
Abstract
Significance:
Metabolic syndrome is a frequent precursor of type 2 diabetes mellitus (T2D), a disease that currently affects ∼8% of the adult population worldwide. Pancreatic beta-cell dysfunction and loss are central to the disease process, although understanding of the underlying molecular mechanisms is still fragmentary.
Recent Advances:
Oversupply of nutrients, including glucose and fatty acids, and the subsequent overstimulation of beta cells, are believed to be an important contributor to insulin secretory failure in T2D. Hypoxia has also recently been implicated in beta-cell damage. Accumulating evidence points to a role for oxidative stress in both processes. Although the production of reactive oxygen species (ROS) results from enhanced mitochondrial respiration during stimulation with glucose and other fuels, the expression of antioxidant defense genes is unusually low (or disallowed) in beta cells.
Critical Issues:
Not all subjects with metabolic syndrome and hyperglycemia go on to develop full-blown diabetes, implying an important role in disease risk for gene–environment interactions. Possession of common risk alleles at the SLC30A8 locus, encoding the beta-cell granule zinc transporter ZnT8, may affect cytosolic Zn2+ concentrations and thus susceptibility to hypoxia and oxidative stress.
Future Directions:
Loss of normal beta-cell function, rather than total mass, is increasingly considered to be the major driver for impaired insulin secretion in diabetes. Better understanding of the role of oxidative changes, its modulation by genes involved in disease risk, and effects on beta-cell identity may facilitate the development of new therapeutic strategies to this disease. Antioxid. Redox Signal. 26, 501–518.
Introduction
A
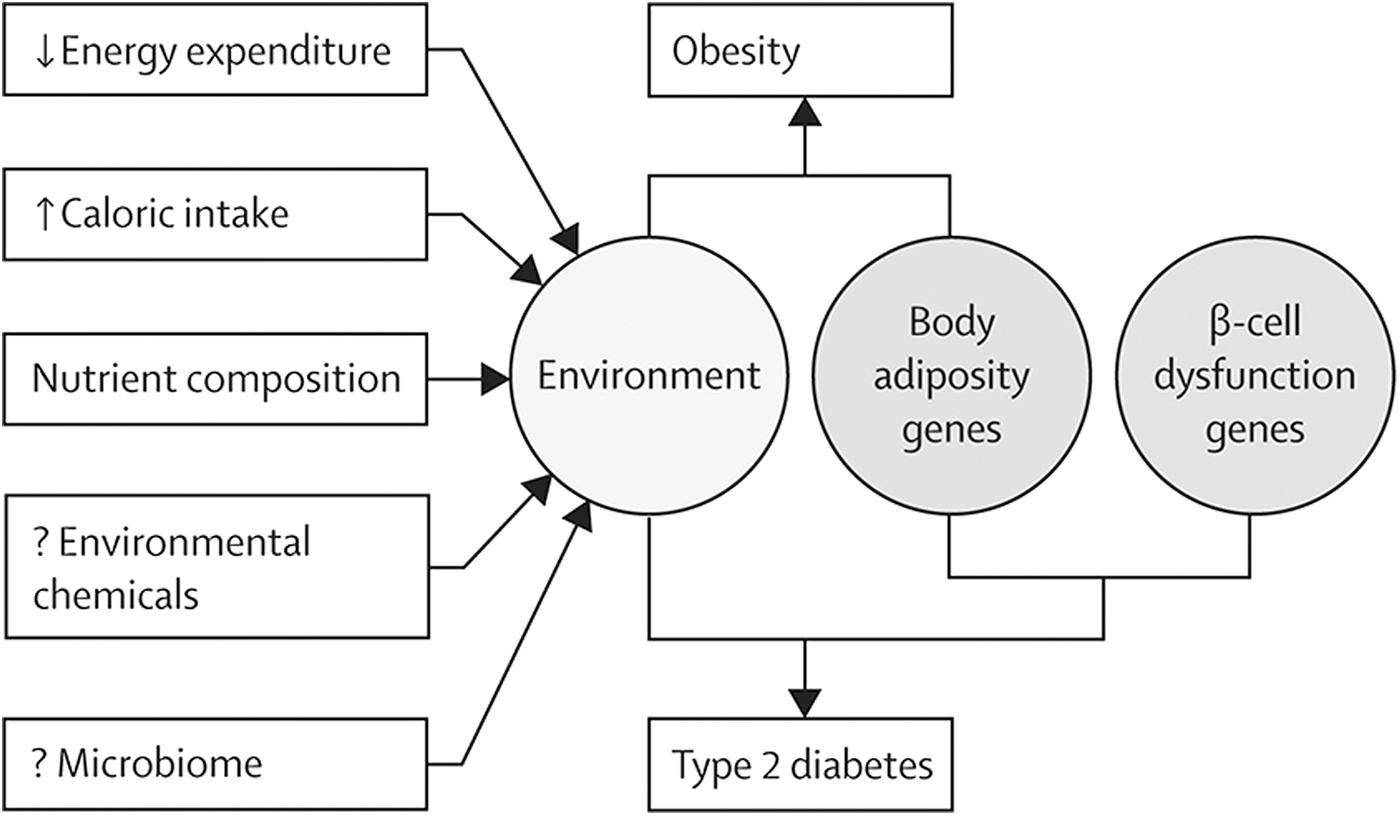
Further demonstrating the importance of disrupted beta-cell function for the development of T2D in the context of the metabolic syndrome, genome-wide association studies (38, 183) indicate that the majority of the known gene variants that increase the risk of T2D affect beta-cell function rather than insulin sensitivity (58, 164). The extent to which decreased beta-cell mass (24) and dysfunction (35) contribute to the impairments in insulin production in T2D is contested (168), although recent estimates of relatively minor changes (∼24% at diagnosis) in the former (112) have placed the onus on dysfunction as the important driver. The interaction of environment and genetic background in the development of obesity and T2D is depicted in Figure 1.
Pancreatic beta cells are among the most metabolically active tissues within the human body, and they are highly dependent on oxidative metabolism for adenosine triphosphate (ATP) synthesis, particularly at elevated glucose concentrations (152, 176). Indeed, elevated oxygen consumption at high glucose levels is central to the stimulation of insulin secretion [(168) and see “Stimulation of the formation of ROS in beta cells by glucose”]. Accordingly, the pancreatic islet is efficiently perfused with blood (76, 107): Although islets occupy only 1–2% of the volume of the pancreas, they receive up to 15% of the pancreatic blood supply (77), and each beta cell is in direct contact with an endothelial cell (17).
Despite this high level of metabolic activity and the fact that reactive oxygen species (ROS) are an unavoidable by-product of mitochondrial respiration during glucose stimulation (and may even be required for normal glucose sensing) (100), enzymes involved in antioxidant defense are present at unusually low levels (103) or encoded by disallowed genes (152) in beta cells. As discussed below, this imbalance may render beta cells highly susceptible for damage induced by either oxidative stress or oxygen deprivation. This hypothesis will be reviewed here. We also discuss the interaction between GWAS genes, hypoxia, and oxidative stress and the possibility that in the metabolic syndrome the latter stressors may reduce functional beta-cell identity and insulin secretion without necessarily causing beta-cell destruction.
Formation of ROS in Pancreatic Beta Cells: The Role of Gluco–Lipotoxicity
Formation of ROS in pancreatic beta cells
The term “ROS” is generally used to describe reactive molecules containing oxygen. Although such molecules share some common characteristics, they also exhibit very different properties regarding their effects in biological systems, which may be either beneficial or toxic.
