
Abstract
The APEX nuclease (multifunctional DNA repair enzyme) 1 (APEX1) has a disordered N-terminus, a redox, and a DNA repair domain. APEX1 has anti-apoptotic properties, which have been linked to both domains depending on cell type and experimental conditions.
Aims:
As protection against apoptosis is a hallmark of vessel integrity, we wanted to elucidate whether APEX1 acts anti-apoptotic in primary human endothelial cells and, if so, what the underlying mechanisms are.
Results:
APEX1 inhibits apoptosis in endothelial cells by reducing Cathepsin D (CatD) cleavage, potentially by binding to the unprocessed form. Diminished CatD activation results in increased Thioredoxin-1 protein levels leading to reduced Caspase 3 activation. Consequently, apoptosis rates are decreased. This depends on the first twenty amino acids in APEX1, because APEX1 (21-318) induces CatD activity, decreases Thioredoxin-1 protein levels, and, thus, increases Caspase 3 activity and apoptosis. Along the same lines, APEX1 (1-20) inhibits Caspase 3 cleavage and apoptosis. Furthermore, re-expression of Thioredoxin-1 via lentiviral transduction rescues endothelial cells from APEX1 (21-318)-induced apoptosis. In an in vivo model of restenosis, which is characterized by oxidative stress, endothelial activation, and smooth muscle cell proliferation, Thioredoxin-1 protein levels are reduced in the endothelium of the carotids.
Innovation:
APEX1 acts anti-apoptotic in endothelial cells. This anti-apoptotic effect depends on the first 20 amino acids of APEX1.
Conclusion:
As proper function of the endothelium during life span is a hallmark for individual health span, a detailed characterization of the functions of the APEX1N-terminus is required to understand all its cellular properties. Antioxid. Redox Signal. 26, 616–629.
Introduction
T
Anti-apoptotic properties of the multifunctional protein APEX1 have previously been described in tumor cells. However, it was not clear, which domain of APEX1 is required for this function. In this study, we describe for the first time that APEX1 suppresses apoptosis in primary human endothelial cells. The N-terminal 20 amino acids of APEX1 are fully sufficient for apoptosis protection and the downstream target is Thioredoxin-1. Thus, small peptide approaches using the N-terminus of APEX-1 could in the future be relevant for disease protection.
Therefore, several studies were performed to identify small-molecule inhibitors specifically targeting the different domains in APEX1 to understand specific functions of this protein in normal and tumor cells [for review see Ref. (31)].
The endothelium plays a central role as a barrier between the blood stream and the surrounding tissue. However, numerous environmental factors have a negative impact on the endothelium by changing the circulating levels of cytokines, chemokines, and/or growth factors. Those mediators influence the endothelium and seem to importantly contribute to endothelial dysfunction (8, 37, 44).
One hallmark of endothelial dysfunction is the damage of endothelial cells leading to apoptotic cell death (3). Several studies have been undertaken to improve endothelial function by inhibiting endothelial cell apoptosis (12, 28, 40, 43, 47). Already decades ago, it was evident that oxygen deprivation for a certain time led to endothelial cell death, which, on the contrary, was also induced by the process of reoxygenation. This was associated with a burst in reactive oxygen species (ROS) damaging the endothelium and resulting in apoptosis induction. One central transcription factor protecting the endothelium under hypoxia and reoxygenation is the Hypoxia Inducible Factor 1 alpha (HIF1α) (5, 23, 42). The regulation of HIF1α under those conditions has been attributed to APEX1 (16, 21), and thus, APEX1 has an anti-apoptotic and prosurvival function when the oxygen concentration is unphysiological. However, till now, the role of APEX1 in apoptosis protection under basal conditions and a potential function of its different domains have not been investigated.
Moreover, several studies suggested that APEX1 acts in concert with Thioredoxin-1 (Trx-1) to regulate the activity of numerous transcription factors and thereby influences cellular functions (2, 4, 9, 16, 22, 41). Indeed, a colocalization between endogenous Trx-1 and APEX1 in primary human endothelial cells has been shown (30).
