
Abstract
Skeletal muscle shows high plasticity in response to external demand. Moreover, adult skeletal muscle is capable of complete regeneration after injury, due to the properties of muscle stem cells (MuSCs), the satellite cells, which follow a tightly regulated myogenic program to generate both new myofibers and new MuSCs for further needs. Although reactive oxygen species (ROS) and reactive nitrogen species (RNS) have long been associated with skeletal muscle physiology, their implication in the cell and molecular processes at work during muscle regeneration is more recent. This review focuses on redox regulation during skeletal muscle regeneration. An overview of the basics of ROS/RNS and antioxidant chemistry and biology occurring in skeletal muscle is first provided. Then, the comprehensive knowledge on redox regulation of MuSCs and their surrounding cell partners (macrophages, endothelial cells) during skeletal muscle regeneration is presented in normal muscle and in specific physiological (exercise-induced muscle damage, aging) and pathological (muscular dystrophies) contexts. Recent advances in the comprehension of these processes has led to the development of therapeutic assays using antioxidant supplementation, which result in inconsistent efficiency, underlying the need for new tools that are aimed at precisely deciphering and targeting ROS networks. This review should provide an overall insight of the redox regulation of skeletal muscle regeneration while highlighting the limits of the use of nonspecific antioxidants to improve muscle function. Antioxid. Redox Signal. 27, 276–310.
C. Time and space orchestration of muscle regeneration: link with muscular diseases
III. Definition of Free Radicals, Oxidative Stress, and Redox Signaling
A. Thiol oxidation as a major mechanism of oxidation-related post-translational modifications
B. From signal transduction to post-translational modifications
VII. Redox Regulation of MuSCs During Skeletal Muscle Regeneration
VIII. Redox Regulation of the Cells Present in the MuSC Microenvironment
IX. Skeletal Muscle Regeneration in Pathophysiological Contexts
I. Introduction
S
Several cell types present in the injured muscle tissue play various roles in the regulation of muscle regeneration (359). Interestingly, functions of MuSCs and their neighboring cells are altered by oxidative stress modulation (44, 255, 348). For instance, macrophages are a major source of reactive oxygen species (ROS) and reactive nitrogen species (RNS) on inflammation (16, 38). Moreover, ROS and RNS have been recently proposed to be crucial actors of stem cell biology through the modulation of various cellular processes (34).
Regarding skeletal muscle biology, oxidative stress has been mainly investigated in skeletal muscle physiology, myofiber adaptation, and contractile properties (22, 156, 206). It is likely that ROS/RNS-dependent modulation of the various cell types involved in skeletal muscle regeneration play important roles that may be altered in pathological contexts and may be the target of the development of therapeutic strategies. The present review focuses on redox regulation of skeletal muscle regeneration. After a presentation of this complex biological process, basics of ROS/RNS and antioxidant chemistry and biology are described in the context of skeletal muscle. Then, the current knowledge related to redox regulation of skeletal muscle regeneration is presented. Finally, specific alterations of redox regulation are described in various pathophysiological contexts, and therapeutic opportunities to improve skeletal muscle repair are discussed.
II. MuSCs and Skeletal Muscle Regeneration
Skeletal muscle is a highly stable tissue that shows a high capacity to adapt to various physiological demands due to the properties of the myofiber, a multi-nucleated cell extending along the length of the muscle, which is capable of atrophy or hypertrophy. Adaptation is also possible because of the properties of different types of muscle fibers, adapted for different purposes, including slow-twitch type I and fast-twitch type II myofibers. In addition to its adaptive properties, skeletal muscle is capable of full functional recovery after an injury through the process of muscle regeneration, which goes beyond healing as the parenchyma recovers its functionality (Fig. 1).
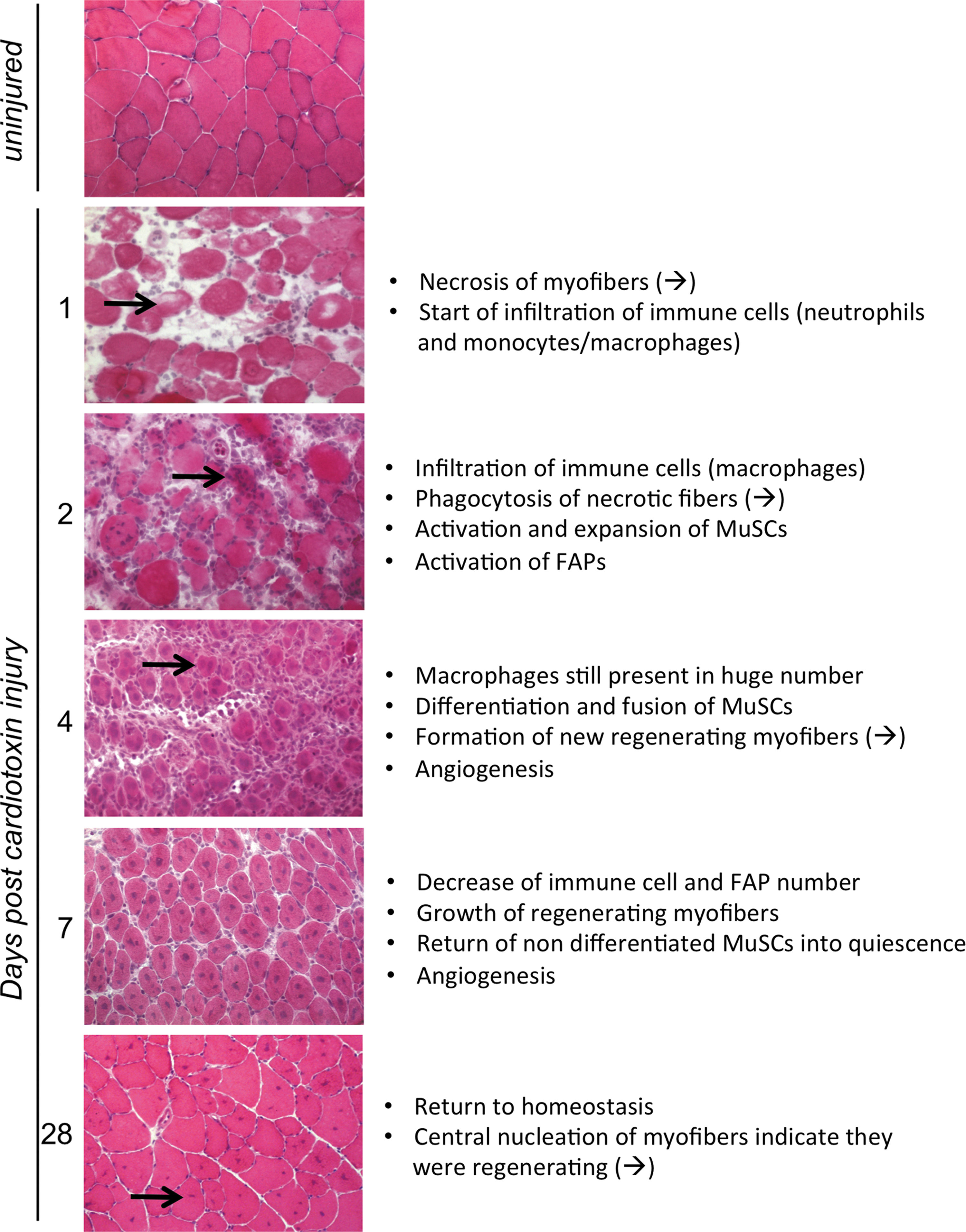
Skeletal muscle regeneration relies on the properties of MuSCs, which are the adult MuSCs that usually reside in a quiescent state between the muscle fiber and its surrounding basal lamina (54). MuSCs are activated on muscle injury and enter the cell cycle to proliferate. After an expansion phase, a large majority of MuSC progeny enters the myogenic differentiation program. The differentiated myogenic cells fuse to each other to form new myofibers and restore the tissue. A small subset of the expanding MuSCs does not commit into the terminal differentiation program but self-renews to replenish the pool of quiescent MuSCs for further needs. A general presentation of the cellular aspects of skeletal muscle regeneration is provided later, since the reader can refer to excellent recent reviews on the topic (92, 359). The molecular aspects of skeletal muscle regeneration will be specifically addressed in the sections dedicated to the redox regulation of muscle regeneration (Sections VII and VIII).
A. MuSCs and adult myogenesis
Although the signaling pathways controlling the activation of MuSCs, that is, their entry into the cell cycle, are still poorly understood and are the subject of intensive investigations, the gene expression program driving activated MuSCs along adult myogenesis is quite well known. MuSCs sequentially express a series of specific transcription factors from their exit from quiescence until their full differentiation and fusion (Fig. 2) (92, 359).
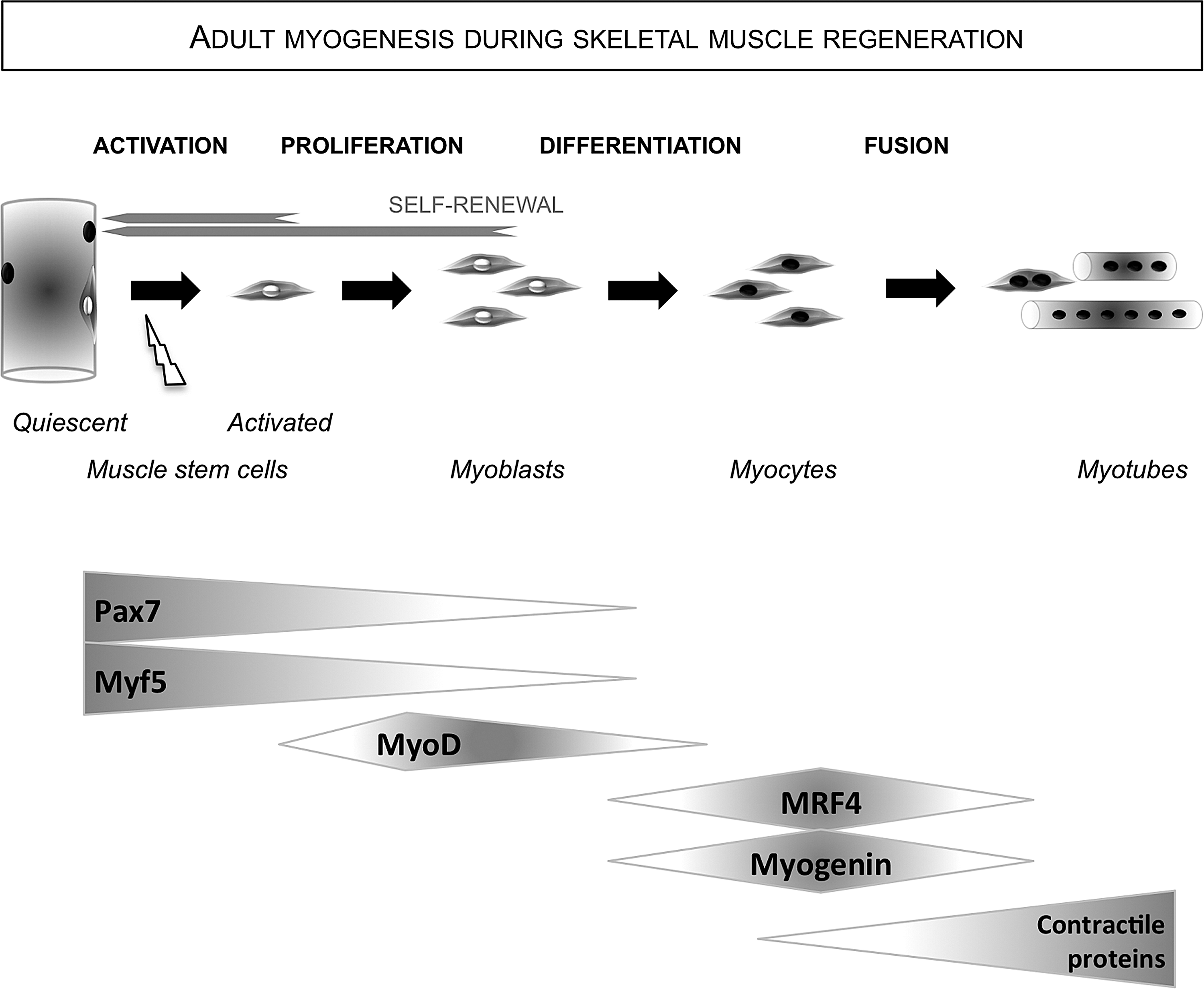
At the quiescent state and during the expansion phase, they express Pax7, a transcription factor that is now considered a canonical marker of MuSCs in various species. Once entered into the cell cycle, MuSCs sequentially express a series of specific muscle transcription factors, the myogenic regulatory factors (MRFs). Structurally, MRFs bind DNA through their basic domain, whereas a helix–loop–helix motif allows their heterodimerization with E proteins, which mediate the recognition of genomic E-boxes, a motif present in the promoters of muscle-specific genes. Although Myf5 is expressed by quiescent/early committed MuSCs, expanding MuSCs express MyoD, which is a strong determinant of their myogenic nature. After the expanding phase, MuSCs exit the cell cycle and downregulate the expression of Pax7 and MyoD while increasing that of Myogenin, which is considered a marker of irreversible terminal myogenic differentiation.
Then, differentiated myocytes are capable of fusing together and with pre-existing fibers to reconstitute functional myofibers. At the time of fusion, differentiated MuSCs start expressing the sarcomeric machinery of proteins required for contraction. Before exiting the cell cycle to differentiate, a subset of expanding MuSCs self-renews by decreasing the expression of MyoD and increasing that of Pax7, therefore returning to a G0 quiescent state. The exact time (at the time of activation, during expansion, or at the end of the expansion phase before differentiation) at which MuSC self-renew in vivo is still debated.
MuSCs are associated with all fiber types, although with unequal distribution (264). Slow-twitch muscles express twofold to threefold higher MuSC density compared with fast-twitch muscles (284, 296). These findings have been recently confirmed in vivo in mice by using genetically labeled MuSCs (164). The phenotype of MuSCs from adult muscle fibers seems to be similar to their fiber of origin and to the fiber they tend to form in terms of myosin expression and contractile capacity (21, 142). Aging is associated with skeletal muscle atrophy and a switch from fast-twitch to slow-twitch fiber type. Concomitantly, the number of MuSCs is reduced in some muscles (gastrocnemius, diaphragm, extraoccular muscles) at 20 months from 16% to 25% in mice (164). So far, no study has reported an impact of the type of myofiber—or the corresponding MuSCs—on the efficiency of muscle regeneration.
B. MuSC cellular neighborhood
MuSCs are absolutely indispensable for adult skeletal muscle homeostasis, since the specific depletion of Pax7-expressing cells completely prevents postinjury regeneration (188, 292). However, numerous studies have recently shown that cells present in the neighborhood of MuSCs are an important support of adult myogenesis (Figs. 3 and 4). One can distinguish between resident and nonresident muscle cells.