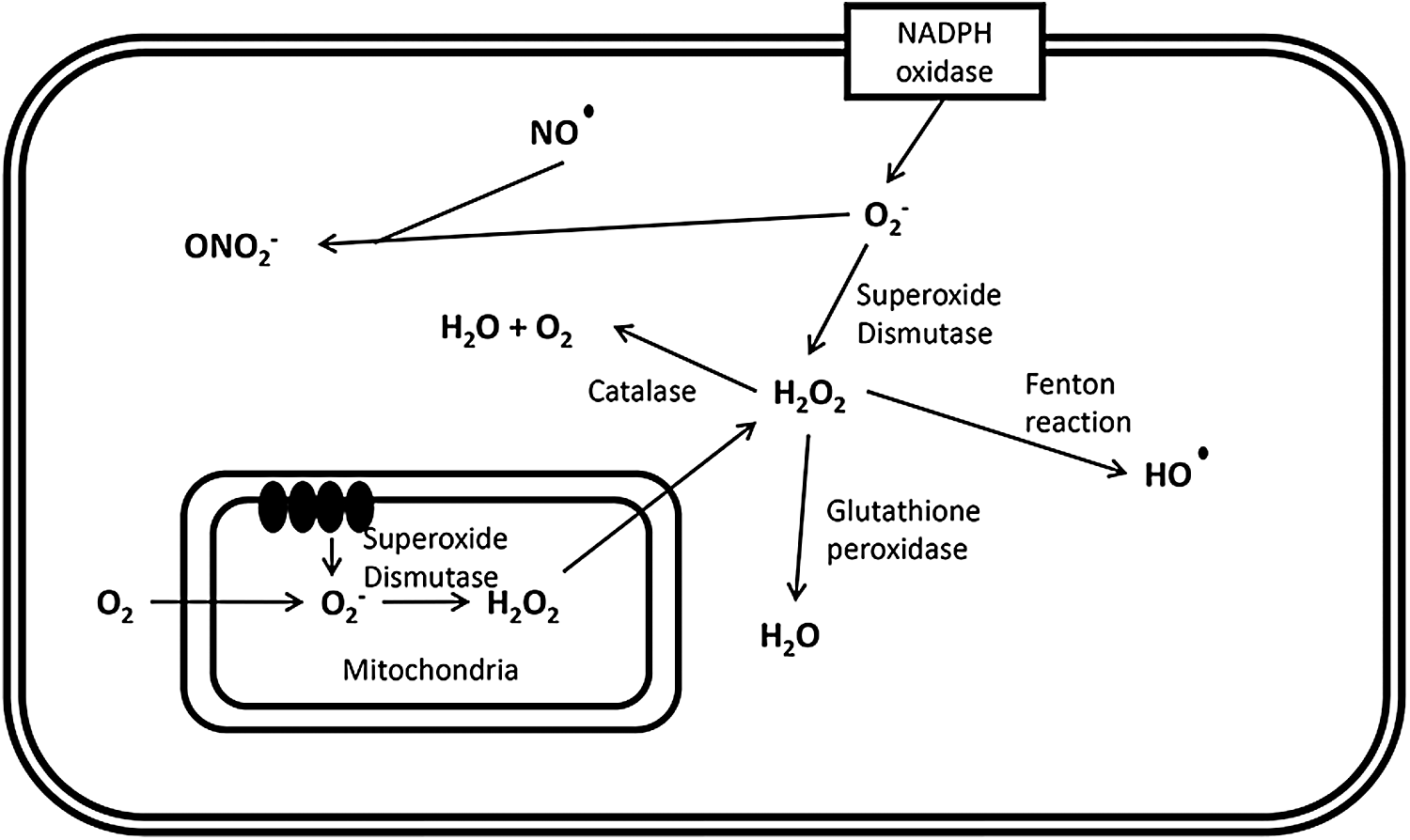
A major source of ROS within the pancreatic beta cell is the mitochondrial respiratory chain, as observed in other tissues (127, 200). Complexes I and III, located within the inner mitochondrial membrane, generate highly reactive superoxide (O2−) ions by single-electron reduction of molecular oxygen (101), which is—as a charged species—not able to freely cross biological membranes. However, it may do so via anion channels. Superoxide is converted to less active hydrogen peroxide (H2O2) by superoxide dismutase (SOD) isoenzymes. Being a less reactive, uncharged species, H2O2 can diffuse across membranes through aquaporins and be converted to highly reactive hydroxyl radicals (HO·). In addition, the formation of peroxynitrite (ONO2−) results from the reaction of superoxide with the free radical nitric oxide (NO) (135).
As well as by mitochondrial production, ROS are also generated by cytosolic and plasma membrane oxidoreductases, which oxidize NAD(P)H and directly produce ROS through the reduction of molecular oxygen (59). The general mechanisms of ROS production within the pancreatic beta cell are depicted in Figure 2.
Stimulation of the formation of ROS in beta cells by glucose
The main task assigned to the pancreatic beta cell is glucose-stimulated insulin secretion, the molecular basis of which is described below (Fig. 3). Briefly, after entering the cell via glucose transporters [with some continued debate about the role of GLUT1 and GLUT2 in human beta cells (118, 196)], glucose is phosphorylated by the high Michaelis–Menten constant (KM) hexokinase, glucokinase (102). Since the capacity for glucose transport is considerably higher than that of glucose phosphorylation, it is usually assumed that glucose concentrations quickly equilibrate across the plasma membrane, leaving glucose phosphorylation as the key step controlling glycolytic flux (168).
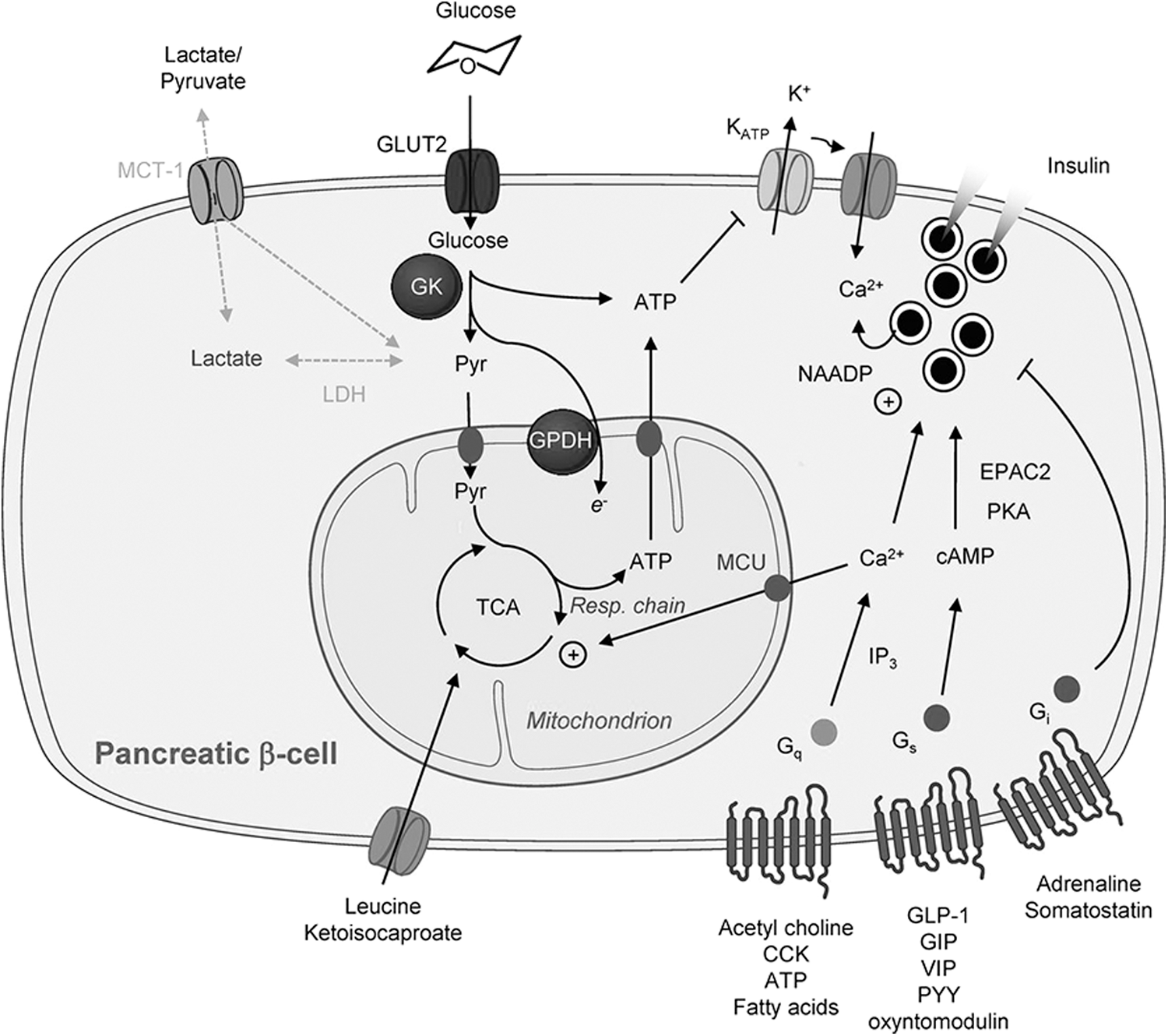
An increase in the glycolytic flux is followed by increased tricarboxylic acid (TCA) cycle activity [as well as increased synthesis of TCA cycle intermediates via anaplerosis (174)], resulting in an increased ATP production in the mitochondria and a rise in ATP-to-ADP ratio within the cytoplasm (195). Subsequently, this promotes closure of the ATP-sensitive K+ (KATP) channels, which in turn decreases the hyperpolarizing outward K+ flux (5). As a result of these changes, depolarization of the plasma membrane occurs, followed by influx of extracellular Ca2+, a rise in intracellular Ca2+, and subsequent insulin secretion (163, 168). Other less well-defined changes, which depend on but do not lead to the closure of KATP channels, also contribute to the stimulation of secretion (67).
