
Abstract
Aims:
Staphylococcus aureus plays an important role in sepsis, pneumonia, and wound infections. Acid sphingomyelinase (Asm)-deficient mice are highly susceptible to pulmonary S. aureus infections. Here, we investigated the role of CD44 as a molecule that mediates important aspects of the infection of macrophages with S. aureus.
Results:
We showed that CD44 activation by S. aureus stimulated Asm via the formation of reactive oxygen species, resulting in ceramide release, clustering of CD44 in ceramide-enriched membrane platforms, CD44/Asm-dependent activation of Rho family GTPases, translocation of phospho-ezrin/radixin/moesin to the plasma-membrane, and a rapid rearrangement of the actin cytoskeleton with cortical actin polymerization. Genetic deficiency of CD44 or Asm abrogated these signaling events and thereby reduced internalization of S. aureus into macrophages by 60–80%. Asm-deficient macrophages also exhibited reduced fusion of phagosomes with lysosomes, which prevented intracellular killing of S. aureus in macrophages and thereby allowed internalized S. aureus to replicate and cause severe pneumonia.
Innovation and Conclusion:
The CD44-Asm-ceramide system plays an important role in the infection of macrophages with S. aureus. Antioxid. Redox Signal. 28, 916–934.
Introduction
S
Staphylococcus aureus infections are clinically very important. We demonstrate that macrophages are activated via CD44 on infection with S. aureus. CD44 triggers a release of reactive oxygen species (ROS) that mediate activation of the acid sphingomyelinase (Asm) and release of ceramide. Ceramide forms distinct membrane domains that cluster CD44 and thereby amplify signaling. Downstream events regulated by this novel pathway are activation of small G-proteins, alterations of the cytoskeleton, internalization of the pathogen, and intracellular fusion of phagosomes with lysosomes. The data show that the CD44-Asm-ceramide system plays an important role in the infection of macrophages with S. aureus.
Acid sphingomyelinase (human gene symbol, ASM; mouse gene symbol, Asm) is a glycoprotein that functions as a hydrolase catalyzing the conversion of sphingomyelin to ceramide (57). Asm is active at an acidic pH not only in lysosomes (10, 57) but also on the outer leaflet of the plasma membrane after surface translocation or secretion of the enzyme (16, 17). The plasma membrane contains domains with acidic pH (66); in addition, Asm functions are also determined by the lipid composition of the membrane allowing the enzyme to be active at almost neutral pH values (14, 55, 56), and thereby to hydrolyze sphingomyelin at the cell surface. Ceramide molecules generated by Asm within membranes tightly interact with each other and spontaneously form distinct domains in the plasma membrane; these small domains then form ceramide-enriched platforms (13, 14, 19, 25). These platforms form a unique microenvironment with biophysical properties that allow them to trap and cluster receptor molecules and intracellular signaling molecules, thereby permitting and amplifying signal transduction (3, 13, 15 –17). Ceramide also directly interacts with cathepsin D, phospholipase A2, kinase suppressor of Ras, and protein kinase C isoforms (21, 24, 27, 68). Recent studies have indicated that the Asm-ceramide system has an important role in regulation of macrophage functions, such as phagosome maturation (64a), cytokine release (28), and apoptosis (53, 69).
The present study was performed to investigate the role of the Asm-ceramide system in the infection of macrophages with S. aureus. We identified that CD44 is critically involved in the infection of macrophages by S. aureus. CD44 induced the generation of ROS that in turn connected CD44 with the Asm, resulting in the formation of ceramide-enriched domains after infection. These domains clustered and thereby amplified CD44 signaling resulting in further activation of the Asm providing a positive forward feedback loop between CD44 and the Asm. CD44 activation by S. aureus stimulates small G proteins, a reorganization of the cytoskeleton, internalization of the pathogen, and fusion of phagosomes with lysosomes, a process that requires again Asm. Deletion of CD44 or the Asm prevents internalization of S. aureus.
Results
Asm is activated upon S. aureus infection and is crucially involved in internalization of the pathogen
The engulfment of bacteria into macrophages is a common mechanism of the host-defense system for eliminating pathogens, but it is also a mechanism used by the bacteria to escape the hostile extracellular environment and the immune system and thus to survive in infected cells. To determine whether Asm is involved in the uptake of S. aureus, we infected wild-type (WT) and Asm-deficient bone marrow-derived macrophages (BMDMs) with S. aureus for 30, 60, or 120 min or left them uninfected and then determined Asm activity. The results show a rapid and strong activation of Asm in WT macrophages, but no Asm activity in Asm-deficient macrophages (Fig. 1A, B). WT macrophages rapidly internalized S. aureus, which was severely reduced in Asm-deficient macrophages (Fig. 1C). Asm deficiency did not affect the adhesion of S. aureus bacteria to macrophages (Fig. 1D). Taken together, these findings indicate that Asm is activated by and is crucial for S. aureus internalization in macrophages.
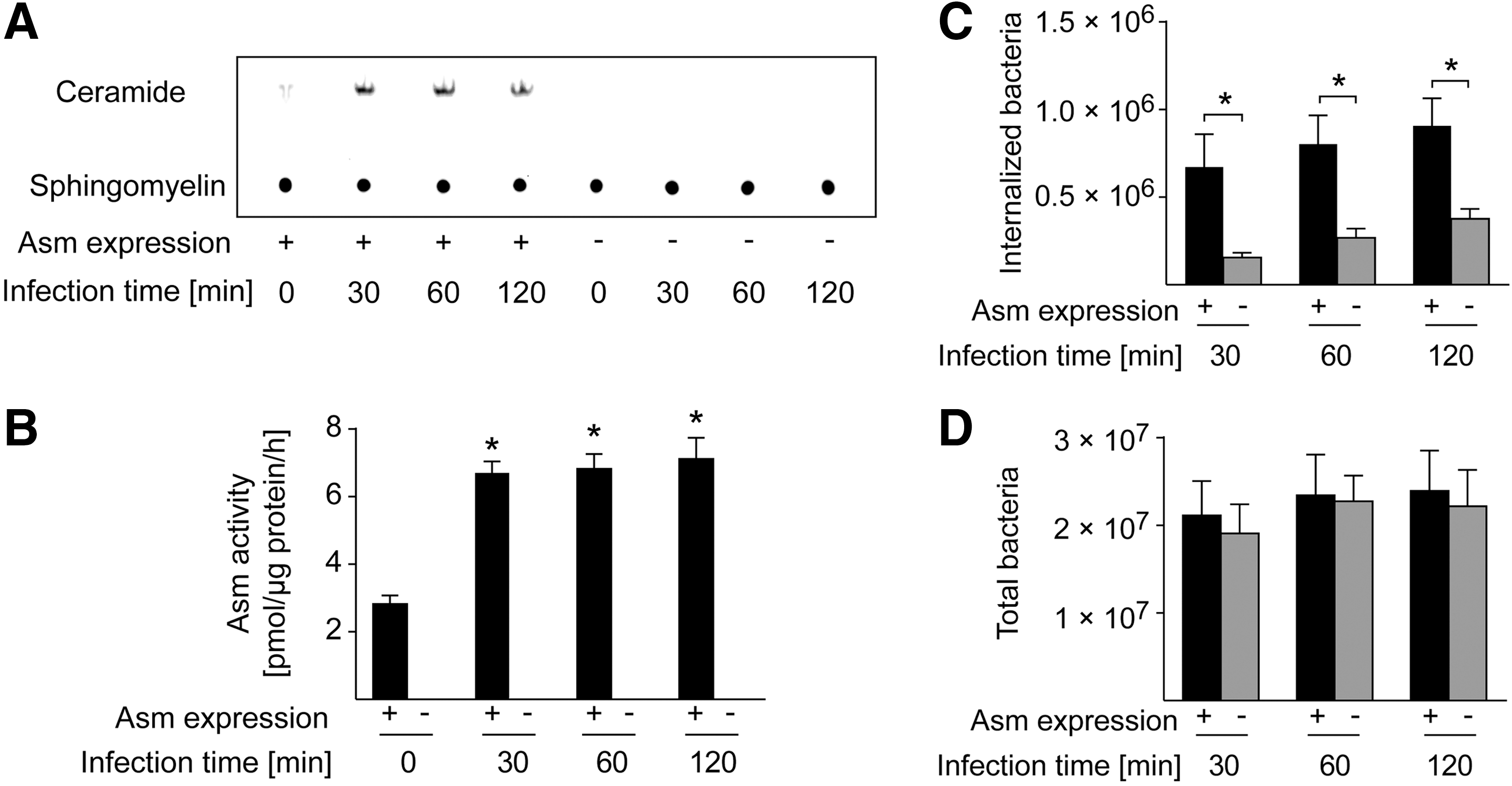
Asm is required for internalization-induced actin polymerization
Actin cytoskeleton dynamics in macrophages is essential for the recognition of pathogens and for phagocytosis (33, 39, 59). Infection of WT BMDMs with S. aureus resulted in dramatic changes of the cell shape, as determined by scanning electron microscopy (SEM) (Fig. 2A). Filopodia, which are thin finger-like actin-rich plasma-membrane protrusions, began to form as early as 30 min after S. aureus infection of WT macrophages and continued to extend at 60 and 120 min after infection. The formation and extension of filopodia were much less prominent and occurred much more slowly in Asm-deficient macrophages infected with S. aureus (Fig. 2A). Immunofluorescence staining of BMDMs (Fig. 2B, C) or of alveolar macrophages (AMs) (Fig. 2D, E) with FITC-phalloidin confirmed these results and showed that actin filaments reassemble and aggregate at the cell surface in WT macrophages after infection with S. aureus, but not in Asm-deficient macrophages.
