
Abstract
Aims:
Radiation-induced normal tissue toxicity is closely linked to endothelial cell (EC) damage and dysfunction (acute effects). However, the underlying mechanisms of radiation-induced adverse late effects with respect to the vascular compartment remain elusive, and no causative radioprotective treatment is available to date.
Results:
The importance of injury to EC for radiation-induced late toxicity in lungs after whole thorax irradiation (WTI) was investigated using a mouse model of radiation-induced pneumopathy. We show that WTI induces EC loss as long-term complication, which is accompanied by the development of fibrosis. Adoptive transfer of mesenchymal stem cells (MSCs) either derived from bone marrow or aorta (vascular wall-resident MSCs) in the early phase after irradiation limited the radiation-induced EC loss and fibrosis progression. Furthermore, MSC-derived culture supernatants rescued the radiation-induced reduction in viability and long-term survival of cultured lung EC. We further identified the antioxidant enzyme superoxide dismutase 1 (SOD1) as a MSC-secreted factor. Importantly, MSC treatment restored the radiation-induced reduction of SOD1 levels after WTI. A similar protective effect was achieved by using the SOD-mimetic EUK134, suggesting that MSC-derived SOD1 is involved in the protective action of MSC, presumably through paracrine signaling.
Innovation:
In this study, we explored the therapeutic potential of MSC therapy to prevent radiation-induced EC loss (late effect) and identified the protective mechanisms of MSC action.
Conclusions:
Adoptive transfer of MSCs early after irradiation counteracts radiation-induced vascular damage and EC loss as late adverse effects. The high activity of vascular wall-derived MSCs for radioprotection may be due to their tissue-specific action. Antioxid. Redox Signal. 26, 563–582.
Introduction
T
Current research efforts are aimed to develop treatment strategies to protect the healthy lung tissue from the toxic effects of ionizing radiation. In this study, we highlight the structures of lung endothelia as key players in the progression of radiation-induced lung disease. In particular endothelial cell (EC) loss as long-term complication after radiation conveys the development of fibrosis. Adoptive transfer of mesenchymal stem cells (MSCs) in the early phase after irradiation limits the radiation-induced EC loss and fibrosis progression by restoration of the antioxidant enzyme superoxide dismutase 1 (SOD1). Thus, adoptive transfer of MSCs early after irradiation is a promising strategy to radioprotect lungs from radiation-induced late damage.
Studies in animal models and patient samples show a complex response of the lung tissue to radiation with multiple interactions between resident cells (epithelial cells, fibroblasts, endothelial cells [ECs]), extracellular matrix molecules, and infiltrating immune cells (1, 32, 69). Among these populations, the nondividing cells, the microvascular endothelial compartment has clearly been shown to play a central role in radiation toxicity in healthy tissues (15, 61).
Particularly microvessels are extremely sensitive to IR: irradiation often results in a rupture of the capillaries, thrombosis, and telangiectasia (27). In small-sized arteries (having a thin muscular wall and measuring up to 100 μm in external diameter), IR causes a neointimal proliferation, thrombosis, fibrinoid necrosis, and acute arthritis, whereas larger blood vessels (ample lumen and thick muscular wall, >500 μm external diameter) seem to be less affected (27). Interestingly, ECs of various organs differ in their sensitivity to IR: in vitro studies show that, for example, sinusoidal EC of the liver are highly radioresistant, whereas microvascular EC of the skin are rather radiosensitive (62).
We and others showed in preclinical studies that radiation-induced normal tissue toxicity in the lung is closely linked to vascular EC damage and dysfunction of the blood–air barrier (9, 25, 31, 84). However, the underlying mechanisms of radiation-induced adverse late effects are still not well understood, and no causative radioprotective treatment is available to date.
Stem cell therapy is a promising option for the prevention or treatment of radiation-induced normal tissue injury as it can promote survival and repair of damaged resident cells (14, 42). However, there is a lack of preclinical and clinical studies of stem cell therapy for radiation-induced adverse effects in the lung, particularly in radiation-induced fibrosis (54, 75). There are also only few ongoing clinical trials with mesenchymal stem cells (MSCs), also referred to as multipotent mesenchymal stromal cells (MPSCs) in chronic lung disease, including their therapeutic application in patients with idiopathic pulmonary fibrosis (77). Importantly, beneficial or adverse effects of stem cell therapy on the pathogenic process seem to depend on the timing of stem cell application after RT. We previously demonstrated that therapeutic application of MSCs has the potential to counteract radiation-induced normal tissue damage when the MSC therapy is performed within 2 weeks after irradiation (44).
We also showed that MSCs derived classically from bone marrow (BM) or from aorta (vascular wall-derived MSCs) have the potential to protect lung EC from radiation-induced vascular leakage observed at 3 weeks postirradiation as well as the associated increased extravasation of infiltrating immune cells and circulating tumor cells. Furthermore, we demonstrated that vascular wall-derived MSCs are particularly well suited for the radioprotection of EC within the processes of radiation-induced lung injury because of their tissue-specific action (42, 44).
Thus, these findings greatly adhere to the concept of the low toxicity multitherapies presented recently in a position article focusing on broad-spectrum approach cancer prevention and therapy (6). To further confirm that MSC therapy is able to downgrade the side effects of radiotherapy in a way that it could be called a low-toxicity approach in the future, we investigated the therapeutic potential of adoptive MSC transfer to protect lung EC from radiation-induced damage, dysfunction, and loss in the long-term follow-up and aimed at defining the mechanisms underlying the protective effects of MSC therapy.