Compared to other mammalian cells, a rapid and proportional increase in glycolytic flux is observed in pancreatic beta cells following a rise in extracellular glucose concentrations. Uniquely in these cells, glycolytic flux is tightly coupled to increased mitochondrial oxidative activity (138, 176), with almost 100% of glucose carbons being fully oxidized to CO2 (174). This in part reflects low levels of lactate dehydrogenase (LDH) (2, 176) and plasma membrane lactate/pyruvate transporter (MCT-1/Slc16a1) (73, 213) activities, as well as the stimulation by high Ca2+ of mitochondrial glycerol phosphate dehydrogenase (167) and intramitochondrial Ca2+-sensitive dehydrogenases (36, 194) at high glucose.
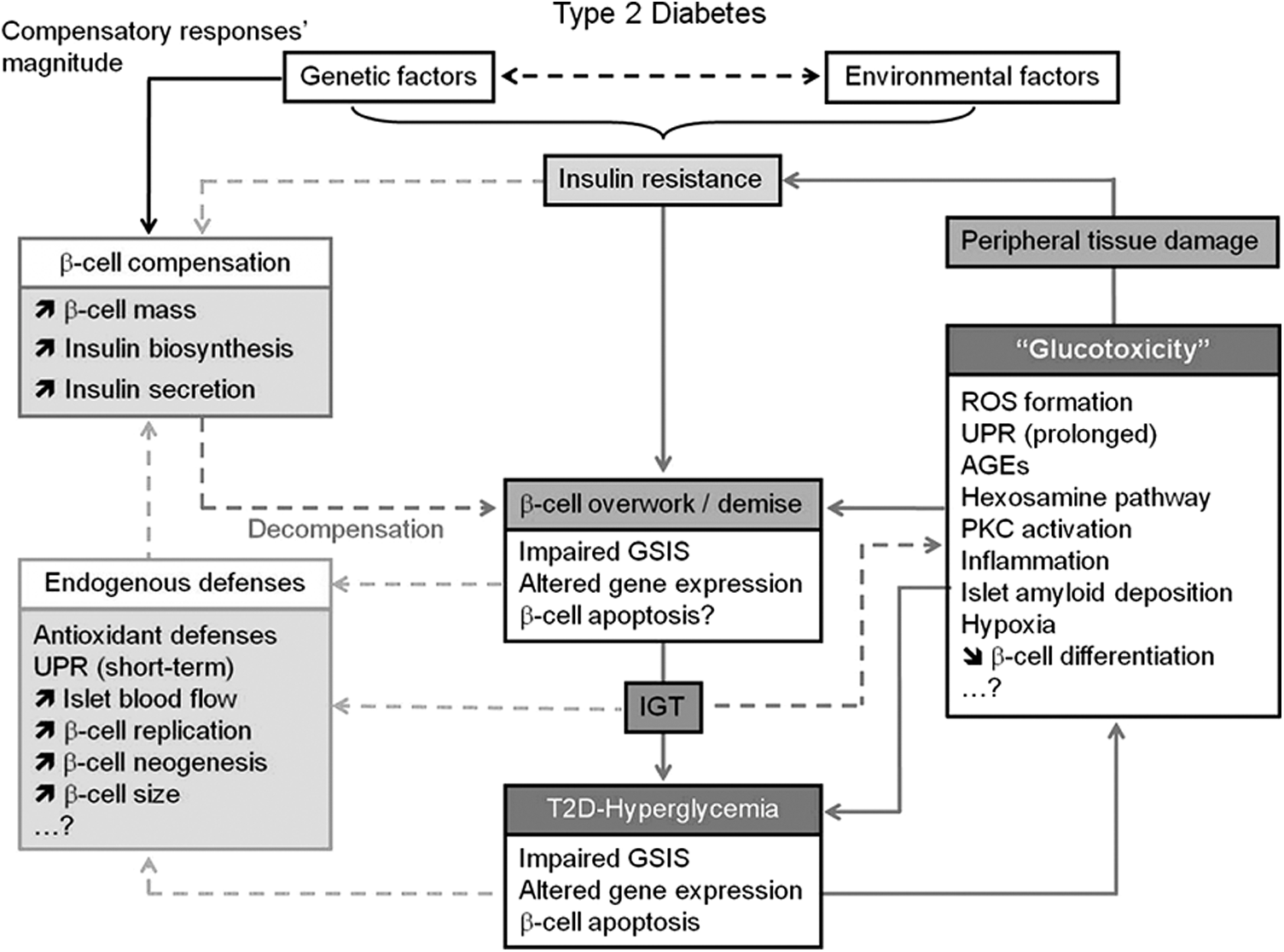
The above increases in both glycolytic and TCA cycle flux are needed to assure adequate insulin secretion as a response to high blood glucose levels. However, when glucose clearance starts to be impaired due to peripheral insulin resistance, the continuous increase in glycolytic flux may also increase ROS production in the beta cell, with potential pathological consequences (Fig. 4). Notably, increases in intracellular Ca2+ are able to both stimulate the mitochondrial generation of ROS (155, 186) and activate protein kinase C (PKC), which in turn leads to NADPH oxidase-dependent generation of superoxide and other species (123, 209). In addition to these effects, there is evidence that not only hyperglycemia but also hyperinsulinemia itself, caused by peripheral insulin resistance, may contribute to the generation of hydrogen peroxide in beta cells (170). This is in contrast to other tissues, where insulin was found to reverse glucose-induced ROS generation (57).
Although high glucose levels are clearly associated with induction of oxidative stress, there is also evidence that low glucose levels promote ROS formation in pancreatic beta cells (70, 171). Furthermore, increasing glucose concentrations suppress the generation of superoxide in these cells (113). Of importance, the suppressive effects of glucose on superoxide production occurred over a low range of glucose concentrations (0–5 mM), consistent with a requirement for basal glucose flux to allow NAD(P)H formation, which in turn is able to suppress superoxide formation by complex I.
Furthermore, recent studies confirmed that lowering of the glucose concentration from 10 to 2 mM reversibly increases, rather than decreases, ROS production in pancreatic beta cells (161). Consecutive studies of the same authors were able to demonstrate that glucose (and other nutrients) acutely reduces the glutathione oxidation ratio in pancreatic beta cells in the mitochondrial matrix but not in the cytosol/nucleus (192). These changes inversely correlated with those in NAD(P)H autofluorescence, suggesting that they indirectly resulted from increased NADPH availability rather than from changes in ROS concentration.
Stimulation of the formation of ROS in beta cells by free fatty acids
Although the relationship between circulating free fatty acid (FFA) levels and obesity, insulin resistance, and dysregulation of fat metabolism is still debated (85), many studies have shown that individuals with insulin resistance exhibit higher FFA concentrations (119), as well as increased fat content in insulin-responsive tissues, including the skeletal muscle or liver (75, 96, 137). Impaired suppression of FFA oxidation and an increased rate of FFA release into the plasma are associated with fat distribution patterns typical of the metabolic syndrome and associated with visceral obesity (78) and hepatic lipid accumulation (nonalcoholic fatty liver disease, NAFLD) (46).
High concentrations of FFA induce ROS production in a variety of different tissues, including the pancreatic beta cell (26, 104). A variety of studies have examined the mechanisms by which FFA may induce this production. In vascular smooth muscle cells, there is evidence that this involves PKC-dependent activation of NAD(P)H oxidase (72). In insulin-secreting BRIN BD11 cells, in contrast, palmitate-induced ROS production was accompanied by an increase in the expression of the p47phox component of the NADPH oxidase (123), although this was also partially sensitive to the actions of the PKC inhibitor, GF109203X. Furthermore, oleate-induced inhibition of the respiratory chain was shown to contribute to enhanced ROS induction in insulin-secreting cells by others (95).
