
Abstract
Significance:
In this review, we discuss the role of nitric oxide (NO) as a key physiological mechanotransducer modulating both local and systemic heterocellular communication and contributing to the integrated (patho)physiology of the cardiovascular system. A deeper understanding of mechanotransduction-mediated local and systemic nodes controlling heterocellular communication between the endothelium, blood cells, and other cell types (e.g., cardiomyocytes) may suggest novel therapeutic strategies for endothelial dysfunction and cardiovascular disease.
Recent Advances:
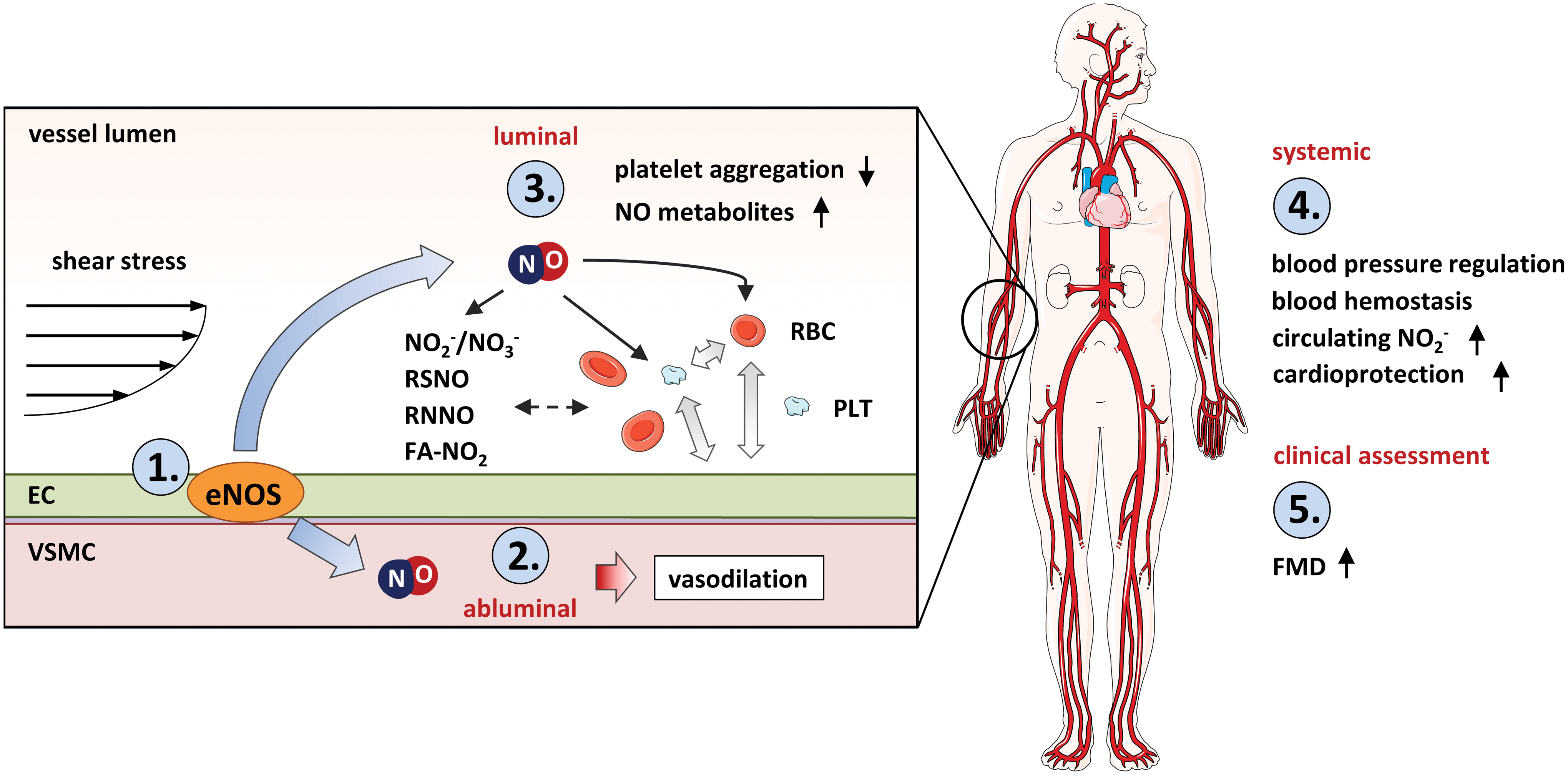
Mechanical forces acting on mechanoreceptors on endothelial cells activate the endothelial NO synthase (eNOS) to produce NO. NO participates in (i) abluminal heterocellular communication, inducing vasorelaxation, and thereby regulating vascular tone and blood pressure; (ii) luminal heterocellular communication, inhibiting platelet aggregation, and controlling hemostasis; and (iii) systemic heterocellular communication, contributing to adaptive physiological processes in response to exercise and remote ischemic preconditioning. Interestingly, shear-induced eNOS-dependent activation of vascular heterocellular communication constitutes the molecular basis of all methods applied in the clinical routine for evaluation of endothelial function.
Critical Issues and Future Directions:
The integrated physiology of heterocellular communication is still not fully understood. Dedicated experimental models are needed to analyze messengers and mechanisms underpinning heterocellular communication in response to physical forces in the cardiovascular system (and elsewhere). Antioxid. Redox Signal. 26, 917–935.
Introduction
N
NO carries physicochemical characteristics, making it an ideal messenger for transferring physiological signals within cells, through cells, and among tissues. Compared with other free radicals participating in redox signaling, for example, superoxide radical anion (O2 −•), NO is more stable and less reactive toward biologically relevant thiols, a such as cysteine and glutathione (53), which are found in millimolar concentrations in cells and tissues. Its peculiar reactivity, together with its lack of charge, allows NO to survive the reducing thiol-rich environment of the cell, to cross cell membranes, and to reach its molecular targets outside in the subluminal and luminal side of the vascular endothelium (Fig. 1). NO has a high affinity for Fe2+-heme centers and rapidly reacts with the soluble guanylate cyclase (sGC; EC 4.6.1.2); sGC catalyzes the conversion of guanosine-5′-triphosphate (GTP) into the second messenger 3′,5′-cyclic guanosine monophosphate (cGMP), which in turn activates its downstream signaling cascade (8, 32). Although the chemical biology of NO-mediated S-nitrosation of biological thiols is still a matter of debate (27, 69), S-nitrosothiols are found in low μM concentrations in vascular tissues, with higher concentrations in rodents than humans (17, 47, 116, 120). In addition, NO may exert pleiotropic cGMP-independent effects via S-nitrosation of key cysteines in enzymes and proteins modifying their activity (6).
In the endothelium, eNOS activity is tightly regulated by different mechanisms, including (i) localization of the enzyme (85, 129); (ii) availability of the substrate L-arginine, of cofactors such as tetrahydrobiopterin, and Ca2+/calmodulin (CaM), which are all essential for NOS activity (63); (iii) phosphorylation at specific amino acids, which may lead to both activation or inhibition of eNOS activity depending on their localization in the protein sequence, as well as on the phosphorylation map (40, 48, 50); (iv) other post-transcriptional modifications, such as S-nitrosation (45, 118), glutathionylation (23), and persulfidation (5), which were shown to modify eNOS activity; and (v) transcriptional and post-transcriptional regulation, including changes of gene expression, messenger RNA (mRNA) stability, or microRNA (miRNA)-dependent mRNA degradation, as extensively revised by Balligand et al. (10).
In addition to the well-known receptor-mediated activation of eNOS, for example, by agonists such as acetylcholine (ACh) or bradykinin (BK) (141), mechanical forces are among the most important physiological regulators of eNOS-dependent NO production in the endothelium (10, 11, 21, 83), and multiple pathways of eNOS activation are involved. The vascular endothelium is exposed to pulsatile flow, conjugating in a temporally defined manner both tangential and circumferential mechanical forces (10, 21, 35, 83). Tangential forces (also defined as shear stress) have been shown to activate tightly regulated biochemical responses by activation of membrane or cytoskeletal proteins (defined as mechanosensors), leading to the activation of biochemical pathways, thus transforming mechanical stimulation into biochemical signal transduction (a process defined as mechanotransduction), which elicits highly regulated physiological responses.
Endothelial NO-mediated mechanosensing and mechanotransduction are mainly considered as local processes. However, it is well known that eNOS-derived NO may exert paracrine effects also via formation of bioactive circulating metabolites (17, 47, 120), including nitrite (31, 38, 61, 101, 117, 137), and thereby participates in systemic heterocellular communication (Fig. 1).
