
Abstract
Significance:
Decrypting the cellular response to oxidative stress relies on a comprehensive understanding of the redox signaling pathways stimulated under oxidizing conditions. Redox signaling events can be divided into upstream sensing of oxidants, midstream redox signaling of protein function, and downstream transcriptional redox regulation.
Recent Advances:
A more and more accepted theory of hydrogen peroxide (H2O2) signaling is that of a thiol peroxidase redox relay, whereby protein thiols with low reactivity toward H2O2 are instead oxidized through an oxidative relay with thiol peroxidases.
Critical Issues:
These ultrareactive thiol peroxidases are the upstream redox sensors, which form the first cellular port of call for H2O2. Not all redox-regulated interactions between thiol peroxidases and cellular proteins involve a transfer of oxidative equivalents, and the nature of redox signaling is further complicated through promiscuous functions of redox-regulated “moonlighting” proteins, of which the precise cellular role under oxidative stress can frequently be obscured by “polygamous” interactions. An ultimate goal of redox signaling is to initiate a rapid response, and in contrast to prokaryotic oxidant-responsive transcription factors, mammalian systems have developed redox signaling pathways, which intersect both with kinase-dependent activation of transcription factors, as well as direct oxidative regulation of transcription factors through peroxiredoxin (Prx) redox relays.
Future Directions:
We highlight that both transcriptional regulation and cell fate can be modulated either through oxidative regulation of kinase pathways, or through distinct redox-dependent associations involving either Prxs or redox-responsive moonlighting proteins with functional promiscuity. These protein associations form systems of crossregulatory networks with multiple nodes of potential oxidative regulation for H2O2-mediated signaling.
I. Introduction
Cell signaling affords organisms a method of delicate control over cellular balance, and enables the rapid coordination of responses to external stimuli and stressors. Intracellular signaling is formed from a variety of mechanisms, from enzymatic posttranslational modifications of substrate proteins, such as acetylation or phosphorylation, to the sensing of mediatory small molecules, such as Ca2+ or cyclic nucleotides. The reversible posttranslational modification of protein thiols presents another axis of cellular signaling, yet despite decades of research, our understanding of oxidative signaling networks remains woefully incomplete. Reactive oxygen species (ROS), such as superoxide (O2 •–), hydroxyl radical (•OH), singlet oxygen (O1), and peroxide (ROOH), are intracellular initiators of oxidative modifications, and of these, hydrogen peroxide (H2O2) is considered most likely to play a role in oxidative signaling due to its relatively longer half-life and specificity toward protein thiols.
For many years, H2O2 within the cell has been regarded as playing a damaging and detrimental role for cell survival, but an appropriate and fine-tuned dose of H2O2 is required for normal cellular functioning (303, 338, 339). H2O2 is highly oxidizing because of the presence of a peroxide bond (O–O), of which the chemical reduction is rather limited by its high activation energy (419), making H2O2 very selective in its reactions with metal centers and specialized protein thiols (404). With the exception of plants, in which the main H2O2 source is thought to be glycolate oxidase, H2O2 is formed predominantly from O2 •– dismutation that either happens in a spontaneous (105 M −1·s−1) or in an activation energy-reduced fashion by the enzyme superoxide dismutase (SOD; 109 M −1·s−1) (98, 99) (Fig. 1). Whereas the cellular electron transport chains are a potential O2 •– source, the contribution of the mitochondrial respiratory chain is generally minor (246, 348). Instead, nicotinamide adenine dinucleotide phosphate [NAD(P)H] oxidases (Fig. 1), also known as rubidium oxidase (RBOH), NADPH oxidase (NOX), and dual oxidase (DUOX), are the main H2O2 sources that transfer electrons from NADPH to molecular oxygen (119). These membrane-spanning enzyme complexes generate H2O2 through two-electron reduction of oxygen or through one-electron reduction to O2 •– and subsequent reduction to H2O2. Their activities are controlled by growth factors and cytokines and have an array of physiological and pathophysiological functions (13, 119). In this study, the oxidative burst used to eliminate invading microorganisms highly dependent on NOX activities and mutations that affect the NOX2 activities lead to recurrent infections and impaired pathogen clearance (140, 376). Besides pathogen removal, tissue wounding induces the generation of NOX-dependent H2O2 that is required for leukocyte recruitment and wound healing (223, 261). Because of the spatiotemporal differentiation of locally high H2O2 concentrations, the H2O2-induced redox signal has to take place in the vicinity of its production, as also recently mentioned in The Redox Code (163). In general, H2O2 is a suitable second messenger, because (i) few protein targets are kinetically relevant for H2O2 reduction, such as thiol peroxidases and metal centers (404); (ii) it is stable enough to diffuse and generate a gradient from the source (100, 404); (iii) its production is fine-tuned and can be controlled by external stimuli, such as growth factors, insulin, and environmental or mechanical stresses (83, 145, 231); and (iv) its scavenging enzyme activity can be regulated, as observed in peroxiredoxin (Prx) overoxidation (65, 350, 375).
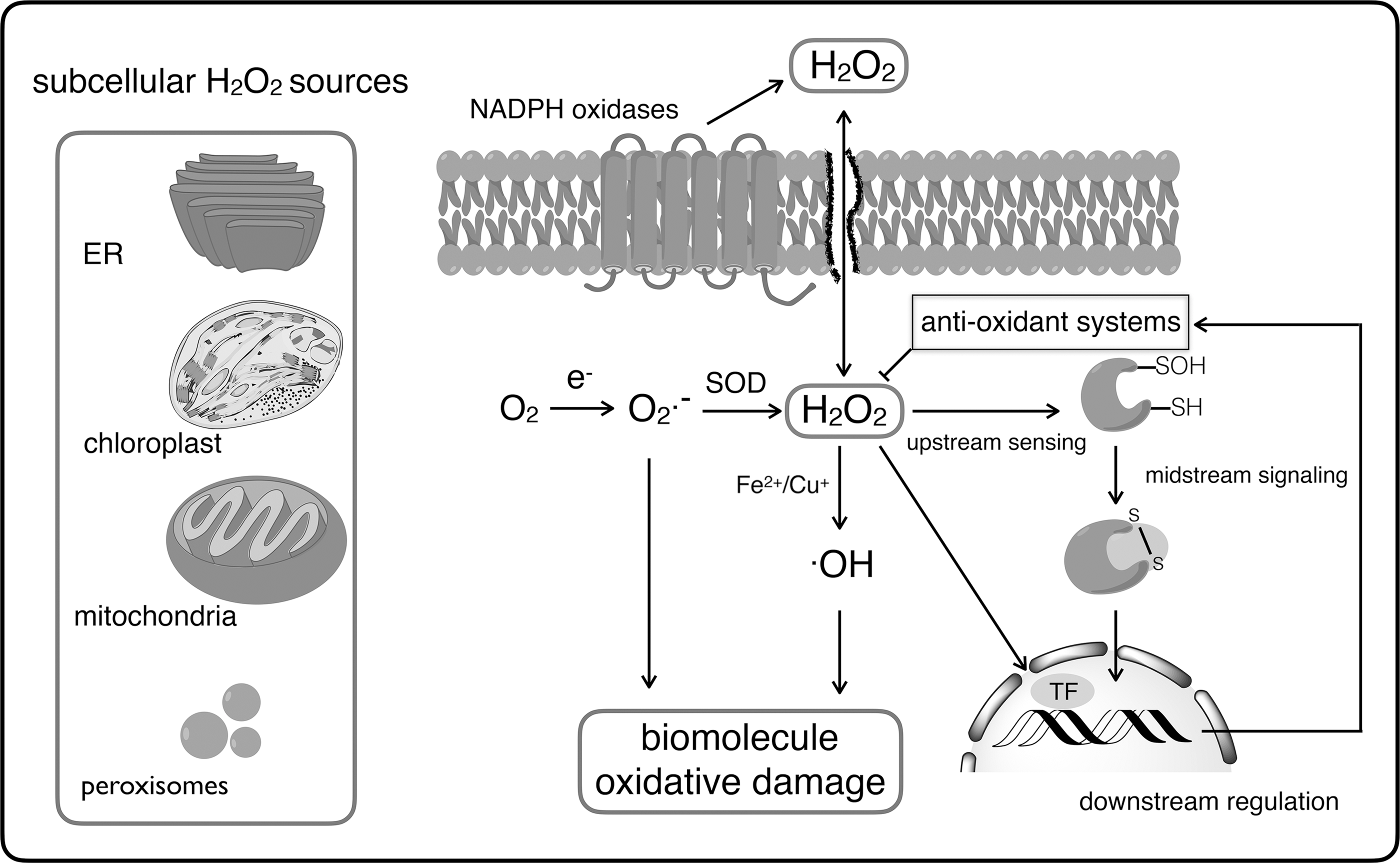
In addition to its role in cellular functioning, intracellular H2O2 production is also induced by biotic and abiotic stresses from the cellular environment, thus, bringing the cells under oxidative stress. The term “oxidative stress” appeared in the literature in the mid-eighties (337). Since then, research on oxidative stress responses and the linked redox switches is flourishing. Oxidative stress is now regarded as an imbalance between oxidants and antioxidants in favor of the oxidants, first triggering a redox signaling response, but in the long run resulting in disruption or blocking of signaling pathways, with molecular and cellular damages as a consequence (340).
Despite the highly oxidative nature of H2O2, the rate constants of its reaction with protein thiols vary over several orders of magnitude (∼0.1–108 M −1·s−1), with some cysteines positioned in pockets catalytically geared toward H2O2 reduction. The proteins most catalytically reactive toward H2O2 are the primary oxidant sensors of the cell and are therefore likely to play a key role in either the initiation of intracellular signaling or rapid induction of a transcriptional response (Fig. 2). Induction of a transcriptional response can be mediated either by transcriptional regulators capable of directly sensing H2O2 (and so contain a “peroxidatic” catalytic cysteine), or through an oxidant signal transduction via interaction with a facilitating enzyme catalytically reactive toward H2O2. Both methods of H2O2-mediated transcriptional regulation are discussed within this review in the context of “downstream redox regulation,” and here it is compelling to compare the more simplistic, direct model of oxidant-responsive transcriptional regulation developed by prokaryotes, to the indirect Prx-mediated mode of oxidative regulation adopted by mammalian transcription factors.
As the enzymes most catalytically reactive toward H2O2, Prxs constitute the entry point of oxidative signaling, and in their recently consolidated role in oxidant transmission, Prxs can be considered as frontline messengers of oxidative signaling, a role discussed within this review in terms of “upstream redox sensing.” Aside from the modulation of transcriptional regulation, signaling through thiol oxidation—Prx facilitated or otherwise—requires specific protein targets to elicit specific functions conducive to the goal of a cellular oxidative response. Considering that H2O2 flux can be both rapid, and random, it appears beneficial for cells to possess constitutively expressed proteins, which can alter function in a redox-dependent manner in response to oxidative signaling. Unlike Prxs, which are obligate antioxidant enzymes with a conserved function, such proteins would effectively be redox-responsive moonlighting proteins, and a selection of such moonlighting proteins are the focus of the section Midstream Redox Signaling by Moonlighting Proteins of this review.
II. Applied Techniques for Redox Biology
A. Redox proteomics
Advances in the sensitivity of mass spectrometry (MS) have facilitated an expanding characterization of the redoxome of reversibly oxidized cysteine residues. Recent reviews have highlighted the importance of this technology in the field of redox proteomics (30, 412). Current techniques for the selective enrichment and/or labeling of redox-sensitive cysteines for MS include the on-resin capture of oxidizable cysteines from soluble cell extracts by means of Thiopropyl–Sepharose beads and the adaption of thiol-reactive alkylating agents, such as iodoacetamide (IAM) or N-ethylmaleimide (NEM), for quantitative techniques, such as isotope-coded affinity tags (ICATs), or iodoacetlyl-based tandem mass tags (iodo-TMTs) (107, 108, 291). These techniques rely on the blocking of free thiols followed by the reduction of oxidized cysteines and subsequent labeling/affinity capture. Whereas ICAT involves the differential labeling of reduced and reversibly oxidized cysteines with “light” and “heavy” ICAT reagents, respectively, iodo-TMT uses a variety of mass reporters, thereby allowing the simultaneous assessment of different oxidant conditions.
A potential drawback to the use of IAM or NEM in the alkylation of free thiols is their crossreactivity with sulfenylated cysteines, although the resulting thioether derivative can be cleaved by reducing agents and, hence, does not necessarily interfere with selective identification of sulfenates (301). A further limitation of IAM-based probes is their potential for toxicity when used at high concentrations (1, 213). In past proteomic approaches, the use of reductants of relative specificity, such as arsenite for sulfenylated cysteines (321) and ascorbate for S-nitrosylated cysteines (153), allowed a partial distinction between oxidative modification types, and iodo-TMT has been resourcefully demonstrated to differentiate sulfenylation, sulfinylation, S-nitrosylation, and S-glutathionylation (355, 405).
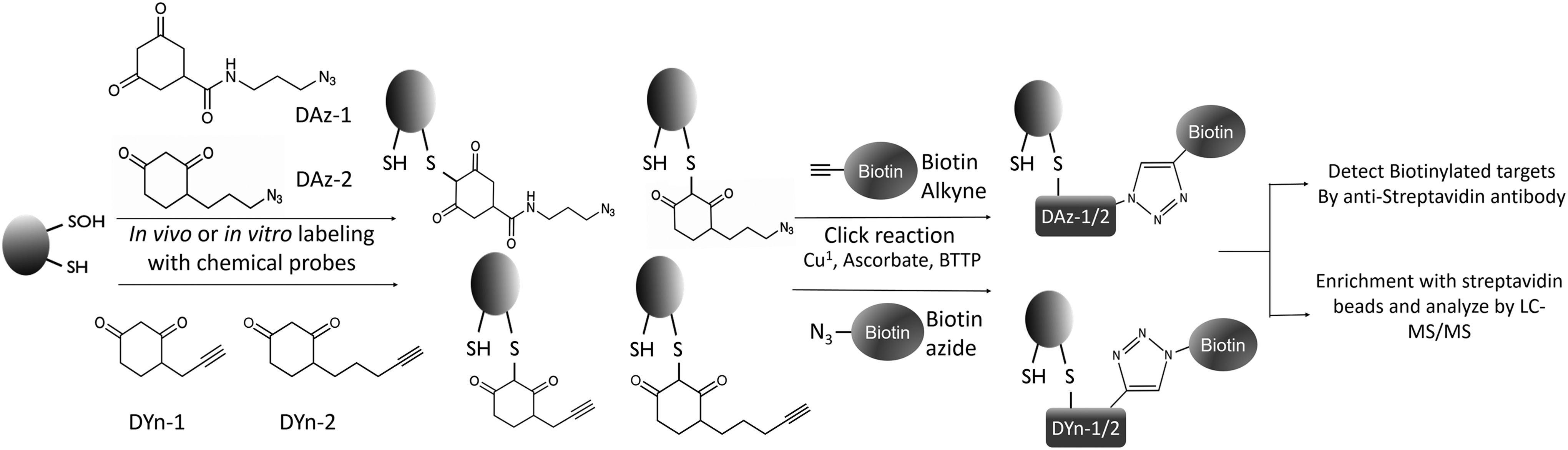
In addition to these approaches that employ electrophiles to target thiols, an array of cyclic carbon nucleophiles with specific reactivity to sulfenic acid have arisen based on 5,5-dimethyl-1,3-cyclohexanedione (dimedone) (124). Azide- and alkyne-functionalized “chemical reporter” analogs of dimedone (DAz-1, DAz-2, DYn-1, and DYn-2) for biotinylation successfully identified the proteomics of sulfenylated proteins in vivo (207, 269, 276, 300, 331), but the biotin tag had to be photocleaved postenrichment to avoid compromising peptide ionization in the MS identification (Fig. 3) (413, 414). However, the specificity of dimedone toward sulfenic acids has recently been put to question (97, 136, 137, 353). There has been experimental evidence of dimedone labeling cyclic sulfenyl amides, which can be formed in a sulfenic acid-independent manner via the reaction of a glutathionylated thiol or disulfide bond with an amide of the protein backbone (97). A recent study has also demonstrated that a large portion of dimedone-tagged proteins are susceptible to cleavage by dithiothreitol (DTT), indicating that the dimedone-bound species was not Cys-SOH, but rather cysteine perthiosulfenic acid (Cys-SSOH), the result of the oxidation of a persulfidated thiol (136). Moreover, there have been some indications of dimedone treatment per se leading to increased intracellular “ROS” levels, as detected by dichlorofluorescein (DCF) fluorescence (288). Hence, it is strongly advised to confirm proteins detected as sulfenylated by dimedone-based probes using other methods.
As an alternative to chemical approaches to sulfenylation trapping, a genetically encoded probe has been developed. This probe, termed YAP1C, is based on the C-terminal region (residues 565 to 650) of the Saccharomyces cerevisiae (baker's yeast) transcription factor yeast AP1-like protein (YAP1) (264, 364, 365, 400), with only Cys598 retained and other cysteines mutated to alanine or threonine (364). Cys598 of YAP1C has proven to specifically form stable mixed disulfides with sulfenylated cysteines, resulting in a protein complex that can be isolated through inclusion of an affinity tag to YAP1C (264, 364, 365, 400). The major advantages of the YAP1C probes when compared with the more common chemical-based approaches to trap sulfenylated proteins are that they are expressed in the cell, thus directly circumventing any permeability issues, and that they can specifically be targeted to tissues or organelles. Potential disadvantages of the YAP1C probes include the necessity for genetic modification of the target organism and the sensitivity of the mixed disulfide to cleavage by endogenous cellular reductants.
For proteomic characterization of reversible disulfides with physiological significance, thioredoxin (Trx) has recently been utilized as a tool for specific capture of cellular redox targets (8, 206, 254, 278, 283, 421). Both the tandem mass tag (TMT) and ICAT proteomic methods have been adapted to identify Trx targets, merely by using Trx as the postalkylation reductant instead of a chemical reducing agent, such as DTT or tris(2-carboxyethyl)phosphine (TCEP) (278, 421). Other in vivo approaches involve replacement of endogenous Trx with a resolving cysteine mutant of Trx, which forms a stable mixed disulfide with oxidized cysteines of the target proteins. The resulting Trx–target complexes have been enriched by immobilized metal affinity chromatography, in which a poly-His tag has been engineered (283), Trx-Sepharose resin (8, 206, 278), or by immunoaffinity (254). Proteins captured in a mixed disulfide with Trx can elute with a reducing agent and can be separated by two-dimensional gel electrophoresis for MS identification (8, 206, 254, 278, 283).