Therefore, we investigated for the first time the effects of APEX1 on basal apoptosis in primary human endothelial cells and showed that its anti-apoptotic activity depends on the N-terminus of the protein, but is independent of the DNA repair domain. Since the N-terminus of APEX1 has recently been described as important for nuclear targeting and interaction specificity (29), we also investigated the functions of the N-terminus of APEX1. Moreover, we also demonstrated a role for Trx-1 in the anti-apoptotic function of APEX1.
Results
The integrity of the endothelium is essential for proper vessel function. APEX1 is a bifunctional protein, which contains two nonoverlapping domains – a redox domain and a DNA repair domain. The redox function – needed for reductive activation of transcription factors – requires the region between amino acids 36 and 127, with the cysteines in position 65 and 93 being critical for redox activity. On the contrary, the DNA repair domain resides in the C-terminus starting at amino acid 162 and extending to the end of APEX1 (39, 45). In contrast to these domains, the functions of the N-terminus of APEX1 are less well defined.
APEX1 acts anti-apoptotic under hypoxic conditions in endothelial cells (21). The underlying mechanisms are supposed to include the activation of HIF1α and NF-κB. However, the role of APEX1 under basal conditions has not been investigated. Therefore, we first cloned an expression vector for APEX1 and transiently overexpressed it in primary human endothelial cells. APEX1 significantly inhibited Caspase 3/7 activity and apoptosis measured by Annexin V-positive/propidium iodide (PI)-negative cells (Fig. 1A, B and Supplementary Fig. S1; Supplementary Data are available online at
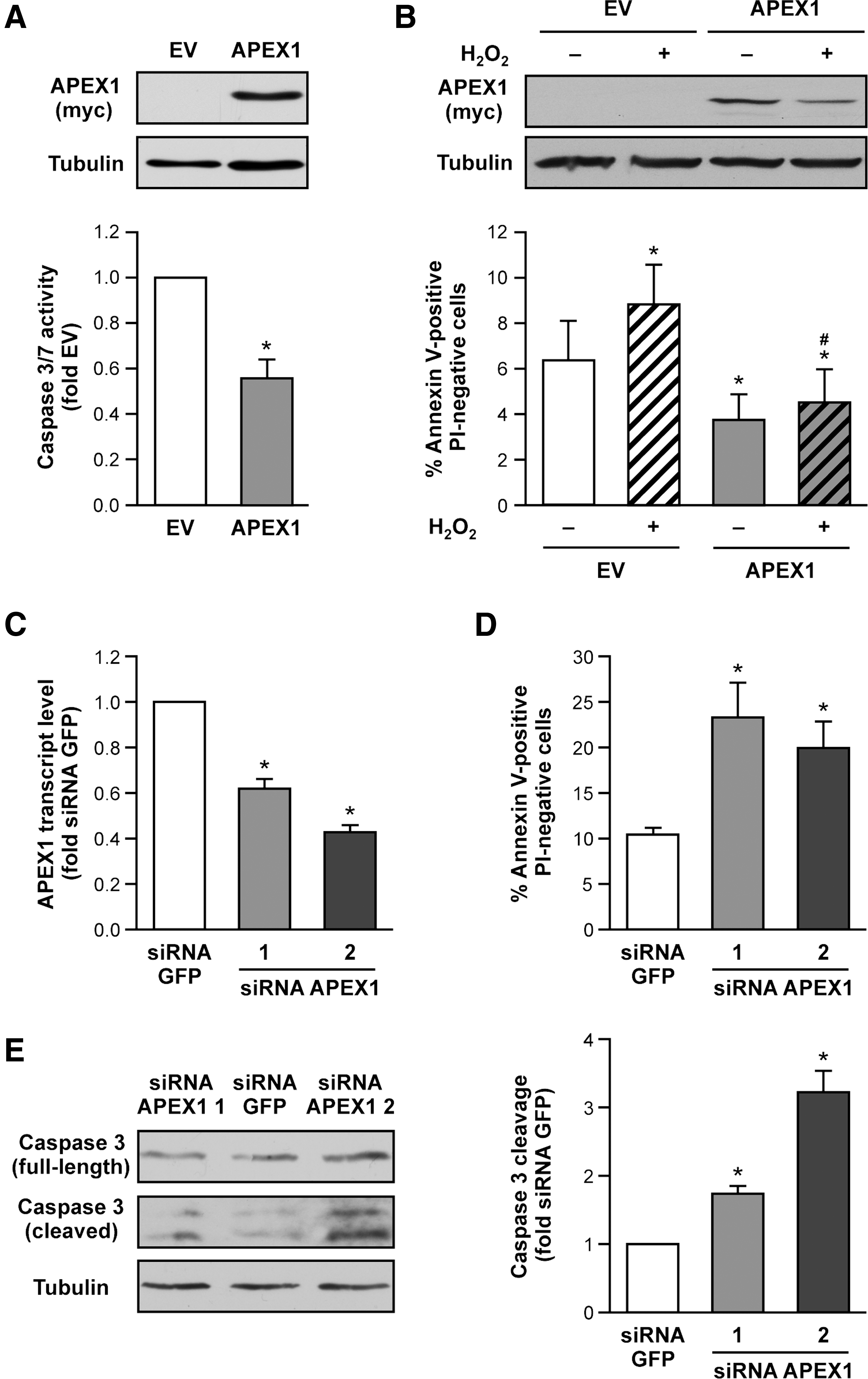
APEX1 deficiency leads to early embryonic lethality with the embryos dying shortly after implantation, indicating a critical role for APEX1 in normal cellular functions. Notably, many cells in the very early APEX1 knockout embryos are characterized by pyknotic nuclei, that is, chromatin condensation, which is a feature of apoptosis (46). Thus, we also examined apoptosis induction after partial knockdown of endogenous APEX1 with siRNA avoiding complete depletion of the protein (Fig. 1C). Reduction of APEX1 levels resulted in increased apoptosis and cleaved Caspase 3 (Fig. 1D, E and Supplementary Fig. S1).
Next, we generated two mutants of APEX1 to understand (i) the role of the DNA repair domain [APEX1 (1-127)] and (ii) of the N-terminus [APEX1 (21-318)] in apoptosis protection (Fig. 2A). Both mutants can be expressed to a similar extent as full-length APEX1 in endothelial cells (Fig. 2B, C and Supplementary Fig. S2). The localization pattern shown in Figure 2B demonstrates that all three proteins can be found in the nucleus and in the cytosol. However, APEX1 (21-318) seems to have increased cytosolic localization. Nevertheless, it can be excluded that the first 20 amino acids in APEX1 are alone responsible for nuclear localization in endothelial cells. With respect to apoptosis protection, APEX1 (1-127) inhibited apoptosis, whereas APEX1 (21-318) significantly increased apoptosis when compared to empty vector control as well as to APEX1 (Fig. 2C). These results lead to the conclusion that the DNA repair domain of APEX1 is dispensable for apoptosis protection in endothelial cells.
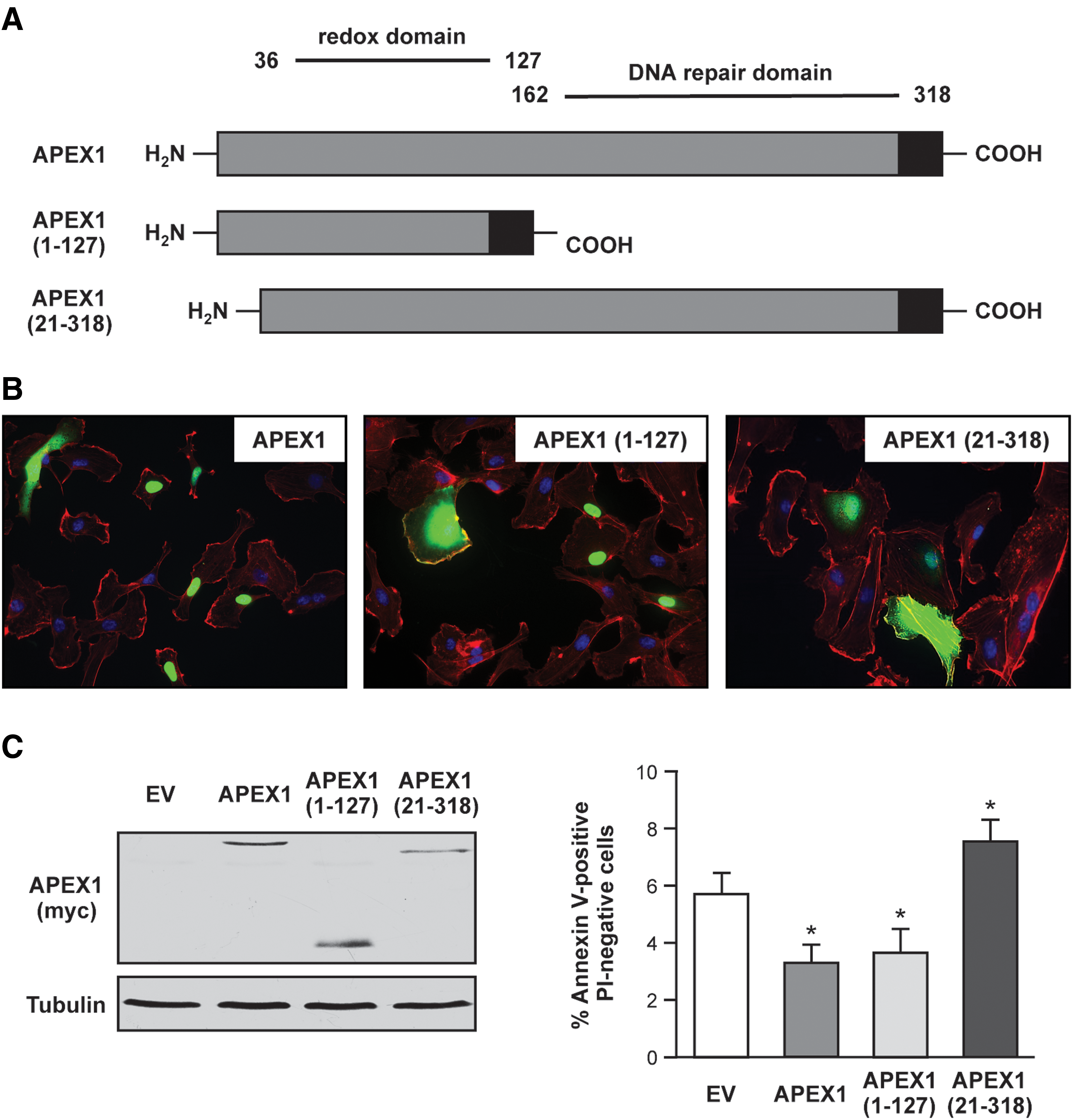
Unexpected and more surprising were the data obtained with the N-terminally truncated mutant APEX1 (21-318). Therefore, we next sought to determine mechanisms underlying the anti-apoptotic properties of the full-length protein and the pro-apoptotic properties of APEX1 (21-318). To reinforce the findings obtained with Annexin V binding, we also measured Caspase 3/7 activity. Indeed, APEX1 inhibited Caspase 3/7 activation (Fig. 3A). In contrast, but in accordance with the data obtained before (Fig. 2C), APEX1 (21-318) significantly increased Caspase 3/7 activity when compared to empty vector control as well as to cells overexpressing full-length APEX1.