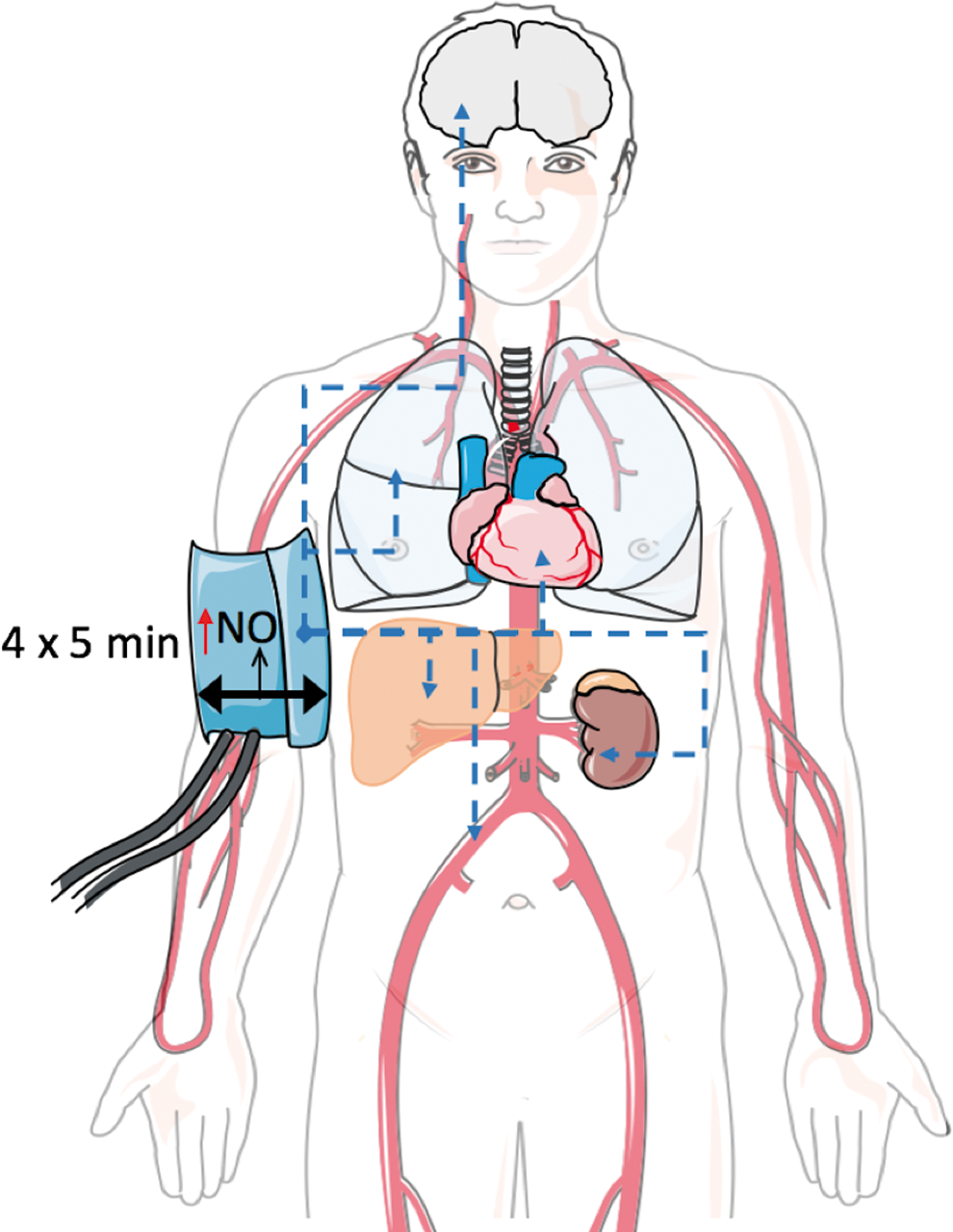
In this review, we aim to discuss the role of NO as a main mechanotransduction messenger in local and systemic heterocellular communication. We will review the role of NO in (i) abluminal heterocellular communication induced by mechanical forces, that is, the mechanisms of mechanosensing and mechanotransduction in the endothelium focusing on flow-mediated eNOS activation, NO production, and vasodilation; (ii) luminal heterocellular communication, that is, the NO-mediated communication among endothelial cells and blood cells; and (iii) systemic heterocellular communication induced by physical forces, that is, the role of NO-mediated mechanotransduction in remote communication involved in organ protection. Moreover, we will discuss (iv) the clinical implication of shear stress-induced eNOS-mediated heterocellular communication (Fig. 1). A deeper understanding of mechanotransduction-mediated local and systemic nodes controlling heterocellular communication between the endothelium and other cell types may provide novel therapeutic strategies to be applied in conditions associated with endothelial dysfunction and cardiovascular disease.
Abluminal Heterocellular Communication Induced by Mechanical Forces
Endothelial cells sense changes in local hemodynamic patterns by the presence of mechanoreceptors localized on their membrane (respectively, on the luminal side of the membrane, in junctional complexes connecting two adjacent cells, or in focal adhesions in the subluminal side of the membrane of cells) as well as inside the cells (i.e., the cytoskeleton). The endothelial cells respond to the mechanical signal by activation of biochemical pathways ( = mechanotransduction), which lead to physiological effects, including changes in arterial wall vasomotion, structure, and gene expression profile. In the following section, we will focus on the shear-induced signaling pathways activating eNOS-derived NO formation and heterocellular signaling between endothelial cells and smooth muscle cells (SMCs), thereby regulating vascular tone and blood pressure.
Shear-dependent vasodilatory response
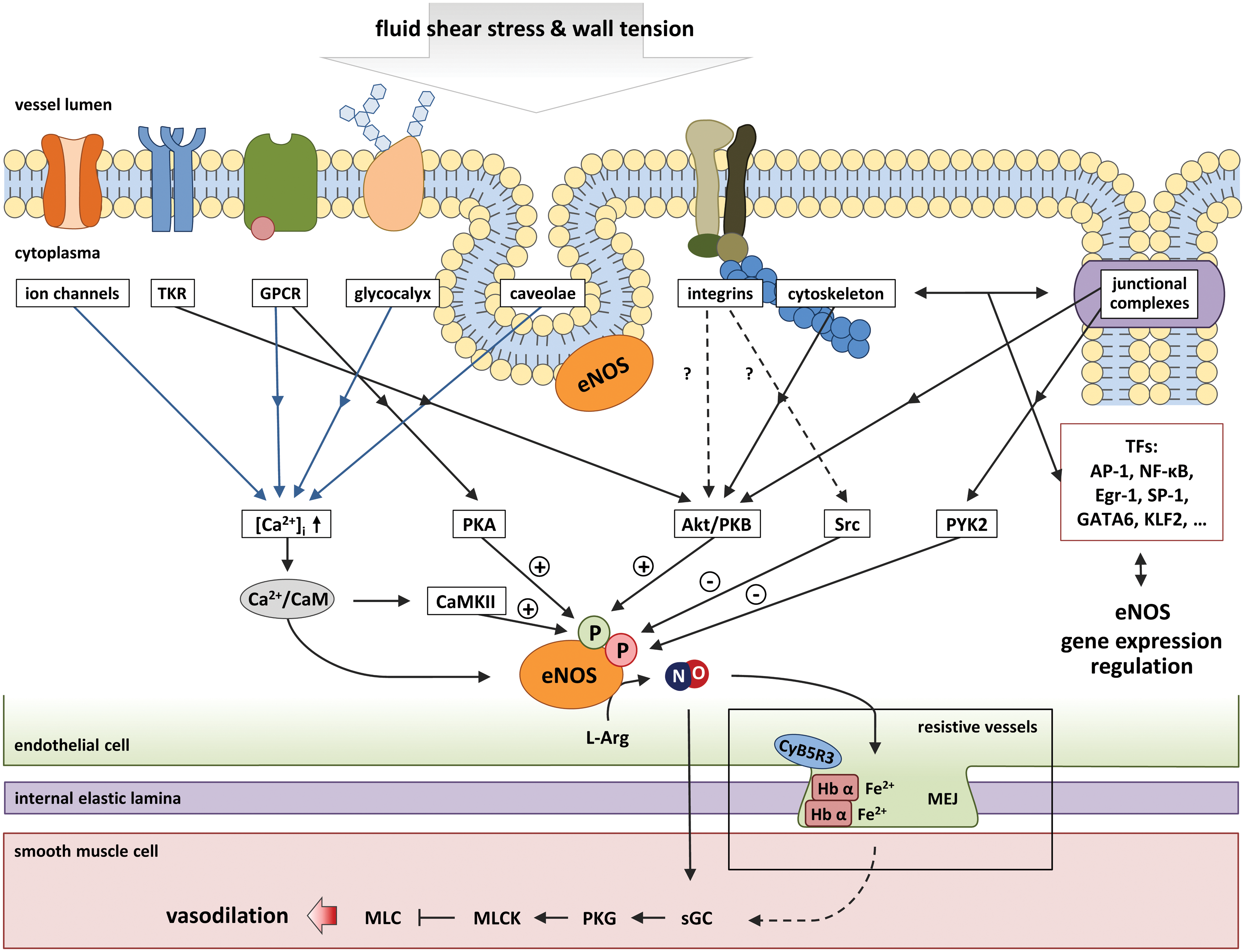
Shear-mediated activation of eNOS activity occurs mainly via three major mechanisms, which are depicted in Figure 2. Activation of mechanosensors can lead (i) to mobilization of intracellular Ca2+ stores, to an increase in intracellular Ca2+ concentration ([Ca2+]i), and to formation of Ca2+/CaM complexes, which activate eNOS by binding to a short regulatory sequence between the two subunits of eNOS (18); (ii) to activation of shear-sensitive protein kinases, phosphorylation of eNOS at specific amino acids, leading to activation or inhibition of the enzyme depending on the phosphorylation site and on the phosphorylation map (50); and (iii) to regulation of expression levels of eNOS, which is mediated by the activation of transcription factors, including activator protein 1 (AP-1), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), early growth response protein 1 (Egr-1), specificity protein 1 (SP-1), GATA-binding protein 6 (GATA6), and Krüppel-like factor 2 (KLF2) (97). In addition, the expression of eNOS is tightly regulated by post-translational mechanisms, including mRNA stability and miRNA-dependent regulation (10). These signaling pathways are deeply intertwined and may act simultaneously and ensure short- and long-term eNOS regulation.
