
Abstract
Significance:
Gastrointestinal (GI) cancer taken together constitutes one of the most common cancers worldwide with a broad range of etiological mechanisms. In this review, we have examined the impact of nitric oxide (NO) on the etiology of colon, colorectal, gastric, esophageal, and liver cancers.
Recent Advances:
Despite differences in etiology, initiation, and progression, chronic inflammation has been shown to be a common element within these cancers showing interactions of numerous pathways. NO generated at the inflammatory site contributes to the initiation and progression of disease. The amount of NO generated, time, and site vary and are an important determinant of the biological effects initiated. Among the nitric oxide synthase enzymes, the inducible isoform has the most diverse range, participating in numerous carcinogenic processes. There is emerging evidence showing that inducible nitric oxide synthase (NOS2) plays a central role in the process of tumor initiation and/or development.
Critical Issues:
Redox inflammation through NOS2 and cyclooxygenase-2 participates in driving the mechanisms of initiation and progression in GI cancers.
Future Directions:
Understanding the underlying mechanism involved in NOS2 activation can provide new insights into important prevention and treatment strategies. Antioxid. Redox Signal. 26, 1059–1077.
Introduction
G
The relationship between GI cancer and nitric oxide (NO) and reactive nitrogen species (RNS) was first proposed in the 1970's (104, 128, 147). These studies postulated that nitrite (
NO is a gaseous-free radical permeable to biological membranes, which works as an intercellular and intracellular messenger playing key roles in a range of processes, including cell metabolism (28), cell death (188), and cell survival (120). The pleiotropic effects of NO in biological systems are also due to its reactivity with different cellular molecules, such as molecular oxygen (O2), superoxide radical anion (
Nitric oxide synthases (NOS) are the main source of NO in mammalian systems. There are three well-characterized NOS isoforms encoded by three different genes: the constitutive and Ca+2-dependent isoforms include neuronal (nNOS) encoded by the gene NOS1 and endothelial (eNOS) encoded by the gene NOS3. The Ca+2-independent and inducible nitric oxide synthase (NOS2) is encoded by the gene NOS2. These isoforms share at least 50% sequence homology, but differ in structure, function, and catalytic activity. They are expressed in a variety of cell types, and isoform-specific domains direct cellular compartmentalization (163).
While NOS1 and NOS3 are constitutively expressed (constitutive NOS, cNOS) (114), NOS2 activity is induced by inflammatory cytokines, endotoxin stimulation, and hypoxic conditions (46). NOS isoforms generate NO at different levels. Under physiological conditions, cNOS generates NO in the picomolar (pM) to nanomolar (nM) concentration range upon transient increases in intracellular Ca2+ levels. The inducible isoform can produce NO at micromolar (μM) concentrations and its activity is sustained (173).
Tumor is not composed exclusively of abnormal cells, but it also contains stromal cells, including cancer-associated fibroblasts, immune cells such as T lymphocytes, tumor-associated macrophages, and polymorphonuclear leukocytes, as well as perivascular cells, endothelial cells, and mesenchymal stem cells. These stromal cells secrete a range of molecules into the extracellular tumor microenvironment, such as growth factors, cytokines, and NO. Tumor tissue can express different levels of NOS according to the disease stage. The tumor growth environment also contributes to establishment of NO steady state levels and activates important cell signaling pathways (48). NO plays an important role in cell signaling and can control cell death and survival in normal tissue and in pathological states.
Inflammatory response in development and progress of GI cancer
In support of a role of inflammation in the initiation of cancer, early studies have shown that aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs), which inhibit cyclooxygenase (COX) enzyme, can delay or prevent the occurrence of colorectal polyps, colorectal cancer, and other GI tumors (59). Inflammation is associated with various cancers (90), including colorectal, gastric, and esophageal cancer (6, 106, 130, 177), and it is a component present in all stages of tumor development, including initiation, promotion, and disease progression (106). Cyclooxygenases 1 and 2 (COX1, COX2) are enzymes that catalyze the formation of paracrine hormones (prostaglandins) from arachidonic acid and play an important role in inflammation. While COX1 is constitutively expressed in most cells, COX2 remains undetectable but is induced upon stimulation by bacterial endotoxins, lipopolysaccharides (LPS), cytokines, grown factors, and hormones that promote inflammation, hence making it a marker of inflammatory response (11).
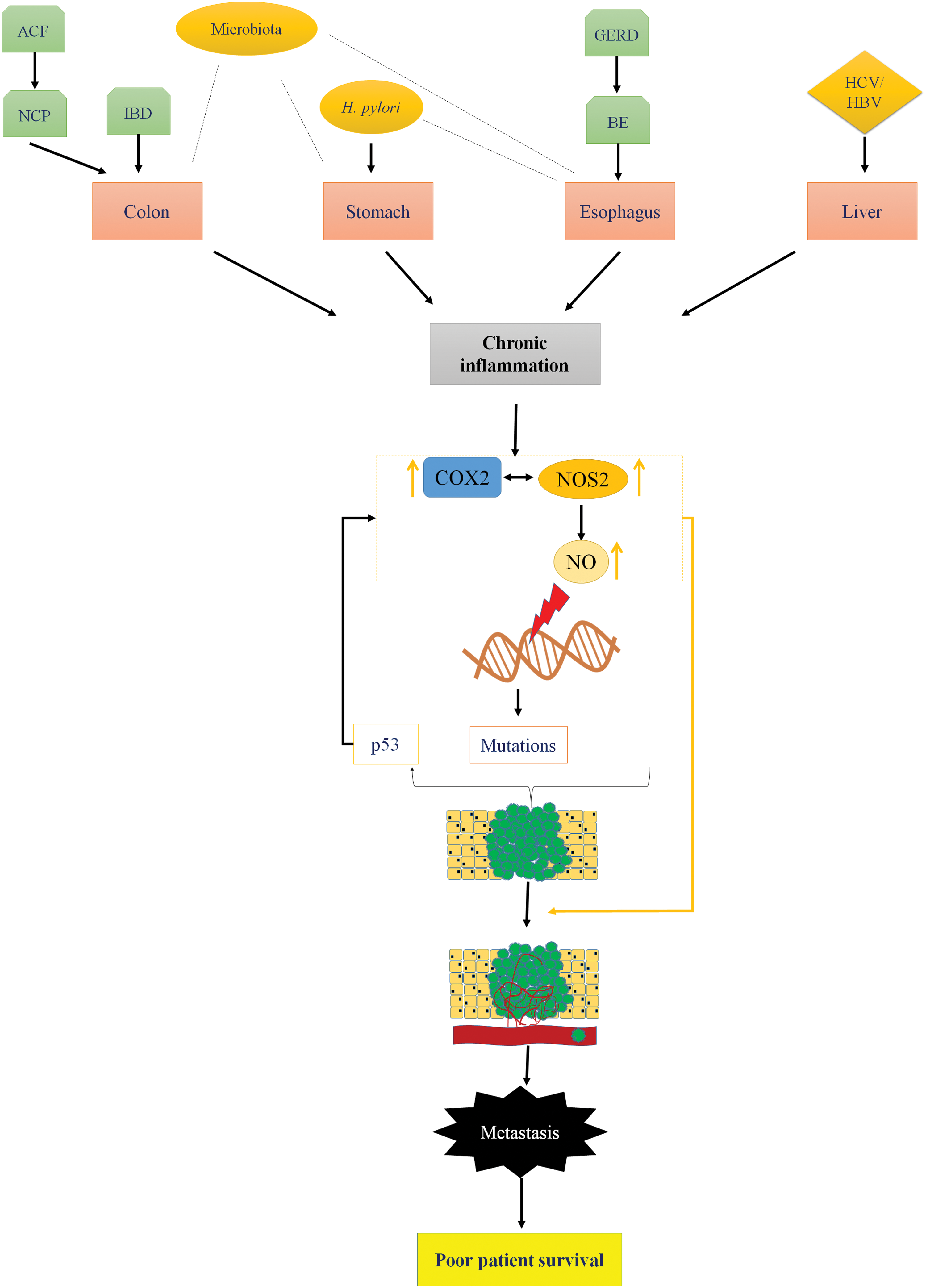
Carcinogenesis is a complex process, but essentially occurs due to a series of mutations leading to altered gene expression and loss of proliferative control (185). Chronic inflammation promotes NO generation through NOS2 and this event is associated with induction of neoplastic transformation. NO and other RNS can induce DNA damage by inhibition of DNA repair activities (187) or by direct modification of the DNA structure, such as nitrosative deamination, nitrosamine formation, DNA alkylation, or DNA strand breaks (168).