Recent studies focused on the subcellular compartment where ROS are produced, in particular with regard to lipotoxicity. It could be demonstrated that H2O2 formation in the peroxisomes rather than in the mitochondria is responsible for toxicity induced by saturated nonesterified fatty acids (NEFAs) (45). Further studies of these authors demonstrated that this H2O2 formation in peroxisomes by saturated NEFAs could be prevented by unsaturated NEFAs (53).
Of note, and via the inhibition of complexes I and III (173), ceramides derived from NEFAs promote ROS generation in heart mitochondria (37, 62). Whether a similar mechanism pertains in beta cells remains to be tested.
Effects of Oxidative Stress and Antioxidant Defenses in the Pancreatic Beta Cell
Positive and negative effects of reactive oxygen and nitrogen species in pancreatic beta cells
The potential deleterious effects of increased reactive oxygen and nitrogen production include oxidative damage to ribonucleic acids, proteins, and lipids by the mechanisms of nitration, carbonylation, peroxidation, and nitrosylation. Consequently, ROS/RNS may impact on the function and survival of the beta cell through a variety of mechanisms, including changes in enzyme activity, ion channel transport, receptor signal transduction, dysregulated gene expression, and apoptosis (88, 128).
Disruption of normal beta-cell function in response to oxidative stress has been demonstrated in multiple studies (110, 215).
For example, oxidative stress appears to be a major contributing factor to the pathophysiology of Friedreich's ataxia (4). Affected individuals are at increased risk of developing diabetes (48), and recent studies confirmed that loss of glucose tolerance is driven by dysfunction and loss of beta cells, as demonstrated by altered glucose tolerance and decreased beta-cell mass (31, 32). Friedreich's ataxia involves lowered expression of the frataxin protein due to the expansion of a GAA repeat in the FXN gene (111). Frataxin is located at the mitochondrial matrix and directs iron–sulfur cluster assembly (125). Deletion of this protein selectively in the beta cell in mice affected oxidative energy flux, impaired glucose tolerance, and led eventually to overt diabetes mellitus (156). Diabetes followed beta-cell growth arrest and apoptosis and was paralleled by an increase in ROS production (156).
Targets for oxidative stress in the beta cell are likely to include duodenal homeobox factor 1 (PDX-1), which plays an important role in pancreas development and differentiation, as well as in maintaining normal beta-cell function (130). Thus, oxidative stress, imposed by the exposure of rat islets to H2O2, reduced the DNA binding activity of PDX-1 and consequently lowered insulin gene expression (83, 114). This effect was mediated by the c-Jun N-terminal kinase (JNK) pathway, and later studies (86) suggested that decreased nuclear accumulation of PDX-1 resulted from the JNK activation and increased nuclear uptake of forkhead box protein O1 (FOXO1) (87).
Changes in the expression or activity of MafA, a member of the basic leucine zipper family of transcription factors involved in insulin gene expression (115), are also implicated in the deleterious effects of oxidative stress on beta cells (66). Thus, beta-cell-selective overexpression of the antioxidant enzyme glutathione peroxidase preserved intranuclear MafA and reversed diabetes in db/db mice (65). p38 MAPK is a major regulator of MafA protein stability under oxidative stress (94), and the prevention of p38 MAPK-mediated MafA degradation ameliorated beta-cell dysfunction under oxidative stress (44). Recent studies in transgenic db/db mice overexpressing MafA conditionally and specifically in beta cells have corroborated these findings (116).
Might there be a positive role(s) of, or a requirement for, reactive oxygen or nitrogen species for the normal function of pancreatic beta cells? Evidence for such a role does exist. Thus, an elegant study by Penicaud and colleagues in 2009 (100) demonstrated that mitochondria-derived ROS are necessary for normal glucose-stimulated insulin stimulation. While antioxidants in this study were able to blunt insulin secretion, insulin release could be induced with mitochondrial complex blockers that generate ROS. More recent studies (109) have confirmed these observations, demonstrating that ryanodine receptor-mediated Ca2+ release induced by ROS is an essential step in glucose-induced insulin secretion.
In addition, the subcellular localization of glucokinase, the key flux-generating step responsible for the stimulation of insulin secretion by glucose (see preceding sections), is also regulated in beta cells by insulin via NO production and S-nitrosylation, the latter leading to an association of the enzyme with secretory granules (157). Similarly, insulin granule exocytosis itself is enhanced by S-nitrosylation of syntaxin 4, a key mediator of granule docking at the plasma membrane (207).
Antioxidant defense strategies of pancreatic beta cells
Given the existence of both beneficial and deleterious actions of reactive oxygen and nitrogen species in the beta cell as described above, it might be anticipated that the antioxidant properties of pancreatic beta cells differ from those of other tissues. Supporting the view that the ability of these cells to counteract oxidative stress is limited, work conducted almost 20 years ago revealed that pancreatic islets exhibit an expression level of the antioxidant enzymes SOD, glutathione peroxidase, and particularly catalase that is substantially lower than most other tissues (103). Indeed, levels of catalase are so low that this enzyme falls into the class of beta-cell disallowed genes as defined by ourselves (151, 152) and others (197). Later work demonstrated that beta cells are particularly rich in other peroxidase-based antioxidant defenses, such as glutaredoxin and thioredoxin (74).
Of interest, but in accordance with the observation that the production of ROS is both necessary and at the same time potentially hazardous for normal beta-cell function, modulation of the different antioxidant systems can elicit either beneficial or adverse effects depending on the context. For example, whereas microinjection of glutaredoxin potentiated the effects of NADPH on exocytosis, thioredoxin antagonized the action of this nucleotide (74).
The role of the uncoupling protein 2 (UCP2) (21) as a possible antioxidant mechanism in beta cells has also been assessed (98, 142, 149, 160, 212). In the report of Zhang et al. (212), UCP2-deficient mice were found to have higher insulin levels and display increased glucose-stimulated insulin secretion which was attributed to increased glucose-stimulated ATP synthesis in these mice. This observation was confirmed in later studies, where beta-cell function was similarly shown to be enhanced in UCP2-null mice after induction of hyperglycemia by multiple low-dose streptozotocin (STZ) injections (98). It was proposed in the latter report that increased chronic ROS signaling in UCP2-deficient mice enhanced beta-cell function but impaired alpha-cell function, leading to an attenuation of STZ-induced hyperglycemia.
However, other studies with UCP2-depleted mice have reached different conclusions. Thus, examined in animals backcrossed for several generations onto highly congenic background, UCP2 deletion led to a significant increase in oxidative stress, as demonstrated by increased expression of antioxidant enzymes and nitrotyrosine staining. Furthermore, on the congenic background, islets from UCP2-null mice displayed impaired glucose-stimulated insulin secretion, although no overt hyperglycemia, hypoinsulinemia, or glucose intolerance was observed in vivo (142).
Further insights into these debated mechanisms were provided by the generation of a beta-cell-specific UCP2 knockout mouse model. Islets from these mice displayed, as expected, elevated intracellular ROS levels but enhanced glucose-stimulated insulin secretion. However, UCP2BKO mice were glucose intolerant, showing greater alpha-cell area, higher islet glucagon content, and aberrant ROS-dependent glucagon secretion under high-glucose conditions. Thus, it was concluded that, in beta cells, UCP2 contributes to the regulation of intraislet ROS signals that mediate changes in alpha-cell morphology and glucagon secretion (160). It should be noted, however, that the Cre deleter strain used in the latter studies (RIP2Cre) (52) is well known to cause deletion in extrapancreatic tissues, including the brain (206), and also to express human growth hormone (23), both of which may complicate the interpretation of the results of the above study.
Transgenic mice with beta-cell-specific overexpression of the UCP2 gene exhibited no significantly modified plasma glucose and insulin levels or glucose-induced insulin secretion (149). Similar results were obtained after UCP2 induction in the pancreatic beta-cell line INS-1, where glucose-induced insulin secretion was not altered. However, increased UCP2 levels resulted in a decreased production of ROS upon cytokine exposure (149), indicating a potential protective effect. This observation was confirmed by UCP2 knockdown experiments in a similar cell line, where it was shown that UCP2 activity prevents a glucose-induced increase in mitochondrial ROS production compared to cells lacking this factor (1).
