
Abstract
Aims:
Inhibition of microRNA-92a (miR-92a) is reported to suppress endothelial inflammation and delay atherogenesis. We hypothesize that miR-92a inhibition protects endothelial function through suppressing oxidative stress in diabetic db/db mice.
Results:
In this study, we found elevated expression of miR-92a in aortic endothelium from db/db mice and in renal arteries from diabetic subjects. Endothelial cells (ECs) exposed to advanced glycation end products (AGEs) and oxidized low-density lipoprotein express higher level of miR-92a. Overexpression of miR-92a impairs endothelium-dependent relaxations (EDRs) in C57BL/6 mouse aortas. Overexpression of miR-92a suppresses expression of heme oxygenase-1 (HO-1), a critical cytoprotective enzyme, whereas inhibition of miR-92a increases HO-1 expression in human umbilical vein ECs (HUVECs) and db/db mouse aortas. Importantly, miR-92a inhibition by Ad-anti-miR-92a improved EDRs and reduced reactive oxygen species (ROS) production in db/db mouse aortas. HO-1 inhibition by SnMP or HO-1 knockdown by shHO-1 reversed the suppressive effect of miR-92a inhibition on ROS production induced by AGE treatment in C57BL/6 mouse aortas. In addition, SnMP reversed miR-92a inhibition-induced improvement of EDRs in AGE-treated C57BL/6 mouse aortas and in db/db mouse aortas.
Innovation:
Expression of miR-92a is increased in diabetic aortic endothelium and inhibition of miR-92a exerts vasoprotective effect in diabetic mice through HO-1 upregulation in ECs.
Conclusion:
MiR-92a expression is elevated in diabetic ECs. MiR-92a overexpression impairs endothelial function and suppresses HO-1 expression in ECs. Inhibition of miR-92a attenuates oxidative stress and improves endothelial function through enhancing HO-1 expression and activity in db/db mouse aortas. Antioxid. Redox Signal. 28, 358–370.
Introduction
D
In the present study, we provide novel evidence that expression of microRNA-92a (miR-92a) was increased in arteries from diabetic mice and patients. Inhibition of miR-92a protects endothelial function and normalizes reactive oxygen species (ROS) generation through enhancing heme oxygenase-1 (HO-1) expression and activity in db/db mice. Thus, targeting miR-92a-HO-1 cascade is effective against diabetic vasculopathy.
The miR-17-92 cluster, comprising six individual miRNAs (miR-17, -18a, -19a, -20a, -19b, and -92a), is abundantly expressed in vascular endothelial cells (ECs) (12). Among them, miR-92a plays an important role in modulation of vascular homeostasis (5, 14, 50). Indirect evidences suggest that miR-92a might mediate vascular dysfunction in diabetes. For example, miR-92a was reported to play a proatherogenic role through upregulation of proinflammatory genes in ECs (7, 28, 49). Another report showed that inhibition of endothelial miR-92a attenuates neointimal lesion formation and improves re-endothelialization and functional recovery following vascular injury in mice (11). However, direct evidence showing the expression pattern of miR-92a in diabetic endothelium is still lacking, and whether miR-92a regulates oxidative stress and endothelial function in diabetes remains unknown.
Upregulation of the expression and activity of antioxidant enzymes in ECs was demonstrated to be vasoprotective in diabetes (8, 22, 39, 43). For example, HO-1 preserves NO bioavailability to restore endothelial function in diabetic mice (27) although how HO-1 expression is regulated in ECs is incompletely understood. Importantly, a genetic study in type 2 diabetic patients showed that length polymorphism in HO-1 gene promoter in vascular cells is associated with oxidative stress and higher incidence of coronary artery disease (6). In the present study, we hypothesize that miR-92a inhibition attenuates oxidative stress through upregulation of antioxidant gene expression in diabetic mouse vasculature.
Results
Oxidative stress contributed to miR-92a upregulation in diabetic mouse aortas and in ECs exposed to advanced glycation end products and oxidized low-density lipoprotein
Using quantitative polymerase chain reaction (q-PCR) analysis, we observed a higher expression of miR-92a in aortas from db/db mice compared with those from db/m+
mice (Fig. 1A), while no difference was detected in aortas with endothelium removed (Fig. 1B). Removal of endothelium was confirmed by significant loss of mRNA levels of EC-specific markers Cdh5, Pecam1, and Vwf (Fig. 1C and Supplementary Fig. S1C, D; Supplementary Data are available online at
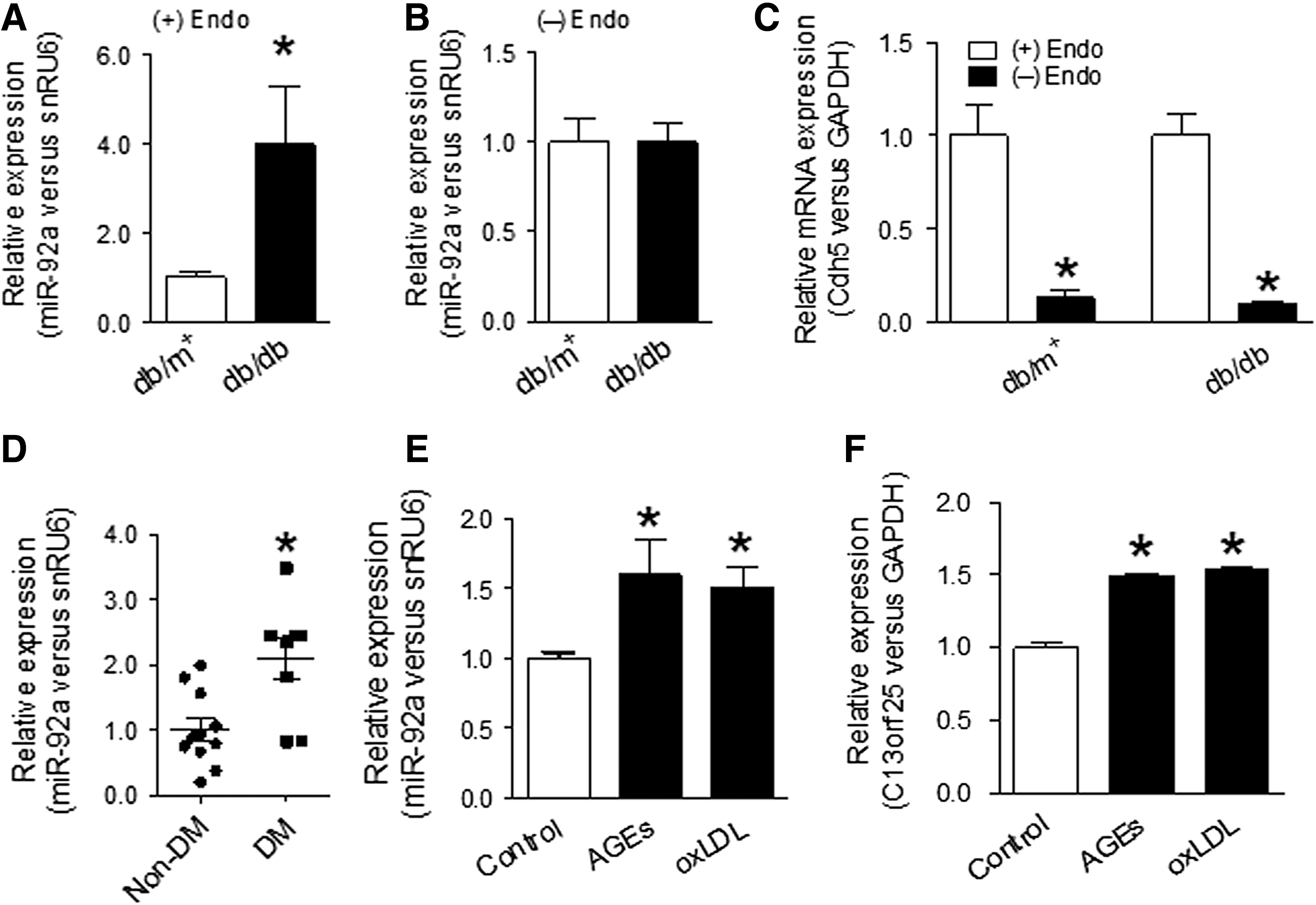
BW, body weight, kg; Cr, creatinine, μmol/L; DM, diabetes; HbA1c, glycated hemoglobin; non-DM, without diabetes.