More than 50% of all human tumors have mutations in the tumor suppressor protein p53 and/or proteins that regulate its function (2). p53 status is a common biomarker in clinical studies of cancers, including breast (29), colorectal (72, 150), gastric (61), and esophageal cancer (63), as well as other cancers (131). As a tumor suppressor, p53 has multiple roles in cell signaling. The activation of p53 induces the expression of a range of genes that can cause cell cycle arrest of damaged cells to facilitate repair or the induction of apoptosis (19). Exposure of cells to an exogenous NO donor or increased expression of NOS2 leads to accumulation of p53 protein (45, 62, 172). Interestingly, p53 also modulates inflammatory response as it negatively regulates NOS2 expression. Accordingly, loss of p53 function has been shown to augment the expression of NOS2 and contribute to increased COX2 expression and increased tumor growth (180). Moreover, Hofseth et al. found that exogenous NO increased the expression and posttranslational modifications of p53 in vitro, in a model compatible with chronic inflammation. In the same study, they observed that samples from patients with colon cancer-prone chronic inflammatory disease had increased p53 phosphorylation, which correlated with enhanced NOS2 expression (62).
Studies performed in the 1990's with NO donors and NOS inhibitors showed a relationship between NO and induction of inflammatory factors. Early observations described the positive effects of NO and RNS on COX2 expression and activity (52). Subsequent studies found that NO-dependent stimulation of COX2 expression in tumor cells was abated by NOS2 inhibitors (71, 146). In human colorectal tumors, NOS2 expression correlates with COX2 expression and NOS2 inhibition reduced COX2 activity (27). Interestingly, NOS2 inhibitors decreased aberrant crypt foci (ACF) in rat colonic mucosa and this effect was coupled with decreased COX2 expression in a model of azoxymethane-induced ACF formation, which is one of earliest changes associated with colon cancer risk (137).
Numerous studies showed that NOS2 and COX2 expression levels are coordinately induced in tumor cells indicating interplay between these enzymes in cancer. Furthermore, NOS2 can augment COX2 expression and vice versa (56, 159). Moreover, both NOS2 and COX2 are regulated by the NFκB pathway (87, 154) and induced by interferon gamma (IFNγ) (113, 166). NFκB is considered to be the primary inflammation-associated transcription factor due to its activation by multiple cytokines and pathogens (126). The relationship between these enzymes in chronic inflammation and their actions as regulators of production of NO in this process is summarized in Figure 1.
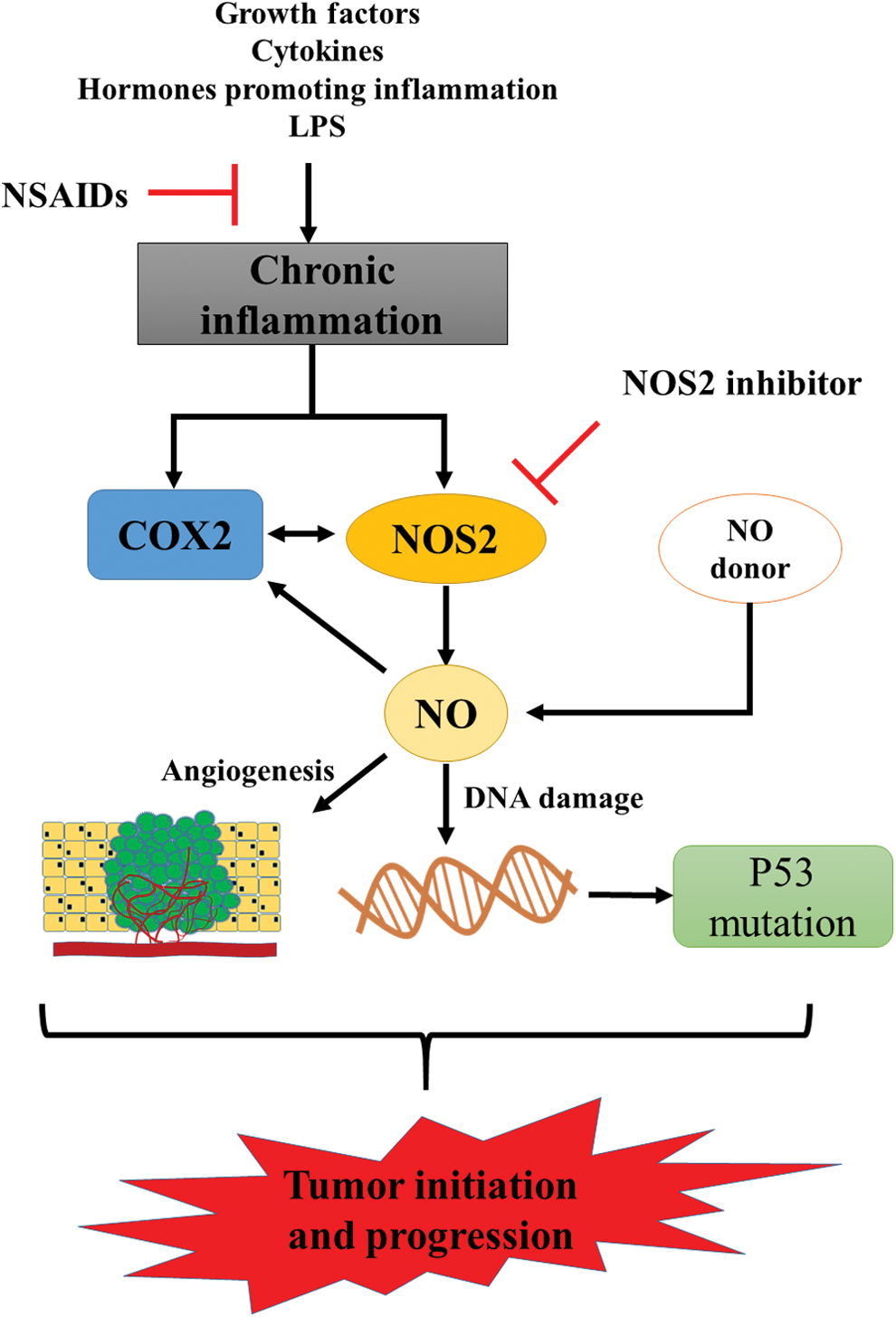
NOS2 expression in GI cancer
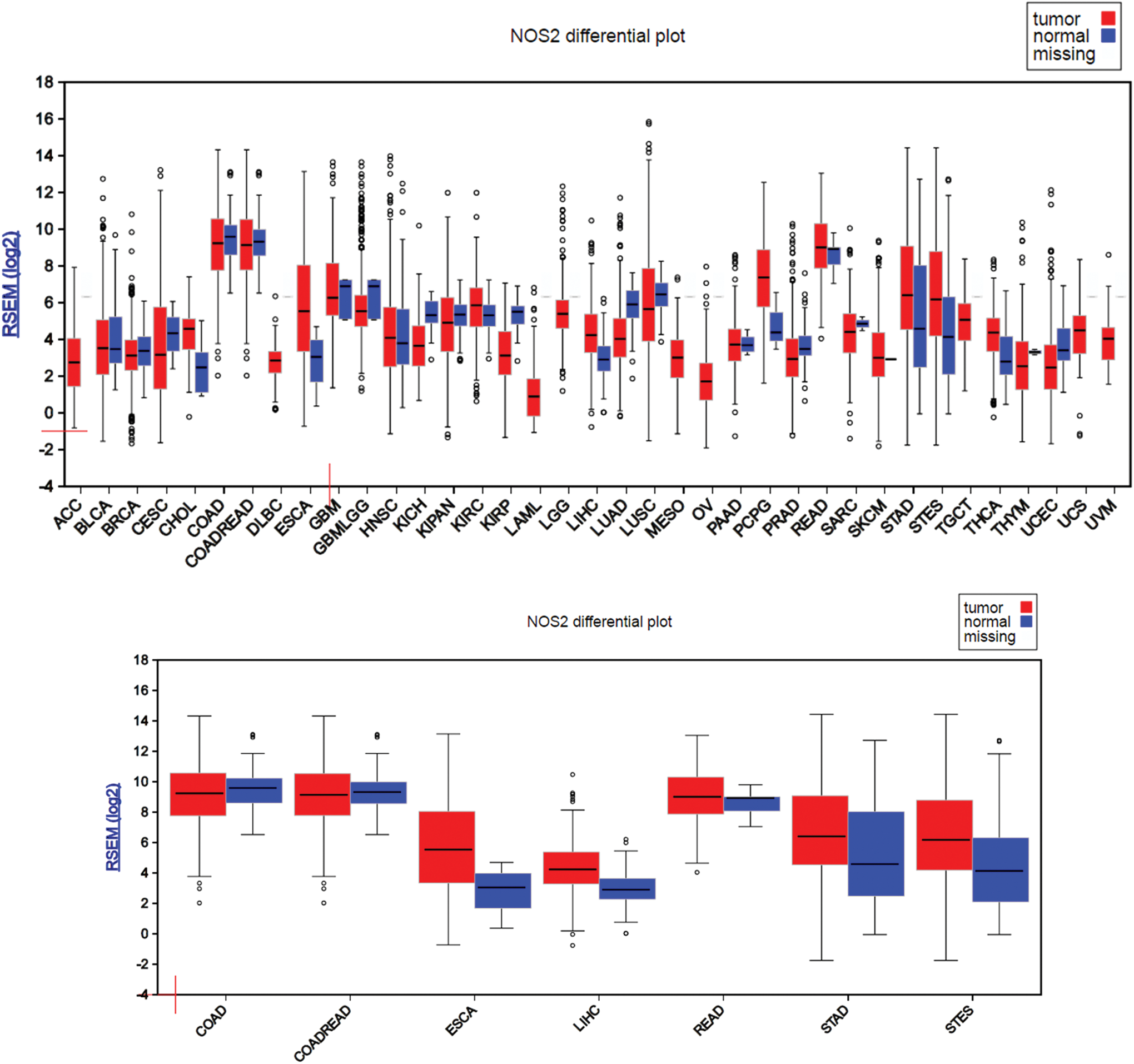