The above results illustrate how a regulator of mitochondrial coupling of oxidative phosphorylation and ATP production, as well as ROS production, may influence beta-cell function in a complex manner, acting by a variety of mechanisms.
There is some controversy regarding possible differences between human and rodent islets regarding antioxidant defenses. Thus, early studies suggested a superior antioxidant capability of human islets compared to rodent islets (42, 43, 205). However, this idea was challenged later by reports indicating low levels of the major antioxidant systems in human islets (33, 159, 198).
Interestingly, differences in antioxidant levels in beta cells between males and females have been observed in studies with mice (33) as well as humans (198), indicating reduced oxidative defense mechanisms in females. This observation is of particular interest in the context of published evidence for a better defense against oxidative stress in women in general (150).
As a possible explanation for this observation, it was proposed that during evolution, beta cells have lost some of their antioxidant defense capacity to guarantee reduced insulin action in situations of increased stress, redirecting glucose disposal toward organs that are not insulin sensitive, in particular the brain (154). The same authors hypothesized that low levels of antioxidants in females facilitate an inhibitory role of ROS on insulin secretion, allowing glucose levels during pregnancy that are sufficient to meet the increasing nutritional requirements of the growing fetus.
Figure 5 outlines the ambiguous role of ROS as well as antioxidants in beta-cell function and oxidative damage.
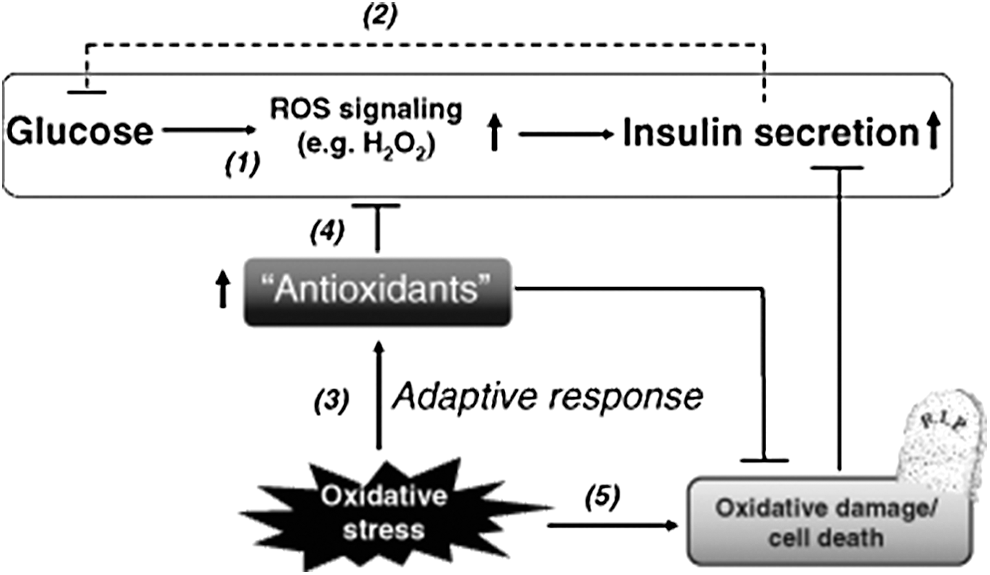
Antioxidant Therapy for Pancreatic Beta Cells
Effects of antioxidant therapy on pancreatic beta-cell dysfunction
Studies in animal models of T2D have, nonetheless, demonstrated beneficial effects of an antioxidant treatment on the course of the disease. Thus, treatment of Zucker diabetic fatty rats with the antioxidant N-acetyl-L-cysteine (NAC) or aminoguanidine prevented hyperglycemia, glucose intolerance, defective insulin secretion, and decrements in beta-cell insulin content (193). Similarly, treatment of diabetic db/db mice with NAC, vitamins C plus E, or both has been evaluated. NAC treatment resulted in retained glucose-stimulated insulin secretion and moderately decreased blood glucose levels. Vitamins C and E were not effective when used alone but slightly effective when used in combination with NAC (82).
In humans, however, beneficial effects of antioxidant treatment on glycemic control and in particular on beta-cell function in T2D have not been observed consistently or when their effects were assessed in comprehensive meta-analyses (190).
Overall, the current data do not, therefore, support the theory that oxidative stress in the pancreatic beta cell is an initial causative element in the development of the metabolic syndrome and T2D but rather a contributing factor initiated by factors, such as gluco–lipotoxicity (as stated above). Thus, any therapy that aims at targeting oxidative stress within the beta cell can only considered being an additional therapy that reduces beta-cell stress arising from a suboptimal therapy for metabolic syndrome, as insulin resistance, hyperlipidemia, and hyperglycemia (158).
Antioxidant treatment in islet transplantation
Although not a useful clinical modality in T2D, transplantation of isolated islets of Langerhans for patients with type 1 diabetes emerged as an alternative to whole-organ pancreas transplantation in 2000 after a study from Shapiro and colleagues demonstrated persistent insulin independence in recipients when a steroid-free regimen was used (180). Subsequent data have confirmed improvement of glycemic control, although with shorter durations of insulin independence (99, 181).
Oxidative stress in isolated and transplanted islets remains a concern that counteracts successful implantation and function of the islet graft (20). Thus, strategies were developed to reduce oxidative stress already before transplantation. Treatment of human islets with SOD mimics by adding these compounds to islets in culture, after isolation, allowed for the survival of a significantly higher islet cell mass. Furthermore, treated islets were superior regarding restoration of glucose control in STZ-treated mice after transplantation of a marginal islet mass (19). Similarly, induction of radical scavenging heme oxygenase-1 (HO-1) was able to protect islets from apoptosis and improve functional performance in vitro as well as in an in vivo model of marginal mass islet transplantation (144).
Further studies aimed at the introduction of antioxidant agents already very early in the process of islet transplantation by treatment of the explanted pancreas with an infusion of L-glutamine through the main pancreatic duct. This led to an increase in islet yield, and the percentage of diabetic mice rendered normoglycemic with glutamine-treated islet transplants was higher than that with control islets (7). Recent work demonstrated that a reduction of ROS in isolated pancreatic islets and improved islet transplantation outcome with increased survival of diabetic recipient mice can also be achieved by administration of exendin-4, a glucagon-like peptide-1 analog (136).
Oxygen Availability and Hypoxia in the Pancreatic Beta Cell
Hypoxia induction in pancreatic beta cells
Similar to their high susceptibility to oxidative stress caused by nutrient excess, pancreatic beta cells are also prone to the stress of oxygen deprivation, which, paradoxically, also leads to ROS production and other signs of the oxidative stress (184). Under normal conditions, the high oxidative phosphorylation rate of pancreatic islets relies on the availability of a constant oxygen supply. However, reduced oxygen content of islets is not necessarily a sign of a pathological process.
In an interesting recent study (132), it was shown that oxygenation differs widely between individual islets within the pancreas at a given time point and that these differences may reflect a mechanism to recruit only a fraction of the available islets into an active (normoxic) beta-cell mass. The remaining less well-oxygenated islets may represent a dormant subpopulation, constituting a functional reserve of endocrine cells. According to this model, the reserve islet pool may be available for recruitment upon reduction of the total islet mass, as elegantly demonstrated by experiments using pancreatectomy in a rat model (132).
In the context of therapies for T1D, oxygen deprivation is always observed in pancreatic islets upon isolation and transplantation. Separation of islets from the exocrine tissue leads to disruption of the vascular access of islets and leaves diffusion as the only way that provides oxygen to the cells. Thus, hypoxia primarily occurs within the core of isolated islets (55) because a gradient in oxygen tension exists between the surface and the center of the islet, which increases with the square of the islet diameter (6, 131). Immediately after transplantation, implanted islets still remain dependent on oxygen diffusion from the surroundings for their survival (97). It is only after 1–3 months that vascularization of engrafted islets develops, a process that depends, among other factors, on implantation location. The liver, currently the main location of human islet transplantation, was shown to be inferior compared to other sites regarding islet revascularization (133).
Of major interest is the question whether processes associated with the development of beta-cell failure in the metabolic syndrome and T2D also involve hypoxic changes in pancreatic islets. Exposure to high glucose levels has been shown to induce hypoxia and hypoxia-induced pathways in pancreatic beta-cell lines and isolated islets (11). Additional studies provided evidence that islets of animals suffering from diabetes are particularly prone to hypoxia (172, 214).