A similar approach has been used to identify intracellular targets of Arabidopsis thaliana 2-Cys peroxiredoxin A (PrxA), with Prx–target complexes isolated by coimmunoprecipitation, followed by separation on sodium dodecyl sulfate–polyacrylamide gel electrophoresis and identification by nano liquid chromatography–tandem MS (42). In addition to the detection of covalently bound partners of Trx or Prx, the determination of transient interaction partners has been attempted through techniques that do not depend on the mixed disulfide stability. A classic and well-established technique is the yeast two-hybrid system that fuses “bait” and “prey” sequences to mutually required domains of a transcriptional activator of a reporter gene (91). Interaction of bait and prey results in nuclear translocation and transcription of the reporter gene, thereby providing a readable output. A yeast two-hybrid system has been used to identify an interaction between the Salmonella virulence-related effector, SlrP (small leucine-rich protein [E3 ubiquitin-protein ligase]), and human Trx (16). Two-hybrid approaches have also been adapted for use in mammalian cell lines, such as the KInase Substrate Sensor (KISS) assay that exploits a kinase pathway, involving glycoprotein 130 (gp130), nonreceptor tyrosine-protein kinase 2 (TYK2), and signal transducer and activator of transcription 3 (STAT3), to control STAT3-dependent reporter gene expression in response to the bait–prey interaction (211).
More recently developed techniques for assessing protein–protein interactions include proximity-based labeling methods, such as proximity-dependent biotinylation. Site-directed mutation of the biotin/bioadenosine monophosphate (AMP)-binding domain of the biotin ligase, BirA, enabled the development of techniques based on promiscuous biotinylating fusion proteins, such as BioID and BioID2 (55, 173, 312, 313). The intracellular expression of a BirA fused with the bait protein allows highly sensitive labeling of any transiently interacting partner proteins in a proximity-dependent manner. Biotinylated proteins can then be efficiently enriched by avidin or streptavidin for MS-based identification. Although this technique provides a sensitive tagging method of noncovalent redox-mediated interactions without omission of any transient mixed disulfide complexes, its evident drawback is the nonspecific nature of the labeling of the proteins in the immediate proximity of the bait, instead of the discerning labeling of direct interaction partners only. In an attempt to overcome this, a derivative of BioID that incorporates protein fragment complementation was developed. In this approach, BirA is split into two fragments that are fused to two interaction partners; hence, BirA-mediated biotinylation of vicinal proteins occurs in a more defined manner, only when and where the two proteins interact (67, 326). Recently, BioID has been applied in the identification of redox-related interactions in the study of interacting partners of Trx-interacting protein (Txnip) in a mammalian cell line (101). Of the 31 interacting partners identified, 17 were found to be independent of mixed disulfides through the additional use of a Txnip-BioID control, in which the redox-active cysteine of Txnip (Cys247) had been mutated to serine.
B. Structural techniques for studying redox-regulated proteins
X-ray crystallography is the preferred technique for the determination of three-dimensional macromolecular protein structures, which can provide specific insight into the molecular features that govern redox sensitivity of thiols/disulfides. Atomic resolution crystallography has enabled the visualization of the intermediate oxidation states of peroxidatic cysteines (280) and facilitated the analysis of disulfide strain energy and other structural determinants of disulfide redox potentials (309, 325). However, practical issues can arise when attempting to crystallize redox-sensitive proteins, as partial oxidation of proteins may result in mixed populations of conformational or oligomeric states with variable stabilities, which obstructs crystallization. The utilization of either reducing or oxidizing agents can successfully drive the protein toward a single redox state, although the choice of the reducing agents can be an important factor, with TCEP generally favored over DTT, because of its enhanced aqueous stability and efficacy over a wide pH range (112). Once crystals of a redox-sensitive protein are obtained, further complications can occur from the use of synchrotron radiation to collect diffraction data. Typical synchrotron diffraction experiments with photon flux in the range of 1011–1012 photons per second subject crystals to a radiation dosage in the order of 107 Gy (J·kg−1), with the absorbed dose depending on the X-ray wavelength and the atomic composition of the protein (249).
One of the first evidence of specific radiation damages to a protein is the electron capture-induced cleavage of disulfide bonds (5), which is particularly undesirable in the case of functional redox-active disulfides. The sensitivity of disulfide bonds toward ionizing radiation varies greatly and depends generally on the local microenvironment and, with the exception of Trx1 from Litopenaeus vannamei (white-leg shrimp), increases with solvent accessibility (36, 297, 401). In a case study of lysozyme crystals, saturated radicalization of disulfide bonds was observed at a cumulative dose of 0.5–0.7 MGy, followed by bond breakage at 1.05 MGy (357). In addition to the radiolytic cleavage of disulfides, oxidation of protein thiols by radiation has also been proposed, although its occurrence in protein crystals is poorly characterized. Oxidation of thiol sulfurs by ionizing radiation has been suggested to proceed via a thiyl radical (RS•) intermediate, which can be formed either by direct electron capture at the thiol or through reaction with a hydroxyl radical (•OH), which is formed upon radiolysis of water. The thiyl radical can then react with molecular oxygen to form a persulfenic acid, which then either rearranges to a sulfinic acid (RSO2H), or condenses to a sulfenic acid (RSOH) (409). Radiation-induced oxidation of cysteine to its sulfenylated form have been proposed to occur in the crystal structures of rat trypsin (402) and isocyanide hydratase (188), and the radiation-induced thiol oxidation of yeast alcohol dehydrogenase in solution has been characterized (307). MS analysis of radiation-exposed cysteinyl peptides suggests a predominant oxidation species of overoxidized R-SO2H/R-SO3H forms over an R-SOH (409). Structural biologists wishing to identify physiologically relevant cysteine oxidation in crystal structures should always consider the possibility of artefactual cysteine oxidation during X-ray diffraction data collection.
A possible alternative to X-ray crystallography for structural studies is nuclear magnetic resonance (NMR) spectroscopy. NMR allows for solution study of small proteins or protein fragments, and has the benefit of bypassing the need for crystallization, although the high concentrations of protein required for this technique can sometimes lead to aggregation. NMR can be applied effectively to study redox-dependent structural dynamics of proteins that adopt different conformations according to the redox status and are prone to partial oxidant-induced unfolding, as seen for heat shock protein 33 (Hsp33) (117, 205). Small-angle X-ray scattering (SAXS), as a complementary biophysical technique, has also provided useful insights into redox-driven structural changes, as for instance, into the large-scale oxidation-induced structural rearrangement of the mycothiol-dependent oxidoreductase, Rv2466c (2), later identified as mycoredoxin-2 (310).
Early applications of NMR spectroscopy enabled the first structural characterization of cysteine sulfenylation (59), disulfide bond formation (332), and thiol pK a determination by monitoring chemical shift changes (96). In view of the increasing number of redox-regulated protein–protein interactions revealed by proteomic studies (42), together with the apparent role of scaffold proteins in facilitating peroxidase redox relays (17), there is a clear need for the structural characterization of transient protein complexes specific to redox processes. Crystallographic studies of such complexes are limited by both the required stability (relatively high affinity) and reasonable conformational homogeneity of the complex. However, with the development in diamagnetic and paramagnetic NMR spectroscopy techniques, characterization of transient protein interactions through NMR may increasingly become an applied field (221, 320).
As cryo-electron microscopy (cryo-EM) has not the same restrictions of conformational homogeneity due to the computational sorting and filtering of sample images, it lends itself well to the structural study of multiprotein complexes (80, 164, 165). In addition to nonnative complexes, cryo-EM is also especially well suited to analyze large native macromolecular assemblies of subunits. Examples of redox-regulated functions characterized by cryo-EM include the observation of the self-assembling filamentous high-molecular-weight (HMW) chaperone form of PrxIII (293, 416) and the involvement of CXXC motifs of the viral chaperone protein UL32 in disulfide bond regulation (3).
Methodological improvements in sample preparation and image processing software and technological advancements in phase plates (for in-focus phase contrast) and detector camera qualities have evolved single-particle cryo-EM into a viable tool for structural study at the molecular level (114). The resolution of electron microscopy density maps has increased remarkably over the years, with a recent determination of the structure of glutamate dehydrogenase at 1.8 Å resolution (236), and will progress even further (390). As an alternative to X-ray crystallography, single-particle cryo-EM also bears the advantage of being more amenable to the study of membrane proteins, as detergent micelles are often obstructive to crystal formation (although this issue is partially overcome by advances in the usage of lipidic cubic phase crystallization) (134). Approximately 30% of proteins are membrane bound (392), yet they comprise only 3% of the crystal structures and 10% of the cryo-EM structures (299). There is an increasing recognition of redox-regulated mechanisms that drive ion channel conductance in response to oxidative stress (26, 398). A distinct lack of structural information regarding redox-regulated ion channels could be addressed by the expanding prevalence of cryo-EM application in the structural biology field, with a shortage of trained users and affordable access to high-quality electron cryomicroscopes as a current bottleneck (390).
C. Genetically encoded redox biosensors
To assess the biological implications of redox-related processes, it is critical to be able to monitor specific redox species and changes in their levels, preferably with a subcellular resolution. One set of methods involves the use of cell-permeable dyes that react with cellular ROS, leading to fluorescence, of which the signal intensity is proportional to the ROS level (116). Traditional dyes such as 2′,7′-dichlorofluorescin diacetate (DCFDA) and its derivatives are plagued by several problems: (i) they lack specificity toward the different forms of ROS, (ii) their subcellular targeting is not specific, (iii) they do not directly target the H2O2 levels, (iv) their reactions are irreversible, (v) the nonspecific reactions of the probes may affect their signal with misleading interpretation of the results, and (vi) as they are intensiometric, differences in dye uptake between different cells could be misinterpreted as differences in ROS levels (27, 399). New generations of H2O2 probes based on boronate caging of the fluorophore have partially helped to overcome these problems. Such dyes are highly specific toward H2O2, can be targeted to different organelles, and display different fluorescence spectra (73 –75, 238), however, reversibility remains a problem.
Redox-sensitive fluorescent proteins have helped to overcome limitations presented by chemical dyes and offer a powerful method for monitoring levels of redox species, as well as investigating the dynamics of protein oxidation and reduction in vivo. Fluorescent proteins, such as green fluorescent protein (GFP), are rendered redox sensitive (roGFP) by the introduction of two cysteine residues onto the surface of the β-barrel. Upon oxidation, the two cysteines form a disulfide bond that induces slight conformational changes, which in turn change the protonation state of the fluorophore, leading to a simultaneous increase of the excitation peak at 400 nm and a decrease of the excitation peak at 480 nm, that is, to a ratiometric response. This property renders the probe readout independent of its concentration or expression levels, which may vary between different cell types and compartments (77,128). Moreover, the fact that the fluorophore is shielded by the β-barrel makes the probe pH independent in the physiological range (328). However, the roGFP probe itself is not specific for the oxidant source and it is not very sensitive or rapidly responding to changes in the cellular redox status.
To overcome these limitations, roGFP is fused, via a short peptide linker, to proteins that are more sensitive and selective to the oxidative source. Mostly, the low-molecular-weight (LMW) thiol-specific redoxins (e.g., glutaredoxins, mycoredoxins, and bacilliredoxins) are used to detect changes in the intracellular LMW thiol redox balance or yeast thiol peroxidases that directly detect changes in the H2O2 levels (19, 125, 126, 222, 246). The intramolecular disulfide formation in roGFP is slightly different in both fusions: for the LMW thiol redox probes, the LMW thiol is transferred from the LMW thiol redoxin to roGFP, whereas for the thiol peroxidases, a disulfide exchange occurs (oxidant receptor peroxidase 1 [Orp1]) or as an yet-to-be-confirmed mechanism (thiol-specific antioxidant 2 [Tsa2]) (19, 125, 126, 222, 246). Furthermore, another application of roGFP2 fusions is seen in a recent study, in which redox catalysis was investigated (351). Here, roGFP2 was fused to the Prx5-type model enzyme antioxidant protein (AOP) from Plasmodium falciparum, and expressed in cells. Kinetic data showed a clear correlation between the roGFP2 readouts and the recombinant PfAOP k cat app values.
Another group of genetically encoded redox probes is fused with a circularly permuted yellow fluorescent protein (cpYFP). By coupling it to Trx and to the stereospecific methionine sulfoxide reductases (MsrA or MsrB), cpYFP is used to detect high stereospecific methionine sulfoxide oxidations (367). The first example of a cpYFP probe (designated HyPer) for detection of changes in intracellular H2O2 levels is HyPer (14). In HyPer, the cpYFP probe is integrated into the C-terminal regulatory domain (RD) of the Escherichia coli oxidative receptor OxyR, in the region between the peroxidatic and resolving cysteine (CR) (14). In contrast to the roGFP probes, the modification in the cpYFP fluorometric properties is not due to an intramolecular disulfide in the probe, but to the conformational changes in OxyR upon disulfide formation that induce structural modifications in cpYFP (14). Upon oxidation with HyPer, the 420-nm excitation peak decreases and the 500-nm peak increases (14).
New versions of HyPer have been developed with an improved dynamic range or with a different fluorescent signal, and in some cases combined with a fully genetically encoded system for localized H2O2 production by the activation of the yeast
The probes described in this study are ratiometric, allowing an efficient readout, regardless of the probe expression level. As of today, the roGFP-Tsa2 probe is by far the most sensitive H2O2-sensing probe reported, in agreement with the high reactivity of Prxs (107 M −1·s−1) (246). HyPer and roGFP-Orp1 have a similar H2O2 sensitivity (105 M −1·s−1), with HyPer responding slightly faster than roGFP-Orp1 (126). In contrast, both HyPer and roGFP-Orp1 return more quickly to the resting state than roGFP-Tsa2 (246). A general disadvantage of HyPer over roGFP-based probes is the intrinsic sensitivity of the cpYFP chromophore to pH changes. A H2O2-insensitive probe, designated SypHer2, has been generated as a control to tackle this problem (233, 336).
A general concern of these probes is their possible interference with H2O2 homeostasis, because they possess an antioxidant activity. However, the roGFP2-Tsa2 probe shows a very low H2O2-scavenging capacity, whereas HyPer does not seem to affect redox-dependent physiological reactions (22, 246). Another concern is related to the role of peroxidases, which act as H2O2 signal transducers (70, 345, 353), as the introduced thiol peroxidase in roGFP probes might lead to the activation of H2O2-signaling pathways. With HyPer, these secondary effects are less likely to happen in eukaryotic cells, due to the bacterial origin of the OxyR-regulatory region. From a more practical approach, usage of genetically encoded redox biosensors may be limited to cells that are difficult to transfect or transduce, such as primary or suspension cells; in these situations, new generations of H2O2 chemical dyes may still be the preferred option of monitoring H2O2 levels. Moreover, currently all ratiometric fluorescent probes (roGFP and cpYFP) have similar excitation spectra, which compromises such applications as the simultaneous monitoring of H2O2 in different compartments of the same cell, for example. However, the recent development of a red fluorescent H2O2 probe, HyPer Red (82) and of a redox-sensitive red fluorescent protein, rxRFP (87) could be employed together with cpYFP and roGFP2-based biosensors for this purpose.
III. Downstream Redox Regulation: Coordinating an Oxidant Response Through Redox-Sensing Transcription Factors
The ability of a cell to survive under oxidative stress conditions depends on its ability to rapidly adapt its transcriptional response to fill itself with antioxidant enzymes (Fig. 1). This responsive capacity is best displayed in prokaryotes and it is the cornerstone that underpins the survival of pathogenic bacteria upon exposure to oxidants released by the mammalian immune system. In this study, we discuss two redox-regulated prokaryotic transcription factors, OxyR and the multiple antibiotic resistance regulator (MarR), which exemplify differing, yet effective, modes of oxidant-induced structural changes that modulate their association to cognate DNA. In comparison, redox-regulated mammalian transcription factors, such as the STAT family, are less effective in their direct response to cellular oxidants and, instead, rely on redox-sensitive signaling effectors, such as kinases or peroxidases to mediate an oxidative stress reaction.
A. Signal transducer and activator of transcription 3
STAT proteins are transcriptional regulators of signaling factors involved in cell survival and proliferation (64). Upon phosphorylation by kinases, such as Janus kinase 2 (JAK2), the cytoplasmic STAT3 dimerizes and translocates to the nucleus to activate transcription (64). Tyrosine-phosphorylated STAT3 plays a role in the control of ROS production through downregulation of mitochondrial proteins of the electron transport chain, thereby reducing ROS leakage (71), and upregulation of the expression of ROS-consuming enzymes, such as mitochondrial SOD (257). STAT3 contains multiple oxidant-sensitive cysteines and under oxidative stress, it oligomerizes through intermolecular disulfides at its DNA-binding domain (DBD) and C-terminal transactivation domain (C-TAD) to form dimers, trimers, and tetramers, which inhibit DNA binding (208, 209). As STAT3 also exerts a regulatory effect on the activities of the electron transport chain complexes, STAT3 oxidation has been postulated to promote its regulatory role in mitochondria (333, 359, 360). Mitochondria-localized STAT3 associates with cyclophilin D (CypD) upon oxidation in an interaction proposed to stabilize the oxidized STAT3 form (234). JAK2 is also sensitive to oxidative inactivation, because oxidation of two cysteines in the “N-lobe” of the catalytic site triggers enzymatic inactivation (344). These cysteines are located 9 Å apart, but whether intramolecular disulfide bond formation or sulfenylation alone mediates the reversible inactivation of this kinase is still unknown (79). As a STAT3 activator, the redox sensitivity of JAK2 adds a supplemental, indirect mode of redox regulation to STAT3. A direct redox relay between a Prx and STAT3 has been identified and will be discussed in the Thiol peroxidase redox relays section (345).