Many studies have shown a relationship between NOS2 expression and cancer risk and development. To better examine and understand the NOS2 gene expression status in cancer versus their normal counterpart, the FireBrowse gene expression viewer was used to visualize expression data collected from various whole genome RNA-Seq studies. The RSEM (RNA-Seq by Expectation Maximization) mRNASeq expression profiles for each The Cancer Genome Atlas (TCGA) disease chart are shown in Figure 2. Using this approach, we found that NOS2 gene expression was detected in all cancer types and most corresponding normal tissues, which represent nearly all organs/systems. Interestingly, majority of cancer data appeared to have larger error bars in their expression levels compared to their normal counterparts, which are likely a reflection of the heterogeneous nature of the cancer itself. The transcriptome data would be more accurate and represent the real expression level of NOS2 gene in cancer if those RNA-Seq studies were performed on laser-captured microdissected cancer samples (50). In GI tumors, our analysis shows that NOS2 gene expression is downregulated in colon adenocarcinoma (COAD) and colorectal adenocarcinoma, but in rectum adenocarcinoma, NOS2 expression shows an upward trend when compared to the corresponding controls. Expression is also upregulated in esophagus adenocarcinoma, stomach adenocarcinoma, and stomach and esophageal carcinoma. In the case of hepatocellular carcinoma (HCC), there is an elevated expression level of NOS2 relative to normal tissue. The high expression of NOS2 in liver cancer is not surprising since high NOS2 levels have been found under a variety of conditions in hepatocytes.
Regulation of NOS2 and genetic alterations in GI
The expression of NOS2 mRNA is induced and regulated by different agents that activate different cell signaling pathways associated with a number of transcription factors. Nevertheless, induction of NOS2 is cell and species specific (86). In fact, mammal's NOS2 promoter possesses transcription factor binding sites for NFκB, NF-IL6, and octamer factor-a transcription factor induced by tumor necrosis factor alpha (TNFα) and IFNγ (86). NOS2 expression may also be modulated by other mechanisms, including protein translation and NOS2 mRNA stability (20, 97), changes in substrate availability (supply and uptake) (117), presence or absence of cofactors (171), posttranslational modifications (119), formation of dimers (1), degradation (42), and localization in various tissues (141) and cell types.
The NOS2 promoter sequence contains binding sites for a range of transcription factors regulated by different pathways. The human NOS2 promoter has a site for binding NFκB and the signal transducer and activator of transcription-1α (STAT1α), which regulates NOS2 expression (49). Interestingly, changes in cell signaling pathways associated with these transcriptional factors are also associated with malignant development (57, 107).