Experimental studies have shown that shear stress affects NO production and heterocellular communication by eNOS in two phases. On applying shear stress, there is a first brief transient Ca2+/CaM-dependent NO burst, which is responsible for short-term activation of eNOS, whereas activation of phosphorylation cascades controls eNOS activity to assure sustained and moderate NO generation (50). NO produced by the endothelium is then released into the subluminal space and reaches the SMC, where NO binds reversibly to the prosthetic ferrous heme group (Fe2+) of the sGC with complex binding kinetics (150), resulting in a conformational change that activates the enzyme. The sGC is a heterodimer, consisting of an α- and a β-subunit, but only the latter contains the prosthetic group (149), and exists in two isoforms both expressed in the vasculature (102, 123). The enzyme catalyzes the conversion of GTP to cGMP, which in turn activates cGMP-dependent protein kinases (PKG) and other targets (115). In vascular SMCs (VSMCs), activation of PKG induces vasorelaxation by multiple mechanisms, including (i) phosphorylation of phospholamban, and sarcoendoplasmic reticulum calcium transport ATPase (SERCA)-dependent decrease in intracellular Ca2+ concentrations; (ii) phosphorylation of IRAG, decrease in IP3, and decrease in intracellular Ca2+ concentrations; or (iii) changes in activity of K+ channels, causing membrane hyperpolarization, inhibition of extracellular Ca2+ influx, and decrease in intracellular Ca+2 concentration, as well as phosphorylation of myosin light chain kinase, which then results in dephosphorylation of myosin light chains (which is also dependent on the activity of a myosin light chain phosphatase), leading to smooth muscle relaxation. Taken together, shear stress keeps eNOS-derived NO production constant, thereby regulating vascular diameter and tone.
Resistance vessels are the main target for peripheral mechanisms controlling blood pressure. Isakson and coworkers (133) discovered that NO-mediated heterocellular communication between endothelium and SMCs in resistance vessels is fine-tuned by the redox state of hemoglobin α within myoendothelial junctions (membrane structures connecting endothelium with SMCs). Thus, if the iron heme of hemoglobin α is oxidized (Fe3+), NO can diffuse through the myoendothelial junction and reach the SMC; however, if the iron heme of hemoglobin α is reduced (Fe2+), NO will bind to hemoglobin α and this will prevent diffusion of NO in the SMC. Regulation of the oxidative state of hemoglobin α is attributed to the enzyme CYB5R3 (133). This work was a new milestone in understanding eNOS-dependent blood pressure regulation involving hemoglobin α; this new role of hemoglobin α within the vascular wall as a direct regulator of NO-mediated control of vascular tone and blood pressure is independent from the role of hemoglobin (αβ) within the red blood cell (RBC), which is known to act as scavenger, transporter, and producer of NO, (discussed in the section “NO-Mediated Luminal Heterocellular Communication: Endothelium, Platelets, and RBCs”).
Mechanisms of endothelial mechanosensing
Accumulating evidence indicates that the vascular endothelium, SMCs, and other cell types, including RBCs and platelets (113, 143), are well equipped with proteins responsible for mechanosensing and mechanotransduction, able to sense shear stress, stretch, and other mechanical stimulations.
Mechanosensors involved in eNOS activation can be found on cell surface, respectively, (i) membrane structures controlling eNOS localization (caveolae), (ii) on the luminal side of the membrane, including the glycocalyx, ion channels, and G protein-coupled receptors (GPCRs) (such as the BK-2 receptor); (iii) in junctional complexes connecting two adjacent cells, including the vascular endothelial growth factor receptor 2 (VGEFR2)/platelet endothelial cell adhesion molecule-1 (PECAM-1) complex; (iv) in focal adhesions in the subluminal side of the membrane of the cells, including integrins (such as α1β1), or (v) inside cells such as the cytoskeleton and intracellular tyrosine kinases (Fig. 2). These are responsible for cells and force specific stimulation and cellular response (Fig. 2). These pathways are studied in the emerging field of mechanobiology (6). Shear stress-dependent stimulation of these receptors leads to the activation of complex, partially interdependent downstream signaling pathways (Fig. 2). In this study, we focus on the mechanosensors, which are likely responsible for eNOS activation in the endothelium mainly by two mechanisms (i) increases in [Ca+2]i or (ii) activation of phosphorylation cascades leading to regulation of eNOS activity.
Caveolae
Caveolae are flask-shaped invaginations of the membrane with a length of 50–100 nm stabilized by the presence of caveolin-1 and play a central role in regulation of cell membrane organization and signaling (97). Sessa and coworkers demonstrated that caveolae regulate eNOS activity in response to shear stress (159). Caveolin-1 is known to directly interact with the eNOS oxygenase domain and inhibit enzyme activity. Shear stress promotes formation of Ca2+/CaM complexes, which bind to eNOS and promote eNOS dissociation from caveolin-1, and eNOS activation (85). Mice, deficient of caveolin-1, have impaired shear stress-dependent regulation of their vessel diameter (159). This impairment was completely reversed by endothelial-specific reexpression of caveolin-1 (159).
Glycocalyx
The glycocalyx is a layer of proteoglycans and glycoproteins on the luminal surface of endothelial cells (119). The glycocalyx has the ability to change its structural conformation in response to flow changes, therefore is an excellent mechanosensor. Since it is directly connected to the cytoskeleton, it enables transduction of the mechanical signal from the outside into the cytoplasm; the mechanical signal can then be redistributed through the cell, influencing other mechanosensors, including intercellular junctions, the luminal surface of cells, abluminal focal adhesion sites, and the nuclear membrane (34). An interesting publication showed that partial degradation of the endothelial cell glycocalyx leads to an impairment of NO production (132). A possible explanation for this phenomenon is that hyaluronic acid, one main constituent of the glycocalyx, is part of a cascade activating Ca2+ influx by interaction with the CD44v10/caveolin complex. Ca2+ influx may on turn activate NO production and affect cell adhesion and proliferation (132). Accordingly, it has been shown that the glycocalyx modulates the motility and proliferative response of endothelial cells (157). Since eNOS-derived NO is known to play a central role in endothelial cell proliferation, arteriogenesis, and vasculogenesis, a part of these effects may be dependent on glycocalyx-dependent eNOS activation.
Ion channels
Shear-sensitive ion channels discovered so far are potassium, chloride, and calcium-permeable channels (76, 107). With the onset of shear stress, transient receptor potential vanilloid 4 (TRPV4) channels open and allow Ca2+ influx into the endothelial cell, leading to the Ca2+/CaM-mediated activation of eNOS. By this signal, calcium-activated potassium channels (KCa) open and release K+ leading to cell membrane hyperpolarization. Likewise, hyperpolarization is increased by activation of shear-sensitive inward-rectifying potassium channels (Kir) (106). Hyperpolarization of the membrane activates hyperpolarization-sensitive Ca2+ channels and Cl− ion channels in a time-dependent manner and results in membrane potential depolarization occurring 35–160 s after onset of shear stress (95, 114).
The mechanism on how blood flow activates these ion channels is yet not fully understood. Three different theories were recently proposed (83). One possible explanation is that ion channels are pushed open due to the drag force. Second, mechanical forces may change the tensional interactions among cytoskeletal proteins (see also the section “the Cytoskeleton”) pulling the channels anchored to the cytoskeleton to open. Third, mechanical forces may affect membrane fluidity by influencing the viscosity of the lipid bilayer. Most likely, it is a combination of these mechanisms that activates ion channels in response to shear stress. An indirect regulation of eNOS activation by mechanosensing channels may also involve shear-dependent activation of ATP release from endothelial cells, leading to activation of the purinergic P2X4 channel (154). P2X4 triggers Ca2+ influx, increases in intracellular Ca2+ concentration, and activates eNOS. The mechanism responsible for ATP release is still unclear (99).