B. Oxidative stress regulator OxyR
OxyR is a prokaryotic transcription factor that regulates several genes important for the cellular redox control, such as catalases and ferritins (148, 423). OxyR is considered to be an important resistance factor against the oxidative burst of innate immune responses due to its influence over virulence factors, such as biofilm formation or secretion systems (33, 162, 229, 330, 394). It belongs to the LysR transcription-type regulator family, of which the members contain a conserved N-terminal DBD, and a C-terminal RD. OxyR contains two conserved cysteines in the RD that are separated by nine amino acids within the sequence and ∼15 Å spatially (53, 161, 315, 358). The N-terminally conserved cysteine is located in a hydrophobic pocket and is highly reactive toward H2O2, with a rate constant of 105 M −1·s−1 (53, 161, 194, 315, 358). Upon oxidation, the nascent sulfenic acid is believed to induce unfolding of the α-helix, allowing the C-terminally conserved cysteine to come in the proximity to form an intramolecular disulfide bond (53, 194, 358). The changes in the tertiary structure of OxyR due to the intramolecular disulfide are translated into the quaternary structure. OxyR is a tetramer in solution and its RDs form tightly associated homodimers (with a buried surface area of >2000 Å2) (53, 358). In the E. coli OxyR, disulfide bond formation provokes a relative rotation of 30° between the protomeric subunits of the RD homodimer, which has an allosteric effect on the DBD subunits and so affects the DNA contacts and the interaction with the RNA polymerase, thereby modulating transcription (Fig. 4) (53).
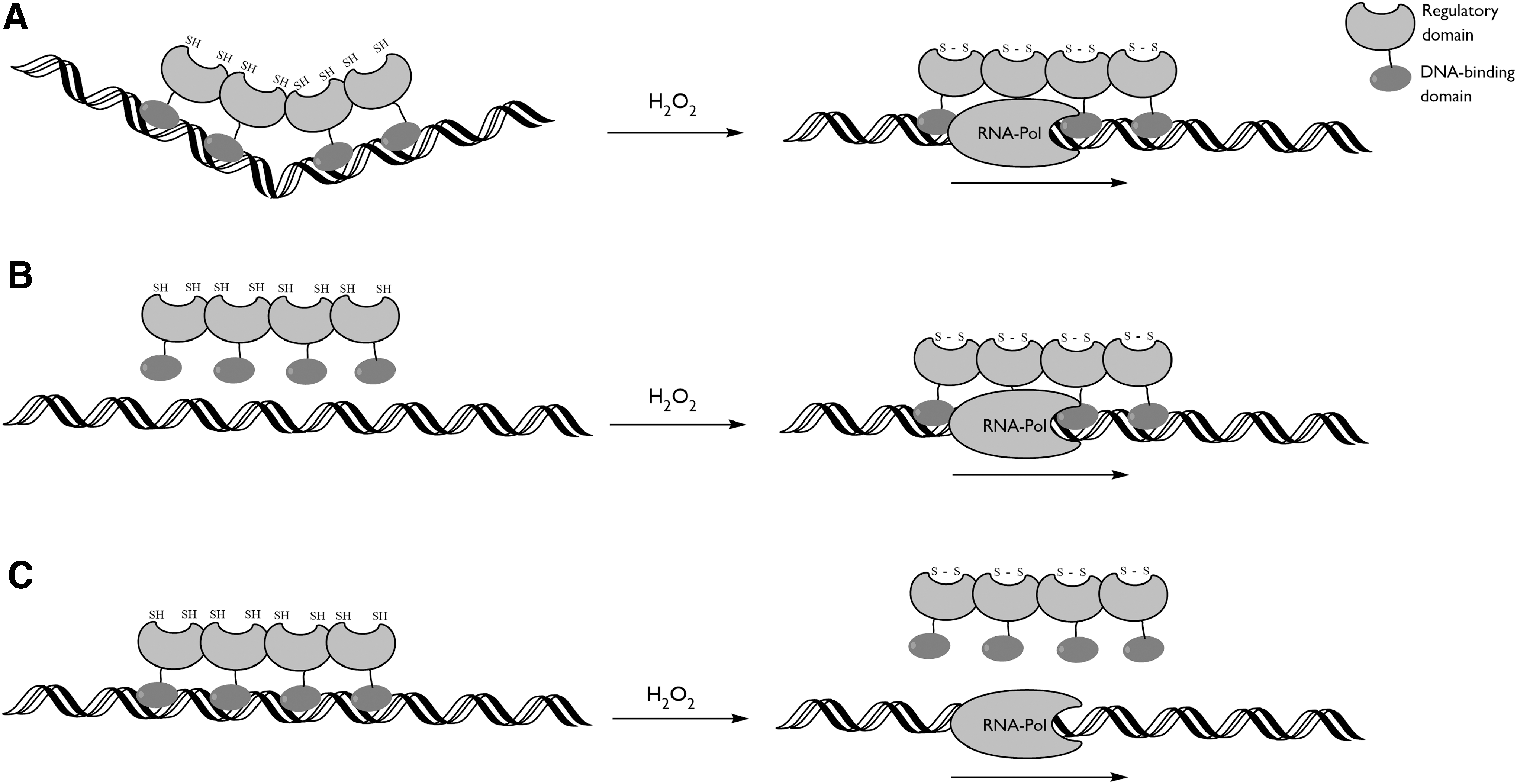
In contrast, the RD of OxyR from Porphyromonas gingivalis displays only a very small interprotomer rotation upon disulfide formation and both the reduced and oxidized conformations resemble the oxidized RD of E. coli OxyR (358). This demonstrates that the redox-induced conformational changes exerted by the peroxidatic cysteine (CP)-CR disulfide switch off are not conserved among OxyRs. Accordingly, the modifications in the OxyR-binding topology and association with RNA polymerase upon oxidation are quite variable among OxyRs from different species. For instance, when binding to the oxyRS promoter region, the reduced E. coli OxyR binds two pairs of adjacent major grooves separated by one helical turn, provoking DNA bending, whereas the oxidized OxyR binds to four consecutive major grooves, eluding DNA bending (374). A similar mechanism is observed in the oxyR2-PrxII promoter region of the bacterium Vibrio vulnificus (178). The genes in oxyRS in E. coli and oxyR2-PrxII in V. vulnificus are divergently transcribed, possibly because these regions share a redox-related transcriptional control. In the binding to the alkyl hydroperoxide C (ahpC) promoter region of E. coli, the reduced and oxidized E. coli OxyR factors have the same DNA contacts, but the oxidized OxyR has an increased affinity and activates ahpC transcription (374). In other cases, OxyR acts as a repressor, although the binding mechanism has not been studied in detail (176, 370, 381). In any case, the conformational changes induced by oxidation and intramolecular disulfide bond formation have dramatic effects on the binding affinity of the OxyR toward its cognate promoter regions, resulting in an altered transcriptome (Fig. 4). In all cases, the allosteric control of the DBD is conferred through oxidative modifications of the RD, with the presently held hypothesis that the localized structural changes produced by intramolecular disulfide formation induce a subtle alteration in the quaternary structure that translates into a change in the relative DBD orientation.
C. MarR family
The MarR family comprises a wide variety of species-specific transcriptional regulators, some of which are responsive to intracellular oxidants. MarR family proteins are homodimers that contain a structurally conserved winged helix-turn-helix (wHTH) DNA-binding motif on each protomer (4). Typically, they act as transcription repressors through steric hindrance of the RNA polymerase to a promoter, although some family members act as activators by either competitive exclusion against repressors or by stabilizing the RNA polymerase (377). MarR proteins possess a dimerization region and form relatively compact homodimers through intercalation of the α1, α5, and α6 helices, all while leaving the wHTH free for DNA recognition (29). In MarR-type structures, the dimerization domain functions as a hinge and any movement in the dimeric interface communicates an equivalent shift in the relative distance between the partnered wHTH DNA-binding motifs, triggering a structural synchronicity in which the MarR protein specificity and affinity toward DNA are modulated (186, 424). Despite their origin from a common ancestor, MarR family proteins have evolved multiple distinct redox regulation mechanisms.
The MarR-type transcription regulator, organic hydroperoxide resistance regulator (OhrR), adopts species-specific modes of redox regulation, depending on the presence of a single (typically N-terminal) cysteine, or two or more cysteines. The 1-Cys variant OhrR of Bacillus subtilis is responsive to oxidative modification by LMW thiol conjugation. Sulfenylation of B. subtilis OhrR does not weaken its DNA-binding affinity, instead, mixed disulfide formation (with bacillithiol, coenzyme A, or cysteine) or condensation with the amide backbone to a sulfenamide is required for derepression (199). The 2-Cys variant of Xanthomonas campestris OhrR also has an oxidant-sensing N-terminal cysteine (Cys22) that, upon oxidation, engages in an intersubunit disulfide bond with a CR (Cys127) of the neighboring protomer (270). It should be noted that referring to such OhrRs as “2-Cys” does not preclude the existence of more than two cysteines; X. campestris OhrR has an additional cysteine in the proximity of Cys127 (Cys131) that is not considered functionally significant. Structural characterization of the reduced and oxidized forms of X. campestris OhrR revealed that, upon oxidation of Cys22 to sulfenic acid, a hydrogen bonding network involving neighboring tyrosines is disrupted allowing a 135° rotation and 8.2 Å translation of Cys127 of the partner protomer to engage in a disulfide (258).
This localized structural transition results in a striking reorganization of the dimerization interface, whereby the stacking arrangement of the α6-helices' interface is effectively swapped, reversing the helix polarity while maintaining the pseudo twofold symmetry and overall triangular shape of the homodimer (Fig. 5). The rearrangement of the dimerization region conveys a 28° rotation of the wHTH domains, weakening the affinity of the oxidized OhrR for its target promoter. Whereas the C-terminal CR of X. campestris OhrR appears to be essential for oxidative regulation of its DNA-binding capacity, the C-terminal cysteines of other 2-Cys orthologs, such as the OhrR of Pseudomonas aeruginosa or Chromobacterium violaceum, are dispensable for oxidative derepression and seemingly act as a protection against overoxidation (9, 61). In the case of the P. aeruginosa OhrR homolog, OspR (bearing 46.5% identity to OhrR), oxidation of the N-terminal cysteine is sufficient to abolish its affinity to the ohr promoter, but both N- and C-terminal cysteines are required for oxidative regulation of its affinity to the glutathione peroxidase (gpx) promoter (9). The usefulness of a C-terminal CR in OhrRs in the protection against overoxidation of the N-terminal Cys has been demonstrated in the conversion of the 1-Cys B. subtilis OhrR into a 2-Cys regulator through introduction of a C-terminal cysteine either at position 120 (G120C) or 124 (Q124C), placing these CR at 14.1 or 13.3 Å Sγ-Sγ distance, respectively, from the N-terminal Cys15 of the sister subunit (347). Both CR variants effectively conferred the reversible oxidative regulation of promoter repression by OhrR. The recently characterized Staphylococcus epidermidis MarR family regulator, the aggregation and biofilm formation regulator (AbfR), senses organic peroxides via the 2-Cys interprotomer mechanism (220), yet AbfR does not undergo the α6-helix swapping that occurs for the X. campestris OhrR, but, instead, a slight “twisting” of the dimer interface is induced (219).
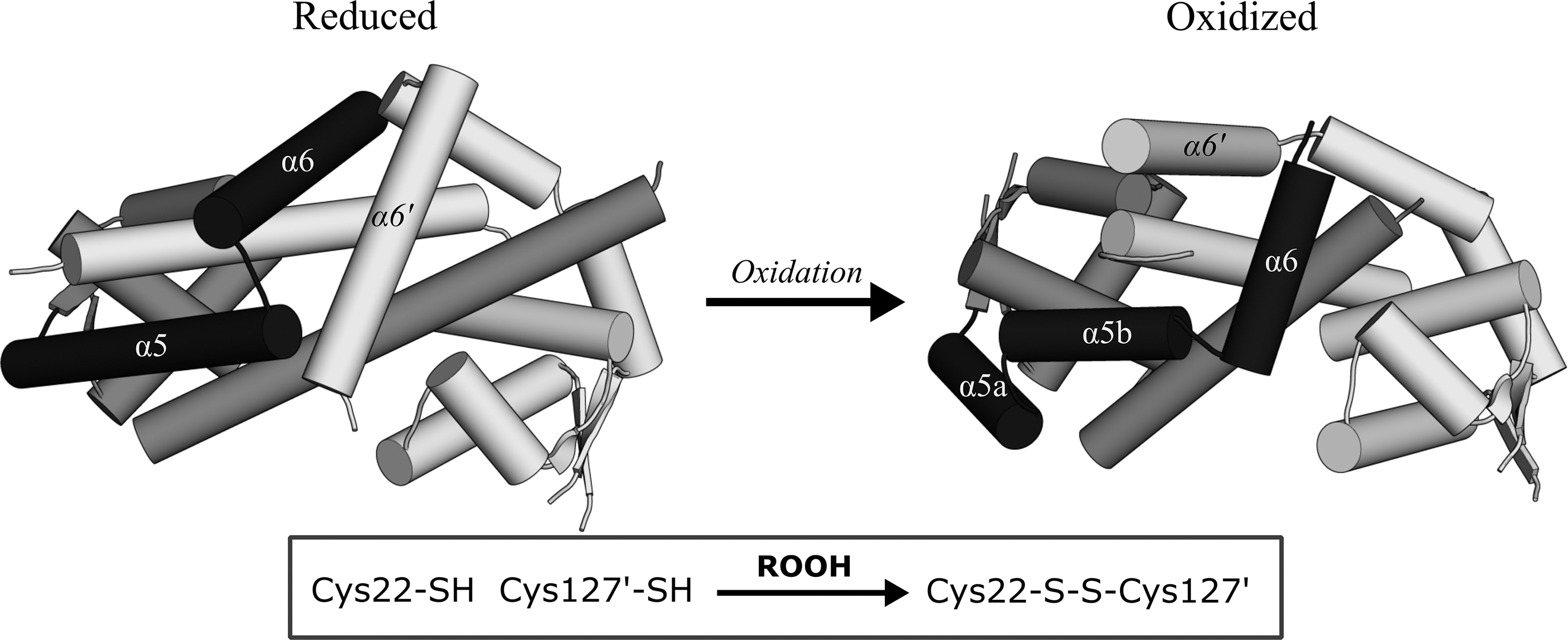
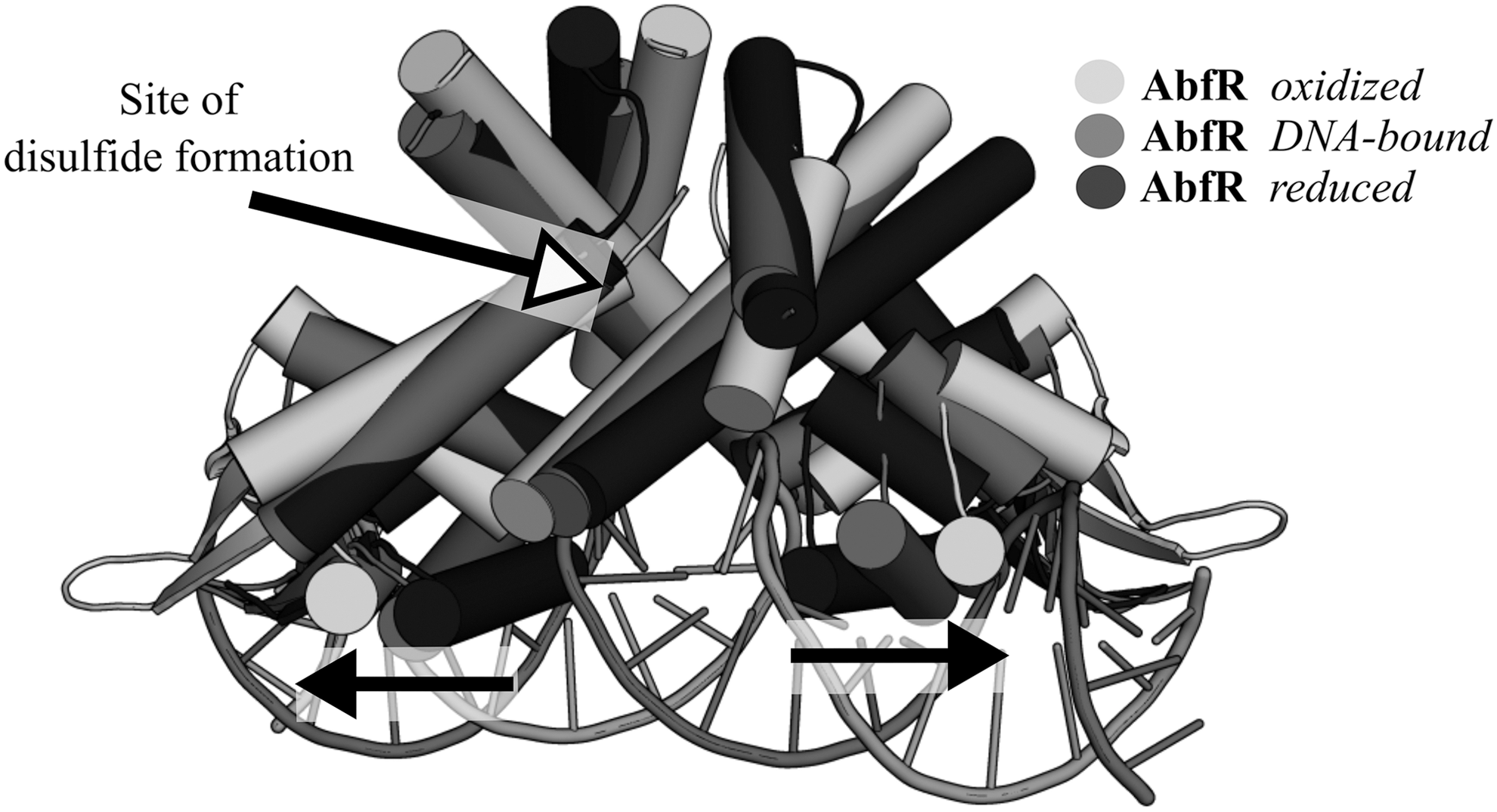
Despite the less dramatic structural changes in the AbfR dimerization region than that of the X. campestris OhrR, a much larger rigid-body transition is transmitted to the wHTH domain of AbfR, with the interprotomer gap between the wHTH DNA-recognition helices (α4) widening by >11 Å in the oxidized form relative to the reduced DNA-bound form due to a marked rigid-body rotation and translation of the wHTH (Fig. 6). AbfR has been shown to sequentially form its cross-subunit disulfides, whereby a population of 1-disulfide crosslinked dimers progresses to a 2-disulfide form with increasing concentrations of organic peroxide (219). AbfR oxidation to a 1-disulfide or 2-disulfide state reduces its affinity toward the abfR operator site by 10-fold and 20-fold, respectively, indicative of two allosteric regulation stages that might be related to independent conformational changes occurring separately in each protomer (219).
