
Abstract
Aim:
Oxidative stress plays a key role in Parkinson disease (PD), and nuclear transcription factor related to NF-E2 (Nrf-2) is involved in neuroprotection against PD. The aim of the present study was to investigate a role for nuclear factor-κB (NF-κB)/Nrf-2 in the neurotherapeutic action of dimethyl fumarate (DMF) in a mouse model of PD and in vitro in SHSY-5Y cells.
Results:
Daily oral gavage of DMF (10, 30, and 100 mg/kg) significantly reduced neuronal cell degeneration of the dopaminergic tract and behavioral impairments induced by four injections of the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Moreover, treatment with DMF prevented dopamine depletion, increased tyrosine hydroxylase and dopamine transporter activities, and also reduced the number of α-synuclein-positive neurons. Furthermore, DMF treatment upregulated the Nrf-2 pathway, increased NeuN+/Nrf-2+ cell number in the striatum, induced activation of manganese superoxide dismutase and heme oxygenase-1, and regulated glutathione levels. Moreover, DMF reduced interleukin 1 levels, cyclooxygenase 2 activity, and nitrotyrosine neuronal nitrite oxide synthase expression. This treatment also modulated microglia activation, restored nerve growth factor levels, and preserved microtubule-associated protein 2 alterations. The protective effects of DMF treatment, via Nrf-2, were confirmed in in vitro studies, through inhibition of Nrf-2 by trigonelline.
Innovation:
These findings demonstrate that DMF, both in a mouse model of PD and in vitro, provides, via regulation of the NF-κB/Nrf-2 pathway, novel cytoprotective modalities that further augment the natural antioxidant response in neurodegenerative and inflammatory disease models.
Conclusion:
These results support the thesis that DMF may constitute a promising therapeutic target for the treatment of PD. Antioxid. Redox Signal. 27, 453–471.
Introduction
P
In this context, it has been proposed that dopamine itself, tyrosine hydroxylase (TH), monoamine oxidase, iron, and/or neuromelanin may play an important role in the death of DA cells (17). Furthermore, oxidative stress (70), mitochondrial dysfunction (51), apoptosis (4), and protein misfolding (20,44) are believed to cause DA cell death or degeneration in PD.
These findings underline that dimethyl fumarate (DMF), as nuclear factor erythroid 2-related factor 2 (Nrf-2)–based therapies, offers a novel cytoprotective modality that at the low dose of 30 mg/kg further augments the natural antioxidant response in a neuronal environment. Moreover, its therapeutic efficacy is also related to the modulation of different parameters related to the Nrf-2 pathways, such as cyclooxygenase-2 (COX-2), nuclear factor-κB (NF-κB), and neurotrophic factors. Furthermore, the confirmation, in in vitro studies, that inhibition of Nrf-2 antagonizes the protective effects of DMF, suggests that DMF-mediated Nrf-2 upregulation could have a possible therapeutic role in other neurodegenerative and inflammatory disease models.
Neurodegeneration in PD is often studied with mouse models based on use of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which presents clinical, biochemical, and neuropathological changes as observed in human PD (60). MPTP is metabolized to 1-methyl-4-phenylpyridinium ion by monoamine oxidase-B in astrocytes, which is then taken up by DA neurons via their dopamine transporter (DAT) (66). Mechanisms of MPTP neurotoxicity include inhibition of complex I in the mitochondrial electron transport chain, inflammation, and the generation of ROS (60).
Fumaric acid esters represent a class of molecules with anti-inflammatory and anti-oxidative activities in a variety of tissues and cell types. Dimethyl fumarate (DMF) is the most pharmacologically effective molecule among the fumaric acid esters. In vitro, DMF and its primary metabolite monomethyl fumarate improved the survival of astrocytes and neurons subjected to oxidative stress (2,43).
Both DMF and monomethyl fumarate covalently modify Kelch-like ECH-associated protein (KEAP1), resulting in accumulation of nuclear factor erythroid 2-related factor 2 (Nrf-2) and upregulation of the transcriptional Nrf-2 signature. Under basal conditions, KEAP1 inhibits Nrf-2-dependent transcription, effectively dampening endogenous expression levels of genes associated with the phase II antioxidant response. However, modification of the cysteine redox sensors on KEAP1, as a result of oxidation or by conjugation with electrophiles, results in Nrf-2 accumulation and transcriptional pathway activation.
Recently, Nrf-2 activation was observed in humans in settings of chronic diseases such as PD (30,32,37), Alzheimer's disease (14), and amyotrophic lateral sclerosis (52). Brennan et al. showed that DMF, by acting as an electrophile, modulates specific KEAP1 cysteine residues and exerts more robust allosteric conformational changes—leading to a diminished KEAP1-dependent degradation of Nrf-2 (10). This allows Nrf-2 to regulate cytoprotective genes associated with the phase II antioxidant response. Indeed, Nrf-2 regulates the expression of antioxidant response element genes (34) such as heme oxygenase-1 (HO-1), NAD (P)H quinone oxidoreductase-1, and glutathione synthesizing enzymes. Loss of Nrf-2-mediated transcription increases vulnerability of DA neurons to oxidative stress (11,29), whereas Nrf-2 activation is neuroprotective (16,30).
DMF is currently used as an oral therapeutic agent for the treatment of relapsing forms of multiple sclerosis (9,32). DMF exerts anti-inflammatory and cytoprotective effects through activation of the Nrf-2 pathway in neuronal cells (2). Further, DMF has beneficial effects in preclinical models of neuroinflammation, neurodegeneration, and toxic oxidative stress, although its precise mechanism of action remains unclear (43,59). In addition, DMF improved lifespan, reduced behavior deficits, and preserved striatal and motor cortex neurons in two different genetic models of Huntington disease (21), suggesting that it may has similar neuroprotective properties in MS. In this study, we hypothesized that DMF could have beneficial effects in an MPTP mouse model of PD.
Results
DMF treatment reduces MPTP-induced degeneration of the DA tract
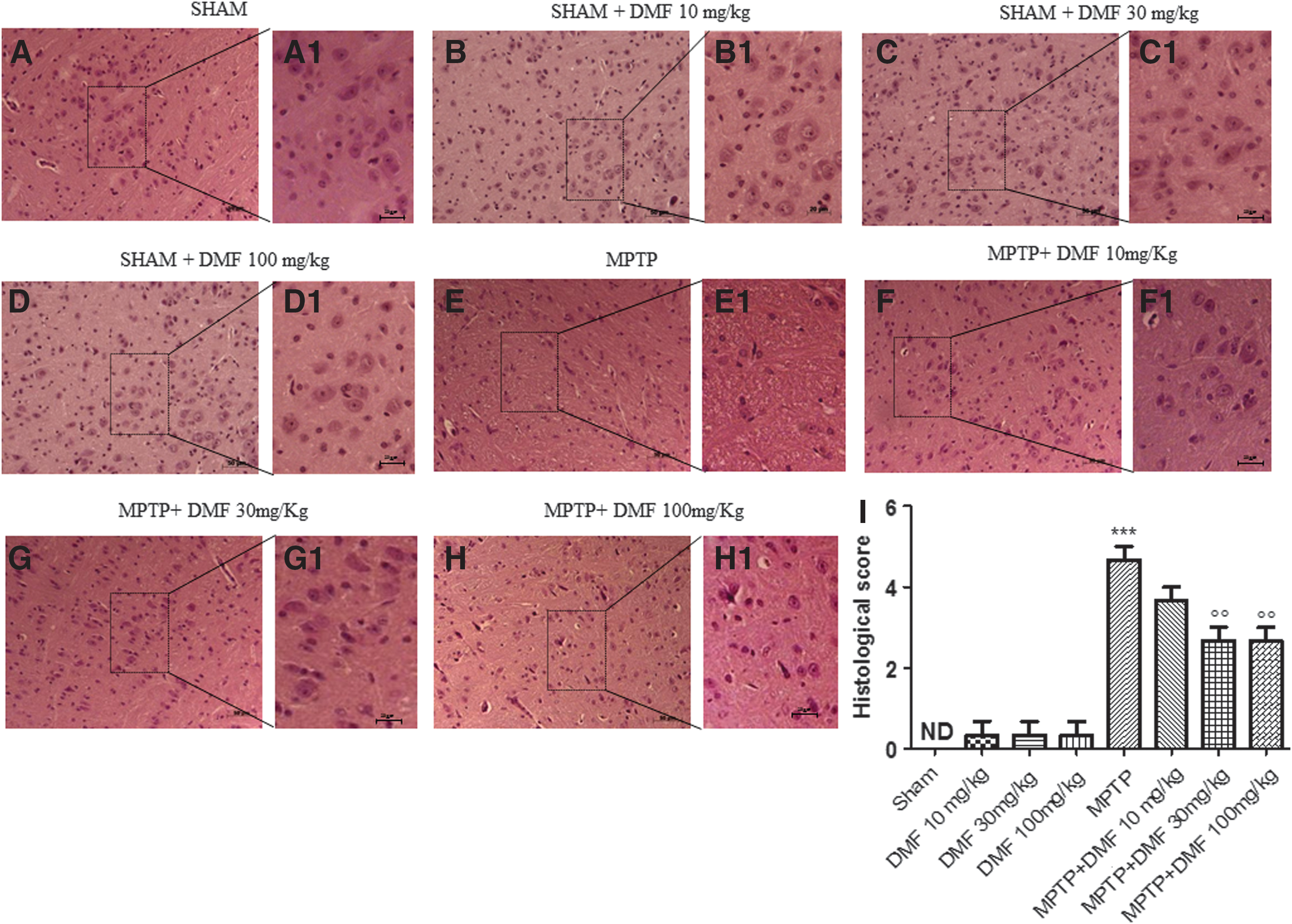
PD predominantly strikes the DA tract. We evaluated the severity of midbrain neuronal degeneration 8 days after MPTP intoxication. MPTP-injured mice were characterized by nigrostriatal DA degeneration, which translated into neuronal cell loss in the substantia nigra pars compacta (Fig. 1E, E1, see histological score Fig. 1I) as compared with the neuronal structure observed in control mice (Fig. 1A, A1, see histological score Fig. 1I) and in the DMF-only group (Fig. 1B, B1, C, C1, D, D1, see histological score Fig. 1I). DMF treatment (10 mg/kg) partly reduced (40% vs. MPTP) this alteration of the DA tract (Fig. 1F, F1, see histological score Fig. 1I), restoring neuronal cell loss (60% vs. MPTP) at doses of 30 and 100 mg/kg (Fig. 1G, G1, H, H1 respectively, see histological score Fig. 1I).
