
Abstract
Significance:
The thioredoxin (Trx) and glutathione (GSH) systems play important roles in maintaining the redox balance in the brain, a tissue that is prone to oxidative stress due to its high-energy demand. These two disulfide reductase systems are active in various areas of the brain and are considered to be critical antioxidant systems in the central nervous system (CNS). Various neuronal disorders have been characterized to have imbalanced redox homeostasis.
Recent Advances:
In addition to their detrimental effects, recent studies have highlighted that reactive oxygen species/reactive nitrogen species (ROS/RNS) act as critical signaling molecules by modifying thiols in proteins. The Trx and GSH systems, which reversibly regulate thiol modifications, regulate redox signaling involved in various biological events in the CNS.
Critical Issues:
In this review, we focus on the following: (i) how ROS/RNS are produced and mediate signaling in CNS; (ii) how Trx and GSH systems regulate redox signaling by catalyzing reversible thiol modifications; (iii) how dysfunction of the Trx and GSH systems causes alterations of cellular redox signaling in human neuronal diseases; and (iv) the effects of certain small molecules that target thiol-based signaling pathways in the CNS.
Future Directions:
Further study on the roles of thiol-dependent redox systems in the CNS will improve our understanding of the pathogenesis of many human neuronal disorders and also help to develop novel protective and therapeutic strategies against neuronal diseases. Antioxid. Redox Signal. 27, 989–1010.
Introduction
T
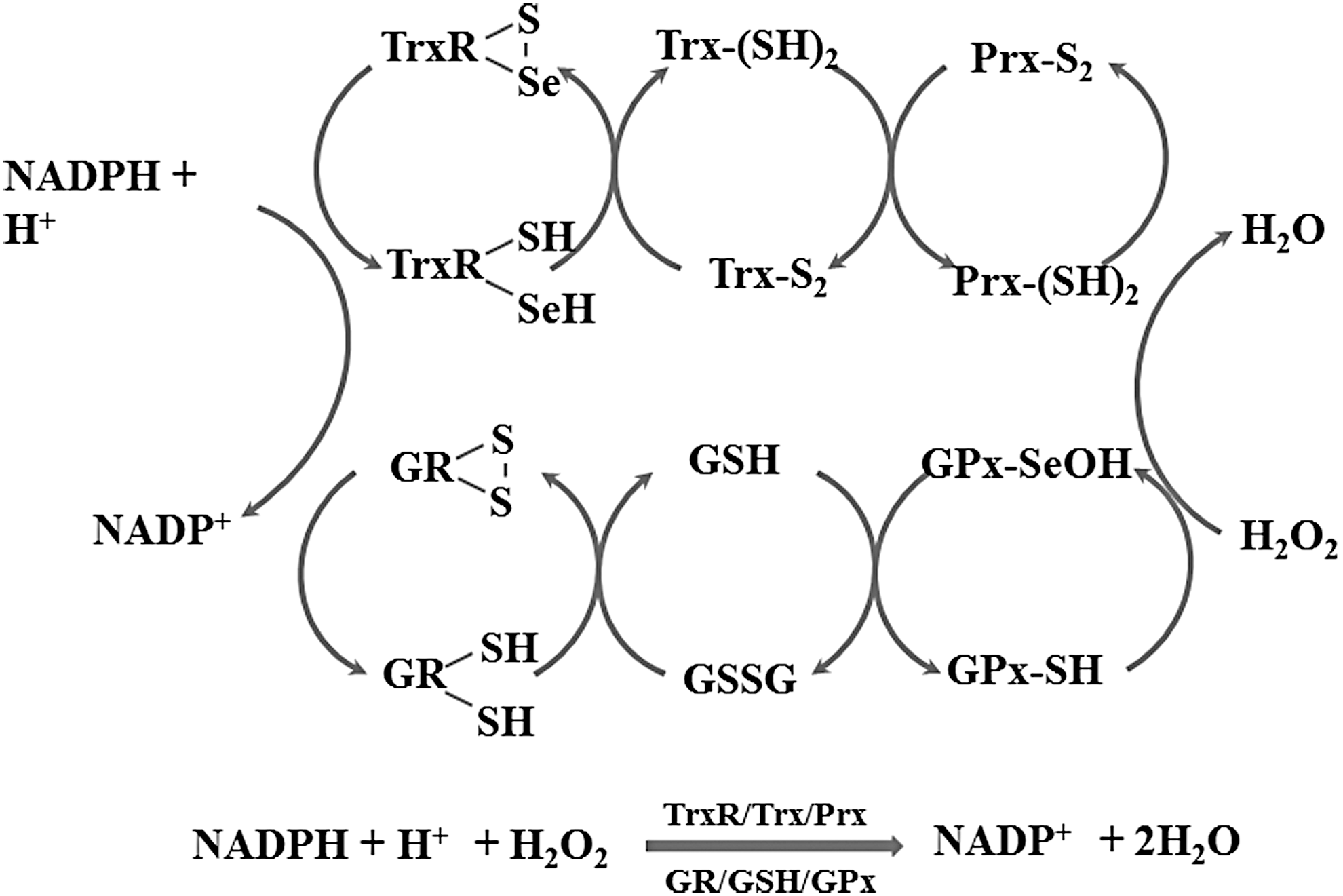
The GSH-glutaredoxin (Grx) system is another disulfide reduction system in cells. Grxs are part of the Trx protein family. As well as a disulfide–dithiol exchange mechanism, Grx can catalyze the reduction of substrates via a monothiol mechanism. There are two major forms of Grx: Grx1, present in the cytosol, and Grx2, located in the mitochondria and nuclei of mammalian cells (124). Grx1 accepts electrons from GSH, while Grx2 can obtain electrons from both GSH and TrxR2 (91). GSH is the most abundant small molecular thiol and is present at a high concentration in the brain (∼1–3 mM). Generally, it is thought that astrocytes produce a high level of GSH, which is released and degraded extracellularly and then subsequently used by other neurons for GSH synthesis (54).
Trx and GSH systems are the two major thiol-dependent antioxidant mechanisms in cells and also participate in DNA synthesis and repair as an electron donor for RNR and protein methionine sulfoxide reduction by providing electrons to methionine sulfoxide reductase (MSR); these processes are described in detail in previous reports (86, 111, 151). This review will focus on the roles of Trx and GSH in regulating redox signaling in the CNS and the effects of redox-active compounds in neuronal disorders via the Trx and GSH systems.
Production of Reactive Oxygen Species in Neuronal Systems
Reactive oxygen species (ROS), including superoxide (O2 •−), hydrogen peroxide (H2O2), and hydroxyl radical (OH•), are produced via the leaking of electrons from the mitochondrial respiration chain or through certain enzymatic reactions, such as NADPH oxidase (NOX) and monoamine oxidase (MAO) under physiological conditions (Fig. 2). Although the brain constitutes only 2% of total body weight, it consumes ∼20% of the total oxygen used by the body. Thus, the high level of glucose metabolism and oxygen consumption continually produce excessive ROS. Besides this, several other factors make brain tissue more susceptible to oxidative stress than other tissues (76): the brain is highly enriched with lipid and about 35% of brain lipids are polyunsaturated fatty acids, which are important for neuronal functions, but sensitive to ROS attack (17); glutamate receptor-mediated excitotoxicity can undergo positive feedback with ROS and cause neuron cell death; high levels of metal ions in the brain can catalyze more toxic free radical production; loss of trophic support can induce activation of NOX; microglia, the resident macrophage-type cells in the brain, can be activated during immune response by cytokines leading to ROS generation and the subsequent expression of inducible nitric oxide synthase (iNOS), which produces nitric oxide (NO).
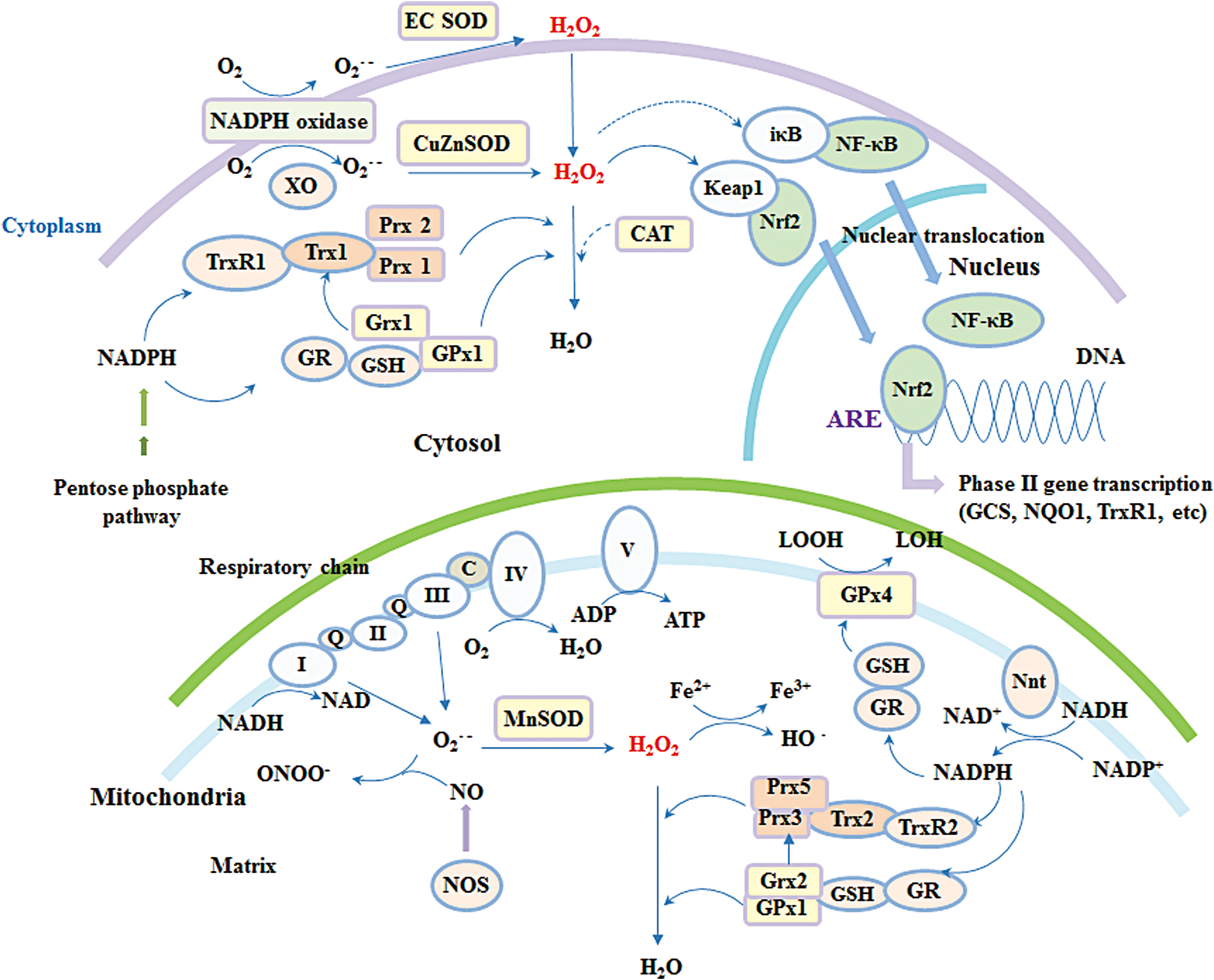
To maintain redox balance, organisms are equipped with various antioxidant enzymes, including superoxide dismutase (SOD), catalase (CAT), GPx, and peroxiredoxin (Prx), and small antioxidants, such as GSH, coenzyme Q, vitamin C, and E (Fig. 3). The antioxidant capacity in most areas of the brain is modest; in particular, there is very low, if any, CAT expression in brain cell mitochondria (76, 180). Human neuronal disorders, such as neurodegenerative diseases, are closely linked to oxidative stress, which is defined as an imbalanced status whereby ROS levels overwhelm antioxidants. However, recently developed methods have provided useful tools to study the role of ROS in a more physiologically relevant context. For example, development of the HyPer probe has enabled real-time imaging of ROS in living systems (24), and the use of Rhodotorula gracilis D-amino acid oxidase allows precise manipulation of ROS production in cells (80). With the help of these new techniques, the definition of oxidative stress has evolved during the last decades, and oxidative stress is now preferably considered to be the disruption of redox signaling and regulation (95). Therefore, it is suggested that the effects of ROS in the brain are most likely mediated through a redox signaling process rather than extensive nonspecific cellular damage. The redox environment is usually determined by several local factors, such as colocalization of oxidant/antioxidant enzymes, the ratio of oxidizing/reducing equivalence, the presence of metal ions, cellular compartmentalization, and so on.