Among the MarR family, alternate 2-Cys mechanisms of redox regulation have also been detected. The P. aeruginosa multidrug efflux regulator (MexR) does not possess the conserved N-terminal cysteine of OhrR, but, instead, forms an interprotomer disulfide between a cysteine at the C-terminus of the α1-helix (Cys30) and a cysteine on a loop following the α3-helix (Cys62) of the opposing protomer (47). Cys62 is thought to form first a sulfenic acid, which leads to localized disruption of hydrogen bonds, allowing an 8 Å translation toward Cys30. The accompanying structural change is relatively localized, leading to a modest conformational shift in the wHTH regions, without changes in the distance between the DNA major-groove recognition helices. The overall conformations of reduced and oxidized forms of MexR remain largely the same and the structures align with a root-mean-square deviation of 1.6 Å (48). The decrease in DNA affinity is proposed to derive mainly from the steric hindrance introduced by the intermolecular disulfide (48). In addition to the intermolecular 2-Cys mechanisms outlined above, intramolecular disulfides have also been found to regulate some MarR family proteins, as in the case of an OhrR homolog of Mycobacterium tuberculosis, oxidation-sensing regulator, MosR.
In MosR, the conserved N-terminal oxidant-sensing cysteine (Cys12) forms an intramolecular disulfide with Cys10 upon oxidation, leading to transcriptional derepression (32). A markedly different mode of oxidation-induced intrasubunit linkage has been suggested for the bleach-sensing MarR family protein of E. coli, the N-ethylmaleimide regulator (NemR). NemR is specifically sensitive to both electrophiles and reactive chlorine species (RCS) and regulates its own expression and that of N-ethylmaleimide reductase (NemA) and glyoxalase (GloA) (195). Only one of the six cysteines of NemR, Cys106, is thought to be responsible for RCS sensing and an oxidation-induced sulfenamide bond has been suggested to form between a sulfenylated Cys106 and the Lys175 amino group (118). In addition to the intersubunit and intrasubunit disulfide/sulfenylamide mechanisms of oxidative regulation of the MarR-type transcription factors, an interdimer system of disulfide regulation has also been characterized for the Burkholderia thailandensis biofilm regulator (BifR) and the E. coli MarR. Both the E. coli MarR and BifR undergo interdimer disulfide linkages to form tetramers upon Cu(II) oxidation, but via distinct, nonconserved cysteines. Whereas disulfide (and subsequent tetramer) formation by MarR inhibits DNA binding, oxidation actually increases the affinity of BifR toward DNA (123, 424).
IV. Midstream Redox Signaling by Moonlighting Proteins
Transcription factors sensitive to oxidants typically regulate the expression of antioxidant enzymes or proteins involved in coordinating cell fate (66, 217). Although such transcription factors exert a significant level of control over a cellular response to oxidative stress, they also limit the potential scale and complexity afforded by a signaling network through acting as simple on/off switches. By possessing an additional “midstream” level of constitutively expressed redox-sensing proteins beyond the transcriptional downstream effectors, cells can form a more complex and sensitive network of redox regulation, complimentary to the core antioxidant response. By virtue of being constitutively expressed, these midstream effectors bypass the need for protein synthesis before their regulatory effects are exerted. To fulfil the role of a redox-regulated midstream effector, the candidate protein should have a cellular function, distinct from any redox-related property, thereby justifying its expression under basal conditions. Furthermore, the candidate protein should be susceptible to reversible oxidation, and oxidation should alter its cellular role or subcellular localization in some way. In this study, we outline candidate proteins which we consider to fulfil this function, and therefore would constitute a level of midstream redox signaling, which shapes the cellular response to oxidative stress. The ability of such proteins to adopt a subfunction in response to redox stimuli compels us to refer to them as redox-regulated moonlighting proteins.
A. Human protein deglycase DJ-1
Human protein deglycase DJ-1 (protein deglycanase-1) is a member of the DJ-1/Hsp31/PfpI superfamily with a wide range of predicted cellular roles, including regulation of RNA–protein interactions (139), glyoxalase activity (200), chaperone function (334), cysteine protease activity (49), RNA binding (384), esterase activity (385), and transcriptional coactivation (410). It occurs preferentially as a noncovalent homodimer of 20-kDa protomers and contains an active site consisting of a catalytic triad of Glu18, Cys106, and His126, which confers glyoxalase and cysteine protease activity (49, 200). In vivo, DJ-1 is responsive to oxidative stress (242, 243) and Cys106 is considered to be the key redox-sensitive residue (37, 181, 317). A 1.2 Å crystal structure of DJ-1 revealed that in the absence of oxidizing agents, Cys106 is overoxidized to a sulfinic acid (37). DJ-1 displays redox-dependent esterase activity that apparently depends on the Cys106 oxidation state (385). Whereas the esterase function of DJ-1 is abolished by thiol alkylation, it is enhanced by exposure to H2O2 or a C106D sulfinylation mimic.
Human DJ-1 contains two additional cysteines, Cys46 and Cys53, and although both are oxidizable upon H2O2 treatment, they are significantly less sensitive to oxidants than Cys106 (181). Cys53 is located at the homodimerization interface of DJ-1 and is purported to form an intermolecular Cys53-Cys53 disulfide under oxidizing conditions to facilitate homodimer stabilization (89); there is also evidence suggesting that this disulfide is a reduction target for Trx (103). A further interaction between DJ-1 and another antioxidant enzyme, PrxII, has been observed, also involving Cys53 of DJ-1 (89). DJ-1 has been demonstrated to form a mixed disulfide with PrxII via Cys53 under oxidizing conditions, possibly resolving itself into an intermolecular disulfide-bridged DJ-1 dimer (89).
DJ-1 oxidation has been found to promote its relocalization to mitochondria, an event associated with neuroprotective effects (37). Null mutation of DJ-1 causes an aberrant mitochondrial phenotype in human cells that can be rescued by infection of the human cells by viral particles expressing the wild-type protein or by administering antioxidants, such as N-acetyl-
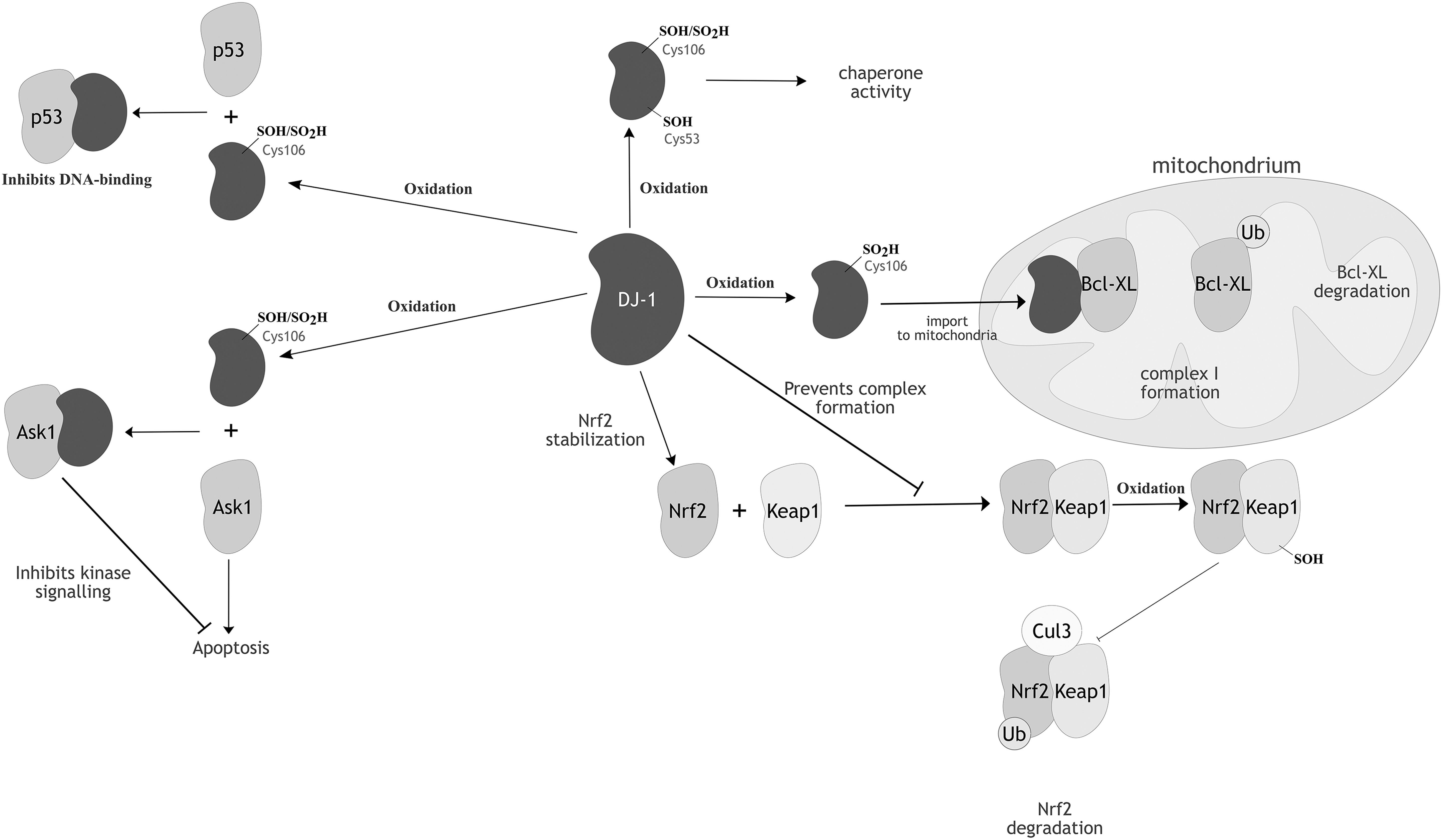
DJ-1 confers cytoprotectivity to oxidatively stressed cells by interfering with the c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) apoptotic pathways, through a redox-dependent interaction with an upstream kinase of the JNK/p38 signaling pathway, the apoptosis signal-regulating kinase 1 (ASK1) (373). Under oxidizing conditions, ASK1 oligomerizes via intermolecular disulfide bridges to form HMW complexes of >3000 kDa, constituting an active ASK1 signalosome (253). ASK1 contains 23 cysteines, of which none is essential for the covalent complex linking. Only the multiple mutation of seven cysteines across the three ASK1 domains (the N-terminal, kinase, and C-terminal domains) abolished the ability of ASK1 to form disulfide-linked multimers (253). Upon oxidation, DJ-1 can engage in a mixed disulfide with ASK1 through its oxidant-sensitive Cys106 (391). Complex formation between ASK1 and DJ-1 inhibits the kinase function of ASK1, thereby suppressing the JNK/p38 apoptosis pathway under oxidizing conditions (Fig. 7). The peripheral Cys53 of DJ-1 might act as a CR, driving the dissociation of the mixed disulfide, but this potential role has not been characterized in vitro. Crucially, whereas mildly oxidizing conditions promote the ASK1-DJ-1 complex formation, overoxidation of DJ-1 leads to its dissociation from ASK1, implying that this regulatory relationship is highly sensitive to cellular redox conditions (38).
DJ-1 also indirectly suppresses ASK1 by sequestering the death-associated protein Daxx and preventing it from ASK1 activation (166). In addition to its redox-regulated ASK1 modulation, DJ-1 also interacts with and inactivates MAPK kinase kinase 1 (MEKK1), another upstream JNK kinase that protects against UV-induced cell death. However, this interaction does not involve the redox-sensitive Cys106 of DJ-1 (10). DJ-1 intersects with another kinase pathway, the phosphatidylinositol 3′ kinase pathway, through its role as a negative regulator of phosphatase and tensin homolog (PTEN) (177). DJ-1 has been suggested to mediate STAT1 dephosphorylation by promoting its interaction with the sarcoma (Src) homology 2 domain-containing protein tyrosine phosphatase 1 (SHP-1) (175).
DJ-1 influences the cellular antioxidant response via its association with the transcriptional regulator, nuclear factor-erythroid 2 p45-related factor 2 (Nrf2) (147). Nrf2 is the transcriptional activator of antioxidant response elements and, hence, mediates the expression of various antioxidant enzymes (151, 268, 388). Although the exact mechanism by which DJ-1 positively regulates Nrf2 activity is not well understood, it might prevent the binding of the inhibitor Kelch-like ECH-associated protein 1 (Keap1) to Nrf2 (Fig. 7) (56). Keap1 is a cysteine-rich (27 cysteines in the human isoform), 70-kDa protein that, under basal conditions, binds to Nrf2 and facilitates the association of the adapter component of the cullin 3 (Cul3)-based ubiquitin E3 ligase complex, leading to ubiquitination and proteasomal Nrf2 degradation (81). Astroglial overexpression of DJ-1 results in upregulation of the antioxidant enzymes, PrxII, Trx, and SOD1, although this could purely derive from the positive regulation of Nrf2 by DJ-1 (102).
DJ-1 has many other reported interaction targets, namely androgen receptor (262, 363), polypyrimidine tract-binding protein-associated splicing factor (PSF) (410), and p53 (86, 335). Among these targets, the interaction with p53 seems to be redox regulated, with oxidation of the Cys106 of DJ-1 essential for the interaction with the p53 DBD and subsequent repression of its transcriptional activity (170). DJ-1 also displays a chaperone activity that prevents α-synuclein aggregation, which is connected to Parkinsonism (115). Cys53 has been found necessary for this particular redox-dependent chaperone activity (334). Characterization of the chaperone properties of the prokaryotic DJ-1 homolog, YajL, revealed an extensive capacity for mixed disulfide formation with a wide range of cellular targets, extending to ribosomal proteins, metabolic enzymes, chaperones, transcription factors, and aminoacyl-transfer RNA (tRNA) ligases (192).
The bacterial ΔyajL mutant displays elevated levels of sulfenylated proteins, further supporting a redox-dependent protective role for YajL (109). Whereas the chaperone function of DJ-1 toward α-synuclein relies on Cys53, the covalent chaperone ability of YajL involves Cys106, consolidating the importance of this residue for the redox-dependent function of DJ-1/YajL. For human DJ-1, Cys106 is probably the predominant residue mediating mixed disulfide complex formation with various cellular partners and the amount of covalently captured complexes is enhanced by a C53A mutation, possibly through promotion of a DJ-1 monomeric state more prone to oxidative interactions (89). One protein identified in separate studies to form a mixed disulfide complex with both human DJ-1 and the bacterial homolog YajL, is another redox-regulated moonlighting protein, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (89, 192). DJ-1 also interacts with GAPDH in an enzymatic manner, deglycating inactive glycated GAPDH (196, 304), and in human neuroblastoma cell lines was observed to form HMW complexes containing GAPDH and proteins related to RNA metabolism (285).
B. Glyceraldehyde 3-phosphate dehydrogenase
GAPDH is a key enzyme of the glycolytic pathway, catalyzing the reversible phosphorylation of glyceraldehyde 3-phosphate to glycerate 1,3-bisphosphate with concomitant generation of reduced nicotinamide adenine dinucleotide (NADH). It is one of the most abundant intracellular proteins and, besides its role in glycolysis, performs many autonomous and often unrelated functions in multiple cellular compartments (342, 343, 378). A conserved cysteine at the active site (Cys152 in human GAPDH) acts as a reactive nucleophile and is essential for the catalytic function (31, 277). The Cys152 thiolate is highly sensitive to oxidative modifications, and, for a nonperoxidatic thiol, has a remarkably high rate constant for its reaction with H2O2 (10–103 M −1·s−1) (215, 403, 418). Mathematical models predict that GAPDH is the first protein to become oxidized, once the dedicated thiol peroxidases are inactivated by overoxidation (above 10 μM H2O2) (403). Experimental observations are in agreement with these in silico predictions, as a 20 μM H2O2 bolus applied to Jurkat cells was demonstrated to be sufficient for GAPDH oxidation (12). H2O2-mediated oxidation of GAPDH decreases its glycolytic activity (215, 277), although it is possible to uncouple the redox sensitivity of GAPDH from the oxidative inactivation of the enzymatic activity through mutations that reduce oxidant sensitivity (e.g., C156S and Y314F) (277). Inactivation of GAPDH disrupts the glycolytic pathway, rerouting the carbohydrate flux to generate pentose phosphate pathway metabolites with concomitant generation of NADPH (295). As NAPDH is the upstream electron donor of the thiol–disulfide regeneration systems, such as thioredoxin reductase (TR) or glutathione disulfide reductase (GR), increasing NAPDH production under oxidative stress conditions is likely to be crucial for cell survival.
As a demonstration, in yeast cells expressing human H2O2-insensitive mutants (C156S and Y314F) of GAPDH, the upregulation of NADPH production under oxidative stress is impaired and susceptibility to cell death is increased relative to wild type (277). In addition to inhibition of the catalytic activity of GAPDH, oxidation also induces aggregation of GAPDH, whereby oxidation of the surface-exposed Met46 of GAPDH is linked to protein misfolding, with subsequent aggregation through intermolecular disulfide formation (318). Neuronal GAPDH aggregation upon oxidative stress has been identified as a marker in neurological diseases (393). Although the Trx/TR system is the main reductant for oxidized GAPDH, tubulin, an abundant neuronal protein, can also engage in thiol–disulfide exchange to reduce GAPDH and, thus, specifically prevent oxidative aggregation/inactivation of neuronal GAPDH (189).