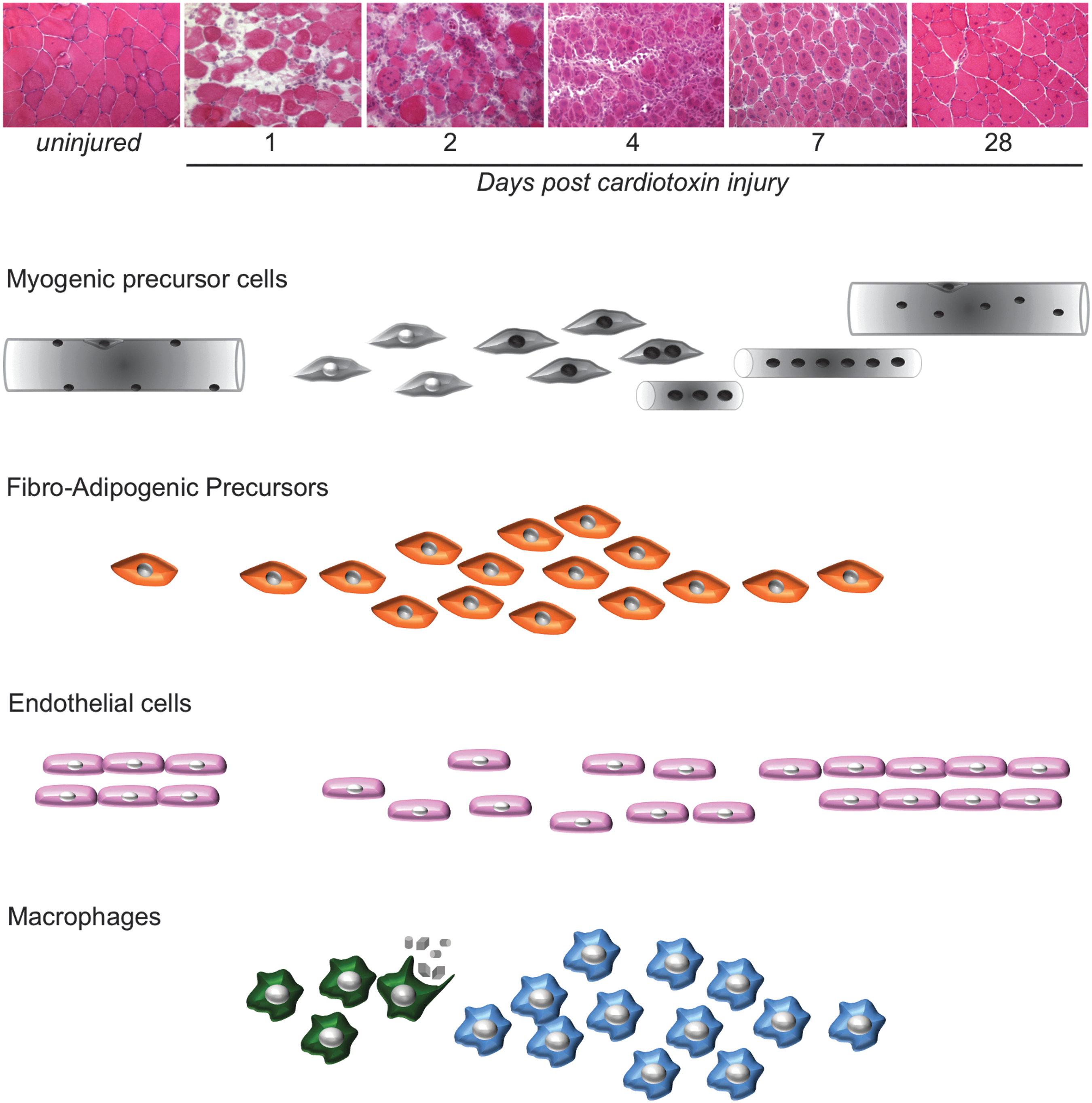
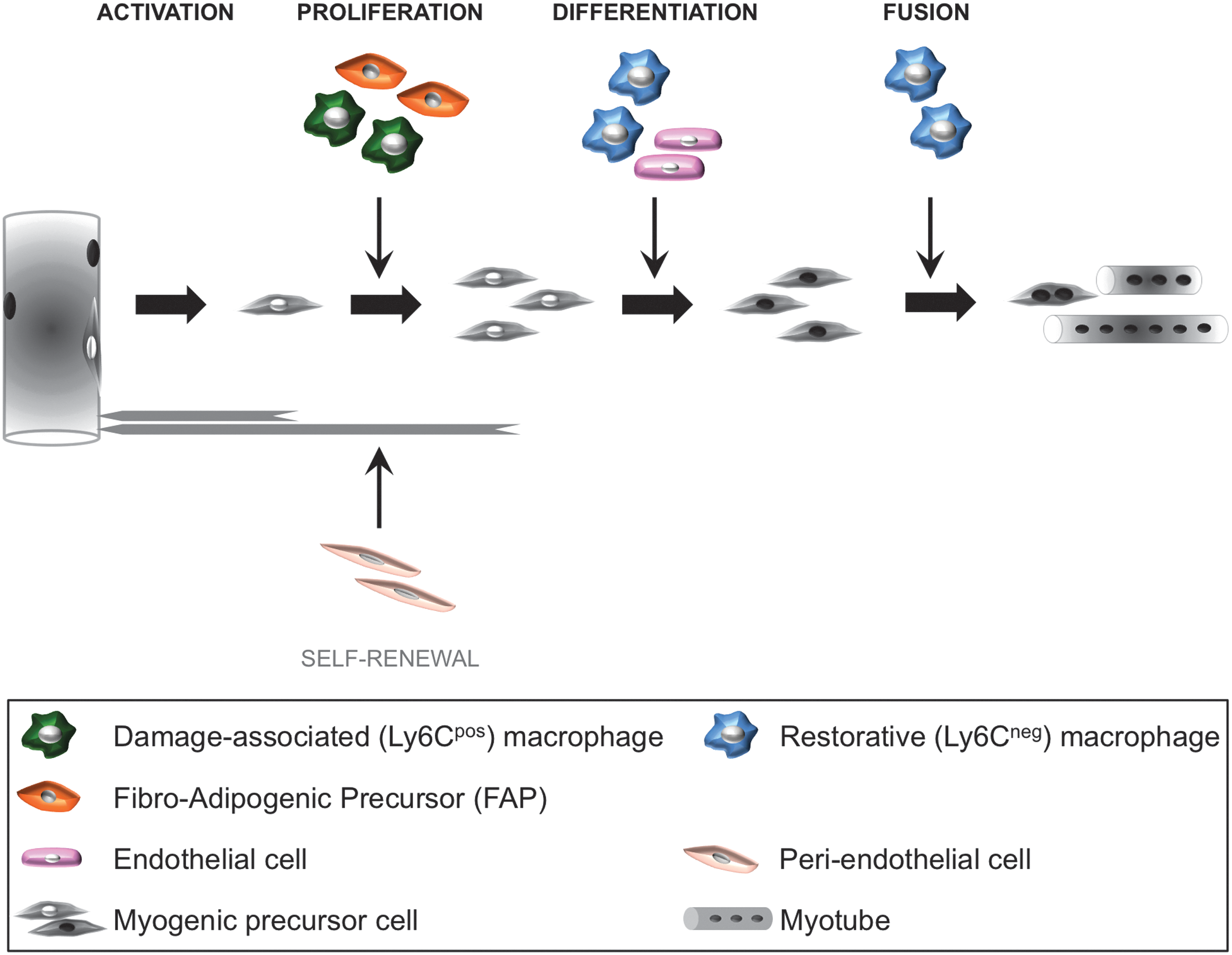
Among the resident nonmyogenic muscle cells that have been shown to participate in muscle regeneration are vascular cells and fibro-adipogenic progenitors (FAPs). Vascular cells comprise endothelial cells (ECs) that specifically interact with expanding MuSCs in a reciprocal way to promote concomitant myogenesis and angiogenesis (63, 177) (Fig. 4). Vascular cells also include peri-ECs such as pericytes and smooth muscle cells that promote the return of MuSCs to quiescence (2, 170). Another cell type that has been recently described in skeletal muscle is the FAP, which also participates in the regulation of MuSC expansion and differentiation (157, 329).
Nonresident muscle cells invade the injured muscle in a tightly organized sequence, as in any other tissue repair process (Fig. 3). Infiltrating leukocytes exert various actions on FAPs and other immune cells, whereas macrophages have been shown to exert specific and direct effects on MuSCs (55, 56). Invading circulating monocytes become inflammatory macrophages that stimulate the expansion of MuSCs while inhibiting their differentiation. Later on, at the time of resolution of inflammation, macrophages skew their phenotype to an anti-inflammatory status. These restorative macrophages exert specific pro-myogenic effects on MuSCs by stimulating their differentiation (expression of Myogenin) and their fusion (Fig. 4).
C. Time and space orchestration of muscle regeneration: link with muscular diseases
Recent studies have shown that asynchronous injuries (76) or desynchronized delivery of anti-inflammatory cues (257) trigger similar defects in skeletal muscle regeneration, resembling what is observed in some muscle diseases. It is, therefore, admitted that the MuSC niche—the quiescence niche, as well as the so-called transient niche for a proper implementation of the myogenic program—is tightly regulated in space and time. It is, therefore, likely that this tight regulation operates through subtle changes in the cues that MuSCs receive all along the regeneration process (56).
Inherited muscular dystrophies are a group of heterogeneous diseases that are characterized by permanent cycles of necrosis/regeneration that lead to the replacement of muscle fibers by connective and fat tissue, and eventually to the loss of muscle function (57, 214). Most of these diseases are caused by a genetic alteration targeting proteins of the dystrophin-glycoprotein complex (DGC), which is located at the sarcolemma and links the myofiber cytoskeleton to the extracellular matrix (6, 214) (Fig. 5).
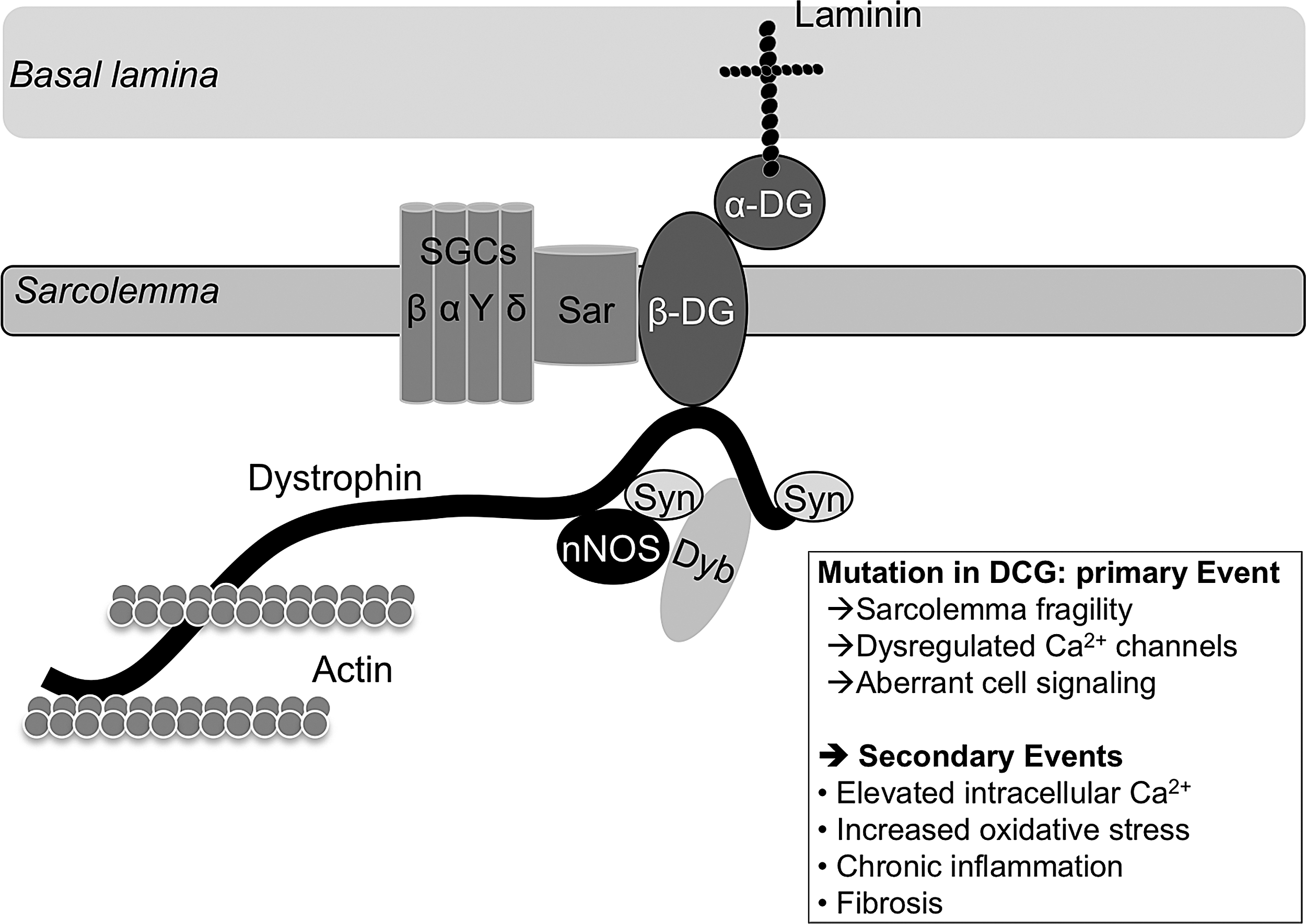
It is believed that a weaker transmission force through the DGC triggers fragility of myofibers, which are, therefore more susceptible to damage on contraction/use (93). This primary event (mutation in a gene encoding for a DGC protein) leads to permanent regeneration processes throughout the tissue, triggering asynchronous delivery of regenerating cues to MuSCs and their close environment. This leads to failure in myogenesis and to secondary events, including elevated oxidative stress, altered calcium homeostasis, chronic inflammation, and endomysial (between myofibers) fibrosis, which eventually replace the atrophied myofibers (6) (Fig. 6).
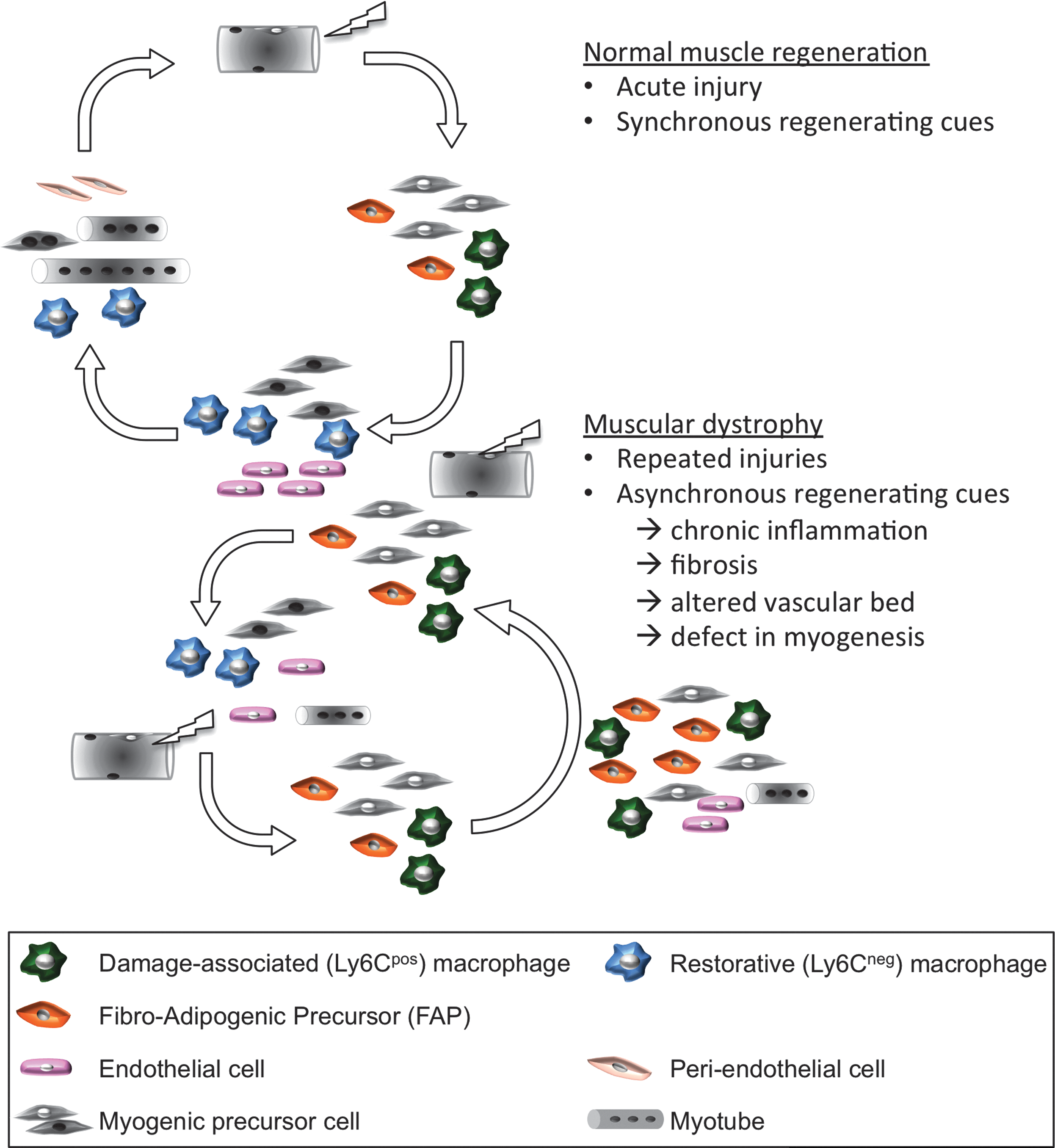
Our knowledge of the cellular events in this context is still fragmented. It has been recently shown that in the Duchenne muscular dystrophy (DMD) mouse model mdx, FAP number is increased and macrophages exhibit altered inflammatory status whereas inhibition of the transforming growth factor-beta (TGFβ) pathway leads to the apoptosis of FAPs, improving the muscle state (187). The molecular pathways that are altered in such an unbalanced tissue environment are only beginning to be defined for each cell type. For instance, nuclear factor kappa B (NF-κB) signaling is deregulated in both myogenic cells and macrophages in DMD (3). Notch signaling, which has been shown to regulate MuSC fate, can be targeted to improve muscle homeostasis in this disease (342). Given that the various cell types in the muscle no longer operate in a synchronized manner, one of the future challenges will be to identify the key pathways controlling the equilibrium of each cell type in degenerative myopathies.