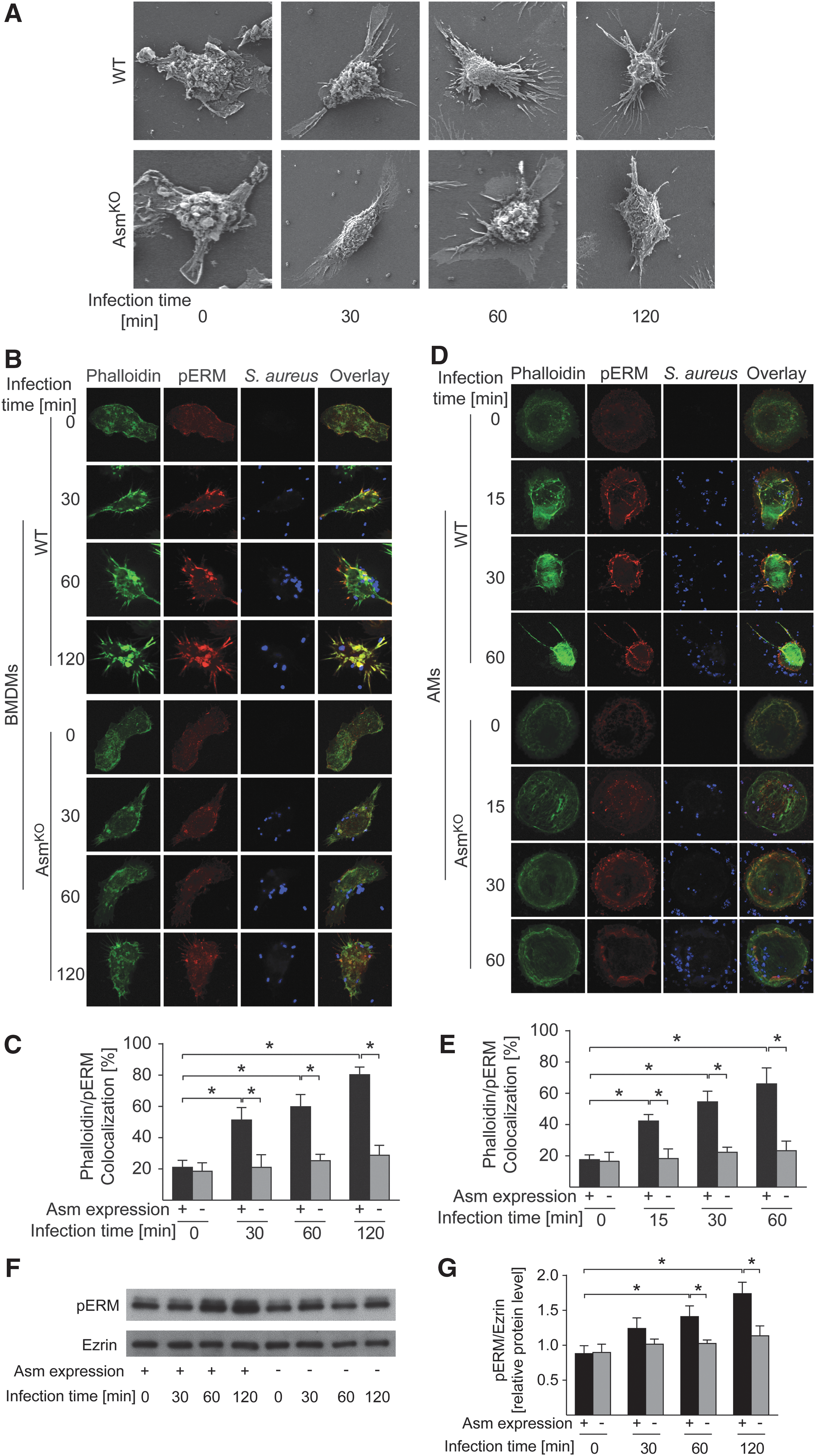
The ezrin/radixin/moesin (ERM) family proteins that bind to actin function as linkers between the plasma membrane and the actin cytoskeleton and transduce signals to mediate cytoskeleton remodeling (9, 22). ERM proteins are activated by phosphorylation (11, 45, 46). To determine whether Asm (indirectly) mediates the phosphorylation of ERM proteins after S. aureus infection, we infected WT and Asm-deficient BMDMs with S. aureus. We then stained the cells with immunofluorescent anti-phospho-ERM antibodies or lysed them, performed Western blots, and measured ERM phosphorylation, respectively. S. aureus infection induced a rapid and marked phosphorylation of ERM proteins in WT BMDMs and AMs, which was almost completely absent in Asm-deficient BMDMs or AMs (Fig. 2B, D, F). In WT macrophages, phosphorylated ERM proteins (pERM) rapidly colocalized with phalloidin in newly formed filopodia at the site of the interaction of S. aureus with the membrane (Fig. 2B, D), whereas pERM or phalloidin did not change in Asm-deficient BMDMs or AMs (Fig. 2B, D).
RhoA, Rac1, and Cdc42 are members of the Rho family of small GTPases, which regulate many aspects of intracellular actin dynamics (51, 52, 58). To determine whether Asm regulates the activity of those Rho GTPases on S. aureus infection, we determined their activity by pull-down assays. RhoA, Rac1, and Cdc42 were activated after S. aureus infection in WT BMDMs (Fig. 3A–C), whereas their activation was almost completely abrogated by Asm deficiency (Fig. 3A–C).
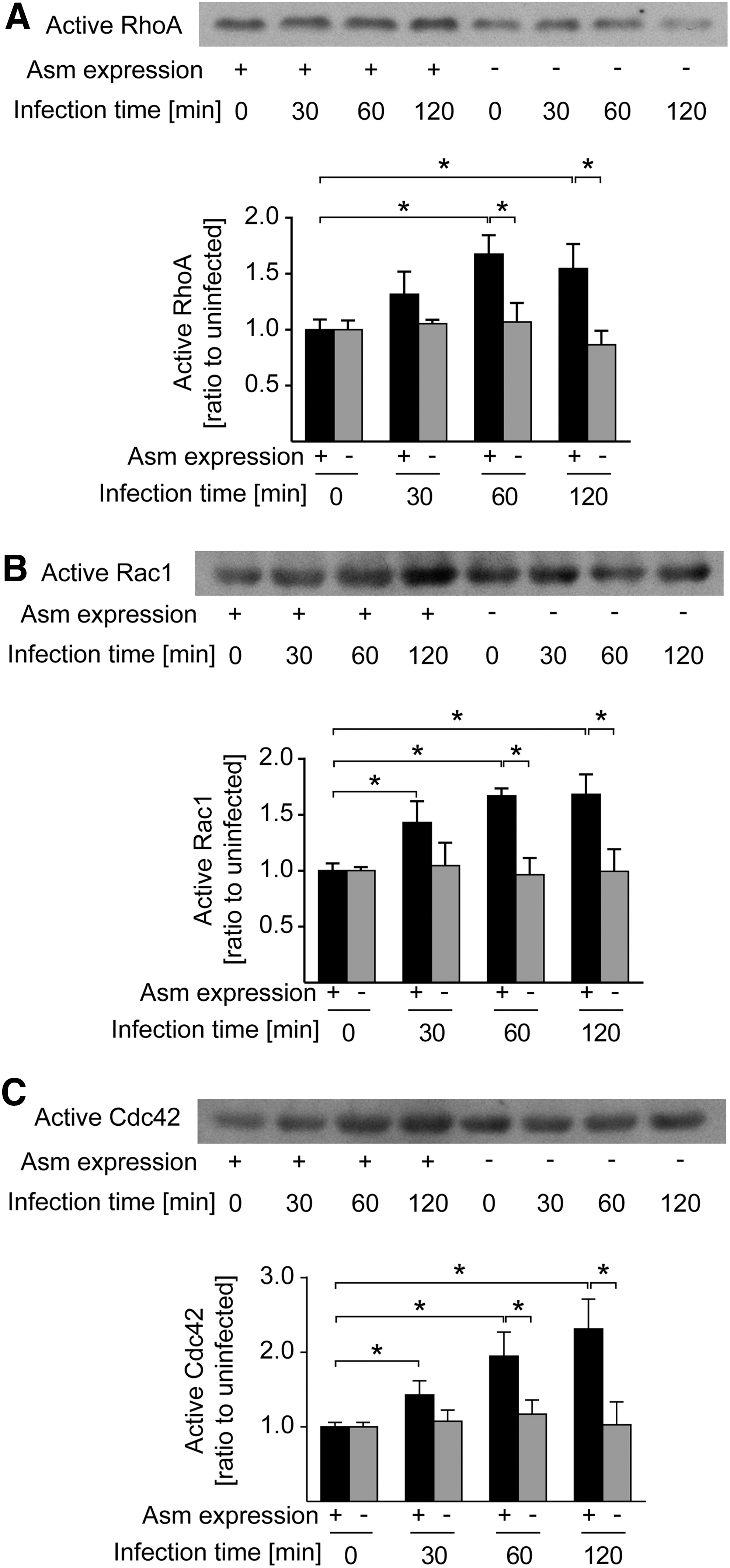
Collectively, these findings indicate that Asm in macrophages is necessary for S. aureus-induced morphology changes, actin cytoskeleton rearrangement, ERM phosphorylation, and Rho GTPase activation.
CD44 links S. aureus to the Asm
CD44 is a glycoprotein that interacts with ERM and links the actin cytoskeleton to the plasma membrane and the extracellular matrix (12, 37). We therefore hypothesized that CD44 might be involved in the activation of the Asm-ceramide signaling pathway by S. aureus. To study this hypothesis, we infected WT and Asm-deficient BMDMs with S. aureus and stained them with specific antibodies to CD44. The results reveal that infection with S. aureus induced a translocation of CD44 to the plasma membrane and colocalization with actin filaments, which were recruited to the infection site at the cell surface. These translocation and colocalization events were almost completely absent in Asm-deficient BMDMs or AMs (Fig. 4A, B, E, F). Furthermore, on infection with S. aureus, CD44 clustered within ceramide-enriched membrane platforms in WT BMDMs or AMs, events that were again absent in Asm-deficient cells (Fig. 4C, D, G, H).