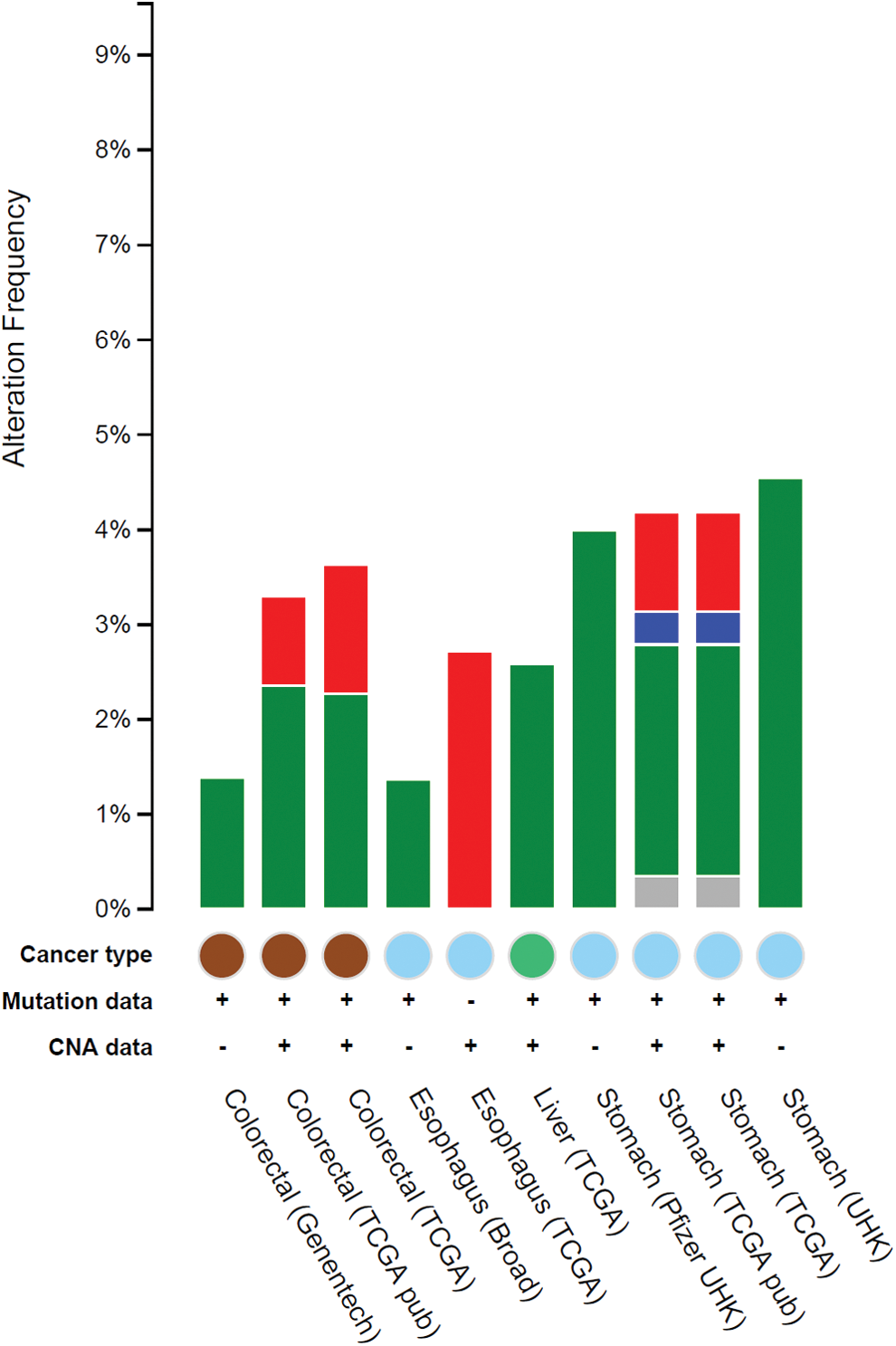
The query interface of cBioPortal enabled us to explore the impact of altered NOS2 genomic sequence in a multidimensional cancer genomics database. The analysis result reveals that low frequency genomic alteration rate of the NOS2 gene still can be associated with GI cancers. Relatively, genomic mutations appeared to be the dominant type of alteration among the studies analyzed here (Fig. 3).
NOS2 and microRNA
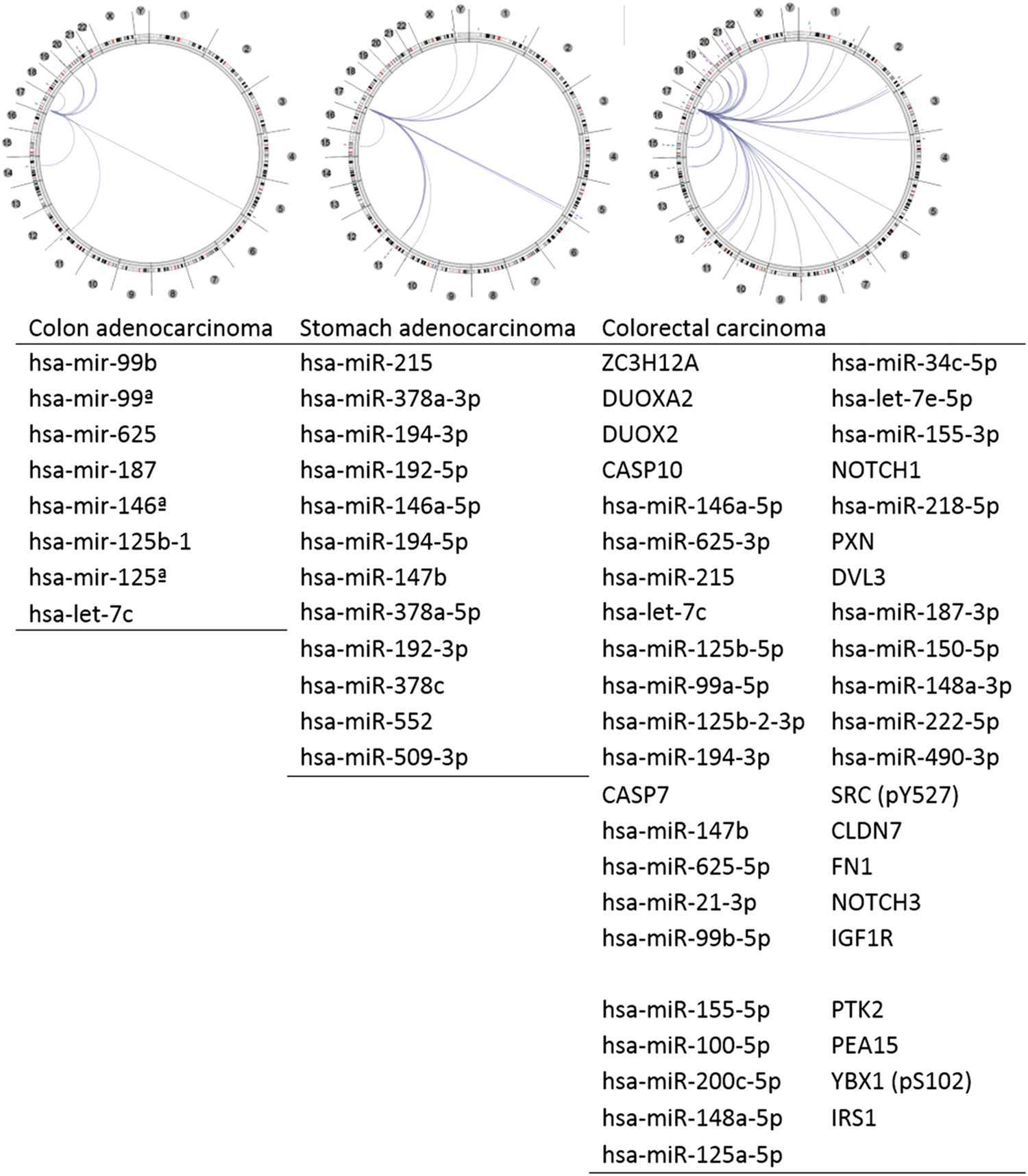
To better understand the NOS2 gene relationship/interaction with other gene(s) in GI tumors, we adopted the Regulome Explorer (Fig. 4) to examine what gene(s) is/are associated with the NOS2 regulome based upon their genomic context. The circular layout displays the associations as edges in the center connecting the features (with genomic coordinates) displayed around the perimeter. The ring displays cytogenetic bands. Using this multiscale association explorer, gene–gene associations are primarily displayed within the context of genomic coordinates. Surprisingly, the analysis result based upon the GI cancer data in the TCGA database revealed most associated genes are microRNA (miRNA). miRNAs are short noncoding RNAs that actively participate in the regulation of virtually all biological pathways. Accordingly, miRNAs influence numerous cancer-relevant processes such as proliferation, cell cycle control, apoptosis, differentiation, migration, and metabolism. We were able to identify eight miRNAs associated with NOS2 gene expression in COAD, 12 miRNAs associated with NOS2 gene expression in stomach carcinoma, and a total of 44 genes/miRNAs associated with NOS2 gene expression in colorectal carcinoma (Fig. 4).