In addition, human umbilical vein ECs (HUVECs) treated with advanced glycation end products (AGEs) and oxidized low-density lipoprotein (oxLDL) also expressed a higher level of miR-92a and the preliminary transcript of miR-17-92 cluster (Fig. 1E, F). These results suggest that diabetic condition might increase the expression of miR-92a derived from miR-17-92 cluster in ECs. Next, we tested whether oxidative stress contributed to upregulation of miR-92a in ECs. Tempol is a superoxide dismutase mimetic limiting the production of ROS (52). Tempol treatment decreased miR-92a expression in aortas isolated from db/db mice (Supplementary Fig. S2A). Also, the induction of miR-92a by AGEs and ROS generator HX-XO was attenuated by tempol treatment in HUVECs (Supplementary Fig. S2B, C). These results indicate that attenuation of oxidative stress can normalize miR-92a expression in ECs exposed to AGEs and HX-XO.
MiR-92a targets at HO-1 in ECs
Imbalanced regulation of antioxidant genes contributes to ROS overgeneration in vascular wall in diabetes. Given that our data show increased expression of miR-92a in diabetic mouse ECs, we hypothesize that miR-92a might regulate expression of antioxidant genes in ECs. To identify the candidates, we determined the expression of several oxidant-related genes in HUVECs transduced with Ad-anti-miR-92a, which inhibits miR-92a expression (Supplementary Fig. S3A, B). We found that HO-1 was upregulated by miR-92a inhibition at the protein level, while others were not changed (Supplementary Fig. S4A–C). HUVECs treated with AGEs, which increases miR-92a expression, expressed lower level of HO-1 protein (Supplementary Fig. S4D). Q-PCR and western blotting analysis showed that miR-92a downregulated the mRNA and protein expression of HO-1 in HUVECs (Fig. 2A, B).
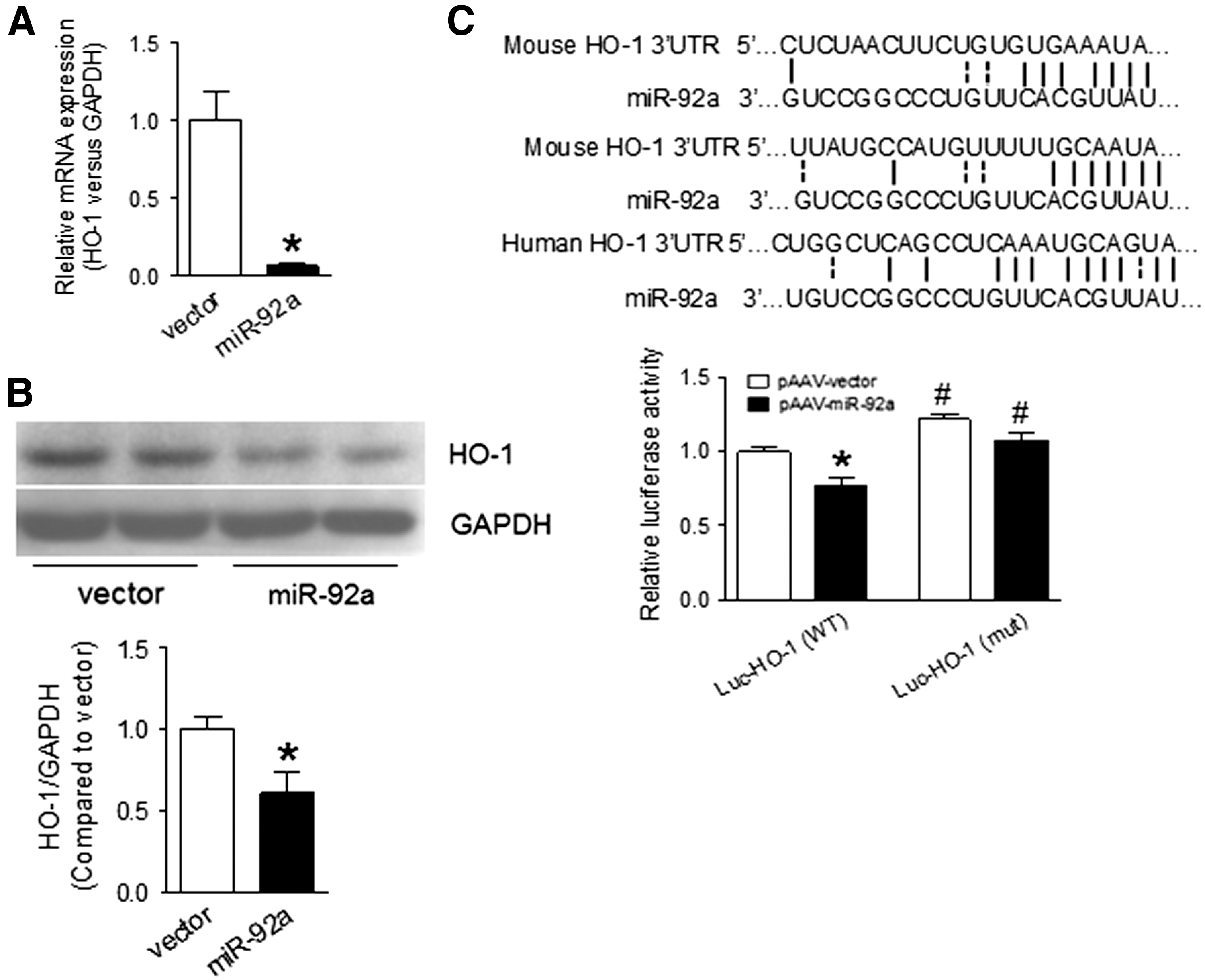
Using bioinformatic algorithms, HO-1 was predicted as a putative target of miR-92a (Fig. 2C). Indeed, miR-92a overexpression reduced wild-type (WT) but not mutated HO-1 3′UTR activity in HUVECs (Fig. 2C). Therefore, modulation of HO-1 expression by miR-92a is highly likely through a direct 3′UTR binding mechanism. By contrast, inhibition of miR-92a increased HO-1 mRNA and protein expression in HUVECs in a time-dependent manner (Fig. 3A–C and Supplementary Fig. S5). In addition, AGE-induced downregulation of HO-1 mRNA and protein was reversed by miR-92a inhibition (Fig. 3D, E). Taken together, our results demonstrate that HO-1 is a previously unrecognized target of miR-92a in ECs.