G protein-coupled receptors
Activation of GPCRs by shear stress was demonstrated for the first time by liposomes carrying isolated GPCRs (64). BK is one of the oldest agonists known to regulate eNOS activity in the endothelium. The classical pathway is characterized by binding of BK to its receptors, activation of protein kinase C, and increase in [Ca2+]i in endothelial cells (20). It was shown that shear stress-dependent activation of BK 2 receptor (B2) results in changes of eNOS activity independent from the presence of BK (84). Under physiological unstressed conditions, the B2 receptor and eNOS form a complex, hindering NO synthesis by eNOS (84). Activation of the B2 receptor leads to dissociation of eNOS from the endothelial B2 receptor, making eNOS available for NO synthesis.
A further GPCR activated by shear stress is the purinergic receptor P2Y2. After activation, this Gq/G11-linked receptor initiates a signal cascade involving the phosphorylation of PECAM-1, VEGFR2, Src-kinase, and Akt, leading to phosphorylation of eNOS at Ser-1177 (154).
Several possible mechanisms of GPCR-mediated shear-induced changes of membrane bilayer are discussed (20). Simulations of the lipid bilayer suggest that changes in intrabilayer pressures may activate membrane proteins (65). One further mechanism proposed is the change in bilayer thickness and thus improved stabilization of the activated receptor conformation (92).
Junctional complex proteins (VEGFR2/PECAM-1/VE-cadherin)
A mechanosensitive complex consisting of VEGFR2, PECAM-1, and the adaptor protein VE-cadherin forms junctional complexes between two adjacent cells. These have multiple functional and regulatory properties, playing a role in endothelial permeability and structure (21). The VEGFR2 has been shown to be activated by shear stress independently of the presence of its ligand VEGF (81) and leads to activation of phosphoinositide 3-kinase (PI3K), the serine-specific protein kinase Akt, which in turn causes phosphorylation of human eNOS at serine 1177 (40, 81). PECAM-1 has no mechanosensing properties, but plays an important role in the delayed Ca2+-independent eNOS activation in response to shear, which seems to involve Akt-dependent phosphorylation of eNOS by serine 1177, as demonstrated in PECAM-1-deficient mice (51, 139).
Focal adhesions and integrins
Integrins are transmembrane proteins regulating cell–cell and cell–matrix interactions and have been shown to influence endothelial response to shear stress in many studies (127). The α1β1 integrin binds to collagen and laminin and was involved in shear-dependent regulation of eNOS activity in resistance vessels and the microcirculation via activation of PI3-kinase-Akt eNOS signaling (100). Mice lacking the α1 integrin gene showed an impairment in flow-mediated dilation (FMD) of mesenteric resistance arteries (100). A study has shown that α1β1 integrin is absent in endothelial cells of conduit arteries (37).
Cytoskeleton
In mechanobiology, the cytoskeleton is considered as a network capable of transmitting forces throughout the whole cell and simultaneously adapts to mechanical forces (77). According to the tensegrity model, the cytoskeleton comprises elements resistant to compression and not directly attached to each other, as well as elements under steady tension (77). In response to shear stress, the cytoskeleton reorganizes its structure by clustering cytoskeletal and membrane molecules directly connected to it, such as cadherins and integrins. These proteins transmit mechanical forces throughout the cell from one mechanosensing locus to the other, leading to structural rearrangements of cytoskeleton-associated proteins (e.g., resulting in their activation) as well as functional redistribution of cell organelles (145).
Cytoplasmic tyrosine kinases
Two cytoplasmic tyrosine kinases have been shown to participate in shear stress-induced mechanosensing and regulation of eNOS activity, the c-Src kinase (82), and the focal adhesion protein proline-rich tyrosine kinase 2 (Pyk2) (48, 51). The Src-kinase increases eNOS transcription via activation of RAS, MEK1/2, and ERK1/2 pathway, as well as mRNA stability by an unknown mechanism (36). Fleming and coworkers have shown that activation of Pyk2 at Tyr 657 in endothelial cells exposed to shear flow results in the decrease or complete loss of eNOS enzymatic activity (35, 48, 51). The mechanisms of shear stress-induced Pyk2 activation remain controversial, but several potential mechanisms, including integrin stimulation (50), Ca2+ influx without integrin activation (158), and shear-dependent increase in vascular oxidative stress (136), have been suggested. The negative regulation of eNOS activity may not only assume a fundamental role for fine-tuning eNOS activity in healthy vessels, but possibly also participate in the pathophysiology of endothelial dysfunction and atherosclerosis (35).
Summary and outlook: mechanosensing and transduction in the vascular wall
Taken together, understanding how mechanosensing and mechanotransduction in the endothelium regulate heterocellular signaling in the vascular wall and blood is a challenging task. The complex crosstalk between the different mechanotransduction and cell–cell communication pathways contributes to adapt and fine-tune the physiological responses and keep the human vasculature in a well-balanced state. A systematical analysis of these pathways will help to identify the common control nodes regulating short-term and long-term regulatory mechanisms in the vasculature induced by mechanical forces.
NO-Mediated Luminal Heterocellular Communication: Endothelium, Platelets, and RBCs
NO-mediated heterocellular communication among endothelium and blood cells plays a central role in maintenance of cardiovascular homeostasis. NO produced by the endothelium can be released into the lumen of the vessels and reach the blood stream, where NO can interact with plasma proteins and cellular blood components, including platelets and RBCs. As a result, NO can participate in short-distance heterocellular communication; as an example, NO produced by the endothelium may activate sGC in platelets and thereby inhibit platelet aggregation (Fig. 3).
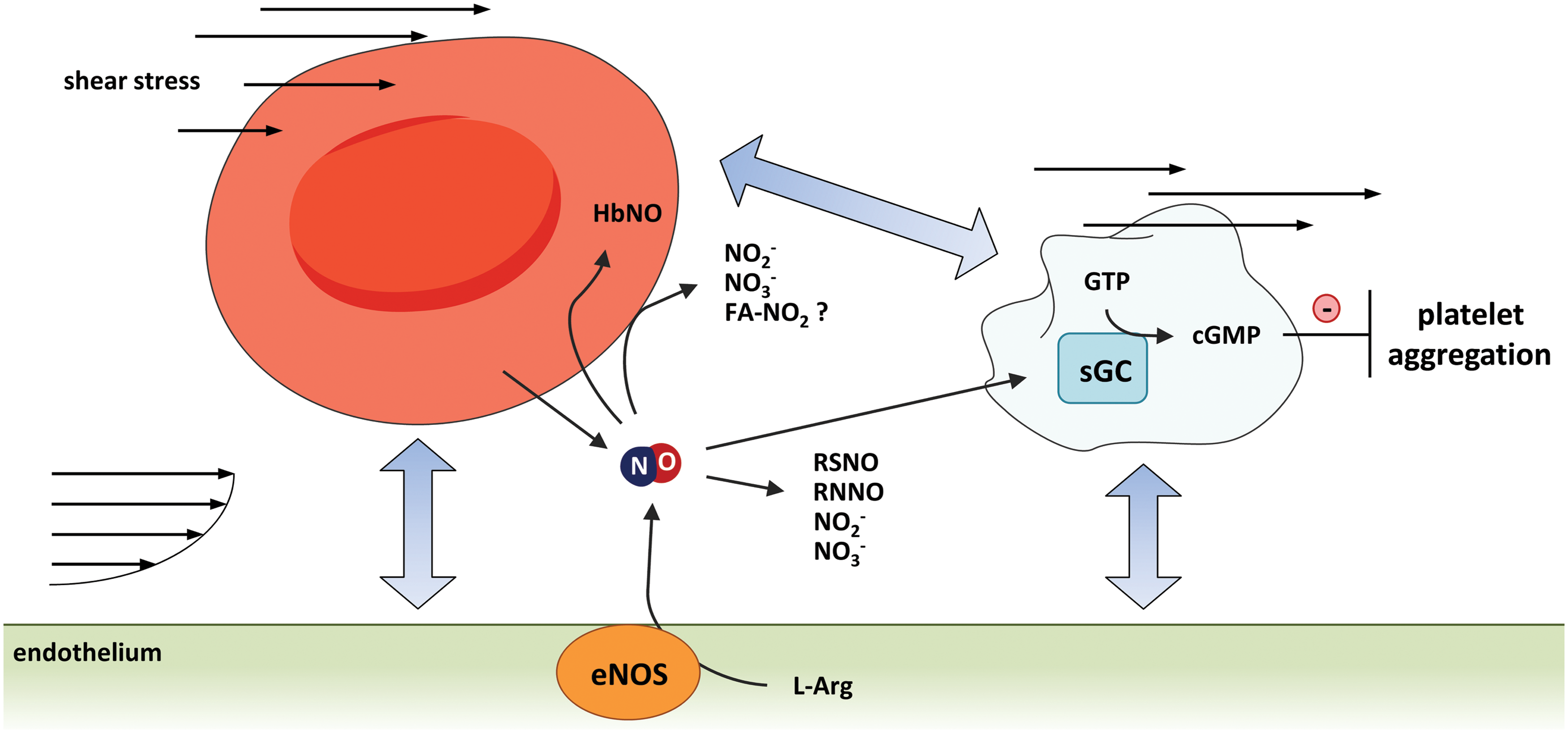
In RBCs, plasma, or tissues, NO can be transformed into bioactive metabolites allowing long-distance heterocellular communication. RBCs and platelets are both mechanosensing cells (113, 144), and changes in flow conditions strongly affect heterocellular communication among them. Shear stress-mediated RBC deformation was shown to cause ATP release (143). Similar to shear-dependent ATP released from endothelial cells (described in the section “Ion Channels”), ATP released from RBCs may bind to purinergic receptors on the surface of the endothelium and activate eNOS to produce NO (99).