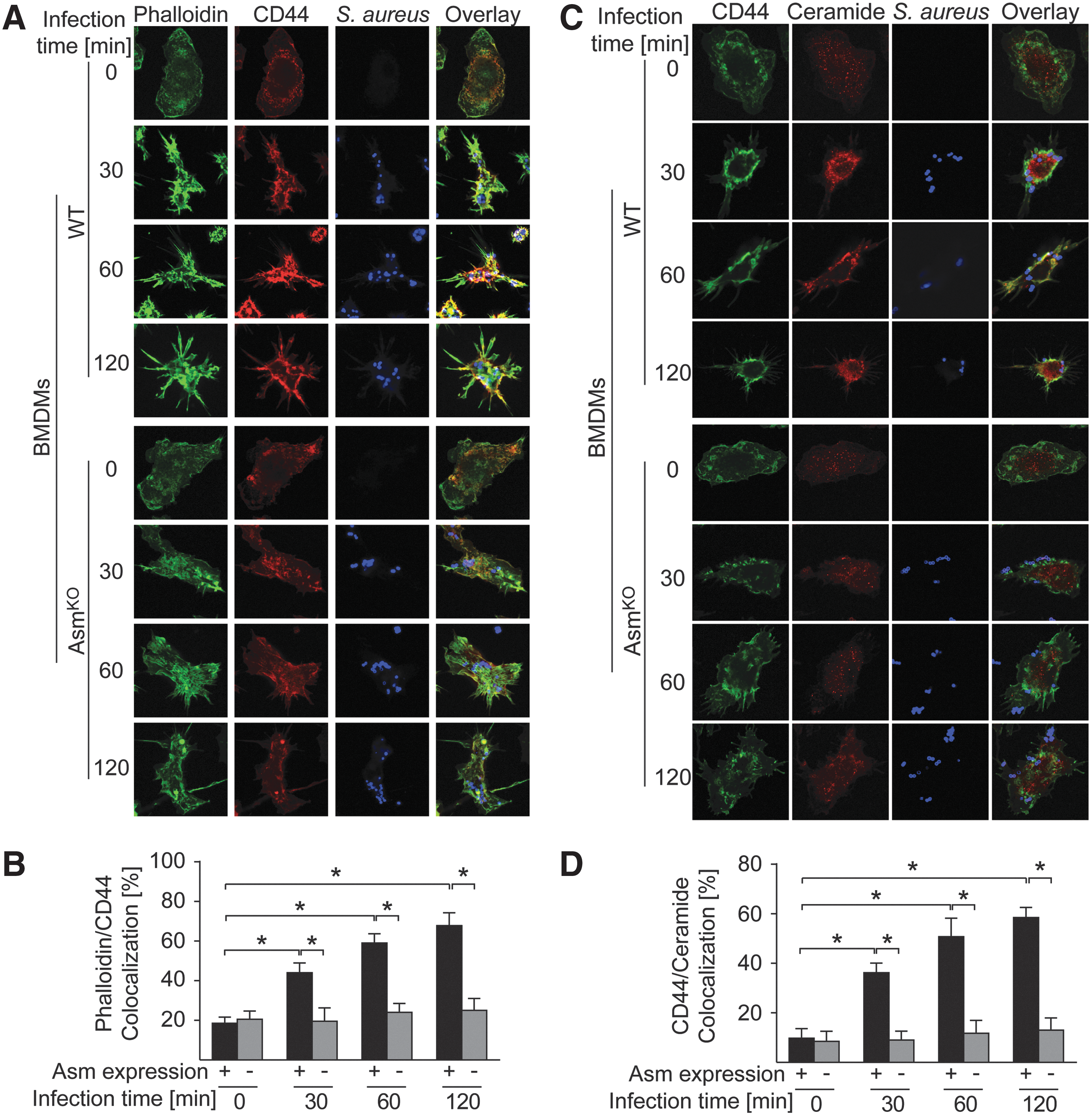
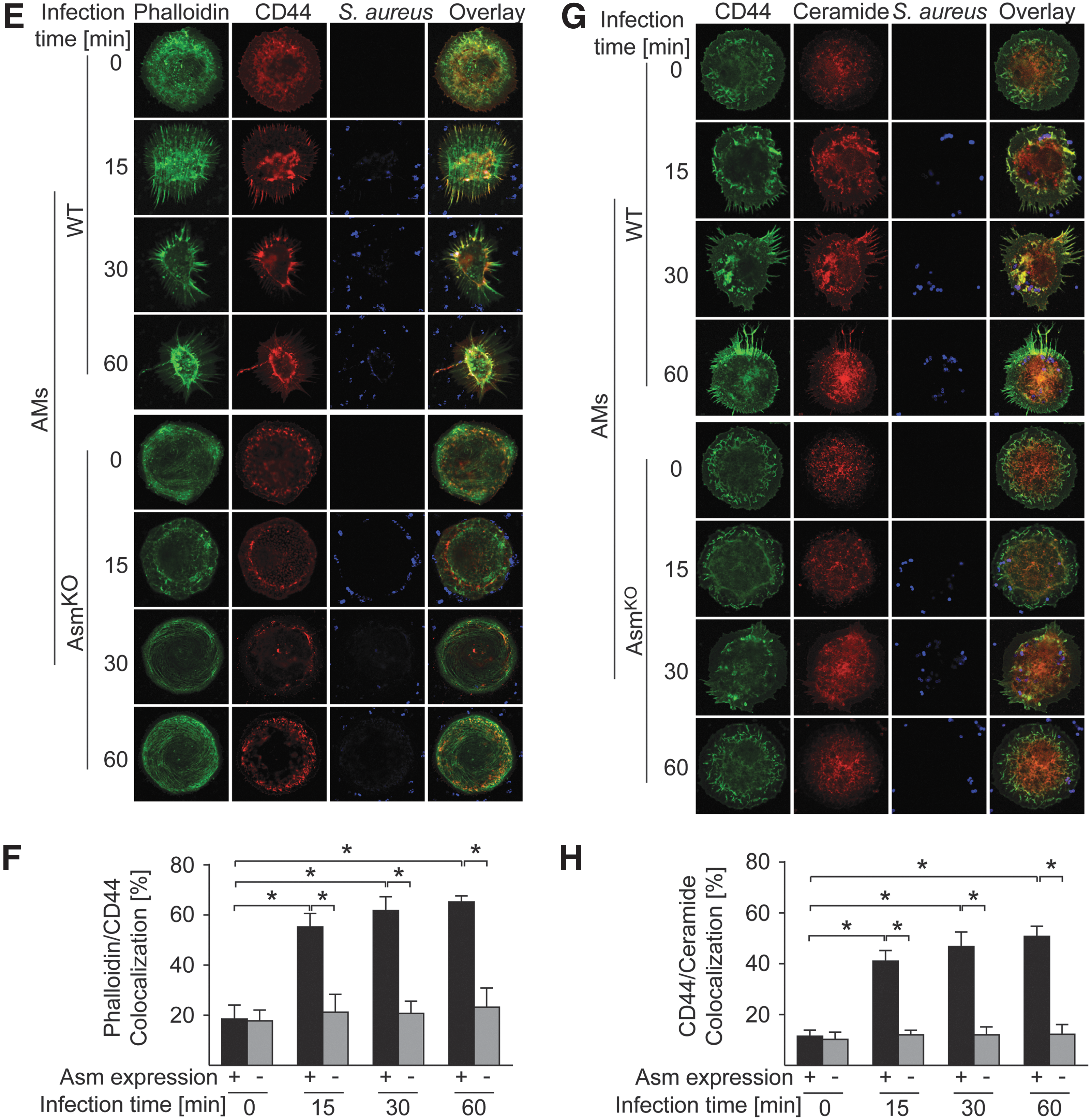
To determine whether CD44 is linked to the Asm-ceramide system, we infected WT and CD44-deficient BMDMs with S. aureus and determined Asm activity. Our results show a marked activation of Asm after infection of WT macrophage with S. aureus (Fig. 5A, B), whereas CD44 deficiency reduced and delayed the activation of Asm, but did not completely prevent the stimulation of the enzyme (Fig. 5A, B).
