
Abstract
Significance:
Cardiovascular diseases are the main cause of death worldwide and pose an immense economical burden. In most cases, the underlying problem is vascular occlusion by atherosclerotic plaques. Importantly, different cell types of the vascular wall and the immune system play crucial roles in atherosclerosis at different stages of the disease. Furthermore, atherosclerosis and conditions recognized as risk factors are characterized by a reduced availability of the vasoprotective molecule nitric oxide and an increase in reactive oxygen species, so-called oxidative stress. Transcription factors function as intracellular signal integrators and relays and thus, play a central role in cellular responses to changing conditions.
Recent Advances:
Work on specific transcriptional regulators has uncovered many of their functions and the upstream pathways modulating their activity in response to reactive oxygen and nitrogen species. Here, we have reviewed for a few selected examples how this can contribute not only to protection against atherosclerosis development but also to disease progression and the occurrence of clinical manifestations, such as plaque rupture.
Critical Issues:
Transcription factors have pleiotropic outputs and often also divergent functions in different cell types and tissues. Thus, in light of potential severe adverse side effects, a global activation or inhibition of particular transcriptions factors does not seem a feasible therapeutic option.
Future Directions:
A further in-depth characterization of the cell- and stage-specific actions and regulation of transcription factors in atherosclerosis with respect to protein-protein interactions and target genes could open up new avenues for prevention or therapeutic interventions in this vascular disease. Antioxid. Redox Signal. 26, 679–699.
Introduction
A
Heart attacks and strokes are acute events, whereas the underlying problem in the majority of the cases is an obstructed blood flow, mainly caused by atherosclerotic plaque buildup over a long time. Plaque development is initiated by recruitment and infiltration of circulating immune cells such as monocytes, into the arterial wall. In the sub-endothelial space, monocytes differentiate into macrophages that scavenge oxidized low-density lipoprotein (oxLDL), eventually becoming lipid-laden foam cells that form fatty streaks. As disease progresses, infiltrating immune cells, buildup of dying cells, and phenotypically switched vascular smooth muscle cells (VSMC), among other mediators, contribute to formation of the atherosclerotic lesion (Fig. 1). As lesions expand, arteries become hardened and occluded (102). In late stages of the disease, plaques can become destabilized and rupture, and subsequent clot formation can lead to heart attacks and strokes.
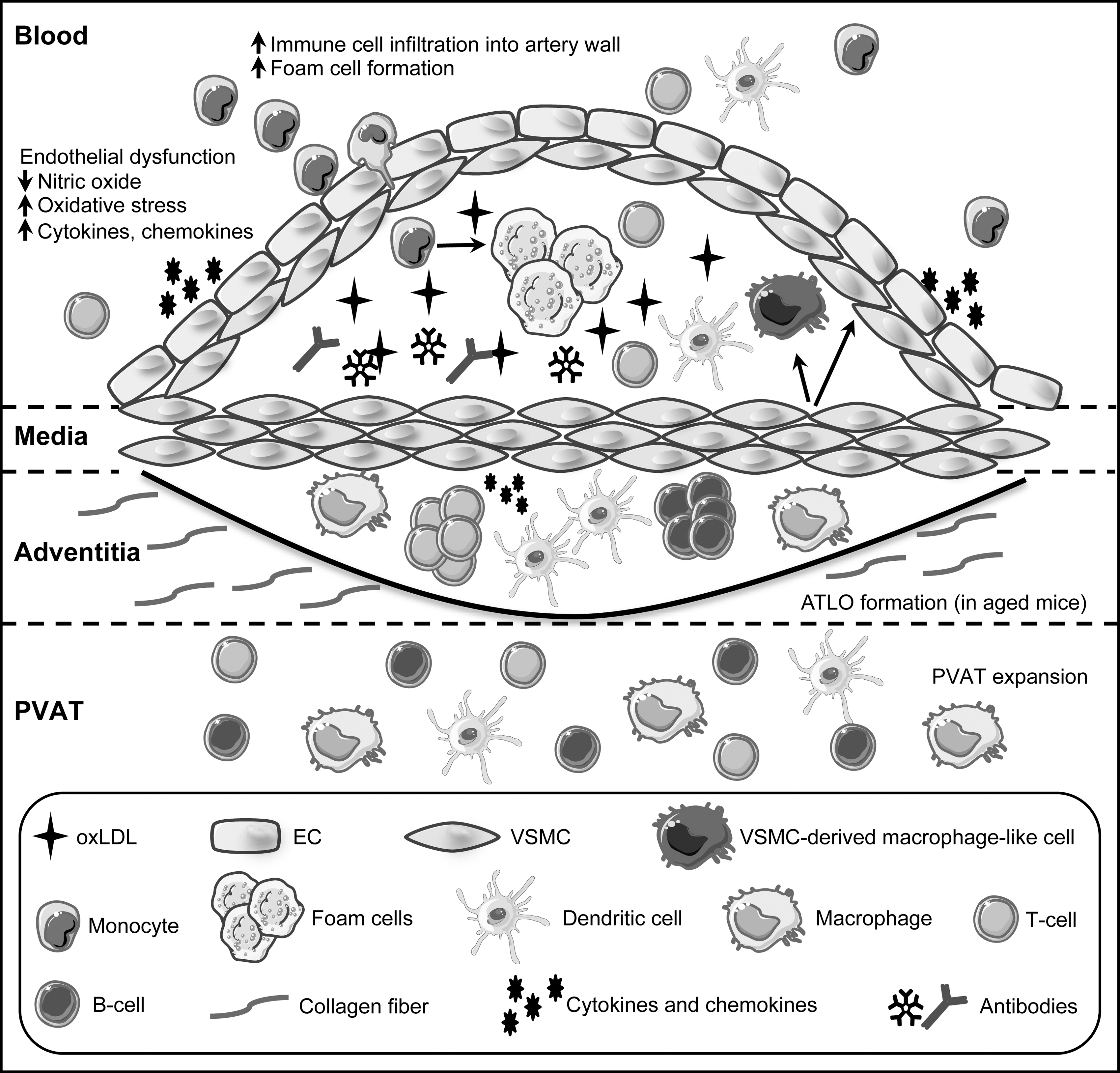
Several diseases or disease conditions are considered as independent atherosclerotic risk factors, among them being diabetes type 2, obesity, and metabolic syndrome, and like atherosclerosis itself, they have an inflammatory component. All these conditions have been linked not only to oxidative stress (55, 72) but also to endoplasmic reticulum (ER) stress (84, 86, 141). Furthermore, they are associated with endothelial dysfunction and senescence (15, 126, 132, 142, 167, 191), the prevalence of which increases with age, underscoring why atherosclerosis is an age-associated disease. Although the site of the final damage is the vessel wall, other cell types are affected and contribute to these disorders. Besides the vascular cells themselves, cells from the immune system play an integral role in such pathophysiological changes (71).
Transcription factors serve as central nodes in intracellular signaling, as they integrate multiple inputs from the environment and translate them into coordinated cellular responses (97). Among these external signals are reactive oxygen species (ROS) and nitric oxide (NO), key mediators of vascular functions.
Instead of discussing transcription factors that have been extensively reviewed, for example, nuclear factor kappa B (NF-κB) (145, 147) or NF-E2-related factor 2 (NRF2) (125), we will exemplarily highlight several facets of how reactive molecules regulate transcription factor functions in selected cell types and how this can promote or alleviate disease development and progression.
The Vascular Wall and Immune Cells in Physiology and Pathology
Healthy arteries consist of a monolayer of endothelial cells facing the blood flow, an adjacent medial layer of smooth muscle cells, and a surrounding collagen-rich layer, termed the adventitia. In addition, the aorta and the coronary arteries are surrounded by a layer of perivascular adipose tissue (PVAT), which is a metabolically active endocrine tissue secreting mediators that can affect the vasculature (13).
The endothelium is a key regulator of vascular homeostasis. It functions not only to transport nutrients, oxygen, and waste products to and from blood and tissue but also as an important metabolic organ, secreting autocrine and paracrine factors that result in the constriction or dilation of vessels (31, 42). Furthermore, it is a critical protective barrier separating inflammatory blood components from tissue. Importantly, factors produced by endothelial cells can be both (i) oxidizing and anti-oxidizing, (ii) vasodilatory and vasoconstricting, (iii) pro-coagulating and anti-coagulating, and (iv) pro-inflammatory and anti-inflammatory, depending on context and stimulus (17, 42). Thus, the endothelium serves an important physiological function of maintaining the balance between these opposing effects at homeostasis. In acute inflammatory conditions, endothelial cells also mediate immune cell trafficking to sites of injury or infection by upregulating adhesion molecules and cytokines, ultimately leading to extravasation and transmigration of immune cells into the tissue (Fig. 1).
NO produced by endothelial cells is a key vasodilatory factor. It regulates vascular tone and redox homeostasis, maintaining VSMCs in a quiescent non-proliferative state, and prevents immune cell recruitment to the artery wall (4). Reduced NO-bioavailability resulting in increased oxidative stress, is a hallmark of endothelial dysfunction, and it is a major contributor in the pathogenesis of atherosclerosis. It is characterized as tipping of the balance of endothelial functions toward vasoconstriction, coagulation, and inflammation. Oxidative stress can trigger endothelial cell activation, leading to a pro-inflammatory state that enhances recruitment and infiltration of circulating immune cells, such as monocytes, into the arterial wall (Fig. 1). Thus, endothelial dysfunction precedes the occurrence of visible atherosclerotic changes (31).
At homeostasis, VSMC in the media primarily serve to modulate blood vessel tone and blood pressure through contraction and relaxation. However, they are highly plastic cells that are able to alter phenotype and function in response to environmental stimuli (7, 140). For example, in response to vascular injury, VSMC can become capable of increased proliferation, migration, and secretion of extracellular matrix components. Furthermore, VSMC can dedifferentiate, losing their typical markers while gaining macrophage markers. Elegant studies by Owens and coworkers using lineage-tracing models have shown that a significant population of lesion macrophages is derived from VSMC (58).