DMF treatment reduces MPTP-induced behavioral impairments
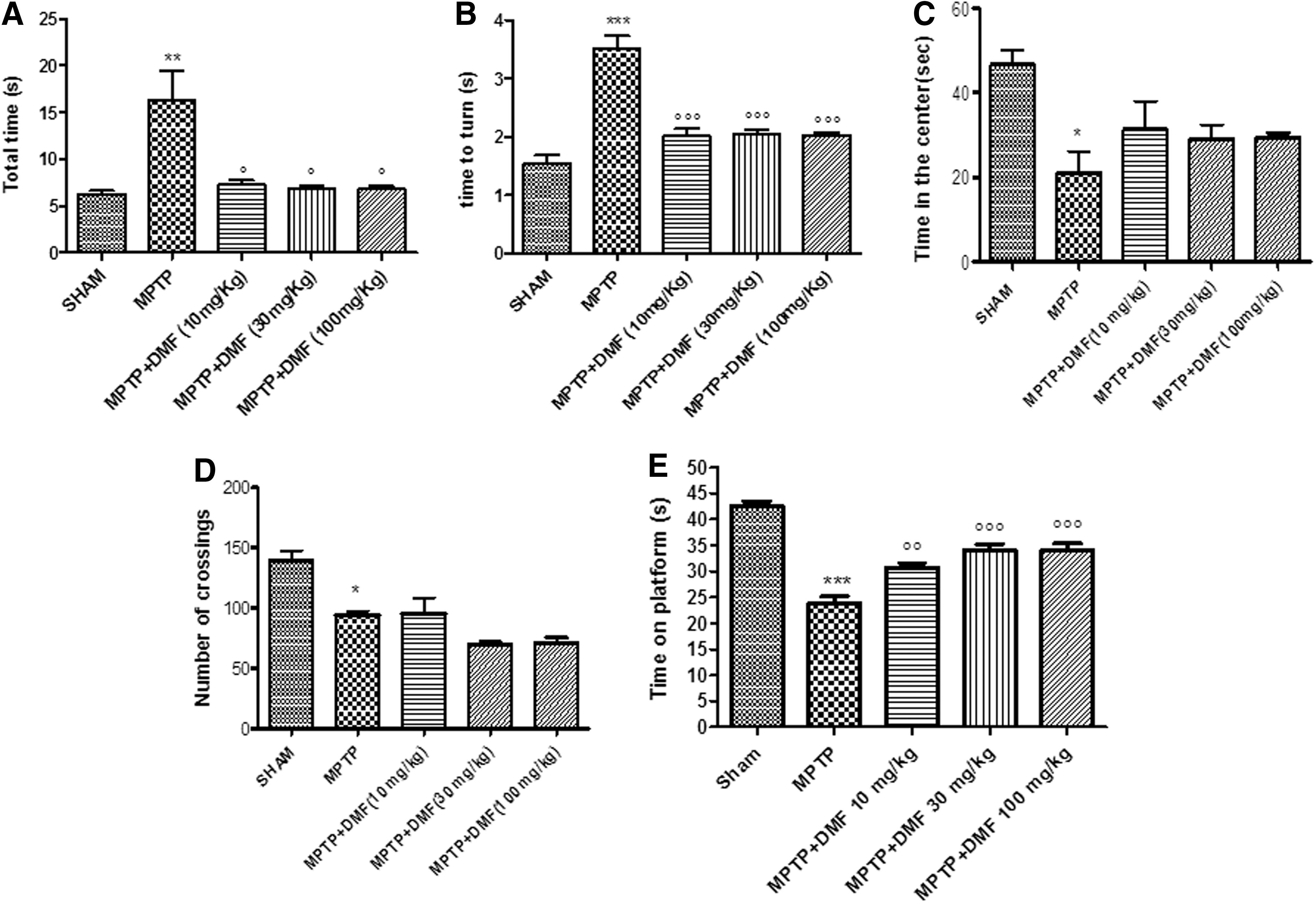
Behavioral deficits in the MPTP-induced mouse model of PD are useful for investigating the relationship between DA neuron degeneration and recovery processes, particularly in terms of motor activity. The pole test was used to assess MPTP-induced bradykinesia and the therapeutic effects of DMF. “Total time” and “time to turn” significantly increased (73% and 65%, respectively) after an injection of MPTP compared with the control group (Fig. 2A, B). DMF treatment, on the seventh day after MPTP administration, significantly reduced “total time” (45%) and “time to turn” (61%) at all doses (10, 30, and 100 mg/kg) (Fig. 2A, B), suggesting a significant reduction of bradykinesia. The rotarod test showed, at 7 days post-MPTP intoxication, a range of impairments in locomotor tasks (Fig. 2E) with respect to control mice (52% vs. sham) (Fig. 2E). DMF treatment, at all doses, improved latency (67% and 75%, respectively), compared with MPTP-lesioned mice (Fig. 2E).
Effects of DMF treatment on emotional state
The open-field test was used to determine whether DMF treatment influences emotional-behavioral disorders in mice after MPTP intoxication. MPTP-lesioned mice showed a pronounced increase in thigmotaxis when compared with sham mice, as indicated by less time spent in the center of the open field (Fig. 2C) and reduced frequency of line crossing (Fig. 2D). However, there were no significant improvements in DMF-treated mice as compared with MPTP-injured mice (Fig. 2C, D).
DMF prevents striatal DA neuron loss and DA depletion in MPTP-injured mice
PD can be considered a TH-deficiency syndrome of the striatum, as TH catalyzes the formation of the DA precursor, L-DOPA. To determine whether DMF might protect against MPTP-induced loss of striatal DA neurons, midbrain sections were stained for TH immunoreactivity. We observed a marked loss (60%) of TH-positive cells in MPTP-injected animals at 8 days after intoxication (Fig. 3B, B1, see densitometric analysis Fig. 3J). DMF treatment (10 mg/kg) provided a significant protection from MPTP-induced TH cell death (Fig. 3C, C1, see densitometric analysis Fig. 3J), whereas 30 mg/kg DMF preserved density of the TH-positive cell population (Fig. 3D, D1, see densitometric analysis Fig. 3J) as compared with control mice (Fig. 3A, A1, see densitometric analysis Fig. 3J). Stereological counting of TH-positive neurons (Fig. 3I) in MPTP-injured mice revealed an ∼75% loss of these cells (Fig. 3I) compared with control mice (Fig. 3I); DMF treatment restored almost completely the neuronal count in a dose-dependent manner (Fig. 3I).