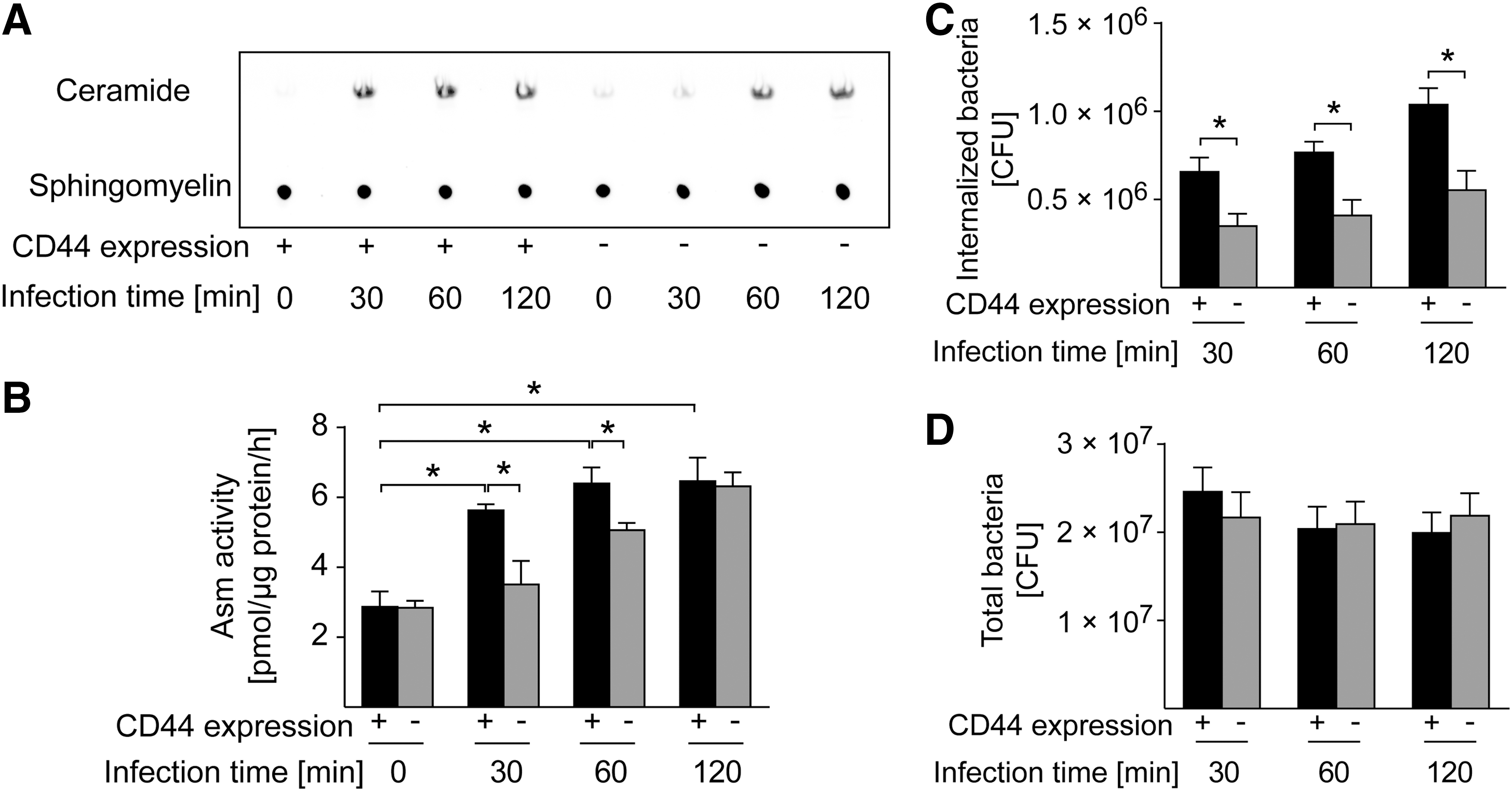
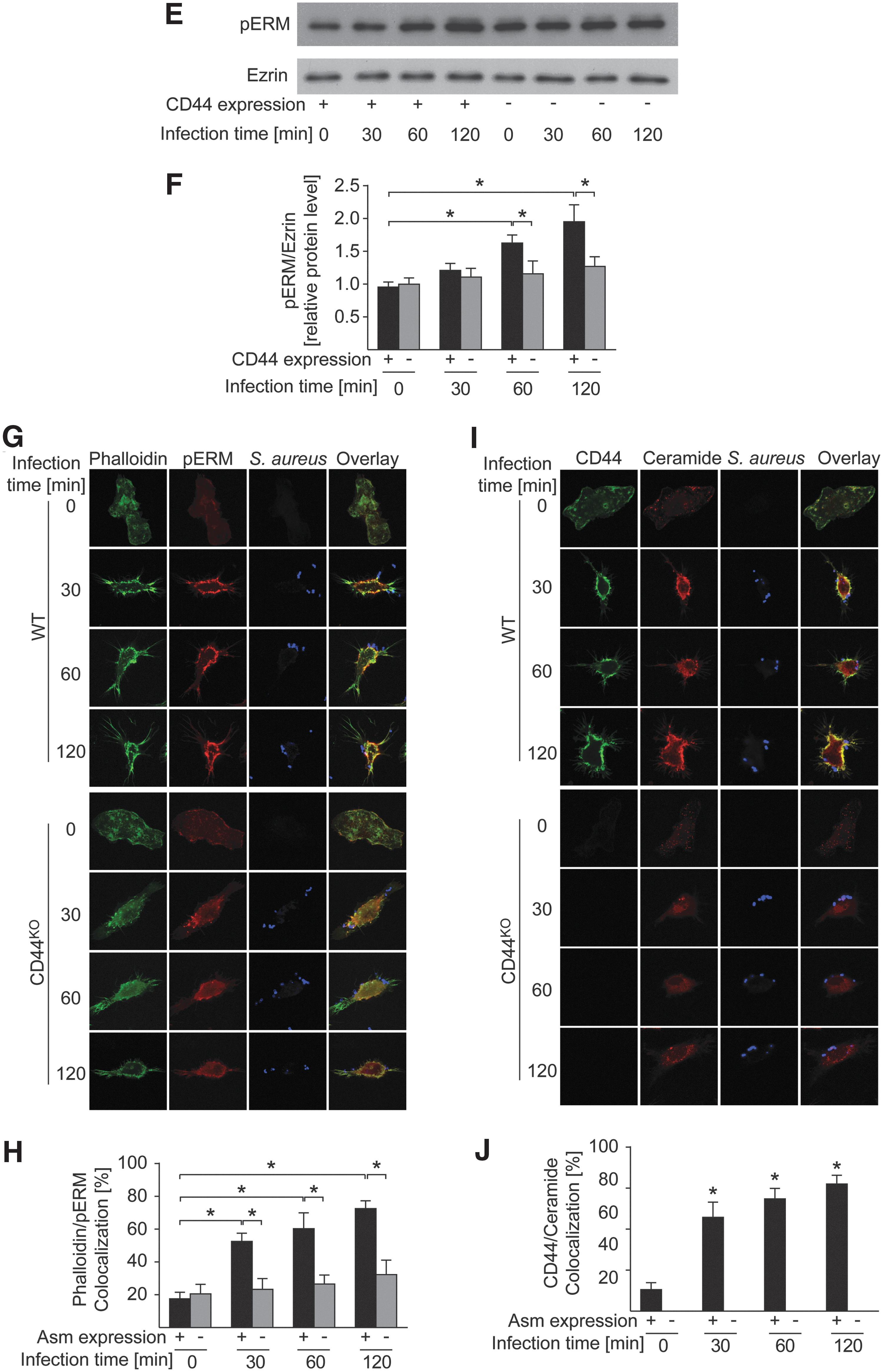
These findings suggest that CD44 functions upstream of Asm. Therefore, we investigated whether CD44 is also involved in the uptake of S. aureus, the rearrangement of the cytoskeleton, and the phosphorylation of ERM proteins. The results show that CD44 deficiency in macrophages prevented S. aureus internalization, whereas adhesion was unaffected (Fig. 5C, D). Like Asm-deficient cells, S. aureus-infected CD44-deficient BMDMs did not rearrange the cytoskeleton to polymerize cortical actin, to cluster actin at ceramide-enriched membrane platforms, or to phosphorylate and translocate ERM proteins (Fig. 5E, G, H) (64). Infection with S. aureus triggered a clustering of CD44 in ceramide-enriched membrane domains. Formation of these domains was abrogated by CD44 deficiency (Fig. 5I, J).
These findings strongly suggest that the Asm-ceramide system serves as a downstream target of CD44 during infection with S. aureus and mediates clustering of CD44 and thereby amplifies signaling via the receptor.
ROS mediate the activation of the Asm by CD44
To gain insight into molecular mechanisms that link CD44 with the Asm-ceramide system, we used anti-CD44 antibody-mediated stimulation of macrophages. As shown by electron spin resonance (ESR) spectra (Fig. 6A), anti-CD44 antibody stimulation triggered a rapid production of superoxide anions (O2 •−), a major species and precursor for other ROS. ROS have been previously shown to mediate activation of the Asm (67). Thus, we stimulated macrophages with anti-CD44 antibodies and tested whether ligation of CD44 also activates the Asm and whether this activation is prevented by pretreatment of macrophages with antioxidants. The results reveal that antibody-mediated ligation of CD44 resulted in a slow and persistent activation of the Asm, very similar to the effects of S. aureus (Fig. 6B). The isotope control antibody had no effect on Asm activity. Activation of the Asm by anti-CD44 antibodies was prevented by pretreatment of the cells with the antioxidants tiron or N-acetylcysteine in BMDMs (Fig. 6B, C) or AMs (Fig. 6D, E). Furthermore, ligation of CD44 triggered a polymerization of actin and a release of ceramide in BMDMs and AMs, events that were again prevented by antioxidants (Fig. 6F–I). These data indicate that ligation of CD44 activates the Asm via a ROS-dependent mechanism.
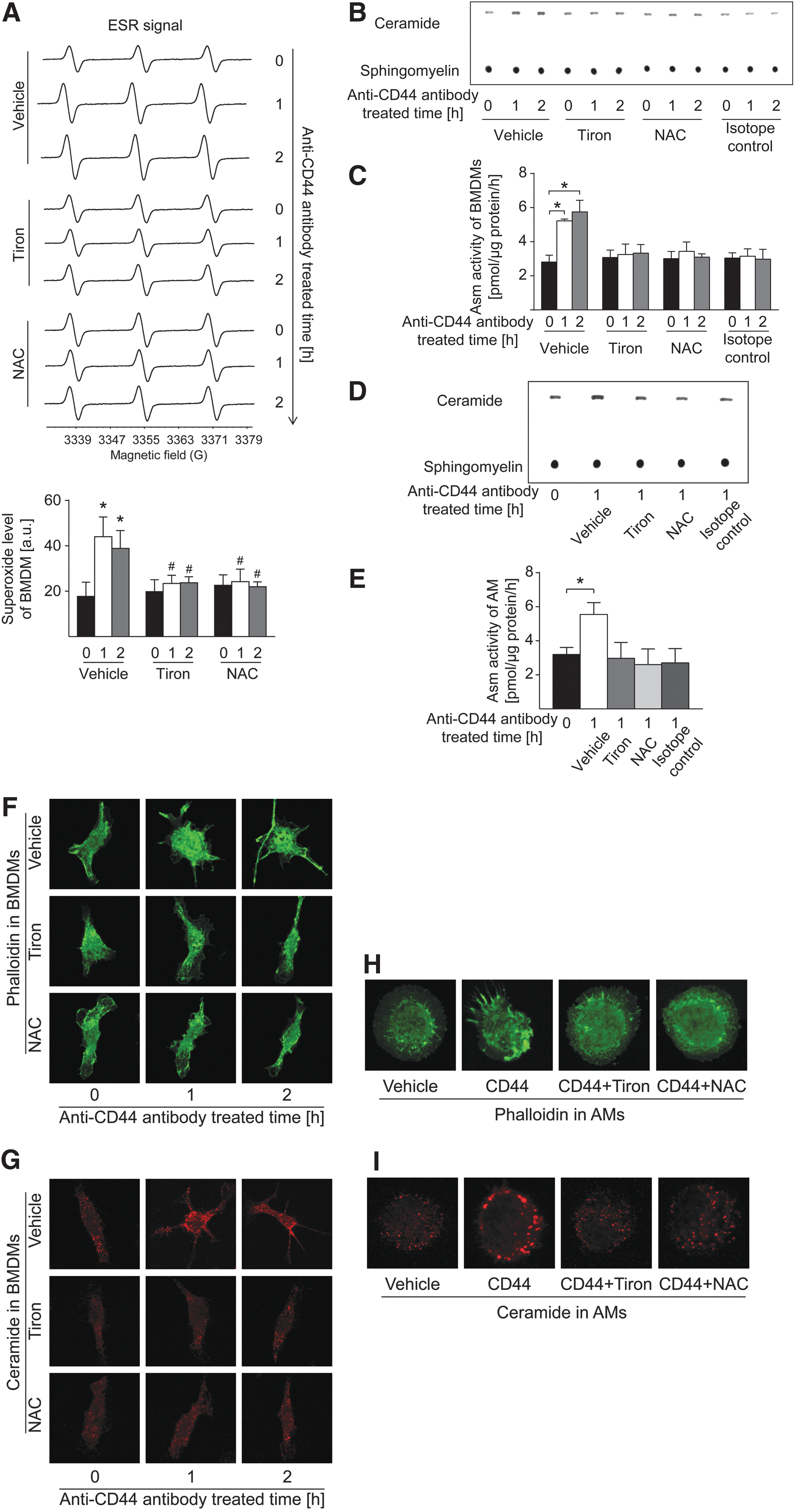
ROS generated from mitochondria and NADPH oxidases play critical roles in both physiological and pathological processes. To specify the source and subcellular compartment of ROS formation on CD44 stimulation, intact macrophages were treated with anti-CD44 antibodies in the presence or absence of different antioxidants, followed by measurements of mitochondrial or intracellular O2
•− using MitoSOX or DHE, respectively (Fig. 7A, B, E, F). As shown in Figure 7A and B, both mitochondrial and intracellular ROS levels were increased in BMDMs after anti-CD44 antibody stimulation, whereas a rat IgG isotope control antibody had no effect on ROS production. Treatment of macrophages with Nox2-specific peptide inhibitor gp91ds-tat, an NADPH oxidase inhibitor apocynin, or a mitochondria-targeted O2