The role of VSMC in the pathological setting of atherosclerosis is conflicting (7). On the one hand, VSMC migration and collagen secretion leads to formation of the fibrous cap, which stabilizes the lesion. On the other hand, VSMC can also produce matrix metalloproteinases that function to erode the fibrous cap. Moreover, VSMC-derived macrophages in lesions differ from monocyte-derived macrophages and demonstrate impaired phagocytic capacity, which may hinder adequate clearance of apoptotic cells and lipids. Finally, VSMC also produce and secrete chemokines that can recruit immune cells to the lesion (Fig. 1).
Atherosclerosis also involves many components of the immune system (Fig. 1). Much like the case for endothelial cells and VSMC, it is the excessive and chronic activation of immune cells, without subsequent resolution of the immune response, that contributes to the pathogenesis of atherosclerosis. Although the role of several types of immune cells has long been recognized, it is increasingly appreciated that their functions are entirely subset- and context dependent. This does not only hold true for macrophages (26) but also for B and T cells. The B-2 cell subset is primarily pro-atherogenic, whereas B-1 cells are largely atheroprotective (148, 181). Similarly, Th1 and regulatory T cells exert opposite effects, either promoting atherosclerosis or being protective, respectively (180).
Although much focus has been given to immune cells within atherosclerotic lesions, less well known is the contribution of immune cells residing in the adventitia and PVAT (18). It has been argued that the PVAT plays a role in atherosclerosis, because it can induce inflammation in the vascular wall (186). Interestingly, the adventitia of aged mice contains arterial tertiary lymphoid organs (ATLOs), which contain distinct B and T cell zones, germinal centers, and vasculature similar to those in secondary lymphoid organs (59). Whether these ATLOs serve a protective or detrimental role in the artery wall or perhaps both is still not resolved (199).
Functions of ROS and NO in the Vasculature
In every cell of the vascular wall, a redox balance exists, which is maintained by endogenous oxidative and anti-oxidative systems. The oxidative systems consist of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, xanthine oxidase, and, to a minor extent, cyclooxygenase and 5-lipoxygenase (51). The major source of superoxide anions under physiological conditions is oxidative phosphorylation, since 2–5% of oxygen molecules consumed by mitochondria undergo one electron reduction (105). To cope with increased ROS, cells possess superoxide dismutases converting superoxide anions to hydrogen peroxide, which is then cleared to water and oxygen by catalase. In addition, the thioredoxin systems 1 and 2 and the glutathione system are mainly used to reduce oxidized proteins (9, 106).
Historically, it was assumed that ROS are only produced to destroy intruders and can damage cellular components such as DNA, lipids, and proteins. Nowadays, it is accepted that ROS also act as signaling molecules and second messengers (160), also in the vascular wall. In this function, they can modulate proliferation of VSMCs (27) and the endothelial transcriptome (161) without inducing damage or cell death. On the other hand, it is well described that increased levels of ROS are involved in CVDs. This so-called oxidative stress has been linked to endothelial dysfunction (17), cellular injury, and tissue damage, as well as to conditions considered as risk factors, such as diabetes (47) or obesity (55), all of which contribute to the pathology of atherosclerosis (72).
Another important second messenger in the vessel wall is NO. Originally identified as endothelial-derived relaxing factor (144), NO plays an important role for all cells of the vascular wall. It is continuously synthesized in endothelial cells by the endothelial NO synthase (eNOS) from the amino acid L-arginine and oxygen to form NO and L-citrulline in nanomolar concentrations (143).
NO is a potent powerful vasodilator and possesses various vasoprotective effects such as inhibition of platelet aggregation, suppression of adhesion of leukocytes or monocytes on the endothelial surface, inhibition of proliferation, and migration of VSMCs and vascular inflammation (104).
NO can exert its downstream effects by binding to soluble Guanylate Cyclase, promoting the formation of cyclic guanosine monophosphate (cGMP) and activating the cGMP-dependent protein kinase (34). However, NO can also act as a signaling molecule through several cGMP-independent pathways, for example by affecting platelet aggregation and immune cell trafficking in the vasculature (195). Moreover, NO as well as different ROS can directly interact with cysteines and metal-containing centers of proteins to modulate the functions of those enzymes.
A reduced NO-bioavailability has been demonstrated to contribute to endothelial dysfunction in cells, animals, and humans. The molecular mechanisms leading to lower levels of NO in the vessel wall are widespread. Experimental evidence exists not only for a reduction of eNOS RNA amount and stability as well as diminished eNOS protein levels but also for a reaction of NO with superoxide anions to form peroxynitrite—all these processes, in turn, reduce NO-bioavailability in the vasculature (51, 80, 94). Unfortunately, eNOS can produce not only NO but also superoxide anions when the enzyme is uncoupled. One major reason for uncoupling is the depletion of the essential cosubstrate tetrahydrobiopterin or its oxidation to dihydrobiopterin, which occurs under conditions of oxidative stress (162).
Due to their highly reactive nature, ROS and NO can interact with several proteins, such as kinases, phosphatases, and receptors, thereby modulating their functions (46, 81). Interestingly, given the nature of transcription factors, several amino acids described as responsible for DNA binding are modified by ROS and/or NO.
Last but not the least, oxidative stress is tightly linked to ER stress, since the latter can disturb the cellular redox homeostasis and conversely, ER malfunction can be accelerated by ROS (39, 159). The ER is required for protein folding and maturation and nearly all proteins, which will be inserted into membranes or secreted, and, therefore, have to pass through this organelle. Associated with these functions is a stringent quality control for properly folded proteins. The oxidative environment and a specific enzymatic repertoire support the formation of disulfide bridges. This oxidative process requires molecular oxygen and hydrogen peroxide as electron acceptors (14).
Intuitively, reductive agents disturb the ER homeostasis; however, it has become evident that this stress response is also triggered by oxidative stress. The first adaptive reaction to reestablish homeostatic conditions is the unfolded protein response (UPR). It principally consists of three arms mediated by proteins anchored in the ER membrane and containing a luminal domain sensing critical levels of misfolded proteins (138, 158).
The inositol-requiring enzyme 1 has two enzymatic functions. A ribonuclease domain is activated by an intrinsic kinase and is necessary for unconventional splicing of the X-box binding protein 1 pre-messenger RNA (mRNA), leading to production of a form of this transcription factor regulating UPR target genes.
The second arm is governed by the PRKR-like ER kinase. It phosphorylates a translation initiation factor to suppress global translation, still allowing translation of specific mRNAs. Among them is the one for activating transcription factor 4 (ATF4), which, in turn, induces the expression of several corrective genes.
The third arm is mediated by another member of the same transcription factor family, ATF6, which on ER stress translocates to the golgi apparatus, where it is cleaved to release its transcriptionally active form, again upregulating compensatory mechanisms. Importantly, sustained ER stress, which occurs on persistent oxidative stress, induces apoptosis (112).
Taken together, ROS and NO are essential for proper function of vascular wall cells. However, chronic overproduction of ROS or long-lasting reduction in NO is detrimental and will inevitably lead to death.
Transcription Factors: Structure and Regulation
Transcription factors serve as central intracellular nodes for signal integration and relay into cellular outputs at the transcriptome level. Numerous signaling cascades converge on these proteins, which themselves activate or repress a plethora of target genes. These pleiotropic responses have convincingly been illustrated through genome-wide characterization of DNA target sites of several transcription factors (89, 111, 154, 197, 212). Those studies revealed several thousand genomic binding sites for each of these proteins; further validation showed in every case the regulation of several hundred genes.
Soon after the characterization of the first transcription factors, it became evident that these proteins have a modular architecture marked by physically separable, interchangeable domains (52).
Pivotal for the function of transcription factors is their DNA binding domain, which is required for sequence-specific DNA recognition. These DNA binding domains fall into different groups, which show strong structural conservation and are used to classify transcription factors into distinct families (202). Even though DNA binding is an intrinsic property of transcription factors, some proteins lacking a DNA binding domain can also be classified as transcriptional regulators. One such family comprises the inhibitor of differentiation (Id) helix-loop-helix (HLH) proteins, which prevent DNA binding and thus, transcriptional regulation by basic HLH (bHLH) proteins (115, 137).
A second crucial feature of transcription factors is the up- or downregulation of transcription, mediated by activation or repression domains. These domains, which unlike the DNA binding domains barely show any structural conservation, serve to increase or decrease the recruitment of RNA polymerase to the transcription start site. This is accomplished by interaction with the basal transcription machinery or recruitment of coactivators and -repressors, respectively (53).
The activity of most transcription factors is regulated to allow cellular adaptation to internal or external signals. As transcription factor activity can be modulated by numerous mechanisms, regulatory domains of transcription factors can serve many functions (Fig. 2). One feature common to many transcription factors is dimerization (1). The formation of homo- and heterodimers within a transcription factor family or with close relatives can have several consequences. If one of the partners lacks a dimerization domain, as in case of the Id proteins (115, 137), this can inhibit DNA binding and thus, provide a passive mechanism precluding transcriptional regulation. Furthermore, dimerization can, depending on the dimerization partner, change DNA recognition, which results in binding to different target sequences (66, 83, 113, 173). Moreover, heteromeric complexes composed of different subunits can serve as activators or repressors of transcription, thereby altering cellular behavior (60, 109). Another mechanism regulating transcription factor activity, which can also be ascribed to a specific domain, is ligand binding, a feature of the majority of nuclear receptors (12, 127).