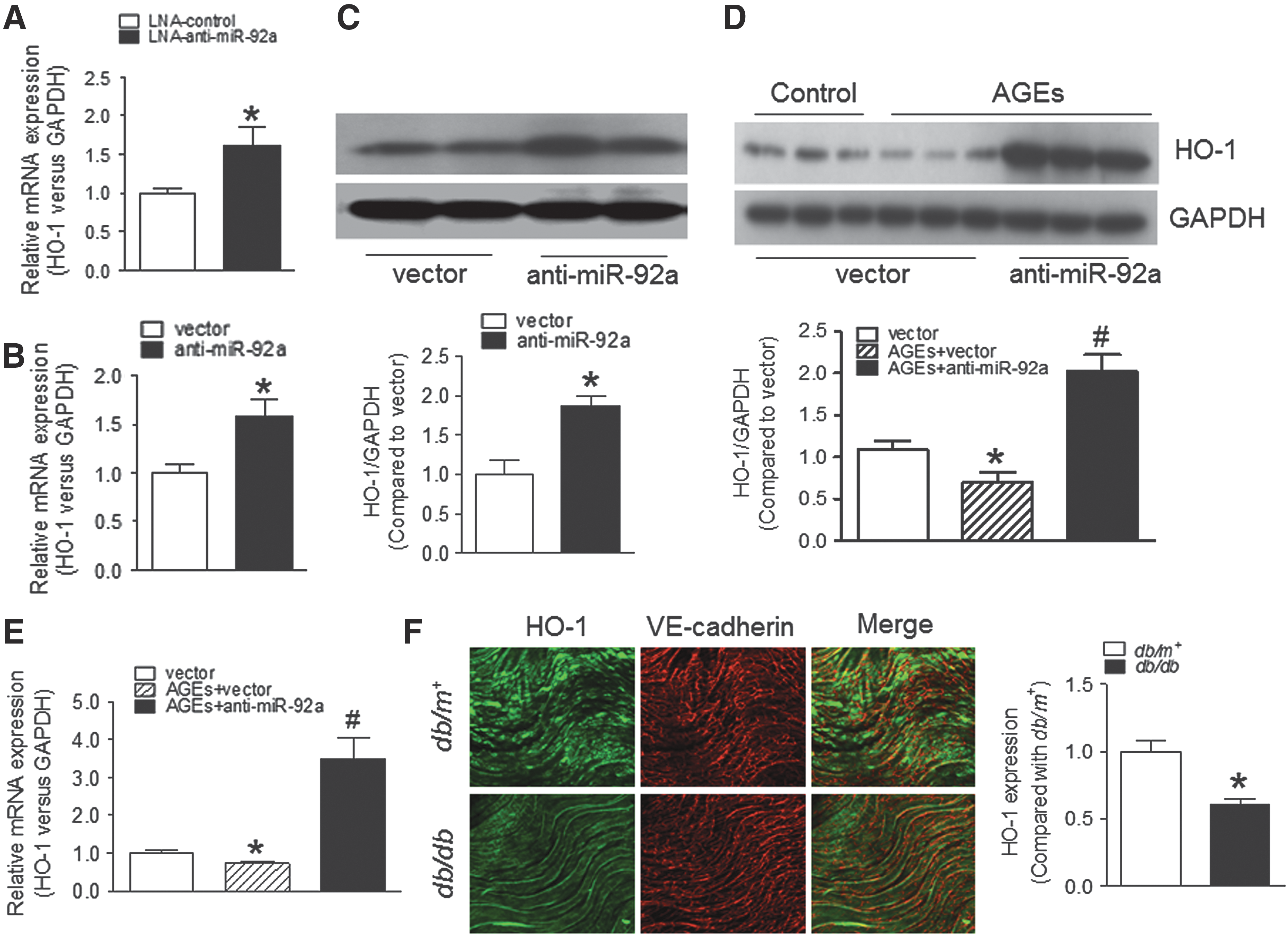
To determine whether HO-1 expression is downregulated in endothelium of db/db mice, we performed en face fluorescence staining of HO-1 in mouse aortas. The results showed that HO-1 level was decreased in aortic endothelium from db/db mice, compared with that from db/m+ mice (Fig. 3F). In addition, HO-1 mRNA level was elevated in aortas without endothelium from db/db mice compared with that from db/m+ mice (Supplementary Fig. S4F). These data suggest that elevated miR-92a is likely to suppress HO-1 expression in vascular ECs in db/db mice.
Overexpression of miR-92a impairs endothelial function
To test whether the elevated expression of miR-92a plays a functional role in ECs, we overexpressed miR-92a in mouse aortas using adeno-associated virus (AAV) vector. Ex vivo transduction of AAV-miR-92a for 36 h increased miR-92a expression in C57BL/6 mouse aortas (Fig. 4A). MiR-92a overexpression impaired ACh-induced EDRs, without affecting sodium nitroprusside (SNP)-induced endothelium-independent relaxations (EIRs) in C57BL/6 mouse aortas (Fig. 4B, C). These results indicate that increased miR-92a level in db/db mouse aortic endothelium impairs endothelial function.