•− dismutase (SOD) mimetic mitoTEMPO significantly decreased both the mitochondrial and intracellular O2
•− in CD44-stimulated macrophages. In contrast, the xanthine oxidase inhibitor oxypurinol or the nitric oxide synthases (NOS) inhibitor Nω-nitro-
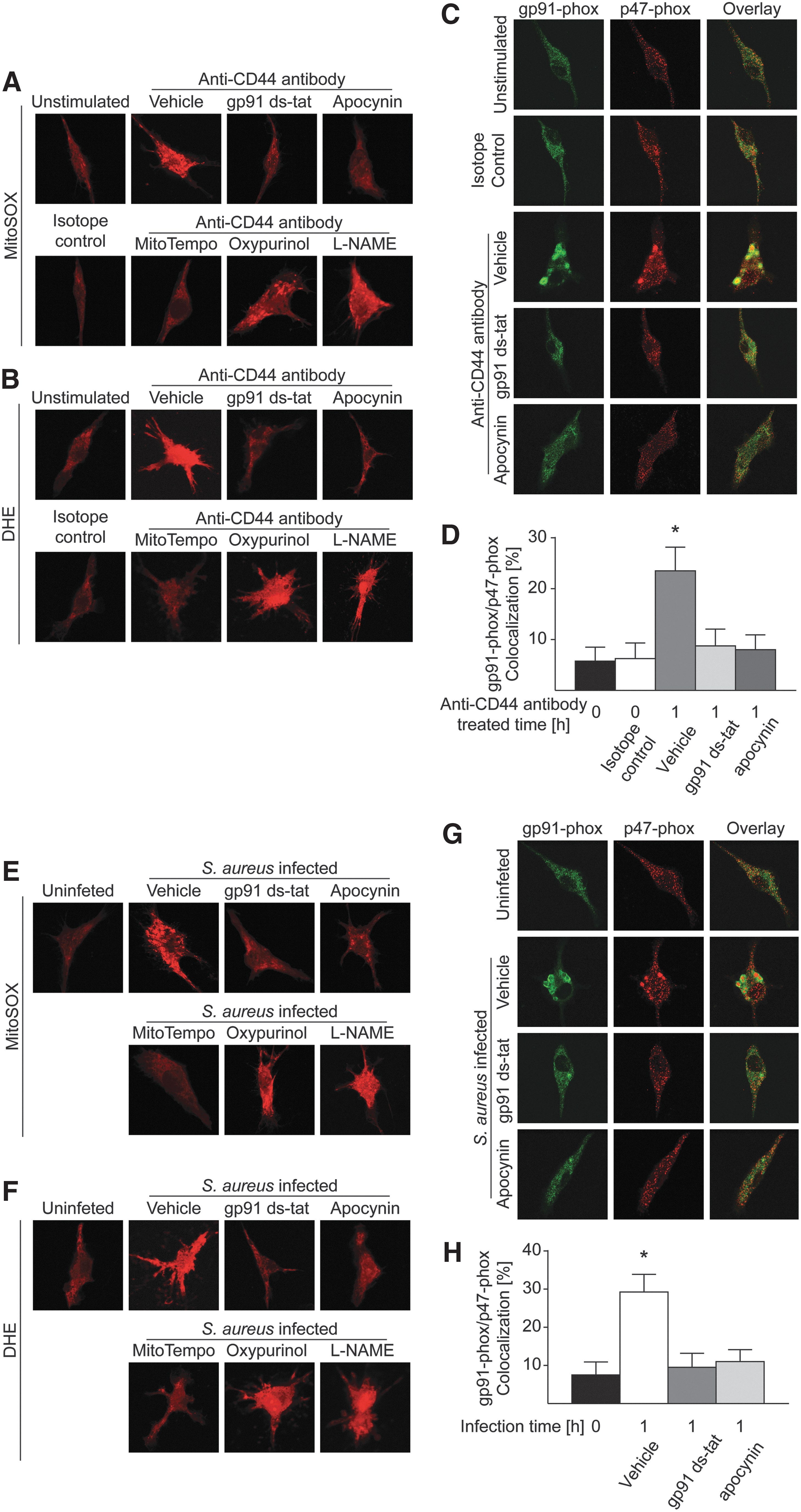
Finally, we studied ROS generation in macrophages infected with S. aureus in the absence or presence of different antioxidants. As shown in Figure 7E and F, similarly to anti-CD44 stimulation, gp91ds-tat or apocynin and the mitochondrial ROS scavenger mitoTEMPO, but not oxypurinol or L-NAME, significantly decreased mitochondrial and intracellular O2 •− production in macrophages after S. aureus infection. In addition, gp91 ds-tat or apocynin prevented the colocalization of the NADPH oxidase subunit gp91-phox with p47-phox on infection (Fig. 7G, H). Taken together, these findings indicate a formation of superoxide via activation of CD44 and the Asm on infection of macrophages with S. aureus.
Asm-deficient mice are highly susceptible to pulmonary S. aureus infections
To study the role of Asm in pulmonary S. aureus infections in vivo, we intranasally infected WT and Asm-deficient mice with S. aureus. Most WT mice rapidly cleared the infection, and almost all WT mice survived (Fig. 8A). In contrast, 80% of Asm-deficient mice died within 5 days (Fig. 8A). To determine killing of bacteria in the lung, we again intranasally infected WT and Asm-deficient mice with S. aureus and determined the total number of colony-forming units (CFUs) in the lung 6 and 12 h after infection. Bacterial numbers were substantially higher in Asm-deficient mice than in WT mice after 6 and 12 h of infection (Fig. 8B).
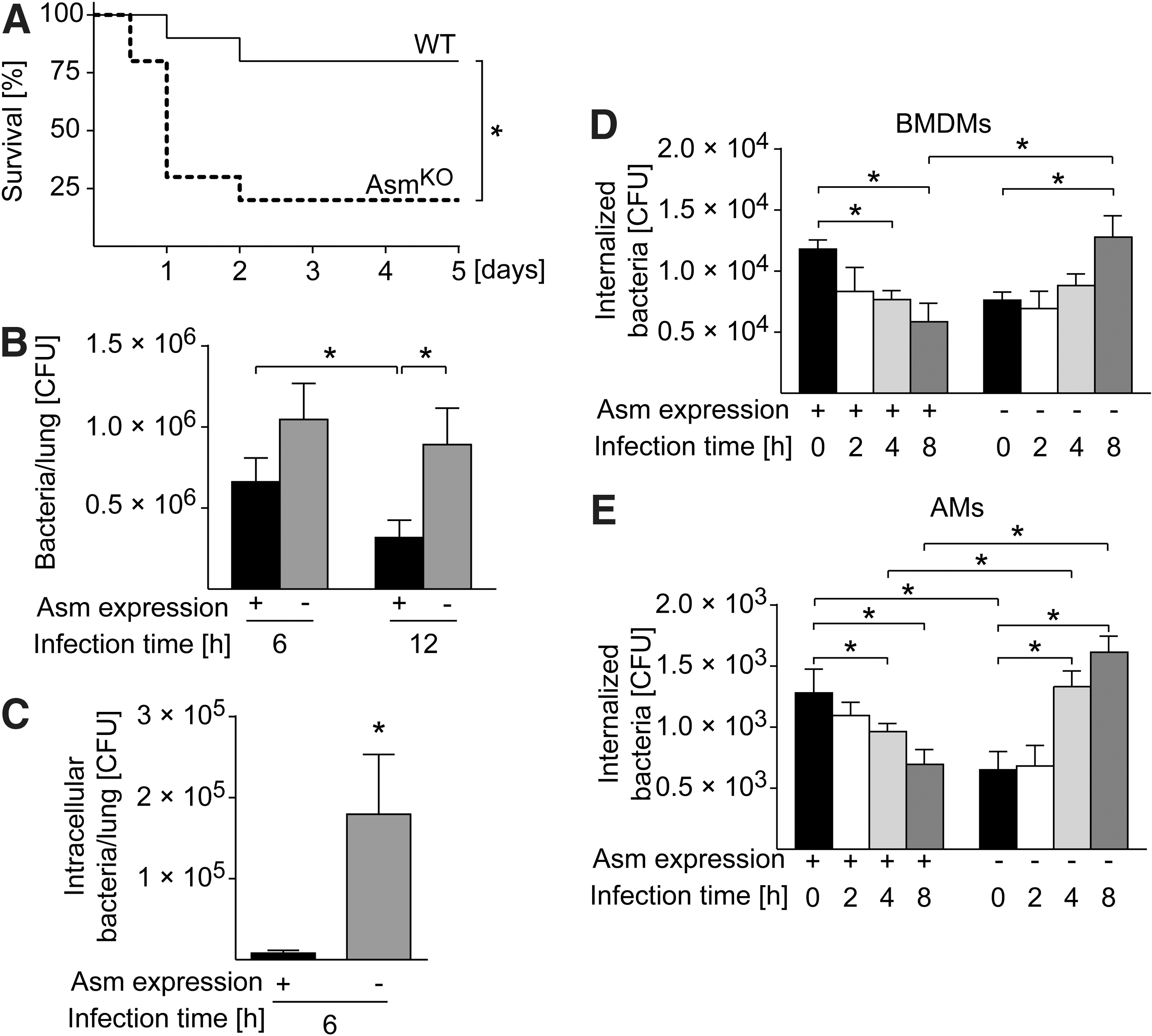
The failure of Asm-deficient mice to kill S. aureus in the lung could be explained by the finding that S. aureus internalization is reduced by 60% to 80% in these mice and that Asm deficiency reduces the release of ROS that kill extracellular pathogens, as previously shown (48, 67). Thus, to determine whether Asm-deficient mice die by an accumulation of extracellular S. aureus, we intranasally infected WT and Asm-deficient mice with S. aureus for 6 h, sacrificed the mice, homogenized the lung tissue without compromising the cell integrity, and incubated the lung homogenates with gentamicin for 1 h. Cells are impermeable to gentamicin, at least for this short time, allowing extracellular bacteria to be killed by gentamicin without affecting intracellular bacteria. The results surprisingly showed that the number of CFUs is still higher in Asm-deficient mice than in WT mice 6 h after infection (Fig. 8C), a finding indicating that S. aureus accumulates within lung cells in Asm-deficient mice. Thus, to determine whether Asm is involved in intracellular killing of S. aureus, we cultured WT and Asm-deficient AMs and BMDMs and determined the killing of intracellular S. aureus 2 to 8 h after infection via CFU assay. WT BMDMs and AMs efficiently killed S. aureus (Fig. 8D, E). In contrast, S. aureus survived and replicated in Asm-deficient macrophages (Fig. 8D, E).