Results
MSC treatment protects irradiated lung from severe radiation-induced vascular EC damage and delayed EC loss
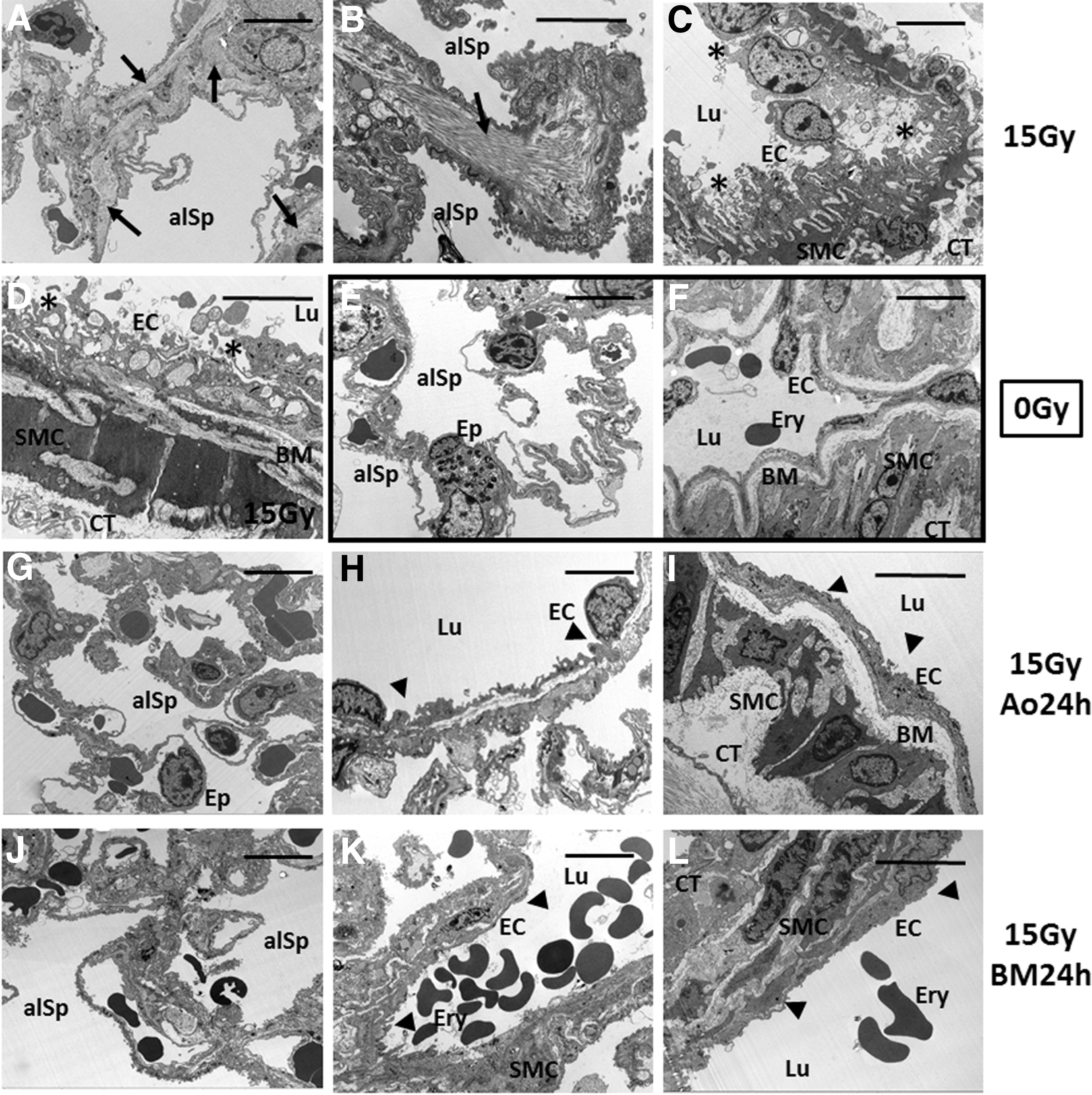
To investigate the adverse late effects of radiation on the lung endothelium, we performed intensive morphological analysis of lungs from mice (C57BL/6) at 25 weeks after whole thorax irradiation (WTI) using electron microscopy (Fig. 1). As expected, a massive collagen deposition in WTI lungs (15 gray [Gy]) confirmed the development of lung fibrosis as a classical long-term complication of WTI (Fig. 1A, B). Moreover, WTI induced multiple signs of severe morphological impairment in EC such as partially degraded mitochondria and numerous vacuoles, as well as a defective and irregular basement membrane lining arterial EC (Fig. 1C, D, and Supplementary Fig. S1; Supplementary Data are available online at
Next, we were interested whether radiation-induced EC damage would result in an EC loss at late time points. Therefore, we quantified the amount of vascular endothelial (VE)-cadherin, a protein specific to endothelial adherence junctions, in whole protein lysates by Western blot analysis (Fig. 2A, B). In addition, we quantified the number of ECs in crude cell extracts of freshly isolated lung tissue using endothelial-specific PECAM1/CD31 expression and flow cytometry analysis (Fig. 2C). WTI induced a significant reduction of VE-Cad expression levels (mean 0 Gy: 1.63 ± 0.11, n = 4; mean 15 Gy: 0.61 ± 0.04, n = 4; 95% confidence interval [CI] of difference 0 Gy vs. 15 Gy: 0.68–1.37), whereas in MSC-treated animals, expression levels were partially restored (mean 15 Gy Ao 24 h: 1.00 ± 0.08, n = 4; 95% CI of diff. 15 Gy vs. 15 Gy Ao 24 h: −0.74 to −0.05 and 95% CI of diff. 0 Gy vs. 15 Gy Ao 24 h: 0.28 to −0.97; mean 15 Gy BM 24 h: 0.76 ± 0.04, n = 4). Furthermore, flow cytometry analysis of the relative number of PECAM1/CD31 expressing cells further confirmed a significant EC loss at 25 weeks after WTI (0 Gy: 35.73 ± 1.62, n = 4; 15 Gy: 16.48 ± 1.10, n = 6; mean difference [MD] 0 Gy vs. 15 Gy: 19.2; 95% CI of diff. 13.7–24.8). Again, the number of PECAM1/CD31 expressing cells was less reduced in animals that received a single MSC injection at 24 h after irradiation (mean 15 Gy Ao 24 h: 23.82 ± 1.26, n = 6; MD 15 Gy vs. 15 Gy Ao 24 h: −7.33; 95% CI of diff. −12.3 to −2.38; and MD 0 Gy vs. 15 Gy Ao 24 h: 11.9 95% CI of diff. 0 Gy vs. 15 Gy Ao 24 h: 6.37–17.4; mean 15 Gy BM 24 h: 20.53 ± 1.07, n = 4; MD 15 Gy vs. 15 Gy BM 24 h: −4.05; 95% CI of diff. −10.1 to 2.01). Immunohistochemistry (IHC) analysis of VE-Cad further revealed a less prominent staining of EC in lung sections of WTI mice, which was restored in lung sections of WTI and subsequent MSC-treated animals (Fig. 2D).