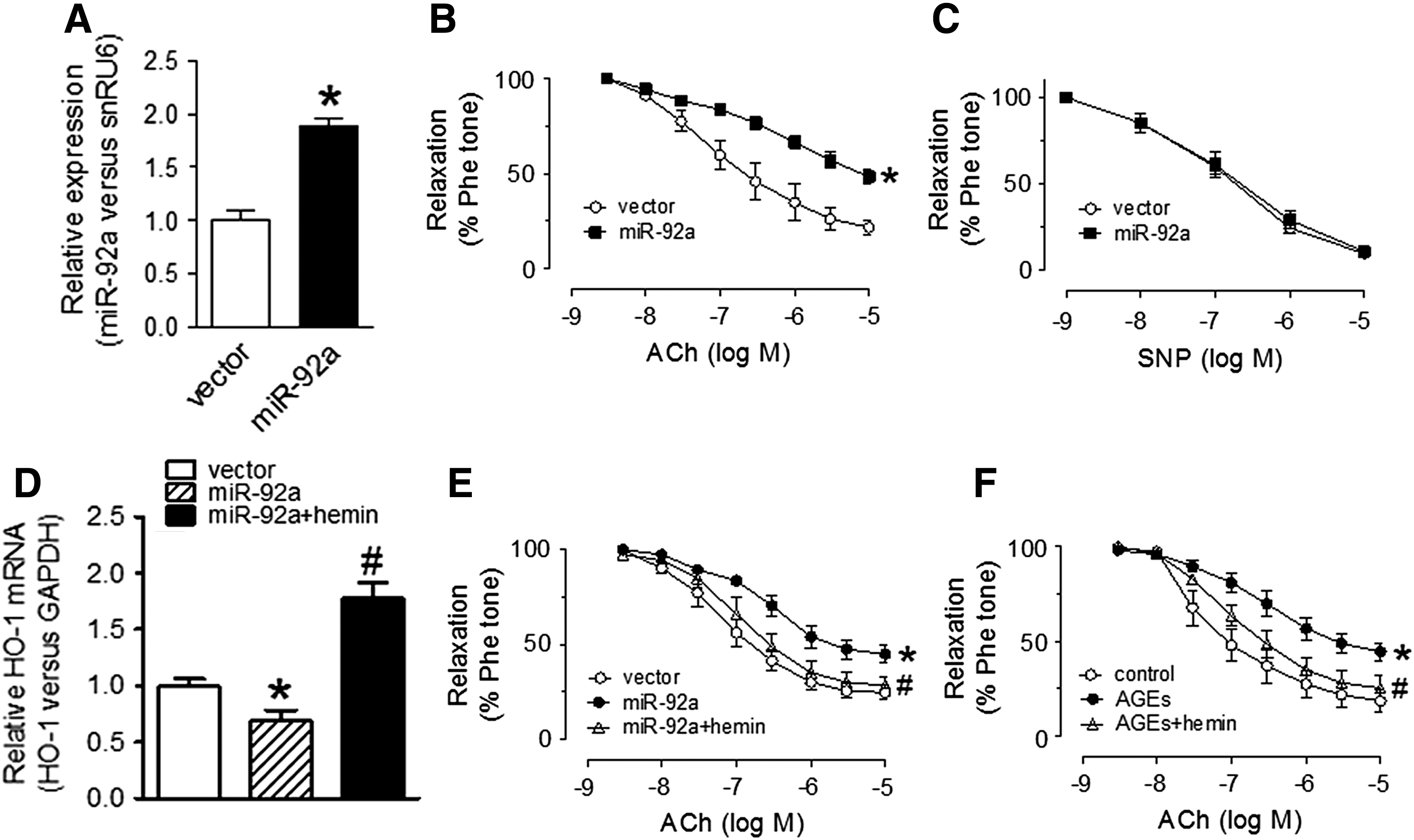
To determine the role of HO-1 in miR-92a overexpression-induced impairment of EDRs, aortic rings were treated with HO-1 inducer hemin to activate HO-1 before AAV-transduction of miR-92a. Detection of gene expression by q-PCR showed that miR-92a overexpression-induced downregulation of HO-1 mRNA level was increased by hemin treatment in mouse aortic rings (Fig. 4D). Hemin treatment also reversed the impairment of EDRs in aortas treated with AAV-miR-92a and AGEs (Fig. 4E, F). These data indicate that HO-1 activation by hemin is vasoprotective to antagonize the harmful effect of miR-92a overexpression.
Inhibition of miR-92a suppresses oxidative stress and improves ACh-induced EDRs in diabetic mice
Increased oxidative stress contributes to endothelial dysfunction in diabetes (17, 34). Based on our finding that the cytoprotective antioxidant enzyme HO-1 is inhibited by miR-92a overexpression, we hypothesize that miR-92a-inhibition-induced HO-1 upregulation might suppress ROS production in diabetic mouse ECs. First, we injected Ad-anti-miR-92a into db/db mice to inhibit miR-92a. The ROS level indicated by dihydroethidium (DHE) staining in aortic endothelium from db/db mice was higher compared with that from db/m+ mice (Fig. 5B). Ad-anti-miR-92a delivery reduced miR-92a expression and normalized ROS overproduction in db/db mouse aortic endothelium (Fig. 5A–C). Likewise, lucigenin chemiluminescence assay showed that miR-92a inhibition reduced superoxide levels in db/db mouse aortas (Fig. 5D). Moreover, comparable to the effect of ROS scavenger tempol, Ad-anti-miR-92a reversed AGE-induced superoxide production in cultured mouse aortas (Supplementary Fig. S6B). In addition, Ad-anti-miR-92a treatment in db/db mice also normalized the elevated levels of 3-nitrotyrosine, the oxidative stress biomarker in aortas (Supplementary Fig. S7). Collectively, these results demonstrate that inhibition of miR-92a suppresses ROS overproduction in ECs under diabetic conditions.
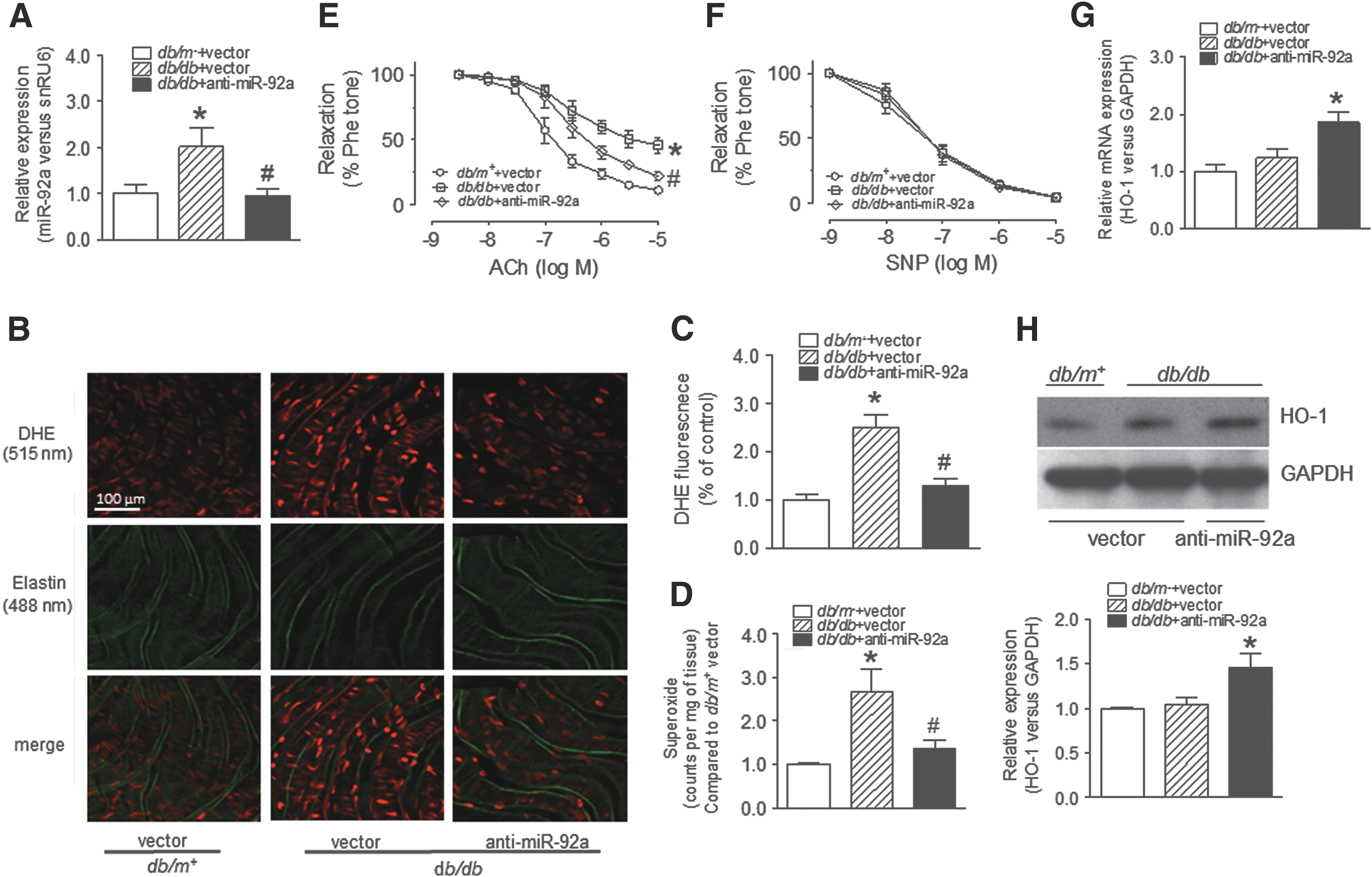
To assess whether miR-92a inhibition improves endothelial function in db/db mice, we measured ACh-induced EDRs in mouse aortas treated with Ad-anti-miR-92a. In vivo delivery of Ad-anti-miR-92a for 1 week improved EDRs, while SNP-induced EIRs were not affected (Fig. 5E, F). In addition, both endothelial nitric oxide synthases (eNOS) and p-eNOS (S1176) levels in db/db mouse aortas were upregulated by miR-92a inhibition (Supplementary Fig. S8A). Moreover, Ad-anti-miR-92a also increased eNOS and p-eNOS protein levels in HUVECs in the presence of AGEs (Supplementary Fig. S8B). On the contrary, miR-92a overexpression decreased levels of both p-eNOS and eNOS in HUVECs (Supplementary Fig. S8C). These results suggest that miR-92a inhibition-induced improvement of EDRs is likely mediated by increased NO bioavailability as reflected by upregulated eNOS and p-eNOS protein in db/db mouse aortas.