These findings indicate that intracellular bacteria, although initially lower in numbers because the ability of Asm-deficient macrophages to internalize S. aureus is reduced, are not killed by macrophages; instead, they proliferate and finally cause severe and even lethal pneumonia.
Asm deficiency leads to a failure in phagosome–lysosome fusion
To understand the mechanisms that allow intracellular S. aureus to survive in Asm-deficient macrophages, we investigated whether the fusion of phagosomes and lysosomes is altered in Asm-deficient macrophages. To this end, we incubated BMDMs with tetramethylrhodamine isothiocyanate (TMR)-labeled dextran and stained the cells after fixation with antibodies to lysosomal-associated membrane protein 1 (Lamp1), a lysosomal marker protein. Lamp1-staining and dextran fluorescence did not differ between WT and Asm-deficient BMDMs before infection. Infection with S. aureus induced fusion of TMR-dextran-positive phagosomes with anti-Lamp1-labeled lysosomes in WT BMDMs (Fig. 9A), while this fusion event was abrogated by Asm deficiency (Fig. 9A).
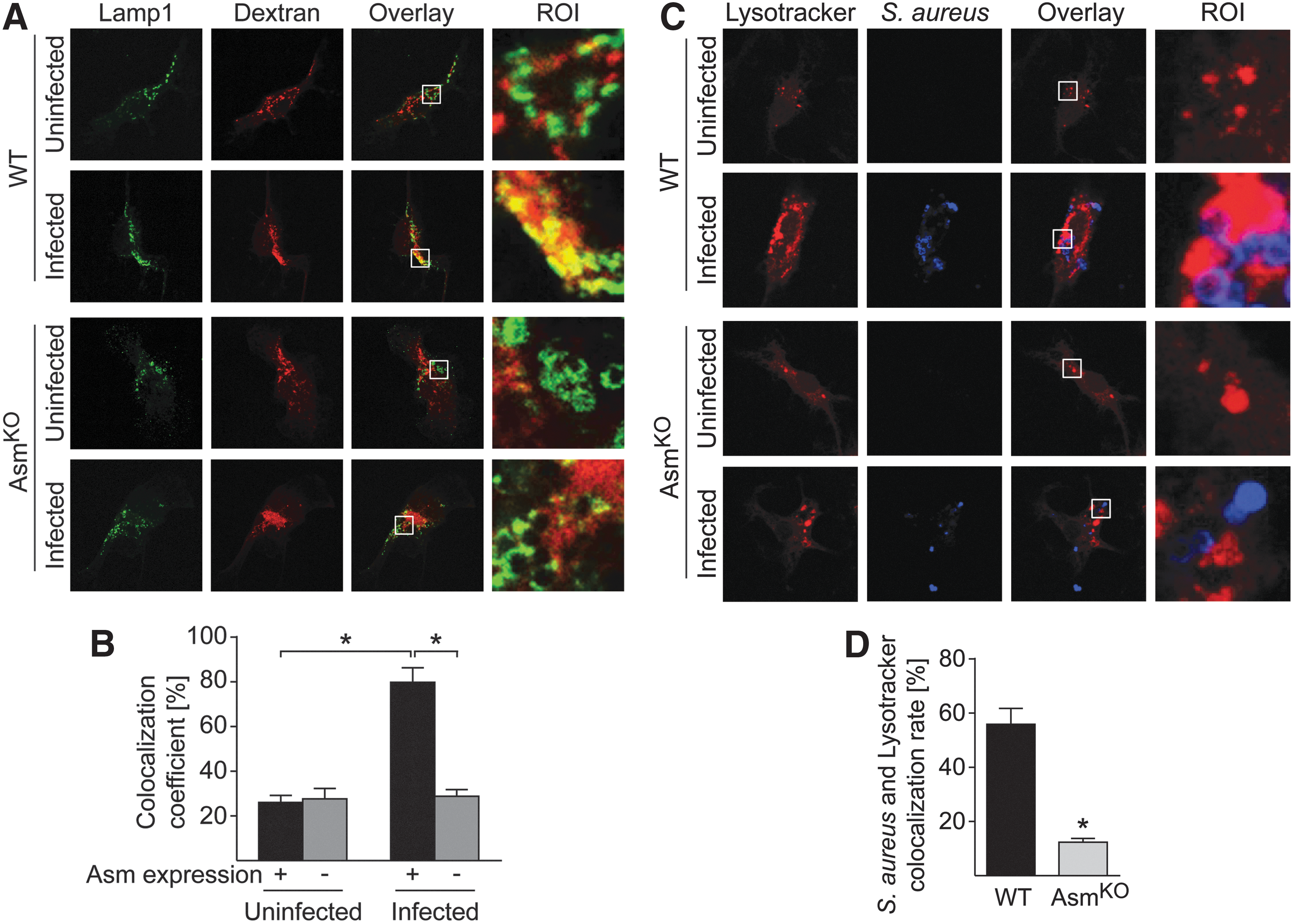
We further stained macrophages with anti-S. aureus antibodies and LysoTracker Red, a fluorescent dye for labeling and tracking acidic organelles in living cells. S. aureus colocalized with LysoTracker-labeled acidic compartments in WT macrophages and most bacteria localized to lysosomes in WT macrophages. In contrast, the fusion of phagosomes and lysosomes was defective in Asm-deficient BMDMs, and intracellular pathogens remained separated from lysosomes (Fig. 9C, D).
Taken together, these findings suggest that Asm regulates phagocytosis and phagosome–lysosome fusion to mediate the killing of S. aureus.
Discussion
The present results identify CD44 as a novel molecule that links S. aureus infection of macrophages to activation of the Asm-ceramide system, resulting in internalization of the pathogen, fusion of phagosomes with lysosomes, and intracellular killing of the pathogen. CD44 and Asm seem to act in a positive feedback loop with Asm activation induced by CD44 binding as an initial event that is then amplified by clustering of CD44 in ceramide-enriched membrane platforms. Additional activation of Asm by the clustered CD44 and further clustering of CD44 finally lead to amplification of intracellular signaling in a positive feedback cycle. The generation of a strong signal by CD44 within a small, defined area of the cell membrane may allow the receptor to transmit the signal into macrophages, resulting in the activation of small G proteins and the rearrangement of the cytoskeleton, an event that finally mediates the uptake of S. aureus and intracellular killing in WT macrophages. The absence of Asm prevents this cascade of events.
Internalization of bacteria into host cells may allow the pathogen to attack the host organism, if the pathogen can survive in the infected cells. On the contrary, internalization may also permit the host to target a pathogen to phagolysosomes, thereby killing the invading pathogen. The present studies show that the internalization of S. aureus is an important part of the defense against the pathogen: Asm-deficient macrophages that exhibit a reduced rate of internalization in vivo and in vitro are also unable to kill internalized S. aureus. Our studies showed that S. aureus accumulates in vivo within Asm-deficient cells, because the pathogen is not killed within Asm-deficient macrophages even if these cells primarily internalize fewer bacteria. These findings strongly suggest that internalization and subsequent killing of the pathogen are an important part of the host defense in WT macrophages and lungs, respectively. The results of recent studies are consistent with this concept, showing that blocking the phagocytosis of S. aureus contributes to increased bacterial survival in human blood, bacterial persistence, and abscess formation both in pneumonia and after intravenous infection in vivo (29, 31).
Our group has previously demonstrated that genetic deficiency of Asm abolishes the extracellular release of ROS after the infection of macrophages with Pseudomonas aeruginosa (67). Thus, activation of the Asm triggers clustering and aggregation of NADPH oxidases in plasma membrane domains and thereby mediates production of O2 •− (1, 36, 67). However, although this mechanism may contribute to the extracellular killing of S. aureus in the lung, our findings show that intracellular killing of the pathogen is also required for its elimination.
Our findings show that intracellular ROS production induced by anti-CD44 antibody stimulation or S. aureus infection is primarily derived from activation of NADPH oxidase and mitochondria respiration chain (Fig. 7B, F). It should be noted that mitochondrial ROS could be blocked by NADPH oxidase inhibitors (Fig. 7A, E). These results may suggest that CD44 ligation or S. aureus infection activates NADPH oxidases in plasma membranes of macrophages resulting in rapid ROS production. Such NADPH oxidase-derived ROS may further stimulate the mitochondrial ROS production via an ROS-induced ROS production mechanism (8). Therefore, our data support the view that both NADPH oxidase and mitochondria-derived ROS contribute to the intracellular killing of the pathogen during S. aureus infections.
Our previous work has shown that Asm-deficient mice are more susceptible than WT mice to septic S. aureus infections (48). These studies demonstrated that systemic infection with S. aureus destroys tight junctions of endothelial cells in the lung, thereby inducing lung edema. Whether destruction of endothelial tight junctions is mediated by intra- or extracellular S. aureus or toxins is presently unknown.