Redox Signaling Regulation in CNS by Trx and GSH Systems
Trx and related antioxidant enzymes are specifically localized to different subcellular organelles in neuronal and glial cells and locally control redox signaling. The Trx1 system, coupled with Prx1/2 and MSR, and the GSH-Grx1 system, coupled with GPx1, regulate redox signaling in the cytosol and nucleus. The Trx2 system, coupled with Prx3/5 and MSR, and the GSH-Grx2 system, coupled with GPx1/4, regulate the mitochondrial redox environment and signaling (Fig. 2) (122). In addition, TrxR1/Trx1 are also present in neuronal synaptic vesicles (160). Generally, the Trx and GSH systems are thought to work in parallel, and in many cases, crosstalk occurs between these two systems, where components of one system serve as a backup for the components of the other (56, 57, 221). Moreover, in the mitochondrial matrix, both Trx2 and Grx2 can transfer electrons to Prx3 to reduce H2O2 (77).
Regulation of H2O2 metabolism via the Trx/Prx and GSH/GPx systems
Prx, GPx, and CAT are the three types of antioxidant enzymes that catalyze the decomposition of H2O2 with high efficiency (122). These enzymes can scavenge H2O2 at a reaction rate of 107–108 M −1s−1, whereas the direct reaction rate of GSH with H2O2 is only 0.89 M −1s−1 (207). Given the notable absence of CAT expression in brain mitochondria, the concentration of CAT in the brain is estimated at 1.2 μM, which is ∼50 times less than its level in the liver (154), whereas the Prx3 concentration in the brain is ∼60 μM, Prx5 is 20 μM, and GPx1 is 2 μM (44, 184).
Mitochondria are the main sites of superoxide and H2O2 production (138). About 0.5–2.0% of electrons leaked from mitochondrial respiration are converted into superoxide. Mitochondrial SOD2 can catalyze the conversion of superoxide to H2O2, a well-known signaling messenger (185). The rate of H2O2 production using NAD-linked substrates in the brain is 50–70 pmol/min/mg mitochondrial protein. Interestingly, mitochondria can scavenge exogenous H2O2 at a rate of 9–12 nmol/min/mg mitochondrial protein in a respiration- and substrate-dependent manner, which is 100 times higher than the H2O2 production rate. Thus, mitochondria act as a cellular source and also as a sink of H2O2 (6, 184).
Several studies have highlighted the essential role of the mitochondrial Trx system in counteracting ROS in the brain, with its importance potentially due to the lack of CAT expression. Inhibition of mitochondrial TrxR2 by auranofin was demonstrated to diminish the H2O2 scavenging capacity of mitochondria, whereas this was not affected by uncoupling of mitochondria, phosphorylation of added ADP, or the genetic ablation of GPx1 (53, 184). Mitochondrial Trx/Prx couple was suggested to reduce H2O2 in a respiration-dependent manner. Nicotinamide nucleotide transhydrogenase (Nnt), which uses NADH and NADP+ to generate NADPH, links the substrate requirement to mitochondrial Trx/Prx as the ultimate electron donor for H2O2 removal. In a previous study, inhibition of Nnt decreased the NADPH level and impeded the Trx system's capacity to scavenge H2O2 in the brain, whereas inhibition of Nnt had no effects on H2O2 removal in liver mitochondria since it is independent of respiration as CAT is highly expressed in mitochondria from liver cells (119).
Reversible regulation of protein thiol modification
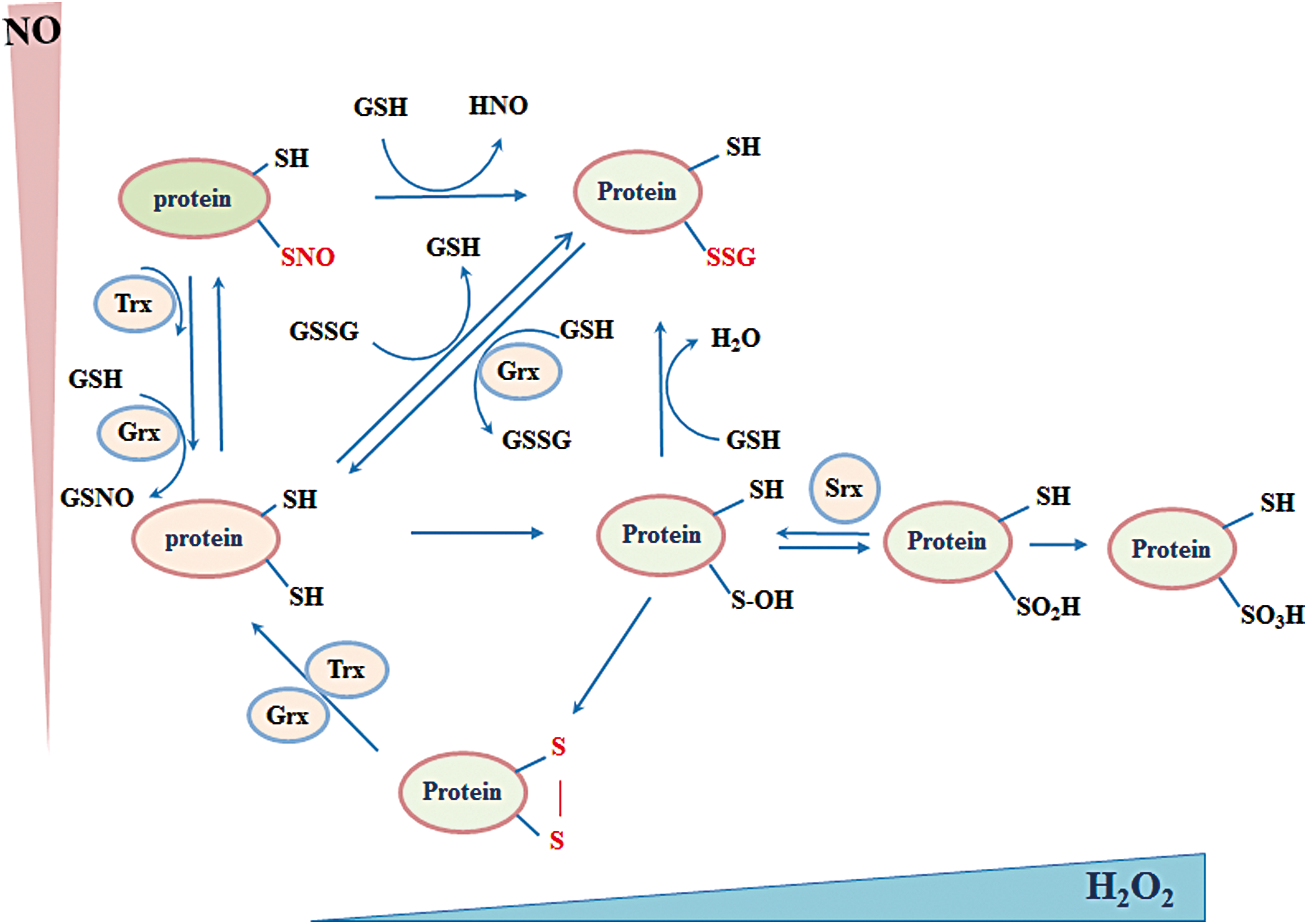
A large part of ROS/reactive nitrogen species (RNS)-mediated signal transduction relies on modifications of cysteine thiol (-SH) within proteins. Irreversible thiol modification (sulfonic acid, -SO3H) has been considered a hallmark for various pathological conditions and usually leads to permanent functional loss and degradation of the protein. Reversible thiol modifications, such as S-nitrosylation (-SNO), S-sulfenylation (sulfenic acid, -SOH), S-glutathionylation (-SSG), disulfide formation (-S-S-), S-sulfhydration (-S-SH), and sulfinic acid (-SO2H), are involved in redox signaling (Fig. 3).