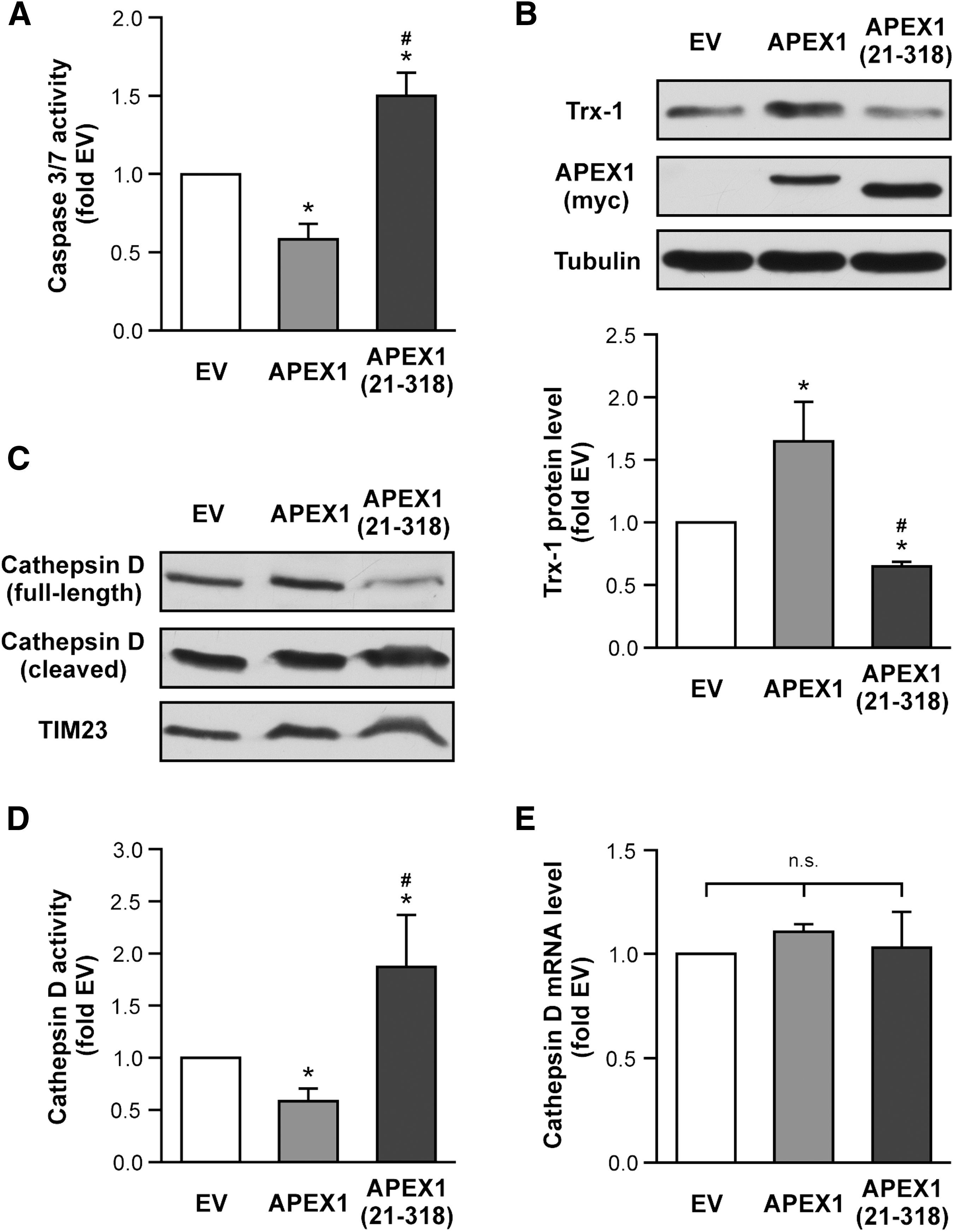
We have previously shown that Trx-1 is essential for the protection of endothelial cells against apoptosis (17, 20, 36, 48). Therefore, we next investigated the effects of APEX1 and APEX1 (21-318) on Trx-1 protein levels. Interestingly, we found that expression of full-length APEX1 increased the amount of Trx-1, whereas APEX1 (21-318) significantly reduced Trx-1 protein levels (Fig. 3B and Supplementary Fig. S3). Since Trx-1 is degraded by the lysosomal protease Cathepsin D (CatD) (15, 19), and microinjection of CatD in fibroblasts leads to Caspase 3 activation and apoptosis induction (34), we next determined the activity of CatD by the amount of cleaved CatD in an immunoblot (Fig. 3C and Supplementary Fig. S3) as well as with a quantitative activity assay (Fig. 3D). Again, APEX1 reduced CatD activity, whereas APEX1 (21-318) drastically increased active CatD.
Because APEX1 and Trx-1 have been implicated in the regulation of transcription factors (22, 41), we also determined CatD RNA levels after APEX1 overexpression. As demonstrated in Figure 3E, neither the full-length protein nor APEX1 (21-318) affected CatD RNA levels. Thus, the reduction in full-length CatD and concomitant increase in cleaved CatD in cells overexpressing APEX1 (21-318) indeed mirror the enhanced CatD activity. Our data indicate that APEX1 (21-318) overexpression reduces Trx-1 protein levels and induces apoptosis. Next, we examined whether we can counteract the pro-apoptotic effect of APEX1 (21-318) by blocking CatD using the cell-permeable CatD inhibitor, Pepstatin A-penetratin. Indeed, inhibition of CatD abrogated APEX1 (21-318)-induced apoptosis and restored Trx-1 protein levels (Supplementary Figs. 1 and S sup1).
Since full-length APEX1 and APEX1 (21-318) have an influence on CatD, we next investigated whether CatD is in a complex with APEX1. Indeed, we found that the unprocessed CatD is associated with APEX1, but not with APEX1 (21-318) (Fig. 4A and Supplementary Fig. S4). Thus, one could speculate that association of APEX1 and CatD inhibits processing and thus activation of CatD. On the contrary, APEX1 (21-318) enhanced CatD cleavage, suggesting that APEX1 (21-318) might affect one or more of the proteases involved in CatD processing, which is required for its activation (33).
