacity of stem cells (167), whereas suppression of the scavenging enzyme Mn-SOD was associated with decreased expression of several pluripotent genes (235). Movement of stem cells to areas of ischemic muscle injury was decreased in Nox2−/− mice (281). As with NO, ROS control of self-renewal appears to be concentration dependent and temporally dependent.
B. TSP1 and CD47 in stem cell self-renewal and tissue regeneration
The first indications that TSP1-CD47 signaling limits stem cell self-renewal came from experiments employing endothelial cell cultures from lungs of young male WT and Cd47−/− mice. Cd47−/− cells could be passaged continuously upward of 6 months with no loss of proliferative capacity. In contrast, lung endothelial cells from WT mice could not be passaged more than three or four times before becoming senescent as assessed by induction of senescence-associated β-galactosidase activity (113). Null cells continued to display normal endothelial cell morphology even after extended passage. A similar proliferative advantage was observed using Thbs1−/− lung endothelial cells.
The absence of CD47 was associated with increased expression of four critical stem cell transcription factors: Oct4, Sox2, Kruppel-like factor 4 (Klf4), and cMyc (Fig. 14A). Treatment with exogenous TSP1 (2.2 nM) decreased cMyc mRNA in WT Jurkat T cells but not in a somatic mutant lacking CD47. Re-expression of CD47 in the CD47-deficient Jurkat cells suppressed cMyc expression. Conversely, in loss-of-function experiments, suppression of CD47 with a morpholino oligonucleotide in isolated endothelial cells and in mice increased mRNA expression of several self-renewal transcription factors (113).
These findings have since been confirmed in murine brain microvascular endothelial cells, as endogenous TSP1 in WT cells was associated with fewer S phase cells than Cd47−/− cells over several passage events (60).The effects of TSP1 on self-renewal and differentiation are not limited to endothelial cells. Bronchioalveolar stem cell lineage-specific differentiation was found to be supported by TSP1 (132). Furthermore, renal tubule epithelial cells harvested from kidneys of young male Cd47−/− mice had constitutive upregulation of stem cell transcription factors compared with cells from kidneys from WT mice (217). However, the implications of these cell studies for self-renewal after injury were not clear. Pretreatment of WT mice with a CD47 antibody preserved organ function after syngeneic survival renal transplant and this was associated with increased expression of Sox2 and cMyc (217).
Surprisingly, Cd47−/− mice given L-NAME in their drinking water (0.5 mg/ml) for 48 h to limit NO production were nonetheless protected from renal ischemia–reperfusion injury, and this was associated with preservation of cell self-renewal as characterized by stem cell transcription factor protein expression and increased Ki67+ cells (217), suggesting that TSP1-CD47 signaling may inhibit self-renewal independently of its established inhibitory effects on NO. Further studies are needed to test whether the advantage in self-renewal found in cells from young mice that lack CD47 persists with advancing age, as other work indicates that the increased number of mitochondria and size in skeletal muscle from young Cd47−/− mice were lost in aged mice (56).
X. Therapeutic Opportunities to Improve Redox Signaling
A. Dysregulation of NO signaling by TSP1/CD47 in clinical disease
1. Association of elevated TSP1 with impaired NO signaling in cardiovascular disease
Hyperactive TSP1 signaling is associated with clinical diseases known to have loss of NO signaling and resistance to NO pathway supplementation. An earlier study of patients with peripheral vascular disease and end-stage critical limb ischemia demonstrated markedly increased (∼20-fold) expression of TSP1 via in situ hybridization of muscle samples from amputated distal limbs compared with those from proximal limb muscle samples (52). Interestingly, TSP1 induction in amputated limb samples was localized to vascular endothelial cells and to macrophages adjacent to the vascular compartment. TSP1 expression was essentially undetectable in muscle samples from the proximal healthier limb areas.
Likewise, plasma TSP1 levels, quantified by an aptamer-based assay, were increased in ST-elevation myocardial infarction subjects with high atherosclerotic plaque burden than in healthy controls (100). Elevated plasma TSP1 levels were independently associated with coronary artery disease in a diabetic population (30). This finding is not unexpected given data showing that TSP1 is upregulated by glucose (151, 195, 201, 292) and increased in individuals with diabetes-associated renal disease (75).