Several properties allow thiols to act as signal switches. First, thiols are susceptible to ROS/RNS challenge that lays a foundation for its sensitivity. Second, in different local environments, thiols exhibit different reactivity, which gives them selectivity (62). Last, most thiol modifications are reversible, which provides flexibility for sophisticated signaling regulation. Thus, great efforts have been made to identify proteins with functional cysteines and understand the regulation of reversible thiol modification in neuronal systems.
S-nitrosylation
NO can be produced endogenously by NOSs, which utilize L-arginine amino acid, oxygen, and NADPH as the electron donor. There are three NOS isoforms identified in mammals, which are named mainly depending on their tissue distribution. Neuronal NOS (nNOS) was first found in the CNS (131). Endothelial NOS (eNOS) is mainly expressed not only in endothelial cells from blood vessels (161) but also in cells from other tissues, such as platelets, smooth muscle cells, hepatocytes, and certain types of neuronal cells (65). The activity of both nNOS and eNOS is regulated by Ca2+ and calmodulin. In contrast to the other two isoforms, iNOS is regulated in a Ca2+-calmodulin-independent manner and is not constitutively expressed in cells. Instead, iNOS expression and NO production can be induced dramatically via stimulation from various factors, such as inflammatory cytokines and lipopolysaccharides. This property is used by immune cells, including macrophages and glial cells, to fight against invading microbes (26). During the last two decades, NO was also identified to be involved in S-nitrosylation, a post-translational modification (PTM) that directly adds an NO moiety to a reactive thiol of cysteine to form S-nitrosothiols (SNOs). Similar to other PTMs such as phosphorylation, S-nitrosylation can alter protein activity, interactions, and redox status and plays an important role in physiological and pathological conditions (5).
S-nitrosylation in neuronal system
In the neuronal system, NO functions as a neurotransmitter, mainly via the classical NO-soluble guanylate cyclase (sGC)-cyclic guanosine monophosphate (cGMP) pathway (69). In addition, accumulating evidence has shown that NO also acts through S-nitrosylation, which can be both beneficial and detrimental.
S-nitrosylation can promote neuron survival by targeting different proteins. For example, overactivation of the N-methyl-D-aspartate (NMDA) glutamate receptors triggers neuron excitotoxicity, which is characterized by uncontrolled cellular calcium influx and cellular damage. NO produced by nNOS can nitrosylate specific thiols within the NR1 and NR2 subunits of NMDA receptors and negatively modulate Ca2+ influx (116). Serine racemase, which catalyzes the formation of D-serine, the coagonist of NMDA receptors, can also be S-nitrosylated and inhibited by nNOS-produced NO (141). Both mechanisms protect neurons from excitotoxicity. Moreover, nitrosylation of the active site cysteine in caspases, the essential executers of programmed cell death, inhibits their activity and therefore plays a protective antiapoptotic role in neurons (191).
Several transcription factors are also regulated by S-nitrosylation. NF-κB has a central role in the immune system. The p50 (46) and p65 (99) subunits of NF-κB are both reported to be S-nitrosylated by iNOS during immune responses. A recent study from Rottenberg and colleagues demonstrated that NO can protect the integrity of the blood–brain barrier (BBB) during trypanosome infection by S-nitrosylating the p65 subunit of NF-κB, thereby inhibiting penetration of immune cells and parasites into the brain (148). Hypoxia-inducible factor 1 (HIF-1) is a master transcriptional factor that mediates the hypoxic adaption that is essential for neuronal development and survival. S-nitrosylation of cysteines in HIF-1α not only promoted its DNA binding (218) but also protected it from proteasomal degradation (114). The nuclear factor E2-related factor 2 (Nrf2) binds to antioxidant response elements and initiates transcription of downstream targets to protect cells from oxidative and nitrosative stress. Under normal conditions, Nrf2 is sequestered by Klech-like ECH-associated protein 1 (Keap1) in the cytosol. Upon NO stimulation, Keap1 can be nitrosylated and release Nrf2, allowing its nuclear translocation and induction of antioxidant functions (200).
Excessive production of NO and aberrant protein S-nitrosylation are often related to pathological changes, especially in neurodegenerative diseases, such as Parkinson's disease (PD), Alzheimer's disease (AD), Huntington's disease (HD), and amyotrophic lateral sclerosis. Oxidative and nitrosative stress are risk factors for neurodegenerative diseases, and S-nitrosylation of antioxidant enzymes deteriorates the situation. Prx2 is predominantly expressed in mammalian neurons (172) and plays important roles in protecting neurons from oxidative damage by degrading H2O2 (104). S-nitrosylation of critical cysteines (Cys51 and Cys172) in Prx2 inhibits its activity, and the level of nitrosylated Prx2 was found to be elevated in the brain of PD patients (59).
Accumulation of misfolded proteins is often observed in the foci of neurodegenerative diseases. Protein disulfide isomerase (PDI), an important protein that regulates endoplasmic reticulum (ER) stress and is involved in protein quality control, was found to be nitrosylated and have reduced activity in sporadic PD and AD (199). Parkin is an E3 ubiquitin ligase that mediates protein degradation and prevents aggregation of misfolded proteins in neurons. Studies have shown that S-nitrosylation of parkin inhibited its activity and contributed to the pathological progression of PD (217). Autophagy is another important cellular mechanism for the degradation of misfolded and aggregated proteins. JNK/Bcl-2/Beclin 1 and IKKβ/AMPK/mTORC 1 are two well-characterized pathways that regulate this process. In an HD model, accumulation of mutant huntingtin was found, and both JNK and IKKβ were found to be S-nitrosylated and have diminished activities (173).
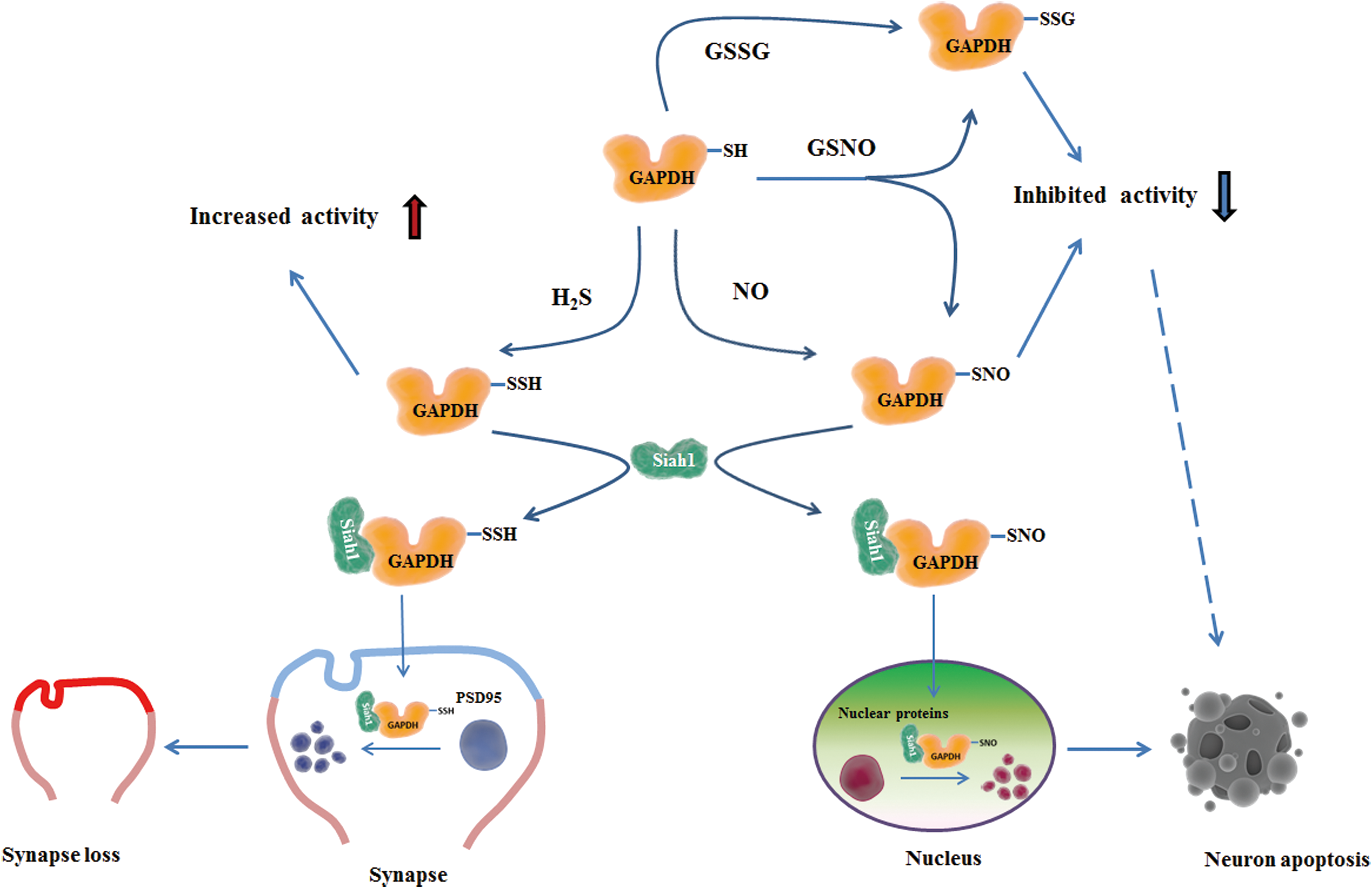
Uncontrolled cell death is another feature of neurodegenerative diseases and can be induced by S-nitrosylation of different molecules. The X-linked inhibitor of apoptosis protein (XIAP), an E3 ligase, directly binds to several members of the caspase protein family, mediating their degradation, and therefore exerts a protective role in neurodegenerative diseases (83). XIAP was found to be S-nitrosylated and have lost caspase-binding ability in PD patients, which allows induction of the apoptosis cascade in PD (196). S-nitrosylation of GAPDH Cys150 was shown to promote its binding to Siah1 and enhanced the nuclear translocation of GAPDH-Siah1 complexes, which initiate the apoptotic pathway, contributing to neuronal cell death (78) (Fig. 4).