Direct comparison of the susceptibility of different islet cell types to hypoxia revealed that hypoxia results in severe functional abnormality in both beta and alpha (glucagon producing) cells, but alpha cells display a significantly lower rate of apoptosis compared to the intensive apoptotic injury of beta cells (13). Gene expression profiling in islets of prediabetic Zucker diabetic fatty rats showed increased expression of hypoxia-related genes, and further investigation revealed a severely disturbed vascular integrity of these islets as a possible reason for the development of hypoxia (106).
Roles of hypoxia-inducible factors in pancreatic beta cells
Hypoxia rapidly activates hypoxia-inducible factors (HIFs). These transcription factors, notably hypoxia-inducible factor 1alpha and 2alpha, are usually present in the cytosol. Under normoxic conditions, they are constantly hydroxylated at conserved proline residues by prolyl hydroxylases (177). This leads to their recognition and ubiquitination by the Von Hippel–Lindau (VHL) E3 ubiquitin ligase (117). In hypoxia, however, degradation of HIFs does not occur, allowing them to dimerize with the HIF-1beta subunit, be transported into the nucleus, and execute their transcriptional functions (79) (Fig. 6).
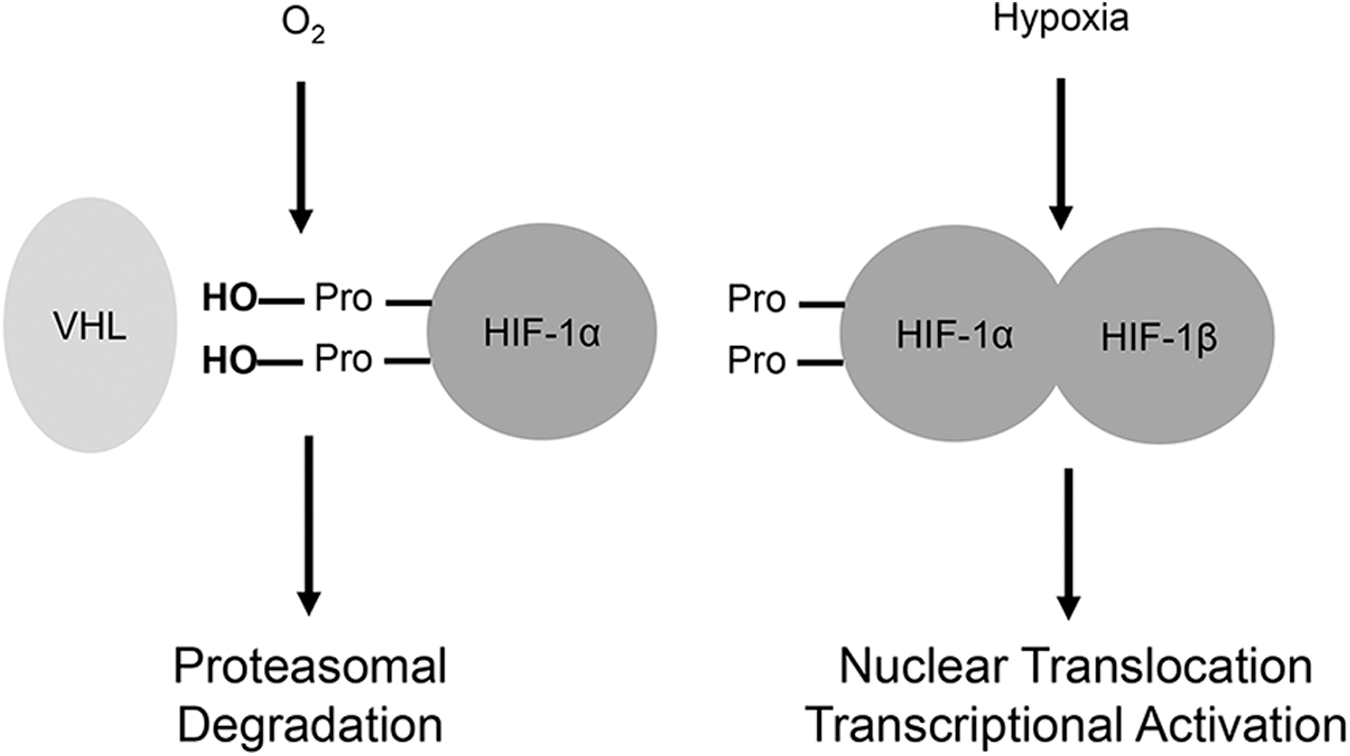
One of the main changes in cellular metabolism promoted by HIFs is a switch from aerobic to anaerobic energy production. Induction of glucose transporter 1 (GLUT1), several glycolytic enzymes, as well as LDH (178), and pyruvate dehydrogenase kinase 1 (PDK1) (91) reprograms the utilization of available glucose into anaerobic metabolism to maintain a high rate of glycolytic ATP production and at the same time downregulating mitochondrial oxygen consumption (139).
The specific effects exerted by this metabolic switch have been explored using mice deleted specifically in the beta cell for VHL. This modification leads to oxygen concentration-independent stabilization of HIFs (25, 153, 210). As might be expected, these animals exhibited stably increased rates of glycolysis, but unaffected or even increased insulin secretion in the fasting state, probably a result of a rise in ATP production. Glucose tolerance in these animals was markedly impaired, a change attributed directly to a disturbed beta-cell function by activation of the hypoxia-inducible pathway, since additional deletion of the factor HIF-1alpha in these VHL-deficient mice was able to rescue the phenotype (Fig. 7).
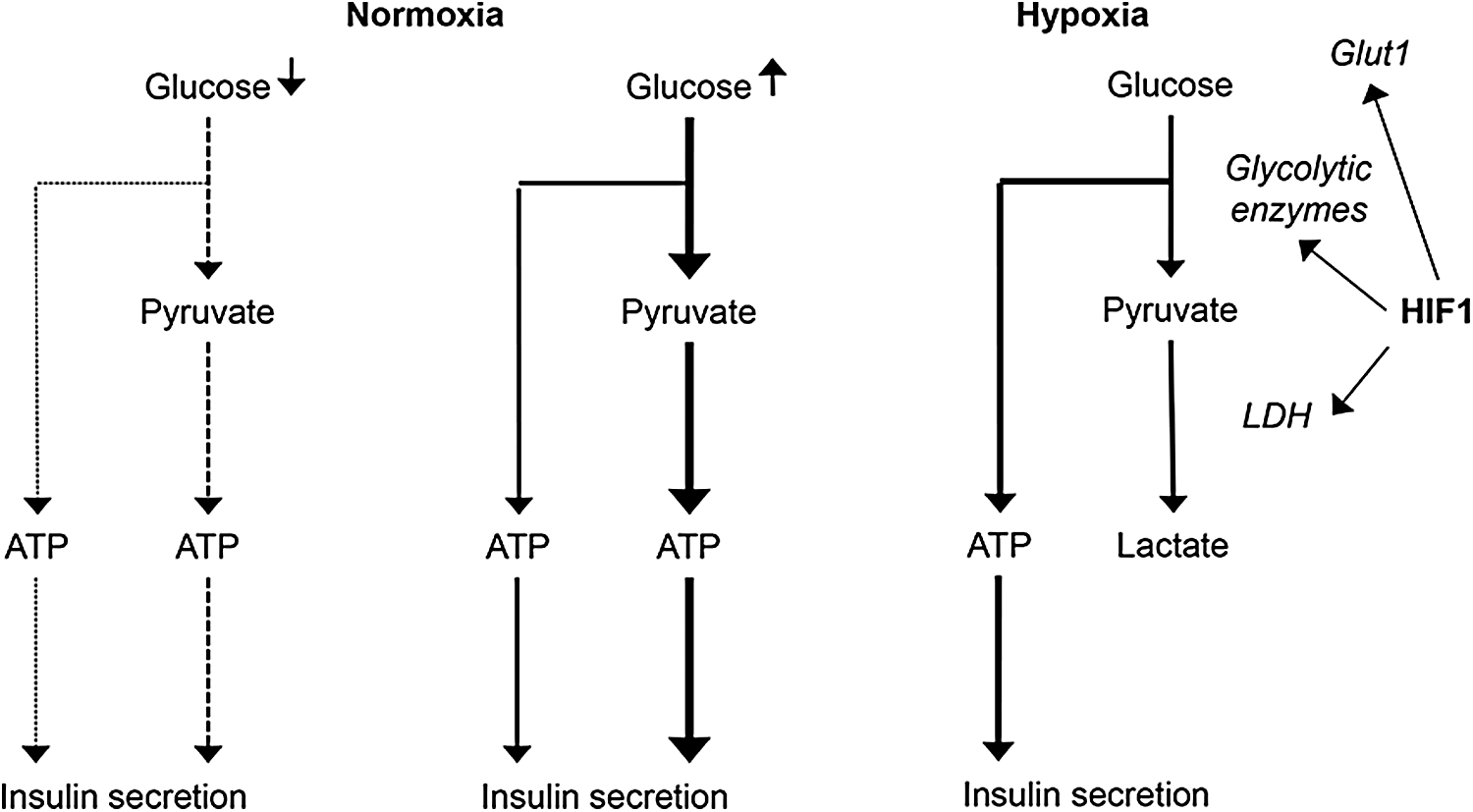
Contrasting with these results, other workers (64) demonstrated that disruption, but not activation, of the hypoxia signaling pathway by knockout of HIF-1beta or HIF-1alpha (29) was also able to cause pancreatic beta-cell dysfunction with consecutive glucose intolerance in mice. The same group also observed a 90% decrease in the expression of HIF-1beta in islets from T2D patients compared to normal glucose-tolerant controls (64).