2. Genetic dysregulation of thrombospondins in cardiovascular disease
A missense mutation of TSP1 (N700S) was associated with increased risk of premature coronary artery disease (276, 317). However, in individuals homozygous for mutation, plasma TSP1 levels were decreased (276), leaving indeterminate whether the mutation provides gain or loss of TSP1 function. However, in vitro studies indicated that treatment with 10 μg/ml of mutant N700S TSP1 enhanced platelet aggregation and surface binding compared with that of nonmutant TSP1, suggesting a gain of function, but was itself more susceptible to proteolysis (177). Assessment of the status of the relevant TSP1 receptors in this population would likely provide further insights.
In a related direction of research, a missense variant of TSP4 (A387P) was associated with myocardial infarction (276), a finding subsequently confirmed (35, 297). Elevated levels of the TSP1 paralog TSP2 were associated with mortality and adverse cardiovascular events in individuals with heart failure (HF) with preserved ejection fraction (114) as well as in individuals with HF and reduced ejection fraction (70). The findings of associations between TSP2 and TSP4 mutations and cardiovascular risk are likely important as both can interact via CD47, although with less affinity as compared with TSP1, to limit NO signaling in vascular cells (84). It is not known whether N700S TSP1 or mutant TSP2 and TSP4 limit NO signaling.
3. Circulating TSP1 as a biomarker of redox dysregulation
Plasma TSP1 is also upregulated in patients with stroke and predicted mortality and poor outcome, as defined as an elevated Rankin score, recurrent stroke, or a new cardiovascular event (59). However, it is not clear what this means in relation to TSP1 expression in the brain or cerebral spinal fluid. TSP1 is too large to cross the normal blood brain barrier, but might do so under inflammation-associated alterations in barrier function. Furthermore, plasma TSP2 levels are elevated in stroke patients than in controls (181). In pediatric sickle cell disease, subjects' elevated plasma TSP1 levels are positively associated with silent cerebral infarction (51).
In adult sickle cell disease subjects, plasma TSP1 is increased in vaso-occlusive events (VOEs) (184). Patients experiencing active VOEs had significantly elevated plasma TSP1 levels [median (IQ range) = 898 ng/ml (381–1657)] versus sickle cell disease patients not in VOEs [303 ng/ml (187–939) and healthy controls (239 ng/ml (125–344) p = 0.001]) (184). As TSP1 is found preformed in platelets, these studies in sickle cell disease cohorts are important in that these authors observed a protocol to mitigate platelet activation during blood collection and processing.
Increased TSP1 signaling concurrent with dysregulation of the systemic vasculature is increasingly demonstrated. In vastus lateralis muscle samples from subjects with essential hypertension, TSP1 protein levels, as characterized by Western immunoblot, trended higher than those in controls but did not reach significance (62). In patients with hypertension, elevated plasma TSP1 was noted in association with decreased forearm-mediated vasodilation (18). However, in this study, TSP1 levels appeared elevated in controls compared to TSP1 levels reported in controls elsewhere (184), suggesting possible confounding by platelet activation. Nonetheless, elevation of plasma TSP1 in individuals with hypertension has been confirmed (80, 241), although interpretation of these other studies is complicated by the presence of other cardiovascular comorbidities. It is not clear whether TSP1 is functioning as a direct presser in hypertension or is elevated secondary to other factors, including deregulation of the angiotensin–renin axis, as angiotensin II stimulates TSP1 production in some cells (175).
4. TSP1 in pulmonary disease
At the same time, a link between TSP1 and dysregulation of the pulmonary circulation has been proposed (116, 212). PH is a rapidly fatal disease that is characterized by loss of NO signaling, increased ROS, pulmonary vascular rarefaction, and remodeling and subsequent right HF. In a prospective cohort of PH patients, elevated plasma TSP1 correlated with the degree of PH and with mortality (103). In a small longitudinal (n = 8) cohort, plasma TSP1 levels increased coincident with the development of scleroderma-associated PH (117). TSP1 and CD47 protein are increased in lungs and distal pulmonary arteries from PH patients compared with samples from non-PH controls (214). Bias-reduced logistic regression analysis found significant positive relationships (p < 0.05) between parenchymal and pulmonary arterial TSP1 protein expression and PH. Interestingly, treatment of healthy human distal pulmonary arteries with exogenous TSP1 (2.2 nM) inhibited NO-mediated vasodilation, whereas treatment with a CD47 antibody (clone B6H12) improved NO-mediated vasodilation in distal pulmonary arteries from end-stage PH lungs (214).