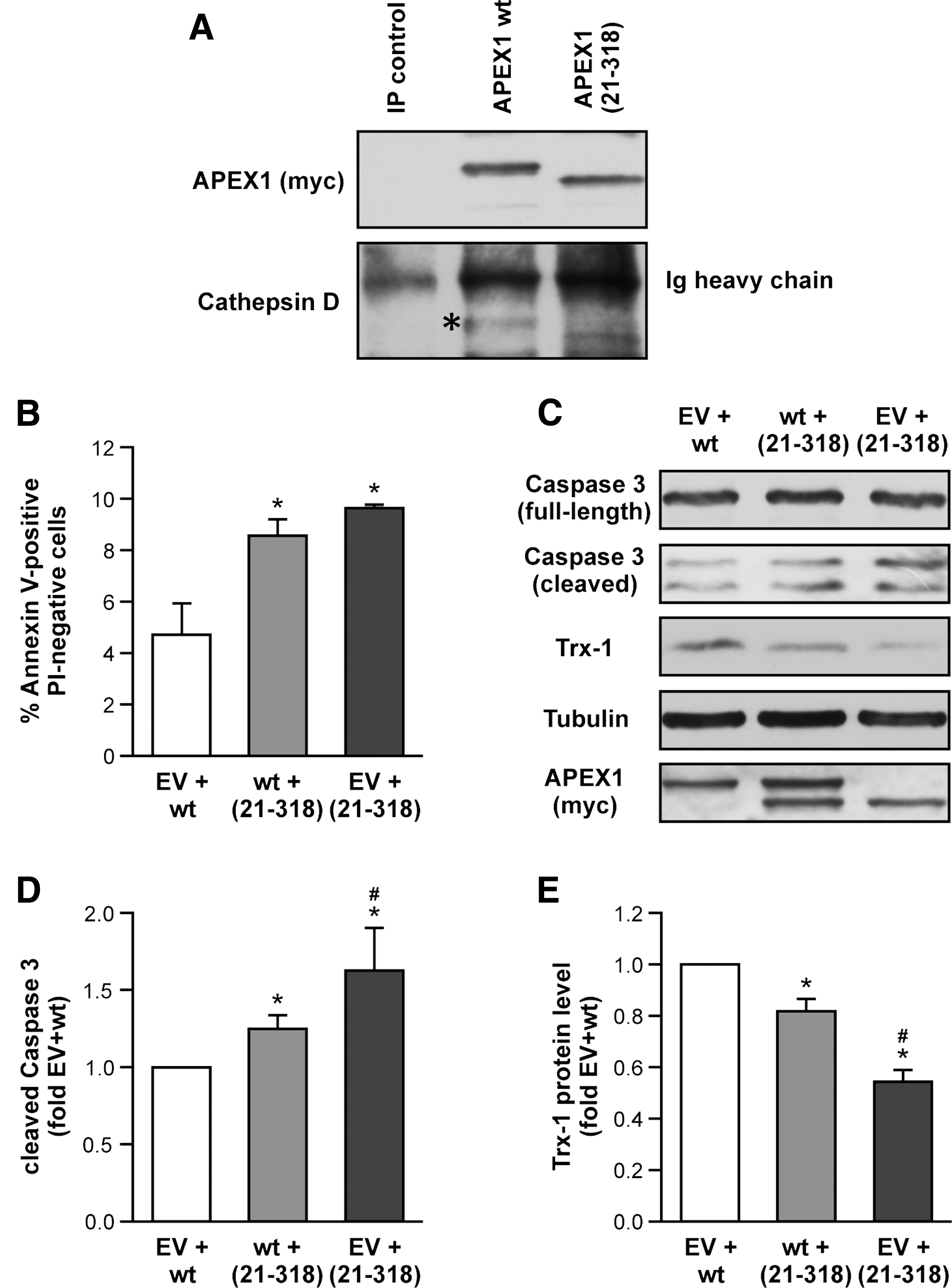
To further understand whether APEX1 wild type (wt) and APEX1 (21-318) compete against each other, we performed cotransfection experiments using equal amounts of empty vector and APEX1 wt, APEX1 wt and APEX1 (21-318), or empty vector and APEX1 (21-318). As expected, cotransfection of empty vector and APEX1 (21-318) displayed the highest apoptosis induction (Fig. 4B). Interestingly, cotransfection of APEX1 wt and APEX1 (21-318) resulted in increased apoptosis induction compared to empty vector and APEX1 wt. Thus, the presence of APEX1 (21-318) seems to overcome the protective effect of APEX1 wt. Along the same line, APEX1 (21-318) increased cleaved Caspase 3 (Fig. 4C, D and Supplementary Fig. S4) and reduced Trx-1 protein levels (Fig. 4C, E and Supplementary Fig. S4).
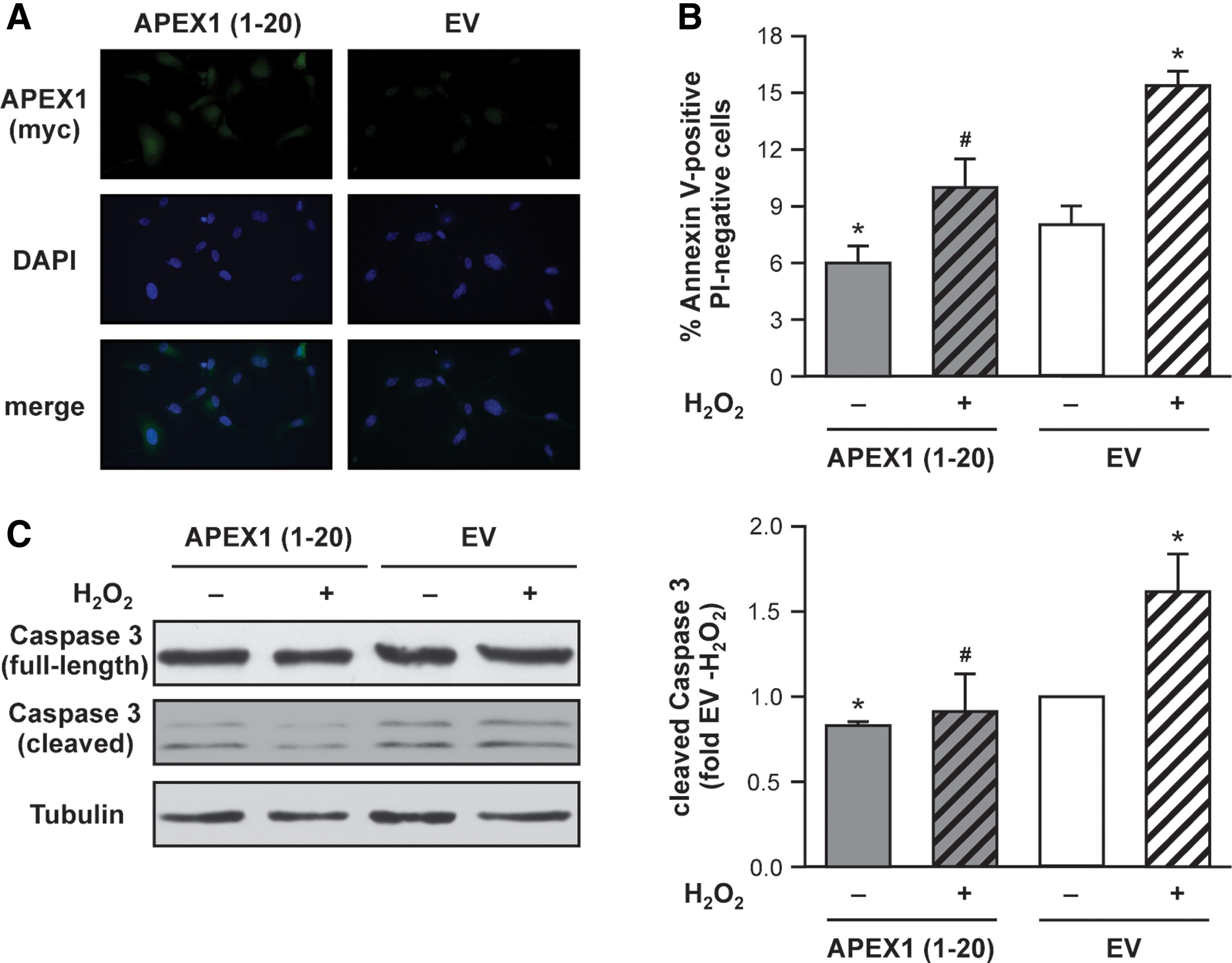
These data further support an important role for the first 20 amino acids of APEX1 in apoptosis protection in endothelial cells. Therefore, we next cloned an expression vector for only the N-terminal region [APEX1 (1-20)] and investigated the impact of APEX1 (1-20) on basal- and H2O2-induced apoptosis and Caspase 3 activation. After expression of APEX1 (1-20), we first analyzed its localization pattern. The small protein can be found in the nucleus and in the cytosol (Fig. 5A). Overexpression of APEX1 (1-20) in endothelial cells resulted in inhibition of basal- and H2O2-induced apoptosis as well as Caspase 3 cleavage (Fig. 5B, C and Supplementary Fig. S5). Of note, the effects of APEX1 (1-20) are less pronounced than those of APEX1 wt (Fig. 1A, B). However, these data clearly demonstrate that the first 20 amino acids of APEX1 have anti-apoptotic properties in endothelial cells.