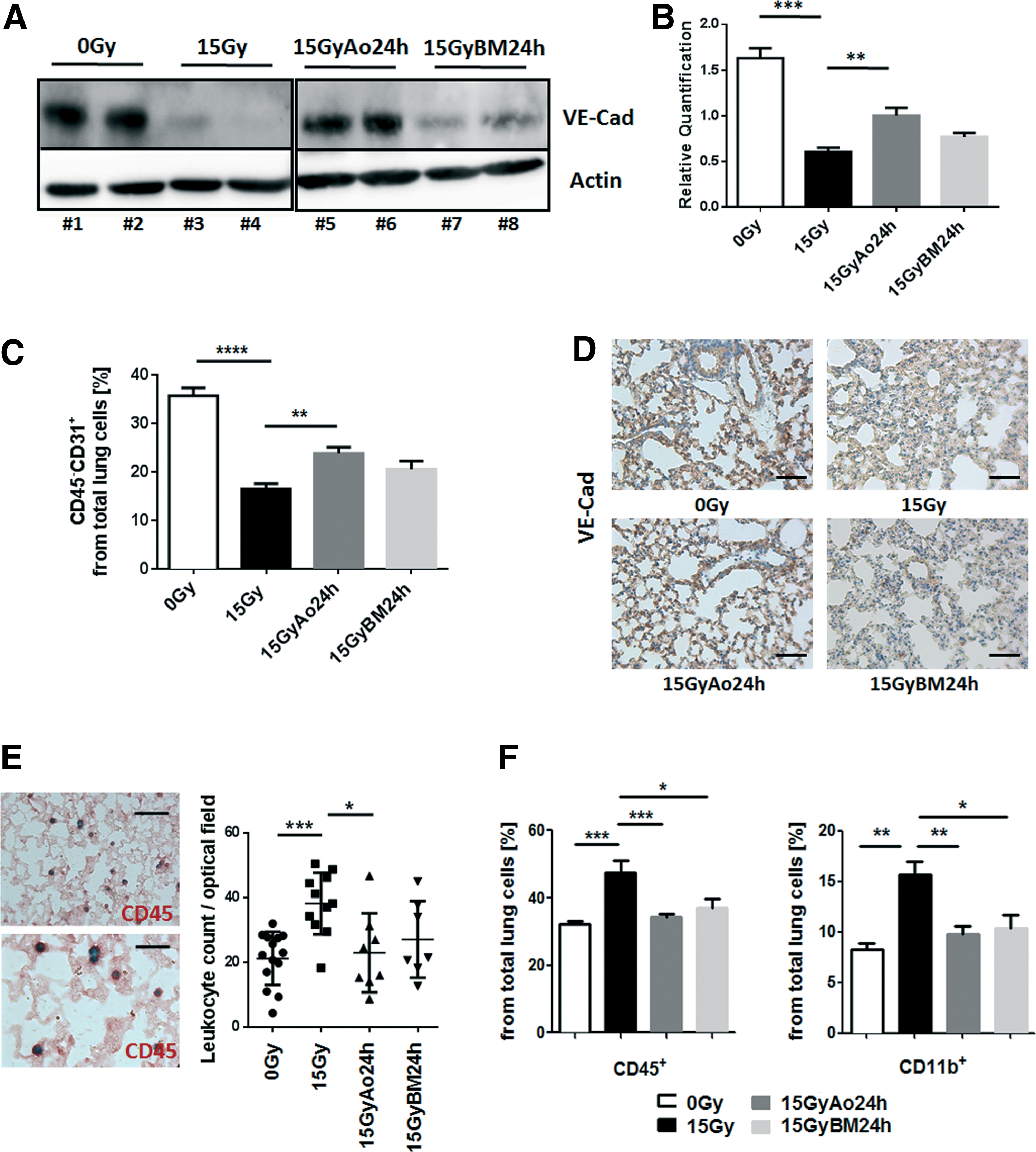
To further gain insight on which vessel structures are affected by IR, smooth muscle cell (SMC)-stabilized vessels were quantified by counting the SMC marker-protein-positive transgelin (Tagln)-immunoreactive vascular structures in whole tissue sections (Supplementary Fig. S2). The amount of Tagln-positive vessels was significantly reduced at 25 weeks after WTI (mean 0 Gy: 19.00 ± 1.38, n = 8; mean 15 Gy: 10.00 ± 1.19, n = 7; 95% CI of diff. 4.08–13.92) and was nearly normalized in animals that received a single AoMSC injection at 24 h after irradiation and by tendency in animals that received BM-MSCs (mean 15 Gy Ao 24 h: 16.60 ± 0.68, n = 5; 95% CI of diff. to 15 Gy: −12.17 to −1.03; mean 15 Gy BM 24 h: 12.00 ± 2.16, n = 4; 95% CI of diff. to 15 Gy: −7.96 to 3.96).
Interestingly, we also observed an increased number of total CD45+ leukocytes in irradiated lungs at 25 weeks after WTI when compared to sham controls (Fig. 2E, F). Particularly the percentage of potential profibrotic CD11b+ myeloid cells and Ly6C+ monocytes (not shown) from CD45+ leukocytes was significantly increased after WTI, potentially as a direct consequence of impaired vascular function and EC loss (Fig. 2F). Importantly, the radiation-induced increase in infiltration of these myeloid cells was significantly reduced in MSC-treated animals at 25 weeks after WTI, which might be due to the protection of lung EC (Fig. 2E, F).