In this paragraph, we will focus on shear-induced NO-mediated heterocellular communication in the vascular lumen, particularly (i) on NO-dependent effects on platelet aggregation and hemostasis; (ii) on the role of RBCs in NO metabolism, transport, and production; and (iii) on mechanosensing and mechanotransduction in RBCs and the effects of endogenous and exogenous NO on their mechanoproperties, including RBC deformability.
Platelets, shear, and NO
Very early, it was observed that endothelium-derived NO inhibits platelet aggregation via activation of the sGC/cGMP/PKG pathway in platelets (142). Interestingly, platelets carry high levels of sGC, and are characterized by the highest NO-stimulated cGMP-forming activity (14) among cells belonging to the cardiovascular system, including vascular cells, cardiomyocytes, and blood cells. The constitutively shear-induced eNOS-derived NO production by the endothelium inhibits platelet aggregation and thereby contributes to the regulation of hemostasis in blood (56).
NO was also shown to inhibit platelet recruitment (142). Recently, a biphasic response to NO in platelets, consisting of a transient stimulatory response inducing platelet aggregation, followed by an inhibitory pathway limiting the size of thrombus formation, was found (13, 160). This work was criticized by others, who never found any proaggregation effects of NO (55); instead, these authors presented compelling arguments demonstrating that the NO/sGC/cGMP/PKG pathway inhibits and does not enhance platelet aggregation (33, 122).
The role of shear stress-mediated eNOS activation in hemostasis and inhibition of thrombus formation was confirmed by observations linking low endothelial shear stress with downregulation of eNOS and prostacyclin, and increased thrombogenicity, or by analyzing atherothrombotic events induced after endothelial injury (22). Similar results were shown in ex vivo studies where platelet aggregation was studied in the presence of cultured endothelial cell monolayers in flow chambers (mainly using human umbilical vein endothelial cells) (130).
NO released by the endothelium in response to shear stress is only one of the mechanotransducers involved in heterocellular signaling among platelets, endothelial cells, RBCs, leukocytes, other blood cells, or microparticles. Platelets are themselves mechanosensing cells, able to feel their mechanical microenvironment and dynamically respond to it (113). Aggregation, disaggregation, adhesion, and recruitment of platelets are strongly affected by both the nature of mechanical forces acting on them (i.e., the flow conditions) and by the presence/absence of adhesion molecules and/or soluble biochemical messengers in blood, released by endothelial cells and by blood cells (94, 130).
RBCs and control of NO metabolism, transport, and production
The role of RBCs in NO metabolism is complex and highly debated. RBCs may participate in luminal heterocellular communication by scavenging, transporting, and releasing NO or its metabolites, as described in the following paragraphs.
NO scavenging by RBCs
In the blood stream, endothelial NO is taken up and inactivated by RBCs by an oxidation reaction catalyzed by oxyhemoglobin (oxyHb or FeIIO2Hb) producing methemoglobin (metHb or FeIIIHb) and nitrate according to the following reaction (Eq. 1) (96):
This reaction is very fast (≈107 M −1s−1) (41, 96) and converts NO into nitrate, which was thought to be biologically inert. b Therefore, this reaction was considered to be responsible for inactivation of endothelial NO signaling (28), and RBCs were considered as the major sink for NO in the circulation. Ex vivo experiments could show that the half-life of NO in plasma alone is about 1.5–6 min (98) and that the addition of RBCs decreases this time to 1.8 ms, which is 1000 times slower than the direct reaction of NO with free hemoglobin. These differences were explained by considering the behavior of RBCs in the flow and observing that particularly in conductance artery the RBCs accumulate in the center of the vessel and create an unstirred layer of plasma, which decrease the diffusion of NO (39).
NO production by RBCs: hypoxic conditions
The observation that under hypoxic conditions, coincubation of RBCs with vascular stripes induced vasodilation (a phenomenon defined as hypoxic vasodilation), which appeared to be dependent on release of NO/NO metabolites from RBCs (109), induced a radical change of view in the field. RBCs were proposed to convert endothelium-derived NO into bioactive metabolites and to release NO on demand under hypoxic conditions when the eNOS activity is impaired. The bioactive NO metabolite acting as NO source in RBCs was proposed to be s-nitrosohemoglobin (HbSNO), produced within the RBCs by the binding of NO to the highly conserved β-chain Cys-93 residue (80). However, the observation that very low levels of s-nitrosothiols are carried by rodent RBCs (146), and even nondetectable levels in human RBCs (116), induced a radical rethink of the proposed mechanism. Later on, it was shown that RBCs lacking the hemoglobin Cys-93 amino acid did not negatively affect hypoxic vasodilation (78); however, some evidence of the role of Cys-93 was presented recently (161). More research is needed to define the role of Cys-93 in hypoxic vasodilation.
Interestingly, it was observed that RBCs carry a bulk of nitrite (38) and that there is a significant circulating arterial-venous nitrite gradient (61). Therefore nitrite, and not S-nitrosohemoglobin, was proposed to be the substrate of nonenzymatic NO synthesis in RBCs, which is converted into NO by a reaction catalyzed by deoxyhemoglobin (38, 61, 116, 146). Iron-nitrosylhemoglobin was also proposed to be responsible for NO bioactivity in RBCs (74). Further mechanisms of nitrite bioactivation were proposed to be responsible for the conversion of nitrite into NO. It was proposed that nitrite reduction into NO can be catalyzed by carbonic anhydrase (1) or by xanthine oxidase carried by RBCs and other tissues (58, 148) and even by eNOS (148). Recent experiments carried out by electron paramagnetic resonance showed that the activity of deoxyhemoglobin is essential for nitrite-derived NO release from RBCs under hypoxic conditions (96) as treatment of RBCs with carbon monoxide completely blocked nitrite reduction, while inhibition of xanthine oxidase, carbonic anhydrase, or NOS did not show any effects (96).
Nitrite-derived NO production participates in heterocellular communication in blood. Thus, studies have shown that nitrite bioactivation by RBCs inhibits platelet activation/aggregation, while nitrite alone has no effects (96, 108). These data show that nitrite-derived NO production may participate in heterocellular signaling in the blood stream. How mechanical forces influence scavenging or production of NO under hypoxic conditions by RBCs and the heterocellular signaling needs to be further elucidated.
Red cell eNOS-dependent NO production: normoxic conditions
Data from our and other laboratories have demonstrated eNOS-dependent NO production from RBCs in normoxia, suggesting that RBCs may contribute to inhibition of platelet aggregation (88), the circulating pool of NO metabolites (88), and thus to overall tissue protection (155). Treating RBCs with eNOS inhibitors decreased accumulation of NO metabolites (75, 88) and L-citrulline in the supernatant (88, 155). By applying a multilevel analytical approach comprising HPLC, LC-MS/MS, flow cytometry, laser scanning microscopy, and enzymatic assays together with functional studies, we have recently demonstrated that human RBCs contain an active eNOS building NO formation under normoxic conditions (29, 30). To biochemically characterize the eNOS expressed in RBCs, we established an immunoprecipitation protocol by using magnetic beads and a purified mouse anti-human eNOS antibody, which was chemically cross-linked to the beads. By applying this protocol, we were able to isolate eNOS from human (30) and mouse RBCs (153). The identity of human red cell eNOS was confirmed by ESI-MS/MS (30). The activity of the protein was assessed by measuring the conversion of 3H- or 14C-arginine to 3H- or 14C-citrulline catalyzed by the immunoprecipitated eNOS (30) and membrane preparations of RBCs (153). We found that the isolated eNOS protein was Ca2+/CaM dependent (30), could be inhibited in the presence of NOS inhibitors (L-NAME, L-NIO) (30), and its activity was absent in RBC membrane preparations from eNOS knockout (KO) mice (153).