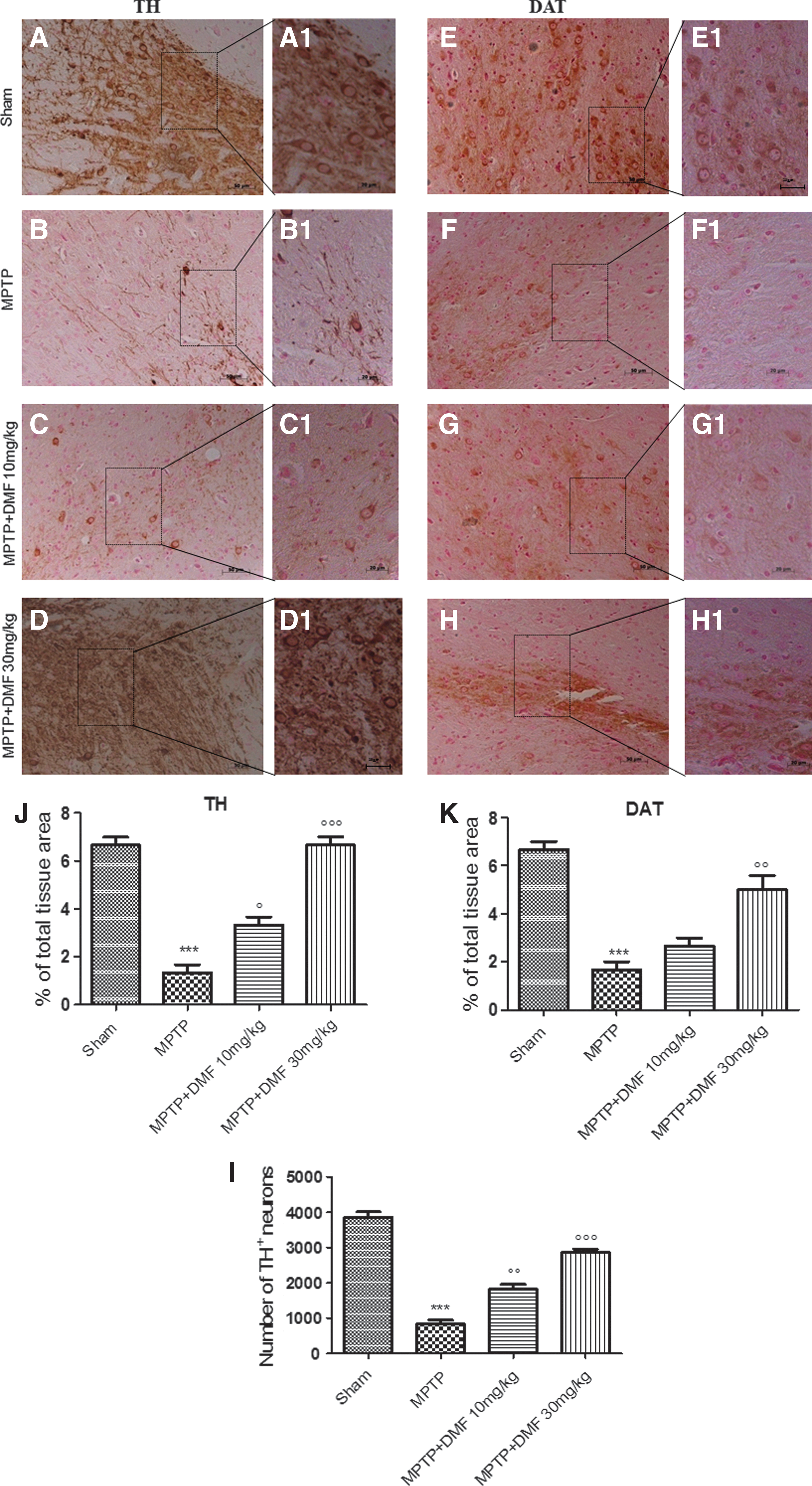
PD is also characterized by a significant loss of striatal DAT. To better study the effects of DMF treatment on the DA pathway, levels of DAT were also evaluated by immunohistochemical analysis. MPTP injection produced a 79% loss of DAT positive staining (Fig. 3F, F1, see densitometric analysis Fig. 3K) compared with sham animals (Fig. 3E, E1, see densitometric analysis Fig. 3K). DMF treatment dose dependently led to recovery of this reduction in DAT staining (69% and 75%, respectively) (Fig. 3G, G1, H, H1, respectively, see densitometric analysis Fig. 3K).
Protective effects of DMF on α-synuclein-induced neurodegeneration
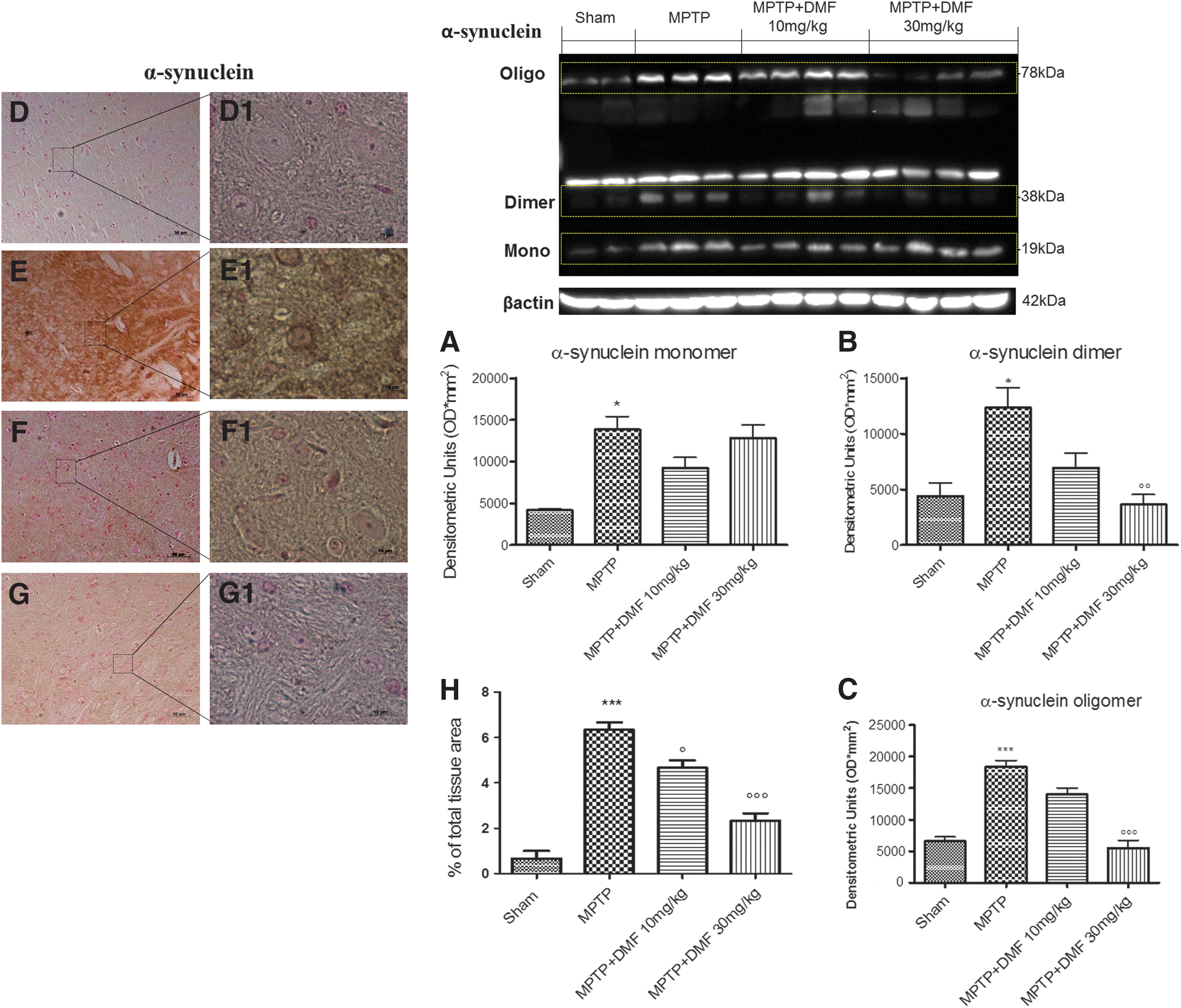
Given the neuroprotective potential of DMF in the mouse MPTP model, we next tested its ability to counteract α-synuclein-induced degeneration. α-Synuclein dimerization can accelerate the formation of neurotoxic aggregates and amyloid fibrils, which may be crucial to PD pathology at the molecular level (56). Western blot analysis revealed a threefold increase of α-synuclein monomer, dimer, and oligomer in MPTP-injured mice (Fig. 4A–C and Supplementary Fig. S1; Supplementary Data are available online at
Effects of DMF treatment on antioxidant response activation
In PD, oxidative stress plays a key role in determining neuronal cell damage. Nrf-2 is a transcription factor with strong antioxidant effects, and it protects neurons from ROS-induced damage. We, thus, evaluated the effect of DMF on the Nrf-2 pathway by Western blot analysis. Nrf-2 expression showed a tendency to decrease (32% vs. sham) after MPTP administration as compared with control mice (Fig. 5A and Supplementary Fig. S2). DMF treatment dose dependently upregulated (90% and 134% respectively) Nrf-2 levels (Fig. 5A and Supplementary Fig. S2). Moreover, in MPTP-injured mice, there were comparable decreases in levels of both HO-1 (79%) and Mn-superoxide dismutase (SOD) (75%) (Fig. 5B, C and Supplementary Figs. S3, S4). Interestingly, treatment with 30 mg/kg DMF increased (30%) Mn-SOD expression (Fig. 5B and Supplementary Fig. S3), whereas in MPTP mice treated with 10 mg/kg DMF, Mn-SOD expression was similar to control mice (Fig. 5B and Supplementary Fig. S3). In contrast, 10 mg/kg DMF showed a tendency to increase HO-1 expression (Fig. 5C and Supplementary Fig. S4), whereas a higher dose (30 mg/kg) returned HO-1 expression to control values (Fig. 5C and Supplementary Fig. S4). To further investigate the actions of DMF associated with oxidative stress, we measured the reduced glutathione (GSH)/oxidized glutathione (GSSG) ratio. MPTP intoxication led to a significant decrease in GSH/GSSG ratio in substantia nigra (Fig. 5J) in comparison to control mice (Fig. 5J). DMF treatment, especially at the higher dose of 30 mg/kg, significantly increased the GSH/GSSG ratio (Fig. 5J).