Taken together, and also considering the fact that hypoxia is probably involved in the regulation of general beta-cell activity as mentioned above, it is tempting to speculate that there might be a basal activity of the hypoxia-inducible pathway, which is necessary for normal beta-cell function but beneficial only on a low level. Disruption as well as prolonged increased activity of this pathway is followed by impaired function of beta cells.
Intermittent hypoxia
The phenomenon of intermittent hypoxia is of particular interest since it is a consequence of obstructive sleep apnea, which is a highly prevalent disorder in obese patients, characterized by repeated episodes of pharyngeal obstruction during sleep that lead to intermittent hypoxia, sleep fragmentation, and excessive daytime sleepiness (140). An association of obstructive sleep apnea with disturbed glucose tolerance and T2D could be shown by a variety of observational studies (18, 148, 162).
Experimental animal models confirmed an increase of insulin resistance in obese mice treated with intermittent hypoxia. In addition, experiments in diabetic mice demonstrated a significant drop in pancreatic tissue oxygenation and impaired insulin secretion as well as an activation of caspase-3 within islets upon treatment with intermittent hypoxia (182). Another study confirmed induction of beta-cell apoptosis by intermittent hypoxia and also demonstrated a protective effect of antioxidant treatment with N-acetylcysteine (47).
The role of hypoxia in beta-cell death
Prolonged hypoxia results in pancreatic beta-cell death, typically by necrosis (55). However, induction of apoptosis-related pathways upon hypoxia exposure has also been described. Thus, activated caspase-3 (146) colocalizes in pancreatic islets with HIF-1alpha (124), suggesting that activation of apoptosis occurs primarily in those parts of the islet where hypoxia is most profound. However, whether hypoxia is a cause or a consequence of apoptosis is still debated (60).
Although induction of proapoptotic pathways by HIFs has been described (185), HIF activation is thought to be mainly an adaptive response that allows cell survival when oxygen levels are low. This conclusion was demonstrated by comparing apoptosis rate and transplantation success between wild-type islets and islets lacking HIF-1alpha (188). The latter exhibited impaired survival and increased apoptosis. The same study presented similar results for human islets where HIF-1alpha was upregulated by chemical agents.
The Role of Zinc Ions in Oxidative Stress and Hypoxic Damage to Pancreatic Beta Cells
Role and regulation of zinc homeostasis in pancreatic beta cells
Only few years after the discovery and successful use of insulin in the treatment of patients with diabetes (8), it was also recognized that zinc ions (Zn2+) may play an important role in insulin crystallization (175). In particular, Zn2+ is present in secretory granules of beta cells, where insulin undergoes a maturation process, aggregating to form 2-Zn–hexameric complexes (14, 121, 187). This dimerization process reduces the solubility of the hexamer, causing crystallization within the granule, and the formation of a dense crystalline core. The process not only increases insulin storage capacity before secretion but also reduces the susceptibility to enzymatic degradation of insulin (40, 69). Reconversion to the monomeric form of insulin is necessary for its biological action. This happens immediately after the exposure of the granule interior to the extracellular milieu, with a concomitant substantial release of Zn2+ (49, 56).
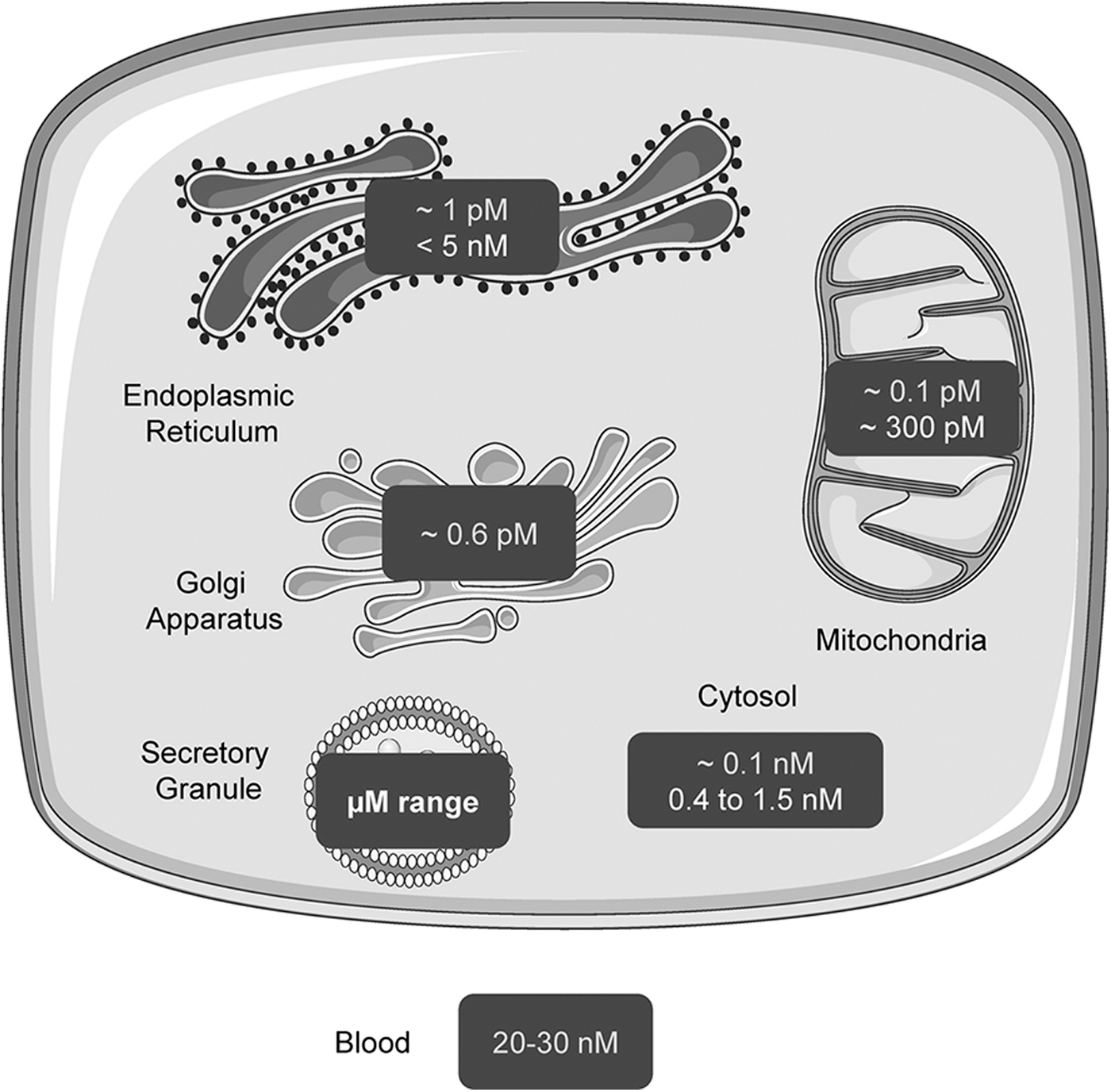
As an important structural and functional component of cells, the intracellular concentration and distribution of Zn2+ are tightly regulated (134, 201). The two main components of Zn2+ homeostasis are Zn2+ transporters and cytoplasmic Zn2+ buffers (34, 41, 81, 165). Whereas cytosolic free Zn2+ concentrations in pancreatic beta cells are comparable to those in other cells and quite low (∼1 nM) (10), they are much higher (∼120 nM) (68, 202) in the secretory granule: Corresponding total Zn2+ contents are in the mM and tens of mM range, respectively (51, 71) (Fig. 8).