Neurons are more vulnerable to mitochondrial dysfunction due to their high-energy demand. In a β-amyloid protein (Aβ)-induced AD model, excessive production of NO led to dynamin-related protein 1 (Drp1) nitrosylation. S-nitrosylated Drp1 resulted in exaggerated mitochondrial fission and fragmentation, which worsened the function of mitochondria (37). In addition, S-nitrosylation of mitochondrial complex I and complex IV was shown to suppress their activity in the respiratory chain and inhibit oxidative phosphorylation (41).
Taken together, these studies demonstrate that S-nitrosylation is an on/off switch signal for the activities of various important proteins that mediate cell death. Through regulation of activities of these proteins, S-nitrosylation may determine neuronal cell viability and subsequently contribute to pathogenesis in neurodegenerative diseases.
S-nitrosylation regulated by Trx
The evolutionarily conserved active site -CXXC- motif in Trxs emphasizes the biological importance of their function. The thiol reduction activity of the Trx system has been intensively investigated during the last few decades and recent studies revealed that Trx is also an efficient denitrosylase. During S-denitrosylation, a cysteine at the active site of Trx may form an intermolecular disulfide with the substrate or be transnitrosylated as an intermediate. The resulting product is nitroxyl (HNO) or NO and oxidized Trxs that can be reduced by TrxR (22). Using a proteomic approach, a broad spectrum of nitrosylated proteins, including some discussed above, were identified as substrates of Trx (23).
It is worth noting that human Trx1 plays different roles in S-nitrosylation regulation under different redox states. In addition to the two cysteines at the active site, human Trx1 contains three structural cysteines, Cys62, Cys69, and Cys73, all of which can be nitrosylated in different contexts (206). However, so far, only Cys69 and Cys73 have been reported to be nitrosylated physiologically (75, 79). Nitrosylated Trx1 acts as a transnitrosylase that can transfer the NO groups to target proteins, including caspases (136), Prx1, and others (210). The formation of active site disulfide or mutated active site cysteines is a prerequisite for the transnitrosylase activity of Trx1 (209). Therefore, under different redox contexts, Trx1 may act as a denitrosylase or a transnitrosylase; Trx1-mediated regulation of apoptosis via caspase 3 is an example of this. Reduced Trx1 and Trx2 denitrosylate S-nitrosylated caspase 3 in the cytosol and mitochondria and facilitate Fas-induced apoptosis, which is important not only for neuronal development but also contributes to pathological progression of neurodegenerative diseases (21). By contrast, nitrosylated Trx1 is able to specifically transnitrosylate the catalytic cysteine of caspase 3 and reduce caspase activity, thus protecting neurons from stress-induced apoptosis (209) (Fig. 5). Using mass spectrum-based bioinformatic analysis in neuroblastoma cells, a study surprisingly found more than 40 proteins that were reversibly regulated via S-nitrosylation by Trx1 (210). Among these targets, GAPDH S-nitrosylation is reported to be closely associated with apoptosis (Fig. 4).
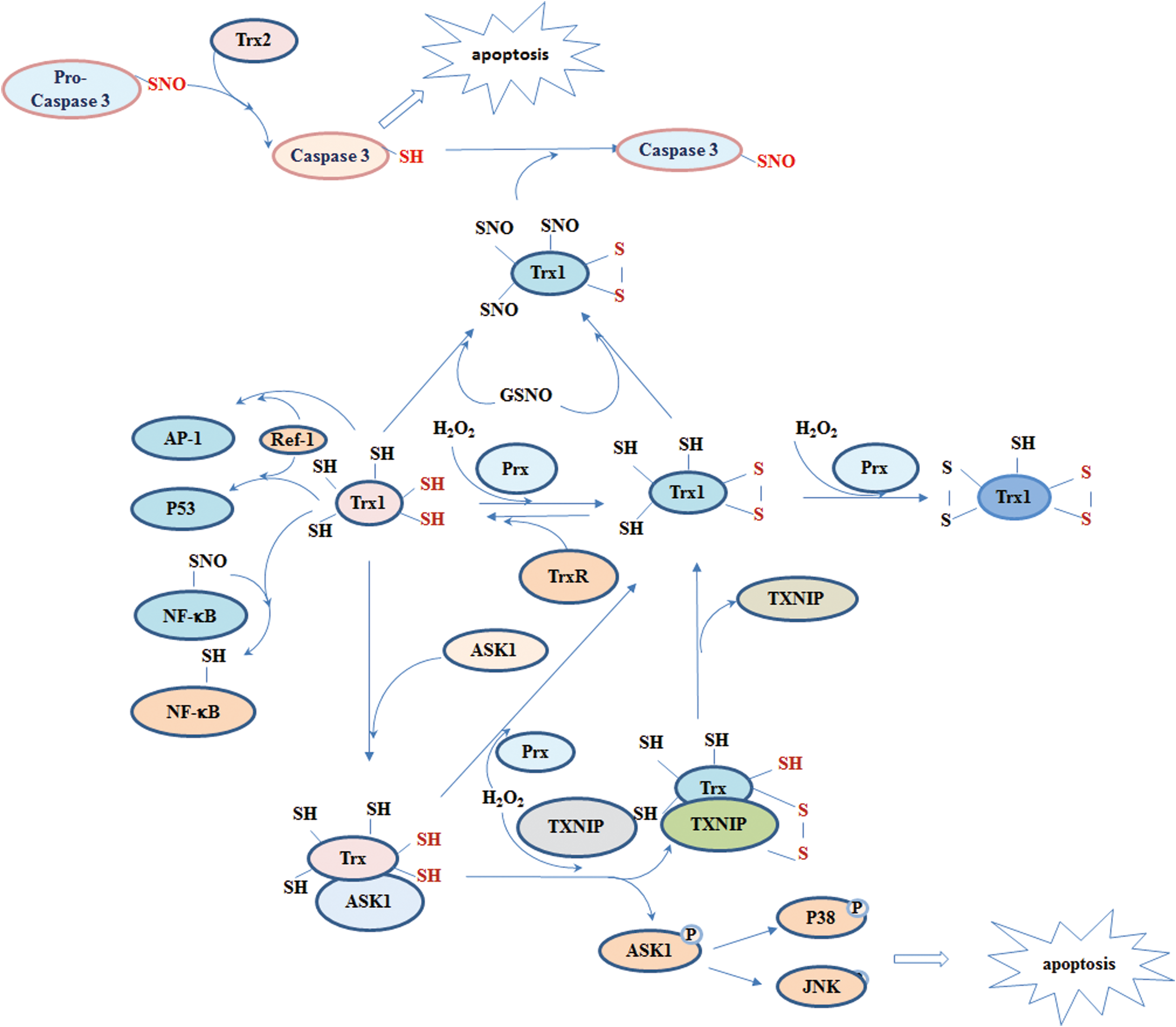
Thus, these results indicate that the Trx system is a sophisticated regulator of neuronal death that alters signaling via the S-nitrosylation of proteins involved in the cell death process.
S-glutathionylation
GSH is the dominant low-molecular-weight antioxidant in mammalian cells. The ratio between GSH and GSSG maintains the cellular redox potential and redox balance. Under oxidative or nitrosative stress, a decreased GSH/GSSG ratio can cause S-glutathionylation: a mixed disulfide formed between reactive thiols and GSH.
S-glutathionylation in neuronal system
Similar to S-nitrosylation, protein S-glutathionylation is a well-controlled reversible PTM that alters protein functions in physiological conditions in the CNS. For example, Ca2+-dependent phosphorylation of ERK and cAMP/Ca2+ response element-binding protein (CREB) are required for synaptic plasticity. A study found that H2O2 increases glutathionylation and activation of ryanodine receptors (RyR), which release Ca2+, enhancing ERK and CREB phosphorylation, and helps maintain long-term potentiation (LTP) in the hippocampus (100).
However, abnormal protein S-glutathionylation has been considered to be a biomarker for neurodegenerative diseases. The pathological foci from patients with AD and PD have been found to contain aggregated GAPDH with inhibited activity and oxidative modifications (143). Analysis of inferior parietal lobule tissues from AD patients revealed a significant increase in the level of S-glutathionylated GAPDH compared with the level in age-matched controls (144) (Fig. 4). Tau, a microtubule-associated protein mainly expressed in the CNS, promotes neurite extension and axonal growth. However, tau is considered to be the major component of the neurofibrillary tangles observed in the brain tissue of patients with AD (70). Electron microscopy analysis suggested that S-glutathionylated tau protein rapidly polymerized to form filaments (50).
The proapoptotic transcriptional factor p53 was also found to be hyperglutathionylated in AD patients, although the biological consequence of this is not clear yet. One explanation is that glutathionylation protects p53 from irreversible oxidation and degradation, therefore promoting neuronal cell death (49). Another speculation is that glutathionylation of p53 is an adaptive response to suppress apoptotic signaling under stress (203).
The mitochondrial NADP+-dependent isocitrate dehydrogenase (IDPm) catalyzes oxidative decarboxylation of isocitrate to α-ketoglutarate and produces NADPH, which supplies electrons for antioxidant systems. Glutathionylation of Cys269 within the active site inhibits IDPm activity. In a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mouse model, an increased level of glutathionylated IDPm was observed in the brain of PD mice compared with the control group (102). One of the mechanisms of MPTP-induced PD is mediated by MPTP inhibition of complex I; decreased complex I activity is often observed in PD with lowered ATP production and elevated ROS formation (195). Glutathionylation of complex I inhibited its activity and increased ROS production in mitochondria, which might play a detrimental role in the brain (190).
The findings discussed above indicate that abnormal protein S-glutathionylation acts as a signal for diverse cellular activities, including protein aggregation, protein degradation, apoptosis, and mitochondrial dysfunction, which are closely linked to neurodegeneration.
S-glutathionylation regulated by Grxs
The reverse reaction of glutathionylation is termed S-deglutathionylation. Although several proteins, including Trxs (73), PDI (155), and sulfiredoxin (63), have deglutathionylase activity under different conditions, Grxs are considered to be the major deglutathionylases due to their high affinity and selectivity for glutathionylated proteins (91).