III. Definition of Free Radicals, Oxidative Stress, and Redox Signaling
ROS are a family of molecules that are continuously generated and consumed in all living organisms as a consequence of aerobic life (85). In 1985, Helmut Sies proposed oxidative stress as “a disturbance in the pro/antioxidant balance in favor of the former” (306). This definition has been widely associated with the view that ROS exert major biological effects as purely deleterious molecules causing damage to DNA, proteins, and lipids, notably during aging (131), chronic diseases (335), or metabolic syndrome (151).
During the early 1990s, ROS-dependent signaling was discovered [review in Schreck and Baeuerle (297)], shedding light on the beneficial role of ROS (316). Thus, oxidative stress has been redefined as “a disruption of redox signaling and control” (158). This notion implies that there is an optimal level of ROS/RNS to sustain both cellular homeostasis and adaptive responses, and that both too low and too high levels of ROS/RNS are detrimental to cell functions (108, 235). This contemporary definition corresponds to a now well-accepted view that ROS production is crucial for cell functions (75, 85, 152, 230, 235). Recent developments in technological tools to detect and quantify ROS and ROS molecular targets, notably in vivo, have helped to better understand the role of ROS in numerous cellular processes (30, 61, 62, 230).
In the present review, the terms “ROS” and “RNS” include the oxygen and nitrogen radicals as well as some nonradical derivatives of O2 and N, respectively (129), and are used when the specific entity is not described, as recently recommended by Forman et al. (104).
IV. Major ROS and RNS in Skeletal Muscle
From its generation to the terminal product, ROS can undergo a wide range of transformations (Fig. 7) that elicit specific effects on cell functions.
A. Major chemical entities
ROS and RNS can be classified depending on their chemical properties (reactivity, half-life) into two groups. Radical compounds (one electron oxidant) form the first group, whereas nonradical compounds (two electrons oxidant) form the second one (17). These chemical entities can also be classified based on the primary or secondary nature of the radicals.
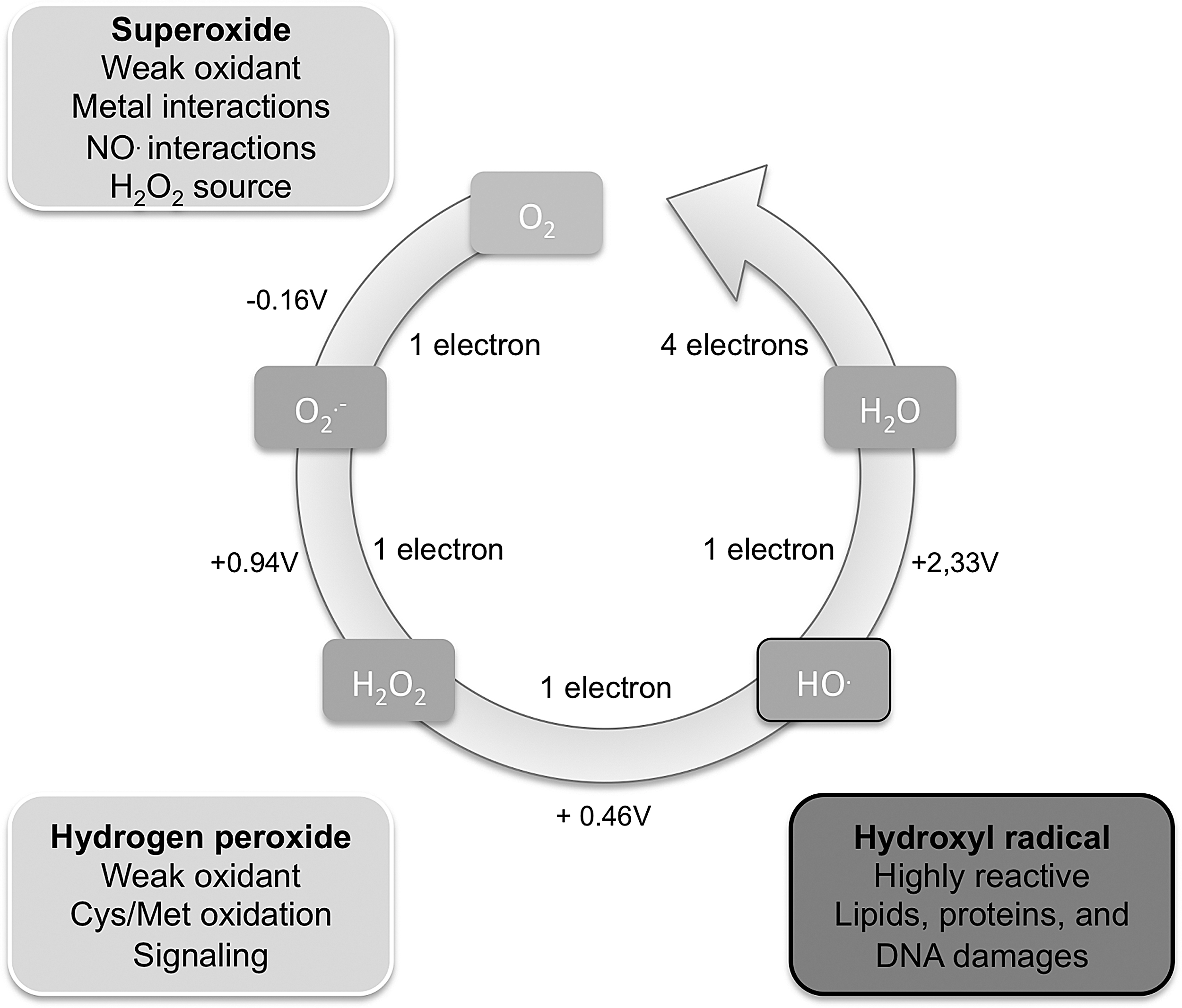
1. Superoxide
The radical superoxide (O2 •−) is an anion that is produced by a monoelectronic reduction of an oxygen molecule (O2 + 1e− → O2 •−). O2 •− can diffuse within the cell but, despite a relatively longer half-life as compared with other radicals, O2 •− is neither a strong oxidant nor a powerful reductant. This is explained by the low standard reduction potential Eh of O2 •−/H2O2 (hydrogen peroxide) above 0.94 V (Eh value, that is, measurement of the tendency of chemical species to acquire electrons and to be reduced; the more positive the potential, the higher the tendency to be reduced). By a dismutation reaction, O2 •− can produce H2O2 [O2 •− + O2 •− + (2H+) → 2H2O2 + O2]. In skeletal muscle as in other tissues, two distinct pools (inner and outer membrane) of mitochondrial O2 •− have been observed (355).
2. Hydrogen peroxide
H2O2 is a two-electron oxidant, nonradical molecule that is generated from the dismutation of O2 •−. This molecule is stable, diffuses within and between cells, and, therefore, acts as a signaling molecule (338). However, it is a weak oxidizing and reducing agent, as evidenced by its inability to directly oxidize DNA and proteins as well as to induce antioxidant defenses in mammals (83). H2O2 is also described as a crucial player in tissue damage and repair [review in van der Vliet and Janssen-Heininger (337)]. It acts as an alarmin to trigger inflammatory cell recruitment that is required for appropriate healing (237). However, H2O2 may also contribute to chronic tissue injury and subsequent inflammation by inducing irreversible oxidative damage (222). H2O2 is now recognized as a key molecule that is involved in the control of skeletal muscle function such as insulin control and glucose transport (135, 338).
3. Hydroxyl radical
Hydroxyl radical (HO•) is a powerful oxidant (Eh value O2 •−/H2O2 = 2.34 V) that is generated by the reaction of metal ions with H2O2. The high reactivity related to its short half-life (10−9 s) restricts its damaging effects to a local area that is close to the generation site (129). In addition, HO• can be formed from H2O2 in the presence of transition metals (iron) through the reactions of Fenton and of Haber-Weiss [O2 •− + H2O2 (+Fe3/Fe2) → HO• + O2 + −OH] to induce oxidative damage (144, 165, 352).
4. Nitric oxide
Nitric oxide (NO•) is a free radical that easily crosses membranes and diffuses between and within cells. NO• is synthesized from
5. Peroxynitrite
The reaction of O2 •− with NO• forms peroxynitrite, which is a strong oxidant that is capable of crossing cell membranes. Cell exposure to peroxynitrite produces many effects such as inhibition or depletion of antioxidant enzymes, modification of receptor functions, lipid peroxidation, and DNA injury. Recently, peroxynitrite has been shown to induce the activation of mammalian target of rapamycin (mTOR) signaling via calcium-dependent signaling, leading to skeletal muscle hypertrophy (147).
B. Major sources of ROS
In 1954, Commoner et al. identified free radicals in skeletal muscle (70). Since then, several sources of ROS production have been identified in various subcellular locations (148) (Fig. 8).
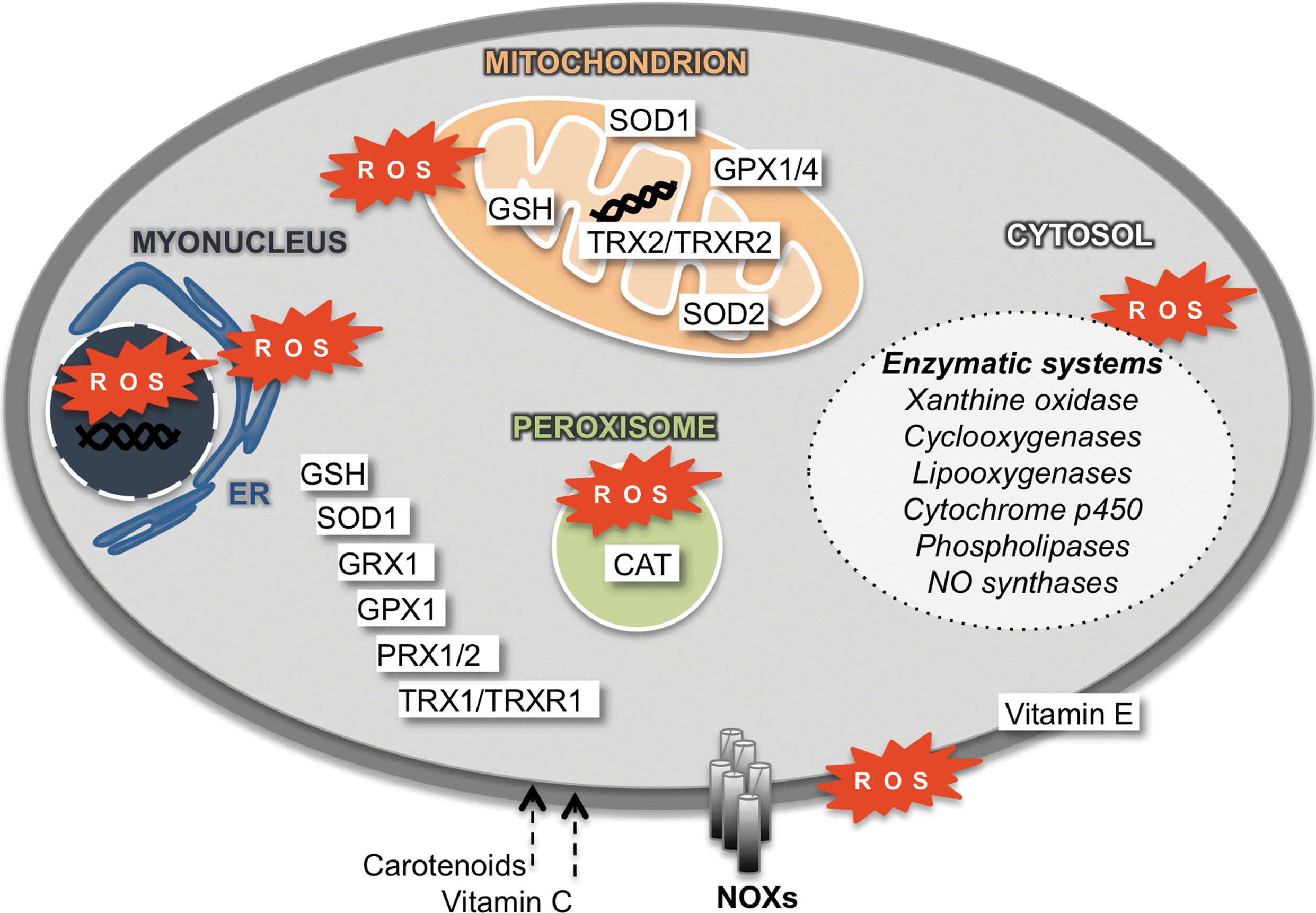
1. Mitochondria
Mitochondria are one of the most important sources of ROS in muscle. At least 10 sites that are capable of producing considerable amounts of ROS have been described (123). Early studies suggested that 2–5% of the total oxygen consumed by mitochondria undergo a one-electron reduction and generate O2 •− (39). However, depending on different mitochondrial states of respiration, the percentage of oxygen that forms superoxide ranges from 0.1% in state 3 to 2% in state 4 (266, 305). Other studies have shown that both complexes I and III of the electron transport chain are particularly involved in ROS production through a leakage of electrons (20, 229). This was demonstrated by the use of specific inhibitors of complex I- and III-derived ROS such as rotenone and antimycin, respectively (364).