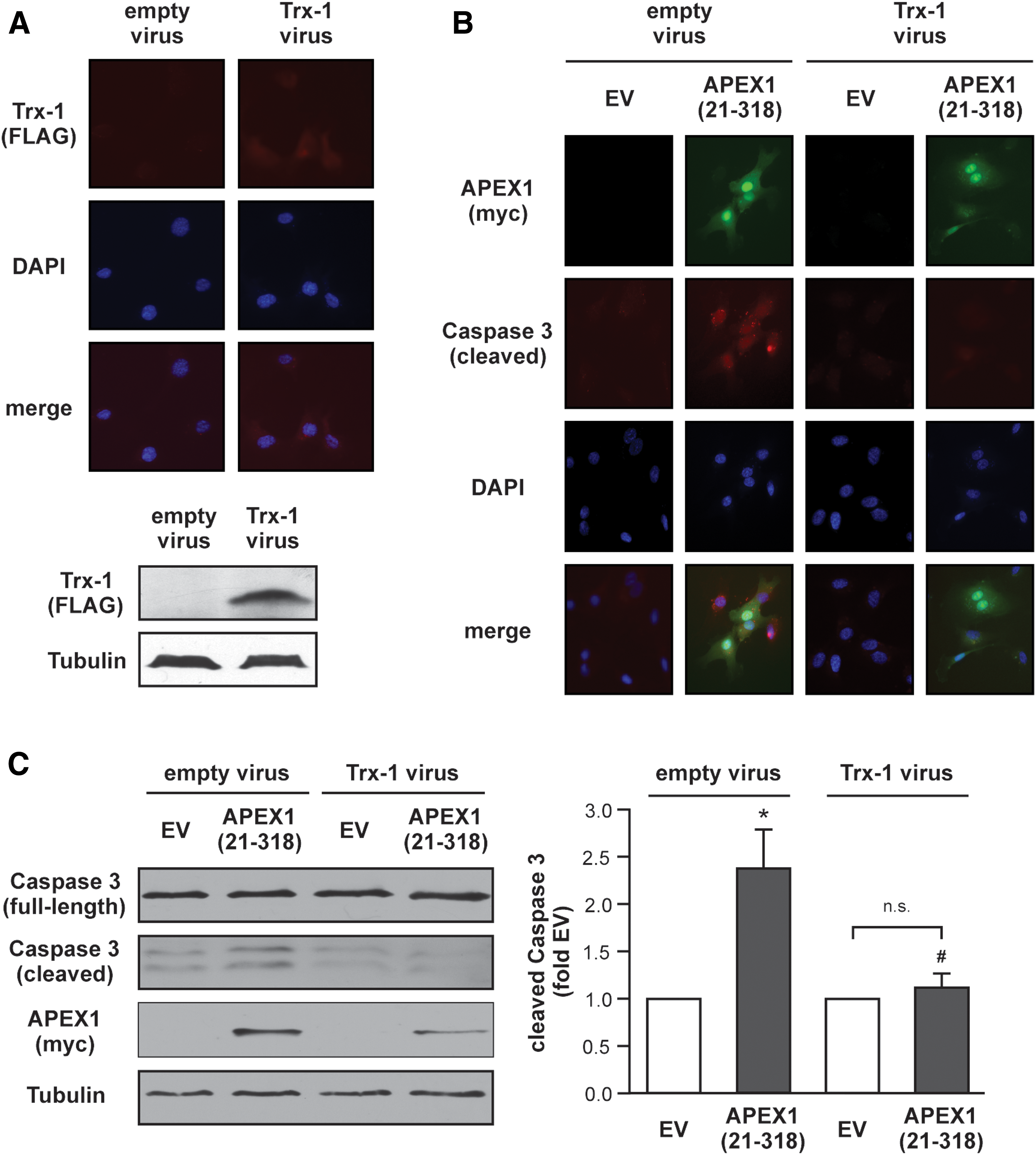
Next, we wanted to determine whether permanent re-expression of Trx-1 can overcome the pro-apoptotic effects of APEX1 (21-318). The stable re-expression of Trx-1 as well as expression of APEX1 (21-318) and cleavage of Caspase 3 were analyzed by immunostaining and immunoblots (Fig. 6 and Supplementary Fig. S6). As demonstrated in Figure 6B, only cells, which overexpress APEX1 (21-318), show an increase in cleaved Caspase 3 under conditions, where the protein levels of Trx-1 are not increased. This effect was completely blunted in cells with increased Trx-1 protein levels. Thus, the amount of Trx-1 protein determines the fate of endothelial cells.