In individuals with sickle cell disease-associated PH, single nucleotide polymorphisms in TSP1 (rs1478605 and rs1478604) are associated with changes in pulmonary arterial systolic pressure, as characterized by Doppler determination of tricuspid regurgitant jet velocity (98). Since the variants localize proximate to the start sight of transcription, it would be important to determine whether they alter TSP1 transcription. The finding of increased TSP1 and CD47 expression in lungs and pulmonary arterial vascular cells from a sickle cell disease patient with end-stage PH (216) underscores the possible importance of this.
5. TSP1 and heart disease
Left ventricular (LV) HF affects >650,000 people in the United Sates annually (115, 253). NO dysregulation contributes to LV HF (221), and Nos3−/− mice experience more LV dysfunction and fibrosis after LV pressure overload than controls (220). TSP1 but not CD47 protein was induced in the left ventricles of WT mice after 4 weeks of transverse aortic constriction (TAC) concurrent with increased histone deacetylase 3 (HDAC3) and calcium/calmodulin-dependent protein kinase II expression, and LV hypertrophy and dysfunction (229). Conversely, Cd47−/− mice did not develop LV hypertrophy and dysfunction and had less LV HDAC3 expression both before and after TAC.
The role of NO in this process is uncertain, as cardiac myocytes treated with exogenous NO ± CD47-binding peptide 7N3 (10 μM) did not show significant changes in either phosphorylated or total HADC3. Nonetheless, WT mice treated with a HDAC3 oligonucleotide morpholino to suppress protein levels or a CD47 antibody responded to TAC with less LV hypertrophy and stiffness (dP/dt min). Together, these findings suggest TSP via CD47 and HDAC3 drives TAC-mediated LV HF.
Other TSPs have been linked to LV HF, but their functions diverge from that of TSP1. Thbs4−/− mice developed more LV hypertrophy and fibrosis than controls after 2 weeks of TAC (57), with null mice displaying maladaptation to TAC within as little as 2 days (32). Isolated cardiac trabecular muscle from Thbs4−/− mice showed minimal calcium flux with sustained stretch (32). Conversely, treatment with the CD47-binding peptide 7N3 (10 μM) stimulated a rapid calcium flux in isolated rat neonatal cardiac myocytes (229). Furthermore, Thbs2−/− mice were more sensitive to doxorubicin-mediated cardiomyopathy than WT mice (284). These results indicate cardioprotective roles for TSPs other than TSP1 under some conditions. Further studies are needed to assess whether TSP1 and CD47 expression and signaling are adversely altered to contribute to disease in Thbs2−/− and Thbs4−/− animals.
The role of other matricellular proteins in pressure overload LV HF from TAC is yet to be studied. In an ex vivo cardiac ischemia–reperfusion model, the absence of CCN1 was associated with greater infarction (311). CCN2 levels were increased in hearts from mutant mice that spontaneously develop cardiac hypertrophy and failure, and CCN2 stimulated increased TSP1 and TSP4 expression in isolated myocytes (278). Mice lacking periostin (Postn−/− ) showed poorer healing after cardiac infarction (238). Ischemia/reperfusion and infarction, although inducing oxidative stress in the heart and contributing to HF, are not comparable with pressure overload. Although they could indirectly regulate NO levels via their effects on ROS production, there is no evidence that CCNs or periostin directly alter NO signaling. Further investigation is needed to define whether these stress-responsive matricellular proteins play a role in pressure overload LV HF.