2. NADPH oxidases
The NADPH oxidases (NOXs) transfer electrons across biological membranes and form O2 •−. The NOX family includes seven NOX isoforms (NOX1, NOX2/gp91phox, NOX3, NOX4, NOX5, DUOX1, DUOX2), two organizer subunits (p47phox, NOXO1), two activator subunits (p67phox, NOXO1), and two DUOX-specific maturation factors (DUOXA1 and DUOXA2). NOX enzymes are primarily known to produce O2 •− in both phagocytic and nonphagocytic cells (26). A small GTPase named Rac1 has been described as an important activator of NOX1 and NOX2 in various cell types, including skeletal muscle (317), and also leads to superoxide production in nonphagocytic cells (140). In skeletal muscle cells, NOX isoforms 2 and 4 are found at the plasma membrane, transverse tubules, and sarcoplasmic reticulum where they modulate calcium release (261). They are described as the major source of ROS during skeletal muscle contraction (217). In this context, pharmacological inhibition of NOX-induced ROS production has been widely used, although these inhibitors are mainly unspecific and not isoform selective [review in Refs. (8, 65)].
3. Xanthine oxidase
Xanthine oxidoreductase (XOR) is an intracellular enzyme that is involved in purine catabolism, which includes two isoenzymes: xanthine dehydrogenase and xanthine oxidase (XO). XOR catalyzes the reduction of hypoxanthine and xanthine in uric acid. The oxidase form is used as an electron acceptor, and both hypoxanthine and xanthine are reduced to uric acid and O2 •− (210). The role of XO as a source of ROS generation has been demonstrated by using allopurinol, a specific inhibitor. In both animals and humans, allopurinol prevents the oxidation of glutathione (GSH) and lipid peroxidation (343). Interestingly, XO is a major source of ROS in skeletal muscle atrophy after unloading and in sarcopenia (82).
4. Other sources
The endoplasmic reticulum is another organelle that is involved in ROS production (50, 199). In addition, various enzymatic systems such as phospholipase A2 induce ROS production by stimulating NOX. Studies reported both calcium-dependent and calcium-independent forms of phospholipase A2 as key actors in ROS generation in the skeletal muscle (124, 236).
V. Major Antioxidants in Skeletal Muscle
Enzymatic and nonenzymatic antioxidant systems exist to detoxify excess ROS, prevent and repair oxidative damage, and maintain redox homeostasis in the cell (127). Halliwell and Gutteridge have defined an antioxidant as “any substance that delays, prevents or removes oxidative damage to a target molecule” (129). This definition suggests that this evolutionary conserved system exerts its antioxidant functions in three distinct ways: by converting ROS into less deleterious molecules, by reducing pro-oxidant molecules such as metallic ions, and by scavenging ROS. Thus, the complex antioxidant network works synergistically to protect cells from redox disturbances and oxidative damage.
Interestingly, there is a growing body of evidence indicating that antioxidants not only are ROS-scavenger molecules but also act as signaling molecules that are required both for the maintenance of cell homeostasis and for adaptive cell responses (277). Human Atlas Proteome (78, 330) and the Redox Atlas of the Mouse (120) provide a global analysis of tissue-specific proteome. Based on these large-scale projects and the available literature, we present hereafter the skeletal muscle tissue-specific antioxidant system.
A. Endogenous
Among endogenous antioxidants, GSH, superoxide dismutases (SODs), catalase (CAT), and members of the thioredoxin (Trx) family contribute to cell protection and regulation of redox signaling.
1. Superoxide dismutases
SODs are enzymes that catalyze the dismutation of O2 •− into H2O2. SODs are divided into three isoenzymes (SOD1, SOD2, SOD3), all of which require redox-active transition metals in the active site (261). SOD1 (CuZnSOD) is a dimer of 32.5 kDa that is associated with one copper ion and one zinc ion, and it is mainly localized in the cytosol and the mitochondrial intermembrane space (Fig. 8). SOD2 (MnSOD) is a tetramer of 24.7 kDa that is associated with one manganese ion. SOD2 is mainly located in mitochondria. SOD3 is an extracellular tetradimer of 30 kDa, and it uses copper-zinc as a cofactor. Deletion of SOD1 in the skeletal muscle induces a phenotype of accelerated age-related loss of skeletal muscle mass and function (291).
2. Catalase
CAT is a homotetramer of 240 kDa that is composed of four subunits, and it requires iron as a cofactor. CAT is mainly localized in peroxisomes (Fig. 8), which were originally named to highlight their content in enzymes either producing (oxidases) or breaking down (CAT) H2O2 (24, 336). Therefore, it was originally believed that the primary function of these organelles was to control H2O2 metabolism [review in Lodhi and Semenkovich (194)]. However, peroxisomes also have an established role in lipid metabolism, as well as emerging functions in cellular signaling that are relevant to metabolism (194). The function of CAT is to catalyze the dismutation of H2O2, which leads to the formation of one molecule of H2O and of O2. It has been shown that a decline of CAT protection, associated with elevated H2O2 levels, is observed at the onset of sarcopenia, which is the loss of muscle mass observed during aging (315).
3. Glutathione
GSH is the most abundant low-molecular-weight component in the cell. It functions as a buffer of the cellular redox status and as an antioxidant in the eskeletal muscle (126). This tripeptide that is synthetized in the liver exerts its antioxidant functions through several features: as a substrate for glutathione peroxidases (Gpxs) and as a reducing agent for vitamin C and E radicals. GSH is predominantly found in the cytosol but is also distributed in various organelles such as the nucleus, mitochondria, and endoplasmic reticulum (107). GSH is involved in NO• metabolism, since GSH and peroxinitrite lead to the formation of S-nitroglutathione (GSNO) that is able to generate NO• (129). In skeletal muscle fibers, GSH is present at millimolar concentrations. Higher levels of GSH are found in type I muscle fibers, which have an oxidative/aerobic metabolism, as compared with type II fibers, which rely mainly on a glycolytic/anaerobic metabolism (185).
4. Family of thioredoxins
The thioredoxin family of proteins encompasses Trxs, Gpxs, glutaredoxins, (Grx), and peroxiredoxins (Prx). These proteins share common structural sequences and are characterized by their active site motifs, containing either one or two cysteinyl residues. They are essential for the reduction of protein disulfides, protein de/glutathionylation, and de/trans/nitrosylation (130, 242).
The Trx system includes two oxidoreductases Trx1 and Trx2 and a thioredoxin reductase (TrxR). Trx1 and TrxR1 isoforms are found in the cytosol, Trx1 is translocated into the nucleus to alter the redox state of transcription factors, and Trx2 and TrxR2 isoforms are located in the mitochondria (283) (Fig. 8). Trxs serve as donors for Prxs to reduce peroxides. In the Trx system, electrons are transferred during a multiple-step process from NADPH to Trxs to ultimately reduce disulfides and generate antioxidants (208). Trx1 and related redox proteins are constitutively secreted by skeletal muscle cells and have been suggested to belong to myokines (203). Moreover, a decreased expression of Trx1 is observed during muscle atrophy that may be responsible for the enhanced oxidative damage during the late phase of disuse muscle atrophy (207).
Grxs also modulate the thiol redox state of several proteins, but no study has reported a role in the skeletal muscle (130). Gpxs catalyze the reduction of H2O2 and organic hydroperoxides to water and alcohol, respectively (42, 136). In the skeletal muscle, Gpx activity is strongly decreased in cancer cachexia (314) and Gpxs have been involved in the regulation of MuSC function (see Section VII). Prxs play multiple roles as peroxidases, sensors, and regulators of peroxides. Interestingly, a role for Prx in H2O2-mediated cellular signaling has been described (354). The expression of Prx3 is decreased during muscle atrophy (207).
B. Exogenous
Exogenous molecules with antioxidant properties are generally classified as vitamins, trace elements, or phytochemicals. Vitamin E is a term encompassing four tocopherols (α, β, δ, γ) and four tocotrienols (α, β, δ, γ) molecules. The α-tocopherol is the most abundant form in nature. Vitamin E, considered a master antioxidant, has the ability to prevent lipid peroxidation and other radical-driven events. It is mainly located in the plasma membrane (Fig. 8) and acts as a chain-breaking antioxidant that prevents the propagation of free radical reactions (43). Vitamin E reacts with oxidants to form tocopheryl radicals that are further reduced by vitamin C into nonradical α-tocopherols. This process requires GSH that gives an electron, leading to the formation of oxidized GSH. Vitamin E is required for skeletal muscle plasma membrane repair (141, 176). However, the underlying cellular and molecular mechanisms of this phenomenon remain to be determined (166).
Ascorbate is the most common form of vitamin C that is found in the organism. This water-soluble molecule, localized in the extracellular compartment, has special functions in redox reactions, as a powerful antioxidant and enzyme cofactor, notably of hydroxylases (204, 325). Vitamin C has the ability to protect against lipid peroxidation by acting as an ROS scavenger and by one-electron reduction of lipid hydroperoxyl radicals via the vitamin E redox cycle (129). The dehydroascorbic acid (oxidized form of vitamin C) thereby formed can be reduced by TrxR and GSH (189), underlying the synergy within the antioxidant network.
Finally, trace elements such as selenium, copper, manganese, and zinc play important roles in supporting antioxidant functions and redox exchanges of many enzymes. Generally, these molecules play a role of substrate or cofactor, and their deficiency leads to oxidative stress (116, 246). They are likely important in skeletal muscle function, as exemplified by the reduced expression of selenoprotein N (sepn1) that triggers a congenital muscle disease (14), highlighting the role of trace elements in skeletal muscle homeostasis.
VI. General Mechanisms of Redox Signaling
In skeletal muscle, ROS/RNS have been identified as playing key roles in numerous physiological processes, including glucose uptake (163), excitation/contraction coupling (261), mitochondrial biogenesis (145), training adaptations (122, 281), and cell death (295). This underlines the need to understand how ROS/RNS act as determinant regulators of biological effectors that are involved in the control of homeostasis (90, 302). It is suggested that oxidation and reduction of the thiol side chain of cysteine amino acids are the major mechanisms by which oxidants integrate signal transduction pathways (85).
A. Thiol oxidation as a major mechanism of oxidation-related post-translational modifications
Oxidants mainly exert their effects by targeted modifications of thiol residue (SH) of cysteine amino acids, resulting in reversible or irreversible oxidation that depends on the state of oxidation (Fig. 9). It results in the formation of sulfenic acid (SOH) that can be further oxidized in sulfinic (SO2H) and sulfonic (SO3H) acids. Additional post-translational modifications involving NO• or GSH are also observed and identified as S-nitrosylation and S-glutathionylation, respectively (17, 35, 85). Such chemical reactions are under the control of pKa, pH, and the local protein environment (127).
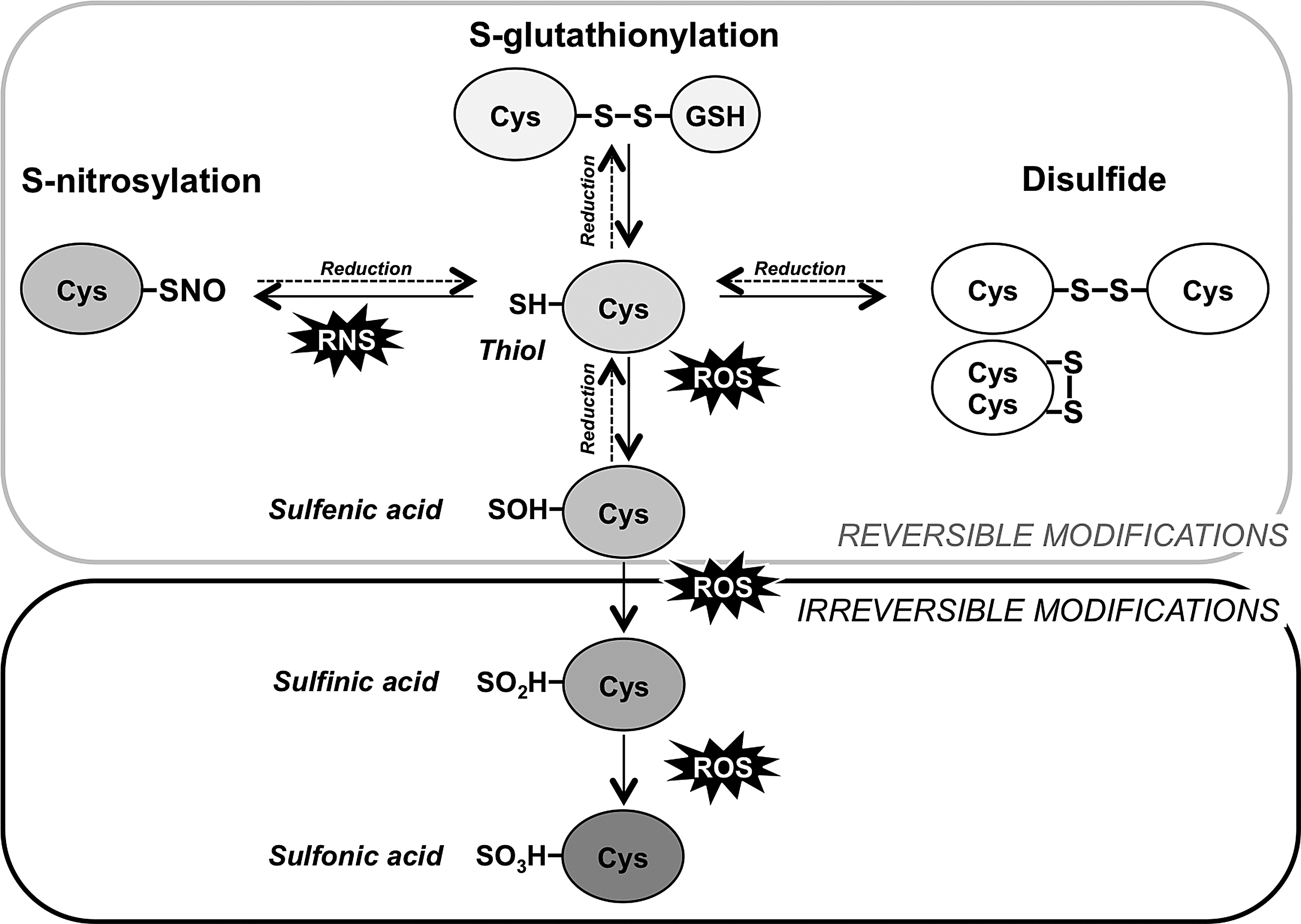
B. From signal transduction to post-translational modifications
The role of ROS in signaling has emerged with the observation that H2O2 production was necessary for normal signaling of growth factors, suggesting that ROS/RNS may act as secondary messengers (316). More recently, Jones highlighted the role of “radical-free” biology of oxidative stress, indicating that the free radical chain reactions are almost completely prevented in biological systems due to the very high concentrations of antioxidant proteins (159). He reported that 10% of unbalanced pro- and antioxidant reactions involve free radical intermediates and that less than 0.1% induce macromolecular damage. Hence, the nonradical H2O2 appears to be an ideal candidate for redox-dependent signal transduction (e.g., large diffusion, weak oxidant, reversible oxidation inductor) (105, 152).
Several mechanisms are described for H2O2-dependent signal transduction. One proposes H2O2-dependent signaling via a relay system involving a redox sensor (Prx2), which oxidizes the target and forms a transient disulfide bond (310). Another mechanism is the floodgate model (354). It is based on the local inactivation of antioxidant defenses, such as Prx1, that allows low H2O2 levels to mediate signaling in a specific cell compartment.
Although less well described, two other mechanisms have been proposed. The first includes an intermediate step involving the oxidation of Trx or GSH to the redox relay mechanism, and the second one likely results from the dissociation and subsequent activation of a target protein after H2O2 oxidation of a scavenger enzyme (272). In addition, NO• and its derivatives (S-nitrosothiols [SNO] and GSNO) are involved in signal transduction (103, 152, 276, 338). These molecules induce modifications of protein structure, thus affecting protein functions as well as the signaling pathways in which these proteins are involved.