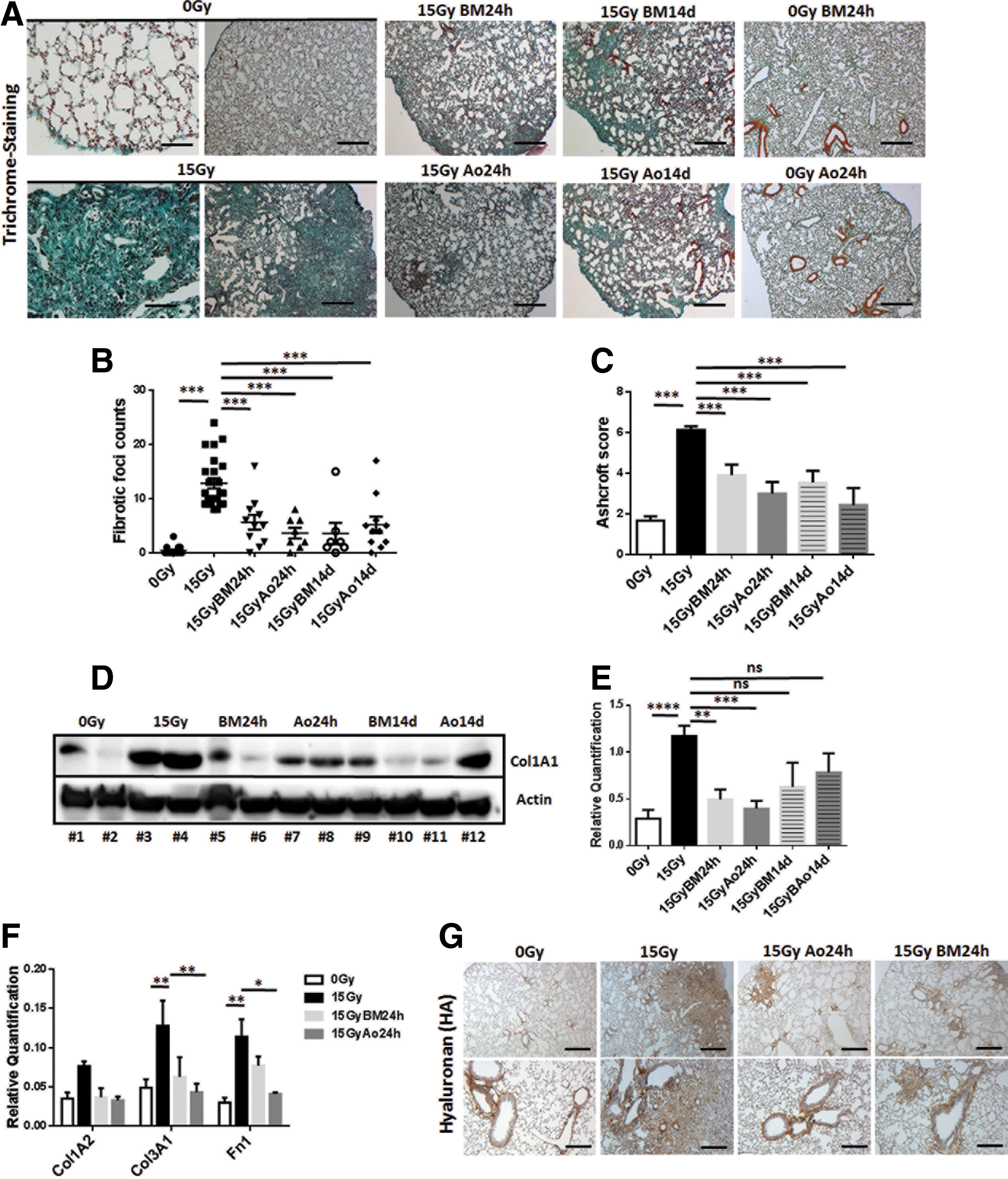
Radiation-induced EC loss at 25 weeks after WTI was accompanied by the development of significant fibrosis as revealed by histological stainings on sections of paraffin-embedded lung tissue with Masson's Goldner Trichrome (Fig. 3A–C and Supplementary Fig. S3), Western blot analysis for significantly increased Collagen (Col1A1) expression levels of total lung lysates (Fig. 3D, E), quantitative reverse transcription polymerase chain reaction (qRT-PCR) quantifications of the extracellular matrix components Col1A2, Col3A1, and fibronectin 1 (Fn1; Fig. 3F), and IHC of the major extra cellular matrix glycosaminoglycan hyaluronan, respectively (Fig. 3G and Supplementary Fig. S3). In all analyses, radiation-induced lung fibrosis was significantly attenuated by MSC treatment (Fig. 3A–G).
Treatment of cultured lung microvascular EC with MSC-derived supernatants rescued radiation-induced endothelial damage
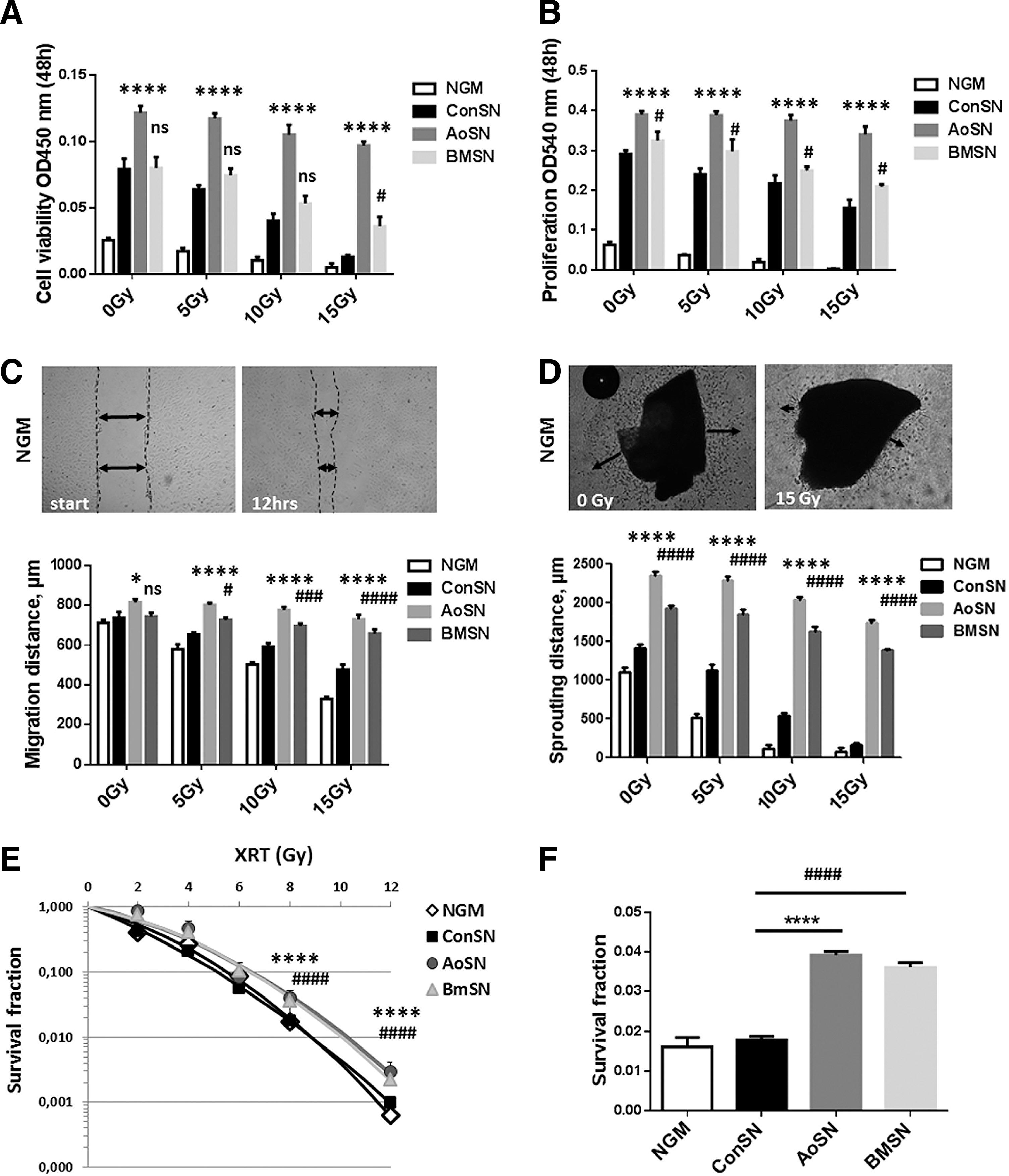
Up to now, our data indicated that adoptive transfer of MSC provides long-term protection of lung EC from radiation-induced damage. To corroborate the assumed protective action of factors secreted from MSCs on EC, we purified lung microvascular EC (LMEC) from ex vivo isolated crude lung cell extracts by PECAM1/CD31 antibody and immunomagnetic separation and compared cell viability and proliferation of irradiated LMEC cultured in normal growth medium (NGM), control supernatant, or supernatants (SN) derived from cultured aortic MSCs (AoSN) and BM-MSCs (BMSN) (Fig. 4A, B). Interestingly, treatment with MSC supernatants rescued the radiation-induced reduction in viability and proliferation of LMEC. Interestingly, the protective effects of AoSN were more pronounced when compared to BMSN. LMEC migration using a wound closure assay and sprouting/invasion using ex vivo isolated lung explants embedded in growth factor-reduced Matrigel were also significantly reduced on irradiation with different radiation doses; again, treatment with AoSN rescued these effects more efficiently than treatment with BMSN (MD 0 Gy: ConSN vs. AoSN: −78.32; 95% CI of diff. −145.8 to −10.79 and MD ConSN vs. BMSN: −6.69; 95% CI of diff. −74.18 to 60.87; MD 15 Gy: ConSN