Chronic Ang II signaling also induces LV hypertrophy and HF in rodents (71, 188), and Ang II can stimulate Nox-associated oxidative stress (43). This is interesting in view of findings that Jurkat cells treated with Ang II display inhibition of sGC through increasing calcium flux (202). The same cells treated with the recombinant domain of TSP1 that targets CD47 showed calcium flux and sGC inhibition (202). Together these findings suggest several reasons for possible resistance to Ang II-mediated vasoconstriction in arteries from Thbs1−/− and Cd47−/− mice, a hypothesis waiting to be tested.
6. TSP1 in aging
Aging is associated with cellular senescence, decreased self-renewal, loss of healing capacity, and altered blood flow. Aging is being reassessed as not only a causative factor in many chronic diseases but perhaps a disease in itself. However, the concept that matricellular proteins may be induced simply by aging is only now being considered (infra vide and Table 2). In associative findings, omentum-derived senescent human mesothelial cells produced significantly more TSP1 than nonsenescent cells (161). That aging alone may induce matricellular expression in people was recently suggested by findings of upregulation of platelet TSP1 in otherwise healthy adults compared with healthy children (33). TSP1 and CD47 expression in the skin of WT mice increased with age and was associated with a loss in tissue blood flow (213), whereas skeletal muscle blood flow in response to ischemia or vasodilators was not diminished in aged Thbs1−/− and Cd47−/− mice (86).
B. Therapeutic targeting of the TSP1 receptor CD47
1. Applications for cardiovascular disease
Because TSP1 is chronically overexpressed in major diseases of aging that are associated with impaired NO production and signaling (Table 2), therapeutics that target the inhibitory receptor CD47 could potentially restore physiological functions of NO as well as limit some sources of ROS production in these diseases. Antibodies can target CD47 efficiently. However, because of potential agonist/antagonist activity, the effects upon NO and ROS signaling will likely need to be determined for each molecule. In contrast, in preclinical studies, CD47 morpholinos have uniformly improved cell and tissue responses to stress. Both antibodies and morpholinos have been administered chronically to people. However, CD47-specific targeting agents should be administered in long-term treatment experiments in mammalian models of cardiovascular and age-related diseases.
Insight into the effects of inhibiting the TSP1–CD47 axis in people may be gathered from current and possible future clinical trials employing CD47 antibodies to suppress tumor growth. Attention to changes in blood pressure, wound healing, and metabolism combined, where possible, with analysis of platelet-poor plasma samples for TSP1 and the blood cell fraction for NO/ROS signaling, would be useful to understanding in human subjects the potential implications of CD47 targeting on homeostasis and cardiovascular health.
2. Therapeutic applications for cancer
The abnormal vasculature of tumors tends to be unresponsive to NO (88), but the ability of CD47 blockade to modulate responses to radiation and redox signaling involved in innate and adaptive antitumor immunity provides therapeutic opportunities for improving the activities of both conventional cytotoxic therapies and immunotherapy of cancer (134, 147, 157, 248). Several companies are developing biologics targeting CD47 that are designed to inhibit its interactions with SIRPα, and several of these have entered human clinical trials for cancer patients (NCT02216409, NCT02678338, NCT02953509, NCT02953782, NCT02367196, NCT02488811, and NCT02641002).
CD47 antibodies have been demonstrated to enhance phagocytic killing of tumor cells by macrophages, but based on the effects of TSP1 and CD47 on ROS production and of TSP1 on oxidative tumor cell killing, complementary therapeutics that selectively inhibit the TSP1/CD47 interaction may also have efficacy. The role of H2S and NO signaling in T cell activation combined with evidence that TSP1/CD47 signaling limits T cell activation and CD8+ T cell killing of target tumor cells also justifies the development of therapeutic inhibitors of TSP1 and CD47 as cancer therapeutics (248). Such TSP1–CD47 targeting therapies are envisioned to have the important added benefit of preventing damaging side effects from radiation and chemotherapy in nonmalignant tissues.