Colorectal Cancer
Colorectal cancer is the third most common cancer worldwide. In 2016, it is estimated that more than 130,000 new cases of colorectal cancer will be diagnosed in the United States alone (153). In Brazil, 34,280 new cases are estimated for 2016 (70). Among human cancers, colorectal cancer is the one most closely associated with chronic inflammation. NOS2-derived NO, generated at inflammatory sites, stimulates colon tumor progression (139). Imbalance in NOS2 expression is associated with events leading to tumor initiation and development. Studies in the early 2000's showed the presence of NOS2 in 50–60% of colon cancer patients and that NOS2 expression correlated with decreased long-term survival and increased incidence of lymph node metastasis and lymphatic invasion (94, 122). These studies suggested that colorectal cancer cells expressing high levels of NOS2 predicted poor outcome. NOS2 expression levels vary within the colorectal tissue, while rectal and lower colon displays low NOS2 expression and upper colon shows increased expression of the enzyme. Although NOS2 expression has been associated with colorectal cancer progression, its role during tumor development is not completely understood. While some studies show a direct correlation between NOS2 expression levels, tumor development, and poor patient outcome, others describe reduced NOS2 expression levels early in colorectal tumor progression. A more recent study shows an interesting dichotomy between NOS2 expression site and patient survival. Unlike that found in tumor cells, increased NOS2 expression in tumor infiltrating macrophages is associated with positive patient outcome. Therefore, the source of NOS2, for example, immune cells versus tumor, is important in prediction of the outcome of colon cancer (121).
Further evidence of elevated levels of NO in colorectal cancer comes from footprints such as nitrotyrosine formation and S-nitrosation of protein thiols. A study showed that abnormal S-nitrosation induced by the activation of NOS2 modulates inflammation and cancer (25). On the contrary, Hao et al. showed that NOS2 protein levels were markedly reduced in ACF or at multiple neoplastic lesions, when compared to normal adjacent tissue (57a). These observations suggest that loss of NOS2 expression in colonic epithelium may be an early event in colorectal cancer development. However, NOS2 expression levels are elevated in the colonic mucosa of inflammatory bowel disease (IBD) and Crohn's disease (CD) (33). However, later in progression, it has been found that there is constitutive expression of NOS2 in colorectal cancer stem cells and this has been correlated with the tumor initiating properties of these cells (132). Interestingly, not all NOS isoforms have the same pattern of expression in inflammation-associated disease. Taken together, NOS2 likely has diverse roles in induction and early progression of colorectal cancer but appears to be a poor prognostic indicator in more advanced stages of the disease. Below, we examine some of the different ways in which NO can affect the progression and prognosis of this disease.
The transformation of a normal epithelial cell to colorectal carcinoma is a complex and multistep process, associated with multiple genetic changes (18). In general, normal epithelial cells first form noncancerous polyps, then progress to adenomas, and finally develop into carcinomas (75). Changes in the gut microbiota and their metabolic products have been linked to inflammatory processes associated with ulcerative colitis (UC), CD (100), and development and progression of colorectal cancer (195). Inflammatory processes found in colitis, such as macrophage activation, are considered a high risk factor for malignancy (39), and NO generation at sites of inflammation has been implicated as one of the causes for colorectal cancer development.
Beyond the etiology of cancer, NOS2 has emerged as an important component of progression to more aggressive tumors. In the late 1980's and early 1990's, NOS2 was shown to be a component of the tumoricidal activity of the immune system. However, in the mid 1990's, studies showed that NOS2 may promote tumorigenesis. Jenkins et al. showed that NOS2 overexpression in a human COAD cell line increases tumor growth and vascularization in nude mice (77). Later, it was shown that NOS2 mRNA, protein, and activity were increased in human colon adenoma and carcinoma (3, 89). These findings suggest that NOS2 expression within the cancer cell promotes progression, while NOS2 expression in the tumor-associated microenvironment could potentially have diametrically opposite effects.
The adenomatous polyposis coli (APC) is a tumor suppressor gene. Mutations in the APC gene are commonly found in colorectal cancers (124) and are also found in other cancers such as breast cancer (111), gastric adenocarcinoma (193), and esophageal cancer (13). APC is a protein member of Wnt signal transduction pathway. The APC complex consists of axin, casein kinase 1 alpha, and glycoprotein synthase kinase 3β (GSK3β) (118), which phosphorylate and direct β-catenin to proteasome degradation. APC mediates and regulates multiple processes, such as cell migration (190), cell adhesion (41), cell proliferation (9), and cell death (38), as well as DNA replication and repair (54). Araki et al. found that activation and overexpression of wild-type APC decrease COX2 mRNA in the human colorectal cancer cell line by reducing β-catenin/T cell factor 4 (Tcf-4) binding to the Tcf-4 binding element (TBE) in the COX2 promoter and this is dependent on K-Ras activation (4).
In 1996, Oshima et al. showed for the first time the relationship between the COX2 and colorectal cancer development using a mouse model of familial adenomatous carcinoma (125). The suppression of COX2 activity either by use of a COX2 inhibitor or by gene knockout decreases the number and size of intestinal polyps in the familial adenomatous polyposis mouse, an APCΔ716 mouse model (125). The increased COX2 expression at sites of inflammation together with augmentation of NOS2 expression could suggest a protumorigenic collaboration between these pathways, which may act in parallel or be redundant in cancer progression.
The regulation of induction of human NOS2 by the transcription factors NFκB and Stat1 is complex, often being cell specific. For example, Ganster et al. showed that in a human fibroblast cell line, the addition of IFNγ represses the induction of NOS2 transcription by NFκB via activation of STAT1 and deletion of STAT1 restores NOS2 expression (49). However, IFNγ has been shown to independently induce NOS2 mRNA in other cancer cells indicating a different regulation of NOS2 in different tumors (60, 170).
Du et al. proposed that cytokine induction of NOS2 depends on the Wnt/APC pathway (36). β-catenin can bind to the TBE in the promoter region of human NOS2, thus increasing its expression in different cell lines, including colon cancer cell lines (35). However, other studies have shown that some cancer cell lines have decreased NOS2 expression when incubated with cytokine mixtures (36, 76). This opposite effect was attributed to the overexpression of β-catenin in some colon cancer cell lines in which the APC gene was defective, leading to inhibition of NFκB activity and diminished NOS2 expression. The response to cytokines was rescued by overexpressing wild-type APC, leading to decreased β-catenin and increased NOS2 expression (35, 36). Thus, induction of NOS2 can be cell genotype dependent as well.