Technological advances such as proteomic and redox proteomics (60) have recently revealed that 500 proteins contain reactive and potentially modulatory cysteine residues (162, 345). Among the targeted proteins detected in the “redoxome” are transcription factors (NF-κB, FOXO), kinases (AMP kinase, mTOR), protein tyrosine phosphatases, proteases (caspases, metalloproteases), and chaperones proteins. This illustrates the broad impact that the oxidation of conserved cysteine residues may have on various physiological processes from signal transduction to gene expression [review in Janssen-Heininger et al. (152)].
In response to oxidative stress, cells modulate their gene expression (7). Several transcription factors have been described as redox sensitive and are involved in various cellular signaling pathways with distinct outcomes. NF-κB is the first eukaryotic transcription factor that has been shown to respond directly to oxidative stress in the L6 skeletal muscle myoblasts on exposure to the nonradical ROS H2O2 and for which activation was controlled by the cell glutathione disulfide/glutathione (GSSG/GSH) ratio (301). In addition, NF-κB is involved in the regulation of a wide array of cellular processes, including immune response, cellular survival, proliferation, and differentiation [review in Bakkar and Guttridge (18)]. Interestingly, NF-κB activity is impaired as a consequence of S-glutathionylation of IKK-β cysteine 179 and is associated with a decrease of pro-inflammatory signals (275).
Redox factor-1 (Ref-1) contains in its N-terminal region a redox regulatory domain that is characterized by two cysteines that are involved in the redox-dependent modification (reduction) of NF-κB and of other transcription factors such as activator protein-1 (AP-1) (Fos and Jun), p53, and hypoxia-inducible factor-1α. The oxidized form of AP-1 exhibits poor DNA-binding activity, due to the cysteine residues within the DNA-binding domain of both Fos and Jun. Inversely, AP-1 is mainly activated by antioxidants (192, 300). Of interest, these redox-sensitive transcription factors NF-κB and AP-1 trigger the synthesis of cytoprotective regulatory proteins such as SODs, CAT, and heat shock proteins after the delivery of ROS generated by the skeletal muscle itself on contractile activity (149).
The transcription factor NF-E2 p45-related factor 2 (Nrf2) is another well-known redox-sensitive master regulator that controls the expression of networks of genes encoding proteins with diverse cytoprotective activities. Nrf2 itself is primarily controlled at the level of protein stability. Under basal conditions, Nrf2 is a short-lived protein that is subjected to continuous ubiquitination and proteasomal degradation. The most common ubiquitin ligases that contribute to the degradation of Nrf2 are Kelch-like ECH-associated protein 1 (Keap1) and a substrate adaptor protein for Cullin 3 (Cul3)/Rbx1 ubiquitin ligase. Keap1 is considered a sensor for a wide array of small-molecule activators of Nrf2, including ROS. These latter block the cycle of Keap1-mediated degradation of Nrf2 by modifying specific cysteine residues within Keap1 (87) or by directly disrupting the Keap1:Nrf2 interaction. As a consequence, Nrf2 is not degraded, and the transcription factor accumulates and translocates to the nucleus, where it forms a heterodimer with a small Maf protein, binds to antioxidant-response elements in the upstream regulatory regions of its target genes, and initiates transcription (133).
In skeletal muscle, which is an active metabolic tissue, insulin signal transduction is crucial. Insulin signaling has been shown to be altered by ROS (such as 4-hydroxy-2-nonenal and peroxynitrate) (13) and to be regulated by the selenoproteins Gpx1 and Sepp1 (through the inhibition of energy metabolic mediators AMPK and Akt) (312).
VII. Redox Regulation of MuSCs During Skeletal Muscle Regeneration
During skeletal muscle regeneration, MuSCs undergo a series of processes called adult myogenesis, which includes activation and entry in the cycle, proliferation, exit from the cell cycle, and commitment into terminal differentiation, followed by fusion into multinucleated cells (myotubes in vitro and myofibers in vivo) (Fig. 2). Various aspects of adult myogenesis have been shown to be or are likely redox regulated (264, 361) (Fig. 10).
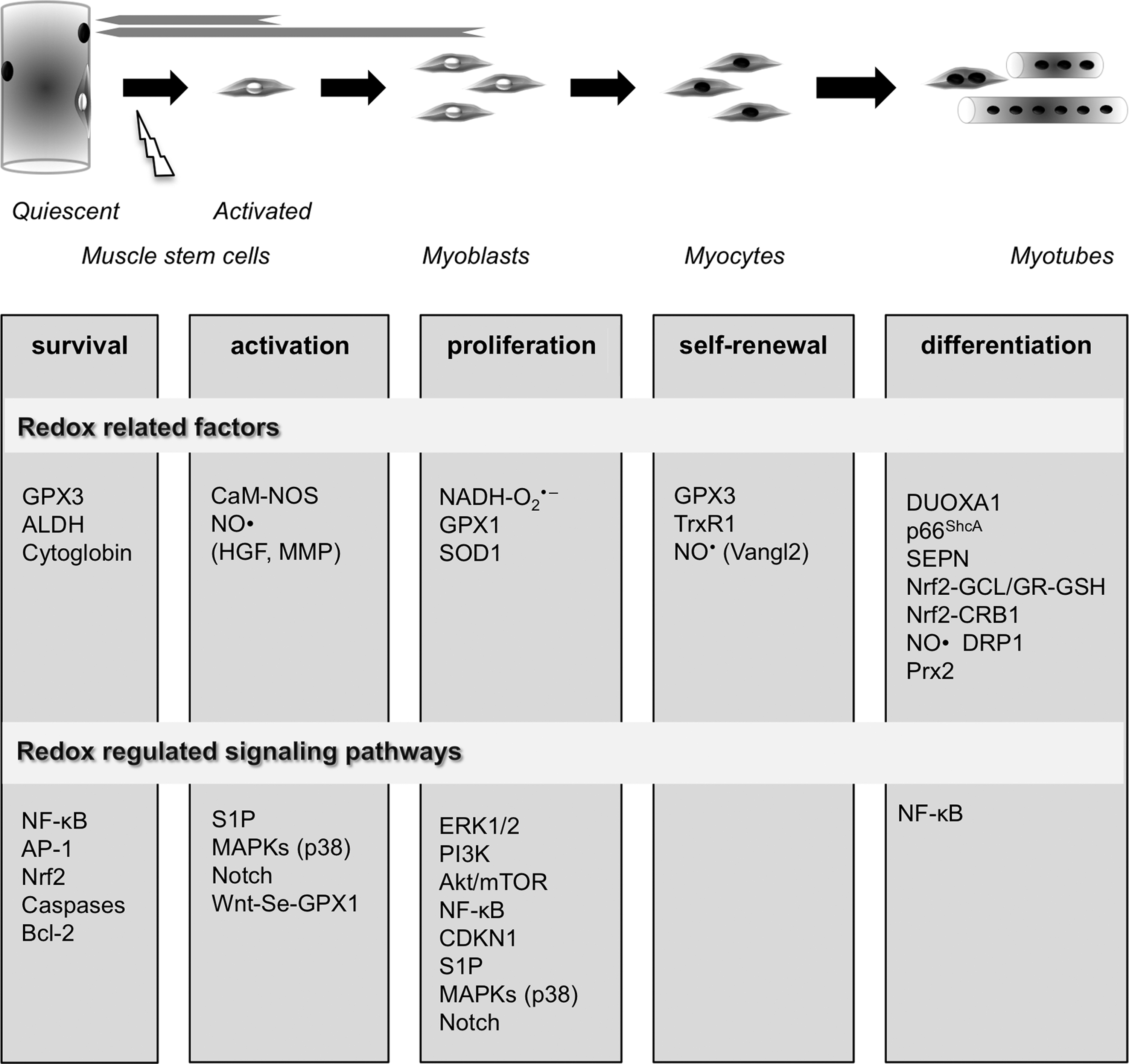
A. Viability: apoptosis
ROS and redox reactions have been described to regulate cell survival through the modulation of transcription factors (NF-κB, AP-1, Nrf2, etc.) and pro-apoptotic proteins (caspases, Bcl-2) (247, 270). The role of oxidative stress in cell death pathways is supported by the fact that antioxidants can protect cells against apoptosis (247). In the skeletal muscle, long-term exercise has been shown to reduce ROS generation, to improve antioxidant status, and to decrease oxidative stress that should serve to attenuate any ROS-induced activation of apoptosis (265).
Elevated apoptotic signaling has been reported in skeletal muscle during aging, stress-induced states, and diseases, a phenomenon that plays a role in muscle dysfunction, degradation, and atrophy (265). During fetal myogenesis, analysis of double conditional pitx2/3 mouse mutants in primary muscle cell cultures reveals excessive upregulation of ROS levels, leading to DNA damage and apoptosis of differentiating cells (175). In adult male rats, Gsh depletion in skeletal muscle increases caspase-independent signaling and increases mitochondrial-associated apoptotic events to subsequent cell death stimuli (77).
Apoptosis in the skeletal muscle is low at rest, whereas skeletal muscle injury causes apoptosis and necrosis (307) and is associated with ROS production (341). Therefore, the protection of MuSCs from cytotoxic damage is crucial to ensure efficient regeneration. MuSC antioxidant levels have been described as a major determinant of MuSC cell therapy efficiency (91). Several antioxidant pathways have been involved in oxidative stress detoxification of MuSCs. Retinoic acid-Gpx3 pathway (96) and aldehyde dehydrogenase activity (153) improve MuSC survival. Recently, cytoglobin, a heme-containing protein, has been involved in the resistance to oxidative stress-induced apoptosis that results in incomplete skeletal muscle regeneration (307). Apoptosis being a highly conserved process playing a critical role in tissue homeostasis (Fig. 10), its putative role in skeletal muscle regeneration remains to be elucidated.
B. Quiescence
At rest, MuSCs are maintained in quiescence (G0 phase) (58). Transcriptomic analysis revealed an antioxidant molecular signature (250) involving the upregulation of Gpx3 and Trxr1, suggesting a protective effect of quiescent cells from oxidative insults. During muscle regeneration, although most MuSCs commit to a myogenic fate and differentiate, a small population self-renews to maintain a stem cell pool for further needs (Figs. 2 and 10). NO• was shown to increase MuSC self-renewal in a cyclic guanosine monophosphate-independent pathway. This requires enhanced expression of Vangl2, a member of the Wnt planar cell polarity pathway that controls the homeostatic number of MuSCs (179).
C. Activation
On muscle injury, MuSCs rapidly enter the cell cycle. Although not fully understood yet, several pathways involved in the control of MuSC activation are redox regulated (Fig. 10).
1. Calmodulin-nitric oxide pathway
Several lines of evidence indicate that NO• is involved in MuSC activation. In vivo inhibition of NOS activity by the pharmacological inhibitor N(G)-nitro-(
2. Sphingolipids
Sphingolipids are a group of lipids playing a crucial role in cell membrane structure and lipid signaling. They may act as second messengers to stimulate oxidant production (11, 241). Notably, ROS production from mitochondria has been identified as a regulator of sphingosine 1-phosphate (254). Interestingly, sphingolipid signaling is required for the entry of MuSCs in the cell cycle and sphingosine 1-phosphate regulates MuSC proliferation both in vitro and in vivo (233). Consequently, inhibition of sphingolipid signaling impairs skeletal muscle regeneration (119).
3. p38 mitogen-activated protein kinase
Mitogen-activated protein kinases (MAPKs) are members of many signaling pathways in eukaryotic cells and are known to be activated in response to oxidative stress both in vitro and in vivo (161). p38δ/β MAPK are required for MuSC activation, their specific inhibition leading to a reduced expression of MyoD and the acquisition of a quiescent state (327). This may support a role of ROS in p38δ/β MAPK-dependent MuSC activation.
4. Notch
The Notch pathway is critical for multiple stages of postnatal myogenesis. Inhibition of Notch signaling abolishes MuSC activation and impairs muscle regeneration (72). In addition to MuSC activation, Notch signaling is also involved in MuSC proliferation, whereas it must be shut down for their differentiation through antagonism with the Wnt pathway (see Section VII.C.5: Wnt) (40, 72). Although there is not yet any evidence in muscle, it was recently shown that Notch is modulated by the ROS-producer NOX1 to control the fate of colonic precursor cells (67), indicating that this important signaling pathway in myogenesis may also be regulated by ROS during muscle regeneration.
5. Wnt
The Wnt/β-catenin pathway plays important various roles in MuSC biology and during regenerative myogenesis [review in von Maltzahn et al. (344)]. Collectively, several Wnts, through the classical pathway and the planar cell polarity pathway, have been shown to control MuSC fate (179), their motility (31), their proliferation (248), and their differentiation (40). In nonmuscle systems, several redox molecules regulate Wnt signaling. Trx nucleoredoxin is a putative interactor of Dishevelled along the Wnt/β-catenin pathway that would suppress the signaling in a redox-dependent manner (115). Another redox molecule, selenium, is associated with the Wnt pathway through Gpx1. Selenium restriction is associated with Wnt activation, whereas high selenium levels inhibit Wnt signaling (41, 169). These studies show that ROS regulate the Wnt/β-catenin pathway. Given its importance in MuSC biology, it is likely that this pathway is also the target of redox regulation in the skeletal muscle.
D. Proliferation
The cell cycle has emerged as a redox cycle [review in Refs. (48, 59)]. Twenty years ago, Burdon stated that low concentrations of oxidants (O2 •−, H2O2) stimulate mammalian cell proliferation (47). Similarly, O2 •− production by MuSCs depends on NOXs (Nox2, Nox4, p22[phox], p47[phox], and p67[phox]), and MuSC proliferation is inhibited by antioxidants (N-acetylcysteine [NAC], apocynin) or Nox silencing by controlling cyclin D1 level through Erk1/2, PI-3 kinase/AKT, and NF-κB pathways (223). Furthermore, it has recently been shown that zinc promotes the MuSC proliferation in a dose-dependent manner through the positive control of PI3K/Akt and ERK signaling pathways, although it is not known whether this occurs through a redox-dependent mechanism (244).
Direct evidence for the involvement of ROS comes from genetically deficient mouse strains. Gpx1-deficient MuSCs exhibit lower proliferation rates and decreased differentiation capacity compared with wild-type MuSCs (183). MuSCs from SOD1-G93A mice (a mutation that leads to amyotrophic lateral sclerosis) demonstrate lower proliferation potential, triggering lower differentiation; whereas the expression of cyclin-dependent kinase inhibitor 1A, which is known to promote myoblast differentiation, is reduced (205).
E. Myogenic differentiation
Myogenic differentiation is an important step in myogenesis that arises from the reprograming of MuSCs to generate differentiated cells that eventually fuse into plurinucleated myotubes/myofibers. Detoxification and DNA repair appear to be crucial steps before the induction of skeletal muscle differentiation to avoid propagation of genomic abnormalities (263). This suggests that antioxidant and cytoprotective genes could be considered crucial players involved in the control of efficient myogenic differentiation, and several studies support this notion (Fig. 10).
In vitro, MuSC differentiation into myotubes elicits a rapid decline in NOX4 and p67phox expression, suggesting a change in ROS production at the time of myogenic commitment (223). Overexpression and inhibition of DUOXA1 in MuSCs reduces and enhances in vitro myogenic differentiation, respectively (293). In vivo, genetic deletion of the p66ShcA protein adaptor that promotes ROS formation results in a lower mitochondrial ROS (mtROS) production and a better muscle regeneration (360). In the same line, differentiation defect is observed in a mouse model that is deleted for sepn1 gene (52), confirming previous reports on selenium deprivation of MuSCs (328).