vs. AoSN: −251.4; 95% CI of diff. −247.0 to −111.9 and MD ConSN vs. BMSN: −71.96; 95% CI of diff. −4.44 to 139.5) (Fig. 4C, D). In line with these findings, a long-term assay measuring the surviving fraction after irradiation revealed that the number of LMEC able to regrow and form colonies after irradiation was significantly increased when cultured in the presence of MSC SN derived from cultured aortic or BM MSCs (MD ConSN vs. AoSN: −0.0214; 95% CI of diff. −0.0267 to −0.0161 and MD ConSN vs. BMSN: −0.0183; 95% CI of diff. −0.0237 to 0.013) (Fig. 4E, F).
Therapeutically applied stem cells secrete superoxide dismutase 1 and restore superoxide dismutase 1 expression in WTI-treated lungs
To identify MSC-secreted factors with radioprotective potential, we compared SN from cultured aortic MSCs, BM MSCs, or control SN by label-free quantitative mass spectrometry (MS) (16). Among the list of identified protein groups, our attention was attracted by superoxide dismutase 1 (SOD1) as this protein was identified in AoSN and BMSN SN, but was below the detection limit in control SN (Fig. 5A; for complete list see Supplementary Table S1).
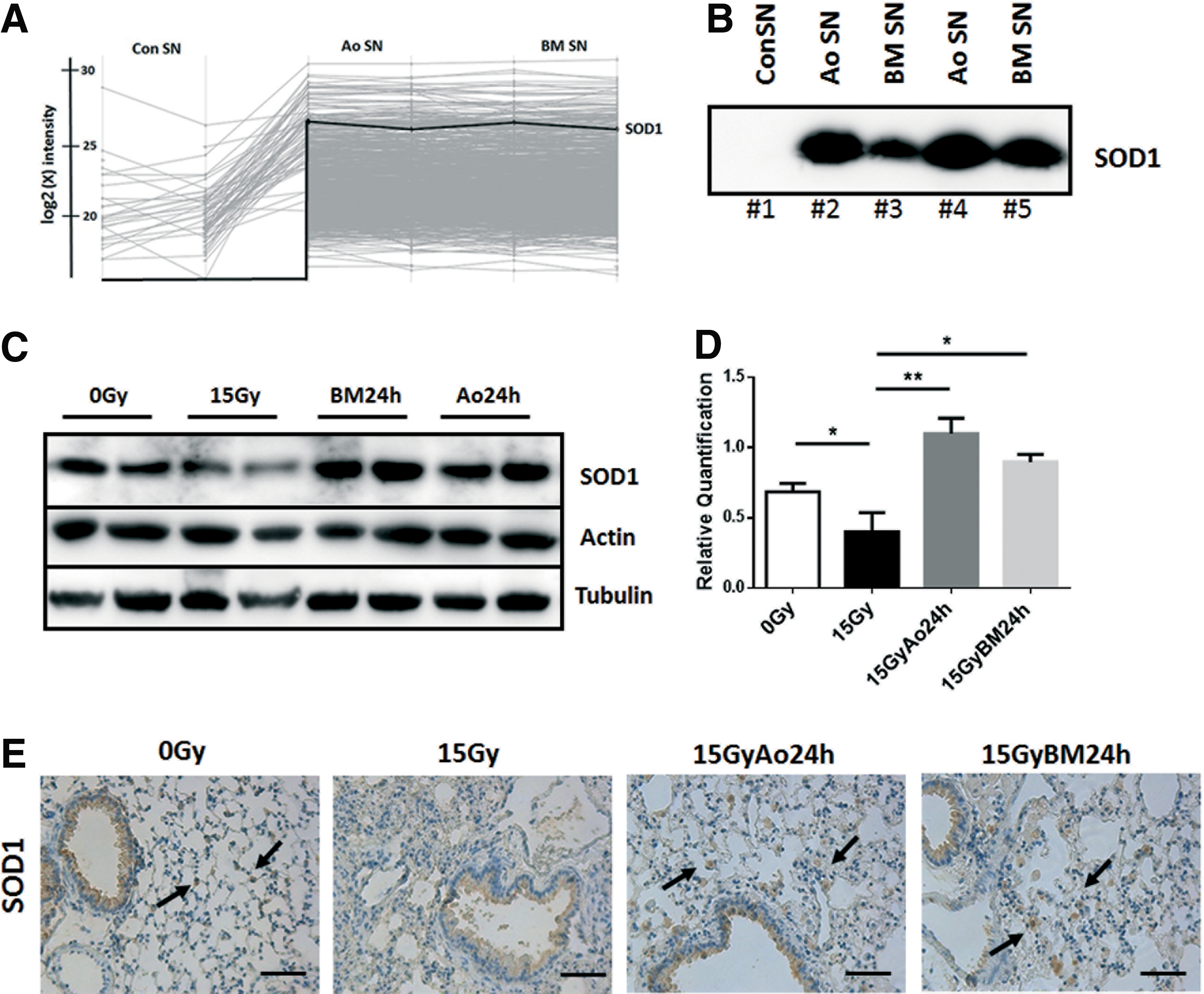
Western blot analysis of SOD1 expression levels confirmed the presence of SOD1 secreted from cultured MSCs in cell culture-derived SN (Fig. 5B). SOD1 protein expression levels were further analyzed in whole protein lysates of control and WTI lungs with and without MSC treatment by Western blot analysis at 25 weeks postirradiation (Fig. 5C, D). Of note, SOD1 expression levels were significantly reduced in lungs on WTI (mean 0 Gy: 0.69 ± 0.058, n = 4; mean 15 Gy: 0.40 ± 0.14, n = 4; MD 0 Gy vs. 15 Gy: 0.285; 95% CI of diff. −0.119 to 0.689) and restored in lungs of irradiated animals that had received stem cell treatment (mean 15 Gy Ao 24 h: 1.2 ± 0.11, n = 4; MD 15 Gy vs. 15 Gy Ao 24 h: −0.699; 95% CI of diff. −1.103 to −0.295 and mean 15 Gy BM 24 h: 0.89 ± 0.05, n = 4; MD 15 Gy vs. 15 Gy BM 24 h: −0.498; 95% CI of diff. −0.902 to −0.009). IHC analysis of SOD1 expression corroborated a less prominent staining of EC in lung sections of WTI mice that was restored in lung sections of irradiated animals with MSC treatment (Fig. 5E and Supplementary Fig. S4).