Oxidation or S-nitrosylation of the catalytic cysteine of GAPDH induces its association with Siah1, an E3 ubiquitin ligase protein involved in proteasome-dependent protein degradation (130). Binding of GAPDH to Siah1 promotes localization of GAPDH to the nucleus, where it influences the apoptosis pathway and the enzymatic DNA repair systems. Nuclear GAPDH promotes acetylation and phosphorylation of p53 in the absence of the poly A-binding protein, stimulating translocation of p53 to mitochondria, where it participates in apoptosis initiation (371). GAPDH has also been implicated in mediating the upregulation of p53 expression (420) and, in turn, p53 has been shown to upregulate the mRNA transcription of GAPDH and Siah1 (50, 93). ASK1 augments the interaction between GAPDH and Siah1 and phosphorylates threonines of Siah1 to activate its nuclear signaling pathway (379), whereas H2O2 treatment has been shown to increase the association of ASK1 to GAPDH-Siah1. Nuclear-translocated GAPDH interacts with the base excision DNA repair enzyme, apurinic/apyrimidinic endonuclease 1 (APE1), and reduces the oxidized APE1 in a manner dependent on the redox-sensitive GAPDH cysteines, Cys152 and Cys156 (10). APE1 itself has redox-dependent functions, capable of reactivating the extracellular signal-regulated kinase (ERK) activity via its oxidant-sensitive cysteine Cys65 (397) and stimulating p53 DNA binding in a redox-dependent manner (60). Oxidation of human GAPDH induces an interaction with the RNA-binding proteins, PSF and 54-kDa nuclear RNA-binding protein (p54nrb), to form a complex that enhances the topoisomerase I activity (146), and this interaction is dependent on the presence of the catalytic Cys152 site of GAPDH. H2O2 treatment of human cell lines has been shown to result in the formation of a mixed disulfide between GAPDH and either PrxI or PrxII (353), with Prx presumably mediating the reversible oxidation of GAPDH thiols, and a proteome-wide study of H2O2-sensitive protein thiols of Arabidopsis chloroplasts confirmed that GAPDH undergoes reversible thiol oxidation (250). Among the 16 other oxidant-sensitive stromal proteins identified were 2-Cys Prx and the peptidyl-prolyl cis–trans isomerase (PPI) enzyme, cyclophilin 20-3 (Cyp20-3) (250).
C. Peptidyl-prolyl cis–trans isomerase
The Arabidopsis PPI, Cyp20-3, is a multifaceted protein, and in addition to its PPI activity, plays a putative role in facilitating protein unfolding and degradation under stress (308). Upon oxidation, Cyp20-3 undergoes a large conformational change to form two sets of intramolecular disulfides, Cys54-Cys171 and Cys129-Cys176 (191). Cyp20-3 oxidation inhibits its PPI activity (191, 248) and has been suggested to induce a regulatory function in cysteine synthesis, as under oxidative stress conditions Cyp20-3 interacts with the cysteine synthase complex to activate cysteine synthesis (76). Despite its low redox potential (E m = −319 mV), Cyp20-3 can be maintained in a reduced state by Trx-m (Em = −357 to 368 mV) (57) and might also be a reductant of 2-Cys Prx A and Prx B (E m of −307 and −322 mV, respectively), although it is unable to enhance the peroxidase activity of 2-Cys Prxs (191, 251).
Mammalian PPIs with redox-responsive functions include the cytosolic cyclophilin A (CypA), cyclophilin B (CypB), which is localized to the endoplasmic reticulum, mitochondrial CypD, and the phosphorylation-dependent parvulin family PPI, Pin1 (peptidyl-prolyl cis–trans isomerase NIMA-interacting 1). Similarly to Cyp20-3, human CypA can also act as an electron donor for Prx II and Prx VI and has been demonstrated to enhance their peroxidase activity in vitro (203). CypA negatively regulates the JNK/p38 signaling pathway in response to oxidative stress by interacting with ASK1 and suppressing its phosphorylation (174). CypA has been observed to be secreted by vascular smooth muscle cells under conditions of oxidative stress and activates ERK1/2 in a manner dependent on its peptidyl-prolyl isomerase activity (160).
Mammalian CypD is a regulator of the mitochondrial permeability transition pore (mPTP) (329), the activation of which leads to loss of the mitochondrial membrane potential and is linked to necrotic and apoptotic cell death. CypD is activated by p53 and, in turn, induces p53 aggregation through its isomerase activity (193). CypD activation occurs in response to oxidative stress (324) and its Cys203 has been identified as the redox-sensing cysteine responsible for oxidative regulation and forms an intramolecular disulfide upon H2O2 exposure in vivo (214, 259). CypD interacts with Trx2 and PrxIII in the mitochondrial matrix (94) and accumulation of oxidized Trx2, either by small interfering RNA (siRNA) knockdown of thioredoxin reductase 2 (TR2), or inhibition of TR2 by auranofin, induces the concomitant oxidation of CypD and PrxIII, implying a level of redox interplay between these enzymes. CypD increases the respiratory activity of Complex III in HEK293 cells by promoting supercomplex formation (85). Overexpression of Cyp22, a CypD homolog of Trypanosoma cruzi, led to increased sensitivity to mitochondrial destabilization through loss of membrane potential in response to oxidative stress (34). Under conditions of oxidative stress CypD binds to mitochondrially localized STAT3 in a manner dependent on the N-terminus region of STAT3, and it is proposed that this interaction relates to reduced mitochondrial ROS production (234).
A relationship between STAT3 and CypB has also been demonstrated in cancer cells, with STAT3 repressing transcription of CypB inhibitors, and CypB in turn promoting the activation of STAT3 (210). The parvulin family PPI, Pin1, specifically targets prolines adjacent to phosphorylated Ser/Thr and makes a junction between PPIases and kinase pathways, whereas oxidation of its Cys113 to sulfenic/sulfinic acid inhibits its catalytic activity (46, 149, 411). Pin1 also intersects with cellular stress responses by targeting JNK (272), p53 (120), and p66 Src homologous-collagen homolog (p66Shc) (88). Pin1-mediated translocation of p66Shc to mitochondria leads to increased levels of mitochondrial ROS and induction of apoptosis (113). The adaptor protein, p66Shc, itself contains multiple oxidant-sensitive cysteines, and can undergo a redox-dependent transition between homodimeric and a homotetrameric disulfide-linked oligomer.
D. Transglutaminase 2
Transglutaminases (TGs) are a family of enzymes that catalyze Ca2+-dependent transamidation and (under acidic conditions) deamidation reactions on a wide range of intracellular and extracellular targets (for a comprehensive list of substrates, see the TRANSDAB database,
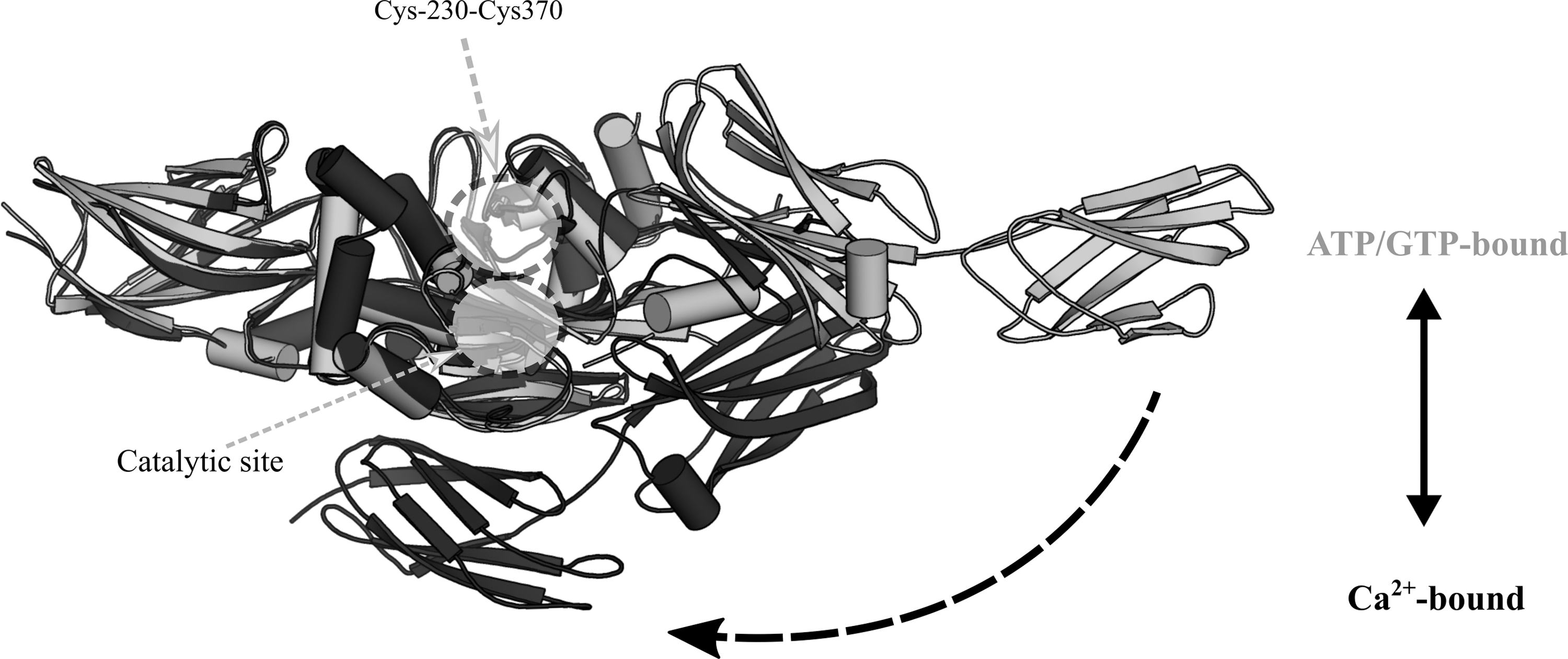
Oxidation of TG2 has been shown to induce intramolecular disulfide formation, either at Cys230-Cys370, or at Cys370-Cys371 (349). Based on site-directed mutagenesis of Cys230, a disulfide relay might occur, with first the formation of the Cys230-Cys370 disulfide and then with Cys371 presumably acting as a CR for the generation of the resulting Cys370-Cys371 vicinal disulfide. Oxidation of TG2 inhibits its transamidase activity, and mutation of Cys230 has been shown to render TG2 insensitive to inactivation by oxidation, although this was attributed to an impaired ability to form the Cys370-Cys371 disulfide (349).
TG2 can adopt either a closed ATP/GTP-bound conformation, in which state its transamidation activity is lost, or an open transamidase-active conformation stabilized by Ca2+ binding (284). Oxidation appears to influence the conformational preference of TG2, as the Cys230-Cys370 disulfide form preferentially adopts the open configuration even in the presence of GTP (Fig. 8) (349). However, the Cys230-Cys370 disulfide itself is not incompatible with the closed configuration of TG2, as proved by its presence in crystal structures of ATP and GTP-bound closed conformation TG2 (127, 156). Just as GTP inhibits the transamidase activity of TG2 by promoting the closed conformation, Ca2+ can decrease the GTPase activity of TG2 (182). TG2 has a binding K D of 1.6 μM for GTP, compared with a K D of 90 μM toward Ca2+ (15). Considering that typical intracellular Ca2+ concentrations are in the range of 100 nM and that free GTP is estimated to be ∼100 μM, the majority of intracellular TG2 is expected to be in a closed, GTP/GDP-bound conformation without transamidase activity (183). In this transamidase-inactive conformation, TG2 has been demonstrated to function as a kinase (239 –241) and as a protein disulfide isomerase (PDI) (131). As the vicinal Cys370-Cys371 disulfide has a redox potential similar to that of bovine PDI [−184 ± 4 mV (159) and −190 ± 10 mV, respectively (226)], this implicates the vicinal disulfide as a potential catalytic site for disulfide isomerization. In terms of an in vivo relevance of the PDI action, a role for TG2 in the correction of defective disulfide bonds in the respiratory chain complexes has been proposed (232).
TG2 can be secreted to the extracellular environment, where it serves as a target substrate for extracellular Trx (7, 159). Trx has been shown to be highly selective in its extracellular TG2 targeting in fibroblasts and the small intestine, although its ability to recognize TG2 in other organ environments has been questioned (7). Surprisingly, chloride intracellular channel protein 3 (CLIC3) has also been proposed to be an extracellular reductant of TG2, utilizing the catalytic cysteine of the CXXC motif of its Trx fold to perform a GSH-dependent reduction of oxidized TG2 (138). CLIC proteins adopt either a soluble globular form, structurally homologous to the glutathione S-transferase (GST) family or an oligomeric integral membrane-associated state for ion conductance. Although CLIC3 has not previously been associated with redox-dependent functions, the structure and/or function of CLIC1, CLIC2, and CLIC4 has been previously demonstrated to be altered depending on the oxidation state of the cysteines of its Trx domain (216). More recently, an extracellular oxidizing protein partner for TG2 has been identified. ERp57 (endoplasmic reticulum-resident protein 57 [also known as protein disulfide-isomerase A3, or glucose-regulated protein 58-kDa]) was observed to colocalize with TG2 in cultured human umbilical vein endothelial cells, and was capable of oxidatively inactivating TG2 at a second-order rate constant >700-fold higher than inactivation by oxidants such as H2O2 or glutathione disulfide (GSSG) (417). The dual axis of Trx and Erp57 in extracellular redox regulation of TG2 may serve to modulate extracellular transamidation, where Ca2+ and nucleotide concentrations favor a transamidase-active form of TG2, and so oxidative inhibition of its activity may have a more significant role.
V. Upstream Redox-Sensing: Prxs as Specific Sensors and Mediators of the Oxidative Stress Response
The high cellular abundance of Prxs and the superior catalytic reactivity of their peroxidatic thiols toward peroxides make them the frontline intracellular oxidant sensor, thereby placing Prx in the upstream section of oxidative signaling pathways. The proposed role of Prx as a sensor and transducer of H2O2 signaling has been consistently strengthened (125, 126, 136, 246, 277, 345, 353, 354), and in this study we consolidate the view of Prx as an instigator of oxidative signaling, and provide a further overview of the potential cellular roles of Prx and its intersection with alternate intracellular signaling pathways.
A. Relationship between redox state and Prx conformation
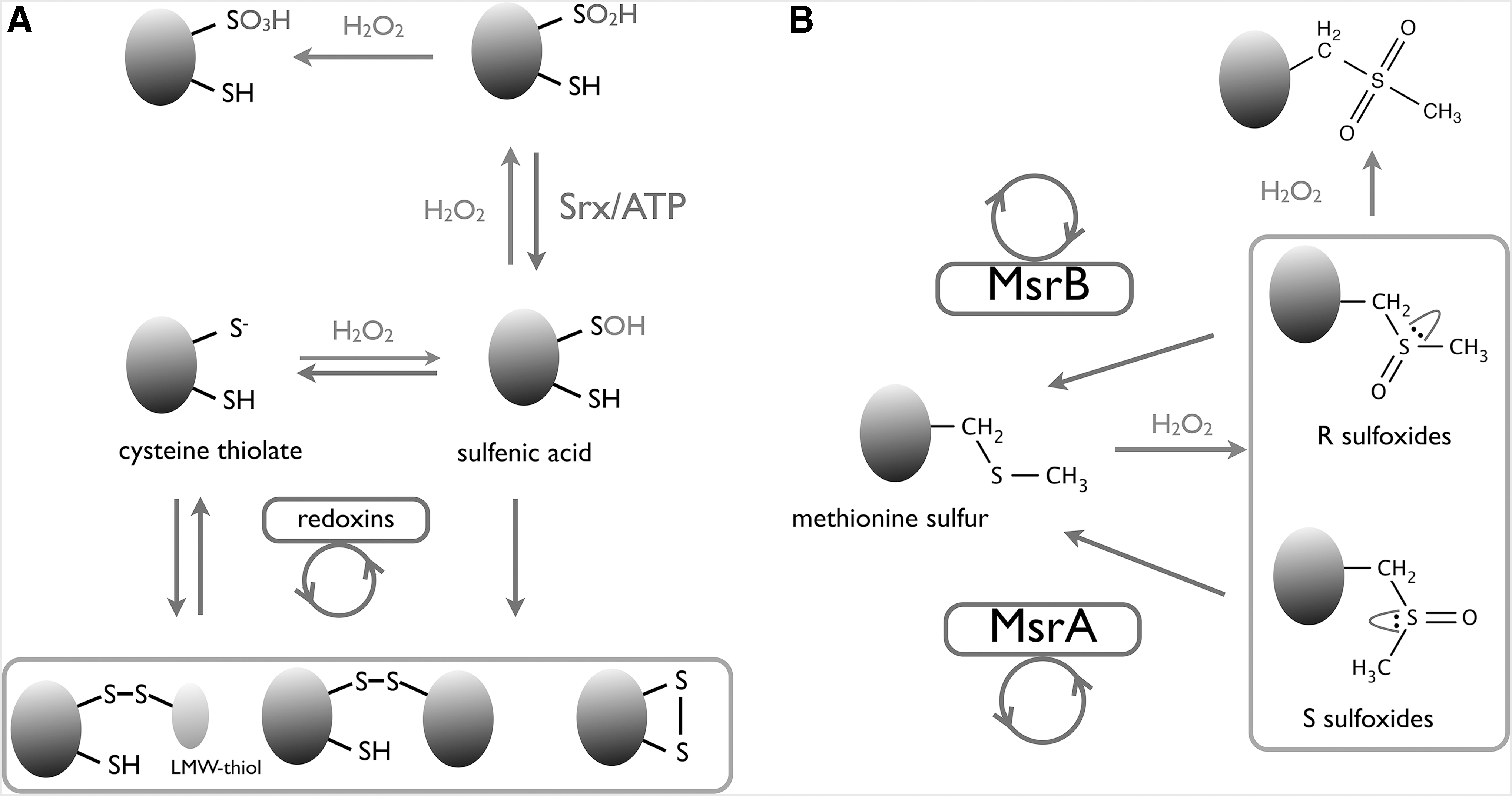
Prxs catalyze the reduction of peroxide to water/alcohol through a nucleophilic attack by a CP thiolate that in the process becomes oxidized to a sulfenic acid. In “typical 2-Cys” Prxs, the CP is spatially conserved in the first turn of α-helix 2, ∼14 Å away from the CR, which is located in the C-terminal region of the neighboring subunit of the Prx homodimer. For a disulfide to form between the CR and a sulfenylated CP, localized unfolding is required of both the α2-helix active site region and the CR-containing C-terminal tail. This process is defined as a transition from a “fully folded” (FF) to “locally unfolded” (LU) state. Recent consensus in the field of Prxs is that the FF-LU transition is in dynamic conformational equilibrium before CP oxidation (167, 168, 260, 279). The structural switch from a FF to LU conformation in Prx is an essential protective mechanism, and stabilization of the FF conformation is widely accepted to promote CP overoxidation to sulfinic or sulfonic acid, inactivating Prx until regeneration of the sulfinylated form by sulfiredoxin (Srx). Prxs that are prone to or resilient to overoxidation are designated “sensitive” and “robust,” respectively. In phylogenetic terms, eukaryotic Prxs are largely sensitive and prokaryotic Prxs robust to overoxidation (408, 415). Eukaryotic Prxs typically contain a conserved YF sequence motif in the C-terminal tail that interacts with the first turn of the α2-helix to support its packing against the active site, while obstructing the local unfolding of the active-site region (168, 322, 395).