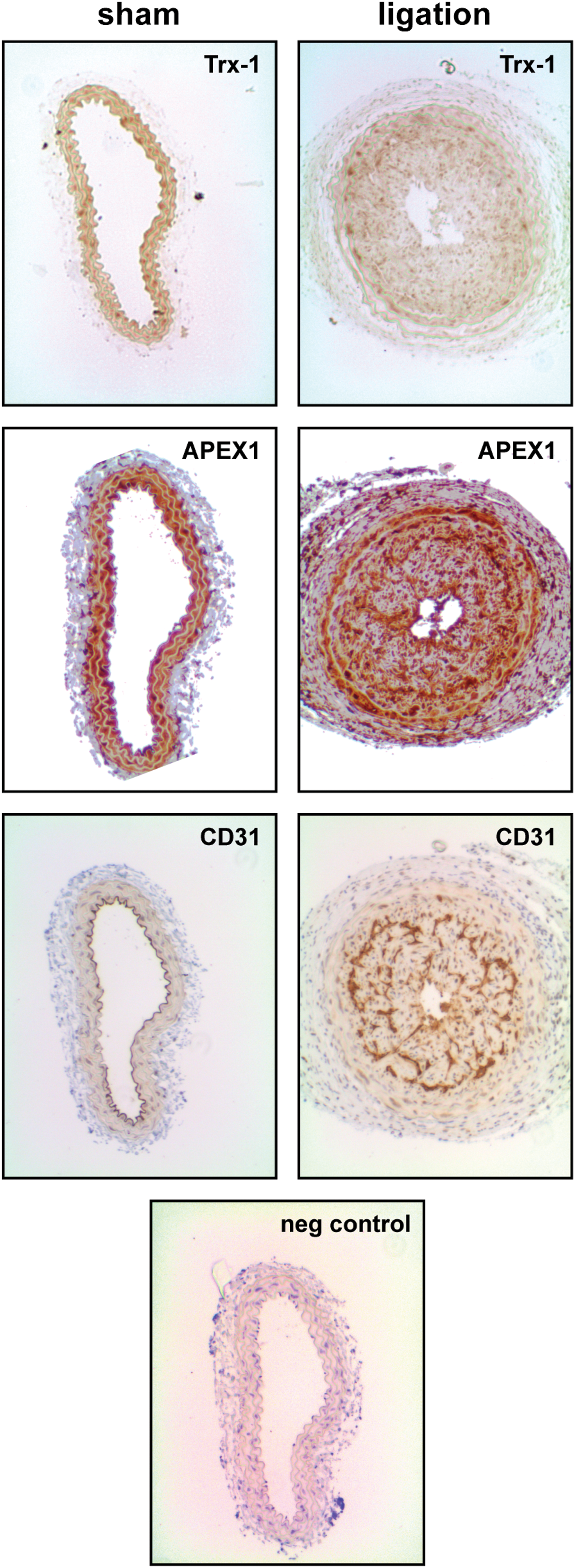
Finally, we used an in vivo model of restenosis induction by carotid ligation. This model is characterized by increased oxidative stress, smooth muscle cell proliferation, and activation of the endothelium (10). The protein levels of Trx-1 and APEX1 in this model have not been determined before. However, activation of the endothelium and smooth muscle cell proliferation have been associated with altered APEX1 pathways like disturbed localization of APEX1, reduced activation of the transcription factor c-Jun, and increased ROS formation (11, 27). Furthermore, oxidative stress has been demonstrated to reduce Trx-1 levels in the endothelium (15, 19). Therefore, we investigated the protein levels of Trx-1 and APEX1 in CD31-positive cells in this model. As demonstrated in Figure 7, ligation leads to a loss of Trx-1 in the endothelium, whereas APEX1 levels seem to be unaltered in the endothelium but increased in smooth muscle cells.
Discussion
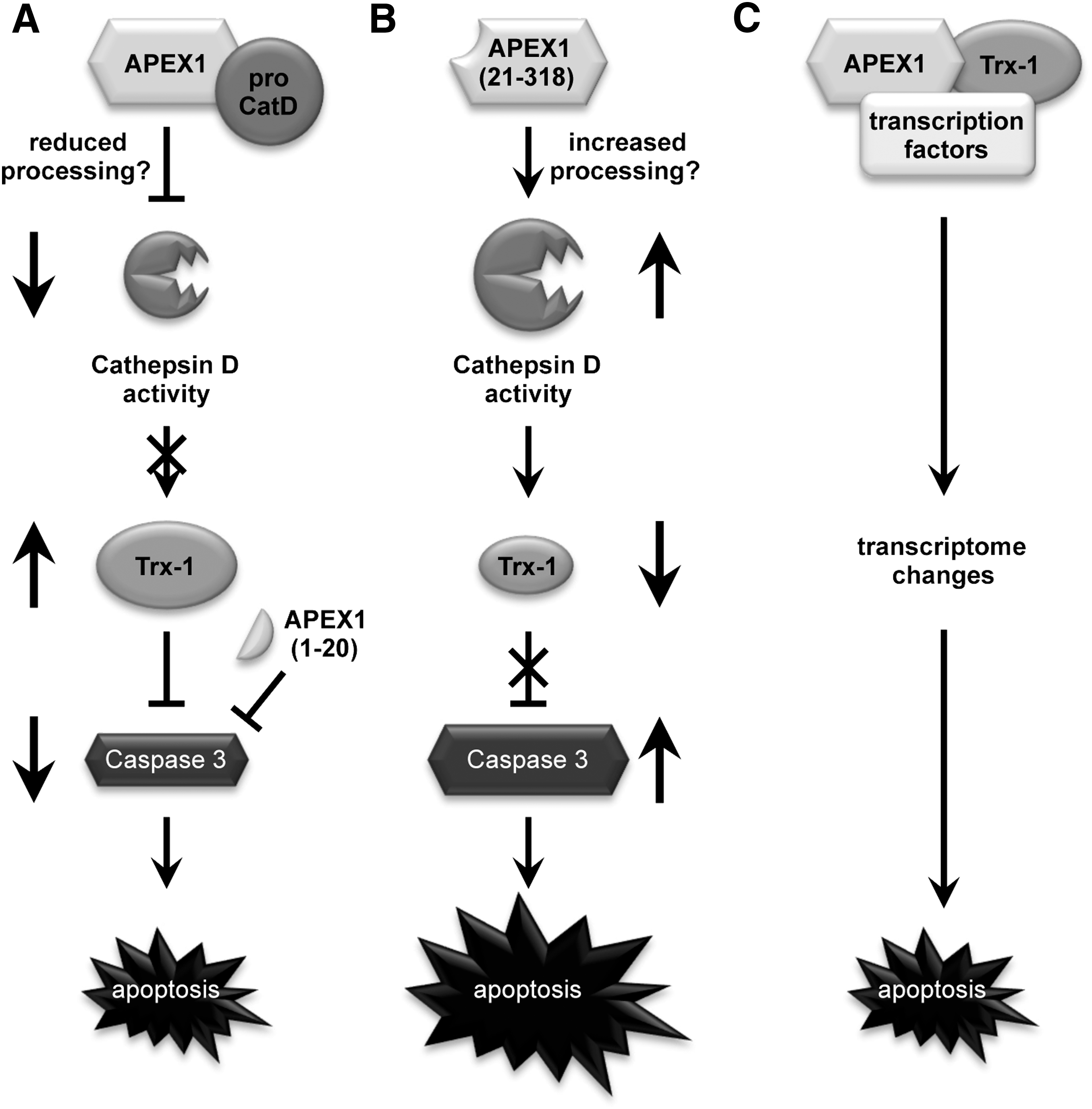
The data of the present study show for the first time that APEX1 acts anti-apoptotic under physiological conditions by inhibiting CatD activity, and thus, Trx-1 degradation and Caspase 3 activation. This anti-apoptotic property depends on the first 20 amino acids of APEX1, since the APEX1 (21-318) mutant increases CatD cleavage and activity and induces Trx-1 degradation and Casapse-3 activation. Moreover, expression of a protein encompassing only these 20 amino acids can protect endothelial cells against apoptosis (Fig. 8). Under conditions of oxidative stress ex vivo and in vivo, Trx-1 is degraded and endothelial function is reduced.
Anti-apoptotic properties of APEX1 have been demonstrated in the hypoxic endothelium by Hall et al. (21). The authors showed that hypoxic conditions reduced APEX1 protein levels and transient overexpression inhibited apoptosis induction. It was speculated that the anti-apoptotic effect of APEX1 under hypoxia is dependent on NF-κB. However, under the hypoxic conditions chosen by Hall et al. (2% O2), NF-κB was not activated, but APEX1 was still anti-apoptotic. In contrast, the anti-apoptotic effect of APEX1 in TNFα-treated endothelial cells was dependent on NF-κB activation. Therefore, it was suggested that APEX1 could act anti-apoptotic by stabilizing HIF1α under hypoxic conditions (21). This is supported by findings, which demonstrated that inhibition of APEX1 redox function reduced the transcription factor activity not only of NF-κB but also of HIF1α (13). Thus, APEX1 can influence transcription factors, but dependent on the conditions, different transcription factors are regulated by APEX1. Under our experimental conditions, APEX1 was neither able to induce the DNA binding activity of the p65 subunit of NF-κB (TransAM activity assay: 98% ± 12% compared to EV transfected cells, n = 4) nor could we find increased HIF1α protein levels (Supplementary Figs. 2 and S sup2).