To confirm this likelihood, we assayed the level of nitrite/nitrate, the breakdown products of NO, and found that Ad-anti-miR-92a treatment increased nitrite/nitrate levels in both the culture medium for HUVECs and the plasma collected from db/db mice (Supplementary Fig. S8D, E). These data demonstrate that miR-92a inhibition protects endothelial function through increasing NO bioavailability in db/db mice.
We measured HO-1 mRNA and protein level in aortas from db/m+ and db/db mice treated with adenoviral vectors. Ad-anti-miR-92a transduction increased HO-1 mRNA and protein expression in db/db mouse aortas (Fig. 5G, H). However, no difference of HO-1 mRNA and protein expression in endothelium-intact aortas was found between db/m+ and db/db mice. While the en face imaging results showed a decreased endothelial HO-1 level in db/db mice (Fig. 3F). It is likely that the increased expression of HO-1 in aortic smooth muscle cells may offset the reduction of HO-1 expression in diabetic aortic ECs when the aortas were assayed for HO-1 levels using q-PCR and western blot (Supplementary Fig. S4F).
HO-1 inhibition by SnMP reversed the effect of miR-92a inhibition on AGE-induced ROS production in C57BL/6 mouse aortas and EDRs in db/db mouse aortas
To determine whether HO-1 mediates the effect of miR-92a inhibition on attenuation of oxidative stress and improvement of EDRs in db/db mouse aortas, we used SnMP (30 μM) to inhibit HO-1 activity as indicated by changes in bilirubin levels in HUVECs (Supplementary Fig. S9A, B). As shown by en face DHE staining, the effect of ex vivo transduction of Ad-anti-miR-92a on AGE-induced ROS production in C57BL/6 mouse aortas was reversed by SnMP, while the expression of HO-1 was not affected (Fig. 6A–D). Consistently, miR-92a inhibition by LNA-anti-miR-92a transfection attenuated AGE-induced ROS production in HUVECs (Fig. 6E, F).
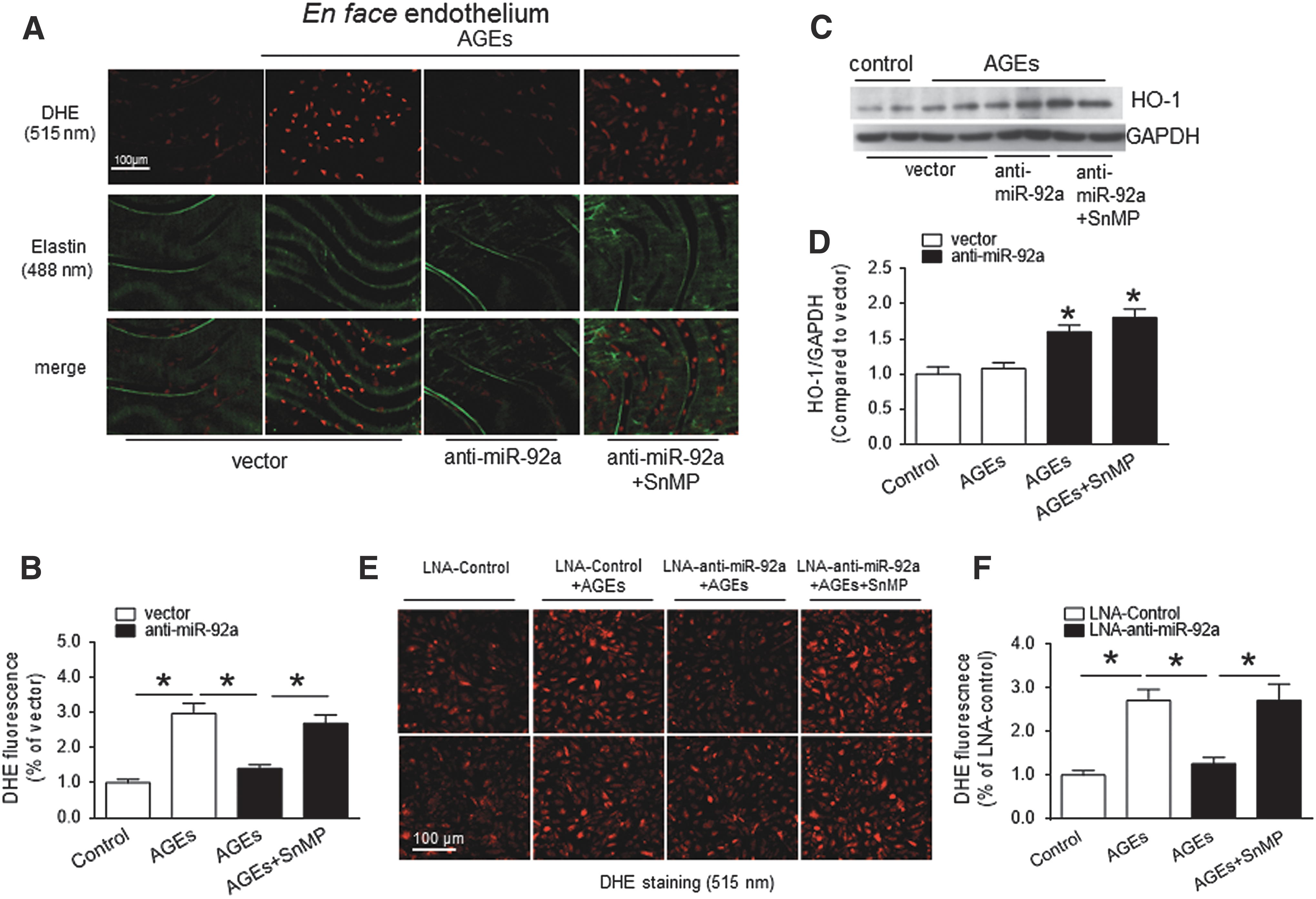
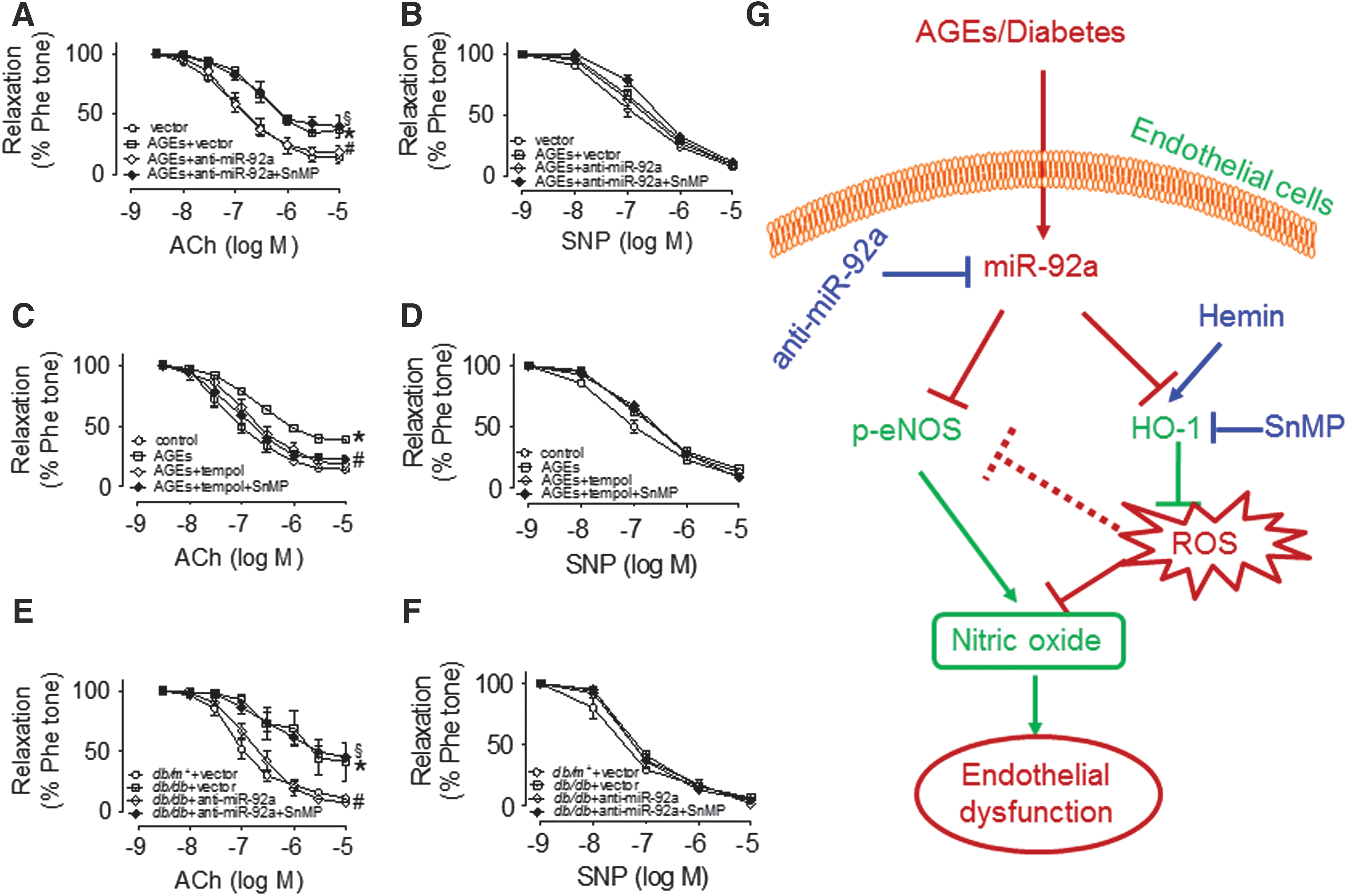
To exclude the possible off-target effect of SnMP, AAV-shHO-1, which specifically knocks down HO-1 expression, was used to confirm that HO-1 silencing also reverses the effect of miR-92a-inhibition on ROS production. The results of en face DHE staining showed that shHO-1 reversed the suppressive effect of anti-miR-92a on AGE-induced ROS production (Supplementary Fig. S10A, B). Functionally, SnMP treatment abolished miR-92a inhibition-induced improvement