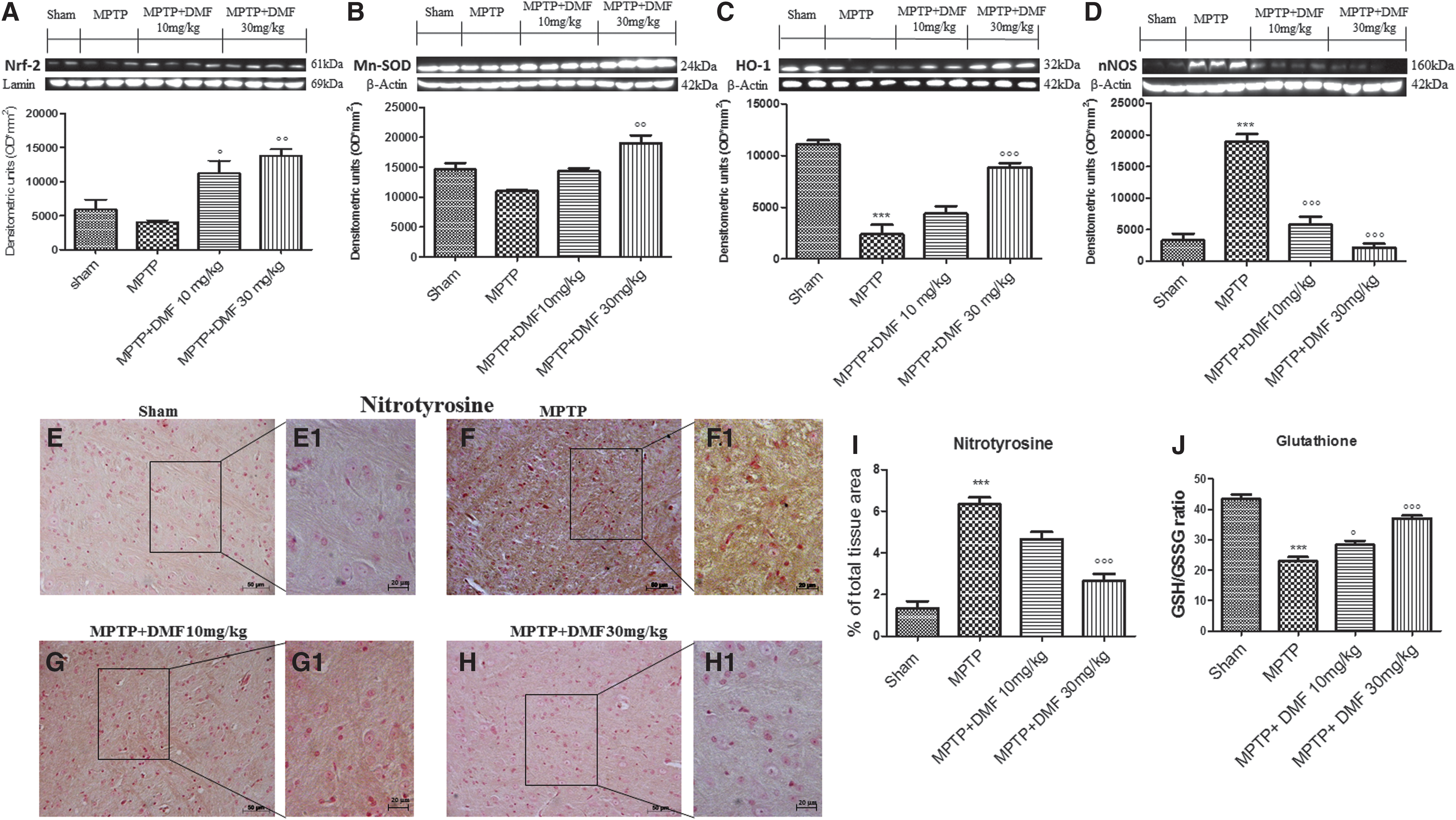
Nitrotyrosine (NT) is an indicator of cell damage, inflammation, and nitric oxide (NO) production. To understand the role of DMF in nitrosative stress, we evaluated NT by immunohistochemical analysis and neuronal NO synthase (nNOS) expression by Western blot analysis. NT-immunopositive cells increased (79%) in MPTP-injured mice (Fig. 5F, F1, see densitometric analysis Fig. 5I) as compared with sham mice (Fig. 5E, E1, see densitometric analysis Fig. 5I). Treatment with DMF protected against nitrosative stress in a dose-dependent manner (41% and 67%, respectively) (Fig. 5G, G1, H, H1 respectively, see densitometric analysis Fig. 5I). Further, a significant fourfold increase in nNOS expression was observed in MPTP-injected mice as compared with controls (Fig. 5D and Supplementary Fig. S5). DMF treatment, especially at the dose of 30 mg/kg, significantly reduced the expression of nNOS (Fig. 7D and Supplementary Fig. S9).
DMF treatment rescues neurons from oxidative stress-induced cell death
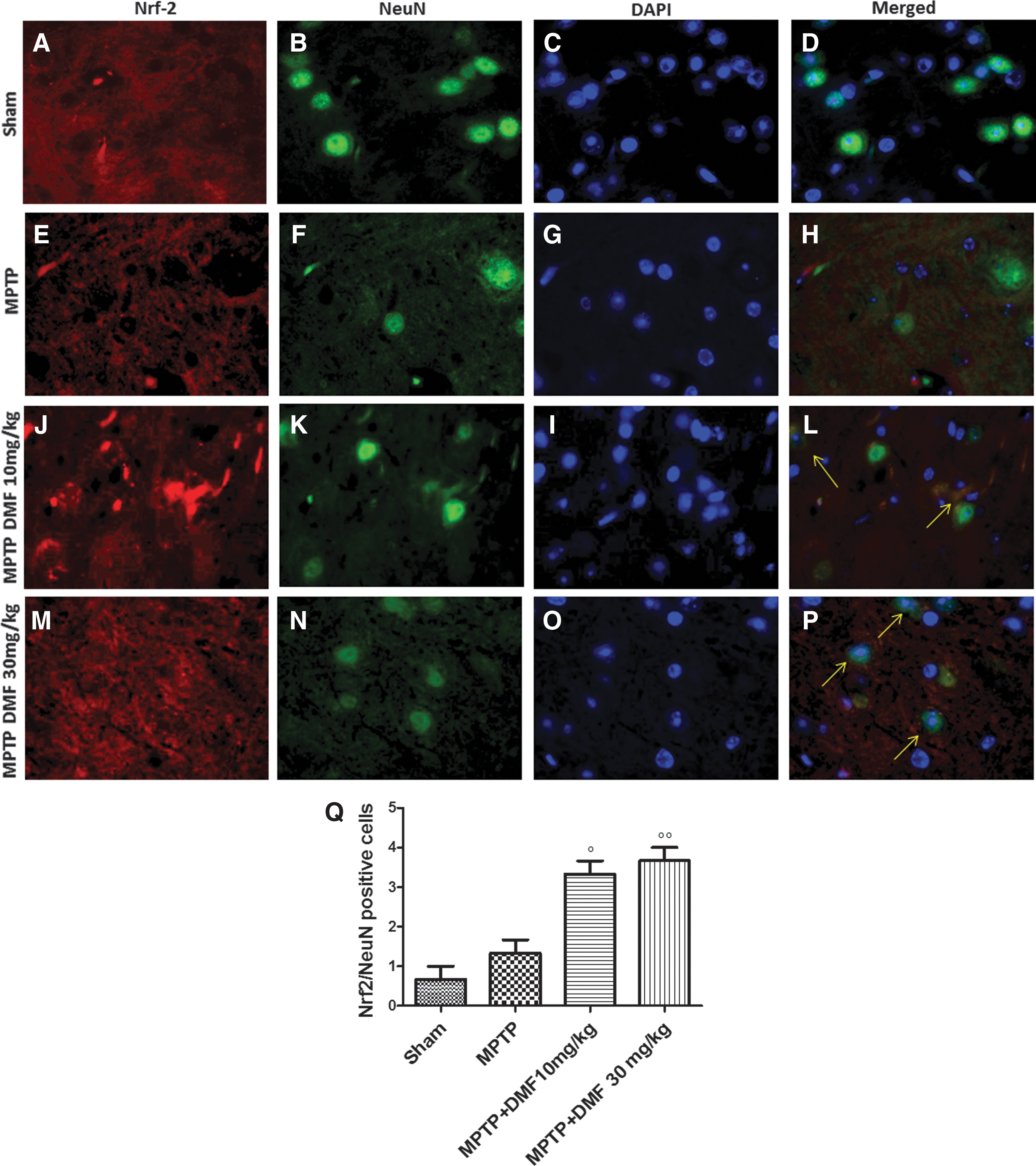
To investigate whether Nrf-2 plays a protective role in rescuing neurons from oxidative stress-induced cell death, midbrain sections were double stained with antibodies against Nrf-2 (red) and NeuN (3), the latter being a DNA-binding neuron-specific nuclear protein that decreases in PD. In comparison with the sham group (Fig. 6B), MPTP decreased NeuN+-positive cell numbers (Fig. 6F); DMF treatment restored this neuronal cell population (Fig. 6H, N). Levels of Nrf-2 were higher in control mice (Fig. 6A) as compared with MPTP-injured mice (Fig. 6E). DMF enhanced Nrf-2 levels in MPTP-treated mice in a dose-dependent manner (Fig. 6J, M). Note the co-localization between Nrf-2 and NeuN (yellow areas in Fig. 6L, P, Q). In MPTP-injured mice, there was a 26% increase (vs. sham) of Nrf-2/NeuN-positive cells, which was augmented with 10 mg/kg DMF (66% vs. MPTP) and 30 mg/kg DMF (72% vs. MPTP). The images shown are representative of three experiments.