Recently, Xia et al. showed that NFκB is a potent transcriptional suppressor of protein tensin homolog (PTEN) (191). PTEN is a protein phosphatase that negatively regulates phosphatidylinositol 3 kinase (PI3K)/protein kinase B (Akt) signaling. Thus, the loss of PTEN increases Akt phosphorylation, which in turn decreases GSK3β levels, thus increasing β-catenin that can serve to augment NOS2 expression. On the contrary, Shaked et al. found that constant activation of NFκB by overexpression of IKβ kinase beta (IKKB) in mouse intestinal epithelium accelerates APC gene loss due to increased NO generation related to NOS2 overexpression (151). IKβ is the inhibitory effector of NFκB that confines it to the cytoplasm, thus preventing its migration to the nucleus where it participates in the transcriptional control of multiple genes (>150) (126). IKKB phosphorylates IKβ and frees NFκB.
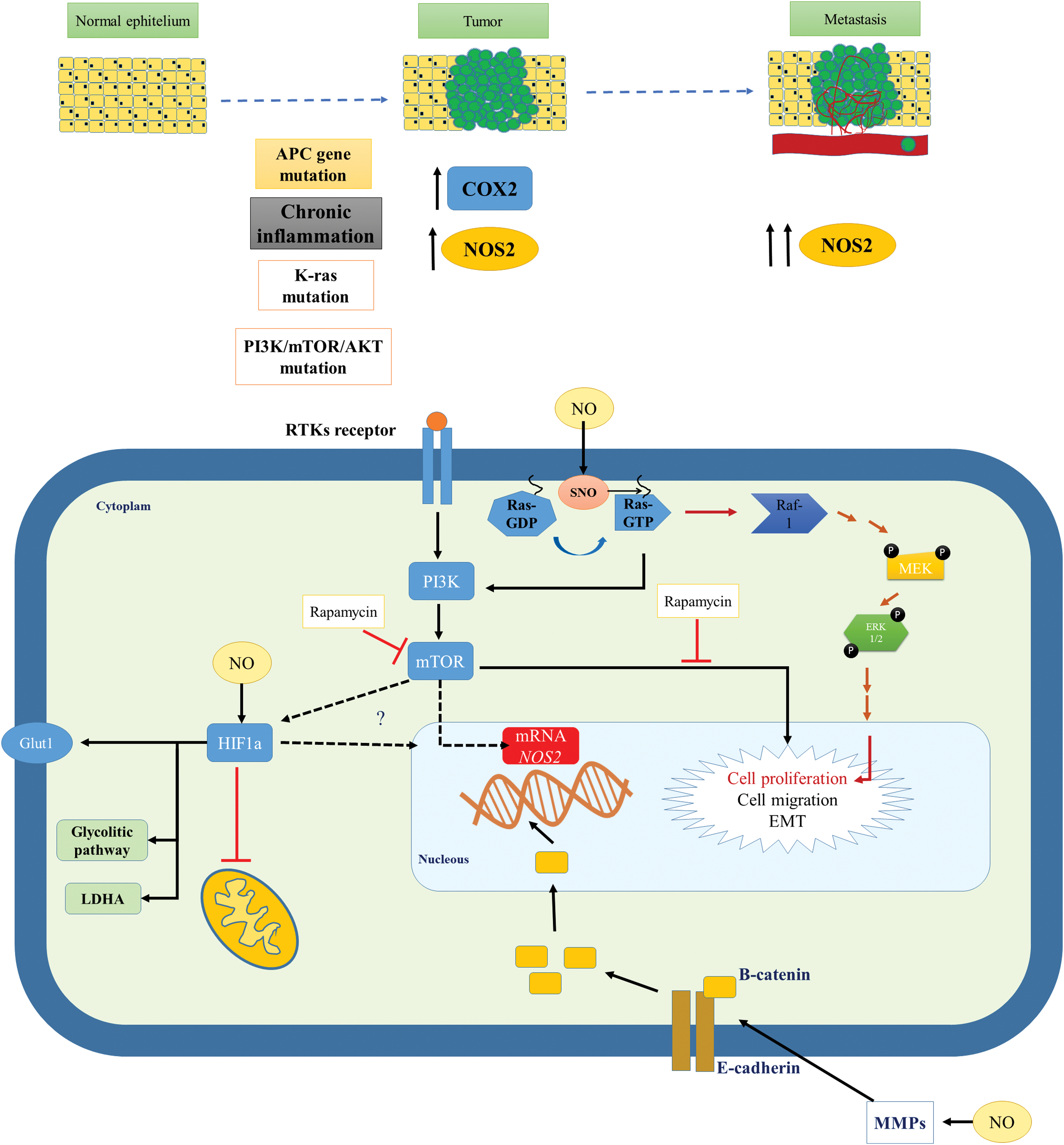
Colorectal cancer development and progress are associated with changes in important cell signaling pathways like the Wnt pathway (37, 73), Notch pathway (179), and PI3K/mammalian target of rapamycin (mTOR)/Akt pathway (55, 78). Despite somatic mutations found in cancers, activation of PI3K pathway can also occur by extra cellular stimuli, via receptor tyrosine kinases, or by stimulation from activated Ras (178). Furthermore, NO can stimulate the Ras/Raf1/MEK/ERK1/2 pathway and promote cell cycle progression (31).
It has been shown recently in colorectal clinical trials with drugs that interfere with PI3K–mTOR pathway extends overall patient survival (127). In response to nutrient availability, the mTOR pathway regulates multiple intracellular processes, such as cell cycle and proliferation, survival, protein synthesis, and actin organization (10). Proteins that belong to mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), and p-Akt (Ser473 one of the downstream targets of mTOR), have been found to be overexpressed in human colorectal cancers (55). The use of rapamycin, an inhibitor of mTOR, or genetic knockdown of the expression of raptor, a component of mTORC1, or knockdown of the expression of rictor, a component of mTORC2, decreases cancer cell migration and invasion. In addition, inhibition of mTOR modulates the expression of proteins associated with the mesenchymal–epithelial transition, E-cadherin, and decreases vimentin, smooth muscle actin, fibronectin, and matrix metalloproteinase 9 (MMP9) (55).
In rodents, inhibitors of the PI3K–mTOR pathway decrease cytokine-induced NOS2 expression in mouse macrophages (145). Lisi et al. showed that rapamycin also decreases NOS2 mRNA stability in astrocytes (97). Furthermore, recent studies have shown that mTOR regulates hypoxia-induced NOS2 expression in microglia (101). However, more studies are necessary to understand the mechanism by which mTOR regulates NOS2 in humans.
Besides biological endogenous production, NO can be delivered exogenously via the breakdown of compounds known as NO donors. These compounds have been helpful in elucidating the mechanisms of NOS-related pathways in cell physiology and pathology. Thomas et al. showed that incubation of cells with a range of concentrations of an exogenous NO donor leads to concentration- and time-dependent cell signaling (172). NO concentrations <50 nM are associated with activation of guanosine 3′5′ cyclic monophosphate (cGMP)-dependent cell signaling pathways in physiological conditions. These include pathways involved in the regulation of vascular tone; concentrations between 50 and 300 nM are associated with activation of ERK, AKT, hypoxia-inducible factor 1α (HIF1α), and p53 signaling; and concentrations higher than 300 nM elicit signaling associated with nitrosative stress (173).