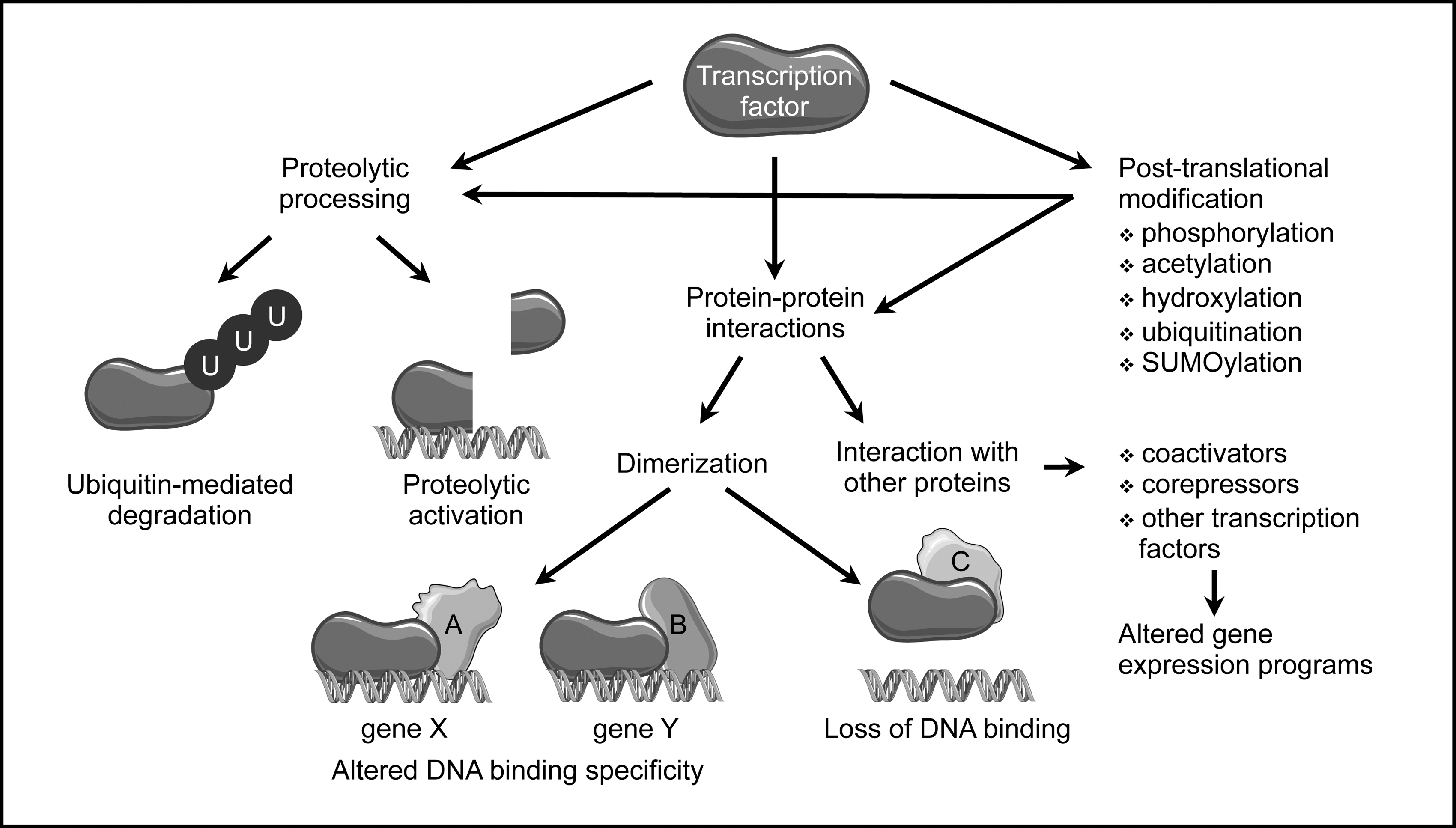
The different functional domains of transcription factors do not necessarily have to be arranged in a particular order, as they seem to adopt their conformation independently of each other. Furthermore, although transcription factors contain—with the exception of non-DNA binding proteins mentioned earlier—only one DNA binding domain directing them to their specific DNA target sequence, the other domains can occur in different copies and flavors within the same polypeptide.
Besides these larger domains, reversible post-translational modifications of single amino acid residues play a major role in controlling transcription factor activity (Fig. 2). They create surfaces recognized by modification-specific interaction domains (164) and thus, act as molecular switches expanding the regulatory networks controlling transcription factor activity even further.
One of the most well-characterized modification is phosphorylation, which can regulate multiple aspects of transcription factor functions, such as stability, localization, DNA binding, and protein-protein interactions (200). A few sequence-specific transcription factors can be acetylated on lysine side chains, resulting mostly in enhanced DNA binding (30). Similarly, attachment of methyl groups to the same amino acid has been described for a small number of transcription factors. However, depending on the number of methylated residues and the site of methylation, the outcomes can be greatly different (19). Addition of the small protein ubiquitin to lysine residues, ubiquitination, usually targets proteins for proteolytic degradation. Interestingly, this modification has been linked to transcriptional activation and might provide a “clock” determining how long a transcription factor can remain active. Moreover, there seem to be non-proteolytic functions of ubiquitination of transcription factors, which are mechanistically not well understood (56). Attachment of small ubiquitin-like modifier (SUMO) proteins is, with a few exceptions, associated with transcriptional repression (57). Prolyl-hydroxylation affects one specific transcription factor, hypoxia inducible factor 1α, where it is required for degradation under normoxic conditions (198). Moreover, several transcription factors can be released from membrane-bound, dormant forms by specific proteolytic cleavage, which allows translocation to the nucleus to exert their actions (82) (Fig. 2).
A direct regulation of transcription factors by the cellular redox status has been shown for members of different families. In most cases, oxidation of the sulfhydryl group of cysteine residues in the DNA binding domain leads to a reduced affinity to DNA (106). Besides these direct effects, numerous redox-sensitive signaling pathways, which are relevant for vascular diseases, can affect transcription factors (110). A few selected examples will be discussed in the following chapters.
Sister-of-Mammalian Grainyhead: Two Messages from One Message
Sister-of-Mammalian Grainyhead (SOM; also known as Grainyhead-like 3; GRHL3) is a member of a highly conserved transcription factor family. It is closely related to two other proteins, Mammalian Grainyhead (MGR or Grainyhead-like 1, GRHL1) and Brother-of-Mammalian Grainyhead (BOM or Grainyhead-like 2, GRHL2) (179, 201). All three proteins contain an N-terminal transactivation domain, a central DNA binding domain, and a C-terminal dimerization domain. Although they share similarity and recognize the same DNA-consensus motif, they are not able to functionally compensate for each other, leading to the suggestion that they regulate different sets of target genes (11). In fact, it was shown that even two isoforms of SOM activate distinct genes (65).
Several studies demonstrated that SOM plays a role in neural tube closure, wound healing, and epithelial cell migration (16, 64, 78, 153, 178, 179). Interestingly, SOM was independently identified in a gene trap screen for genes upregulated during apoptosis induction and then shown to be expressed in primary human endothelial cells. In addition, it was demonstrated that SOM promotes migration of these cells (63). In line with the original screen, later investigations uncovered an anti-apoptotic function of SOM (107).
Further complexity arises from the existence of human-specific protein isoforms of SOM. Usage of an additional first exon not present in rodent genomes and alternative splicing of the primary transcript containing this exon yield a total of three SOM proteins (Fig. 3). They are identical except for their N-terminus. SOM2, the homolog of murine SOM, resembles SOM1, with the only difference being the N-terminal 11 amino acids. The third isoform, SOM3, is derived from a transcript lacking the second exon of SOM1 and encodes an N-teminally truncated protein originally described as a putative repressor (179). All three SOM isoforms are coexpressed in human endothelial cells and are transcriptional activators (65, 107).
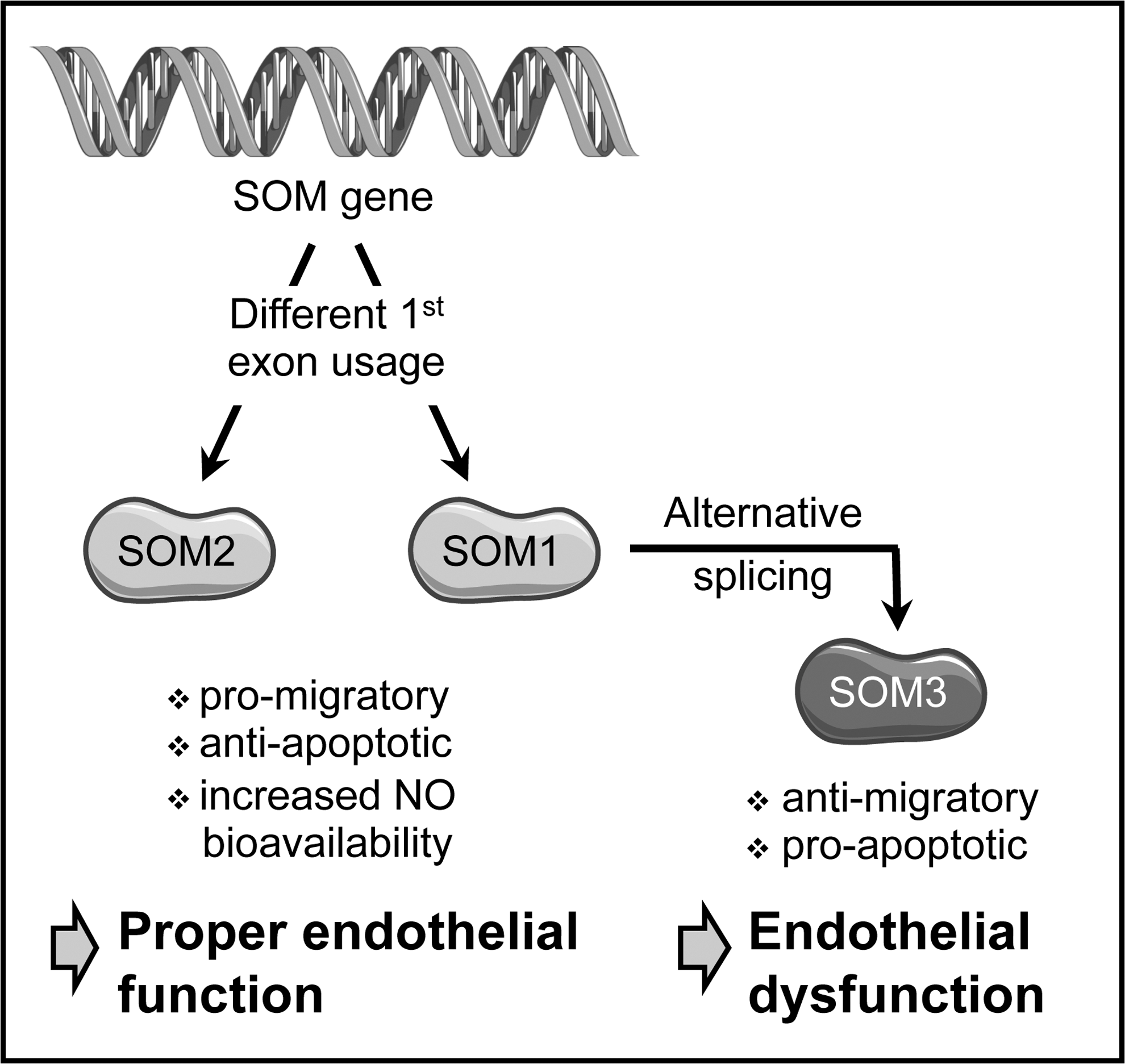
SOM1 and SOM2 are pro-migratory and anti-apoptotic in endothelial cells, whereas SOM3 has opposite effects (Fig. 3). An explanation for these different cellular outcomes is provided by the different target gene spectrum activated by the SOM isoforms (65). Moreover, these data suggest that the balance between the isoforms of this transcription factor could be disturbed in endothelial dysfunction.