DMF treatment exerts anti-inflammatory effects
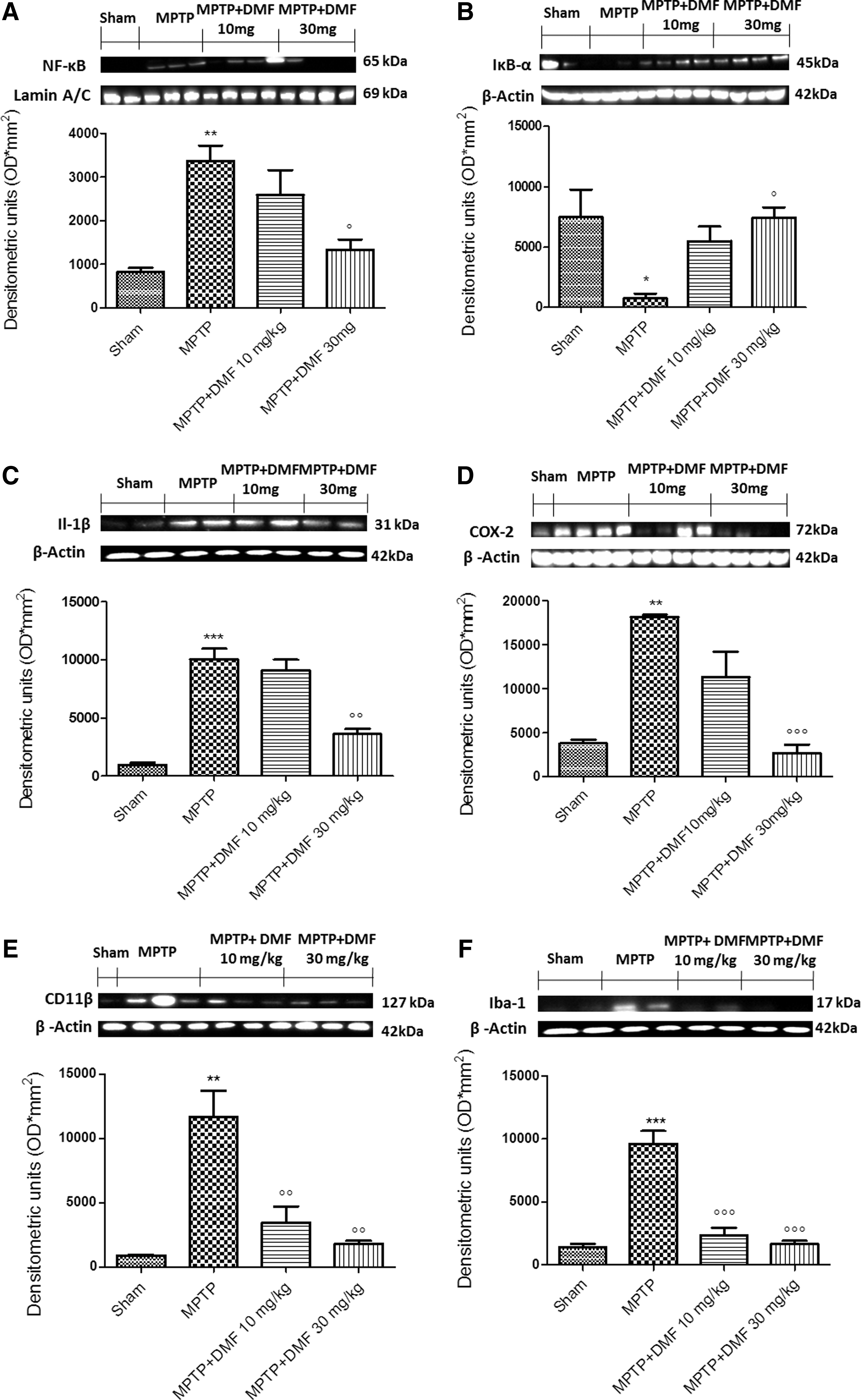
To evaluate the anti-neuroinflammatory activity by which DMF treatment may attenuate the development of PD, we assessed the expression of nuclear factor kappa-light-chain-enhancer of activated B cells (nuclear factor-κB [NF-κB]), IκB-α, interleukin-1beta (Il-1β), cyclooxygenase-2 (COX-2), CD11β, and ionized calcium-binding adapter molecule 1 (Iba-1) by Western blot analysis in midbrain homogenates 8 days after MPTP administration. Because the NF-κB pathway plays a key role in MPTP-induced DA damage, we evaluated levels of NF-κB and IκB-α expression by Western blot analysis. Treatment with MPTP significantly increased NF-κB DNA-binding activity in the substantia nigra as compared with control mice (Fig. 7A and Supplementary Fig. S6). DMF (10 mg/kg) treatment attenuated NF-κB nuclear translocation in MPTP-injured mice by 31%, whereas 30 mg/kg DMF reduced NF-κB DNA-binding activity by 60% (Fig. 7A and Supplementary Fig. S6). Conversely, IκB-α degradation was markedly diminished (90%) in MPTP-injured mice as compared with sham animals (Fig. 7B and Supplementary Fig. S7), whereas DMF treatment dose dependently acted to maintain IκB-α cytosolic activity (Fig. 7B and Supplementary Fig. S7). Moreover, expression of both Il-1β and COX-2 was significantly elevated in MPTP-injected mice in comparison to controls (Fig. 7C, D and Supplementary Figs. SF8, and SF9 respectively). The rise in Il-1β expression induced by MPTP (99%) was considerably reduced (65%) by treatment with 30 mg/kg DMF (Fig. 7C and Supplementary Fig. S8), whereas COX-2 expression was robustly blocked (71%) by this dose of DMF (Fig. 7D and Supplementary Fig. S9). Furthermore, microglial cell numbers and activation were assessed by Western blot analysis of expression levels of CD11β and Iba-1, respectively. Increased numbers (+95%) of microglia (Fig. 7F and Supplementary Fig. S11) as well as activated microglia (>99%) (Fig. 7E and Supplementary Fig. S10) were observed in MPTP-injured mice, when compared with control mice. DMF treatment, at 10 and 30 mg/kg, significantly reduced microglial cell expression (77% and 84%, respectively) and activation (70% and 85%, respectively) (Fig. 7E, F and Supplementary Figs. S11, and S10, respectively).
DMF reduces alteration of microtubule-associated protein 2 and restores neurotrophic factors levels in MPTP-intoxicated mice
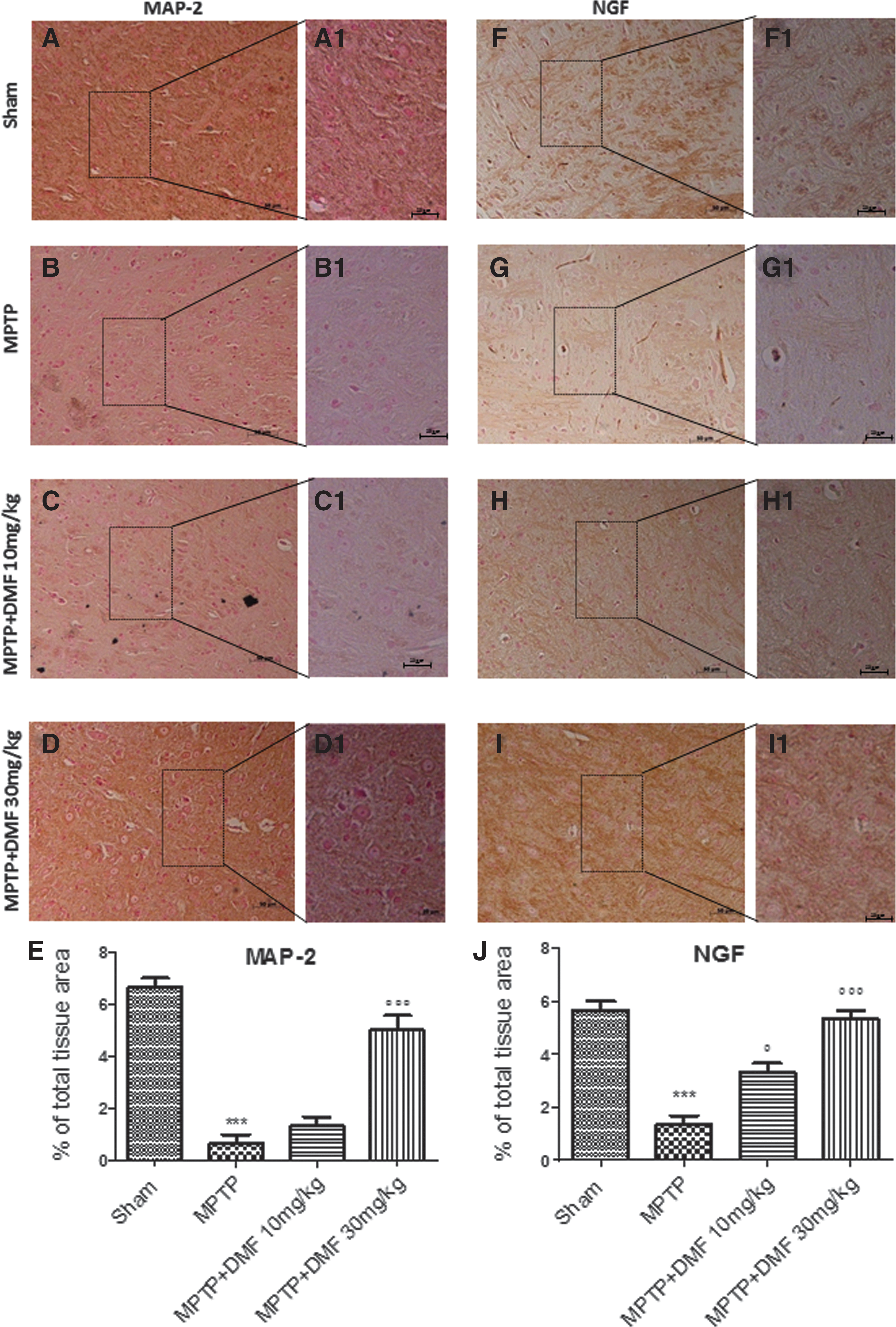
To understand the effect of DMF treatment on microtubule assembly/stabilization in neuronal dendrites, microtubule-associated protein 2 (MAP-2) expression levels were evaluated by immunohistochemical analysis. MAP-2 is a cytoskeleton protein mainly localized in neuronal dendrites that stabilizes microtubule assembly and mediates their interactions with other neuronal cell components, whose expression decreases in PD. MPTP administration significantly decreased (91%) MAP-2 expression (Fig. 8B, B1, see densitometric analysis Fig. 8E) compared with control mice (Fig. 8A, A1, see densitometric analysis Fig. 8E). DMF treatment at 30 mg/kg was more efficacious in protecting (62%) against MPTP-induced MAP-2 loss (Fig. 8D, D1, see densitometric analysis Fig. 8E) as compared with a dose of 10 mg/kg (Fig. 8C, C1, see densitometric analysis Fig. 8E). We also assessed the role of DMF treatment in restoring nerve growth factor (NGF) levels after MPTP intoxication, by immunohistochemical analysis. NGF is a neurotrophic factor that regulates the development and maintenance of the sympathetic and sensory nervous systems, whose expression is lower in PD. In midbrain sections collected 8 days after MPTP, NGF expression levels were significantly reduced (84%) (Fig. 8G, G1, see densitometric analysis Fig. 8J), in comparison to sham mice (Fig. 8F, F1, see densitometric analysis Fig. 8J). DMF treatment significantly restored NGF expression (41% and 66% respectively, at 10 and 30 mg/kg) (Fig. 8H, H1, I, I1, see densitometric analysis Fig. 8J).