With the exception of the monomeric PrxQ/bacterioferritin comigratory protein (BCP) (158, 212), the dimerization of Prxs is a highly conserved feature, with two distinct dimerization interfaces, designated A-type and B-type. Typical 2-Cys Prxs form homodimers through the B-type interface with protomers interacting in a head-to-tail fashion to bring adjacent β-sheets together in an antiparallel alignment (54), whereas the atypical 2-Cys Prxs dimerize via the A-type interface. Through the A-type interface, typical 2-Cys Prxs form higher-order oligomeric structures that occur most commonly as a toroidal decamer of five homodimers and in a few rare cases as dodecamers (121, 327). In the FF conformation, the CP-containing active-site loop buttresses the decameric interface, thereby stabilizing the oligomeric toroid. CP sulfenylation from the catalytic reduction of H2O2 leads to disulfide formation between the CP and CR, locking the active site in an LU conformation and destabilizing the decamer-building interface (407).
The strong relationship between the structural environment of the CP and the oligomeric state of Prxs has been explored by site-directed mutagenesis, with mutation of the conserved active-site threonine (Thr44 in S. cerevisiae thiol-specific antioxidant 1 [Tsa1]) to valine or serine, modulating the oligomeric state toward dimer or decamer, respectively (362). The Thr44-flanking phenylalanines (Phe43 and Phe45) form hydrophobic interactions at the A-type interface and the oxidation-induced switch to the LU conformation is thought to disrupt these stabilizing interactions, leading to decamer dissociation (245, 407). As an exception of such redox-dependent oligomerization in 2-Cys Prxs, PrxIV keeps a stable decameric arrangement in the absence of a reducing agent (40). The oligomeric structure of PrxIV might be maintained by its distinct N-terminal extension, a feature lacking in other Prx subtypes. In the 2-Cys Prx of Salmonella typhimurium (AhpC), a threonine mutation at the decameric interface either stabilizes (T77V) or destabilizes (T77I) the oligomeric structure without direct interference with the active site (275). Destabilization of the AhpC decamer greatly reduces the catalytic efficiency of the enzyme for H2O2, but also makes the enzyme more resistant to overoxidation at the CP. The decreased sensitivity of the decamer-destabilizing mutants to peroxide-mediated inactivation is attributed to an increased flexibility of the active-site region, evidenced by high temperature factors in the crystal structure of AhpCT77I (275). This phenomenon could imply a dynamic mechanism linking redox-dependent oligomerization to the catalytic Prx function, in which the decameric state of the enzyme reacts with peroxide and then dissociates to dimers to allow the rapid formation of a CP-CR disulfide bond, thereby preventing enzymatic inactivation by the CP overoxidation.
Two possible mechanisms for the role of Prxs in oxidative signaling have been proposed; a direct transfer of oxidative equivalents from the oxidized Prx to a reduced target protein via a redox relay involving a temporary interprotein mixed disulfide (Fig. 9) (95), or the “floodgate” hypothesis, whereby Prx inactivation by hyperoxidation to a sulfinylated form leads to a localized build-up of H2O2 (408).
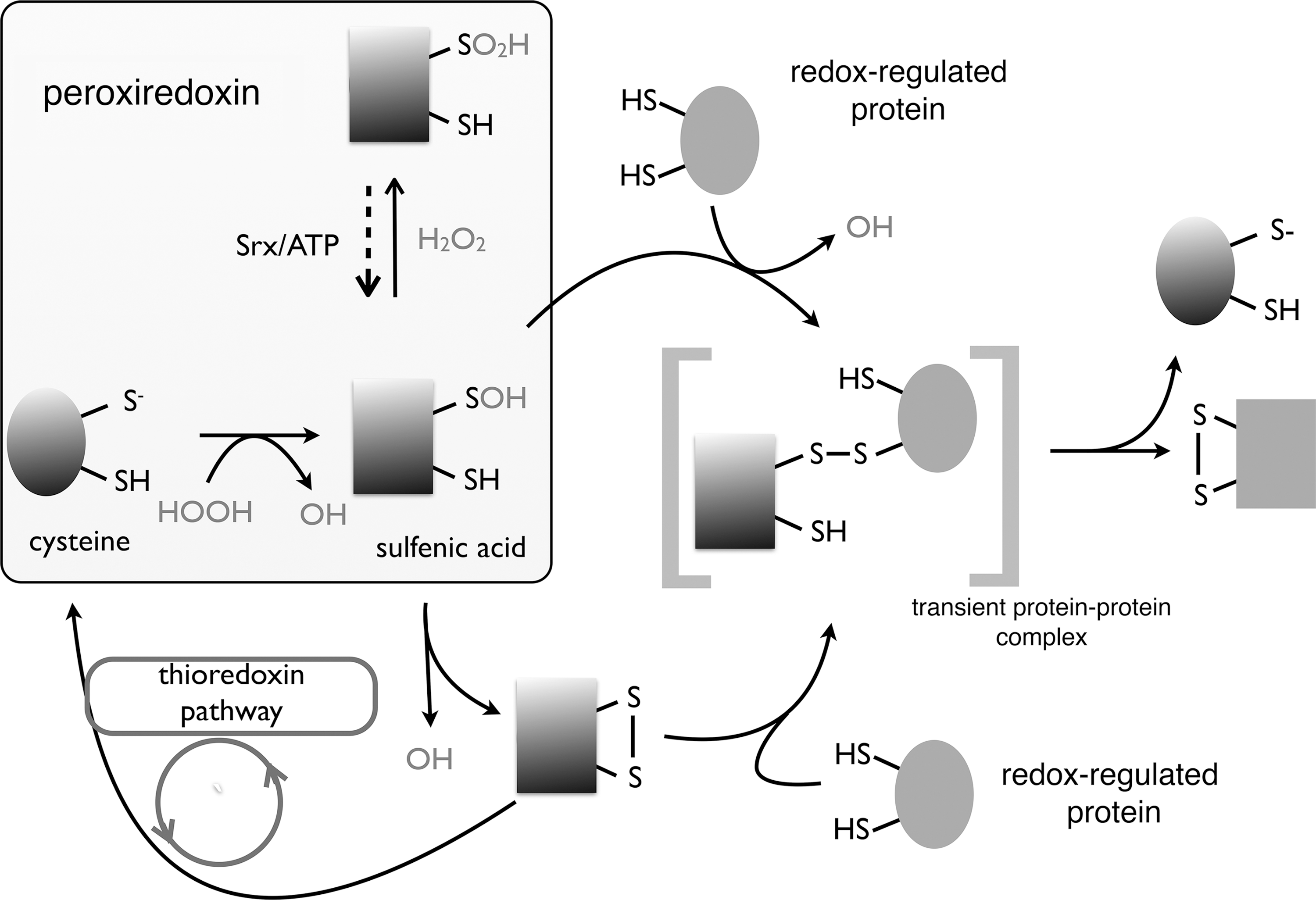
B. Thiol peroxidase redox relays
The concept of a Prx-based redox relay first gained credence after a ground-breaking study had revealed that the yeast thiol peroxidase Orp1 (previously termed GPx3) transferred oxidative equivalents to the transcription factor Yap1 (70, 354). The critical significance of this redox relay is that Yap1 is not directly responsive to H2O2, and oxidation can occur only with peroxidase involvement (228). Upon oxidation by H2O2, a sulfenic acid is formed on the CP of Orp1, Cys36, which can be resolved into a mixed disulfide with the Cys598 of the C-terminal cysteine-rich domain of Yap1. In turn, Cys303 of the N-terminal cysteine-rich domain of Yap1 reacts with Cys598 to produce an intramolecular, interdomain disulfide and to release Orp1. Unless oxidized, Yap1 is rapidly reduced and further sequential interdomain disulfides can occur, resulting in a final triple disulfide oxidation state, involving Cys310-Cys629 and Cys315-Cys620 in addition to the initial Cys303-Cys598. The single Cys303-Cys598 interdomain disulfide is sufficient to mask the C-terminal nuclear export signal of Yap1, leading to nuclear localization and transcriptional activation of oxidative stress response genes (69, 70), but the peak of transcriptional activity is reached only upon generation of the triple disulfide form that has an enhanced resistance to reduction (as evidenced by a decrease in redox potential relative to the single disulfide form) and, thus, can probably confer a more sustained activation in the presence of reductants (266).
Functionally equivalent redox relays have been described for Yap1 homologs, the Candida albicans Cap1 that is also oxidized by Orp1, and the Schizosaccaromyces pombe (fission yeast) Pap1 that is oxidized by Tpx1, a homolog of S. cerevisiae Tsa1 (35). Whereas the redox relay between Orp1 and Yap1 proceeds via an Orp1-SOH intermediate, the Tpx1 relay is proposed to proceed via a thiol–disulfide exchange, involving the CP-CR disulfide of the Tpx1 oxidized form (17, 35). As proteomic studies have revealed an abundance of proteins with surface-exposed thiolates, along with millimolar concentrations of LMW antioxidants, such as GSH, and submicromolar concentrations of Trx, the question arises of why Orp1 specifically oxidizes Yap1, especially considering the presence of a CR in Orp1 that should rapidly react to form a CP-CR disulfide.
This issue has been addressed by the identification of a Yap1-specific scaffold protein, the Yap1-binding protein 1 (Ybp1) (122, 387). In the absence of Ybp1, Orp1 forms a mixed disulfide with Yap1 at a predicted rate constant of 10−3 s−1, too slow for effective Yap1 oxidation in the cellular environment. Ybp1 recruits Orp1 and Yap1 into a ternary complex, effectively increasing their respective local concentrations and the oxidative relay rate constant to 1 s−1 (17). The binding of Ybp1 to Orp1 also prevents the CR of Orp1 (Cys82) from reacting with the sulfenylated CP before a mixed disulfide can be formed with the Cys598 of Yap1 (17). In yeast strains that produce a truncated Ybp1 gene product, Tsa1 takes over the function of Orp1 (265, 361). Tsa1ΔCR is able to form a mixed disulfide with Yap1 via its CP (Cys48), but only the wild-type Tsa1, not the ΔCR variant, can activate Yap1 in response to oxidative stress. Thus, either a thiol–disulfide exchange mechanism is the preferred mode of oxidant transduction between Tsa1 and Yap1, or a Tsa1 homodimer with only one intermolecular disulfide linkage, leaving the CP of one subunit free for sulfenic acid condensation, might be the Yap1-activating form (361).
The highly efficient redox relay between Orp1 and Yap1 inspired the development of a fluorescent probe based on roGFP2. By linking Orp1 and roGFP2 together as a fusion protein construct, the proximity dependence of the relay facilitated by Ybp1 can be enabled artificially. By comparing the redox relay characteristics of a wild-type and an Orp1 CR mutant (C82S) fusion construct, the roGFP-Orp1wt was demonstrated to transduce oxidation by a thiol–disulfide exchange mechanism, whereas the roGFP-Orp1ΔCR relays oxidative equivalents directly via a sulfenic acid intermediate (126). Besides the redox relay systems described above, the occurrence of oxidant transduction between the yeast Ahp1 and the Yap1 homolog Cad1 has been suggested and an oxidant relay between PrxII and the transcription factor STAT3 has been described. Direct STAT3 oxidation by H2O2 is very slow, but in the presence of PrxII, the oxidation rate is significantly enhanced (345). PrxII oxidizes STAT3 via a mixed disulfide, although it is not known whether via thiol–disulfide exchange, or direct sulfenic acid condensation. STAT3 oxidation leads to the formation of intermolecular disulfide-bonded STAT3 dimers and tetramers, and the involvement of different cysteines of STAT3 in dimer and tetramer formation implies independent PrxII oxidation pathways, resulting in different oligomerization states (345).
Another mammalian transcription factor found to be regulated by Prx-mediated oxidation is forkhead box protein O (FOXO) 3, which has been demonstrated to form mixed disulfides with PrxI in vivo, and formation of a PrxI-FOXO3 mixed disulfide appears to induce nuclear localization of FOXO3 (141, 287). PrxI has also been reported to facilitate oxidation of the APE1, and although the mechanism of oxidation is not fully characterized, it appears to involve a PrxI-APE1 mixed disulfide intermediate (256). The resulting oxidized form of APE1 is also unknown, although studies of its oxidation states show that it can form a mixture of disulfide-bonded multimeric species through Cys65, Cys93, Cys99, and Cys138, and that it is also capable of forming a mixed disulfide complex through Cys99 with Trx (227).
Further to its role as a moonlighting redox sensor, DJ-1 is suggested to engage in a thiol–disulfide exchange mechanism with PrxII (72), whereby oxidized PrxII with a CP-CR disulfide forms a mixed disulfide with Cys53 of DJ-1, which resolves into an intermolecular disulfide involving the Cys53 of another monomer to generate a covalent homodimer (89). All-in-all, peroxidase-based redox relays have been established to have both a functional relevance and technological applications, but until recently, the number of truly validated redox relays has been exceptionally low, especially relative to the vast number of oxidizable protein thiols identified in proteomic studies. However, the recent findings that the simultaneous depletion of PrxI and PrxII massively decreases the levels of H2O2-induced protein thiol oxidation (353) provide compelling evidence in favor of oxidative signaling via redox relays.
C. Nonperoxidatic roles for Prx
The floodgate hypothesis was proposed based on the observation that some eukaryotic Prxs appear to have evolved a specific sensitivity to hyperoxidation (408). However, there is presently a lack of experimental support for this mechanism and the proportion of hyperoxidized 2-Cys Prx in mammals is usually quite low, estimated to be <1.6% in mice erythrocytes (52). Accordingly, specific cellular situations might occur in which overoxidized 2-Cys Prx can sufficiently accumulate to facilitate floodgate-based signaling. For example, in the adrenal cortex, 10–20% of PrxIII is found to be hyperoxidized (172). Increased ROS leakage caused by overstimulation of steroid hydroxylation by CYP11B1, a cytochrome P450 family protein, in mitochondria of adrenal gland cells has resulted in PrxIII hyperoxidation and a corresponding increase in p38 MAPK phosphorylation (172). In this study, hyperoxidation was proposed to allow the build-up of mitochondrial H2O2 and subsequent overflow to the cytosol, whereby oxidative ASK1 activation, an upstream kinase of p38, leads to p38 phosphorylation (172). However, another study has highlighted a steep concentration gradient of H2O2 across the mitochondrial membrane, hence, changes in mitochondrial H2O2 levels are expected to have relatively little impact on the cytosolic concentration of H2O2 (246). A redox relay mechanism for ASK1 regulation has also been suggested based on the observation that PrxI and PrxIII form a mixed disulfide with ASK1 in mammalian cell lines, thereby inferring a direct ASK1 oxidation mechanism by Prx (157).
An alternative to a hyperoxidation-driven floodgate is the transient inactivation of Prx by reversible phosphorylation that would influence H2O2 gradients independently of H2O2 itself. The peroxidase activity of PrxI can be inactivated by Tyr194 phosphorylation (which is positioned ∼9 Å from the CP) by Src kinases upon cell stimulation by growth factors (406), Thr90 and Thr183 phosphorylation by the mammalian sterile20-like kinase 1 (MST1) (298), and by Thr90 phosphorylation by the cyclin-dependent kinase 1 (CDK1) (45). The peroxidase activity of PrxII decreases upon Thr89 phosphorylation by the CDK5/p35 complex (290). Conversely, phosphorylation of Ser32 of PrxI by the T-Lymphokine–Activated Killer (LAK) cell–originated kinase (TOPK) enhances the peroxidase activity (425), thereby demonstrating that location of phosphorylation is critical for its influence on Prx function. Prx susceptibility to phosphorylative inactivation or hyperoxidation can be independent of the isoform, with PrxI relatively insensitive to hyperoxidation, but readily inactivated by phosphorylation, whereas PrxII is typically not susceptible to inactivation by phosphorylation, but more prone to hyperoxidation (406). A number of kinases and phosphatases are becoming regarded as being redox regulated themselves, thereby adding an additional level to phosphorylative Prx regulation. Oxidation of a conserved cysteine of the Src kinases promotes their activation, increasing the likelihood of PrxI phosphorylation and inactivation.
Furthermore, several members of the protein tyrosine phosphatase (PTP) family, which could potentially dephosphorylate (and therefore activate) PrxI, are susceptible to inactivation through the reversible oxidation of cysteine residues (235, 267), culminating in a positive feedback loop that favors PrxI inactivation by phosphorylation and prevention of dephosphorylation. As PTPs have a relatively low reactivity toward endogenous H2O2 levels (k = 9–18 M −1·s−1) (72, 90), it is tempting to consider the possibility of direct oxidation of PTP by Prx as an additional level of redox-regulatory control. However, in the case of PTP1B and PrxII, a redox relay was not found to occur upon H2O2 exposure (62). Although possible redox relay systems between Prx/peroxidases and kinases have been proposed for ASK1 (157) and the S. pombe p38/JNK stress-activated protein kinase (SAPK), Sty1 (386), many other regulatory, redox-related interactions with Prx and kinases/phosphatases have been characterized without involvement of a mixed disulfide intermediate. PrxI engages in regulatory interactions with the phosphatases, PTEN, MAPK phosphatase 1 (MKP1), and MKP5, in a manner which is redox dependent, yet does not involve mixed disulfide formation (39, 382). PrxI binds to PTEN and MKP1 with similar affinity (K d = 247.3 ± 33.6 and 261.0 ± 21.2 nM, respectively) and dissociates upon oxidation, but binds MKP1 with an increased affinity (2.4 ± 0.2 nM) to form a complex that does not dissociate upon oxidation (382). PTEN, MKP1, and MKP5 reside in a class of PTPs that possess a nucleophilic catalytic cysteine, sensitive to oxidation. PTEN oxidation leads to the formation of an intramolecular disulfide, involving its active-site cysteine, whereas oxidation of MKP1 and MKP5 induces the formation of disulfide-linked oligomers; in both cases, oxidation results in loss of phosphatase activity. PrxI preserves the lipid phosphatase activity of PTEN by protecting it against oxidative inactivation, and also protects MKP5 against oxidation-induced oligomerization and subsequent inactivation.