Treatment with superoxide dismutase mimetic (EUK134) counteracts radiation-induced EC loss
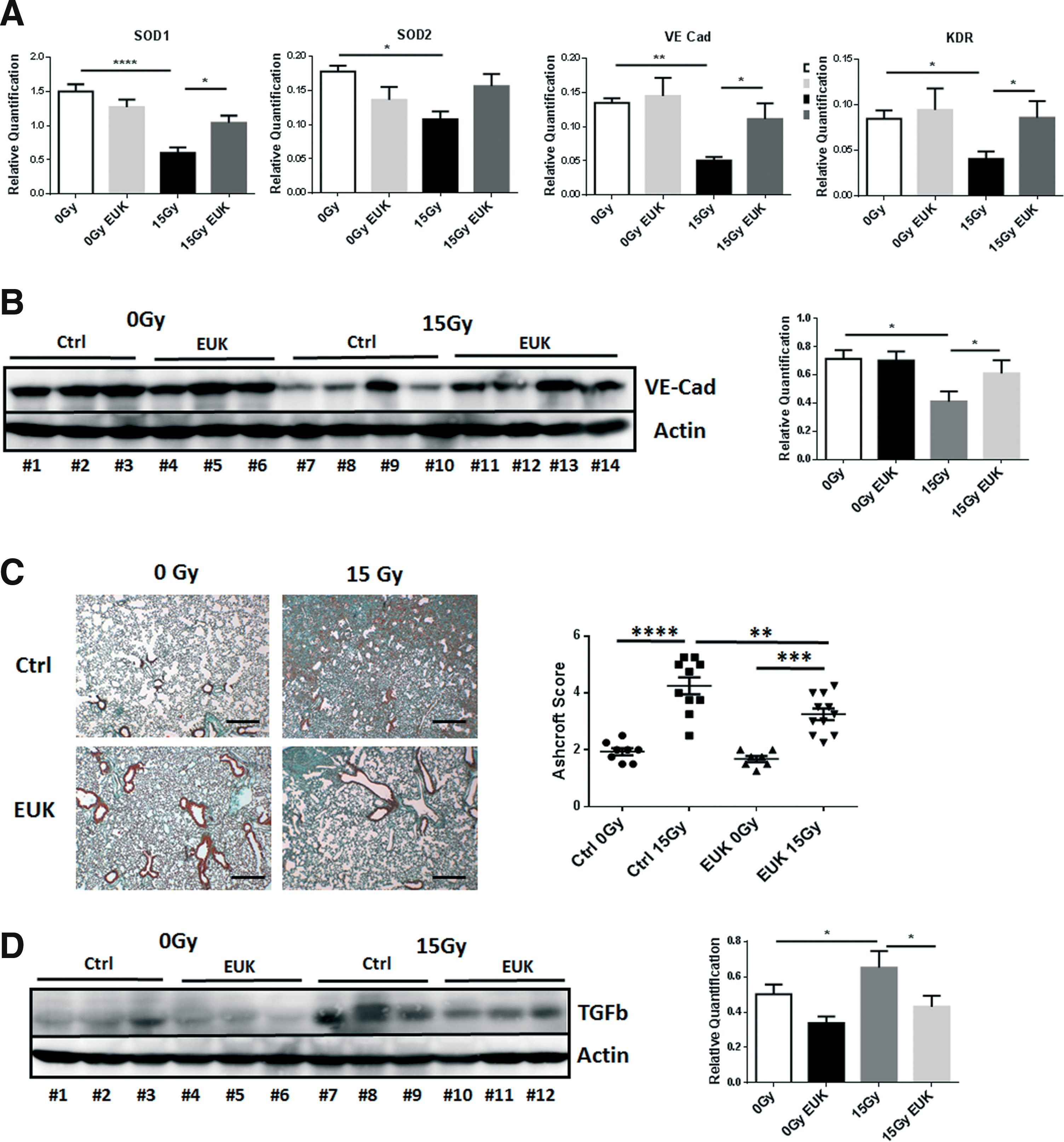
To investigate whether restoration of SOD1 may contribute to the protective MSC action and counteract the RT-induced EC loss as an adverse late effect, we applied the superoxide dismutase mimetic EUK134 during the first 3 weeks postirradiation. Mice were sacrificed at 25 weeks postirradiation, and lung tissues were collected for further analysis. qRT-PCR quantification of SOD1 and also SOD2, as well as for the EC markers VE-Cad and VEGFR2/KDR, demonstrated normalized expression levels of the genes in EUK134-treated animals compared to a significant reduction after WTI (Fig. 6A). To further determine restoration of EC levels by EUK134 treatment, we quantified the VE-Cad in whole protein lysates by Western blot analysis 25 weeks after irradiation (Fig. 6B). WTI induced a significant reduction of VE-Cad expression levels (mean 0 Gy: 0.71 ± 0.06, n = 6; mean 15 Gy: 0.38 ± 0.05, n = 6; MD 0 Gy vs. 15 Gy: 0.33; 95% CI of diff. 0.068–0.606) 25 weeks after irradiation, whereas in EUK134-treated animals, these expression levels were restored to the levels of sham controls (mean 15 Gy EUK: 0.64 ± 0.08, n = 7; MD 15 Gy vs. 15 Gy EUK: −0.263; 95% CI of diff. −0.523 to 0.127). Furthermore, while radiation-induced EC loss at 25 weeks after WTI was accompanied by a significant fibrosis progression, RT-induced fibrosis was significantly reduced by an early treatment with the antioxidant EUK134 as revealed by Masson's Goldner Trichrome staining (Fig. 6C), as well as determination of the expression of the profibrotic cytokine transforming growth factor-beta 1 (TGFb), respectively (Fig. 6D).