However, we found here an additional mechanism, by which APEX1 stabilizes Trx-1 protein levels through inhibition of CatD processing, and thus, activation of this protease. Trx-1 is known to be essential for endothelial function as well as for survival of cells and organisms. Trx-1-deficient mice die shortly after implantation (32), demonstrating that Trx-1 is essential for early development of the mouse embryo. Along the same line, partial knockdown of Trx-1 in endothelial cells leads to cell death (20). Furthermore, senescence induction in endothelial cells is accompanied by loss of Trx-1 and can be rescued by lentiviral re-expression of Trx-1 (15). Therefore, our newly discovered mechanism, which increases Trx-1 protein levels in endothelial cells, could be of therapeutic interest.
Unfortunately, it has to be noted that APEX1 as well as Trx-1 have been well described to be essential for tumor growth and both proteins are expressed in a variety of different tumors. One of the most important functions of APEX1 and Trx-1 in tumor development is the anti-apoptotic property of both enzymes. Interestingly, in cancer biology, the DNA repair domain of APEX1 seems to play an important role in protecting the tumors [for reviews see Refs. (6, 25)], whereas in endothelial cells, the DNA repair domain is dispensable for its anti-apoptotic properties – as we demonstrate here. Moreover, a huge effort has been undertaken to also characterize the redox domain within APEX1. This resulted in the design of small molecules to specifically inhibit the redox activity of APEX1. Those small molecules inhibited cancer cell growth as well as network formation of tumor endothelial cells (26). Thus, inhibition of the redox function of APEX1 could also be important for cancer therapy.
However, our experiments clearly show that the N-terminal 20 amino acids, and thus, most likely not the redox domain of APEX1, are required for apoptosis protection in endothelial cells. Structural analyses of APEX1 show that the first 62 amino acids of APEX1 seemingly do not adopt an ordered structure, while the remainder of the protein is characterized by extensive α-helices and β-sheets (PDBsum entry 3u8u at
Moreover, the 33 N-terminal residues of APEX1 are required for complex formation with nucleophosmin, and it has been proposed that this interaction regulates the ability of the endonuclease to remove damaged RNA molecules (38). Damaged RNA molecules can be taken up by lysosomes (14), and thus, it is conceivable that a dysregulation of RNA metabolism can interfere with lysosomal activity. Therefore, impairment of RNA quality control might play a role in the regulation of cell death. Interestingly, in a model of Helicobacter pylori infection of gastrointestinal epithelial cells, it has been demonstrated that APEX1 can be acetylated at the lysine residues in position 6 or 7. This modification is important for its recruitment to a multiprotein complex on so-called negative calcium response elements. However, the acetylation status of APEX1 did not affect the transcriptional activity of p53 (7).
Notably, we did not observe changes in the CatD mRNA levels after overexpression of APEX1 (21-318), suggesting that the extreme N-terminus of APEX1 might not be involved in the activation of transcription factors regulating CatD expression, but may rather be involved in the processing of the enzyme precursor(s). Our data indicate that APEX1 interacts with unprocessed CatD (Fig. 4A), which might inhibit its processing. Interestingly, this interaction is not observed with the mutant APEX1 (21-318), which increases CatD cleavage and activity (Fig. 3C, D). Therefore, one could speculate that the N-terminally deleted APEX1 protein enhances the processing of CatD. However, the activation of CatD requires multiple proteases, not all of which have been identified so far (33), and therefore, uncovering the mode of action of APEX1 (21-318) requires further studies, including complete characterization of the CatD processing cascade. However, other mechanisms could also be imagined, and future studies will hopefully uncover the mechanism by which APEX1 regulates CatD activation.
Innovation and Conclusion
In conclusion, we present here that characterization of the redox domain and the DNA repair domains of APEX1 is probably not sufficient to understand the full spectrum of its functions, because the N-terminus of APEX1 is required for the anti-apoptotic properties in the normal endothelium. Moreover, the N-terminus of APEX1 is crucial for the maintenance of Trx-1 protein levels. Re-expression of Trx-1 can block the pro-apoptotic properties of APEX1 (21-318), demonstrating that a certain Trx-1 level is essential to protect the endothelium. This can be also seen in vivo in the ligation model, in which loss of Trx-1 is present in the restenosis areas.
Besides the post-translational mechanisms demonstrated here, the regulation of transcription factors by APEX1 in concert with Trx-1 should not be overlooked (Fig. 8). Thus, depending on the cellular conditions, multiple APEX1- and Trx-1-dependent pathways might contribute to protection of the endothelium.
Materials and Methods
Cell culture
Primary human endothelial cells were cultured as previously described (17). After detachment with trypsin, cells were grown for at least 18 h as described (17).
Transient transfection of endothelial cells
Cells were transiently transfected with plasmid DNA using SuperFect (17) and with siRNAs using HiPerFect as described previously (18). Transfection reagents and siRNAs were purchased from Qiagen (Hilden, Germany).
Apoptosis assay
Detection of cell death was performed by flow cytometry using Annexin V-APC binding and PI staining as described previously (36); early apoptotic cells are Annexin V positive and PI negative.
Caspase 3/7 activity assay
Caspase 3/7 activity was measured using the Caspase 3 activity assay (Cell Signaling Technology, Frankfurt, Germany) with the following modifications: samples were lysed in CHAPS buffer (50 mM HEPES pH 7.5, 10% Sucrose, 0.5% Triton X-100, 0.1% CHAPS), including protease inhibitor cocktail (BioTool, Munich, Germany). Treatment of identically transfected endothelial cells with ZVAD (20 μM; Enzo Life Sciences GmbH, Lörrach, Germany) served as a negative control for the Caspase 3/7 activity measurements and was always subtracted from the corresponding sample. The remaining activity thus reflects solely Caspase 3/7 activity. DMSO was used as solvent control. Caspase 3/7 activity was normalized to protein content.
CatD activity assay
CatD activity was measured using CatD activity fluorimetric assay (BioVision, Milpitas, CA) according to the manufacturer's instructions. Human recombinant CatD (Sigma, Deisenhofen, Germany) was used as a positive control. Negative control samples were incubated with 1 μg/ml pepstatin A. The CatD activity was normalized to protein content.
Determination of NF-κB p65 DNA binding activity
The DNA binding activity of the p65 subunit of the transcription factor NF-κB was determined with the TransAM® NFκB p65 Transcription Factor ELISA Kit (Active Motif, La Hulpe, Belgium) according to the manufacturer's instructions.