Previous studies have shown that the Asm-ceramide system is involved in infection with several pathogenic bacteria, such as pathogenic Neisseria gonorrhoeae, P. aeruginosa, S. aureus, and Salmonella species (23, 42, 48, 60, 67). However, the molecular mechanisms of Asm activation by bacteria are still poorly understood. Our current studies identify for the first time a receptor, CD44, that couples a pathogen with activation of Asm.
Our findings do not show a complete absence of Asm activation, ceramide formation, cytoskeletal changes, and internalization in CD44-deficient cells. This result may be explained by the binding of the pathogen to additional receptors, such as intercellular adhesion molecule 1 (ICAM-1) that may also couple with Asm (47).
Similarly, Asm deficiency does not completely abrogate the internalization of S. aureus. Because cells contain only a single Asm gene, it might be possible that other pathways independent of Asm are also involved in the uptake of the pathogen.
Asm hydrolyzes sphingomyelin to ceramide, which has been shown to spontaneously form ceramide-enriched platforms in the plasma membrane (14). These platforms trap and cluster specific proteins, thereby inducing and amplifying signal transduction (3, 17). This mechanism may also apply to the positive feedback between CD44 and Asm activation. CD44 consists of three regions: an extracellular domain, a transmembrane domain, and a C-terminal cytoplasmic domain (12). The results of structural studies of CD40 indicated that the transmembranous domain of CD40 determines clustering within ceramide-enriched membrane domains (3). It is tempting to speculate that a similar mechanism applies to CD44, although the exact mechanisms by which receptors cluster in ceramide-enriched membrane domains are unknown.
CD44 isoforms were previously shown to participate in host infections (6, 20). Thus, it was shown that CD44 functions as a receptor molecule for group A Streptococcus. This was mediated by a group A Streptococcus capsular polysaccharide that seems to mimic the physiological ligand of CD44, that is, hyaluronic acid (6). Binding of group A Streptococcus to CD44 resulted in cytoskeletal rearrangements, membrane ruffling, and disruption of intercellular junctions, similar to the present findings with S. aureus (7). In addition, CD44 was demonstrated to function as a binding site for Mycobacterium tuberculosis, a process that was shown to be involved in macrophage recruitment and protection against the pathogen (35). Escherichia coli also uses CD44 as receptor, although indirectly by binding to hyaluronic acid that is a ligand for CD44 (54). CD44v6 was shown to promote the internalization of the food-born pathogen Listeria monocytogenes on binding of the virulence factor Internalin B to the receptor tyrosine kinase MET (30). CD44 was also described to interact with IpaB, a protein secreted by Shigella (62). The formation of this complex appeared important for invasion of epithelial cells. The interaction between CD44 and IpaB was described to occur within lipid rafts thereby promoting infection (34). More recently, peptides targeting CD44 were shown to block Helicobacter pylori-induced proliferation and subsequent gastritis (2). Most importantly, CD44 has been shown to function as a coreceptor among CD2, CD11a, CD18, CD28, CD58, and ICAM-1 for staphylococcal enterotoxin B (SEB) (32). It was shown that CD44 plays an important role in the cellular response to SEB and is involved in the induction of apoptosis in mononuclear cells (43). These studies indicate that CD44 is involved in the infection of host cells by many pathogens. However, a link from CD44 to the Asm, ceramide, ROS, and further downstream signals has not been reported so far.
In summary, we describe a novel mechanism by which the participation of CD44 in the invasion of S. aureus via macrophages leads to elimination of the bacteria: Interaction of S. aureus with CD44 on macrophages activates via ROS the Asm-ceramide system, resulting in the formation of ceramide-enriched membrane platforms that in turn cluster and amplify CD44 signaling and thereby act as a positive feedback loop. CD44 and Asm are crucially involved in the activation of small G-proteins, the phosphorylation of ERM proteins, and the rearrangement of the cytoskeleton, culminating in the phagocytosis of S. aureus. Phagosomes containing the pathogen fuse with acidified lysosomes in an Asm-dependent manner to kill intracellular pathogens, which is a requirement for successful host defense in the lungs. Asm deficiency reduces or prevents all of these events, allowing the intracellular survival of internalized pathogens.
Materials and Methods
Ethics statements
All procedures performed on mice were approved by the Bezirksregierung Düsseldorf, Düsseldorf, Germany (project license number: AZ 84-02.04.2013.A348). All experiments were done according to the rules of the Federation of Laboratory Animal Science Associations (FELASA). The S. aureus strain was obtained from adult patients within the EudraCT 2006-002259-33 study, which was approved by the Institutional Review Board of the University Hospital Tubingen. All patients provided written informed consent.
Mice and cells
Asm-deficient mice and wild-type (WT) littermates (sphingomyelin phosphodiesterase 1 knockout; Smpd1−/− ) and CD44-deficient mice and wild-type littermates WT mice were maintained on a C57BL/6 background (26). The genotype was verified by polymerase chain reaction. We used Asm-deficient mice and WT littermates aged 6 to 8 weeks to avoid sphingomyelin accumulation (38). Mice were bred in the animal facility of the University of Duisburg-Essen under specific pathogen-free conditions according to the criteria of the Federation of Laboratory Animal Science Associations (FELASA).
The culture of BMDMs has been previously described in detail (67). Briefly, mice were sacrificed, femurs and tibias were flushed with minimum essential medium (MEM; Gibco) supplemented with 10% fetal bovine serum (Gibco), 10 mM HEPES (pH 7.4; Roth GmbH), 2 mM
AMs were isolated from bronchoalveolar lavage fluid from WT or Asm-deficient mice (67). Mice were sacrificed, the trachea was exposed, catheterized with a polyethylene tube, and lavaged with a total of 15 mL ice-cold phosphate-buffered saline (PBS). Cells were centrifuged for 5 min at 300 g at 4°C, supernatants were discarded, and the pellets were resuspended in MEM/HEPES. Cells were then seeded and washed for further experiments. To characterize the population of AMs, cells were stained with anti-Siglec F antibodies and anti-CD11c antibody, which are specific antibody for AMs (not shown) (44).
Infection and stimulation experiments
The S. aureus strain used in the present study was isolated from a patient with sepsis. The strain produces alpha toxin and enterotoxin D but not the Panton-Valentine leukocidin or the toxic shock syndrome toxin. S. aureus was grown overnight on trypticase soy agar (TSA) plates with 5% sheep blood (BD), resuspended in 40 mL tryptic soy broth (BD) at an optical density of 0.2 to 0.25, and incubated at 37°C with shaking at 125 rpm for 75 min. Bacteria were pelleted during the early logarithmic growth phase by centrifugation at 2800 rpm for 10 min and washed with RPMI 1640 (Gibco) supplemented with 10 mM HEPES (Roth GmbH). The bacteria were finally resuspended in HEPES/saline (H/S) buffer consisting of 132 mM NaCl, 20 mM HEPES (pH 7.4), 5 mM KCl, 1 mM CaCl2, 0.7 mM MgCl2, and 0.8 mM MgSO4. Cells or mice were then infected within the next 10 min. Infection was facilitated by a 5-min centrifugation (1000 rpm) of the bacteria onto the cells. The end of the centrifugation was defined as the starting point of infection.
To determine Asm activity, ezrin/radixin/moesin (ERM) phosphorylation, and Rho family GTPase activity, we infected ∼106 macrophages in a six-well plate with S. aureus at a multiplicity of infection (MOI) of 100 bacteria per macrophage. For all immunofluorescence studies or intracellular bacteria killing assay in vitro, macrophages were cultured in 24-well plates, and cells were infected with S. aureus at an MOI of 100 bacteria per BMDM or an MOI of 50 bacteria per AM. Macrophages were left uninfected or were infected with S. aureus for the indicated times.
For in vivo experiments, we infected mice intranasally with 8 × 108 CFUs per mouse (67). S. aureus was prepared and resuspended in saline plus 10 mM HEPES to a final concentration of 8 × 108 CFU in 20 μL. The mice were then inoculated with S. aureus via a plastic-coated 30-gauge needle, which was inserted 2 mm into the nose. The mice were observed for 5 days or were sacrificed at 6 or 12 h to determine the number of CFUs in the lungs.
To perform bacterial killing and phagocytosis assays in vitro, cells were infected as described above. The infection was terminated by gently washing the cells with sterile PBS followed by incubation of the macrophages with or without 100 μg/mL gentamicin (Sigma) for 1 h at 37°C in MEM/HEPES to determine the number of internalized bacteria or the total number of intra- and extracellular bacteria. The macrophages were then extensively washed, lysed in 5 mg/mL saponin (Serva Electrophoresis GmbH, Heidelberg), resuspended in PBS for 10 min to release intracellular bacteria, pelleted by centrifugation at 3200 rpm, resuspended in PBS, aliquots were plated on Luria broth agar plates, and the CFUs were counted after growth overnight at 37°C.
For in vivo assays, mice were infected with S. aureus and sacrificed at 6 or 12 h for detecting total bacteria in the lung. The lung tissue was collected, homogenized into very small pieces, lysed with 5 mg/mL saponin/PBS, and total bacteria were determined after overnight growth at 37°C as described above. The counts represent the number of bacteria in whole-lung samples. To determine intracellular bacteria, mice were sacrificed 6 h after infection and lung homogenates were incubated with 100 μg/mL gentamicin in PBS for 1 h or left untreated. The samples were then extensively washed, lysed with 5 mg/mL saponin/PBS, and the CFU of internalized bacteria were determined as described above.
To determine the activation of macrophages by anti-CD44 antibody, BMDMs were washed and left unstimulated or incubated with 1 μg/mL anti-CD44 antibodies for 1 or 2 h. AMs were left untreated or stimulated with anti-CD44 antibodies for 1 h. To inhibit the formation of ROS, cells were pretreated with several antioxidants, that is, the ROS scavenger disodium 4,5-dihydroxybenzene-1,3-disulfonate (tiron, 10 μM; Sigma-Aldrich), N-acetylcysteine (NAC, 10 μM; Sigma-Aldrich), the mitochondrial ROS scavenger mitoTEMPO (25 μM; Sigma-Aldrich), the ADPH oxidase (NOX) inhibitor gp91 ds-tat (10 μM; AnaSpec), apocynin (10 μM; SantaCruz), the NOS inhibitor L-NAME (100 μM; Sigma-Aldrich), or oxypurinol (100 μM; Sigma-Aldrich). The stimulations were terminated by shock freezing the samples for Asm activity assays and ROS measurements, or by 4% PFA/PBS fixation for immunofluorescence study.