The transcription factor HIF1α has an important role in adaptation of tumors to hypoxia. Under normoxia, HIF1α is hydroxylated by prolyl hydroxylase domain enzymes (PHDs) and targeted for proteasomal degradation (143). PHDs are dioxygenases dependent on 2-oxoglutarate and iron and they have O2-dependent activity. PHDs are also sensitive to competitive inhibition by some citric acid cycle metabolites known to accumulate in some cancer types, such as succinate and fumarate. Decreased levels of 2-oxoglutarate due to mutations in isocitrate dehydrogenases also decrease the activity of PHDs (135). Therefore, HIF1α can be stabilized under hypoxic conditions and normoxic conditions via accumulation of citric acid metabolites.
HIF1α plays a role in a metabolic change observed in tumorigenesis by regulating mitochondrial metabolism, mitochondrial biogenesis, and mitophagy (149). It also regulates the expression of glucose transporters and lactate dehydrogenase (108). NO modification of the nonheme iron protein leads to stabilization of HIF1α. This mechanism is a major component of HIF1α stabilization and signaling via NO. Moreover, HIF1α protein is a target of S-nitrosation, which may further enhance its stability and signaling (164). NO can modulate HIF1α function and its role changes according to the availability of O2. Under normoxia, endogenous NO or exogenous NO donors can lead to HIF1α accumulation. In contrast, under hypoxia, NO attenuates HIF1α accumulation (15). Increased stabilization of HIF1α in turn can lead to increased vascular endothelial growth factor (VEGF) production, angiogenesis, and metabolic changes.
NO activates soluble guanylate cyclase to produce cGMP in different tissues (5). The level of this second messenger has an important role to play in cell physiology. cGMP plays a critical role in regulating normal GI function (88, 103). Colorectal cGMP-dependent protein kinase (PKG) expression levels are decreased in samples from colorectal cancer patients and its overexpression in colon carcinoma cells decreases tumor growth and invasiveness in nude mouse models (64). Furthermore, some studies have shown that PKG1 downregulates β-catenin (92) and in metastatic colon carcinoma decreases tumor angiogenesis (91), inhibits cell migration, and increases apoptosis (32, 175), possibly due to a concurrent decrease in NOS2 expression.
Degradation of the extracellular matrix is associated with tumor invasion and metastasis. This process is mediated by endopeptidases called MMPs (14). These enzymes are capable of digesting components of the extracellular matrix, including collagens, laminins, fibronectin, and proteoglycans (123). Increased expression levels of MMP-2 and MMP-9 are associated with poor survival outcome in colorectal cancer (17, 144). The activity of MMP is dependent on removal of Zn-thiolate, which leads to a conformational protein change that allows substrate binding. NO can regulate expression of MMPs and their endogenous inhibitor tissue inhibitor matrix metalloproteinase by changing the level of the NO second messenger cGMP. NO and RNS can also disrupt the zinc-cysteine in MMPs, leading to their activation (138). Babykutty et al. found that activation of MMP-2/9 is accompanied by increased expression of Rho and Rac1 and phosphorylation of Erk-1/2 (8). The activation of this signaling axis results in stimulation of colon cancer cell migration and invasion. Furthermore, MMP activation releases β-catenin from interactions with E-cadherin, leading to translocation of β-catenin to the nucleus, where it can activate the transcription of genes, including NOS2 (99).
The relationship between NO, COX2, and tumor survival is not always linear. Liu et al. found that human colon cancer cells incubated in the presence of S-nitroglutathione increase both COX2 expression and PGE2 in a dose- and time-dependent manner, although they inhibit the growth of cells (98). Furthermore, the metastatic human colon cancer cell line SW620 when incubated in the presence of an NO donor was more sensitive to proapoptotic treatment through induction apoptosis-inducing factor (AIF) and protein expression, suggesting a mitochondrial apoptotic pathway associated with this process (65).
The processes associated with colorectal tumor initiation and progressions are complex and associated with diverse cell signaling pathways. NOS2 and its product NO play a role in many cellular responses, including inflammation, genomic changes, tumor development, and prognosis. The mechanisms associated with these changes are not clearly understood and are summarized in Figure 5.
Inflammatory bowel disease
Chronic, remittent, or progressive inflammation of colon and rectum is classified as IBD, and this includes CD and UC. CD may affect the entire GI tract, whereas UC is related to inflammation of the colonic mucosa (83). IBD, being derived from a chronic inflammatory process, is associated with increased risk of colon cancer (158).
The increase of both forms of IBD is associated with multiple factors, including genetic susceptibility, and environmental factors such as microbial flora and immune dysregulation (83). Very early onset inflammatory bowel disease (VEO-IBD) is described as IBD in children under the age of 10 years. Dhillon et al. found that NOS2 has increased activity in VEO-IBD associated with a NOS2 variant (33).
Studies conducted in the early 1990s with samples from patients suffering from IBD showed increased activity of NOS2 (12), levels of its substrate,
Hofseth et al. showed that colon tissues from patients with colon cancer-prone chronic inflammatory disease had increased phosphorylated and total p53 associated with increases in NOS2 expression. Furthermore, they showed that NO derived from decomposition of NO donors promotes DNA damage, phosphorylation, and accumulation of p53 (62). Moreover, tissues from patients with UC and CD had shown increased macrophage infiltration associated with increased inflammation (158). The increase in general inflammation and prolonged increase in NO concentrations contribute to the rise and development of colon cancer.
Gastric Cancer
The American Cancer Society estimated more than 26,000 new cases of gastric cancer and 10,700 deaths resulting from this disease in 2016 (153). The majority of gastric cancers are adenocarcinomas, that is, it begins in glandular cells responsible for the release of mucus and other fluids. The etiology of gastric cancer is unknown, but several risk factors have been associated with the development of this disease, including advanced age, male gender, diet, gastric adenomatous polyps, chronic inflammation, family history of gastric cancer, and Helicobacter pylori infection.
One of the strong risk factors in the development of gastric cancer is H. pylori infection, which affects more than half of the world's human population (51). H. pylori is a gram-negative bacterium and it has in its outer membrane LPS, which can interact with Toll-like receptors (TLRs) leading to initiation of innate immune responses. Inflammation caused by infection with H. pylori has an important role in development and progress of gastric and esophageal cancers. Approximately 10% of infections are associated with the development of peptic ulcers, chronic atrophic gastritis, gastric cancers, or mucosa-associated lymphoid tumors (58).
Gastric epithelium expresses TLR5 and TLR4 (148) that can recognize different components from H. pylori and initiate multiple cell signaling responses against the infection. H. pylori, through activation of TLR4, which has a site that recognizes LPS as a ligand, can induce the expression of host inflammatory genes, such as TNFα, which in turn activate NFκB leading to increased COX2 and NOS2 expression (84). H. pylori infection also augments proliferation associated with activation of MEK1/2-ERK1/2 pathway (196) and Ras-mediated activation of AP-1, a transcription factor associated with regulation of COX2 and NOS2 expression (26). Furthermore, levels of NOS2 and nitrotyrosine in the gastric mucosa are significantly higher in H. pylori-positive than in H. pylori-negative patients (53) with gastric cancer, showing that high NOS2 is an important determinant of carcinogenesis of gastric cancer following H. pylori infection.