A molecular mechanism may be through NF-κB, which has been shown to regulate myogenic differentiation (18). In addition to NF-κB, the Nrf2-glutamate-cysteine ligase (GCL)/glutathione reductase (GR)-GSH signaling pathway is involved at the time of myogenic differentiation to regulate the GSH redox status after endogenous H2O2 is produced (259). Moreover, it has been shown that the Nrf2 target the gene encoding for carbonyl reductase 1 (CBR1), an NADPH-dependent cytosolic enzyme that reduces highly reactive aldehydes and prevents both protein carbonylation and DNA damage, is upregulated during MuSC differentiation. CBR1 inhibition is associated with an increase of ROS production, and with impaired differentiation and skeletal muscle regeneration in vivo (190). Consistently, skeletal muscle regeneration is strongly delayed in Nrf2-deficient animals, where alteration in the time course of myogenesis is observed and has been attributed to an alteration of MuSC proliferation/differentiation balance (4, 304).
Similarly, in vitro experiments showed that MuSCs cultured with dithiothreitol (a reductant agent) display a higher expression of the contractile protein myosin heavy chain as compared with those cultured under oxidized conditions (H2O2 in medium) (341). Similar results were observed on myogenic differentiation in vitro by using the antioxidant resveratrol (160, 224).
However, other studies support pro-differentiating effects of ROS in skeletal muscle. MuSC differentiation is accompanied by an increase of mtROS production (200), especially H2O2 (184, 353). As a response, an upregulation of antioxidant enzymes, mainly with peroxidase function such as Prx2, is also observed (109, 184, 353). In addition, the treatment of MuSCs with mitochondria-targeted antioxidants such as MitoQ, MitoTEMPOL, or MCAT alters their differentiation (184), as does treatment with nordihydroguaiaretic acid (NDGA) (146).
Particular attention was given to NO• effects on myogenic differentiation. First, experiments using chick MuSCs showed that NOS activity transiently increases at the time of differentiation and requires Ca2+, calmodulin, and NADPH (182). This transient activity is associated with the activation of NF-κB sites in the promoter region of the gene encoding iNOS. Interestingly, only differentiating, and not proliferating, cells exhibit these properties (182). Consequently, an NO• donor (sodium nitroprusside) induces precocious MuSC fusion, whereas an NO• competitor (NG-monomethyl-
Collectively, the major concern of all the studies cited earlier is the lack or inappropriate characterization and/or quantification of ROS entities. Moreover, opposite conclusions may result from the nonlinear dose–response relationship of oxidative stress on biological effects. Indeed, pioneer studies showed that NO• exerts positive or negative effects on MuSC proliferation in a dose-dependent way in vitro (331). Moreover, several studies showed transient ROS upregulation at the time of differentiation (181, 195), suggesting a tight regulation of their release in the MuSC neighborhood.
A recent study, using muscle conditional mutants of the homeodomain transcription factors Pitx2/3, highlights the importance of ROS levels in myogenesis (175). Double pitx2/3 mutants show excessive upregulation of ROS levels, leading to DNA damage and apoptosis of differentiating MuSCs, during both fetal and adult myogenesis. Pitx2/3 deficiency triggers the downregulation of antioxidant genes (such as Nrf1) and the impairment of mitochondrial function. Interestingly, premature muscle differentiation occurs in single Pitx3 mutants. These various effects are directly linked to dose-dependent changes in ROS levels and can be reversed by lowering ROS with NAC. Thus, a controlled increase in ROS is required for MuSC differentiation (175).
Although the impact of oxidative stress on MuSC fate is recognized, only a few studies have explored the regulation of the master genes MyoD and Myogenin by the redox balance. Cellular redox status is reflected in the balance between a number of reduced and oxidized molecular pairs, including NADH/NAD. Some transcription factors and DNA-modifying enzymes are sensitive to NADH/NAD ratio, implying that cellular redox status regulates gene transcription (192). At the time of MuSC activation, reprograming of cellular metabolism decreases intracellular NAD+ levels, decreasing, in turn, the activity of Sirtuin 1, an NAD+-dependent histone deacetylase. This leads to elevated H4K16 acetylation and activation of MyoD transcription. SIRT1 forms a complex with the acetyltransferase P300/CBP-associated factor or KAT2B (PCAF) and MyoD and, when overexpressed, delays muscle differentiation (113, 286). These results indicate that the redox status can control the myogenic gene expression program.
Later on, during myogenesis, exposing differentiating MuSCs to oxidative stress increases the nuclear localization of focal adhesion kinase, which interacts with MBD2 (methyl CpG-binding protein 2). This leads to a decreased MBD2 association with HDAC1 (histone deacetylase complex 1) and methyl CpG sites in the Myog promoter, thus inducing myogenin expression (198). Finally, heme oxygenase-1 (HMOX1), which is a cytoprotective enzyme and whose expression is induced in response to oxidative stress, inhibits miRNA processing, leading to a decrease of several myomiRs (miR-1, miR-133a/b, miR-206), which triggers a reduced expression of MyoD, myogenin, and myosins. However, it is important to note that this activity is independent of the antioxidative properties of HMOX1 (172).
ROS are clearly involved in the numerous signaling pathways that control the sequential steps of MuSC fate after skeletal muscle injury (Fig. 10). The recent development of new tools (redox sensors, redox proteomics) will allow for a more accurate identification and quantification of these ROS and for the identification of which entity, at which level, and during which time period, controls a specific stage of MuSC fate. Finally, MuSCs evolve in a continuously changing environment during muscle regeneration, and receive cues from their immediate microenvironment, which has been shown to be necessary for efficient regeneration, and which is also subjected to redox regulation.
VIII. Redox Regulation of the Cells Present in the MuSC Microenvironment
Skeletal muscle regeneration requires the involvement of many cell types, including vessel cells, interstitial cells, and inflammatory cells, such as monocytes/macrophages (55) (Figs. 3 and 4).
A. Myeloid cells
After skeletal muscle injury, leukocytes rapidly invade the damaged area. Neutrophils rapidly disappear in a few days, whereas monocytes convert into macrophages that exert various roles on MuSCs and on the regeneration process (289) (Figs. 4 and 11). Numerous data on redox signaling in macrophages are available in the context of infection where they are shown to actively produce ROS (44, 106). Inversely, only a few studies have investigated the redox status and redox regulation of macrophage biology during tissue injury/repair. In wound-healing models in zebrafish and Drosophila embryo, leukocyte recruitment depends on an H2O2 signal gradient generated by DUOX activation (237, 271).
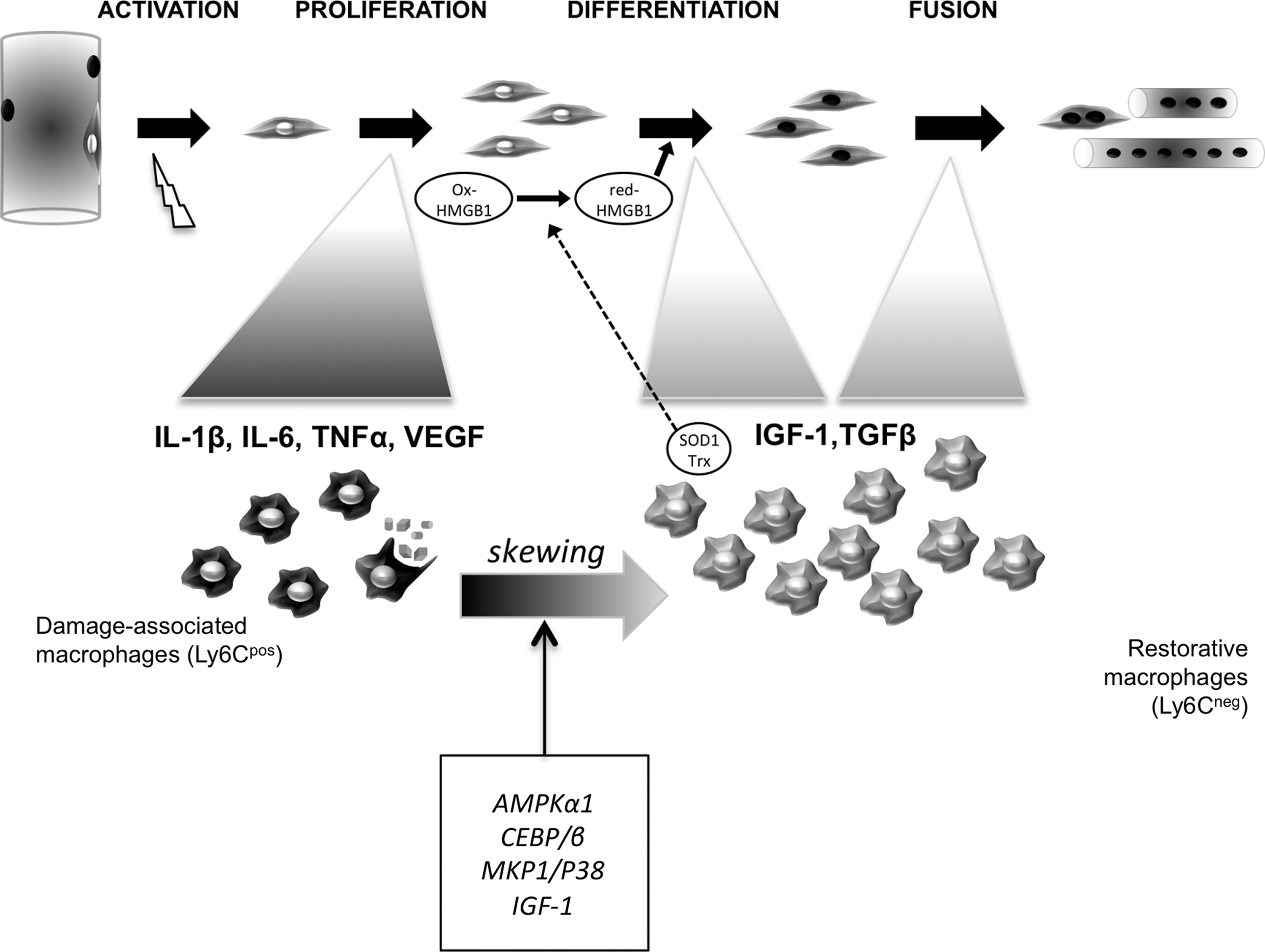
In skeletal muscle, as in other tissues, entering monocytes/macrophages exhibit a damage-associated inflammatory profile and release pro-inflammatory cytokines (TNF-α, IL-1β, IL-6). In skeletal muscle, the most studied redox-regulated factor is high-mobility group box 1 (HMGB1), a ubiquitous nuclear protein acting as a Damage-Associated Molecular Pattern to signal tissue damage when released extracellularly. When cysteine residues are reduced, HMGB1 promotes the recruitment of inflammatory cells via its interaction with the CXCL12 chemokine to bind the CXCR4 receptor. When cysteine residues are oxidized, HGMB1 promotes the release of proinflammatory cytokines (339). After injury, an oxidized form of HMGB1 is detected in the muscle within 6 h (339). Then, an antioxidant response is observed in the regenerating muscle, notably in leukocytes, which express high levels of SOD1 and Trx, suggesting that a biphasic expression of oxidized/reduced forms of HMGB1 is required for skeletal muscle regeneration. In accordance, HMGB1 oxidation abrogates migration of MuSCs and their ability to differentiate into myotubes in vitro (341).
To date, no study has characterized ROS production in macrophages in the context of skeletal muscle regeneration or tissue repair. iNOS is expressed by inflammatory macrophages that infiltrate a tissue on infection. It is also expressed by inflammatory macrophages during the first steps of muscle regeneration, its expression declining with time, and notably in restorative anti-inflammatory macrophages (227). It has been shown that mice deficient for inos (which is mainly expressed by macrophages infiltrating the injured muscle) fail to regenerate properly because of a defect of MuSC proliferation and differentiation, associated with an increase of inflammatory markers (280), indicating a role of macrophage-derived iNOS in skeletal muscle regeneration.
The pro-inflammatory phase ends with phagocytosis of muscle debris, at which point macrophages switch their phenotype to give rise to anti-inflammatory/restorative macrophages (15, 227). These restorative macrophages produce soluble factors (IL-10, TGFβ, VEGF, IGF-1) to dampen inflammation and stimulate tissue repair, notably triggering myogenic differentiation (Fig. 11) (15, 227, 290).
The switch (skewing) of the macrophage phenotype occurring during skeletal muscle regeneration is a key feature of efficient repair (227). Interestingly, all effectors identified so far as regulators of this process are redox sensitive. The MKP-1/p38 MAPK axis, which belongs to the redox-sensitive MAPK family (7), regulates macrophage phenotype skewing in a timely way to promote the resolution of inflammation (257). Another redox-sensitive pathway, CREB-C/EBPβ (27) is required for the expression of the anti-inflammatory program in macrophages (285). A third effector is AMPKα1, which is required in macrophages for the acquisition of the anti-inflammatory phenotype, ensuring efficient muscle regeneration (227). AMPK is linked to redox-based signals, notably it is targeted by pro-oxidant species, suggesting a role for AMPK in the tuning of redox-dependent processes [review in Cardaci et al. (51)].
A fourth molecule has been involved in the skewing of macrophages during skeletal muscle regeneration. Insulin growth factor-1 (IGF-1), which is a master regulator of myogenesis and skeletal muscle growth, is expressed mainly by macrophages during muscle regeneration and is required for their skewing in an autocrine way (324). One study reported that ROS suppression (UK-134 and α-lipoic acid) restores IGF-1 sensitivity of diabetic fibroblasts and improves their healing functions both in vitro and in vivo (37).
In vitro studies have shown that ROS/RNS production is needed for macrophage skewing. Inhibition of O2 •− production specifically blocks the differentiation of alternatively activated (G-CSF+IL-4) (so-called M2) macrophages. In vivo, continuous administration of an ROS inhibitor (butylated hydroxyanisole) efficiently blocks the occurrence of tumor-associated macrophages, which exhibit an M2 phenotype in mouse cancer models (362). Similarly, macrophages issued from Sod1 −/− mice display a pro-inflammatory profile as compared with WT macrophages (134). Inversely, in Sod1-Tg mice, where Sod1 is overexpressed and H2O2 generation is enhanced, macrophages exhibit an anti-inflammatory status, associated with an increased activation of STAT6 (134), which is known to trigger the expression of anti-inflammatory genes in macrophages (178).
B. Endothelial cells
Skeletal muscle regeneration is associated with the angiogenic process, to rebuild the vascular bed after injury. Privileged interactions have been described between MuSCs and ECs. MuSCs are located close to capillaries, and their number increases with the capillarization of myofibers (63). MuSCs express several angiogenic factors, including vascular endothelial growth factor (VEGF), and exhibit angiogenic properties (278). Similarly, ECs release factors supporting MuSC growth. Thus, MuSCs and ECs interact to support skeletal muscle regeneration (63, 177, 226).
Although the role of ROS in these processes has not yet been investigated in skeletal muscle, there is a considerable amount of evidence coming from other models that ROS modulate angiogenesis through the regulation of redox-sensitive signaling pathways. This results in the control of EC growth, proliferation, and migration [review in Refs. (95, 111)]. ECs generate ROS such as O2 •− and H2O2. NOX is the main source of ROS in ECs and is activated by growth factors, cytokines, hormones, and hypoxia (125).