The regulatory networks affecting SOM expression and function are only poorly understood, and it remains unclear whether the cellular redox status has an influence on this transcription factor. NO, however, plays an important role in SOM functionality in endothelial cells. It was shown that SOM induces activation of eNOS and that the pro-migratory effect of both SOM and its anti-apoptotic function are dependent on NO (107). Furthermore, SOM expression is upregulated by physiological concentrations of NO, suggesting the existence of a positive feed-forward loop (Fig. 4).
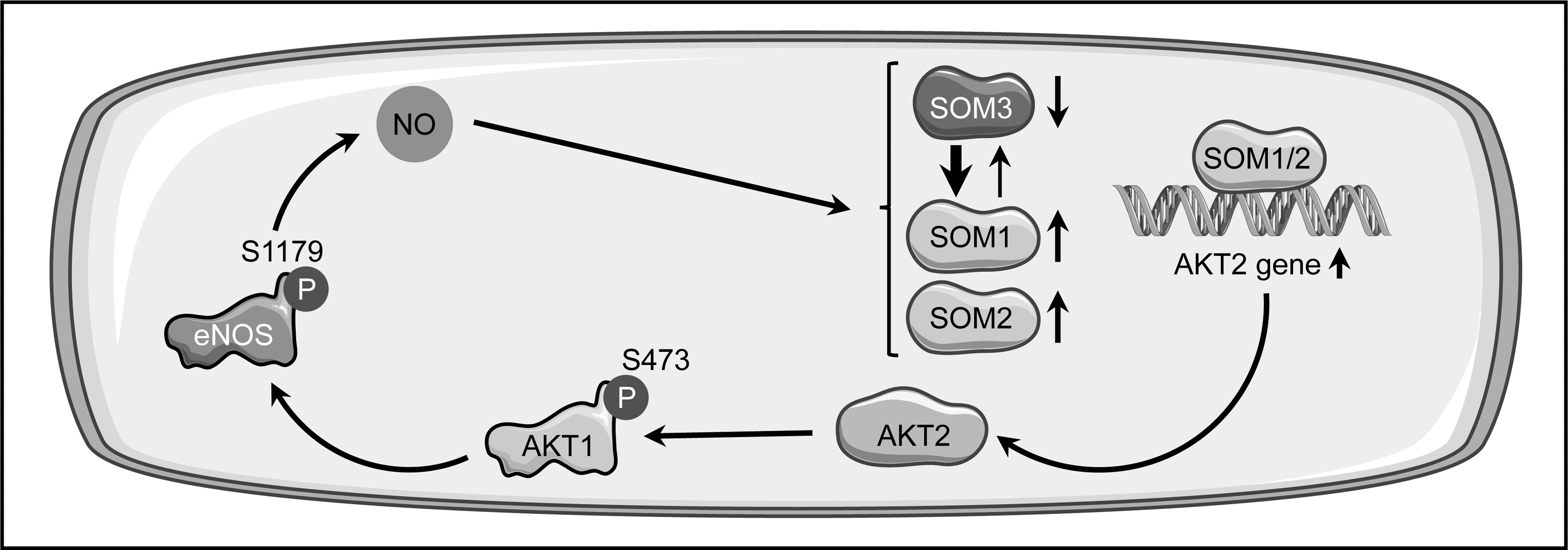
On the molecular level, it was shown that SOM1 and SOM2 increase NO-bioavailability in endothelial cells via eNOS phosphorylation on serine 1179 by protein kinase Bα/AKT1 (65, 107), resulting in increased eNOS activity (54). This is most likely due to the upregulation of protein kinase Bβ/AKT2 expression, a master regulator of all AKT isoforms (150), which was demonstrated after overexpression of SOM1 (65) (Fig. 4).
Remarkably, NO not only induces an increase in SOM2 expression but also has a profound influence on the balance between the two other isoforms. Treatment of endothelial cells with an NO-donor led to an elevated steady-state level of the SOM1 transcript and a concomitant decrease in the SOM3 mRNA (65) (Fig. 4). This suggests that not only transcription and post-translational modifications are regulated by stimuli affecting endothelial functions, but also RNA splicing. Interestingly, it has been described that expression of the splicing factor pre-mRNA Processing Factor 19 is downregulated in endothelial senescence, which goes along with endothelial dysfunction, and that overexpression of this protein extends the life span of endothelial cells (190). This is not an isolated phenomenon, as another splice regulatory protein, serine/arginine-rich splicing factor 1, has been described as a marker for endothelial senescence (10).
The data presented here show an intimate involvement of SOM in endothelial function and dysfunction and, therefore, it will be of interest to better understand its regulation and target genes in disease settings and aging.
Krüppel-Like Factor 2 and 4: Go with the Flow
The family of Krüppel-like factors (KLFs), named according to their homology to the Drosophila Krüppel protein, consists of 17 members. They are characterized by Cys2/His2 zinc fingers in their DNA binding domain, the most abundant structural motif in transcription factors, estimated to be present in ∼900 different transcriptional regulators (183). Characteristic for the KLFs are three contiguous zinc fingers at the extreme C-terminus and additional, conserved residues between these fingers (174). In addition, three amino acids close to the histidines, which are involved in coordination of a zinc ion, and thus, critical for the three-dimensional structure, determine target site interactions (203). This results in a similar DNA recognition of G/C-rich sequences by all KLFs (8, 174). The variable N-terminal part is much less conserved and contains activation as well as repression domains (8). Within the KLFs, the proteins KLF1, KLF2, and KLF4 form a closely related subgroup (8, 174).
A systemic knockout as well as endothelial-specific deletion of KLF2 is embryonic lethal (96, 194), demonstrating a requirement for this transcription factor during embryogenesis. However, hemizygous KLF2-deficiency augments diet-induced atherosclerosis on an apolipoprotein E (ApoE) negative genetic background (3), clearly indicating that KLF2 is atheroprotective. Mechanisms involved in atheroprotection by KLF2 are the upregulation of eNOS expression in the endothelium and the interference with cytokine-induced expression of adhesion molecules such as vascular cell adhesion molecule 1 (VCAM1). VCAM1 is a marker for endothelial cell activation, which occurs early in atherosclerosis development (168).
The expression of KLF2 itself is controlled by hemodynamic forces, which could explain why specific regions in the blood vessels are more prone to atherosclerotic lesion development than others, even if they are exposed to the same systemic risk factors. A steady, pulsatile blood flow is atheroprotective; however, disturbed flow in the form of oscillatory or turbulent flow creates an atheroprone environment (22). Interestingly, oscillatory shear stress also enhances ROS production by NADPH oxidases and monocyte adhesion in cultured endothelial cells (87, 88, 119). Along these lines, treatment with the antioxidant N-acetylcysteine can abrogate the upregulation of the adhesion molecule VCAM1 by oscillatory shear stress (21). In vivo oxidative stress has been detected in atherosclerosis-susceptible regions of the mouse aorta, even in a strain with a different genetic susceptibility to atherosclerosis (68), demonstrating that oxidative stress is independent of the disease per se.
The fact that, depending on blood flow type, specific regions of the aorta are more vulnerable to atherosclerosis development than others is mirrored in the shear stress dependence of KLF2 expression. In endothelial cells, KLF2 is downregulated by atheroprone flow (73) and upregulated by laminar shear stress (168). Of note, the latter effect was much stronger when prolonged unidirectional pulsatile flow was applied instead of steady flow (33). This dependence of KLF2 expression on flow type was also shown in vivo in different regions of the rat aorta (192). Furthermore, experimental manipulation of shear stress conditions has an impact on KLF2 expression, with disturbed flow reducing KLF2 expression (207). In addition, in zebrafish mutants with a non-contractile heart and thus, no blood flow, vascular expression of KLF2 is lost (146).
On the mechanistic level, it was shown that steady laminar shear stress leads to phosphorylation of histone deacetylase 5 (HDAC5) coupled with its nuclear export and to disruption of the interaction with myocyte enhancer factor 2C (MEF2C), which, in turn, can then activate KLF2 transcription (Fig. 5).