Conversely, PrxI-associated MKP1 is more susceptible to oxidative inactivation upon exposure to H2O2. As MKP1 dephosphorylates both p38α MAPK and JNK, whereas MKP5 dephosphorylates only p38α MAPK, the involvement of PrxI indirectly activates JNK signaling while inactivating p38α (190). A direct regulation of p38 MAPK by PrxI has been suggested based on the coimmunoprecipitation of PrxI with phosphorylated p38 MAPK, but the nature of this interaction has still to be characterized in vitro.
The HMW oligomeric form of PrxI has been proposed to promote MST1 activity by association with the MSTI1 C-terminal kinase-inhibitory domain (247). Significantly, phosphorylation of PrxI at Thr90 by CDK1 or MST1 induces the formation of HMW oligomers of PrxI (Fig. 10), hinting to a positive feedback loop promoting PrxI phosphorylation by MST1 (155). Phosphorylation is not, however, a general mechanism for the stimulation of Prx oligomerization, because phosphorylation at Tyr194 of PrxI by Src kinases does not influence the oligomeric state of PrxI (406). The conversion of PrxI to a HMW oligomer confers a chaperone function, but reduces the peroxidase activity (179). Prx hyperoxidation is known to trigger the formation of HMW oligomers; therefore, under the conditions to which the floodgate signaling hypothesis applies, a corresponding increase in Prx chaperone action should also be considered (263). The conversion to HMW oligomers involves stacking of decamers via “R-interface” interactions to create filamentous structures. Stacking of the decamers into HMW oligomers prevents the proper folding of the C-terminal tails, leaving them as disordered protrusions (314). As the unstructured, hydrophobic regions of other chaperone proteins are known to be involved in substrate binding, these unfolded C-terminal regions of HMW oligomeric Prxs have been postulated to play a role in conferring holdase activity (314).
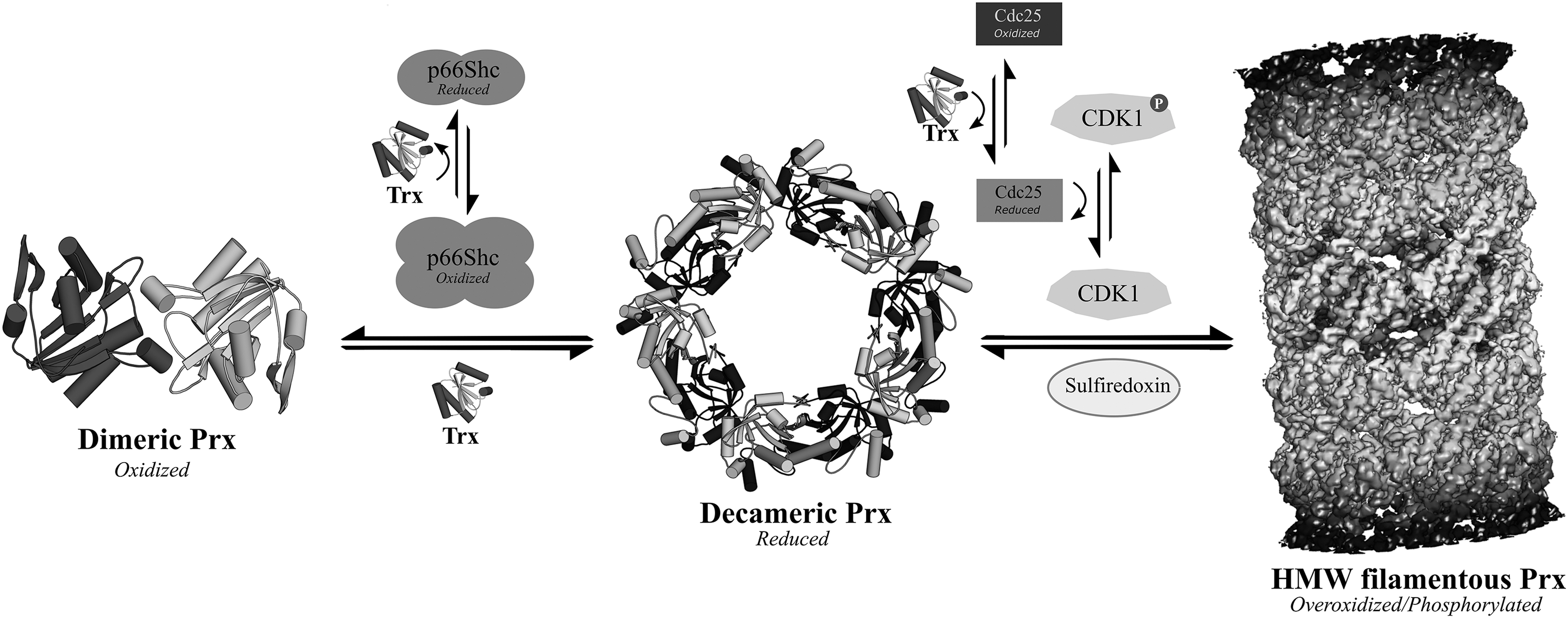
Currently, the connection between the oligomeric state and regulatory interactions of Prx is unclear and seems to vary depending on the protein substrate. In the case of mitochondrial Prx of the protozoan parasite Leishmania infantum, overoxidation and oligomerization to HMW species is not necessary for Prx to act as a holdase on mitochondrial proteins under heat stress (369). PrxI is proposed to bind PTEN as a monomer, as preservation of PTEN phosphatase activity reaches its maximal under equimolar ratios of PrxI and PTEN (39). A relationship between the oligomeric state of PrxI and its regulation of MKP1/MKP5 has not been fully characterized. PrxI binds to Toll-like receptor 4 (TLR4) in the presence of myeloid differentiation protein 2 (MD2) and cluster of differentiation 14 (CD14) to stimulate cytokine release, in a manner independent of peroxidase activity, but dependent on PrxI oligomerization (305). Nevertheless, whether the decameric form or HMW chaperone form of PrxI is necessary for its interaction with TLR4 is still unclear. Chloroplast 2-Cys Prx can enhance the activity of chloroplast fructose-1,6-bisphosphatase (CFBPase), although not through function as a chaperone, as it is unable to prevent CFBPase denaturation, but via protection against CFPase autoxidation and alteration of its affinity to its substrates (41).
In some cases, a specific physiological function of Prx can require the specifically directed regulation of the balance between hyperoxidized HMW oligomers and the reduced decameric form. Recently, the potential role of the yeast Prx, Tsa1 has been demonstrated in preventing the accumulation of H2O2-induced aggregates in stressed yeast cells (129). Decameric Tsa1 was found to physically associate with the Hsp70-type protein, Ssa1, in a redox-independent manner and, upon hyperoxidation, localized along with Ssa1 or Ssa2 to inclusion aggregates, leading to a decrease in aggregate formation by the promotion of proteasomal degradation or refolding of substrate proteins (Fig. 11). Tsa1 was also able to trigger Hsp104- and (Hsp40-type) Sis1-mediated disaggregation in a Srx-dependent manner, implying a necessary conversion of the hyperoxidized form of Tsa1 to its reduced form, possibly driving Tsa1 dissociation from aggregates to allow disaggregation (129), thus providing a eukaryotic example of redox-dependent regulation of the proteostasis machinery through the control of the oxidative state of a Prx.
Considering the significant influence of the oligomeric state of Prx on its cellular function, it is important to note that the presence of fusion tags can affect oligomerization and hyperoxidation sensitivity. A C-terminal Myc-tag on PrxII conferred resistance to hyperoxidation (169), whereas an N-terminal His6-tag on Arabidopsis 2-Cys PrxA increased its sensitivity to hyperoxidation (184) and an N-terminal Myc-tag on yeast Tsa1 destabilized its oligomeric forms (263). The oligomeric state of Prx can also be influenced by the presence of other proteins. In Arabidopsis, Cyp20-3, an abundant stromal protein, interacts with 2-Cys Prx with a K d of <0.5 μM, in a pH- and redox-dependent manner. The Cyp20-3 association drives the oligomeric state of 2-Cys Prx toward the dimer. Conversely, titration of chloroplast stromal proteins with isolated Arabidopsis 2-Cys Prx shifted the oligomeric equilibrium from dimer toward decamer.
The influence of proteins on the oligomeric Prx state has also been characterized in eukaryotic systems, for instance the interaction between PrxI and p66Shc (110). In a coincidental link, p66Shc is also regulated by a cyclophilin, Pin1 (88). The apoptosis-inducing role of p66Shc is activated by a redox-dependent conversion from homodimers to disulfide-linked tetrameric p66Shc, which constitutes the proapoptotic form (111). The interaction with reduced homodimeric p66Shc induces the dissociation of PrxI decamers to dimers, purportedly due to the enhanced affinity of p66Shc for the dimeric form of PrxI (Fig. 10) (110). The association of oxidized tetrameric p66Shc with the decameric PrxI also generates a decamer-to-dimer conversion, although this time through a thiol–disulfide relay whereby oxidative equivalents are transferred from p66Shc to PrxI (110). It is still unclear whether PrxI can act as a reductant of p66Shc in vivo, whereas a role for Trx as a reductant of oxidized p66Shc has been described (111).
Finally, it is important to consider the common characteristics that define the specifity of Prx for its binding partners. Data-mining studies have unearthed recurrent motifs conserved among the interaction partners of human PrxI and PrxII, regardless of the interaction mode or oligomeric state of Prx (18). Besides the redox-active CXXC motif, many binding partners of PrxI/PrxII have been found to possess a LXXLL and/or PXXP motif. Among the functional Prx associations described above, both the LXXLL and PXXP motifs are conserved among STAT3, ASK1, and PTEN, and the PXXP motif alone for p66Shc, and the LXXLL motif alone for TLR4.
D. Complementary roles of Prx and Trx
Significantly, many of the physiolocally interacting partners of Prx also interact with Trx. Most typically, the role of Trx in relation to its target proteins is that of a disulfide reductant that “flips the disulfide switch,” by reduction of either an intermolecular disulfide, as for p66Shc, or an intramolecular disulfide, as for example, cell division cycle 25 (Cdc25). Cdc25 is a PTP that is reversibly inactivated by H2O2-mediated oxidation, leading to intramolecular disulfide formation between its catalytic Cys and a backdoor Cys (346). Cdc25 participates in the activation of CDK1, a kinase that, as detailed above, phosphosphorylates PrxI (45), thereby providing an indirect link between Trx action and phosphorylative Prx regulation. A more direct connection between kinases and Trx and Prx is the mutual interaction with MST1. Trx1 binds to the SARAH domain of MST1 and inhibits autophosphorylation by blocking its homodimerization (44). Although the association of Trx1 to MST1 does not involve a mixed disulfide, it is still redox sensitive, as mutation of the CXXC motif cysteines to serine abolishes the interaction and Trx1 oxidation also induces the dissociation from MST1 (44). Whereas PrxI is a phosphorylation target of MST1, MST1 does not phosphorylate Trx1.
In addition to its redox-regulated interaction with PrxI, the phosphatase, PTEN, is also regulated in a redox-dependent manner by Trx. Remarkably, Trx can both activate and deactivate PTEN through distinct mechanisms. In its capacity as a disulfide reductase, Trx reduces the intramolecular disulfide that is formed in PTEN between the active-site Cys (Cys124) and the backdoor Cys (Cys71) upon oxidation by H2O2, converting PTEN from an inactive to an active state (204). PTEN inactivation by Trx might occur through the formation of a mixed disulfide between the catalytic Cys32 of Trx1 and Cys212 of the C2 domain of PTEN, inhibiting the lipid phosphatase activity (237). The inhibitory interaction between Trx and PTEN could be reversed by TR. Notably, this interaction involves the reduced form of Trx, with the oxidized form unable to interact with PTEN in an inhibitory fashion, thereby precluding a mechanism of thiol–disulfide exchange between Trxox and PTEN.
A final example of the dual regulation of a kinase by both Prx and Trx is in the case of ASK1. Whereas Prx is proposed to act as an oxidant transducer that oxidizes ASK1 to active, disulfide-linked multimers (180), Trx1 instead suppresses ASK1 activity through binding its N-terminal Trx-binding domain (TBD) and preventing the homophilic interaction of ASK1 required for its functionality (104, 185). Upon oxidation, Trx1 dissociates and ASK1 can convert into its active kinase form and participate in the initiation of the programmed cell death (316). The catalytic cysteine of Trx1 is essential for the maintenance of its affinity for ASK1 and of the seven conserved cysteines of the TBD of ASK1. Cys250 has been identified as the key residue modulating the association between ASK1 and Trx1 (185, 187, 252).
In all the cases of protein coregulation by Trx/Prx outlined above, an antagonistic system of regulation can be seen. Whereas PrxI acts as an activator of ASK1, MST1, and PTEN, Trx conversely acts as an inhibitor. Additionally, the redox stimuli for the interactions of Prx and Trx with these proteins are also in opposition. Under oxidizing conditions, an association of ASK1 and MST1 with PrxI is promoted, whereas under reducing conditions an interaction with Trx is created. A further significant characteristic of these interactions is that they are redox sensitive, but do not involve the oxidoreductase or chaperone functions of either Prx or Trx.
Although the role of Prx as a chaperone is focused on in the Nonperoxidatic roles for Prx section, Trx also has notable cellular chaperoning functions. The molecular chaperone capabilities of Trx and TR were first observed in E. coli (171). Since then, a critical role for Trxs as stress-responsive chaperones in plants has been established. Tobacco (Nicotiana tabacum) Trx-m and Trx-f and Arabidopsis Trx-h act as chaperones depending on their oligomeric states (273, 319). The monomeric form of Trx functions as a disulfide reductase and, under heat stress or oxidative stress, Trx oligomerizes and gains chaperone activity (273). A similar duality of function has been observed for plant NADPH-TR type C (a fusion of TR and Trx) that has a holdase and a disulfide reductase activity in HMW and LMW forms, respectively. Mutation of the active-site cysteines abolished disulfide reductase and foldase activity, but the effect was less pronounced on the holdase activity (43). Recently, a novel bipartite protein with both designated reductase and chaperone domains has been identified, the Arabidopsis thaliana tetratricopeptide repeat domain-containing thioredoxin (AtTDX). AtTDX has an N-terminal Hsp70-interacting protein (HIP)-like domain that contains three tetratricopeptide repeat domains and a C-terminal Trx domain conferring disulfide reductase activity. In response to heat stress, AtTDX switches from a LMW form with disulfide reductase activity to a HMW form with holdase function (198). AtTDX has been demonstrated to act as a cochaperone of the yeast Hsp70, Ssb2, modulating its ATPase activity and recruiting it to the Hsp90 complex (286). AtTDX associates via its HIP-like domain with the ATPase domain of Ssb2 in a redox-dependent interaction. The complex between AtTDX and Ssb2 dissociates under oxidative conditions and mutation of either the Cys20 of Ssb2, which is conserved by many other Hsp70 proteins, or of both cysteines of the catalytic CXXC motif of Trx, renders the association redox insensitive (389). AtTDX can also function as a general chaperone, acting as a foldase in its LMW form and, under heat-shock conditions, oligomerizes to a HMW form with holdase activity (198).
Thus far, beyond the respective peroxidase and reductase activities of Prx and Trx, a functional promiscuity relating to redox-related interactions and chaperoning is exhibited by both enzymes. Although Prx has been demonstrated to participate in oxidative signaling through redox relays (265, 345), its preferred oxidation target is undoubtedly Trx. The reduction of Trx by TR is expected to prevent Trx from further participation in an oxidative relay, but, under stress conditions, sufficient quantities of oxidized Trx may possibly accumulate to allow the Trx-mediated oxidation of target proteins in a relay system mirroring that displayed by Prx. In S. pombe, exposure to moderate oxidative stress (0.2 mM H2O2) overwhelms the TR system, resulting in high amounts of oxidized Trx1 either in an intramolecular disulfide-bonded form, or in mixed disulfides with various cellular proteins (65). A model of Trx-mediated oxidation of Yap1 has been suggested in S. cerevisiae (294) and the action of Trx as an oxidant has also been characterized in Δtrr E. coli (68, 352). Proteomic studies of S. pombe have revealed extensive thiol oxidation (31% of cysteines oxidized) in Δtrr cells, whereas only minor thiol oxidation (1.5% of cysteines oxidized) occurred in Δtrx1 cells, further confirming the possible role of Trx as a cellular oxidant when the electron flow from the TR/NADPH system is limited (106).
In Arabidopsis, a physiologically relevant oxidative relay has been characterized between the atypical chloroplast Trx (ACHT) and a cellular substrate protein. ACHT has a noncanonical Trx active site and a higher redox potential than that of typical plant Trxs. The redox potentials of ACHT1 and ACHT4 at pH 7 are −237 and −240 mV, respectively (63), whereas other plant Trxs, such as Trx-m, has an E m of approximately −300 mV (57). However, ACHTs are still able to effectively act as electron donors for 2-Cys PrxA during peroxide reduction, despite the relatively low E m of PrxA (−307 mV) (63, 142). A specific redox relay in Arabidopsis has been defined, whereby PrxA oxidizes ACHT4, which in turn oxidizes a redox-sensitive subunit of the starch synthesis enzyme AGPase (APS1). This relay depended on the C-terminal domain of ACHT4 and could not be substituted by ACHT1, thereby demonstrating that for such an oxidative relay to occur, regions of the protein outside the redox-active site are required to confer binding specificity. Whereas Trx is kept in a reduced state by the TR/NADPH system, the rate of Prx-mediated oxidation of ACHT exceeds its reduction rate under normal growth conditions. Therefore, a significant fraction of ACHT can be expected to be oxidized, lending further relevance to its role as a transducer of oxidative signals.