of EDRs in AGE-treated C57BL/6 mouse aortas, with no effect on tempol-rescued EDRs (Fig. 7A–D). Furthermore, administration of SnMP to db/db mice for 1 week reversed the protective effect of miR-92a inhibition on EDRs in db/db mouse aortas, and SNP-induced EIRs were not affected (Fig. 7E, F). Collectively, these results demonstrate that HO-1 mediates the vasoprotective effect of miR-92a inhibition in db/db mouse ECs.
Discussion
MiRNAs play important roles in regulating cardiovascular diseases through targeting a broad network of related genes (3, 9, 33). Increased expression of endothelial miR-92a is involved in the atherosclerotic progression (7, 28). In the present study, we found an increased miR-92a expression in db/db mouse aortic ECs (MAECs) and in renal arteries from diabetic patients. Our data showed that the upregulated miR-92a in diabetic mouse ECs is possibly oxidative stress dependent, and miR-92a inhibition normalized ROS level in vascular wall and improved EDRs in diabetic db/db mouse aortas.
Mechanistically, miR-92a targets at endothelial HO-1, and the upregulated HO-1 mediates miR-92a-inhibition-induced beneficial effect on endothelial function in db/db mice. The results highlight the potential importance of miR-92a in mediation of vascular dysfunction in diabetes, and normalization of the upregulated miR-92a level might hold therapeutic promise against diabetic vasculopathy.
One mechanism responsible for the upregulation of miRNA expression under diabetic condition is oxidative stress. For example, ROS-dependent elevation of miR-200c in ECs increases COX-2 expression to impair endothelial function in diabetic mice (54). Existing evidences show that miR-92a is abundantly expressed in ECs and exhibits antiangiogenic function (12). Oxidative stress-dependent activation of sterol regulatory element binding protein 2 (SREBP2) is reported to transactivate miR-92a expression in ECs (7). Despite the importance of miR-92a in regulation of vascular health, the expression pattern and how miR-92a expression is regulated in diabetic mouse ECs remain unclear. Our results showed that diabetes promotes miR-92a expression, which is likely mediated by increased oxidative stress in vascular wall of diabetic mice. The results were supported by the observation that tempol treatment normalized the increased miR-92a expression in db/db mouse aortas and in HUVECs treated with AGEs and HX-XO.