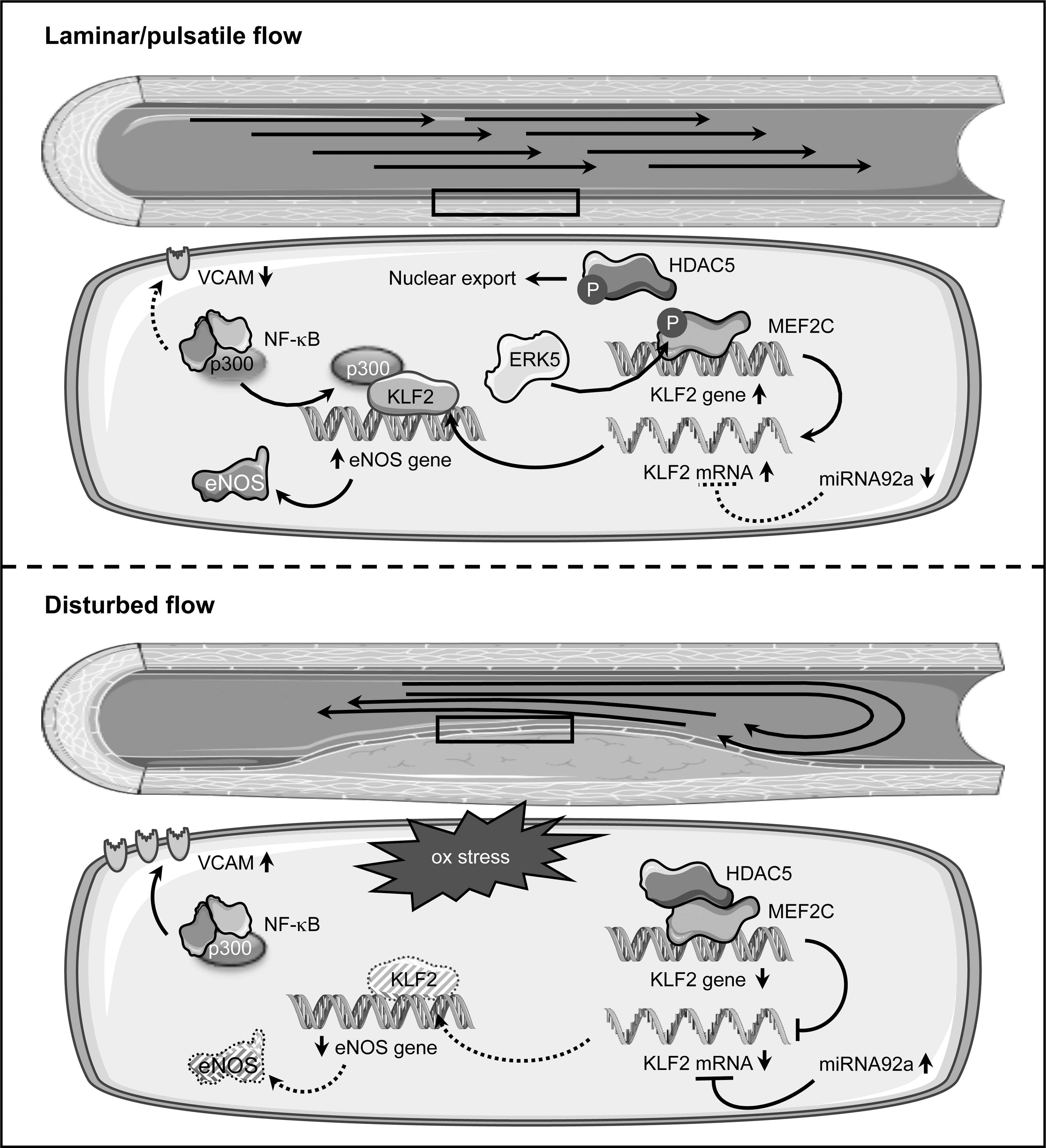
The relevance for HDAC5 phosphorylation was demonstrated by adenoviral overexpression of a non-phosphorylatable form, which downregulated flow-mediated KLF2 and eNOS expression and enhanced adhesion of monocytes on the endothelial cell surface even under laminar flow (193). Full activation of MEF2C requires its phosphorylation by the extracellular-signal regulated kinase 5 (ERK5), which itself is activated by constant flow. A critical role for this phosphorylation in flow-induced KLF2 upregulation has been demonstrated by expression of a permanently active form of ERK5, which induced KLF2 expression in endothelial cells even under static conditions. Conversely, a non-phoshorylatable, dominant-negative ERK5 mutant suppressed KLF2 induction by laminar flow (146). It has to be noted that ERK5 is not the kinase responsible for nuclear export of HDAC5.
In addition, laminar shear stress leads to stabilization of the KLF2 mRNA (185). One explanation for the change in the KLF2 transcript stability is microRNA-dependent degradation (Fig. 5). The microRNA 92a (miRNA92a), which, compared with KLF2, is reciprocally regulated by different flow conditions, targets the 3′ untranslated region of the KLF2 transcript. Expression of a miRNA92a precursor in endothelial cells reduced the levels of KLF2 and eNOS and, conversely, a knockdown increased them (204). In vivo, higher levels of miRNA92a in the atherosusceptible aortic arch of swine aortas were associated with decreased KLF2 expression, and the reciprocal situation was found in the thoracic aorta, which is much less vulnerable to plaque formation (41). Interestingly, this microRNA is upregulated not only by oscillatory flow but also by oxidative stress (25), providing a link between these two phenomena culminating on KLF2.
The upregulation of eNOS by KLF2 is simply explained by binding of the transcription factor to the eNOS gene promoter (168), whereas the suppression of the cytokine induction of adhesion molecules is based on a different mechanism. Upregulation of endothelial VCAM1 in inflammatory conditions is controlled by NF-κB both ex vivo and in vivo (28, 124, 171). One coactivator critical for NF-κB function is p300 (170, 205). Seemingly, KLF2 sequesters this coactivator independent of its own binding to DNA, thereby compromising NF-κB functions and upregulation of VCAM1 (168).
The closest relative of KLF2, KLF4, also has atheroprotective functions. However, the functions of these two proteins, although overlapping, are not redundant, as shown by the different phenotypes of knockout animals. Unlike KLF2-deficiency, the lack of KLF4 does not lead to embryonic lethality. Instead, the animals die postnatally due to a defective skin barrier, resulting in dehydration (165). Endothelial-specific deletion of KLF4 resulted in enhanced atherosclerosis development. Conversely, transgenic overexpression in the endothelium reduced the atherosclerotic lesion area and inflammatory cell infiltration (213).
Like KLF2, KLF4 is induced by laminar shear stress in endothelial cells. There, its overexpression induces eNOS expression and anti-thrombotic responses, for example suppression of VCAM1, whereas knockdown enhances tumor necrosis factor α-induced expression of this adhesion molecule (70). KLF4 upregulates eNOS and suppresses the expression of VCAM1 by the same mechanisms as KLF2 (213). In addition, it is also regulated by the same upstream regulators ERK5 (139) and miRNA92a (25, 41).
Taken together, KLF2 and KLF4 have important atheroprotective functions in the endothelium by enhancing NO-production via the upregulation of eNOS and interfering with the expression of surface molecules that are critical for monocyte adhesion. Furthermore, they are co-regulated by the same pathways. These two closely related transcription factors cannot fully compensate for each other as demonstrated by the different phenotypes of the knockout mice. Nevertheless, they seem to have not only overlapping and partially redundant but also distinct functions in the endothelium (69), which have to be dissected further in the future.
Id3: Guilty by Association
The four Id proteins (Id1–4) interfere with the class I bHLH transcription factors, known as E proteins. E proteins contain an HLH domain, which facilitates dimerization with other bHLH proteins, and a basic DNA binding domain. Id proteins lack the latter, but retain the capacity to dimerize with E proteins via the HLH region, resulting in E protein sequestration and inhibition. This dominant negative effect of Id proteins can, depending on the E protein bound, lead to activation or suppression of gene transcription. All Id proteins have been implicated in growth control, which is most likely exerted through the suppression of cyclin-dependent kinase inhibitor (CDKI) expression (157, 211).
One of the Id proteins, Id3, is implicated in different aspects of vascular pathologies (49). It has been shown that ROS can directly induce proliferation of VSMC (152), which is also promoted by angiotensin II (Ang II), an inducer of oxidative stress in the vasculature. The Ang II effect is more pronounced in neointimal VSMC than in medial cells or the normal vessel wall (29). Interestingly, Id3 is upregulated by Ang II in VSMC in a superoxide-dependent manner. Furthermore, it has been shown that Id3 represses expression of p21 and p27, two members of the CDKI family, after Ang II treatment (131, 136), placing it in a central position in redox-sensitive growth control of VSMC. Along these lines, Id3 is profoundly upregulated in neointimal VSMC in vivo after wire injury of the carotids (136) and strongly expressed in human atherosclerotic plaques, but not in normal coronary arteries (196).
A splice variant of human Id3 has been identified, which possesses a unique C-terminus of 60 amino acids generated by read-through into a coding intron. This isoform, Id3 L, seems to have impaired dimerization capacity toward E-proteins (32). A similar intron inclusion can occur in rats, leading to an mRNA coding for Id3a, which also has a unique C-terminus, in this case of 29 amino acids (117). Both isoforms are coexpressed in cultured VSMC; however, the expression of fully spliced Id3 is much higher. A change was noticed after denudation of the carotid artery, when the Id3a RNA, which was undetectable in uninjured carotid arteries, was upregulated beginning 6 days after injury. Expression was detected mainly in the growing neointima and inner media and was sustained for the whole observation period of 28 days. During this time window, no Id3 transcript was observed (Fig. 6). Upregulation of the alternative isoform is not unique to the animal model, as the corresponding Id3 L transcript was also detectable in human carotid atherosclerotic plaques (117).