The protective effects, NF-κB/Nrf-2 mediated, of DMF treatment in vitro
To evaluate the effect of DMF treatment on cell viability, SH-SY5Y cells were incubated with increasing concentrations of DMF (1–10–30–50–100 μM). Cell viability assessed after 24 h showed that only DMF concentrations of 1, 10, and 30 μM lacked cytotoxicity (Fig. 9A). Incubation of SH-SY5Y cells with 3 mM MPTP significantly reduced viability, whereas pretreatment with 30 μM DMF, 2 h before MPTP, significantly reduced cell death compared with the MPTP-only and the 1–10 μM DMF pretreatment groups (Fig. 9B), thus demonstrating 30 μM DMF to be the most effective concentration. To confirm that the in vitro DMF protective effect is Nrf-2 dependent, SH-SY5Y cells were stimulated with MPTP 2 h after pretreatment with 30 μM DMF. Moreover, SH-SY5Y cells were also stimulated with the Nrf-2 antagonist trigonelline (TR) (1 μM), 30 min before MPTP. TR notably inhibited the DMF cytoprotective effect compared with the MPTP +30 μM DMF group (Fig. 9C).
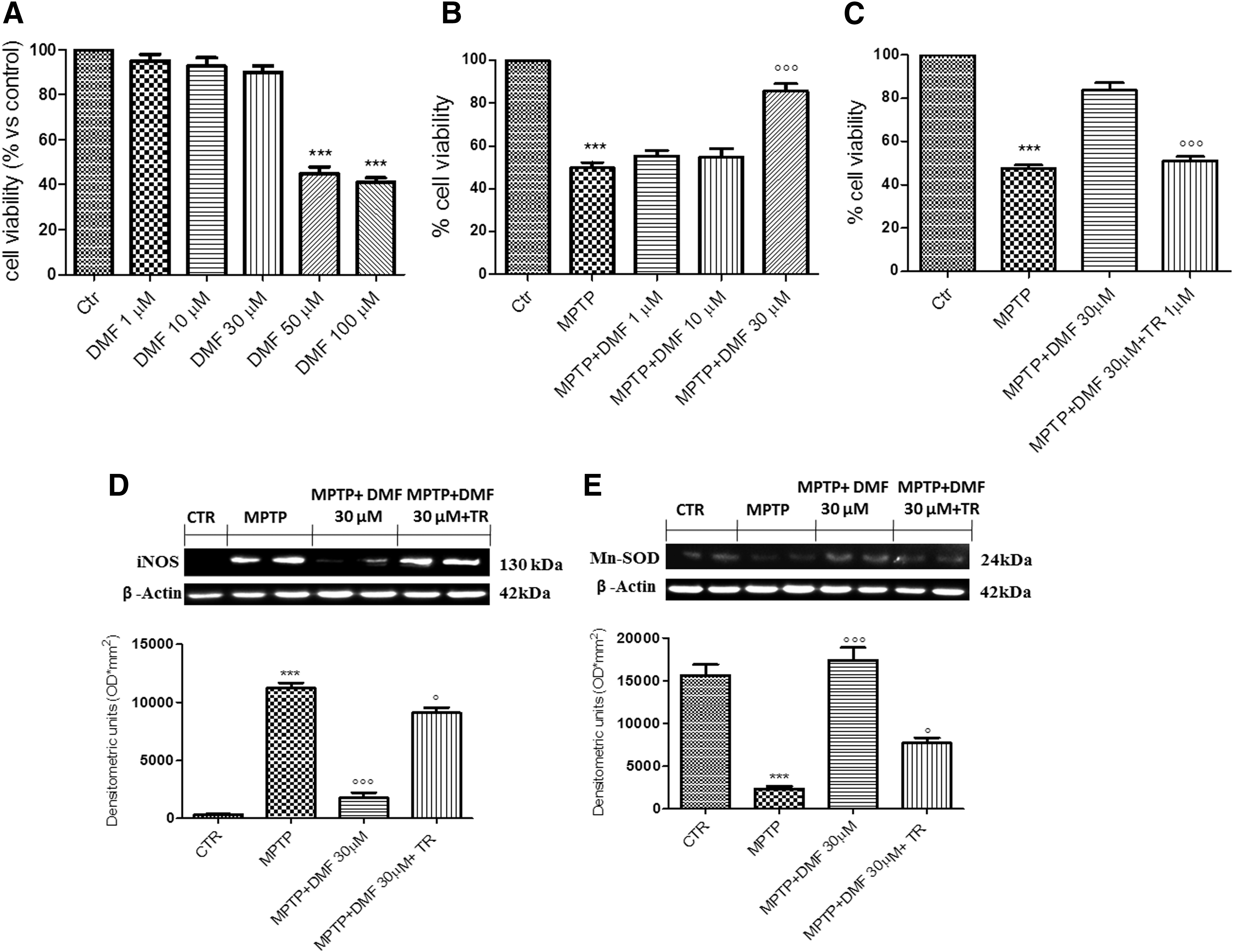
To corroborate that these anti-inflammatory and antioxidant effects of DMF treatment are NF-κB/Nrf-2 mediated, the action of DMF treatment in the regulation of inducible NO synthase (iNOS) and Mn-SOD expression in SH-SY5Y cells was examined. Western blot analysis demonstrated that iNOS expression was significantly higher (fourfold increase) in the MPTP group compared with the control, whereas pretreatment with 30 μM DMF lowered iNOS expression by 84% (Fig. 9D and Supplementary Fig. S12). Under these conditions, 1 μM TR abolished the DMF protective effect against MPTP, resulting in a threefold increase in iNOS expression (Fig. 9D and Supplementary Fig. S12). Conversely, Mn-SOD expression decreased by 85% after MPTP stimulation, whereas DMF treatment restored this parameter to control levels (Fig. 9E and Supplementary Fig. S13). Here as well, incubation with 1 μM TR increased SHSY-5Y susceptibility to MPTP damage, lowering Mn-SOD expression (Fig. 9E and Supplementary Fig. S13)—thus adding support to the view that Nrf-2 inhibition antagonizes the protective effect of DMF.
Discussion
PD is the second most common chronic neurodegenerative disorder, characterized by both motor and nonmotor symptoms, resulting from a pathophysiologic loss or degeneration of DA neurons in the midbrain substantia nigra pars compacta and the development of neuronal Lewy bodies (6). Recent years have witnessed an increase of therapies for the treatment of PD, including pharmacotherapy, nonpharmacological alternative approaches, and innovative surgical interventions (1,7). Despite these breakthroughs, effective disease-modifying therapy for PD has not yet been approved (41,64). Modification of the disease course by neuroprotective therapy is an important unmet clinical need; toward this end, understanding the cellular mechanisms involvement in PD is the subject of intensive research (26). Although the pathogenesis of neuronal degeneration in PD is yet to be fully understood, oxidative stress is an underlying mechanism that leads to cellular dysfunction and demise in a number of neurodegenerative diseases, including PD (67).
The Nrf-2 signaling cascade is a promising therapeutic pathway via transcriptional modulation of both inflammation and oxidative stress, and it has generated increasing clinical interest in using Nrf-2 activators in inflammatory diseases (32). A number of drugs that activate the Nrf-2 system are already in clinical use, including the fumaric acid ester DMF (57). DMF is a U.S. Food and Drug Administration–approved oral agent for the treatment of multiple sclerosis (24) and an effective and safe drug for the treatment of psoriasis (47). Formulations of this Nrf-2 inducer have been tested, in in vivo and in in vitro, for different therapeutic indications, including autoimmune myocarditis (45), inflammatory bowel disease (13), HIV-associated neurocognitive disorders (18), and other biomedical applications (19). DMF exerts beneficial effects in preclinical models of neuroinflammation and neurodegeneration (59); protects SH-SY5Y cells against 6-hydroxydopamine-induced neurotoxicity and the brain from oxidative stress via Nrf-2-dependent mechanisms (31). Moreover, DMF was shown to reduce MPTP-induced degeneration of the DA tract (38). In light of what has been mentioned earlier, our study was designed to evaluate the ability of DMF to protect mouse nigrostriatal neurons from MPTP-induced neurotoxicity and neuroinflammation. Another aim was to investigate the possible therapeutic effects of DMF on the modulation of parameters related to Nrf-2 pathways, such as COX-2, NF-κB, NGF, and behavioral impairments.