NO was proposed to regulate RBC deformability (15). NOS inhibitors were shown to decrease RBC deformability in ektacytometry (15), while low concentrations of NO donors increase RBC deformability (15), membrane fluidity (138), and RBC filterability (88). However, the role of NO in human RBC deformability is controversial (12). In the microcirculation of the chorioallantoic membrane of the chicken egg, eNOS inhibition and NO donors were shown to affect RBC deformation and velocity independently of changes of the vascular diameter (75). The role of red cell eNOS in RBC signaling is still unknown.
RBCs as NO producers: how does NO survive the reaction with hemoglobin in RBCs?
According to the findings reviewed in the previous paragraphs, there is compelling evidence that RBCs produce NO under both hypoxic and normoxic conditions. However, taking into account the chemical biology of the reactions between NO and hemoglobin, one may conclude that any NO produced within an RBC will react with hemoglobin under any physiological condition under consideration, that is, with oxyhemoglobin to form nitrate and methemoglobin (Eq. 1), as well as with deoxyhemoglobin to form nitrosylhemoglobin (Eq. 2) (73):
Thus, the main open question still to be answered is how NO produced within the RBC may survive the reaction with hemoglobin. It was proposed that compartmentalization of NO production in RBCs by formation of protein complexes on the RBC membrane (e.g., deoxyHb, AE1/band 3, and Rh-protein channels) facilitates NO production under hypoxic conditions and its export (60). Recently, it was shown that deoxyhemoglobin forms a complex with band 3 on the membrane of RBCs and that the stability of the complex depends on the oxygenation state of hemoglobin (124). Another possibility proposed by us is the presence of specific protein targets within the RBCs, leading to activation of downstream signaling and or protection of NO by local conversion of hemoglobin into methemoglobin (28). More research is needed to understand how NO may survive scavenging by hemoglobin as well as to understand the role of NO production in RBC signaling and heterocellular communication.
Mechanosensing and mechanotransduction in RBCs
RBCs are mechanosensing cells that respond to mechanical forces by changing their shape. RBC deformation is a complex dynamic process mediated by aggregation and disaggregation of interactions among cytoskeletal and membrane proteins (52, 143, 144). RBC deformability c is required for an efficient delivery of oxygen and nutrients to the tissues (19) and contributes to define the rheological properties of blood (143). RBC deformability may modulate the viscosity of blood and importantly allows the cells to dynamically participate in the flow by adapting their shape to the different flow conditions found in conductance and resistance vessels and in the microcirculation (143). Moreover, RBCs need to deform to be able to enter and transit the narrowest capillaries of the microcirculation, which may be even narrower than their own diameter (143). Capillary blood flow and RBC velocity in the microcirculation strongly depend on vascular tone and RBC deformability (143).
It was shown that in response to shear stress, RBCs release ATP, which may act as a mechanotransducer and bind to the purinergic receptors on the endothelium, activating eNOS-derived NO formation and heterocellular signaling leading to vasorelaxation and increased blood flow (43, 99, 143). Recently, Stone and coworkers applied a microfluidic approach to study the time-dependent dynamics of deformation-induced ATP release from RBCs (144) and demonstrated a link between shear stress-mediated mechanical deformation of RBCs and release of ATP (52, 144). Several key elements in the mechanotransduction pathways of ATP release from RBCs, including GPCR and hemichannels, such as the ubiquitous ATP-releasing channel pannexin-1, have been proposed (144), but not investigated in detail. Recently, it was found that shear stress activates PIEZO-1 cation channel inducing Ca2+ influx and ATP release from RBCs (26). Noteworthy, the absence of Ca2+ in the incubation medium did not fully block ATP release from RBCs, indicating that Ca2+-independent mechanotransduction pathways are present in RBCs (26). A methodological article by Sikora et al. proposed that the experimental conditions described in the literature to induce ATP release caused hemolysis, and therefore hemolysis and not the activation of signaling pathways in RBCs (including mechanosensing and mechanotransduction) was causing the increase in ATP in the supernant of RBC suspensions (131). This article started a lively discussion (87). In our opinion, the conclusions of this article are not fully justified by the results provided because of some methodological issues, including the use of a vortex for inducing shear stress on RBCs. As pointed out by many authors in the field (26, 143), controlling for hemolysis and effects of vehicle in drug application is fundamental before driving conclusions about signaling in RBCs. Indeed, research with RBCs is not without its pitfalls as we already discussed in a recent review (28).
Considering the central role of eNOS activation in mechanotransduction within the endothelium, it is tempting to speculate that red cell eNOS may participate in mechanotransduction in RBCs, inducing intracellular signaling and NO-mediated regulation.
Summary and outlook: mechanical forces and luminal heterocellular communication
The role of mechanical forces in NO scavenging, transport, and synthesis in RBCs and their interaction with platelets and endothelium is not understood yet. The rheological behavior of RBCs in the flow of conduit vessels (where RBCs accumulate in the center of the vessel leaving a cell-free zone near the endothelium) was proposed as one of the mechanisms for limiting endothelial NO scavenging by RBCs allowing NO to control vascular tone and blood pressure. Mechanical forces and flow conditions are known to regulate aggregability and activation of platelets. Similarly, the action of mechanical forces on the cytoskeleton of RBCs may regulate their deformation, e.g. via modulation of aggregation/disaggregation of protein complexes regulating deformability, as well as erythrocrine function, including synthesis, signaling, or export of NO from RBCs or ATP release. Therefore, investigations on the role of mechanical forces on RBC-mediated NO production may be of fundamental importance to fully understand the role of RBCs in NO metabolism and transport under flow conditions.
Systemic Heterocellular Communication Induced by Physical Forces
In this paragraph, we will describe how increase of vascular shear may modulate systemic heterocellular signaling, leading to improvement of cardiovascular function, and will highlight the role of endothelial NO in initiation and induction of these favorable systemic changes. The effects of exercise training and remote ischemic preconditioning are described as examples of complex systemic physiological responses to changes in shear (Figs. 4 and 5).
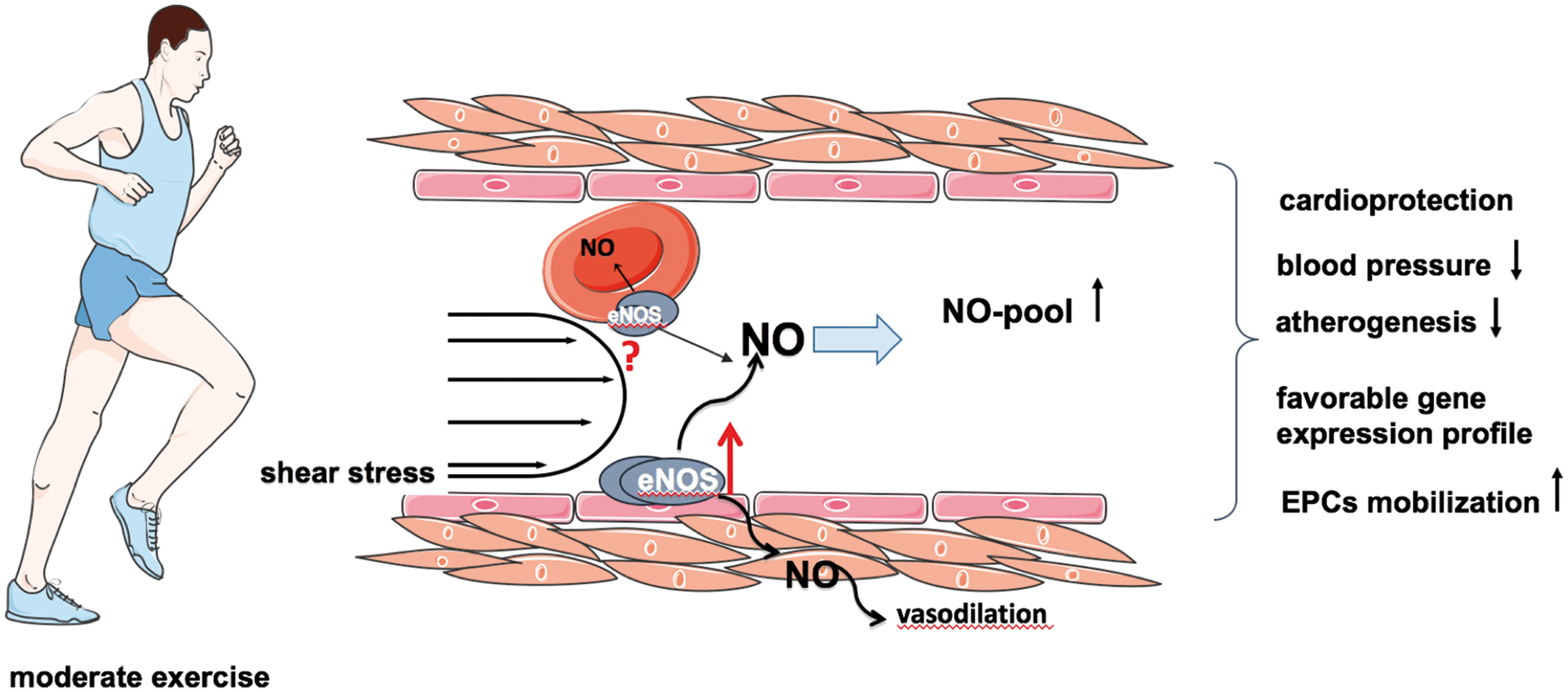
Role of eNOS in vascular adaptation to exercise
Moderate exercise training has proven to be efficient to prevent cardiovascular disease conditions in a multitude of observations and studies (126). Regular physical activity promotes a variety of favorable changes and adaptations of cardiovascular function, including reduction of blood pressure (16) and inhibition of atherogenesis (111). Exercise training acutely increases heart rate and blood flow, thus changing frequency and magnitude of hemodynamic forces (Fig. 4). During exercise, vascular cells are simultaneously exposed to very heterogeneous spatial and temporal stresses, such as shear stress, cyclic strain, and transmural pressure. In the long-term view, regular exercise training results in alterations of endothelial gene expression pattern, that is, upregulation of atheroprotective and downregulation of potentially atherogenic genes (147), which likely lead to protection of vascular cells against apoptosis, inflammation, and oxidative stress.