VI. Conclusion
In considering the concept of intracellular redox signaling networks, we impose the simplification of separately defining downstream, midstream, and upstream redox-responsive proteins. Transcription factors of prokaryotes provide an example of direct redox regulation with immediate and measurable positive responses, namely upregulation of antioxidant enzymes. Important efforts have been made to understand the structural determinants that (i) allow some transcription factors to display oxidant sensitivity in the presence of ultrafast scavenging peroxidases, and (ii) modulate conformational changes mediating affinity and specificity of macromolecular associations (in this case toward DNA). A demonstrated asset of understanding the mode of oxidative structural regulation of the prokaryotic transcription factor, OxyR, is the development of a series of genetically encoded ratiometric H2O2 sensors, known as HyPer (22). The architecture of oxidative regulation of yeast transcription factors, such as Yap1, has been clearly established (387), with the first identification of a peroxidase redox relay mechanism in lieu of the direct peroxide responsiveness exhibited by prokaryotic transcription factors, such as OxyR or OhrR (258, 374). Our view of redox regulation of mammalian transcription factors can be partially obscured by additional regulatory mechanisms, such as phosphorylation by upstream kinases, which themselves are susceptible to redox regulation; examples being the p38 MAPK/JNK activation of FOXO transcription factors (84), or JAK2 activation of STAT3 (372). The additional axis of phosphorylative regulation of proteins can to some extent complicate the interpretation of oxidative signaling pathways.
Therefore, we have partially attempted to assess this issue through the discussion of some of the interregulatory interactions between antioxidant enzymes and kinases/phosphatases. A clear relationship between phosphorylation pathways and oxidative signaling has been demonstrated in vascular smooth muscle cells, in which a correlation between the H2O2 concentration and the tyrosine phosphorylation levels has been observed upon oxidative stimulation (356). As Prxs consume the vast majority (>99%) of the intracellular H2O2 (403), the ability of kinases/phosphatases to modulate their activity is a key intersect of the oxidative and phosphorylation signaling pathways and the significance of this has been emphasized previously (271). We have also outlined the redox-dependent noncovalent associations between kinases/phosphatases and Prx/Trx that do not involve a direct oxidation or reduction. It is important to note that Prx and Trx often play opposing regulatory roles in these associations and are differentially responsive to redox conditions. The emerging role of Prx as an intermediary transducer of oxidation through interprotein redox relays derives from the observation that typical redox-sensitive protein thiols are 4–7 orders of magnitude less reactive to peroxides (approximately 1–100 M −1·s−1) than the peroxidatic Prx thiol (∼105 to 107 M −1·s−1) (296).
Although several operational redox relays between Prx/peroxidases and substrate proteins have been characterized, there are still a multitude of reversibly oxidized thiols identified by proteomic studies, for which a functional redox relay has yet to be established. Redox relays have been shown to occur through either thiol–disulfide exchange or direct sulfenic acid condensation, with the former mechanism in competition with reduction by Trx and the latter mechanism in some cases seemingly depending on a scaffolding protein (such as Ybp1) to prevent condensation to a CP-CR disulfide. When the Prx redox relay mechanism of direct CP sulfenyl condensation to a mixed disulfide is considered, it is important to highlight that the mammalian PrxII has a significantly slower CP-CR disulfide formation rate than that of other Prxs, correlating sensitivity to hyperoxidation (PrxII, 1.7 s−1, PrxIII 22 s−1, and PrxV 15 s−1) (281, 380). Therefore, redox relay through direct sulfenyl condensation could preferentially occur via PrxII. Prokaryotic Prxs are remarkably robust against overoxidation and undergo self-condensation of the CP sulfenyl at a rate almost 50-fold that of PrxII, suggesting that, if present, the relay mechanism is likely via a disulfide exchange (274).
The remarkable complexity and functional promiscuity of mammalian antioxidant enzymes in comparison to their prokaryotic counterparts is evident, and several ground-breaking studies over the past decade have advanced our understanding of the role of eukaryotic antioxidant enzymes in oxidative signaling. In this study, we support the previous description of Prx as a “redox relay hub,” but wish to also draw focus to the role of redox-regulated moonlighting proteins as additional hubs, providing intersects between pathways of oxidative signaling and other cellular processes. In this review, we have touched upon a few of these moonlighting proteins, namely DJ-1, GAPDH, TG2, and PPIs, and have attempted to present their functional promiscuity in the context of redox regulation. Despite not being enzymes of the cellular antioxidant system, GAPDH, DJ-1, TG2, and CypA are all differentially expressed in response to oxidative stress (78), and there is some evidence to support crossregulatory influences in transcription of these moonlighting proteins in response to stress conditions. In human neuroblastoma cell lines expressing a mutant overoxidant mimic of DJ-1 (C106DD), expression of TG2 was downregulated and ASK1 upregulated relative to cells containing wild-type DJ-1 (285), and astroglial DJ-1 overexpression has been shown to relate to upregulation of CypA (102). The “polygamous” influence of these moonlighting proteins cannot be understated and a further spectrum of complexity can arise from functional interactions between the moonlighting proteins themselves, such as TG2 transamidating GAPDH (152) and DJ-1 deglycating the glycated form of GAPDH (304). The association of redox-responsive moonlighting proteins with regulatory phosphorylation also provides additional intersects between oxidative signaling and kinase pathways. DJ-1 disrupts the p38 MAPK pathway by inhibiting ASK1 kinase activity (244); the PPI, Pin1, enzymatically targets phosphorylated proteins (88); TG2 plays a role in downregulating phosphatases, such as PTEN (241, 396), and this function is regulated by the phosphorylation status of TG2; and the cellular localization of GAPDH is altered upon phosphorylation (144).
It is also useful to highlight cases where a single kinase is targeted by multiple redox-responsive proteins, such as the regulation of ASK1 (the upstream signaler of the JNK/p38 pathway) by DJ-1, CypA, PrxI, and Trx, as this is indicative of a potential redox-regulated hub for kinase signaling (Fig. 12). All four of the moonlighting proteins discussed associate with p53, a tumor suppressor protein regarded as a key coordinator of the response to stress and cellular aging (Fig. 13) (218). DJ-1 engages in a redox-dependent inhibitory interaction with p53 (170), nuclear GAPDH forms a complex with p53 and promotes phosphorylation and expression of p53 (420), TG2 phosphorylates p53 when acting as a kinase (241), and p53 forms a complex with mitochondrial CypD to activate the CypD-dependent apoptotic pathway (21, 289).
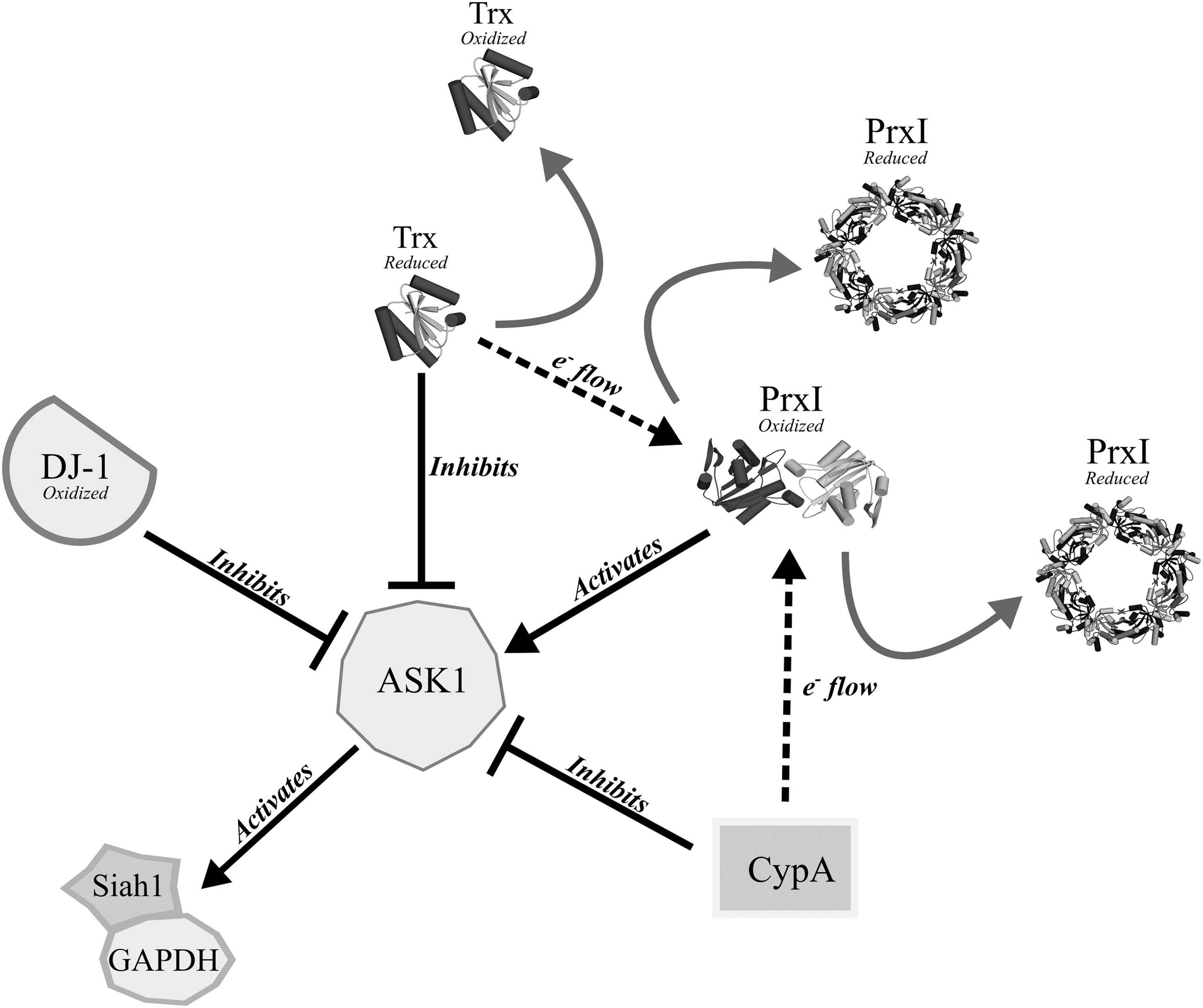
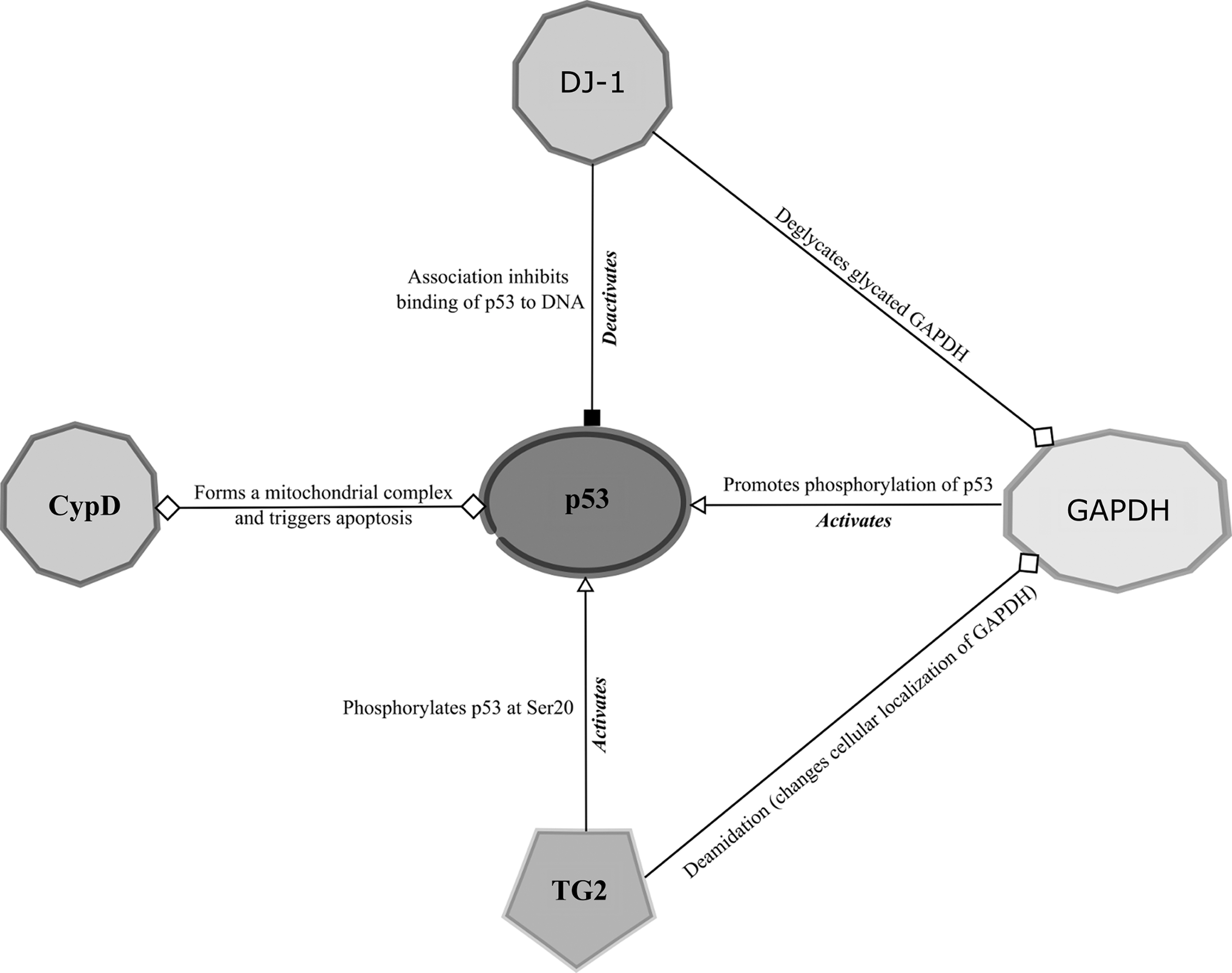
Cellular localization is also likely to be a key factor in the cooperative roles of redox-responsive moonlighting proteins and obligate antioxidant enzymes. As a significant intracellular generator of ROS, and an influential coordinator of cell death, mitochondria present a subcellular space, wherein controlled redox regulation of processes is highly beneficial (306). Mitochondria-localized DJ-1 has been proposed to prevent degradation of the antiapoptotic mitochondrial protein, Bcl-XL, and preserve the respiratory Complex I activity to limit mitochondrial ROS generation. CypD both acts to modulate Complex III, and forms part of mPTP, which mediates the collapse of the mitochondrial membrane potential as part of the cell death pathway. Other components of the mPTP include the voltage-dependent anion channel (VDAC), and adenine nucleotide translocase (ANT), which itself can be redox regulated through cysteine oxidation (58). GAPDH translocates to mitochondria during apoptosis and interacts with the VDAC to promote pore opening (368). TG2 promotes both the upregulation and mitochondrial translocation of GAPDH, and is also able to act on ANT as both a transaminase and as a PDI (230). Overexpression of TG2 in the mitochondria leads to GSH depletion, increased ROS levels, and membrane hyperpolarization in a mechanism distinct from mPTP opening (282), and TG2 plays a critical role in autophagic clearance of damaged mitochondria in a manner dependent on its transaminase activity (311). Even proteins of this review not labeled as moonlighters appear to adopt a subfunction within mitochondria. STAT3, for example, has been recognized to act in mitochondria to partially suppress electron flow through Complex I and has a cytoprotective role independent of its function as a transcription factor (359).
Due to the limited scope of this review, several elements of cellular redox regulation have not been covered. Discussion of oxidative modifications, such as S-nitrosylation, glutathionylation, and persulfidation, were mostly excluded, although they provide an important spectrum of redox control that influences the fate of oxidative signaling. Furthermore, other posttranslational modifications, such as acetylation, provide a separate level of control over redox pathway enzymes (92), and redox control of deacetylases, such as sirtuins, links this regulatory level to redox conditions (292). Direct oxidative regulation of ion channels has been identified (26, 341, 383), and redox control of ion channels will likely have a marked influence over the cellular response to oxidative stress, as Ca2+ influx for example can influence both redox-dependent and redox-independent signaling pathways alike. Understanding of the molecular mechanisms of ion channel redox regulation is relatively limited, in part due to the inherent difficulties of the structural characterization of membrane proteins. Inevitably, technical and methodological improvements in cryo-EM will facilitate the structural study of more membrane proteins that will translate in a greater access to the molecular mechanisms of redox-regulated ion channels.
The role of redox signaling in disease development is continuously validated (143), and, as recently stated, “An improved understanding of the sophisticated workings of redox biology is imperative to defeating cancer” (51). In this study, we hope to have emphasized that statement and encouraged consideration of moonlighting redox-regulated proteins as key players in deciding cellular fate under oxidative stress. CypA, TG2, CLIC (mentioned within the Transglutaminase 2 section), and DJ-1 are all associated with increased invasion and metastasis of cancer cells (133, 138, 197, 422), with both DJ-1 and CypA exerting their influence through the regulation of kinase pathways (133, 422). As cancer cells tend to block mitochondrial apoptotic induction, developments in cancer therapeutics often target the mitochondrial apoptosis signaling pathway, for example at p53 or the mPTP (105), and a direct link has been established between redox regulation of p53 and tumor progression (323). All of the moonlighting redox sensors described in this review play some role in regulating either p53, the mPTP, or both in response to redox stimuli. It is troubling that the precise molecular mechanisms governing the redox regulation of DJ-1, TG2, CLIC, and cyclophilins are still poorly understood.
Footnotes
Acknowledgments
The authors thank Martine De Cock for help in preparing the article. This work was supported by Vrije Universiteit Brussels (Strategic Research Program 34), the Research Foundation-Flanders (Excellence of Science project no. 30829584 [to F.V.B and J.M.], the Research Foundation-Flanders (G0D7914N [to F.V.B.] and G0D70149 [to J.M.]), and the European Cooperation in Science and Research (COST Action BM1203/EU-ROS). B.P. and B.D.S. are indebted to the Agency for Innovation by Science and Technology, A.L. to VIB (through the International PhD Program in Life Sciences), and N.B. to Indian Council of Agricultural Research for predoctoral fellowships. M.-A.T. is a predoctoral fellow of the Research Foundation-Flanders.