Two mechanisms have been proposed for Grx-catalyzed deglutathionylation: (i) the dithiol mechanism requires both cysteines in the active site—initially, the N-terminal cysteine forms a disulfide with the target protein and releases GSH, and subsequently, the other active site cysteine resolves the disulfide and leaves deglutathionylated protein and a resulting disulfide at the active site of Grx that can be later reduced by GSH; (ii) the monothiol mechanism suggests that the N-terminal cysteine first takes over the -SG group, becoming glutathionylated and reducing the target protein, and subsequently, the glutathionylated active site cysteine is reduced by GSH or forms a disulfide with the other active site cysteine (133) (Fig. 6). Although both mechanisms were reported by different studies, the monothiol mechanism is considered to be prevalent due to its universal recognition of glutathionylated targets (61).
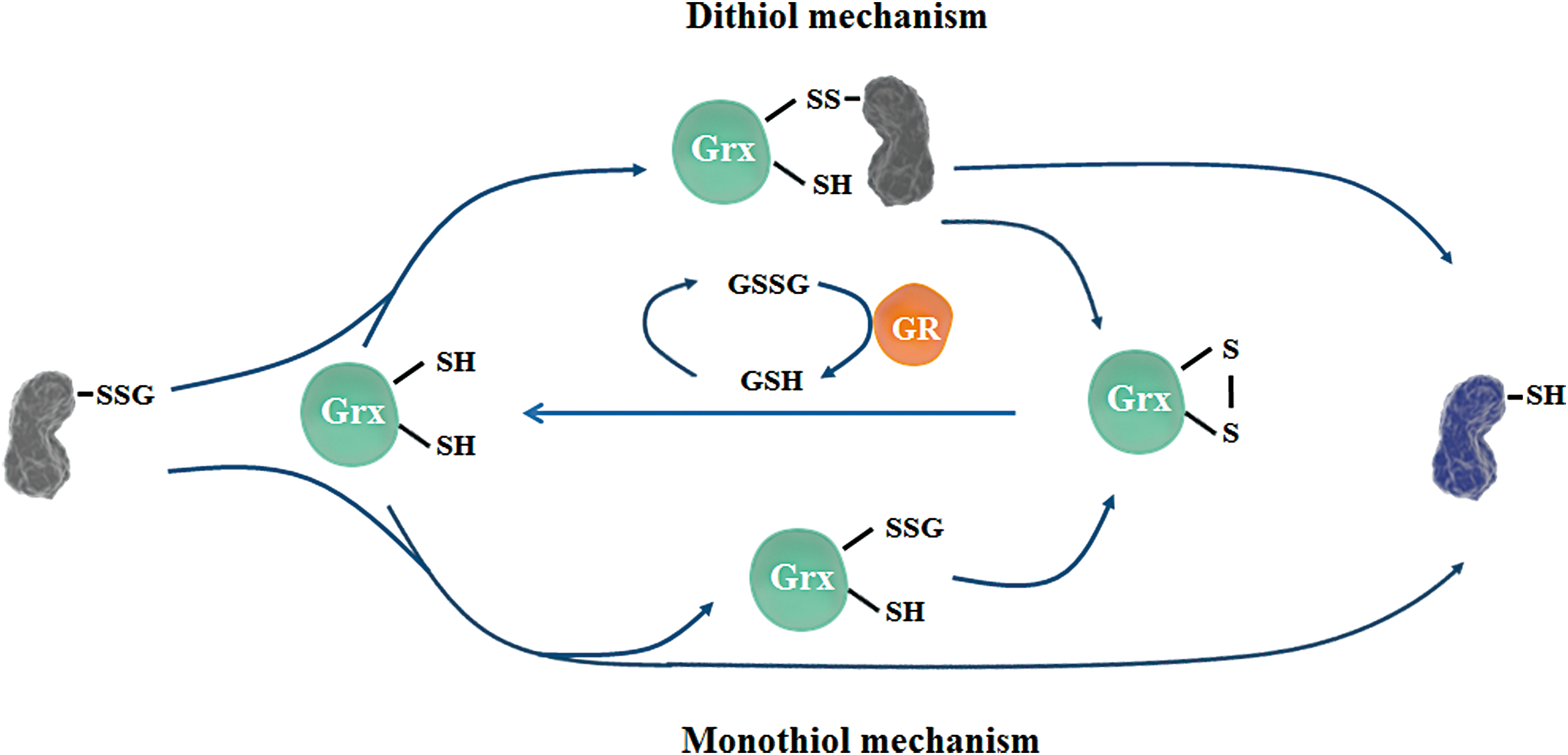
Grxs have an important influence on the nervous system through their ability to catalyze deglutathionylation. For example, actin was reported to be constitutively glutathionylated in the human CNS (182). Studies have shown that Grx1-catalyzed deglutathionylation of actin is crucial for its polymerization, which is a key event required for cellular dynamics, and protects neurons from accumulation of disarranged actin filaments (153). In an MPTP-induced PD model, upregulation of Grx restored complex I activity, which is most likely mediated via deglutathionylation of its catalytic cysteines (101). Studies also revealed that Grx2a, the human mitochondrial Grx, exhibited higher affinity for glutathionylated substrates and electron donors compared with Grx1, suggesting it has an important role in regulating mitochondrial redox homeostasis by deglutathionylation of target proteins, including complex I (19, 91). Glutathionylation of cysteine in the active site of Prx2 was also reported in Grx1 knockout cells treated with H2O2, suggesting that Grx1 has a protective role in guarding Prx2 activity under oxidative stress (158). Mutation of the Parkinsonism-associated deglycase (PARK7/DJ-1) gene or loss of DJ-1 protein function is associated with autosomal recessive early-onset PD. An in vivo study suggested that the DJ-1 protein level was regulated by Grx1-mediated deglutathionylation, which protected DJ-1 from proteasome degradation and exerted beneficial effects in PD (93). A recent study using a Caenorhabditis elegans PD model reported that global deficiency of GLRX-10, the homolog of mammalian Grx1, aggravated neurodegenerative symptoms in worms (94).
The results described above indicate that protein S-glutathionylation is an important signal for neuron cell death, for example, via the effects on abnormal protein polymerization and protein degradation. Grxs, including Grx1 and Grx2, are important mediators of protection against oxidative stress in the CNS by reversal of S-glutathionylation.
S-sulfhydration
Similar to NO, hydrogen sulfide (H2S) also targets reactive protein cysteines and forms persulfide bonds (-SSH). This cysteine-based PTM is termed S-sulfhydration, which has recently drawn attention for its biological functions. Compared with S-nitrosylation, which only occurs in a small fraction of proteins, sulfhydration is a more abundant PTM that has been reported to modify 10–25% of the total proteins in liver lysates, including actin, tubulin, and GAPDH (140). S-nitrosylation usually inhibits the activity of proteins, whereas sulfhydration typically enhances the reactivity of modified cysteines.
S-sulfhydration in the neuronal system
S-sulfhydration regulates various biological events in the CNS and some of the modifications are protective. For example, sulfhydration of parkin enhances its E3 ligase activity, and depleted parkin sulfhydration was detected in brain samples from PD patients, suggesting a beneficial effect on the clearing of misfolded proteins in the brain (202). H2S also boosted GSH production by enhancing the activity of the rate-limiting enzyme, γ-glutamyl cysteine synthetase (γ-GCS). Although the mechanism is not clear yet, it is possible that γ-GCS activity is enhanced through sulfhydration (103). Sulfhydration of Keap1 led to activation of Nrf2 that increases cellular antioxidant capacity and protects against senescence (215). In another oxidative stress-induced neuronal senescence model, sulfhydration of p66Shc, a protein that controls mitochondrial ROS production, hampered its translocation into the mitochondria and lowered ROS production (212). Sulfhydration of the p65 subunit of NF-κB was shown to promote its binding to coactivator ribosomal protein S3 (RPS3) and facilitated its antiapoptotic effects (176).
Excessive sulfhydration is harmful to the nervous system. Chronic inflammation is one of the hallmarks of neurodegenerative diseases. Inflammatory cytokines can cause sulfhydration of GAPDH and enhance its binding to Siah1 (Fig. 4). This binding promotes Siah1 activity, which subsequently leads to degradation of postsynaptic density 95 (PSD95), an important protein involved in neuron maturation and synaptic plasticity. Exaggerated loss of PSD95 is seen in several neuronal diseases, including dementia and depression (135). As the gene that encodes cystathionine β-synthase (CBS), the enzyme that produces H2S in cells, is located on chromosome 21, patients with Down's syndrome (DS) or trisomy 21 have much higher H2S production, which may cause hypersulfhydration. This hypersulfhydration has been hypothesized to be one of the contributors to the effects in the CNS during early-onset AD in adults and impaired brain growth in children (89). However, further studies and more conclusive evidence are needed to consolidate the link between hypersulfhydration and DS-related symptoms.
Regulation of S-desulfhydration
Several studies have reported that the Trx system catalyzes desulfhydration, the reverse reaction of sulfhydration, under different conditions (105, 134). Recent studies using different methods have suggested that the Trx system globally regulates cellular desulfhydration (96, 205). The Grx system and Trx-related protein of 14-kDa (TRP14) are also able to reduce protein sulfhydration (52). Our overall understanding of desulfhydration remains incomplete and more effort is required to elucidate the whole picture.
Redox regulation of receptor signaling
Activation of protein tyrosine kinases (PTKs) to phosphorylate target proteins is a common mechanism to transduce ligand–receptor-based signals, which are important for growth factor-mediated neural development. Therefore, protein tyrosine phosphatases (PTPs), which negatively regulate the activity of PTKs and downstream cascades, are important to maintain the balance of total protein phosphorylation.
Although members in the PTP superfamily share limited sequence similarity, the presence of a -CXXXXXR- (X can be any amino acid) motif in the active site is a common catalytic hallmark for PTPs (194). Due its low pKa value (4–6.5), the cysteine in the active site of PTPs exists as a thiolate (-S−) at physiological pH and it is sensitive to ROS or RNS modification in vivo (159). In particular, upon growth factor stimulation, ROS transiently and locally produced by NOX are required to activate PTKs and downstream cascades (187). During the last two decades, redox regulation of PTPs in signal transduction has received much attention due to its implication in various physiological and pathological settings in the CNS. Details have been discussed in recent reviews (81, 112).