Exercise-induced eNOS upregulation and increased endothelial-dependent vasodilation were first observed in dog coronary arteries (54) and then confirmed in many other studies (59, 66, 135). Likewise, experiments conducted in different animal models and humans revealed that exercise activates a complex pattern of intracellular eNOS regulatory systems, including changes in phosphorylation state, and intracellular localization (59). Furthermore, beneficial effects of training on vascular endothelial function attributed to the increased vascular NO bioavailability and upregulation of eNOS were reported in patients with coronary artery disease (66) and chronic heart failure (151). Recent studies demonstrated that high-density lipoprotein (HDL) can modulate the phosphorylation of eNOS and thereby its activity; interestingly, HDL-mediated eNOS activation was significantly impaired in cardiovascular disease patients, but can be restored by regular exercise (2). Therefore, mild to moderate exercise training may be beneficial even in conditions previously considered as contraindications (e.g., heart failure after coronary bypass grafting).
The effects of exercise training on eNOS expression and phosphorylation profile are influenced by a number of factors such as training time, exercise intensity, basal level of physical activity, and eNOS gene polymorphisms (89). Furthermore, since blood flow and hemodynamic shear forces strongly depend on the localization and morphology of the vessels along the vascular tree (22), the effect of exercise on vascular eNOS may differ in various regions (35). These effects are particularly evident for regions near arterial branches and curves, which are characterized by disturbed flow conditions (22). In contrast to the laminar flow in large conductance arteries, oscillatory shear can reduce eNOS expression and thus elicit a proinflammatory endothelial phenotype (35, 89). In fact, these regions are known to be more prone to development of atherosclerosis (22, 25). The impact of exercise training on vascular responsiveness and endothelial function in different regions of the circulation needs to be investigated in more detail.
Numerous studies provided evidence for exercise-induced mobilization of endothelial progenitor cells (EPCs) from the bone marrow to the circulation (93) and thus its contribution to the vascular regeneration and angiogenesis (9). The mechanisms of EPC mobilization from the bone marrow are highly complex (4) and depend on the activation of eNOS in the presence of numerous mobilizing factors (e.g., VEGF, placental growth factor). Experimental studies have shown that mobilization of stem and progenitor cells in vivo from the bone marrow stromal cells is substantially eNOS dependent (3) and due to activation of matrix metalloproteinases 2 and 9 (79). Interestingly, exercise-induced EPC mobilization is abolished in the eNOS-deficient mouse strain (91).
Remote ischemic preconditioning
Already in 1986, Murry at al. observed that repetitive brief periods of occlusion of a coronary artery before cardiac ischemia can reduce the infarct sizes in a canine model, a phenomenon defined as ischemic preconditioning (105). Przyklenk et al. found that the effects of ischemia were cardioprotective, even if carried out in another area of the heart (112). Further studies demonstrated that a preconditioning maneuver is cardioprotective even if performed in noncardiac tissues (57). It was proposed that brief reversible episodes of ischemia and reperfusion may result in release of a protective signal from the site of ischemia and result in cardioprotection (67). Proposed mechanisms include neuronal transmission (152) [although criticized by others (90)] as well as may involve humoral mediators such as adenosine, BK, opioids, angiotensin I, and endocannabinoids (68). Rassaf et al. observed that nitrite levels in plasma are increased as a result of the preconditioning maneuver and that preconditioning is ineffective in eNOS KO mice (117). They have suggested that preconditioning-activated eNOS-derived formation of NO in the endothelium (probably via changes in shear stress) leads to increase of circulating nitrite levels and cardioprotection via myoglobin-dependent conversion of nitrite into NO and nitrosation of complex 1 (117). These studies demonstrate that shear stress-induced NO production within the endothelium may result in local heterocellular communication within the vasculature or to the blood cells upon biotransformation of NO into stable bioactive metabolites transported within the plasma or RBCs.
Summary and outlook: systemic signaling induced local and hemodynamic forces
Taken together, there is evidence that changing frequency or magnitude of hemodynamic forces by systemic changes (such as in exercise training) or locally (such as by temporary arterial occlusion in remote ischemic preconditioning) may induce short-term and long-term changes in vascular NO production and lead to protection of heart and vessels, as well as other tissues (Figs. 1, 4, and 5).
Critical Issues for Experimental Studies
Mechanical forces affect a number of different cell types in the human body. Our understanding of the complex heterocellular communication induced by mechanical forces in the organism has evolved from investigations using in vitro, ex vivo, and in vivo methods. Each of these approaches can provide only partial information about the complex and intertwined effects of mechanical forces and the induced local and systemic heterocellular signaling in the organism.
The cellular effects of physical forces were mainly studied usually by applying nonpulsatile shear stress and by analyzing the behavior of endothelial cell monolayers in culture in the absence or in the presence of blood cells in microfluidic devices (140). These in vitro experiments revealed, for example, that the laminar flow modulates endothelial cell morphology, adhesion molecules, and gene expression patterns, secretion of extracellular matrix proteins, and cell–cell and cell–matrix adhesions and orchestrates collective behaviors of adherent cells (10). Similar devices were also applied for studying platelet aggregation in response to vascular injury involving exposure of the extracellular matrix found below the endothelium (113, 130).
Microfluidic technology enables studies of cell behavior from single- to multicellular organism levels. The true potential of the use of microfluidics has emerged recently with the advent of hydrogel systems, offering increased throughput, multicellular interactions, substrate functionalization on three-dimensional (3D) geometries, and simultaneous control of chemical and mechanical stimulation (140). Applications of 3D cell culture systems for analysis of cell–cell interactions under flow conditions may provide a proper environment to study 3D cell–cell interactions and heterocellular communication in response to mechanical forces.
The isolated/perfused Langendorff heart is traditionally applied to analyze coronary function and changes in coronary flow in response to brief ischemia/reperfusion periods or to pharmacological stimuli. This technique was also applied to analyze the effects of RBCs on tissue oxygenation conditions, coronary vascular response, and myocardial performance and oxygen consumption (24). Likewise, endothelium-dependent and -independent coronary flow responses were increased in RBC-perfused hearts (24). Recently, the cardioprotective effects of red cell eNOS (via activation of arginase-1) toward ischemia were demonstrated by application of this method (156). Foaming, hemolysis, and isolation of the heart, as well as stability of the preparation, make this technique technically demanding. Therefore, it is necessary to integrate this set of heterogeneous information to improve our understanding of these mechanisms.
In vivo investigations in animal models are the best available tools for evaluation of systemic hemodynamics and blood cells/luminal components in shear stress-induced heterocellular communication. Modulation of systemic heterocellular communication can be studied by combining genetic approaches (e.g., eNOS KO mice) with physiological stimuli, which are known to increase shear/hemodynamic forces, for example, forced and voluntary exercise (134) and remote ischemic preconditioning (117). To untangle the role of vascular and blood components responsible for eNOS-mediated heterocellular signaling, we created chimera mice by bone marrow transplantation (103, 153). Applying this approach, we found that blood cell eNOS participates in regulation of blood pressure and cardioprotection. Although useful, this approach presents some methodological limitations, including irradiation-dependent activation of inflammatory pathways (which may increase the expression of an inducible NOS), the possibility of protein transfer from the blood to the endothelium (due to housing of circulating EPCs), the presence of low levels of circulating blood cells from the recipient, and—most importantly—the lack of blood cell lineage targeting specificity.
Development of inducible and conditional transgenic mice with tissue-specific target expression or ablation of genes important for regulation of NO bioavailability and mechanotransduction may overcome these limitations.
Taking together, investigations in tissue-specific transgenic mice applying a wide range of in vitro, ex vivo, and in vivo approaches represent a future strategy to unravel the complexity of NO-mediated, shear-induced heterocellular signaling and will help to identify new potential molecular targets and strategies for therapeutical, pharmacological, and nonpharmacological interventions aiming to reduce cardiovascular disease.