NO has an important role in normal gastric function by controlling gastric blood flow and maintenance of gastric mucosal barrier integrity (105). NO controls the gastric blood flow through the activation of its second messenger cGMP. Changes in NO generation and NOS expression driven by various agents have been associated with gastric cancer onset and progression (21, 133). Mutations within the NOS2 gene or its promoter are associated with NOS induction and correlate with increased rates of cancer, including gastric cancer (80, 152), colorectal cancer (47), and esophageal cancer (43).
Studies of resected tumors showed a correlation with increased NOS2 expression and reductions in patient survival (197) and disease stage (184). Furthermore, NOS2 expression was associated with increased metastasis and angiogenesis in patients with gastric cancer (160). Zhang et al. showed that in over 50% of gastric cancer patients, increased NOS2 expression was associated with elevated rates of lymph node metastasis, vascular invasion, distant metastasis, and tumor node metastasis stage (197). Moreover, NOS2 is also associated with increased density of intratumor microvessels and angiogenesis in gastric cancer (67, 161). In addition, NOS2 has been shown to be positively correlated to lymphangiogenesis and lymphatic metastasis in gastric cancer (82).
GI microbiota and cancer
Microbiota participate in the production of NO from
Recent studies have shown a relationship between pretumor microbiota composition and development of gastric cancer. Interestingly, some studies found differences in microbiota composition between healthy subjects and patients affected by digestive diseases (7, 194). Furthermore, chronic H. pylori infection can shift the gastric microbiota by altering stomach pH. Besides H. pylori, other bacteria, such as Escherichia coli, Fusobacterium nucleatum, and Bacteroides fragilis, are associated with colon cancer pathogenesis (129). Presence of bacteria can change the GI tumor microenvironment, thus effecting the carcinogenic process. E. coli colonization of tumor can influence the protumor activity of macrophages by sustaining increased levels of COX2 expression (136). Furthermore, commensal microbiota can modulate the tumor response by altering the levels of cytokine (TNF) production by myeloid cells within the tumor (68). Commensal microorganisms are important regulators of the immune system and inflammation and may be addressed in therapies for cancer treatment (156).
Esophageal Adenocarcinoma
In 2016, the American Cancer Society estimated more than 16,900 new cases and 15, 600 deaths associated with esophageal cancer in the United States (153). The majority of esophageal adenocarcinomas arise from Barrett's metaplasia, in which reflux-damaged esophageal cells are replaced by mucus-secreting columnar cells (162). Changes in microbial flora were found in patients with esophageal cancer (40), including infection by human papillomavirus (183) and Epstein–Barr virus (192). In South America, a study found relationships with increased fungal infection (176). In contrast, other studies showed a relationship between inflammatory disorders after cancer treatment and increases in fungus infections, such as candidiasis (95, 142). Either way, these studies suggest an important role for the microbiome in esophageal cancer. In fact, new studies have shown changes in the distant esophageal microbiome in Barrett's esophagus (BE) and gastroesophageal reflux from an oropharyngeal-like microbiome to a more distinct microbiome with increased levels of gram-negative bacteria (157). Chen et al. found that specific changes in the salivary microbiota are also associated with increased rates of esophageal squamous cell carcinoma (24).
Gastroesophageal reflux disease (GERD) is one of the most common chronic diseases affecting up to 20% of United States population (115). BE is a complication of GERD that affects ∼10% of patients with GERD and that increases the predisposition for esophageal cancer (74).
NOS2 expression is increased in all associated stages of disease: GERD (96), adenoma (186), and esophageal carcinoma (110). The evolution of metaplasia to adenocarcinoma is associated with inflammatory processes and augmented the production of PGE2, COX2 mRNA, and COX2 protein expression (130, 186).
Esophageal cancers also show increased expression (23) and mutation of p53 (63). A recent article showed that esophageal squamous cell carcinoma (ESCC) is associated with decreased content of long noncoding RNAs (lncRNAs) in tumor of patients. Furthermore, overexpression of lncRNA in ESCC cell line induces apoptosis and inhibits proliferation and invasion by regulating the activation of p53 (182).
The Mutated in Colon Cancer (MCC) gene mutation is implicated in sporadic colorectal cancer and located in the same chromosome region as APC. Boynton et al. found that deletions of one allele of the tumor suppressor gene APC and/or MCC occur in more than 75% of analyzed samples from patients with esophageal cancer (13). Furthermore, the loss of heterozygosity at the APC and MCC loci was also found in patients with primary breast carcinoma (111), indicating that this type of tumor could share same pathways and mechanisms seen in esophageal and colorectal cancer.
The transcription factor HIF1α was found increased in esophageal carcinoma (109). Furthermore, some studies had shown a direct correlation between increases in HIF1α, VEGF (85), and NOS2 (167) in squamous cell carcinoma from samples of patients with esophageal cancer. Moreover, increased VEGF is associated with deep invasion in esophageal carcinoma (167).
Liver Cancer
Liver cancer includes HCC and intrahepatic bile duct cancer. Worldwide, 700,000 new cases were diagnosed at 2008 (44). The American Cancer Society estimates more than 39,000 new cases of liver cancer just within the United States. The major cause of HCC in Asia and sub-Saharan Africa is infection by hepatitis B virus (HBV), where HBV accounts for more than 70% of all cases. The risk factor associated with development of HCC in North America, Europe, and Japan is with hepatitis C virus (HCV), which accounts for 50–70% of cases (44).
Nitrosamines that are metabolized by liver P450 enzymes were shown to be powerful carcinogens in rodent models through alkylation of DNA.
In liver tumor, several studies have shown that NOS2 is present and correlated with HIF1α expression and increased oxidative damage. Some studies have shown a relationship between hepatocellular cancer severity and elevated NOS2 and COX2 expression (69, 134). Increased NOS2 is also correlated with poor outcome (134).
HCC is usually associated with vascular invasion and metastasis due to the high vascularization of this tissue. NOS2 has been shown to induce the activity of COX2, which has been proposed to increase vessel density in other cancer types (79, 197). These studies also suggest that NOS2 is associated with increased metastasis. Sun et al. showed that resected tissue from patients with hepatocellular cancer had increased level of MMP9 associated with increased expression of NOS2 (165). Although there is little predictive value of NOS2 and COX2 in HCC, in cancers initiated from HCV and HBV, these are predictive of poor outcome, suggesting that there is causal relationship. Generally, liver cancer is discovered at an advanced stage, suggesting that multiple factors can be driving the tumor.
Conclusions
Chronic inflammation plays a key role in initiation and progression of GI cancers. NOS2 and COX2 are important mediators of inflammation-driven cancer progression. The role of COX2 in various GI cancers is well established and NSAIDs have been shown to be a viable chemopreventive option. Recent observations show that majority of patients with colon, gastric, esophageal, and liver cancers have elevated expression of NOS2 in their lesions as well. Furthermore, NOS/NO levels are often associated with increased metastasis, leading to poor patient prognosis. The association of elevated NOS2 expression with cancers arising due to bacterial, viral, and fungal infections suggests an important relationship of the same with tumor immune response and chronic inflammation. The cross talk between NOS2 and COX2 may increase risk and determine patient survival (Fig. 6).
Footnotes
Acknowledgments
This research was supported, in part, by the Intramural Research Program of the NIH, Cancer and Inflammation Program. G.A.d.O. is supported by the program Science Without Borders–CNPq process number 205342/2014-0.