Inactivation of PTPs by other oxidants, such as H2O2 (48), oxidized GSH (GSSG) (14), lipid peroxide (42), NO (13), and H2S (105), results in reversible cysteine modifications, such as sulfenic acid, glutathionylation, disulfide bonds, nitrosylation, or sulfhydration. The oxidized PTPs can be reduced and restored by the Trx and GSH systems (113). Interestingly, the Trx system was reported to exhibit certain specificity toward different PTPs. A recent study showed that both Trx1 and Trp14 coupled with TrxR and NADPH can reactivate oxidized PTP1B, but not SHP2, another member of the PTP family (45). Grx was reported to reduce glutathionylated PTP1B and restore its phosphatase activity (14).
Prx (38) and GPx (42) also contribute to receptor signaling due to their high efficiency for removing peroxides and protection of PTPs from oxidation. Prx can protect PTEN from oxidation by a direct protein–protein interaction (33). Interestingly, under certain conditions, Prx acts as an oxidase of PTPs, in contrast to its typical well-characterized role as an H2O2 scavenger. During H2O2 burst, the cysteine at the active site of Prx is initially oxidized into sulfenic acid or intra-/intermolecular disulfide, then the oxidized Prx may selectively transfer the equivalent oxidation to target proteins via specific protein–protein interactions (74). However, more evidence is needed to understand this process in mammalian cells. It was also reported that growth factor stimulation inactivated Prx via phosphorylation and that dephosphorylation by PTPs may restore Prx activity (208).
Trx and GSH Systems in CNS Disorders
Several human CNS disorders, such as ischemia/reperfusion (I/R) damage and neurodegenerative diseases, are characterized by dysregulated apoptosis and accumulation of damaged proteins, DNA, and membranes. Dysfunction of many cellular processes, including protein degradation, ROS removal, mitochondrial function, and neuroinflammatory responses, contributes to pathological processes (115). Failure to respond correctly to oxidative stress, generation of ROS, and protein aggregation may be involved in pathogenesis (90). Since GSH and Trx are the major players in redox signaling, it is not surprising that changes in these two systems have been found in the aforementioned diseases (2, 10, 120).
Aging is a major risk factor for neurodegenerative disorders and is associated with higher production of ROS and the progressive loss of certain neuronal populations (129). Downregulated expression of Trx and GSH system proteins resulting in attenuated antioxidant activity has been observed during aging (171), and overexpression of components of these two systems was shown to rescue the aging-related phenotype in several animal models (67, 137). The Trx1 and Grx1 levels in cerebrospinal fluid are correlated with the expression of the established AD markers, tau and phospho-tau, in AD patients (10).
Regulation of redox-sensitive apoptosis induced by ER stress
Accumulation of misfolded proteins caused by ER stress is a hallmark of neurodegenerative diseases. ER is an important organelle for protein quality control and ensures that proteins are synthesized, folded, and post-translationally modified correctly. Disturbance of ER function leads to ER stress, which may sequentially trigger undesired apoptosis (213).
Trxs are critical mediators of ER stress-induced apoptotic signaling. Trx1 has been found to be a physiological inhibitor of apoptosis-signaling kinase 1 (ASK1) via a direct protein–protein interaction. ASK1 is a mitogen-activated protein kinase kinase kinase (MAP3K) and activates MAP2K-JNK/MAPK-p38 signaling cascades, which are essential for ER stress-induced apoptosis (146). Trx1 inhibits ASK1 kinase activity via the formation of a disulfide between its active site Cys32 or Cys35 and Cys250 in the N-terminal portion of ASK1. When cytosolic Trx1 is oxidized in response to proinflammatory stimuli, ROS, and cellular stress, ASK1 is released from Trx1-ASK1 complexes and subsequent ASK1-dependent apoptosis is activated (Fig. 6). Trx2 associates with mitochondrial ASK1 via binding with the ASK1 Cys30 and activates a JNK-independent apoptosis pathway (222).
The activity of Trx is also regulated by Trx-interacting protein (TXNIP), its endogenous inhibitor (147), which is also induced during ER stress (149). The interaction between Trx and TXNIP involves disulfide formation between Cys32 in Trx and Cys247 in TXNIP, which may be regulated by an internal disulfide switching mechanism between Cys63-Cys247 and Cys63-Cys190 in TXNIP (87).
Regulation of redox-sensitive apoptosis in I/R
ROS production is involved in ischemic brain damage, thus overexpression of antioxidant enzymes, such as Trxs and Prx2, attenuates cerebral infarction following ischemia and I/R. Prx2 protects brain tissue from I/R-induced injury by suppressing the ASK1/JNK-mediated mitochondrial signaling pathway (68). Prx2 also attenuates the poly(ADP-ribose) polymerase 1 (PARP1) and p53-dependent cell death pathway (110).
The NMDA receptor is a critical mediator during ischemic brain damage. The TXNIP pathway has been shown to be involved in neuroprotection mediated by the synaptic NMDA receptor in rat neurons. Synaptic activity blocks TXNIP expression and activates the expression of sulfiredoxin and sestrin 2, thus enhancing reduction of Prx by the Trx system and facilitating intrinsic antioxidant defenses in neurons (150). Although the Trx system has a protective role in cerebral I/R, the mitochondrial Trx system has been indicated to be involved in caspase 3-mediated neuronal apoptosis in rat hippocampus (186). Cerebral I/R can activate glutamate receptor 6 (GluR6), which upregulates the expression of FasL, and thus enhances the expression of TrxR2 and Trx2. The mitochondrial Trx2 system then denitrosylates and activates procaspase 3 (186) (Fig. 5).
GSH in health and diseases of human CNS
GSH is the most abundant thiol-containing molecule and one of the most important antioxidants involved in neuroprotection (8). The GSH level in the brain is ∼2–3 mM (43); it is highest in the cortex, followed by the cerebellum, hippocampus, and striatum, and is lowest in the substantia nigra (97).
GSH reacts nonenzymatically with oxidants to inhibit oxidative stress in the cell, including superoxides, NO, hydroxyl radicals, and peroxynitrite. In addition, GSH reacts enzymatically with GPx and GSH-S-transferase (GST) against neurodegeneration (9). In neurons, GR is sufficiently active to allow the rapid reduction of accumulated GSSG (55). Under oxidative stress, GR maintains the equilibrium of the GSH/GSSG redox state in the cell. Most importantly, GSH depletion induced by oxidative stress can exacerbate oxidative injury in the brain (126).
An age-related decline in GSH level has been observed in humans, and the GSH concentration in the cerebral spinal fluid decreases with age. Such findings suggest that aging-associated decreases in GSH may underlie changes to antioxidant capacity, which occur during aging and during the onset of various aging-related diseases (193). Previous works have suggested that GSH depletion is involved in neurodegeneration (175). Indeed, the GSH levels are decreased in the brain in some neurodegenerative diseases, but it remains unclear whether the decreased GSH level is a cause or an outcome of neurodegeneration (163). In PD, although there is a 30–40% decrease in GSH concentrations, no corresponding increase in the level of GSSG has been detected, thus the reason for GSH depletion is not clear (178). In AD, the total GSH level in brain is not affected, whereas GPx and GR were found to be either elevated or unchanged in different brain regions (157). In clinically isolated syndrome and relapsing–remitting multiple sclerosis, there is evidence that GSH content and GPx activity are decreased, and the former might serve as a marker closely correlated with the neurological scoring of acute CNS inflammation (117). The low concentration of GSH during inflammation is likely to be a consequence of accelerated turnover linked to elevated oxidative stress. In a beta-N-oxalyl amino-L-alanine (L-BOAA)-induced mouse model of neurolathyrism (a motor neuron disease involving the pyramidal system), GSH loss and inhibition of mitochondrial complex I were observed in the lumbosacral cord of male mice (51). Other CNS toxicants such as ethanol also cause a decrease in GSH concentration, particularly in the cerebellum, striatum, and cortex, which may be associated with an increase in the concentration of acetaldehyde, which is normally removed from cells by GSH (11).
Modification of Trx and GSH Systems by Small Molecules in the CNS
Since the Trx and GSH systems are so important in the CNS, many small molecules can exert their effects in the CNS via modification of the two thiol-based signaling pathways. Several examples are discussed below.
Mercury
Exposure to mercury (Hg), whether it is in the form of methylmercury (MeHg) in ingested fish, mercury vapor (Hg0) released from dental amalgams, or ethylmercury (EtHg) derived from thimerosal-containing vaccines, results in its accumulation in the CNS, which eventually leads to neurotoxicity. The neurotoxic effects are particularly serious if exposure to Hg takes place during fetal development, or in early infancy, due to the immaturity of the CNS and BBB (40).
Systemic distribution of MeHg and its accumulation in the CNS are facilitated by binding to -SH groups in cysteine due to the molecular similarity between the MeHg-Cys complex and the amino acid methionine, which enables MeHg-Cys transport across the BBB and into the brain via neutral amino acid transporters (39). On the other hand, Hg0 has no charge and can freely diffuse across membranes and reach the CNS. Both the organic species of Hg and Hg vapor are converted to the highly reactive Hg2+. In fact, Hg2+ is the most abundant Hg species found in the brain following chronic exposure to MeHg, EtHg, or Hg0 (32, 201).
One of the known mechanisms by which Hg exerts neurotoxicity is through the disruption of glutamate (Glu) transport, either by increasing its release from the presynaptic terminal or by inhibiting its reuptake from the synaptic cleft by astrocytes. Impairment of glutamate uptake is associated with increased ROS production and subsequent oxidative stress (60). The Hg electrophilic nature underlies its oxidative effects and its high binding affinity for thiols and selenols (35, 169). As a result, both the GSH and Trx systems are targets for mercurial compounds (Fig. 7).