Clinical Aspects: Mechanotransduction in Endothelial Dysfunction
Alterations in endothelial function and the functional integrity of the endothelium are associated with a variety of pathological conditions, including hypertension, diabetes mellitus, hypercholesterolemia, and heart failure (49). Likewise, primary and secondary risk factors such as age, smoking, or hypercholesterolemia have been correlated with a decrease in endothelial function (Fig. 6 and Supplementary Table S1).
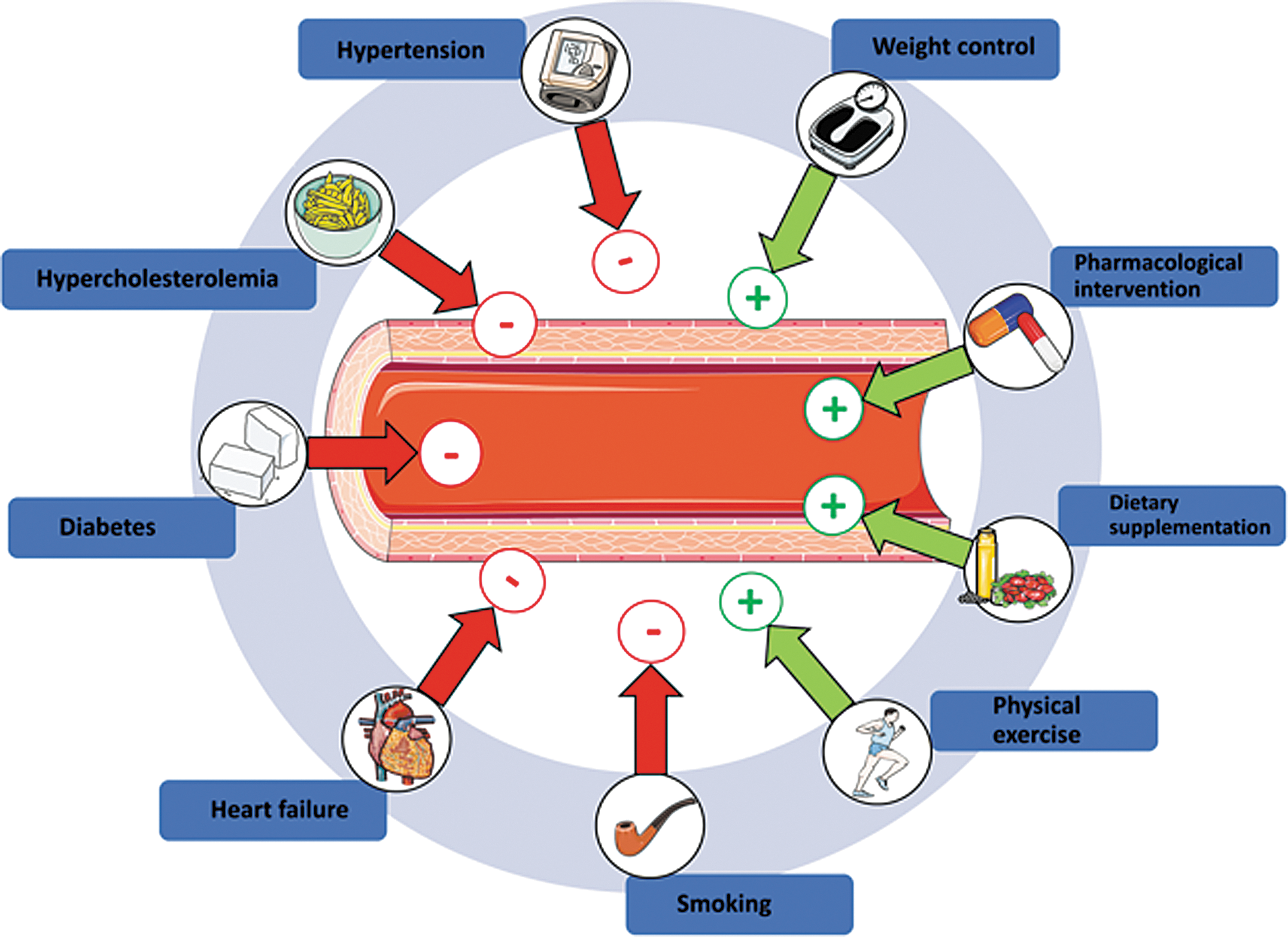
Measurement of endothelial function allows assessment of the patients' risk of cardiovascular events, progression of atherosclerosis-related conditions, efficacy of lifestyle changes/pharmacological intervention, and patient stratification (Fig. 6 and Supplementary Table S1). Interestingly, shear-induced eNOS-dependent activation of vascular heterocellular communication constitutes the molecular basis of all methods applied in the clinical routine for evaluation of endothelial function. Assessment of FMD of the brachial artery by high-resolution ultrasound is one of the most widely applied methods for noninvasive evaluation of endothelial function in clinical setting (72). Analysis of changes in reactive hypermemic response by laser Doppler perfusion imaging was proposed for evaluation of microcirculatory function (86). Clinical studies based on administration of specific NOS inhibitors have shown that the FMD response is strongly dependent on eNOS activity (72). Indeed, FMD correlates with levels of circulating nitrosospecies in plasma (70), and mice lacking eNOS show no FMD response (44, 125). Interestingly, the same experimental setting allowed showing that low flow conditions (achieved during occlusion) induce eNOS-independent vasoconstriction, as demonstrated in man (62) and in mice (44). These data provide a further experimental proof of responsiveness and sensitivity of the endothelium to different acute changes of flow conditions and highlight the fundamental role played by eNOS in physiological and pathophysiological shear-induced heterocellular signaling in the human vasculature.
According to the guidelines of the American Heart Association (42) and the European Society of Cardiology (110), the most effective treatments preventing the development or progression of endothelial dysfunction are lifestyle changes, including a low-calorie and low-fat diet, as well as regular exercise, reduction of alcohol consumption, and smoking cessation. These strategies, together with pharmacological interventions aiming to reduce blood pressure or/and hypercholesterolemia, have been shown to decrease morbidity and mortality of cardiovascular disease (121). Understanding how mechanotransduction and changes in NO bioavailability contribute to local and systemic heterocellular communication will help in identifying novel diagnostic and therapeutic targets for prevention of cardiovascular disease.
Summary and Future Directions
NO produced by the vascular endothelium is a key mechanotransducer for local and systemic heterocellular communication. The endothelium is well equipped with different mechanosensors found on the luminal side of the membrane (such as glycocalyx, ion channels, and GPCRs), in the junctional structures responsible for cell–cell interactions (such as the VEGFR2/PECAM-1/VE-cadherin complex), in focal adhesion points (such as integrins), or inside cells (such as the cytoskeleton or tyrosine kinases such as Pyk2). Mechanosensors are activated by local changes in blood flow and elicit transformation of flow-induced mechanical stimulation into biochemical signals, which are then transduced within the cells and across cells, finally leading to activation of local and systemic responses.
Laminar shear stress is now considered as one of the most important physiological stimuli for activation of eNOS within the endothelium and it is responsible for NO-mediated local and systemic heterocellular communication leading to regulation of vascular tone, blood pressure, and blood hemostasis. Interestingly, not only vascular cells but also RBCs and platelets are capable of sensing mechanical forces and inducing specific signaling mechanisms to modulate heterocellular communication in the blood stream and in the vasculature. NO produced by the endothelium in response to mechanical forces can be metabolized in RBCs, plasma, and other tissues and can be transported in the form of different metabolites in the blood stream; in turn, bioactive NO metabolites allow shear-dependent systemic heterocellular signaling (Fig. 1).
Exercise training and remote ischemic preconditioning represent two beneficial examples of effective systemic NO-mediated heterocellular communication in response to physiological shear. Systemic shear-induced NO-mediated communication between the endothelium and other cells is highly complex and new experimental approaches are needed to differentiate the specific role of each cell population in modulation of systemic cell–cell communication and signaling. Under pathological conditions, changes in mechanical properties of the vessel endothelium may limit NO production/bioavailability affecting systemic hemodynamics, vascular tone, and overall cardiovascular homeostasis.
Future studies are needed to unravel mechanisms underpinning changes in heterocellular communications among endothelium and other cell types in response to mechanical forces. These studies will help to identify novel therapeutic strategies, including pharmacological and nonpharmacological interventions, to modify mechanosensing and mechanotransduction-dependent heterocellular signaling in health and disease.
Footnotes
Acknowledgments
The authors are grateful to the German Research Council (DFG CO 1305/2-1 to M.M.C.-K., SFB1116 TP B06 to M.M.C.-K. and M.K., and IRTG1902 to M.K. and M.M.C.-K.), to the Forschungskommission of the Universitätsklinikum Düsseldorf (to M.M.C.-K. and to T.S.), and the Susanne-Bunnenberg-Stiftung of the Düsseldorf Heart Center (to M.K.) for financial support.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.