Radiation of cultured tissue-resident MSCs results in decreased expression levels of SOD1 and induces a fibroblast-like phenotype
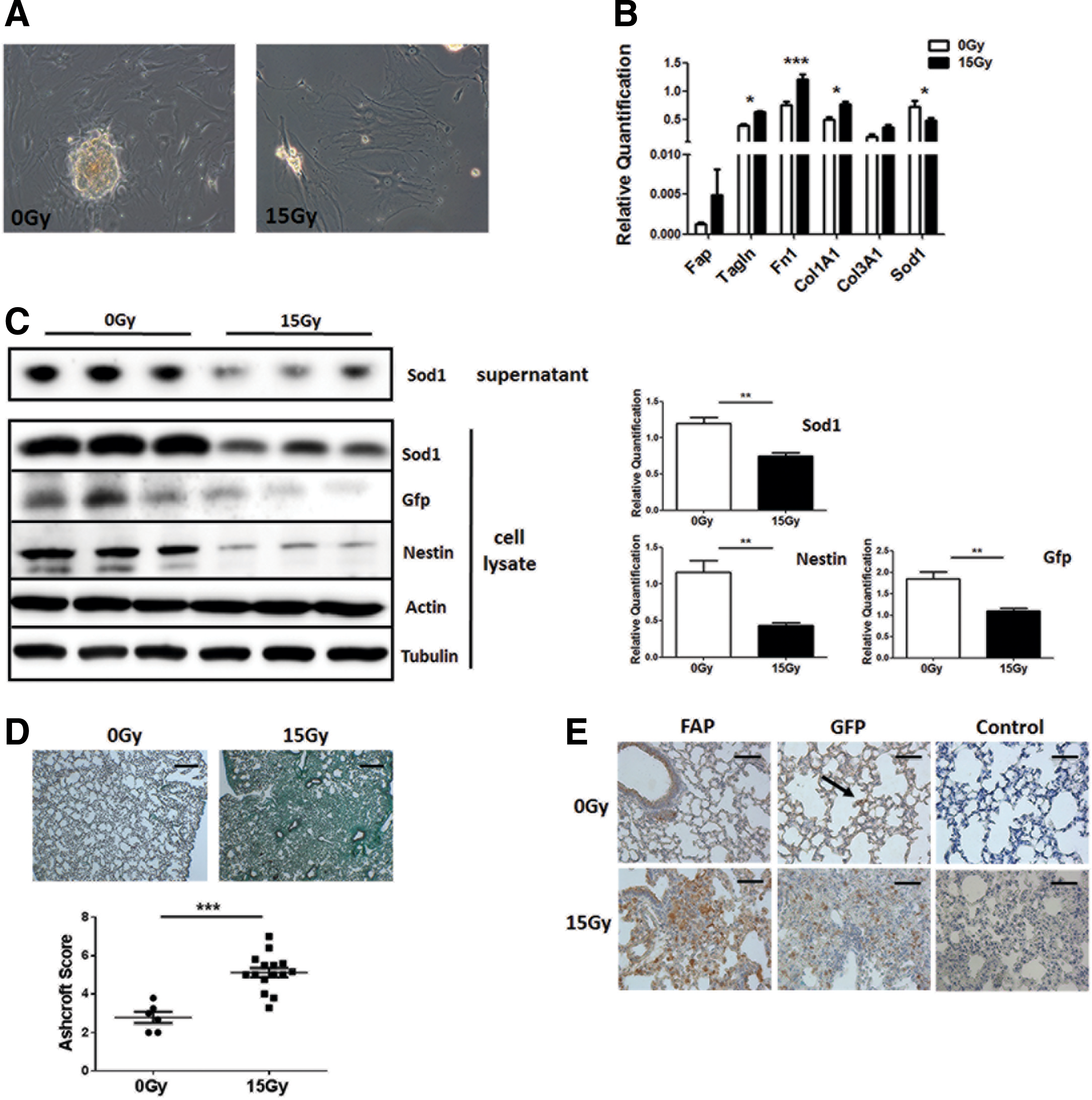
To investigate whether RT affects SOD1 expression of endogenous lung resident MSCs, cultured MSCs derived from the aorta of Nestin-GFP (NestGFP)-transgenic mice were used as a model for tissue-resident MSCs. Phase contrast microscopy revealed morphological alterations of cultured AoMSCs after irradiation with 15 Gy when a more enlarged and flattened fibroblast-like phenotype became prominent (Fig. 7A). qRT-PCR analysis was performed to confirm the acquired fibroblast-like phenotype after irradiation. The fibroblast marker genes, FAP for activated fibroblasts and transgelin (Tagln) for contractible fibroblasts (myofibroblasts), as well as genes encoding for extracellular matrix proteins (collagens and fibronectin), were upregulated after irradiation, whereas SOD1 was significantly downregulated (Fig. 7B). Western blot analysis for SOD1 protein expression levels revealed a downregulation of SOD1 in cell lysates as well as of secreted SOD1 in cell culture SN of irradiated AoMSCs 96 h after 15 Gy irradiation and a significant downregulation of the MSC marker protein Nestin (Fig. 7C). To further investigate the role of tissue-resident MSCs after WTI directly in the lungs, NestGFP mice were left untreated or received a 15 Gy WTI. Histological evaluations confirmed the development of fibrosis 25 weeks after WTI as visualized by a massive collagen deposition (mean 0 Gy: 2.78 ± 0.29, n = 6 vs. 15 Gy: 5.12 ± 0.25, n = 15; p ≤ 0.001) (Fig. 7D), as well as a significant downregulation of SOD1 (Supplementary Fig. S5). Lung sections were further stained for the activated fibroblast marker FAP and the MSC marker GFP, which is under the regulatory control of the Nestin promotor (Fig. 7D). FAP immunoreactivity confirmed the presence of activated fibroblasts in the fibrotic areas of WTI lungs, and, interestingly, an increase of GFP expressing cells was also observed within this fibrotic area, whereas in not irradiated control lungs, only single GFP-positive MSCs could be detected (Fig. 7D, arrow). Western blot analysis of GFP expression in whole lung protein lysates confirmed a significant increase of NestGFP (Supplementary Fig. S4).