Mice treated with the DA neurotoxin MPTP develop a variety of behavioral deficits (61) and a longitudinal decrease in locomotor performance, as evidenced by akinesia, rigidity, tremor, gait, and posture disturbances (35). The present study demonstrates, for the first time, that DMF significantly improves locomotor agility, stability, and latency. Nonmotor features of PD represent important challenges in the clinical management of this disorder, with nigrostriatal circuit degeneration playing a role in anxiety-like behaviors in animal models of PD (58). We, therefore, also assessed whether DMF could affect not only sensorimotor performance but also emotional aspects. DMF did not significantly alter the emotional behavior component, suggesting a possible role of DA medication for DMF, because nonmotor features of PD are usually unresponsive to DA treatment (15). Because histological and behavioral studies showed a DMF dose of 30 mg/kg to be as effective as 100 mg/kg, we performed all remaining experiments with 30 mg/kg DMF as the upper dose limit to avoid possible side effects. Furthermore, histological evaluation showed neither toxicity of DMF between 10 and 100 mg/kg nor improvement with respect to sham mice. For this reason, we decided to complete the study excluding DMF-alone groups.
The characteristic decline in DA level in PD has been believed to arise solely from the severe loss of nigrostriatal DA neurons. However, DA deficit in the brain affected regions clearly exceeds the loss of DA neurons (5), suggesting that DA synthesis is impaired before cellular demise. In fact, studies on experimental models of PD demonstrate that the reduction in DA metabolism-related markers such as TH and DAT is far greater than the loss of neuronal cell bodies (28). In this study, we demonstrated that acute treatment with DMF, especially at 30 mg/kg, protects against MPTP-induced loss of TH+ neurons in the substantia nigra. Despite the fact that DAT expression depends on striatal extracellular DA concentration (69), DMF (30 mg/kg) increased DA release. MPTP-induced neuronal degeneration was associated with the redistribution of α-synuclein from its normal synaptic location to aggregates in degenerating neuronal cell bodies, the early stages of Lewy bodies formation (36). Under pathologic conditions, α-synuclein progresses from monomers to inclusions through a multistep process, leading to the formation of soluble oligomer species that cause neuronal cell toxicity (33). DMF treatment, especially at 30 mg/kg, significantly reduced α-synuclein-positive neuron number, mainly by inhibiting formation of dimers and oligomers.
Oxidatively damaged α-synuclein is reported to mimic some of the abnormal behaviors of mutant α-synuclein (25). Nrf-2 is a key regulator of endogenous inducible defense systems in the body that, in response to oxidative stress, translocate to the nucleus and bind to specific DNA sites termed “anti-oxidant response elements” (57). The properties of fumarate, combined with our data, suggest that DMF upregulated the Nrf-2 transcriptional system, dysregulated in brains of individuals with neurodegenerative diseases such as PD, thereby playing a major role in neuronal cell and tissue defense against oxidative stress. Nrf-2 induces phase II detoxifying and antioxidant enzymes (23) such as HO-1 and Mn-SOD, which work together to contest oxidative stress and inflammation. DMF treatment, via the Nrf-2 pathway, upregulates Mn-SOD and HO-1 expression, conferring resistance against neurodegenerative insults. These findings provide new insights into the biochemical properties of DMF as an Nrf-2/HO-1/Mn-SOD activator that is relevant for the control of inflammatory processes in neuronal cells. The anti-oxidative pathway activated by DMF involves downstream GSH regulation. GSH levels are reduced in PD within the substantia nigra pars compacta, and GSH depletion is an early index of oxidative stress in PD (8). DMF treatment, through Nrf-2-dependent regulation of GSH redox, is essential for neuronal cell survival during oxidative stress in PD disease. DMF treatment increased Nrf-2/NeuN immunoreactivity in neuronal subpopulations, highlighting a protective effect in terms of preservation of morphologically intact striatal neurons.
Inflammation is clearly involved in the response to MPTP, as indicated by microglial cell activation. Microglia are persistently activated in PD and with elevated production of cytokines and ROS (21). Activation of the Nrf-2 system occurs in response to anti-neuroinflammatory effects (43); furthermore, neuroinflammatory agents, such as COX-2 and Il-1β, have been documented in the DA neurodegeration in PD (65), where they mediate neuronal cell damage presumably by forming excessive amounts of harmful prostanoids and free radicals. DMF treatment, especially at 30 mg/kg, restored COX-2 and Il-1β to baseline levels, highlighting the role of DMF in moderating neuronal cell damage under these conditions by forming excessive amounts of harmful prostanoids and free radicals (50). Recent findings have also linked activation of the Nrf-2 system to anti-inflammatory effects, and they may also be involved via interactions with NF-κB (49). The relationship between Nrf-2 and NF-κB is not well characterized, but the identification of NF-κB binding cites in the promoter region of the Nrf-2 gene suggests cross-talk between these two regulators of inflammatory processes. Thus, DMF treatment, in particular mostly at the dose of 30 mg/kg, inhibiting the nuclear translocation of NF-κB and promoting the transcription of IκB-α, points toward representing a functional system for regulation of neuroinflammation in response to oxidative stress in the brain. However, oxidative stress is intimately linked as well not only to inflammation but also to other components of the neurodegenerative process, such as nitrosative stress (68). Various isoforms of the nitric oxide (NO) producing enzyme nitric oxide synthase (NOS) are elevated in PD, indicating an important critical role for NO in disease of the pathophysiology mechanisms, considering that increased expression of glial and neuronal NOS isoforms in astrocytes and neurons contributes to the synthesis of peroxynitrite synthesis, which leads to the generation of NT (nitrotyrosine) (27). We demonstrated that DMF treatment may be considered a selective inhibitor of neuronal nitric oxide synthase (nNOS), producing dose-dependent protection against MPTP-induced NT increase. Then, DMF treatment significantly downregulated the MPTP-induced oxidative and nitrative stress, pointing toward indicating its useful antioxidant properties against MPTP insults. To confirm, in in vitro confirmation, that the anti-inflammatory and antioxidant effects of DMF are NF-κB/Nrf-2 dependent, the ability of DMF to protect against SH-SY5Y cell death after MPTP treatment was examined after stimulation in SH-SY5Y cells, highlighting that DMF treatment loses its ineffectiveness in presence of the Nrf-2 inhibitor TR. Therefore, DMF-mediated Nrf-2 upregulation could be considered a possible therapeutic target to explore in terms of a tool to be exploited for counteracting PD progression.
PD may be associated with low production of neurotrophins, such as NGF. These proteins migrate into the brain at the site of disease, where they contribute to the resolution of neuroinflammation (42) and decreased expression of MAP-2 in the striatum and substantia nigra (48). Here, DMF treatment restored the MPTP-induced loss of MAP-2 and raised NGF levels.
In summary, our results indicate that Nrf-2 function is required for the therapeutic effect of DMF in PD. The central nervous system–protective effects of DMF could involve regulation of Nrf-2-mediated oxidative stress and inflammatory response mechanisms implicated in a variety of pathological conditions. Given its excellent pharmacokinetic profile, further studies with DMF as a potential neuroprotective approach in PD are warranted. However, DMF at high doses displays undesirable side effects (9), such as reduced motility, ataxia, dyspnea, cyanosis, muscular hypotonia, and an increase in nonglandular stomach and kidney tumors in animals. The present findings point to the therapeutic efficacy of DMF at lower doses, for example, 30 mg/kg, suggesting that its application to further augment the natural antioxidant response in a neuronal environment could be extended to other neurodegenerative and inflammatory disease settings.
Methods
In vivo studies
Animals
Male CD1 mice (30–34 g; Envigo, Italy) were accommodated in a controlled environment and supplied with standard rodent chow and water. Mice were housed in stainless steel cages in a room maintained at 22°C ± 1°C with a 12-h light, 12-h dark cycle. The study and experimental protocols were approved by the University of Messina Review Board for the care of animals. Animal care was in conformity with regulations in Italy (D.M.116192), Europe (O.J. of E.C. L 358/1 12/18/1986), and the United States (Animal Welfare Assurance No. A5594-01, Department of Health and Human Services).
MPTP-induced PD and treatment
Eight-week-old male CD1 mice received four intraperitoneal injections (i.p.) of MPTP (20 mg/kg; Sigma-Aldrich, St. Louis, MO) in saline at 2-h intervals in 1 day: The total dose per animal was 80 mg/kg. For DMF treatment (Sigma-Aldrich) (10, 30 and 100 mg/kg in 10% carboxymethylcellulose [CMC]), mice received by oral gavage (o.s) DMF starting 24 h after the first MPTP injection and continuing through 7 additional days after the final MPTP dosing. Mice were anesthetized with ketamine and xylazine (2.6 and 0.16 mg/kg body weight, respectively) administered i.p. and then decapitated with large bandage scissors; their brains were harvested, sectioned, and processed. The dose of MPTP (20 mg/kg) used was based on previous in vivo studies (22), whereas doses of DMF were based on our previous colitis study (13) as well as on prior dose-response and time-course studies in our laboratory.
Experimental group
Animals were randomly distributed into the following groups:
Group 1: Sham = vehicle solution (saline) was administered i.p. during the first day, as in the MPTP protocol (N = 20).
Group 2: Sham+DMF 10 mg/kg administered o.s for 7 days (N = 20).
Group 3: Sham+DMF 30 mg/kg administered o.s for 7 days (N = 20).
Group 4: Sham+DMF 100 mg/kg administered o.s for 7 days (N = 20).