Exogenous ROS, as well as overexpression of NOX4, increase the expression of VEGF, VEGFR2, and angiopoietin 1 and stimulate EC proliferation, migration, and angiogenesis in vitro (1, 64, 79, 334). Furthermore, angiopoietin 1-induced angiogenesis is enhanced in CAT-deficient mice, suggesting an involvement of H2O2 in angiogenesis (168). Conversely, in Tie2-driven Cat transgenic mice (where Cat is constitutively expressed in ECs), impairment of neovascularization and reduced vessel sprouting after injury are observed (333). In the same line, silencing the 66ShcA protein adaptor, which promotes ROS formation and by which phosphorylation is enhanced by VEGF, inhibits both VEGF-induced ROS generation and EC proliferation and migration (245).
A few studies have directly investigated the role of ROS in the environment of MuSCs during skeletal muscle regeneration. However, evidence from other models of tissue repair argues for the redox regulation of both immune cells and ECs, which likely takes place in the skeletal muscle milieu.
IX. Skeletal Muscle Regeneration in Pathophysiological Contexts
A. Exercise-induced muscle damage
In a pioneering paper, Davies et al. demonstrated an increase in free radical concentrations in rat skeletal muscle after exercise to exhaustion (80). Since then, aerobic and anaerobic exercises have clearly been shown to increase ROS/RNS production in humans (102). The relationship between ROS and contractile activity has also been widely investigated. NO• derivatives and ROS modulate contractile function of skeletal muscle (273). At rest and during exercise, skeletal muscle produces both species (261) and antioxidant defense systems as an adaptive response (154). It is now acknowledged that low and physiological levels of ROS are required for force production, whereas high levels of ROS promote contractile dysfunction, as a result of oxidative damage of proteins and lipids, triggering muscle weakness and fatigue (261).
Exercise-induced muscle damage (EIMD) is particularly induced by eccentric contractions, which require lengthening of the muscle (112, 143, 262). After physical exercise with repeated eccentric contractions, such as long-distance or downhill running, soreness appears within a few hours after the effort, a phenomenon called delayed-onset muscular soreness (DOMS). Despite an unclear etiology, DOMS is characterized by various markers such as histological changes (Z-line disruption), biochemical changes (increased creatine kinase and lactate dehydrogenase in plasma/serum), and altered clinical scores (increased soreness, reduced specific force, decreased range of motion). Several studies showed that damaging exercises are temporally associated with an increase in ROS production (66) and oxidative stress markers (239). Lipid peroxidation markers such as malondialdehyde (MDA), thiobarbituric-acid reactive substances, or protein carbonyls are increased in the blood (99, 240) and in the skeletal muscle (218).
In parallel, no meaningful alteration of antioxidant enzyme activity is reported in the blood [review in Lee et al. (180)]. In whole muscle lysates, the GSH/GSSG ratio is slightly decreased at 2 and 4 days postexercise, whereas the levels of antioxidant enzyme activitity only slightly increase, most notably when compared with markers of oxidative stress/damage (218, 240, 313). Moreover, as insulin receptor substrate-1 oxidation is induced after a strenuous exercise, it was proposed as a possible explanation for the impairment of insulin-induced glucose uptake observed in damaged muscle, through the alteration of insulin signaling (12).
A difficulty of such skeletal muscle physiology studies is to evaluate the level of myofiber damage, which is dependent not only on the type of exercise but also on the status of the subject (healthy/pathologic, trained/sedentary, young/old, etc.) (253). Thus, the link between EIMD and MuSC activation is still unclear. Several levels of EIMD, classified according to the loss of muscle force, have been proposed and linked to the relative extent of inflammation (253). Similar comparative investigations should help to determine the role of ROS in EIMD.
B. Aging
Aging is a process that is characterized by an impairment of functional capacities. Many studies on ROS during aging were inspired by Harman's “free radical theory of aging” (131), which reports that “aging and the degenerative diseases associated are attributed basically to the deleterious side attacks of free radicals on cell constituents and on the connected tissues.” Later on, the identification of mitochondria as the main source of ROS and oxidative damage during aging strengthened this free radical theory of aging (221). Interestingly, aging is associated not only with an increase of oxidative damage but also with an upregulation of antioxidant enzyme levels in skeletal muscle. Indeed, aging increases the skeletal muscle content of Trx1, Trx2, TrxR2, cytosolic and mitochondrial SOD, CAT, and Gpxs (86, 155).
Aggregation of damaged proteins, mitochondrial dysfunction, and epigenetic alterations and accumulation of toxic metabolites (ROS) and of DNA damage are hallmarks of aged stem cells (243). In the skeletal muscle, proteomic investigations revealed that aged skeletal muscle is targeted by oxidative protein post-translational modifications such as carbonylation that alters protein functions (19). Moreover, aging causes a reduction in redox-sensitive proteins that are involved in energy metabolism, indicating a loss in the flexibility of the redox energy response (211). Although numerous studies have investigated the impact of ROS and its regulation on the aged skeletal muscle [review in Refs. (12, 148)], few experimental data establish a link between MuSC aging and ROS (Fig. 12).
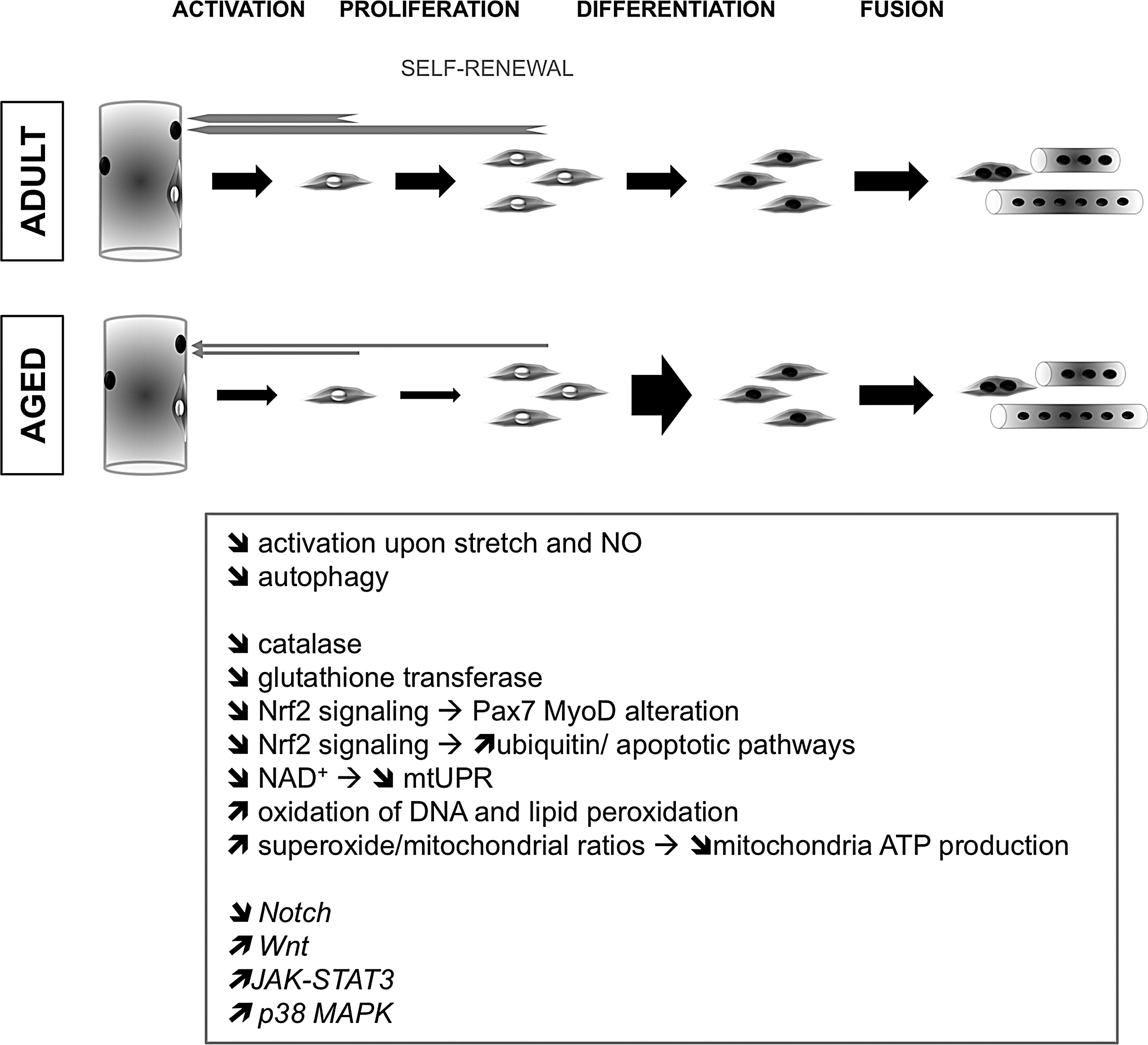
In aged muscle, MuSCs exhibit a reduced capacity to proliferate and self-renew, whereas they are prone to differentiation. This is explained by a deregulation of several signaling pathways, including decreased Notch signaling (71), increased Wnt signaling (36), increased JAK-STAT3 (322), and p38 MAPK (32) signaling pathways. All these pathways are redox sensitive as discussed earlier (see Section VII.C).
Only a few studies directly investigate ROS in aged MuSCs. In vitro, old MuSCs are refractory to stretch and NO•-induced activation, a property that is rescued by NO• donors (33). An increase in oxidation of DNA and lipids is observed in the elderly MuSCs, together with a reduction in CAT and glutathione transferase activities (114). Similarly, an increase in lipid peroxidation is observed in myotubes derived from aged MuSCs, and is associated with a defective status of the excitation-contraction coupling apparatus (25). Cultured aged MuSCs exhibit increased O2 •−/mitochondrial mass and H2O2/mitochondrial mass ratios. The increased ROS production correlates with the corresponding decreased mitochondrial ATP production (220). The antioxidant Nrf2 signaling is impaired in aging muscle (219). Of note, in Nrf2-deficient mice subjected to acute endurance exercise stress, an alteration in the expression of the MuSC markers Pax7 and MyoD is observed along with the activation of ubiquitin and apoptotic pathways (234).
More recently, studies demonstrated that autophagy has an important role in sustaining the higher bioenergetic demands required for MuSC activation (318) and in maintaining MuSC stemness by preventing the senescence caused by mitochondrial dysfunction and oxidative stress during aging (117). Hence, it is clear that ROS and autophagy participate in skeletal muscle homeostasis (193, 268) and are involved in muscle dystrophies (249); however, it is still unclear whether ROS are crucial for the induction of autophagy and whether up- or downregulation of these processes induces a detrimental or a beneficial response (282). Another redox-sensitive mechanism has recently been involved in the maintenance of aged MuSCs. Reduction of the cellular NAD+ pool (which is required for mitochondria function) blunts the adaptive mitochondrial unfolded protein response pathway that leads to the loss of MuSCs and a decrease of their regenerative capacities (361). Although the precise mechanisms are still to be deciphered, these studies show that in aged skeletal muscle, higher ROS provide an unfavorable environment, which can adversely affect MuSC survival and functions and limit skeletal muscle repair (9).
C. Muscular dystrophies
The increase of oxidative stress in muscular dystrophy, which was acknowledged a long time ago (132, 228), has been shown to correlate with the severity of the pathology (274) and is considered to contribute to the pathology of several muscular dystrophies (173). Interestingly, mutations in the antioxidant sepn1 gene cause SEPN1-related myopathy, a different class of muscle disease (14). Although oxidative damage is assumed to participate in the pathogenesis of the disease, the exact mechanisms are still poorly understood, particularly at the level of the sarcolemma, where there is no direct experimental evidence of myofiber damage by oxidative stress [review in Tidball and Wehling-Henricks (321)].
Calcium homeostasis is perturbed in muscular dystrophies (6). In this context, it has been shown that S-nitrosylation of the ryanodine receptor 1 (RyR1), an intracellular calcium channel that is essential for the excitation-contraction coupling of the myofiber, induces a leak of sarcoplasmic reticulum Ca2+, contributing to muscle weakness (28, 29, 94). Nevertheless, cellular and molecular mechanisms involving ROS signaling in the MuSC defects observed in muscular dystrophies are still poorly understood (Fig. 13).
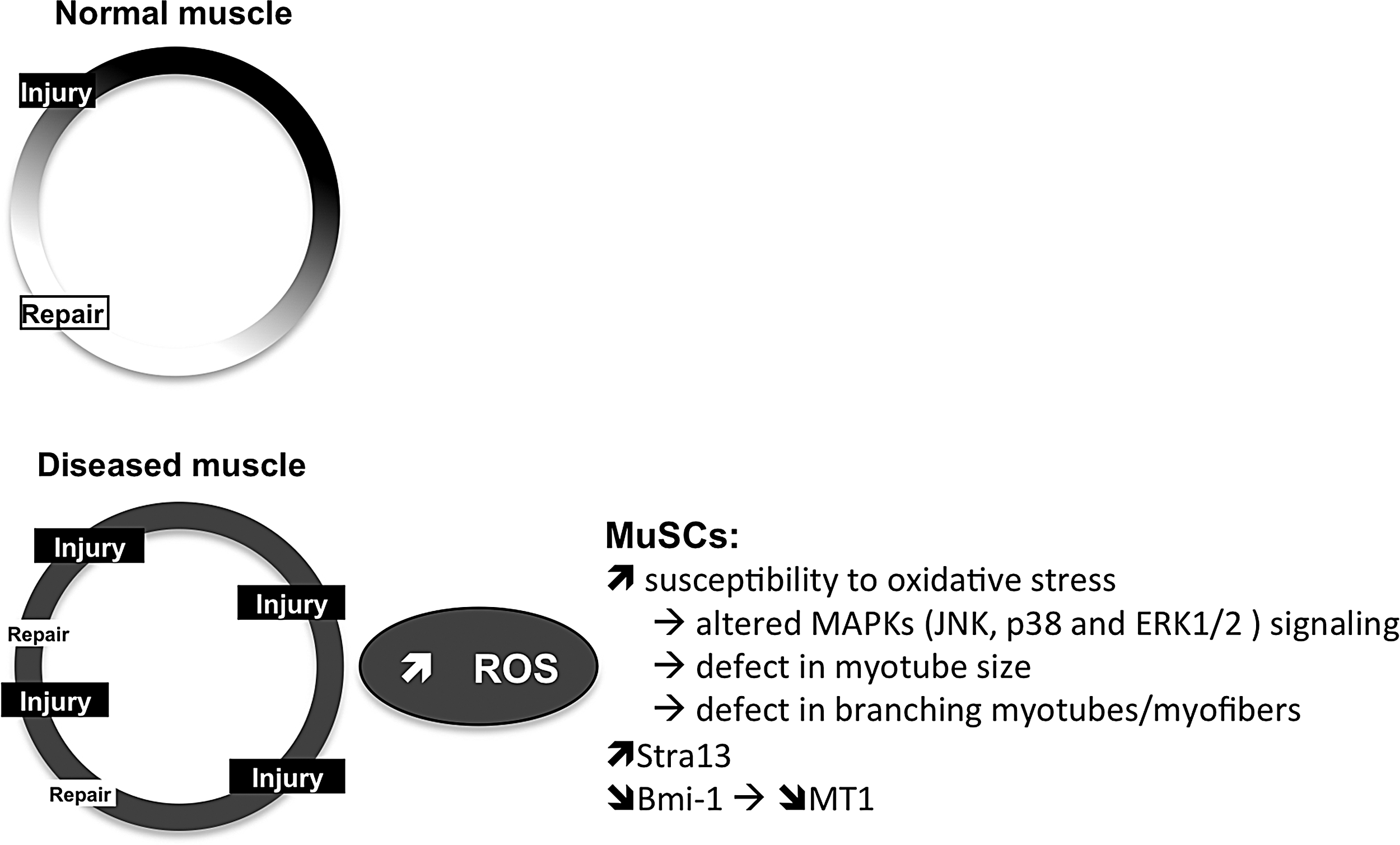
The first evidence came from cultured MuSCs derived from either normal or dystrophic muscles. In mice, dystrophin-deficient MuSC-derived myotubes are more susceptible to free radical-induced injury (269). The relative susceptibility of MuSC-derived myotubes to oxidative stress is associated with the severity of the dystrophy (assessed by the expression of different lengths of the dystrophin protein), and the extent of protein oxidation, suggesting that the DCG complex has important functions in antioxidant defense (88). It was shown that nNOS, which binds to DGC, has no impact on MuSC susceptibility to oxidative stress (363). Inversely, increased susceptibility of dystrophin-deficient-derived myotubes to oxidative stress is associated with altered phosphorylation of MAPKs, including JNK, p38, and ERK1/2 (309), which are both targeted by ROS and involved in myogenesis (see Section VII).