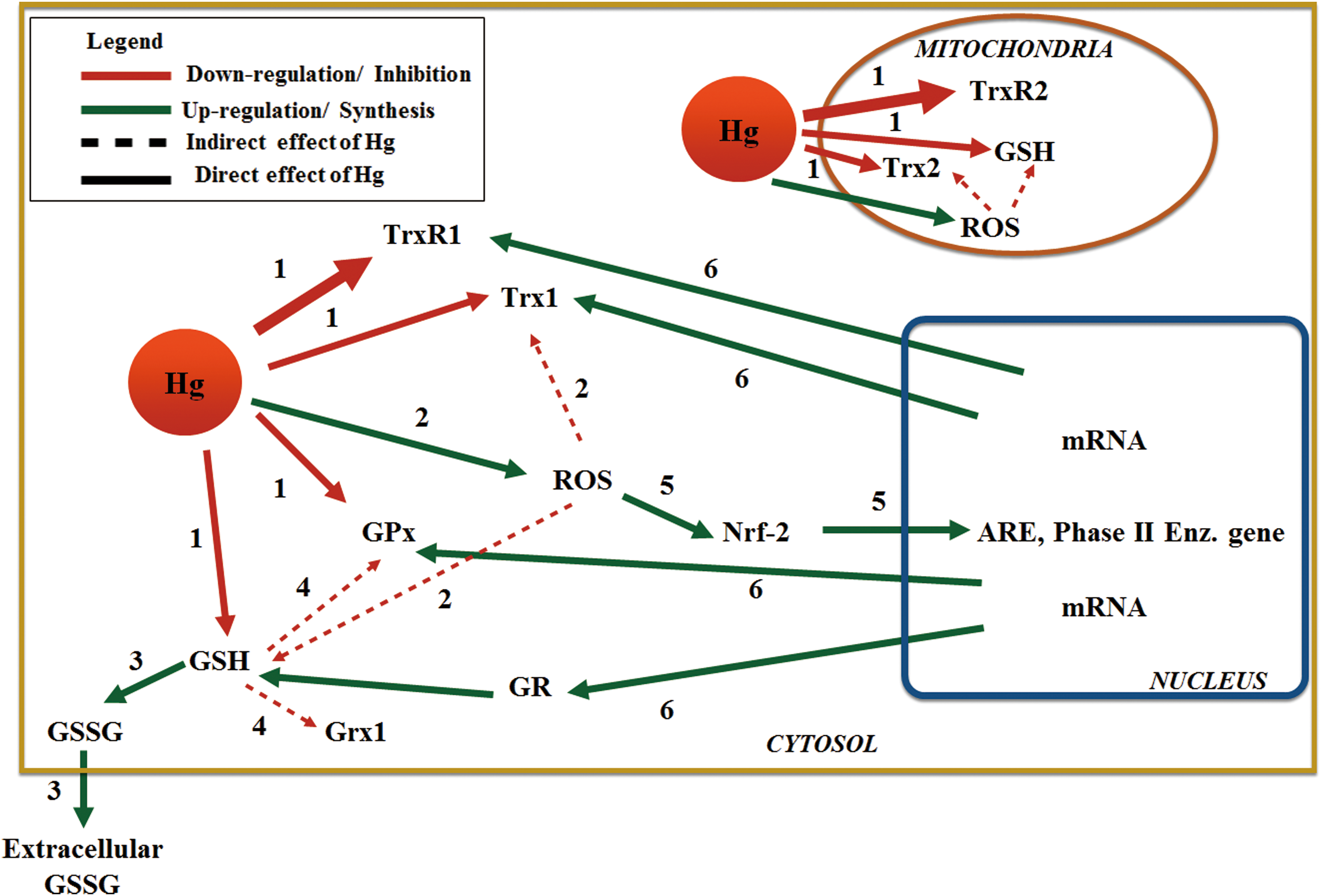
Depletion of GSH has been shown to occur after exposure to Hg compounds in vitro and in vivo using experimental animals (47, 204). In primary cultures of neurons and astrocytes, GSH was decreased by 30–50% following a 30-min treatment with 5 μM MeHg (98). This abrupt decrease in GSH levels cannot be explained on the basis of a 1:1 complex formation. Therefore, it is likely that upon exposure to Hg, ROS generation plays an important role in GSH oxidation with the subsequent formation and excretion of GSSG. Additionally, decreased synthesis of GSH, due to impaired uptake of its precursor Glu, leads to higher ROS generation (47) (Fig. 7). Concomitantly, in the study by Kaur et al., pretreatment with diethylmaleate depleted cellular GSH and further aggravated MeHg toxicity, whereas pretreatment with N-acetylcysteine increased the intracellular GSH level and offered additional protection against toxicity (98). Most interestingly, in vivo data suggest that GR activity in the brain is increased by exposure to MeHg, which may function as a compensatory response following GSH oxidation (27).
The pivotal role of GSH in modulating Hg toxicity cannot be separated from the fact that it is an important cofactor for enzymes involved in antioxidant defense, such as GPxs and Grxs. GPx1, an important scavenger of H2O2, is affected by exposure to mercurials, in particular MeHg. Likewise, membrane-located GPx4 presented decreased activity and expression in the cortex and cerebellum of mice exposed to MeHg (220). Besides dependence on GSH as an electron donor, GPx activity relies on the reducing ability of Sec residue in the active site. This Sec is prone to direct interaction with MeHg (28), but not Hg2+, as shown in vitro by Bulato et al. (31).
Data on the interaction between Hg and Grxs are scarce. We have previously reported that both MeHg and Hg2+ target the thiols in purified Grx1; however, in HeLa cells, inhibition of Grx activity is only achieved at very high concentrations. To the best of our knowledge, there are no reports concerning the interaction between Hg and Grx in CNS models or in experimental animals (34).
The Trx system has been proven to be particularly sensitive to Hg. TrxR is a prime target for mercurials due to the high reactivity and location of the Sec residue at the open C-terminal (34, 35). Several in vitro and in vivo studies have shown a significant drop in TrxR activity in the brain following exposure to different Hg compounds, which is both time and concentration dependent (27, 28, 166, 220). In liver cells, inhibition of cytosolic TrxR1 by Hg2+ is counteracted by upregulation of TrxR1 synthesis via the Nrf2 pathway (29). This Nrf2 response is weaker during exposure to MeHg, resulting in decreased expression of TrxR1. It has been shown that a fast Nrf2 response occurs upon MeHg exposure in microglia and neurons (145); however, the relationship between Nrf2 signaling and TrxR1 in the CNS has not been fully elucidated. Contrary to its cytosolic counterpart, TrxR2 is not regulated by Nrf2 (29) and thus it might be a very important early target in the development of Hg toxicity. In fact, TrxR2 and the overall mitochondrial Trx system, via Prx3, play a vital role in the detoxification of H2O2 in brain mitochondria, and TrxR2 inhibition increases the sensitivity of CNS cells to other environmental stressors such as paraquat (PQ) (118).
Trx1 is also inhibited by mercurials in vitro with a 1:2.5 and 1:5 enzyme:inhibitor ratio for Hg2+ and MeHg, respectively (34). In SH-SY5Y cells exposed to EtHg (166) and in the brain of experimental animals exposed to MeHg and Hg2+, Trx activity is inhibited in a dose-dependent manner similarly to the effect on TrxR, although the reduction in activity is less significant (27, 28).
Paraquat
PQ (1,1′-dimethyl-4-4′-bipyridinium dichloride) is a typical quaternary ammonium herbicide, which kills plants nonselectively by targeting the chloroplasts in green plant tissues. In plants, PQ mainly exerts its toxic effect by acting as a redox cycler, which can constantly consume NADPH and disturb the electron transfer photosystems (25). In humans, PQ is not easily absorbed from the skin or gut. Once absorbed, PQ mainly accumulates in lungs and causes pulmonary fibrosis. Acute PQ poisoning results in respiratory failure and death within a few days or weeks. Chronic exposure to PQ leads to elevated oxidative stress in the plasma, including increased lipid peroxidation, decreased antioxidant capacity (affecting the ferric-reducing ability of plasma), and decreased blood thiol content (164). Although it is still under debate whether PQ can pass through the BBB and exert its toxic effect directly in the brain (15, 16, 30, 177), many epidemiological and etiological studies have shown a clear association between low-dose exposure (absence of acute toxicity) to PQ and neurodegenerative diseases, especially PD. Mechanistic studies also demonstrated that PQ can induce apoptotic cell death in several types of neuronal cells in association with PD, including in cerebellar granule cells and other dopaminergic neurons (71).
In mammalian cells, the mechanism of PQ-induced toxicity is very similar to the mechanism in plant cells. PQ ions (PQ2+) are reduced to the free radical form of PQ (PQ•+) by NADPH. PQ•+ can spontaneously react with oxygen, resulting in the formation of superoxide anion (O2 •−), which then leads to the generation of other types of ROS, including H2O2 and hydroxyl radicals (25). The overproduction of ROS then causes damage to proteins, lipid membranes, and DNA (Fig. 8). Furthermore, PQ constantly consumes NADPH and disrupts essential NADPH-dependent biochemical processes (167) (Fig. 8). Besides acting as a redox cycler, the role of mitochondria in PQ neurotoxicity has been addressed. ROS generation following PQ exposure is dependent on respiratory chain components, especially complex III (36).