Group 5: MPTP+vehicle administered i.p. during the first day, as in the MPTP protocol; vehicle solution (saline+CMC) was administered o.s for 7 days after the last MPTP dosing (N = 20).
Group 6: MPTP+DMF 10 mg/kg administered o.s for 7 days after the last MPTP dosing (N = 20).
Group 7: MPTP+DMF 30 mg/kg was administered o.s for 7 days after the last MPTP dosing (N = 20).
Group 8: MPTP+DMF 100 mg/kg was administered o.s for7 days after the last MPTP dosing (N = 20).
Experimental data regarding groups 2, 3, and 4 are related only to histological evaluation, as DMF administration did not result in either toxicity or improvement in comparison to sham controls (revised Fig. 1). Furthermore, the MPTP+DMF 100 mg/kg group was only subjected to histological and behavioral analysis, as it did not differ from the DMF 30 mg/kg group. We, therefore, decided to continue analyzing only DMF 10 and 30 mg/kg treatments.
Western blot analysis of cytosolic and nuclear extracts from midbrain
Tissue samples from brain were processed as previously described (22). The expression of Mn-SOD, HO-1, neuronal NO synthase (nNOS), IκB-α, COX-2, Il-1β, CD11β, Iba-1, and α-synuclein was quantified in the cytosolic fraction from brain tissues. Nrf-2 and NF-κB expression was quantified in the nuclear fraction. The membranes were probed with the following primary antibodies: anti-MnSOD (1:500; Millipore), anti-HO-1 (1:500; Santa Cruz Biotechnology), anti-nNOS (1:500; Cell Signaling), anti-NF-κB (1:500; Santa Cruz Biotechnology), anti-IκB-α (1:500; Santa Cruz Biotechnology), anti-COX-2 (1:500; Cayman), anti-Il-1β (1:500 Santa Cruz Biotecnology), anti-CD11β (1:500; Biorad Antibodies), anti-Iba-1 (1:500; Santa Cruz Biotechnology), anti-α-synuclein (1:500; Santa Cruz Biotechnology), or anti-Nrf-2 (1:500; C-20 sc-722; Santa Cruz Biotechnology) in 1 × PBS, 5% w/v nonfat dried milk, 0.1% Tween-20 at 4°C, overnight. Protein loading was controlled by probing with either an anti-β-actin antibody (1:500; Santa Cruz Biotechnology) or an anti-lamin A/C antibody (1:500; Sigma-Aldrich Corp.) for cytosolic and nuclear fractions, respectively. Signals were detected with an enhanced chemiluminescence system reagent according to the manufacturer's instructions (Thermo). Relative expression of protein bands was quantified by densitometry with BIORAD ChemiDoc™ XRS+software and standardized to β-actin and lamin A/C levels. Images of blot signals (8 bit/600 dpi resolution) were imported to analysis software (Image Quant TL, v2003).
Histology
Brain tissues were taken 8 days after the MPTP injection. Tissue sections were stained with hematoxylin/eosin and studied by using light microscopy connected to an imaging system (AxioVision; Zeiss, Milan, Italy) as previously described (62). Histological assessment was made by blinded observation, and slides were scored for severity of pathological profiles after hematoxylin/eosin staining by using a semiquantitative 5-point rating: 0 = no pathology; 1 = mild pathology; 2 = moderate pathology; 3 = severe pathology; 4 = more severe pathology (63).
Behavioral testing
Behavioral assessments on each mouse were made 7 days after the MPTP injection.
Pole test
The test consists of a 50-cm high, gauze-taped pole (1 cm in diameter). Mice are placed with their head upward right below the top. Two parameters were assessed: time until the animal turned by 180°, time until the animal descended to the floor (61).
Open-field test
The test consists of open-field boxes (80 × 80 cm) into which mice were placed for 5 min, after which activity was observed (54).
Rotarod test
The rotarod treadmill (Accuscan, Inc., Columbus, OH) assesses motor balance and coordination. For testing, the animals were subjected to three trials; the average score was used as the individual rotarod score, as previously described (12).
Immunohistochemical localization of TH, DAT, NT, MAP-2, NGF, and α-synuclein
Immunohistochemical localization was performed as previously described (22). Sections were incubated overnight with one of the following primary antibodies (all dilutions in PBS): polyclonal anti-TH polyclonal antibody (1:250; Merck-Millipore), polyclonal anti-DAT (1:00; Santa Cruz Biotechnology), polyclonal anti-TH (1:250; Merck-Millipore), anti-MAP-2 (1:250; Merck-Millipore), polyclonal anti-NGF (1:250; Santa Cruz Biotechnology), and polyclonal anti-α-synuclein (1:50; Santa Cruz Biotechnology). Photomicrographs were assessed by densitometric analysis using an Imaging Densitometer (AxioVision; Zeiss).
Stereological nigral DA neuron counts
Unbiased counting of TH+ DA neurons within the substantia nigra was performed as previously described (40). Every fourth free floating section was incubated with polyclonal rabbit anti-TH (1:400; Merck-Millipore) overnight and processed with the ABC method (Vector Laboratories, Burlingame, CA). The stereologist was blinded to the treatment received. For each mouse brain, five representative sections of the substantia nigra pars compacta were analyzed with StereoInvestigator software (Microbrightfield, Williston, VT).
Immunofluorescence staining of Nrf-2 and NeuN
Sections were processed for immunofluorescence staining as previously described (63). Sections were incubated with rabbit anti-Nrf-2 (1:100; Santa Cruz Biotechnology) or mouse monoclonal anti-neuronal nuclei (anti-NeuN) (1:100; Merck-Millipore) in a humidified chamber overnight at 37°C. Sections were observed and photographed at × 20 magnification by using a Leica DM2000 microscope. All images were digitalized at a resolution of 8 bits into an array of 2560 × 1920 pixels. Optical sections of fluorescence specimens were obtained by using a HeNe laser (543 nm), a laser UV (361–365 nm), and an argon laser (458 nm) at a 1-min, 2-s scanning speed with up to 8 averages; 1.5-μm sections were obtained by using a pinhole of 250. Contrast and brightness were established by examining the most brightly labeled pixels and applying settings that allowed clear visualization of structural details while keeping the highest pixel intensities close to 200. The same settings were used for all images obtained from the other samples that had been processed in parallel. Digital images were cropped, and figure montages were prepared by using Adobe Photoshop 7.0 (Adobe Systems, Palo Alto, CA).
Measurement of GSH and GSSG
GSH and GSSG were measured in the substantia nigra and cerebral cortex by using a GSH assay kit (Cayman) with enzymatic recycling, as previously described (39). Absorbance was measured at 405 nm. GSH concentration of each sample was calculated as nmol/mg protein.
In vitro studies
Cell culture
Neuroblastoma SH-SY5Y cells (ATCC® CRL-2266™) were cultured as previously described (62). In preliminary experiments to assess cell viability, 3 × 104 cells were plated in a volume of 150 μl in 96-well plates. Increasing concentrations of DMF (1, 10, 30, 50, 100 μM) were used to determine the effective concentrations with minimal cytotoxicity. The DMF concentrations chosen were 1, 10, and 30 μM. In another set of experiments, 8 × 105 cells were plated and incubated with retinoic acid (100 nM) for 24 h to induce differentiation. After this time, cells were pretreated for 2 h with 1, 10, and 30 μM DMF (based on previous 4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide colorimetric cell viability assay), followed by addition of MPTP to a final concentration of 3 mM. The protective concentration chosen was 30 μM DMF; 1 and 10 μM DMF were not protective after MPTP damage. Cells were then preincubated for 30 min with 1 μM TR, an Nrf-2 inhibitor, as previously described (55) before treatment with DMF (30 μM). The concentration of MPTP (3 mM) used was based on previous in vitro studies (62). After 24 h, cell lysates were prepared for Western blot analysis.
SH-SY5Y cultures were divided into four groups: 1. Control group (Ctr): differentiated cells were cultured with normal medium; 2. MPTP group: differentiated cells were treated with 3 mM MPTP; 3. MPTP+DMF 30 μM group: differentiated cells were treated with 30 μM DMF for 2 h before addition of 3 mM MPTP; 4. MPTP+DMF+TR group: differentiated cells were pretreated with 30 μM DMF for 2 h and with 1 μM TR for 30 min, before addition of 3 mM MPTP.
Western blot analysis
Western blot analysis was performed as previously described (62). The membrane was incubated overnight at 4°C with: anti-iNOS (1:500; Transduction Labs) and anti-Mn-SOD (1:500; Merck-Millipore). To ascertain that blots were loaded with equal amounts of protein lysate, they were also incubated with β-actin antibody (1:500; Santa Cruz Biotechnology). Signals were detected as previously described (62).
Materials
Unless otherwise stated, all compounds were acquired from Sigma-Aldrich. All other chemicals were of the highest commercial grade available. All stock solutions were prepared in nonpyrogenic saline (0.9% NaCl; Baxter, Milan, Italy).
Statistical evaluation
All values are given as mean ± SEM, and they are representative of at least three independent experiments. Data were examined by one-way analysis of variance followed by a Bonferroni post hoc test for multiple comparisons. A p-value of <0.05 was considered significant.
Footnotes
Acknowledgments
The authors would like to thank Antonietta Medici for excellent technical assistance during this study and Miss Valentina Malvagni for editorial assistance with the article.
Author contributions
E.E. and S.C. planned the experiments, MiC and MaC performed the experiments, R.C. analyzed the results, and F.B. and G.C. performed the biochemical analysis and prepared the article. All authors read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.