Assay for Asm activity
Asm activity was measured with green fluorescent BODIPY-FLC12-sphingomyelin (Thermo Fisher Scientific) as a substrate. Briefly, cells were infected or left untreated, harvested, and lysed in 250 mM sodium acetate (Sigma) and 1% Nonidet P-40 (pH 5.0; Sigma) for 5 min on ice. Cells were further disrupted by sonification for 10 min in an ice bath sonicator (BANDELIN electronic). The protein concentration was measured by a Bradford protein assay (BioRad), and 5 μg of protein in 20 μL lysis buffer was added to 250 mM sodium acetate (pH 5.0) containing 100 pmol BODIPY-FLC12-sphingomyelin. The samples were incubated at 37°C for 1 h with shaking at 300 rpm. The reaction was stopped by the addition of 1 mL chloroform:methanol (2:1, v/v) followed by centrifugation for 5 min at 14,000 rpm. The lower phase was dried in a SpeedVac Concentrator (Thermo Fisher Scientific) and resuspended in 20 μL of chloroform:methanol (2:1, v/v). The samples were spotted on a 5 × 20 cm thin-layer chromatography plate (Merck) in 3 μL steps, waited until all spots were dry, separated with chloroform:methanol (80:20, v/v), scanned with a Typhoon FLA 9500 laser scanner (GE Healthcare Life Sciences), and analyzed with ImageQuant software (GE Healthcare Life Sciences).
Activation of macrophages with anti-CD44 antibodies was achieved by incubation of the cells with 1 μg/mL anti-CD44 (Calbiochem). Rat IgG2b isotope antibodies served as controls. Cells were then lysed and the Asm activity was determined as above. To block the effects of ROS, the cells were preincubated for 30 min with tiron (10 μM) or N-acetylcysteine (10 μM) that remained on the cultures during the stimulation time.
Electron spin resonance spectrometric detection of superoxide
Superoxide production was determined by ESR as previously described (4). After stimulation or infection, respectively, of macrophages for the indicated times, 20 μL of cell homogenates was resuspended in modified Krebs-HEPES buffer containing deferoxamine (100 μM; Sigma) and diethyldithiocarbamate (5 μM; Sigma). The reaction was started by addition of the spin trap 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH, 1 mM final concentration; Noxygen Science Transfer & Diagnostics, Elzach, Germany) in the presence or absence of polyethylene glycol (PEG)-conjugated superoxide dismutase (SOD, 200 U/mL; Sigma-Aldrich). The reaction mixture loaded in glass capillaries was immediately analyzed for O2 − production for 10 min using a MiniScope MS400 ESR spectrometer (Magnettech, Germany) with the following ESR settings: biofield, 3360; field sweep, 60 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 3 G; points of resolution, 4096; receiver gain, 50; and kinetic time, 10 min. The SOD-inhibited signals were normalized for protein concentration and expressed as the fold change of treatment group versus control.
Immunocytochemistry
Macrophages were grown on coverslips and were left uninfected or infected with S. aureus. Infection was stopped by adding 4% paraformaldehyde (PFA; Sigma) in PBS (pH 7.4) for 10 min. Cells were washed three times with H/S. For confocal microscopy, macrophages were permeabilized with 0.1% Triton X-100/PBS for 5 min at room temperature. Cells were washed again with PBS and incubated for 1 h with goat anti-Armenian hamster immunoglobulin G (IgG) antibodies (dilution 1:100; Jackson ImmunoResearch) to block nonspecific binding. Samples were washed and incubated for 1 h with rabbit monoclonal IgG anti-phospho-ezrin (phospho Thr567)/anti-phospho-radixin (phospho Thr564)/anti-phospho-moesin (phospho Thr558) antibodies (1:200; Cell Signaling Technology), rat monoclonal IgG2b anti-CD44 antibodies (1:200 diluted; Abcam), mouse monoclonal IgM anti-ceramide antibodies (1:100 diluted; Glycobiotech), rabbit polyclonal IgG anti-S. aureus antibodies (1:200 diluted; Abcam), mouse monoclonal IgG anti-S. aureus antibodies (1:200 diluted; Abcam), mouse IgG anti-gp91 phox antibodies (1:200 diluted; BD), rabbit IgG anti-p47 phox antibodies (1:200 diluted; millipore), or fluorescein isothiocyanate-conjugated (FITC) phalloidin (25 μg/mL; Sigma-Aldrich). All antibodies were diluted in 5% FCS/PBS. Cells were then washed three times for 5 min each with 0.05% Tween-20/PBS and incubated for 45 min with secondary antibodies corresponding to the primary antibodies, that is, FITC-conjugated F(ab′)2 fragments of goat anti-rat IgG; Cy3-conjugated F(ab′)2 fragments of donkey anti-rat IgG, donkey anti-mouse IgM, and donkey anti-rabbit IgG; DyLight 649-conjugated F(ab′)2 fragments of donkey anti-mouse IgG; and Alexa Fluor 647-conjugated F(ab′)2 fragments of donkey anti-rabbit IgG (all antibodies from Jackson ImmunoResearch; final concentration of all antibodies, 1.5 μg/mL; diluted in 5% FCS/PBS). To confirm the specificity of fluorescent stainings, samples were incubated with rat IgG isotype control antibodies to CD44, rabbit IgG antibodies to phospho-ERM, mouse IgM isotope control antibodies, or just with secondary antibody controls. The samples were then washed three times with PBS/0.05% Tween 20 and once in PBS and were mounted with Mowiol (Kuraray Specialities Europe GmbH). Samples were visualized with a Leica TCS SP5 confocal microscope using a 100 × oil lens, and images were analyzed with Leica LCS software (Leica Microsystems).
For SEM, macrophages were treated as above. After fixation and washing with H/S, cells were dehydrated in a graded series of ethanol followed by critical-point drying (CPD 7501; Polaron) and sputtered with platinum/palladium (208HR high-resolution sputter coater; Cressington). Specimens were analyzed with an S-4000 SEM (Hitachi), and images were obtained with a DISS5 (Point Electronics) analysis system.
MitoSOX and DHE staining
Macrophages were infected with S. aureus or stimulated with anti-CD44 antibodies as described above. Cells were then incubated with 2 μM MitoSOX™ Red mitochondrial superoxide indicator (Invitrogen) or 5 μM dihydroethidium (DHE) (Abcam) in MEM supplemented with 10 mM HEPES. Cells were incubated for 15 min at 37°C protected from light, then washed with ice-cold PBS, and rapidly fixed with 4% PFA/PBS for 10 min. The samples were then washed three times with PBS/0.05% Tween 20, once in PBS, and then mounted with Mowiol. Samples were stored at 4°C for further microscopy study.
Western blots and pull-down assay
Cells were lysed in 100 μL 0.1% sodium dodecyl sulfate (SDS), 25 mM HEPES, 0.5% deoxycholate, 0.1% Triton X-100, 10 mM ethylenediaminetetraacetic acid (EDTA), 10 mM sodium pyrophosphate, 10 mM sodium fluoride, 125 mM NaCl, and 10 μg/mL aprotinin/leupeptin (Sigma). Samples were centrifuged at 14,000 rpm for 5 min at 4°C, and supernatants were collected. Proteins were separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes, followed by blocking with 5% bovine serum albumin in Tris-buffered saline supplemented with 0.05% Tween 20. Blots were incubated overnight at 4°C with anti-phospho-ERM (1:2000; Cell Signaling Technology) or anti-Ezrin antibodies (36a) as loading control (1:1000; Cell Signaling Technology). Blots were washed and developed with alkaline phosphatase-coupled secondary antibodies (1:20,000; Santa Cruz Biotechnology, Inc.) using the Tropix chemiluminescence system (Amersham Pharmacia Biotech, Inc.).
Rho family GTPase activity was detected using the RhoA/Rac1/Cdc42 Activation Assay Combo Biochem Kit (Cytoskeleton, Inc.) according to the manufacturer's instructions. Briefly, cells were infected and lysed in 50 mM Tris (pH 7.5), 10 mM MgCl2, 0.5 M NaCl, and 2% Igepal. Equivalent amounts of protein were added to a predetermined amount of rhotekin Rho binding domain (for RhoA activation assay) or PAK-PBD beads (for Rac1 and Cdc42 activation assays) and incubated at 4°C on a rotator for 1 h. Beads were washed with a buffer consisting of 25 mM Tris (pH 7.5), 30 mM MgCl2, and 40 mM NaCl. Finally, 20 μL of Laemmli sample buffer was added to each sample. Proteins were separated on a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane. Blots were incubated with monoclonal anti-RhoA (ARH04), monoclonal anti-Rac1 (ARC03), or monoclonal anti-Cdc42 antibodies (ACD03) (all antibodies from Cytoskeleton, Inc.) as described by the vendor and developed as described above.
Phagosome–lysosome fusion
For detection of phagosome–lysosome fusion, macrophages were preincubated with 1 mg/mL TMR dextran (10 kDa; Sigma-Aldrich) for 1 h in HEPES/MEM at 37°C. Cells were then washed with HEPES/MEM and infected with S. aureus as above. Cells were fixed and permeabilized as above. Lysosomes were stained with antibodies against anti-Lamp1 (Abcam) followed by incubation with FITC-coupled anti-rat antibodies. For observation of intracellular acidic organelles, macrophages were incubated with 60 nM LysoTracker Red DND-99 (Thermo Fisher) in MEM. Macrophages were then infected with S. aureus, washed, and analyzed with confocal microscopy, as described above.
Statistics
Data are expressed as arithmetic mean ± standard deviation unless otherwise indicated. The Shapiro–Wilk test was used for testing for normal distribution of the data. One-way analysis of variance followed by the Student–Newman–Keuls test was used to test between-group and within-group differences; pairwise comparisons were performed with Student's t-test. Comparisons of survival variables were performed with log-rank tests. Statistical significance was set at the level of p < 0.05. All data were obtained from independent measurements. The GraphPad Prism statistical software program (GraphPad software) was used for the analyses.
Footnotes
Acknowledgments
The authors thank Prof. Dr. Elke Winterhager for excellent support with the electron microscopy. The study was supported by DFG grant HG 1697/2-2 and GU 335/33-1 to H.G. and E.G.
Author Disclosure Statement
No competing financial interests exist.