Dyslipidemia, mainly characterized with elevation of circulating cholesterol and triglyceride levels, is recognized as a major risk factor for diabetic complications (20, 44). In addition, increased AGE formation contributed to endothelial dysfunction in diabetes (4, 42). Both oxLDL and AGEs are able to trigger oxidative stress in ECs (16, 18). These previous findings suggest that lipoprotein- and AGE-associated oxidative stress is highly possible to mediate the upregulation of miR-92a in diabetic ECs. Further experiment using tempol treatment in db/db mice is needed to test this hypothesis. Moreover, diabetes increases SREBP2, which is a transcriptional activator of miR-92a in renal tissues (21). Therefore, whether the increased miR-92a expression in diabetic ECs is mediated by SREBP2 is worthy of further investigation. In addition, upregulation of nuclear factor-κB (NF-κB) activity increases miR-92a expression in ECs (7). However, in our preliminary test, IL-1β treatment failed to increase miR-92a expression in HUVECs (Supplementary Fig. S2D). Although NF-κB is known to be activated in diabetic vasculature, it is less likely to account for miR-92a elevation in diabetes based on our preliminary results.
Recently, miRNAs are recognized as important regulators of multiple cellular processes, including oxidative stress (51). In diabetic cardiomyocytes, miR-144 overexpression increases ROS production, while miR-144 inhibition abates excessive ROS levels (53). Furthermore, downregulation of miR-137 reduces high glucose-induced cellular oxidative stress in ECs (25). In contrast, augmenting the expression of miR-200c in HUVECs leads to increased oxidative stress and cellular injury (29). These reports highlight miRNAs as critical regulators of cellular ROS production, which can be targeted by miRNA inhibitors or activators. In this study, our results revealed that miR-92a inhibition reduces endothelial oxidative stress in vivo and in vitro. To our knowledge, this is the first report describing the functional significance of miR-92a inhibition in combating against oxidative stress in diabetes. In addition, our findings further support the notion that miRNAs are functional regulators of oxidative stress in pathological conditions.
ROS overproduction critically contributes to diabetic endothelial dysfunction. This adverse effect is likely through the mechanisms involving decreased NO bioavailability and direct damage to ECs (34, 37). In contrast, normalization of oxidative stress using antioxidants reduces vascular injury in diabetes, suggesting the beneficial effect of antioxidant treatment on diabetic vasculopathies (23, 47). Our results showed that miR-92a inhibition increased p-eNOS and plasma nitrite/nitrate level and improved EDRs in db/db mouse aortas. This suggests that the improvement of endothelial function by inhibition of miR-92a in ECs is most likely through enhancement of eNOS-NO signaling in diabetes. MiR-92a inhibition also increased basal eNOS protein levels. However, whether the improvement of EDRs is secondary to the suppressed endothelial ROS levels by miR-92a inhibitor is currently unsolved. Therefore, further experiments using ROS scavengers to treat db/db mice before anti-miR-92a administration are needed to determine the relative contribution of ROS suppression and eNOS upregulation to miR-92a inhibition-induced preservation of NO.
We identified the target genes that might contribute to the protective effect of miR-92a inhibition on diabetic ECs. We performed a screening on expression of oxidant-related enzymes in HUVECs treated with miR-92a inhibitor. MiR-92a inhibition increased HO-1 protein expression, while others were not changed. Dual luciferase assays with WT and mutated 3-′UTR of the HO-1 gene support the prediction that miR-92a inhibition may directly upregulate HO-1 expression in ECs. As a critical inducible antioxidant and cytoprotective enzyme, few studies investigated the mechanisms underlying the regulation of HO-1 expression in ECs, especially under diabetic conditions. In the present study, we found that AGEs downregulated the expression of HO-1, which was increased by miR-92a inhibition in HUVECs. Consistently, the en face staining data showed a downregulation of HO-1 level in aortic ECs from db/db mice. Importantly, we also demonstrate that HO-1 mediates the vasoprotective effect of miR-92a inhibition in diabetic mouse ECs.
HO-1 is critically involved in regulation of organ function, such as the heart, kidney, and brain (2, 10, 19, 35). Carbon monoxide and bilirubin, two molecules downstream of HO-1 signaling, are the major mediators of the regulatory function of HO-1 in tissue homeostasis and redox signaling (38, 40). Recent studies have implicated that HO-1 polymorphisms are associated with a high risk of vascular diseases (26, 30). Induction of HO-1 exerts arterial antioxidant actions and causes vascular relaxations (1, 13, 27, 45). We showed that miR-92a inhibition increased HO-1 expression, and HO-1 inhibitor SnMP reversed the beneficial effect of miR-92a inhibition on endothelial function in db/db mouse aortas. Induction of krüppel-like factor 2 (KLF2), a target gene of miR-92a, also enhances HO-1 expression in ECs (15). We found that miR-92a inhibition increased KLF2 expression in ECs (Supplementary Fig. S4E), which was consistent with previous reports (50). Therefore, it is also valuable to study whether induction of HO-1 by miR-92a inhibition can be contributed by KLF2 in ECs under diabetic conditions.
In conclusion, our study provides the first line of evidence that increased expression of miR-92a in arteries from diabetic mice and patients. MiR-92a inhibition improves endothelial function and normalizes excessive ROS levels through enhancing HO-1 expression and activity (Fig. 7G). Thus, targeting miR-92a-HO-1 cascade can be an alternative therapeutic strategy for ameliorating diabetic vasculopathy.
Materials and Methods
Animals
C57BL/6 mice, db/db, and db/m+ mice were provided by the Chinese University of Hong Kong (CUHK) Laboratory Animal Service Center and maintained at controlled temperature (22–23°C, 55% ± 5% humidity) with a 12-h light/12-h dark cycle with free access to standard mice diet (Research Diet, Inc.) and water. All animal procedures were approved by the CUHK Animal Experimentation Ethics Committee.
EC culture
HUVECs (Lonza, Walkersville, MD) were cultured in EC growth medium (CC3024; Lonza) with 10% fetal bovine serum (FBS) and antibiotics. Cells from passage 4 to 8 were used for experiments. Primary MAECs were cultured in the method of previous reports (48).
Construction and production of recombinant virus
The full human miR-92 sequences were amplified by PCR from human genomic DNA and cloned into pAAV-MCS digested by BglII and HindIII. The RGDLRVS tagged AAV (serotype 9, a gift from Dr. O.J. Müller, University Hospital Heidelberg, Heidelberg, Germany), which has higher infection efficiency in ECs and induces long-lasting expression, was used to packaging recombinant AAV (46). Empty AAV-vector was used as control in corresponding experiments. Ad-anti-miR-92a against mature miR-92a was purchased from Vector Biolabs (USA). Empty Ad-vector was used as the control in corresponding experiments.
Transfection with LNA-anti-miR-92a
Apart from Ad-anti-miR-92a, LNA-anti-miR-92a was used to inhibit miR-92a function in HUVECs. LNA-anti-miR-92a and LNA-control were purchased from Exiqon Inc. with sequences as follows: 5′-AGGCCGGGACAAGTGCAAT-3′ (LNA-anti-miR-92a) and 5′TAACACGTCTATACGCCCA-3′ (LNA-control). Lipofectamine® RNAiMAX Reagent (Invitrogen) was used as the transfection reagent to transfect LNA-anti-miR-92a into HUVECs. Six hours after transfection, culture medium was changed to fresh medium and HUVECs cultured for additional specified time.