Although CD47/SIRPα inhibitors show antitumor efficacy in a number of xenograft models in Nod/SCID mice, and two SIRPα decoy receptors are in clinical trials (NCT02663518 and NCT03013218), it is important to recognize that Nod encodes a variant SIRPα with supraphysiological affinity for human CD47 (122). Therefore, this model creates a strong bias for agents that block the CD47/SIRPα interaction, which is enhanced by interspecies markers of nonself that remain intact in the Nod/SCID model. Indeed, antibody blocking or knockdown of CD47 in immune competent syngeneic mouse tumor models generally does not significantly inhibit tumor growth (160, 242, 246, 268). Thus, it seems unlikely that the CD47 targeting biologics currently in clinical trials will have significant antitumor activities as single agents in human patients who all also bear syngenic tumors.
In contrast, combining CD47 knockdown or blockade with tumor irradiation or cytotoxic chemotherapy can effectively control tumor growth (134, 147, 160, 246). Although autophagy is required for the cytoprotective effect of CD47 blockade in nonmalignant tissue (244), mechanisms by which blockade may sensitize tumor cells to radiation and chemotherapy remain to be identified. Generation of free radicals is central to the antitumor activities of anthracyclines and ionizing radiation. Therefore, understanding how blocking TSP1/CD47 signaling alters the redox environment in tumor versus nonmalignant tissue is important for advancing the clinical applications of such combination therapies.
Studies using hepatocellular and breast carcinoma stem cells have identified genes that exhibit altered expression when CD47 is knocked down or ligated by a therapeutic antibody (109, 134). These include regulators of NF-κB in the hepatocellular carcinoma (134), which control oxidative stress responses. The same study reported CD47-dependent expression of thioredoxin-interacting protein (TXNIP), and TXNIP was upregulated by treatment with the CD47 antibody B6H12 in triple negative breast cancer stem cells (109). In part by inhibiting the antioxidant function of thioredoxin-2 (Trx2) in the mitochondria, TXNIP limits resistance to oxidative stress, and deletion of TXNIP-protected mice from ischemia–reperfusion injury (308, 309). Further studies are needed to test the hypothesis that CD47-mediated upregulation of TXNIP in cancer stem cells could sensitize them to the oxidative stress caused by radiation or anthracyclines.
XI. Conclusions and Outlook
Matricellular proteins play important roles in higher animals to mediate communication between cells and their microenvironment. Redox signaling is both a target of matricellular protein signaling and a regulator of the expression of several matricellular proteins. TSP1, and to some extent osteopontin and ADAMTS1, regulates the production of NO in specific cell types, although by different mechanisms. Controlling the production of NO is one mechanism to regulate NO signaling, but to date only TSP1 is known to also regulate the ability of cells to respond to exogenous NO (Fig. 5).
Conversely, NO signaling controls the expression of many matricellular proteins, including TSP1, TSP2, SPARC, SMOC1, ADAMTS-2, and ADAMTS5. H2S and ROS signaling likewise control the expression of many matricellular and extracellular matrix proteins, but only TSP1 has been demonstrated to control H2S production and responses to exogenous H2S. TSP1, CCN1, and periostin are the known matricellular regulators to date of ROS production (Fig. 2), although the latter functions to suppress rather than increase oxidative stress.
Our understanding of function of matricellular proteins has evolved from their initial roles in altering cell adhesion to recognizing matricellular proteins as physiologically relevant activators of maladaptive redox signaling involved in cellular responses to stress. Dysregulated redox signaling contributes to the pathogenesis of many acute and chronic diseases. NO, H2S, CO, and ROS each play roles in mediating intracellular and paracellular signaling that contributes to these processes. Impaired NO signaling is a key contributor to age-related deficiencies in tissue repair and maintenance of vascular perfusion.
Matricellular proteins play important roles in redox homeostasis in acute injury and chronic disease. Dysregulation of TSP1 and other matricellular protein expression and function increases with age (Table 2) and coincides with progressive impairment of NO signaling. The improved recovery from ischemic injury and elevated tissue cGMP in aged Thbs1−/− and Cd47−/− mice provides strong genetic evidence that this pathway plays a major role in age-related loss of NO/cGMP signaling. The ability of a CD47 antibody and antisense morpholino suppression of CD47 to restore normal ischemic injury responses to aged WT mice is proof of principle that therapeutics targeting this pathway could have broad clinical applications (86, 213).