Immunofluorescence staining
Endothelial cells were fixed in 4% paraformaldehyde, permeabilized for 15 min using 0.3% Triton X-100 and 3% bovine serum albumin in phosphate-buffered saline (PBS). For a direct immunostaining, cells were incubated with an anti-FLAG antibody directly conjugated with Alexa Fluor 594 (1:25; New England Biolabs, Frankfurt, Germany), an anti-myc-tag antibody directly conjugated with Alexa Fluor 488 (1:25; New England Biolabs, Frankfurt, Germany), or an anti-cleaved Caspase 3 antibody directly conjugated with Alexa Fluor 594 (1:25; New England Biolabs, Frankfurt, Germany) in PBS overnight at 4°C. Actin filaments were stained with Alexa Fluor 568-coupled phalloidin for 30 min at room temperature (1:100; Invitrogen, Karlsruhe, Germany). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Karlsruhe, Germany) for 5 min at room temperature. Cells were washed and mounted with ProLong Gold antifade mounting medium (Invitrogen, Karlsruhe, Germany). Fluorescence images were taken with an AXIOVERT 200 M microscope (Zeiss, Jena, Germany, 40 × magnification, oil).
Preparation of protein lysates
Endothelial cells were washed with PBS and scraped off the dishes on ice. After centrifugation, the resulting pellet was lysed in RIPA-buffer for 30 min on ice. After removing cellular debris by centrifugation (16,000 × g, 15 min, 4°C), protein concentrations were measured using the Bradford protein assay (BioRad, Munich, Germany).
Immunoblotting
Immunoblotting was performed as previously described (18). Blotting membranes were incubated with primary antibodies directed against myc-tag, Caspase 3, HIF1α (1:500 and 1:300, respectively, all Cell Signaling Technology, Frankfurt, Germany), Trx-1, CatD (both 1:500; Abcam, Berlin, Germany), TIM23 (1:500; BD Biosciences, Heidelberg, Germany), Topoisomerase I (1:250; Santa Cruz Biotechnology, Heidelberg, Germany), PTP1B (1:1000; ECM Biosciences, Cologne, Germany), or Tubulin, FLAG (1:50,000 and 1:300, respectively, both Sigma, Deisenhofen, Germany) overnight at 4°C, before they were washed and incubated with secondary antibodies according to standard procedures. Detection was performed by enhanced chemiluminescence using the ECL reagent (GE Healthcare, Freiburg, Germany) and standard X-ray films. Semi-quantitative analyses were performed on scanned X-ray films using ImageJ 1.42q (1).
Immunoprecipitation
Lysates (500 μg) were immunoprecipitated with 5 μg anti-myc-tag antibody or 5 μg anti-FLAG antibody overnight at 4°C. After incubation with protein A and G Sepharose (GE Healthcare, Solingen, Germany) for 2 h at 4°C, the beads were washed, resuspended in SDS-PAGE sample buffer, and subjected to gel electrophoresis on a 10% SDS-PAGE gel.
Cloning of APEX1 expression vectors
The APEX1 coding sequence without the translation termination codon was amplified from human endothelial cell cDNA using the primers hAPEX1 NT for1 (5′-CTCGAGAATTCCACCATGCCGAAGCGTGGGAAAAAG-3′) and hAPEX1 CT rev1 (5′-GCGCAAGCTTCAGTGCTAGGTATAGGGTGAT-3′). The resulting amplification product was cut with EcoRI and HindIII and inserted into pcDNA.3.1™/myc-His(-) (Invitrogen, Karlsruhe, Germany) opened with the same enzymes, thereby creating a contiguous reading frame from the APEX1 coding sequence into the the C-terminal myc/His6-epitope-tag. An identical approach was chosen for the two APEX1 deletion mutants APEX1 (1-127) and APEX1 (21-318). For the mutant APEX1 (21-318), an ATG initiation codon was introduced directly 5′ to the codon for proline in position 21. To create the expression vector for the mutant APEX1 (1-20) the downstream primer contained the coding sequence for a myc-epitope tag, and the resulting amplification product was used to replace the myc-His6-epitope tag in pcDNA.3.1/myc-His(-). The identity of all plasmids was confirmed by restriction enzyme digestion and DNA sequencing of the APEX1 coding region.
Semiquantitative reverse transcriptase polymerase chain reaction
RNA isolation and cDNA synthesis were performed as previously described (18). Gene-specific mRNA levels were determined by semiquantitative real-time polymerase chain reaction using a Rotor-Gene Q (Qiagen, Hilden, Germany). For the detection of the CatD cDNA, the primers hCTSD Ex07/08 for1 (5′-TCAGGGCGAGTACATGATCC-3′) and hCTSD Ex09 rev1 (5′-ATGTCCATGCCCATGAAGCC-3′) were used, for the APEX1 cDNA hAPEX1 Ex04 for1 (5′-ACAAGGAAGGGTACAGTGGC-3′) and hAPEX1 Ex05 rev1 (5′-GCCTTCCTGATCATGCTCCT-3′). The ERK2 and RPL32 cDNAs were amplified as controls for normalization using the primers hERK2 Ex08 for1 (5′-TGCTAGATTCCAGCCAGGAT-3′) and hERK2 Ex09 rev1 (5′-ACGGCTCAAAGGAGTCAAAG-3′) or hmRPL32 Ex02 for1 (5′-GTGAAGCCCAAGATCGTCAA-3′) and hmRPL32 Ex03 rev1 (5′-TTGTTGCACATCAGCAGCAC-3′), respectively. All primer pairs were designed such that the two different primers are separated by an intron. Relative expression of APEX1 and CatD was calculated by the ΔΔCt method (35).
Production of lentiviral particles and transduction of endothelial cells
Virus production, concentration, and titration, as well as transduction of endothelial cells were performed as previously described (15). Endothelial cells were transduced with a multiplicity of infection of 15.
Animals
Wild-type mice were purchased from Jackson Laboratories (Bar Harbor, ME). Fourteen age-matched male mice aged 12 weeks on a regular chow diet were used for the carotid ligation procedure. The University of Virginia Animal Care and Use Committee approved all procedures and protocols used in this study.
Carotid ligation
Twelve-week-old mice received a daily heparin dose of 600 U/kg/day beginning 30 min before surgery and continuing for 4 days postsurgery. Mice were anesthetized with an intraperitoneal injection of 80 mg/kg Ketamine. The ventral surface of the neck was shaved, wiped with betadine and 70% ethanol, and a midline incision was made through the skin. The right common carotid artery was exposed and completely ligated just proximal to the carotid bifurcation. The left carotid artery served as an uninjured control. Both carotid arteries were harvested 21 days after injury and embedded transversely in paraffin. Six micrometer cross-sections of vessels were prepared and stained for Trx-1, APEX1, and CD31; for the negative control, the primary antibody was omitted. Paraffin sections were removed from xylene, deparaffinized, and rehydrated with dH2O. Briefly, the staining involved endogenous peroxidase staining for 30 min, antigen retrieval for 20 min, blocking with avidin/biotin blocking kit (Vector Laboratories, Burlingame, CA), secondary antibody staining for 1 h, followed by immunoperoxidase staining with Vectastain Elite ABC Kit (PK-6100; Vector Laboratories), and finally visualizing the stain using DAB (3,3-diaminobenzidine) HRP substrate. Hematoxylin was used as the counterstain for all sections imaged.
Footnotes
Acknowledgments
P.J. is a scholarship holder of the IRTG1902. This work was, in part, supported by the following grants: Deutsche Forschungsgemeinschaft (HA2868/9-1, HA2868/10-1, IRTG1902 P2) and Forschungskommission of the Medical Faculty, University of Duesseldorf (28/2014) to J.H.; NIH grant R01 DK096076 to N.L.; Deutsche Forschungsgemeinschaft (AL288/2-1, IRTG1902 P1) to J.A. V.S. was supported by a training grant NIH T32 GM007055-42 and an American Heart Association predoctoral fellowship 15PRE25560036.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.