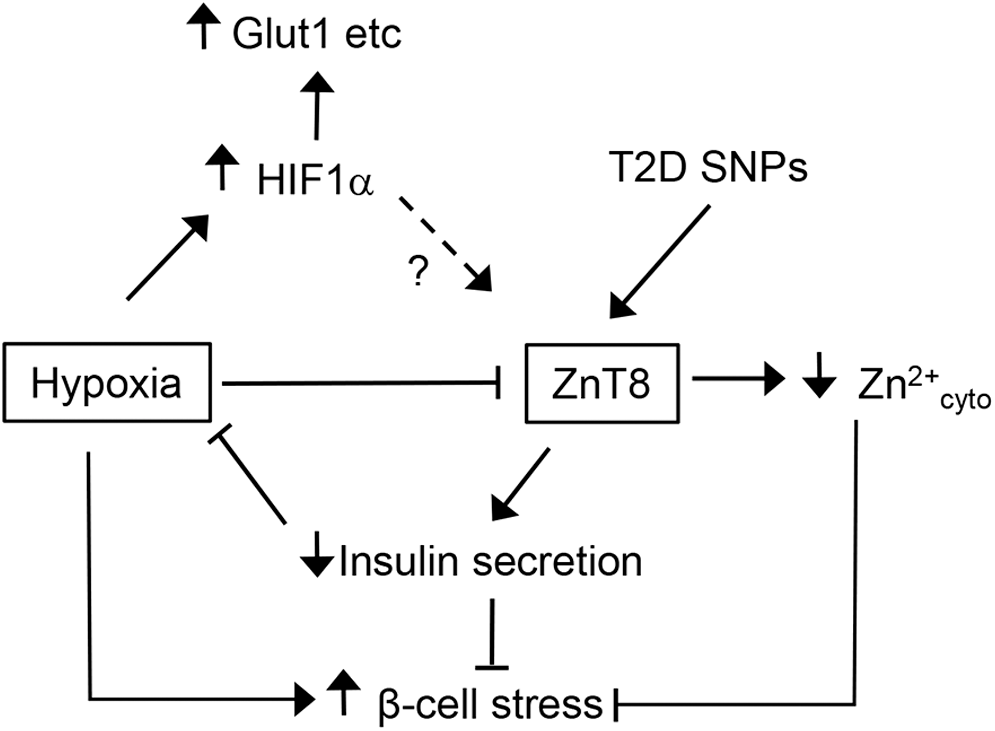
One of the most important regulatory transporters in beta cells, expressed almost uniquely in these cells and pancreatic alpha cells, is the zinc transporter ZnT8, responsible for zinc transport from the cytosol into the secretory granules (30). Paradoxically, however, deletion of ZnT8 from beta cells lowers both cytosolic and granular free Zn2+ (54, 122, 129). This zinc transporter became of major interest when genome-wide association studies (GWAS) revealed that a nonsynonymous single-nucleotide polymorphism (rs13266634) in the SLC30A8 gene, which encodes ZnT8, is associated with an ∼20% increase in T2D risk per allele (16, 92, 183). However, it is noteworthy that recent studies show that rare loss-of-function mutations in the SLC30A8 gene are associated with protection against T2D. The potential reasons for this apparent discrepancy are discussed elsewhere (166).
The role of Zn2+ in oxidative stress and hypoxia
Besides its specific role in insulin storage and secretion in the pancreatic beta cell, zinc is known for its antioxidative properties (22, 27, 147) and has been studied extensively as a possible treatment option in the context of diabetes in animal models (3, 208, 211) as well as human patients (50, 63). However, the results have been inconclusive possibly because changes in zinc intake are often too small to alter intracellular levels of Zn2+ to an extent that allows a measurable effect on oxidative stress. Nonetheless, zinc supplementation was found to lower fasting glucose levels in carriers of the common T2D risk allele at rs11558471 (84), and studies in rodent models of diabetes have indicated an action to improve insulin sensitivity (9).
A further challenge in the use of zinc supplementation as a therapeutic or prophylactic strategy in the metabolic syndrome is that the range of intracellular concentrations offering beneficial effects may be limited: From studies in neuronal tissues, it is well known that high levels of intracellular Zn2+ are able to induce production of ROS and that this effect can be attenuated by antioxidant treatment (145, 169). The dose dependency of the antioxidant versus oxidative stress-inducing effects of zinc was also demonstrated using isolated brain mitochondria (179).
Similarly, Zn2+ has “Janus-faced” properties regarding its effects in hypoxia. As described above, hypoxia induces apoptosis in pancreatic islets. Zinc is known to be a potent inhibitor of the apoptotic protease, caspase-3 (141), improves cell survival after hypoxia exposure (15), and protects tissues from ischemia-induced damage (203). In contrast, based on experiments performed with neuronal tissues, it is believed that rising zinc levels increase hypoxia-mediated cell death occurring during tissue ischemia (120, 189). Correspondingly, this effect can be reduced by the Zn2+ chelator N,N,N',N'-tetrakis(2-pyridylmethyl)ethane-1,2-diamine (TPEN) (108, 204).
In pancreatic beta cells, the effect of zinc on hypoxia-mediated effects has not been studied in detail. In general, it is known that high levels of zinc are cytotoxic and enhance pancreatic islet cell death (28, 89). In contrast, zinc increases the expression of metallothioneins (39, 93, 126), which are protective factors in pancreatic islets and other tissues vis-à-vis the deleterious effects of hypoxia and oxidative stress (90, 105).
Our own studies (54) have revealed that hypoxia has profound effects on cytosolic Zn2+ in pancreatic beta cells, downregulating zinc transporter ZnT8 and decreasing cytosolic zinc levels. When comparing the response to hypoxia of islets lacking ZnT8 completely, we detected a reduced rate of cell death in subpopulations of these islets in older mice. Thus, we concluded that reduced expression of ZnT8 and reduced cytosolic Zn2+ levels induced by hypoxia may reflect an adaptive response of beta cells to permit survival under hypoxia by reducing possible deleterious effects of high levels of zinc in this situation of acute stress (Fig. 9).
Concluding Remarks
Availability of oxygen, aerobic and anaerobic glycolysis, oxidative phosphorylation, and the production of ROS are closely linked to the normal function of the pancreatic beta cell since these processes are directly involved in glucose sensing and insulin release. Although hypoxia and oxidative stress are potentially harmful, and even lethal, processes for these metabolically highly active cells, evidence gathered during the past two decades indicates that both ROS and hypoxia-induced pathways have an important regulatory function in pancreatic beta cells. It became evident that these factors belong to the regulatory network of the beta cell and that changes in their availability and activation may lead to beta-cell failure and death.
There are still many open questions regarding the precise role of these processes in the development of disturbed glucose tolerance and T2D in the context of the metabolic syndrome, and further research is necessary to understand their contribution to the pathophysiology as well as possibilities to modulate them in a therapeutic way. To what extent, for example, does oxidative stress affect beta-cell identity—and consequently function—independently of cell survival (168)? Do T2D-associated genes in addition to SLC30A8 also modulate beta-cell responses to hypoxia or oxidative stress? Future studies will be needed to explore this and other questions and may lead to the identification of new ways to modulate the impact of oxidative stress for therapeutic benefit.
Footnotes
Acknowledgments
GAR is supported by grants from the Wellcome Trust (Senior Investigator Award, WT098424AIA), an MRC Programme Grant (MR/J0003042/1), Biotechnology and Biological Sciences Research Council (BB/J015873/1) and Diabetes UK (11/0004210) project grants, an Imperial Confidence in Concept (ICiC) grant, and a Royal Society Wolfson Research Merit Award. PG was supported by the Foundation of Diabetes Research at the University Hospital Zurich. This work was also supported by the Innovative Medicines Initiative Joint Undertaking under grant agreement 155005 (IMIDIA), resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007–2013), and European Federation of Pharmaceutical Industries and Associations (EFPIA) companies.