Construction of HO-1 3′UTR reporter
The 3′UTR of HO-1 containing the putative binding sites of miR-92a was cloned into pmirGLO (Promega) digested by Pme1 and Xho1. The HO-1 3′UTR was cloned using primers as follows: forward primer: CGGGTTTAAACGGCT CCCAGGGCCATGAACTT and reverse primer: CCGTCTAGACAAGCTACT ATCAGACAATGTT. In addition, mutated HO-1 3′UTR was cloned with a site-directed mutagenesis method. The primers were used as below:
Upper primers,
forward primer: CGGGTTTAAACGGCTCCCAGGGCCATGAACTT and
reverse primer: ATGTCATGTTTGAGGCTGAGCCAGG
Lower primers,
forward primer: AACATGACATTTTTGTTGTGTTCTGTTGTTTTTATAGC and
reverse primer: CCGTCTAGACAAGCTACTATCAGACAATGTT
Construction and production of HO-1 shRNA AAV
shRNA sequence targeting mouse HO-1 was generated from Sigma-Aldrich and has been used previously (27). Briefly, the complementary oligonucleotides (oligos) of HO-1 shRNA were annealed and ligated into the pAAV-ZsGreen shuttle vector. The RGDLRVS-tagged AAV (serotype 9) was used to package AAV-shHO-1 in HEK-293T cells.
Organ culture of mouse aortas and functional assay
Mouse aortas were dissected in ice-cold Krebs solution containing (in mmol/L) 119 NaCl, 4.7 KCl, 2.5 CaCl2, 1 MgCl2, 25 NaHCO3, 1.2 KH2PO4, and 11
ROS production in aortas and ECs
En face staining of ROS generation using DHE fluorescence was performed in en face endothelium from mouse aortas according to the previous report (55). In brief, isolated rings from mouse aorta were incubated with DHE in extracellular medium (ECM, in mmol/L: 121 NaCl, 5 NaHCO3, 10 Na-HEPES, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 2 CaCl2, 10
Western blotting
Cells and aortic tissues were homogenized in an ice-cold RIPA lysis buffer composed of RIPA solution, complete protease inhibitor cocktail, and PhosSTOP phosphatase inhibitor cocktail (Roche). After lysis for 30 min on ice, the lysates were centrifuged at 20,000 g for 20 min at 4°C. After concentration measurement, the proteins were mixed with 4 × loading buffer, following by denaturation for 10 min. Subsequently, equal amounts of protein samples were electrophoresed on a 7.5%–12.5% sodium dodecyl sulfate/polyacrylamide gel together with the prestained protein molecular weight marker. The resolved proteins were electrophoretically transferred onto an immunoblot polyvinylidene difluoride membrane (Bio-Rad) in the Bio-Rad Trans-Blot cell at 110 V for 100 min. The blots were blocked with 3% Bovine serum albumin (BSA) dissolved in TBST buffer for 1 h, following incubation with primary antibodies, including anti-eNOS (1:1000; BD Biosciences, San Jose, CA), anti–phospho (p)-eNOS Ser1177 (1:1000; Abcam; Cambridge, United Kingdom), anti-HO-1 (1:500; Abcam), anti-NQO1 (1:1000; Abcam), anti-NOX4 (1:1000; Abcam), anti-NOX2 (1:1000; Abcam), anti-p67pox (1:500; Cell Signaling, Danvers, MA), anti-SOD1 (1:1000, Santa Cruz, California), anti-SOD2 (1:1000, Santa Cruz), and anti-nitrotyrosine (1:500; Millipore). Then, the membrane was washed three times in TBST and subsequently incubated with corresponding secondary antibodies conjugated to horseradish peroxidase (DakoCytomation) at the dilution of 1:3000 for 1 h at room temperature. After washing with TBST for three times, the membrane was developed with ECL detection solutions before exposure to X-ray films. Densitometry was performed using a documentation program (Flurochem). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was selected as internal control for checking equal loading of each sample.
En face immunofluorescence staining
Aortic segments were fixed in 4% paraformaldehyde, followed by permeabilization with 0.01% Triton X-100 for 15 min at room temperature. After washing with PBS for three times, the aortic rings were blocked with 5% normal donkey serum overnight at 4°C, and primary antibody incubations were performed for an additional 24 h at 4°C using mouse anti-HO-1 (1:100 dilution; Abcam) and goat anti-VE cadherin (1:100 dilution; Santa Cruz) antibodies. Subsequently, the aortic segments were incubated in a corresponding fluorescent second antibody, including Alexa Fluor 488 anti-mouse and Alexa Fluor 546 anti-goat (1:500 dilution; Molecular Probes), for 2 h at room temperature. Finally, the specimens were cut open and imaged by a confocal system as described (56).
Q-PCR analysis
For mRNA detection, total RNA was extracted from ECs or mouse aortas using RNAiso plus (TAKARA) according to the manufacturer's instruction. MicroRNA detection using q-PCR analysis was performed using NCode™ SYBR® Green miRNA q-PCR Kit (Invitrogen). The primers were purchased from GeneCopoeia (Guangzhou). U6 small nuclear RNA (snRU6) was used as endogenous control. In addition, TaqMan miRNA assay (Life Technology) was used to confirm miR-92a alterations in ABI ViiA7 system (Applied Biosystems). snRU6 was used as the internal control for miRNA detection.
HO-1 activity and nitrite/nitrate assay
Changes of HO-1 activity were measured as the bilirubin production in HUVECs. Forty-eight hours after SnMP treatment, cultured medium from HUVECs was collected for bilirubin assay. The bilirubin level was determined using an assay kit (BioVision) according to the manufacturer's instructions. NO level was measured as the accumulation of accumulated nitrite/nitrate. Briefly, NO level in cell culture medium and plasma of db/db mice was detected using the nitrite/nitrate colorimetric assay kit (Cayman Chemicals).
Luciferase assays
To identify whether miR-92a has a potential direct regulation on HO-1 expression, we examined whether miR-92a overexpression decreased the firefly luciferase activity in HUVECs transduced with HO-1 3′UTR-containing luciferase plasmids. In brief, pAAV-miR-92a was cotransfected with WT pmirGLO-HO-1 3′UTR, or mutant pmirGLO-HO-1 3′UTR using a Neon Transfection kit (Lonza). Luciferase activity was measured using a Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol.
Statistical analysis
All data are presented as mean ± standard error of mean. GraphPad Prism software (Version 5.0) was used for all data analysis. Concentration/response curves were analyzed using nonlinear regression curve fitting. Statistical significance was evaluated by Student's t-test or one-way ANOVA. p < 0.05 was interpreted to denote statistical significance.
Footnotes
Acknowledgments
This study is supported by the Hong Kong Research Grants Council (C4024-16W, CUHK14105814 and 14124216), the Natural Science Foundation of China (81561128017), the CUHK Vice Chancellor's Discretionary Fund, and the CUHK Lui Che Woo Foundation.
Author Disclosure Statement
There are no competing financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.