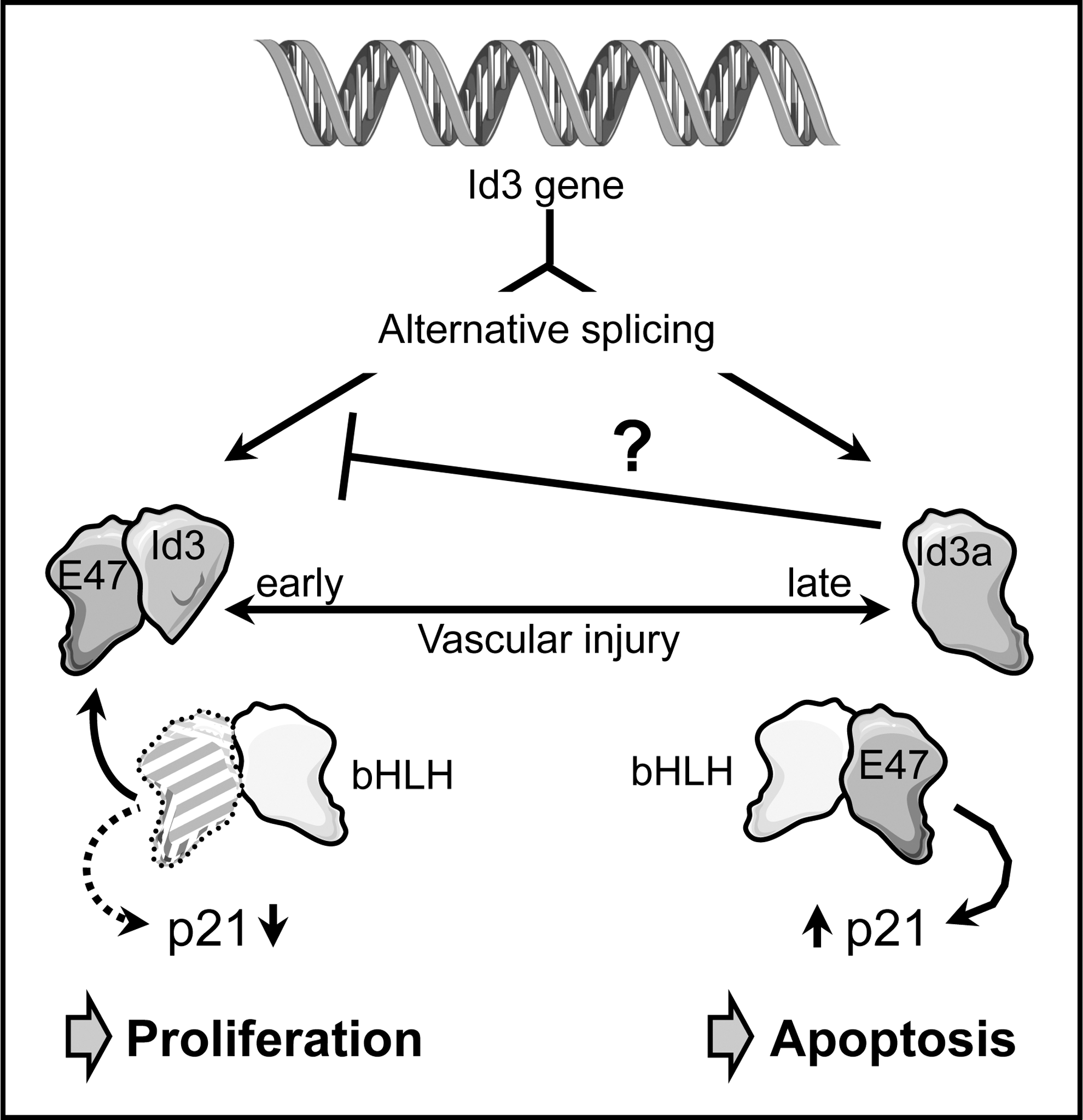
A similar switch in Id3 isoforms was also observed on the protein level. Although Id3 levels increased in carotid arteries up to 7 days after balloon denudation before they returned to baseline levels, Id3a protein accumulated later, but upregulation was sustained (50).
The functional outputs of the two Id3 isoforms in VSMCs are strikingly different. Overexpression of Id3 induced proliferation, whereas Id3a decreased the number of viable cells by apoptosis induction (Fig. 6). Adenoviral expression of Id3a immediately after balloon injury reduced neointima formation in vivo. Furthermore, Id3, in contrast to Id3a, inhibited transcription of the cell cycle inhibitor p21 (50, 117). This was corroborated by the reduced proliferative capacity of VSMC from Id3 knockout mice and their inability to suppress transcription from the p21 promoter (50).
Inline with the notion that dimerization of human Id3 L with ubiquitous bHLH proteins is impaired (32), Id3a cannot interfere with E47-mediated transcriptional upregulation of p21 (50), which might, in turn, induce apoptosis (118). One of the most interesting points in this study was the observation that Id3a can downregulate Id3, suggesting that upregulation of Id3a establishes a feedback loop controlling a switch in VSMC phenotype (Fig. 6). Which signals initiate the switch in Id3 pre-mRNA splicing is currently not known.
Taking this evidence together, it is likely that the switch in Id3 pre-mRNA splicing induces a transition from VSMC proliferation toward apoptosis. This change also occurs in atherosclerotic plaques, where advanced lesions are characterized by a low proliferative index and increased apoptosis of VSMC finally leading to plaque rupture (5).
Besides these roles in VSMC proliferation and apoptosis, Id3 has been shown to mediate protection against atherosclerosis in vivo. This was demonstrated by an increase in atherosclerotic plaques in ApoE/Id3-double knockout mice fed a Western diet exhibiting increased atherosclerosis compared with ApoE-deficient/Id3-proficient animals. This protective role of Id3 was also demonstrated in LDL receptor (LDLR)/Id3 double knockout mice. In both LDLR and ApoE knockout mouse models, Id3-deficiency led to decreased numbers of aortic B cells (103). B cells can localize to atheroprone regions, and Id3 mediates B cell homing to the aorta by upregulating the expression of the chemokine (C-C motif) receptor 6 (CCR6). CCR6 has been implicated in homing to sites of disease, and its expression is downregulated by the bHLH protein E12, which is antagonized in the presence of Id3 (38) (Fig. 7).
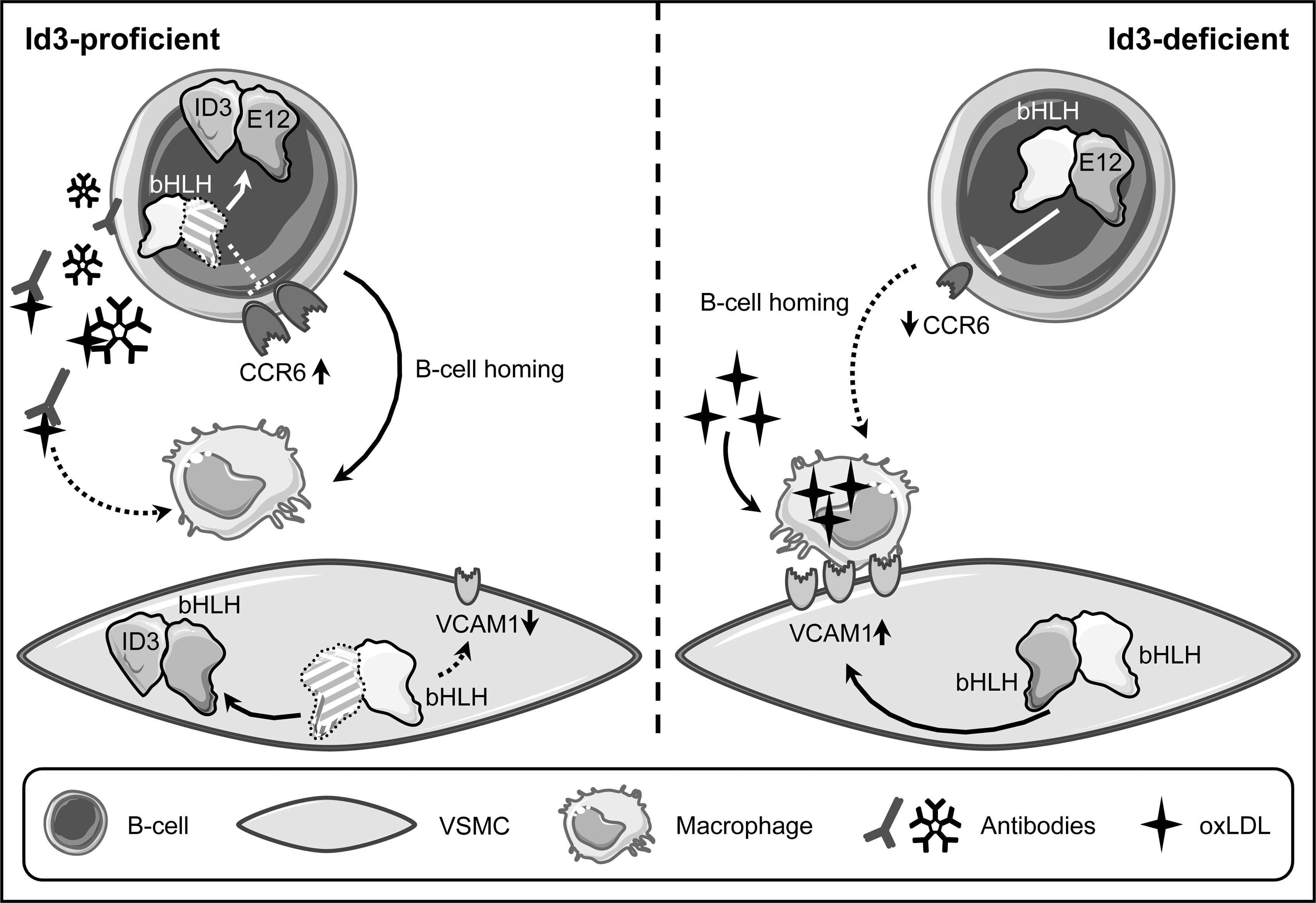
Another consequence of Id3-defciency was an increased macrophage burden in the plaque areas, most likely mediated by an increase in VCAM1 expression in VSMC, again due to insufficient interference with E proteins (103) (Fig. 7). Although endothelial VCAM1 is required for initial monocyte recruitment, its expression on VSMCs in the developing plaque might be necessary for macrophage retention.
Furthermore, B cells, primarily the B1a subtype, produce IgM-type antibodies against oxLDL that can block oxLDL uptake by macrophages, thereby inhibiting foam cell formation (93, 169) (Fig. 7). Therefore, the reduced aortic B cell homing in Id3-deficient mice may also contribute to increased macrophage numbers in lesions (38). These data illustrate that Id3 is critical for B cell-mediated protection in early stages of atherosclerosis development.
Interestingly, a non-synonymous single-nucleotide polymorphism (SNP) in Id3 has been associated with subclinical atherosclerosis in humans (37, 114). This SNP, which does not affect expression, leads to an amino acid exchange in the C-terminus of Id3, which is essential for dimerization with bHLH proteins (23). The mutant protein has reduced affinity to E proteins, resulting in impaired interference with transcriptional activation by these bHLH transcription factors (37). Together with the mouse experiments, this supports a role for Id3 in atheroprotection, mediated in large parts via regulating macrophage and immune cell content in the plaque, which, in the long run, might open up avenues for immune system-directed therapeutic approaches.