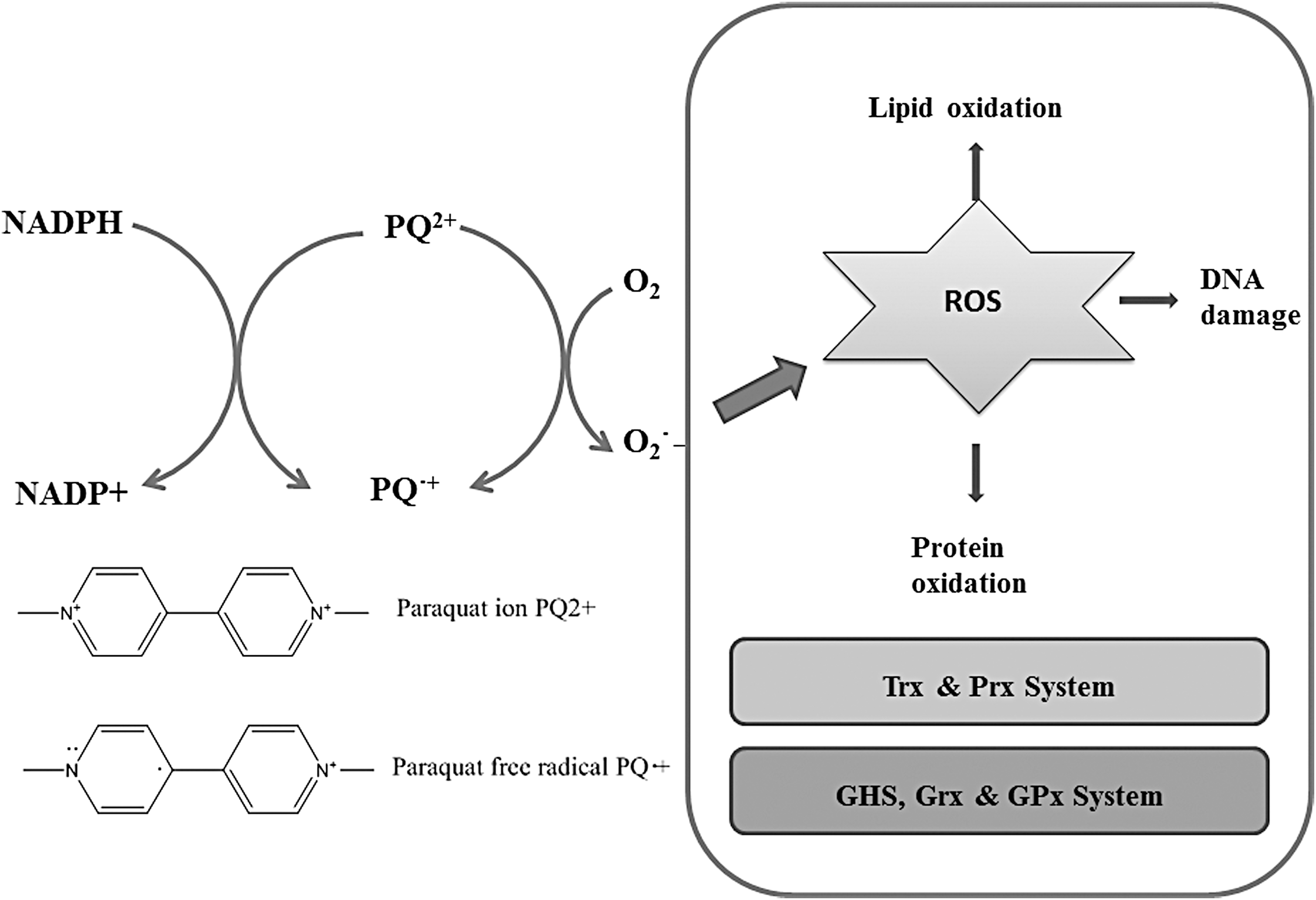
Trx and Grx systems are not only very important for the defense against PQ toxicity but are also disrupted by PQ toxicity as both systems use NADPH as the ultimate electron donor. In SH-SY5Y and SK-N-MC human neuroblastoma cell lines, PQ was found to oxidize Trx1 and activate the ASK1/JNK/caspase 3 cascade. PQ treatment also reduced the activity of TrxR and GPx (188, 198). Dopaminergic N27 cells were sensitized to PQ treatment by disruption of mitochondrial Trx2 and TrxR2 (118). Drosophila with Trx2 loss of function were sensitized to PQ toxicity (197). In Drosophila cells, Prxs also had a cytoprotective effect following PQ treatment (162). The expression of certain Trx and Grx system proteins is regulated by the transcription factor Nrf2. Although PQ treatment was found to decrease the protein level of Nrf2, it can also activate the Nrf2 pathway and increase the transcription of target genes (167, 216). TrxR appears to have a dual role in PQ toxicity. Several studies in different species have reported that TrxR has a protective role following PQ treatment (118, 142). However, in the presence of NADPH, TrxR was found to reduce PQ and generate ROS that may contribute to its toxicity (72, 189). Several known antioxidants or proteins with antioxidant properties, including vitamin C, vitamin E, Se, N-acetylcysteine, and coenzyme Q10 (CoQ10), were found to have protective roles against PQ treatment (12, 71, 132, 188, 198, 219). These findings also suggest the importance of antioxidant enzymes for protection against PQ toxicity.
Coenzyme Q
CoQ10 (ubiquinone) is an electron carrier lipid of the mitochondrial respiratory chain and an endogenously synthesized antioxidant. CoQ10 can accept electrons from complex I and complex II and transfer them to complex III, which is considered to be the rate-limiting step in ATP production (130). CoQ10 is able to protect lipids from peroxidation in cell/organelle membranes and in the circulation (168). As well as effects on lipid membranes, in vivo administration of CoQ10 can also protect membrane proteins (64) and DNA (192) from overoxidation. CoQ10 is also able to regenerate other antioxidant molecules, such as vitamin E (66). Additionally, oral administration of CoQ10 has been proven to be safe even at very high concentrations (3600 mg/day) (88).
The two major functions of CoQ10, oxidation and regeneration, both depend on changes in its redox state. In the mitochondrial matrix, CoQ10 is regenerated by the respiratory chain enzymes and the majority of CoQ10 in mitochondria is in its reduced form (1). In the other cellular compartments and plasma, several enzymes with reductase activity can regenerate CoQ10, including lipoamide dehydrogenase, TrxR, and GR. Compared with lipoamide dehydrogenase, full-length TrxR (in the presence of selenocysteine) is more effective, whereas the truncated form of TrxR does not have the ability to regenerate CoQ10 (211). This may also explain why several studies reported that the combined administration of Se and CoQ10 yields better cardiovascular protective effects than administration of CoQ10 only (3, 4).
It has been well established that mitochondrial dysfunction is associated with neurodegenerative diseases. In neuronal cells, pretreatment with CoQ10 was found to reduce ROS generation in mitochondria and protect neuronal cells from cell death induced by neurotoxins such as kainic acid (181). A decrease of CoQ10, or the reduced form of CoQ10, in SH-SY5Y neuroblastoma cells resulted in increased oxidative stress (58). In rats, oral administration of CoQ10 can significantly increase the CoQ10 level in plasma, cells, and mitochondria and decrease oxidative damage to proteins (107). Several trials in humans and in rats have validated the beneficial role of CoQ10 in neurodegenerative diseases, such as PD and motor dysfunction disorders (18, 174).
Selenium
Se is an essential micronutrient with a narrow range of optimal concentrations. Disease can be caused by both Se insufficiency (e.g., Keshan disease) and an excessive Se intake (e.g., selenosis) (165). Upon entering cells, Se-containing compounds, organic or inorganic, are reduced to selenide (Se2−) and then integrated in proteins in the form of selenocysteine by a fairly complex synthesis mechanism [reviewed in detailed by Papp et al. (151)]. At least 25 genes that encode selenoproteins have been identified in mammals, including genes encoding TrxR isoforms 1, 2, and 3 (TGR) and GPx isoforms 1, 2, 3, 4, and 6 (151). At physiological pH, the selenol (-SeH) in the selenocysteine residue is in the form of selenolate (-Se−), providing selenoenzymes with high reactivity and sensitivity to electrophilic agents (as previously discussed for Hg) (35).
The brain is a prioritized organ for Se distribution, meaning that during periods of insufficient intake, the Se stored in visceral organs (e.g., liver) is redistributed to the brain (20). In agreement, TrxR activity was unchanged in the brain of rats fed an Se-deficient diet, whereas activity was reduced in the kidney and liver (82). In fish, supplementation of water with sodium selenite did not change TrxR activity in the brain, contrary to what was observed in the liver (28). However, SH-SY5Y cells treated with 2.5 μM sodium selenite had a 40% increase in TrxR activity (166), indicating that Nrf2-mediated gene transcription was induced, as previously observed in the liver (29). The selenite amount used in these experiments is considerably higher than the reported 100 nM necessary to optimize TrxR1 synthesis physiologically (156), and thus, it is expected that redox cycling and additional ROS formation will occur. However, it should be noted that the aforementioned experiments were performed with the aim of using Se as an antagonist to the toxicity of Hg compounds and to effectively recover the TrxR activity following inhibition by mercurials (Hg2+; MeHg; EtHg), thus Se needs to be in a molar excess relative to the inhibitors (29, 35, 166). In fact, selenite treatment is able to remove Hg2+ from the active site of TrxR1 and fully restore TrxR1 activity in vitro (35). The protective effect of selenite following Hg-induced inhibition of TrxR1 has also been observed in SH-SY5Y cells (166).
Generally, GPx1 activity in the brain is not enhanced to the same extent as is TrxR1 upon Se supplementation, indicating its low ranking in the selenoprotein hierarchy (139). Nevertheless, GPx4 is essential for neuronal development and survival and has been reported to be highly expressed in 90% of the mouse brain (223). The use of the small organoselenium compound Ebselen, a mimetic of GPx activity, has been reported to offer antioxidant protection in the CNS (214). Interestingly, human subjects exposed to Ebselen exhibited reduced levels of brain GSH (128), which could be due to its GPx-like activity and GSH consumption. This contrasts with the upregulation of GSH observed in rat brain following selenite treatment (108, 183).
Concluding Remarks
Our knowledge of ROS has been renewed during last two decades. Instead of simply considering ROS as toxic molecules that are closely associated with various diseases, the physiological function of ROS as signaling messengers has been increasingly emphasized. Correspondingly, the Trx and GSH systems, as the two major antioxidant mechanisms, have also been highlighted for their pivotal roles as regulators of redox signaling. Redox signaling heavily relies on reversible modification of key thiols in proteins. Although the modifications are usually transient and ubiquitous, they surprisingly exhibit both tissue and target specificity. The CNS produces relatively high levels of ROS due to the large energy demand and, concurrently, is more susceptible to the damaging effects of ROS compared with other tissues. The Trx and GSH systems are the key factors required to maintain redox balance in the CNS. It is not surprising that both systems are dysfunctional in various neuronal disorders, especially in neurodegenerative diseases. Trxs can exert their antiapoptotic effect via binding ASK1 and thus protecting neurons from cell death. By closely evaluating several small compounds, we found that they all interact with the Trx and GSH systems and their beneficial or detrimental effects on CNS were further elucidated. However, further effort is needed to fully understand this complex system. New methods to measure ROS in a more physiologically relevant context should be developed, and biomarkers that can precisely predict and monitor ROS-related therapies are required. Furthermore, the misconception that ROS only act as damaging factors should be altered. It is likely that more redox signaling pathways regulated by the Trx and GSH systems, and the mechanisms involved, will be revealed. We believe that the field of redox signaling will attract more scientific efforts and be beneficial to patients suffering from various neuronal disorders.
Footnotes
Acknowledgments
The authors are thankful for the financial support of China Scholarship Council to X.R. and L.Z. C.C. and V.B. are supported by Fundação para a Ciência e Tecnologia, Portugal (