Several of the CD47 redox signaling targets discussed here coincide with genes that exhibit significantly altered expression in a global transcriptome analysis of WT versus Cd47−/− mouse lung endothelial cells, including Nos, Txnip, and NADPH oxidase genes (113). However, examination of the resulting regulatory network reveals a larger number of CD47-dependent genes for which we currently have no understanding of how TSP1 and CD47 regulate their expression (Fig. 15). Therefore, many important questions remain to be addressed to achieve a comprehensive understanding of even one of the signaling receptors that mediates responses to TSP1. It is safe to assume that a similar number of important questions remain to understand the function of the other matricellular proteins in the context of redox signaling.
TSP1 signaling through CD47 has also emerged as an inhibitor of stem and nonstem cell self-renewal and the genes that coordinate this process (112, 113, 217), which has implications for age-related loss in healing capacity and for tissue regeneration using stem cell therapy and in tissue bioengineering. It will be necessary to fully define how CD47 functions to balance and, when activated by ligands such as TSP1, imbalance these multiple pathways. Because other matricellular proteins can also regulate redox signaling, it will be important to determine whether each protein acts alone or in cooperation with each other and with TSP1. The knowledge gained from such studies could lead to additional therapeutic possibilities.
Dysregulation of matricellular protein expression is also a common feature in cancer and has been linked to risk for carcinogenesis, malignant progression, and metastatic disease (88, 174, 236, 247, 250). Although correlations have been clearly established with altered redox signaling, a causal relationship between aberrant matricellular protein expression and impaired redox homeostasis in cancer remains largely unknown. Given the multiplicity of matricellular protein receptors, it is likely that redox-dependent and redox-independent signaling contributes to the observed changes in any specific cell type, and experimental design must account for this complexity to advance our understanding of mechanism and enable successful translational applications for treatment.
Two classes of biologics targeting CD47, humanized anti-CD47 antibodies, and bivalent recombinant SIRPα are advancing in clinical trials for cancer patients (144, 192, 194). The intended/expected activity of both classes of therapeutics is to enhance phagocytic clearance of tumor cells by macrophages by inhibiting the CD47–SIRPα interaction (157), but understanding the role of CD47 in redox signaling can illuminate potential side effects of these therapeutics.
Preclinical data for CD47 antibodies that inhibit CD47–SIRPα interaction have generally found that TSP1–CD47 interactions are also inhibited, and such antibodies consequently alter blood pressure by an NO-dependent mechanism when injected intravenously (10) and, by enhancing red blood cell clearance, can induce anemia (144, 194, 239). Thus, systemic cardiovascular side effects need to be considered and monitored. Based on the sensitivity of Cd47−/− mice to anesthesia (91), surgical procedures may be contraindicated for patients enrolled in these clinical trials. Platelet aggregation is another concern for patients treated with humanized CD47 antibodies and antagonists (192, 194), and the CD47 antagonism of antithrombotic NO signaling in platelets provides a mechanistic basis for this side effect (95).
More broadly, CD47-targeting drugs for cancer should be viewed as immune checkpoint inhibitors that enhance adaptive as well as innate antitumor immunity (248). Regulation of NO and H2S signaling in T cells by CD47 likely contributes to their adaptive immune checkpoint function, and assessment of these pathways in circulating T cells could be useful surrogate biomarkers to evaluate CD47-targeting therapeutics in clinical trials.
Footnotes
Acknowledgments
This work was supported by the Intramural Research Program of the NIH/NCI (D.D.R.) and by 2P01HL103455, R01 HL-108954, and 1R01HL112914 (J.S.I.). This work was also supported by the Institute for Transfusion Medicine, the Hemophilia Center of Western Pennsylvania, and the Heart, Lung, Blood, and Vascular Medicine Institute of the University of Pittsburgh School of Medicine (J.S.I.).
Author Disclosure Statement
J.S.I. serves as Chair of the Scientific Advisory Board of Radiation Control Technologies, Inc. (RCTI, Garden City, NJ) and has equity interest in RCTI and Tioma Therapeutics (St. Louis, MO) that have licensed CD47 technology for development. The other authors have no competing financial interests to disclose.