ATF6: When Stress Kills
The seven activating transcription factors (ATF1–7) belong to one of the largest classes of transcriptional regulators, the basic leucine zipper proteins identified nearly 30 years ago (67). The members of this superfamily can form homo- and heterodimers within specific subfamilies (66). They are characterized by a contiguous bipartite α-helical stretch encompassing a DNA binding domain and a dimerization region termed leucine zipper, the latter forming an amphipatic helix characterized by a periodic repetition of leucine residues. The leucine zippers of dimerization partners are arranged as a coiled coil of two parallel helices positioning the DNA binding domains such that the dimers form a “scissors-grip”-like structure (189). The ATFs are closely related to Cyclic AMP Response Element Binding Proteins (188).
ATF6 is unique among the ATF transcription factors, as it is synthesized as a 90 kDa transmembrane protein that is inserted into the ER membrane. Induction of ER stress leads to appearance of a 50 kDa proteolytic cleavage product that is localized in the nucleus (76). Processing encompasses reduction of disulfide bridges linking the luminal domains of individual ATF6 molecules, thereby creating monomers. Only these monomers are transported to the golgi apparatus, where they are sequentially cleaved by two enzymes, site-1- and site-2-protease, leading to the release of the transcriptionally active 50 kDa form, which is imported into the nucleus (134).
The induction of ER stress provides a compensatory, atheroprotective mechanism. However, in pathological settings such as atherosclerosis, sustained activation of this pathway can promote cell death (163). Endothelial cell apoptosis is an early event in atherosclerosis development, whereas death of VSMCs (6) and especially foam cells (77, 214) seems to occur only in advanced stages of disease and contributes to plaque instability and the risk for rupture. An involvement of ER stress in plaque morphology and stability was suggested by the observation that an increase in ER stress markers was observed in VSMC and foam cells from thin-cap atheromas and ruptured plaques of human atherectomy specimens, but not in the fibrous caps of thick-cap atheromas (133).
A mechanistic link between ER stress and macrophage apoptosis was provided by the fact that 4-phenylbutyrate (PBA) can reduce lipid-induced ER stress as well as apoptosis in macrophages. Furthermore, treatment of ApoE-deficient mice with PBA during the last 2 weeks of an 8-week period on Western diet reduced atherosclerosis and the percentage of apoptotic cells in the lesion area (40). Interestingly, ER stress is involved in oxLDL-induced lipid accumulation in macrophages through upregulation of the scavenger receptor CD36 (208) (Fig. 8), one of the two principal receptors responsible for oxLDL uptake (92). In advanced atherosclerotic lesions, where apoptosis of foam cells is observed, these cells show—consistent with early studies demonstrating increased levels of free cholesterol in advanced plaques (108)—an accumulation of free cholesterol (176). This increase in free cholesterol content is linked to the ER, as this organelle is the site of cholesterol toxicity in these cells (44). In addition, cholesterol-induced macrophage apoptosis requires ER stress pathways (35).
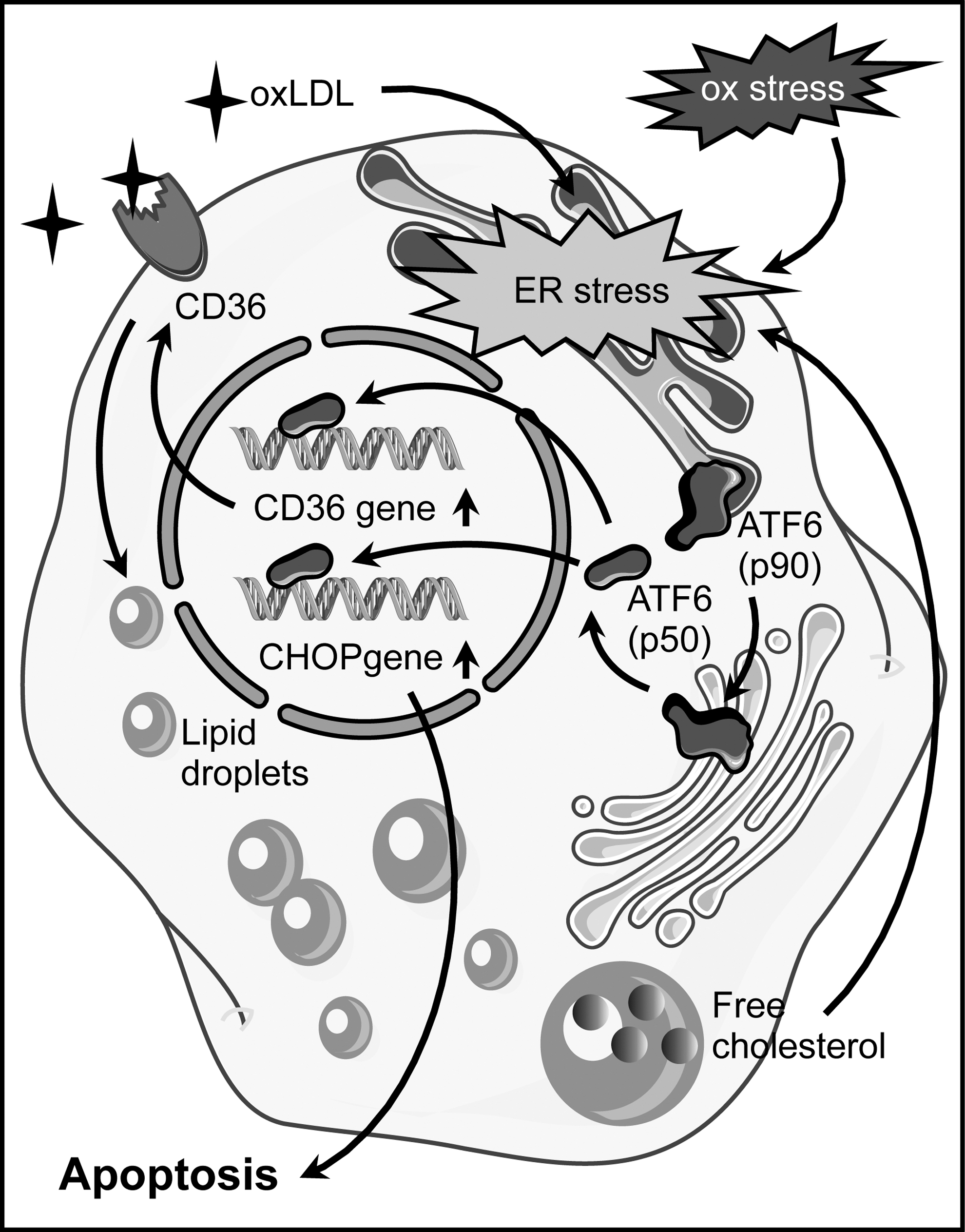
ATF6 has been implicated in multiple aspects of foam cell differentiation and apoptosis. An involvement of ATF6 in ER stress induction in macrophages treated with oxLDL was demonstrated by the upregulation of its proteolytically activated form. Furthermore, downregulation of ATF6 by RNA interference reduced cholesterol accumulation, most likely due to impaired upregulation of CD36, and attenuated apoptosis (208, 210). Conversely, CD36 silencing interfered with nuclear translocation of ATF6 (208). These findings are indicative of a feed-forward loop with CD36-mediated oxLDL uptake triggering the ER stress response, which, in turn, is required for CD36 upregulation (Fig. 8). CD36 plays an essential role in atherosclerosis, as CD36-deficiency results in reduced atherosclerotic lesion size, and it was speculated that macrophages are the cells that are critical for this effect (43, 91). Interestingly, macrophages from CD36 knockout mice showed reduced intracellular ROS generation on oxLDL stimulation (91).
Another downstream effector of ATF6 in ER stress is the pro-apoptotic CCAAT/enhancer-binding protein homologous protein (CHOP). The levels of CHOP in macrophages increase in response to oxLDL, and this upregulation is abrogated by downregulation of ATF6 (210) (Fig. 8). In vivo, CHOP contributes to plaque instability and plays a crucial role in cholesterol accumulation in the ER of macrophages and their apoptotic death (182). Furthermore, CHOP-deficient mouse models for atherosclerosis are characterized by a reduction in the size of aortic root lesions and necrotic areas. In addition, they show a decrease in macrophage apoptosis in the lesions (177). This pathway seems to be also activated in humans, as macrophages in instable or ruptured plaques are characterized by increased CHOP expression and the CHOP-positive cells show DNA strand breakage that is indicative of apoptosis (133).
A direct link between oxidative stress and the activation of ATF6 has been demonstrated ex vivo in a cell line, where it was shown that ROS upregulate site-2 protease (62). Furthermore, the flavonoid quercetin, which has anti-oxidative properties, inhibits the oxLDL-triggered nuclear translocation of ATF6 in macrophages. This is associated with reduced expression of CHOP and attenuation of oxLDL-induced apoptosis (209).
The cumulative evidence indicates that oxidative stress and the ER stress response with ATF6 as one of its effector arms are involved in lipid overloading of macrophages, foam cell apoptosis, and plaque destabilization and could thus be of relevance in the treatment of advanced atherosclerosis.
Estrogen Receptors: Who Does the Job in Atheroprotection?
In women, clinical manifestations of atherosclerosis occur approximately a decade later than in men. These gender differences are apparent not only in clinical presentation but, also in subclinical structural changes in the different vascular beds, as well as in plaque morphology (116, 120). Although the progression rate of age-related arterial dysfunction at younger age is higher in men, menopause demarcates a point of turnaround. There is a roughly tenfold increase in the onset of CVD—the major manifestation of atherosclerosis—in postmenopausal women, whereas the increase in men of the same age group is only about half of that (98). This menopausal transition in cardiovascular risk points toward a critical role for the primary female sex hormone estrogen in cardiovascular protection and indeed, it has been shown more than 60 years ago that estrogens can inhibit atherosclerosis (149).
Originally, estrogen actions had been attributed to proteins belonging to the superfamily of nuclear receptors (155), namely the estrogen receptors α and β (ERα and ERβ). After a ligand-induced conformational change, dimers of these receptors either act as sequence-specific transcription factors or modulate the activity of other transcription factors without binding to DNA. However, a fraction of these classical ERs is present in a larger complex outside the nucleus, which induces signal transduction events through various downstream kinases, leading to transcriptional and non-transcriptional outputs (99). Moreover, estrogen does not only act through nuclear receptor type sensor proteins but also induces very rapid responses via the G-protein-coupled estrogen receptor 1 (GPER1) (151).