Susceptibility to oxidative stress seems to be a common feature in several muscular dystrophies. Indeed, MuSCs derived from affected muscles in facioscapulohumeral muscular dystrophy (FSHD) patients exhibit higher susceptibility to an induced oxidative stress than MuSCs derived from nonaffected muscles of the same patients (350). This is associated with a defect in the size and branching of the differentiated myotubes (23). At the regulatory levels, it was shown that the transcriptional repressor Stra13, which is elevated in dystrophin-deficient mdx mice, protects MuSCs from oxidative damage whereas Stra13 deficiency exacerbates muscular dystrophy (340).
Epigenetic regulation has also been reported to control redox signaling and MuSC fate. The Polycomb group protein Bmi1, which is an essential epigenetic regulator of stem cell function, is upregulated during normal skeletal muscle regeneration, whereas the number of MuSCs expressing high Bmi1 level is reduced in mdx muscle. Upregulation of Bmi1 in MuSCs in mdx triggers the increase of metallothionein that confers MuSCs with the ability to resist oxidative stress and to limit DNA oxidative damage, thus improving the pathology (84). These data confirm that ROS are elevated in muscular dystrophies and that MuSCs are more sensitive to oxidative stress, altering their functions and participating in the skeletal muscle deterioration in these progressive diseases.
Only a few studies have investigated the role of oxidative stress in the nonmyogenic cell types in muscular dystrophies. It is assumed that NO deficiency in DMD contributes to the pathogenesis of the disease (69). In transgenic mdx animals expressing normal NO levels (through the constitutive expression of nnos), an improvement of the muscle state is observed with a decrease of inflammation and of sarcolemma injury (346). In this context, long-term treatment of dystrophic mice with HCT 1026, a nitric oxide-releasing derivative of flurbiprofen, ameliorates the morphological, biochemical, and functional phenotype of the diseased muscle (45) through a decrease of the number of inflammatory cells and FAPs, thus reducing fibrosis (74).
X. Therapeutics Opportunities
Various strategies have been used to improve the recovery of skeletal muscle function. Here, we present data related to exercise and antioxidant supplementation as approaches to improve skeletal muscle homeostasis.
A. Exercise as therapy
Skeletal muscle is a secretory organ. On contractile activity, it releases myokines that are known to control hypertrophy (LIF, IL-4, IL-6, IL-7, IL-15) [review in Pedersen and Febbraio (256)]. Some myokines have also been identified as crucial molecules that are involved in skeletal muscle regeneration (290). Few studies have investigated the role of exercise/contractile activity after skeletal muscle injury. It appears that exercise (running) increases skeletal muscle mass and the cross-sectional area of regenerating muscle as well as the recovery of metabolic properties (171, 279). These effects can be explained by a higher activation of the mTOR pathway, the master regulator of cell growth and protein synthesis previously described as a redox-regulated molecule (294) and of PGC-1α (171), the master regulator of oxidative metabolism (191, 196).
Exercise, through ROS production, initiates the transcription of antioxidant genes and specific signaling pathways (122, 154). Thus, an increased production of ROS may act as an activator of the adaptive response in skeletal muscle, resulting in the increased production of antioxidant enzymes that could facilitate rapid muscle remodeling and provide protection against subsequent periods of damaging exercise (209). However, no study specifically investigated the effect of exercise on ROS in MuSCs. In contrast, endurance exercise of long duration and/or high intensity appears to generate much higher levels of ROS that can overwhelm cellular antioxidant defenses, and may cause tissue damage. This paradoxical effect of physical exercise refers to the hormesis theory (267). Such ROS-induced damage requires effective repair systems, and sufficient recuperation, before exercise adaptation can recommence (288).
However, the over generation of ROS observed during acute exercise is blunted in endurance-trained subjects, and most studies demonstrate that postexercise oxidative stress is reduced after endurance exercise training (267). The main mechanism for this reduction in oxidative stress is likely the upregulation of enzymatic antioxidants in response to an exercise-training program (261). SOD, one of the initial antioxidant enzymes expressed in response to oxidative stress, has been regularly reported to increase in response to exercise training (185), whereas activity of Gpx demonstrates a muscle fiber-specific pattern with type IIa being the most responsive to training (260). On the contrary, the measurement of CAT activity in the skeletal muscle in response to exercise training gives inconsistent results.
Aged skeletal muscle is more susceptible to damage, particularly after exercise, and is unable to rapidly adapt after sequential periods of exercise. However, the elderly who are physically active benefit from exercise-induced adaptation in cellular antioxidant defense systems (326). Improved skeletal muscle mechanics, strength, and endurance make them less vulnerable to acute muscle injury and chronic inflammation (138). Therefore, many critical questions remain regarding the relationship of aging, ROS, and exercise. At this time, there is no recommendation for exercising in dystrophic patients. It could be hypothesized that patients receiving pharmacological/gene/cell therapy could practice adapted exercise with exclusive concentric contraction to release beneficial myokines and adaptive responses to free radicals.
B. Antioxidant supplementation
1. In EIMD
The use of antioxidants in the field of physical exercise is still a controversial issue [review in Refs. (122, 212, 258)]. Some groups have reported that antioxidant supplementation has a negative effect on training-induced adaptations and health (73, 121, 201, 202, 252, 281). On the contrary, other groups reported no alterations of training-induced performance improvements (225, 357, 358). These conflicting results have to be considered in the light of training status, type of exercise, and qualitative and quantitative supplementation. Further studies are needed to elucidate this complex relationship. A wide range of studies has used nutritional antioxidant supplementation as a countermeasure to improve recovery after EIMD (212, 231). Indeed, studies have investigated the role of vitamin C, vitamin E, combined antioxidant supplementation, or NAC as potent protectors against skeletal muscle damage after eccentric contraction. Antioxidant supplementation before and 7 days after intensive knee maximal eccentric contraction reduces the loss of force after exercise as compared with the control group (303).
However, some data indicate that antioxidants may delay muscle recovery (66, 218). Indeed, prolonged vitamin C supplementation 2 days before and 14 days after downhill running exercise reduces muscle soreness and MDA levels, but it is associated with a delayed recovery of muscle function (66). In 2013, a double-blind study suggested that the modulation of redox biology by NAC supplementation for 8 days after quadriceps eccentric contractions at low velocity may interfere with skeletal muscle repair and myogenic program through the modulation of transcription factors involved in inflammation and myogenic program (218). In view of these results, there is little evidence to support a beneficial role of antioxidant supplementation in response to skeletal muscle damage (231).
As for the investigation of the ROS mechanisms in MuSCs, few studies explored the effects of antioxidant treatment on MuSC behavior after EIMD. In fact, to our knowledge, only one group has investigated the effects of antioxidant administration during postcontusion skeletal muscle regeneration at the cellular level. In vivo administration of polyphenol in rats 2 weeks before and after the injury induces an accelerated activation of MuSCs, and consequently, a rapid neofiber formation that is completed earlier than in the control group (232). Furthermore, polyphenol treatment blunts the entry of neutrophils into the injured muscle, whereas the kinetics of macrophages and of their skewing to restorative macrophages is accelerated, and the production of the anti-inflammatory cytokine IL-10 is increased (174, 232).
2. In aging
So far, most studies performed to counteract oxidative stress in aged muscle have targeted sarcopenia and muscle atrophy. Results consistently report significant improvements of antioxidant biomarkers after antioxidant supplementation, whereas the effects on muscle function are more mitigated [review in Cerullo et al. (53)] (Fig. 14). However, increased mitochondrial antioxidant activity, using transgenic mice with targeted overexpression of the human cat gene to mitochondria (mtCAT mice), restores age-dependent skeletal muscle dysfunction (332). This suggests that the preservation of muscle function in aging may be specific to the type of antioxidant and their site of action.

Studies investigating the functions of MuSCs in aging muscle are rare and relatively new, due to the new development of tools allowing the specific analysis of these cells. In vitro treatment of human senescent MuSCs with tocotrienol rich fraction (vitamin E) reduces their senescent phenotype and increases their proliferation capacity (190). In aged mice, antioxidant treatment (using Trolox, a vitamin E analog) prevents the appearance of senescence markers and restores the expansion capacity of MuSCs, and therefore their regenerative properties, through the recovery of an effective autophagy (117). Similarly, treatment with the NAD precursor nicotinamide riboside restores MuSC concentration of NAD+, improves mitochondrial homeostasis, and maintains MuSC functions in aging mice (110, 361). Moreover, in mice deficient for SIRT1 (an NAD+-dependent deacetylase that increases mitochondrial biogenesis) in MuSCs, nicotinamide riboside beneficial properties are dampened in regenerating muscle, making a link between the NAD+-SIRT1 pathway, mitochondrial activity, and MuSC function (361) (Fig. 14).
3. In muscular dystrophies
More than 30 years ago, thus before the discovery of the dystrophin gene, supplementation with antioxidants (SOD precursors, vitamin E, selenium) was carried out in DMD patients, since vitamin E deficiency triggers similar skeletal muscle damage as that observed in DMD patients (6). Ever since, antioxidants have been shown to reduce necrosis in the skeletal muscle of dystrophin-deficient mdx mice (215). NAC administration results in decreased numbers of regenerating myofibers (349). FSHD patients supplemented with vitamin C, vitamin E, zinc gluconate, and selenomethionine increase their antioxidant level and improve some physical performance (251). However, it should be acknowledged that most of the results obtained with antioxidant treatments are inconsistent (167), highlighting the complexity of redox biology (Fig. 14).
Few studies have explored the effects of antioxidant treatment on MuSC behavior (Fig. 14). In vitro, NDGA (inhibitor of Lysyl oxidase) treatment of primary MuSCs issued from a DMD patient inhibits their proliferation while stimulating their differentiation and myotube formation in a dose-dependent manner (118). Green tea polyphenol blend and its major polyphenol (EGCg) efficiently protect mdx myotubes from oxidative damage, since mdx MuSCs express sevenfold more 67LR, a receptor for EGCg, than normal MuSCs (89). In desminopathies, mutations trigger the formation of desmin aggregates in MuSCs on stress conditions, including redox-associated (H2O2 and cadmium chloride) stress. Antioxidants (fisetin, NAC, α-tocopherols, Trolox) prevent desmin-aggregation during stress in MuSCs expressing specific desmin mutants (49, 299). Still in vitro, it was shown that supplementation of MuSCs with α-tocopherol promotes plasma membrane repair on oxidant challenge (141).
In vivo, various antioxidant treatments trigger an improvement of the muscle phenotype of mdx or sgca KO (α-sarcoglycan null mice, a severe mouse model of dystrophy) mice, together with an increase in the muscle function. Some studies evaluated the impact of such treatments on MuSCs. Mdx treatment with flavocoxid for 5 weeks increases the number of myogenin-positive cells (216). Similarly, oral administration of the flavonol quercetin during 6 months to adult mdx mice increases the number of new fibers as well as the number of MuSCs, besides a general improvement of the muscle (139). In sgca KO mice, NCX 320 (cyclooxygenase-inhibiting NO donator) treatment for 8 months leads to an increase in the number of newly formed fibers, as well as an increase of the number of MuSCs. Moreover, MuSCs exhibit an increased ability to differentiate in vitro and to express the differentiation markers myogenin and myosin heavy chain, indicative of their good regenerative potential (298). Finally, mdx mice supplemented for 10 weeks with nicotinamide riboside exhibit an increased number of MuSCs while muscle regeneration is improved, indicating that nicotinamide riboside also prevents MuSC senescence in muscular dystrophy (287, 361), as it does in aging muscle (see Section X-B-2: In aging) (Fig. 14).
On the whole, these studies indicate that antioxidant supplementation can benefit muscle health in muscular dystrophies. It is worth mentioning that the importance of inflammation and inflammatory cells in the establishment and maintenance of the pathology during muscular dystrophies is recognized, whereas the phenotype and functions of these cells have been overlooked. In this context, it is interesting to mention that antioxidant treatment has been shown to reduce the amount of inflammatory cells in these diseases, and this decrease is associated with the improvement observed in the muscle (298).
XI. Perspectives
Although redox signaling has long been known to play a role in skeletal muscle homeostasis, it has recently been established that the cellular and molecular processes at work during skeletal muscle regeneration are redox regulated. Numerous signaling pathways that control MuSC biology as well as their close environment, including immune cells, are targeted by ROS/RNS and are altered in physiological (EIMD, aging) or pathological (muscular dystrophies) situations. Attempts to counteract oxidative stress in such contexts have led to inconsistent results that highlight the complexity of redox biology (6, 167). This complexity has begun to be deciphered at the cellular and molecular levels. Measuring ROS concentrations, as well as redox modifications is difficult, notably in vivo, given the short half-lives, high specificities, and high reactivity of the involved compounds.
Very recent years have seen the development of tools to decipher redox complexity. Given the current knowledge, the field should move toward accurate identification of sources of ROS and their molecular targets. Indeed, sensor reporters will help to precisely identify and quantify the ROS/RNS that are produced in a tissue and in a given cell (Fig. 15). In addition, techniques of redox proteomics will help to decipher molecular targets of ROS/RNS and the nature of their post-translational modifications (Fig. 15).
Such technological improvements already form the basis of new therapeutic advancements. For example, the development of selective antioxidant probes has led to therapeutic efficiency in preclinical animal studies, and in some cases, such as with the mitochondria-targeted antioxidant MitoQ, in phase II human trials (308). The design of a specific mitochondria-selective S-nitrosating agent, MitoSNO, has been found to be cardioprotective in vivo in mice after ischemia/reperfusion, through the S-nitrosation of mitochondrial complex I, which is the entry point for electrons from NADH into the respiratory chain (62).
A genome-wide Cas9-mediated screen enabled the identification of the hypoxia response as protective during mitochondrial respiratory chain inhibition. Genetic or small-molecule activation of the hypoxia response is, therefore, protective against mitochondrial toxicity (150).
NOX are also potent targets for therapeutic developments. New NOX inhibitors, identified through a high-throughput screen after structure–activity relationship analysis, showed improved specificity for NOXs and were validated in in vivo animal studies, although they require further developments to improve isoform selectivity. For instance, the NOX4 inhibitor GKT137831 has been shown to reduce fibrosis in several animal models of chronic diseases and is under clinical development (8). Inversely, after an acute injury, inducing a compartment-specific increase of the NAD+/NADH ratio (using LbNOX water-forming NOX from Lactobacillus brevis) allowed to decipher the NAD+/NADH ratio-driven signaling and to improve proliferative and metabolic defects in mitochondrial-deficient cells (323). Several studies recently confirmed that the redox couple NAD+/NADH is a privileged target to improve cell and tissue homeostasis, including in aged and diseased skeletal muscle (see Section X.B) (110, 287, 361).
Therefore, such an integrated approach would be an interesting option to better understand the underlying mechanisms linking redox regulation and skeletal muscle regeneration. Thus, the identification of redox-regulated networks will be useful to design specifically targeted antioxidant probes to improve skeletal muscle phenotype and function.