It has been shown that systemic administration of 17β-estradiol reduces the inflammatory response (123), leukocyte infiltration (206), and neointimal proliferation (24) after vascular injury. These observations suggest that more than one cell type in the vascular wall is affected by estrogen. All three receptors, ERα, ERβ, and GPER1, are expressed in endothelial cells and VSMCs (90), but the data as to which type of receptor is involved in atheroprotection are conflicting. ERα has been described as the major mediator of the atheroprotective effects of 17β-estradiol in ApoE-deficient mice (79), whereas estrogen can attenuate early atherosclerotic lesion development even in ERα-deficient, ovariectomized animals (187). Furthermore, there is evidence that GPER1 protects against atherosclerosis (122).
On the mechanistic level, estrogen supports endothelial NO-production by activating eNOS (Fig. 9). An involvement of a membrane-associated receptor was suggested by the observation that a membrane-impermeable estrogen conjugate can rapidly activate eNOS (75). Later on, it was demonstrated that an N-terminally truncated variant of ERα, ERα46 is responsible for this effect, while at the same time it inhibited transcriptional activation by full-length ERα (45, 100). This activation of eNOS requires the tyrosine kinase c-Src at the cell membrane (74), which upon estrogen treatment becomes phosphorylated on tyrosine 419. It then phosphorylates ERα46, favoring the assembly of a complex containing c-Src, ERα46, and PI3-kinase, which results in the successive activation of Akt1 and eNOS (74, 101). Interestingly, treatment of endothelial cells with specific GPER1 agonists and antagonists also demonstrated an influence of this receptor on eNOS activation, again via the PI3-kinase pathway (122) (Fig. 9).
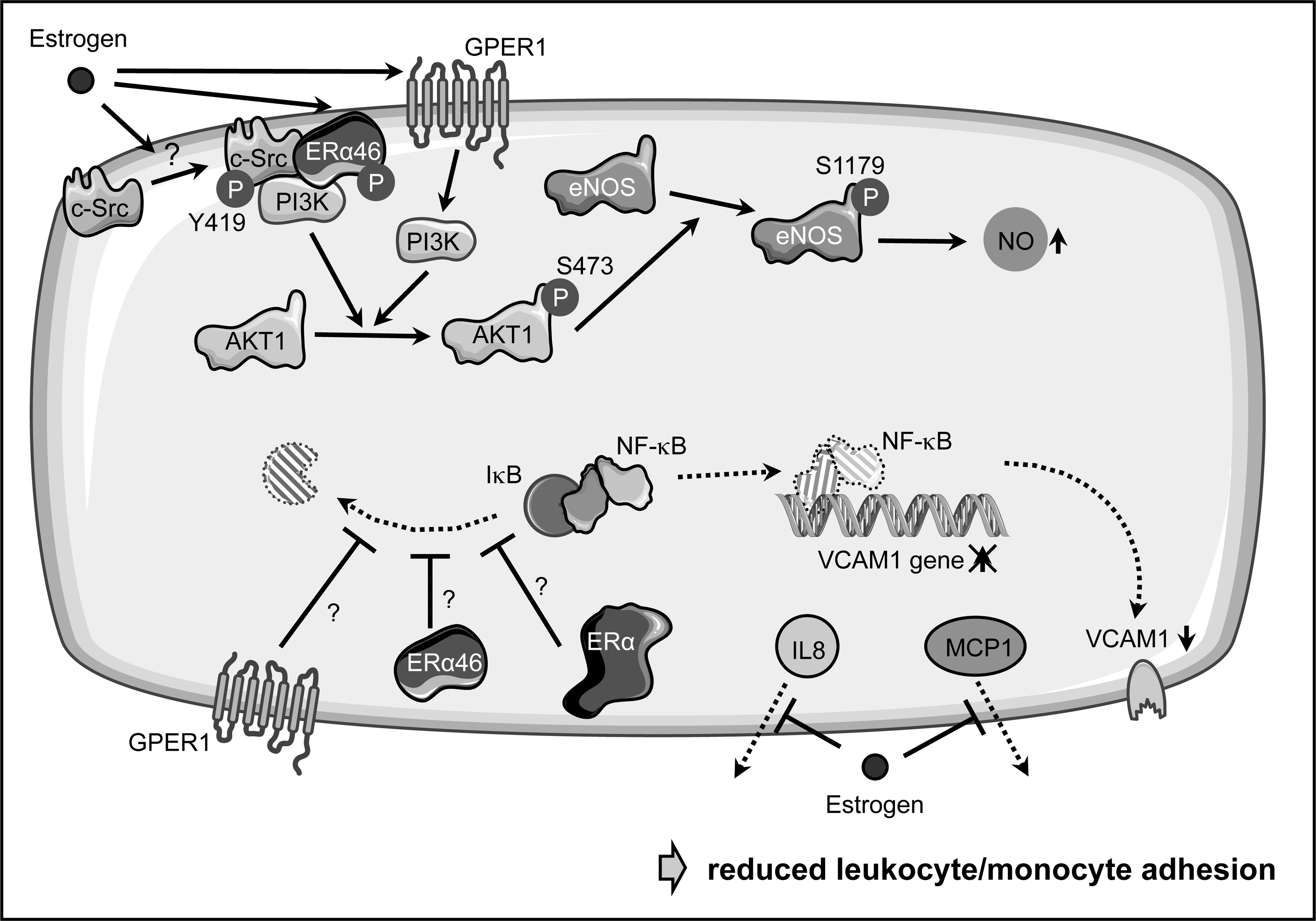
Besides activating eNOS, estrogen suppresses monocyte adhesion to endothelial cells (128, 172); however, the detailed pathways are less well described. On one hand, this is achieved by suppressing the upregulation of adhesion molecules such as VCAM1 by interfering with NF-κB activation through stabilization of its inhibitor IκBα (172) (Fig. 9). Again, it is debatable as to which ER mediates this effect, as it has been shown that a selective GPER1 agonist can attenuate the upregulation of VCAM1 (20). In addition to its effects on gene expression, 17β-estradiol can inhibit leukocyte adhesion by blocking secretion of monocyte chemoattractant protein-1 and interleukin 8 without affecting their expression (156) (Fig. 9).
With respect to neointima formation, it has been shown that ovariectomy induces aortic intimal thickening, which is abrogated by 17β-estradiol by preventing basic fibroblast growth factor accumulation, thereby inhibiting the mitogenic response of VSMC to this pro-atherogenic factor (166, 175). Later on, it was shown that inhibition of VSMC proliferation and migration is due to non-nuclear estrogen receptor signaling involving ERα and protein phosphatase 2A (184). Interestingly, a recent study indicated that GPER1 also plays a critical role in maintaining the differentiated phenotype of VSMC and suppression of their migration by activating the ERK1/2 pathway (85).
In addition to its impact on endothelial and smooth muscle cells, estrogen can reduce oxidative stress in the vasculature by regulating various ROS-producing and -removing enzymes (2). However, mechanistic insights into which estrogen receptors are involved are currently missing.
The complex network of estrogen receptors and actions indicates that simple hormone replacement therapies should be taken with caution. Along these lines, the American Heart Association has advised in 2011 that hormone therapy and selective estrogen receptor modulators should not be used for primary or secondary prevention of CVD in postmenopausal women (129).
A further delineation of the exact pathways contributing to the atheroprotective effects of estrogen and the use of receptor-specific agonists and antagonists (90, 121) affecting non-genomic responses as well as the timing of their delivery (61) might in the future help to develop new therapeutic approaches toward reducing the cardiovascular risk in postmenopausal women.
Conclusions/Future Directions
The examples discussed earlier illustrate how transcription factors are involved not only in the maintenance of a healthy vasculature but also in specific disease-associated processes. As eluded to in the different chapters, their activity in health and disease is regulated at all levels, from expression, RNA splicing, and stability to post-translational modifications, proteolytic activation, and protein-protein interactions of various kinds.
Furthermore, the different transcription factors reviewed exert their functions at different stages of atherosclerosis development and progression. At one end of this process are different types of blood flow providing an atheroprotective or an atheroprone environment, which either preserves endothelial function or induces endothelial activation and dysfunction. In addition, the control of vascular smooth muscle proliferation after vascular injury is important for neointima formation. During disease progression, B cells and macrophages play a role in the cellular composition inside the plaque. In final stages, plaque destabilization and rupture occur. One fatal consequence is vascular occlusion, leading to myocardial infarction or stroke.
Although we have described several aspects of how modulation of the activity of specific transcription factors in different cell types affects the various facets of atherosclerosis, this does not exclude that they regulate completely different processes in other cell types and other organs. Therefore, and due to pleiotropic outputs owing to their large number of target genes, global therapeutic activation or inhibition of specific transcription factors does not seem feasible. This goes along with the view of these proteins as being “undruggable,” since such interventions are prone to have adverse side effects as exemplified in the case of peroxisome proliferator-activated receptors (135). An in-depth analysis of cell type-, disease-, and perhaps also stage-specific protein-protein interactions or target genes might open up avenues that are more promising. The increasing knowledge about the structural basis of transcription factor interactions with each other and other proteins might in the long run allow the design of drugs blocking such contact sites with high specificity (48).
Furthermore, the CRISPR/Cas system can be used not only for genome editing but also for the creation of transcriptional activators or repressors (36). The guide RNA directing the engineered Cas-derived transcriptional regulators to their target site is longer than most transcription factor recognition sites on DNA, which allows a much more specific regulation, perhaps also of single genes that are relevant for disease.
Footnotes
Acknowledgments
S.K. is a scholarship holder of the IRTG1902. This work was, in part, supported by the Deutsche Forschungsgemeinschaft (AL288/2-1 and IRTG1902 P1) to J.A., by NIH R01 HL107490 and P01 HL55798 to C.A.M., and by an NIH Immunology Training Grant to A.U. (2 T32 AI 7496-21). Single elements for figures were taken from the Powerpoint image bank of Servier Medical Art (