Tissue-resident NestGFP(+) cells and not BM-derived MSCs contribute to fibrosis development
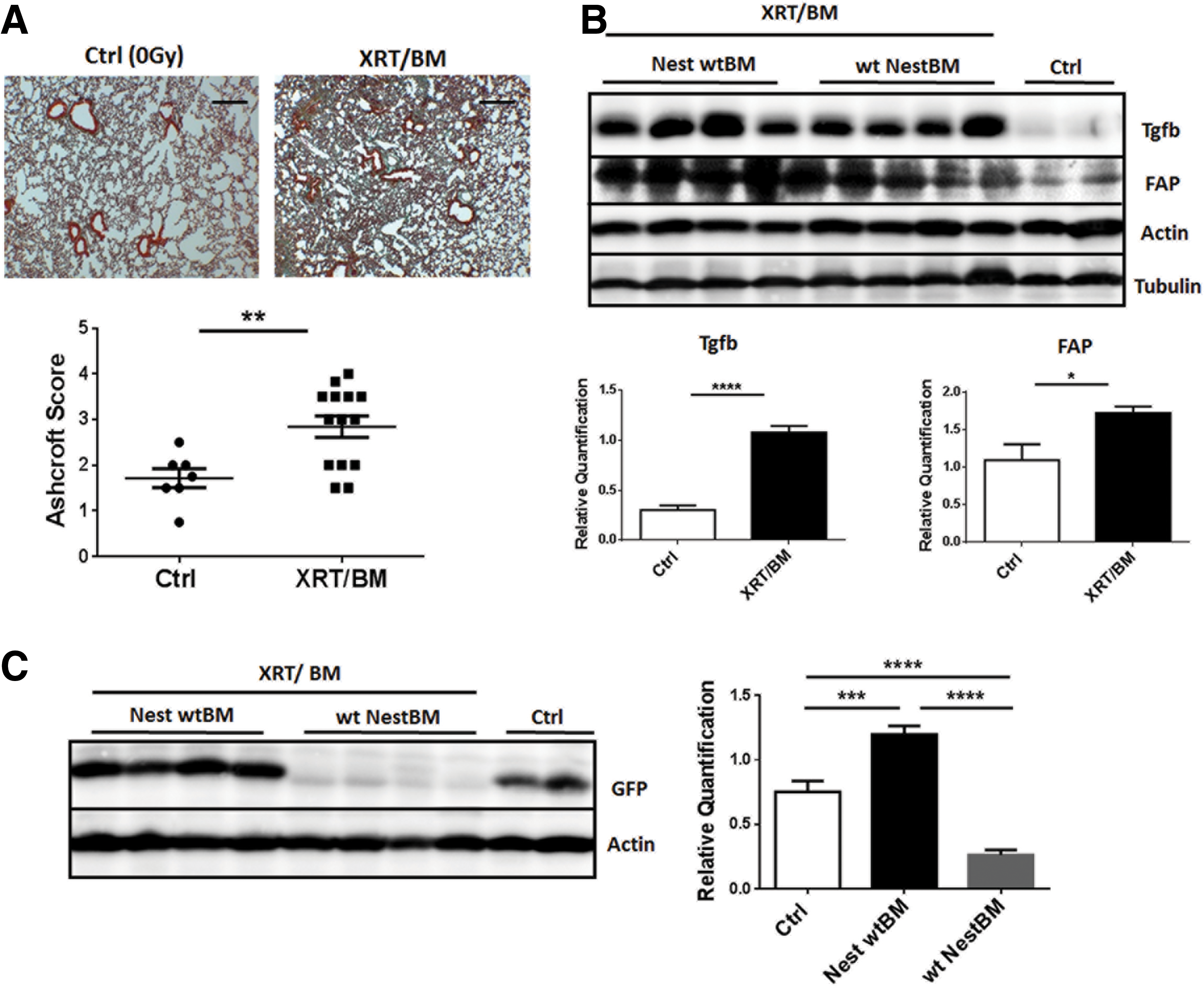
Next, we analyzed the putative contribution of tissue-resident lung MSCs compared to BM-derived MSCs to fibrosis development. Therefore, NestGFP transgenic mice were lethally irradiated with a split dose of 7 + 3 Gy total body irradiation (TBI) and subsequently adoptively transferred with BM cells from C57BL/6 donor mice into the tail vein (Nest wtBM) and vice versa (wt NestBM). Unirradiated NestGPF mice were used as control. Histological evaluations with Masson's Goldner Trichrome on paraffin-embedded lung sections at 25 weeks after WTI confirmed fibrosis development in the lungs of TBI mice, although the degree of fibrosis displayed a mild phenotype as revealed by the decreased Ashcroft scores compared to WTI NestGFP mice (mean 0 Gy: 1.71 ± 0.21, n = 7 vs. 7 + 3 Gy: 2.85 ± 0.23, n = 14; 95% CI of diff. −1.97 to −0.30) (Fig. 8A). Significantly increased Tgfb1 and FAP protein expression by Western blot analysis in whole protein lung lysates further confirmed the fibrosis phenotype after TBI (Fig. 8B). Interestingly, determination of GFP expression levels corroborated that lung-resident and not BM-derived GFP(+) MSCs contribute to fibrosis development as revealed by significantly increased GFP expression levels in Nest wtBM lungs (Fig. 8C).
Discussion
In this study, we show for the first time that adoptive transfer of MSCs during the early phase after irradiation efficiently counteracts vascular damage and EC loss as adverse late effects of IR in a preclinical murine model of radiation-induced pneumopathy. Furthermore, MSC-derived cell culture SN efficiently rescued cultured lung EC from the radiation-induced toxic effects in short-term and long-term survival assays, indicating the contribution of MSC-secreted factors. Finally, we identified MSC-derived SOD1 as a potential paracrine mediator of the protective MSC action: (i) MSCs secreted SOD1 into the culture SN; (ii) therapeutically applied MSCs isolated either from the BM or the aorta have the potential to restore SOD1 expression in previously irradiated lung tissue when applied early after WTI; (iii) irradiation of cultured AoMSCs as an in vitro model for endogenous lung MSCs led to a decrease in the expression and secretion of SOD1; and (iv) the protective effect of MSCs was mimicked by therapeutic application of the SOD-mimetic EUK134 and may involve, besides protection of lung EC, also the protection of endogenous lung-resident MSCs. These findings contribute to an improved understanding of radiation-induced normal tissue toxicity as the main dose-limiting side effect of RT and can be of direct clinical relevance, since they suggest SOD1 as a potential therapeutic target for the radioprotection of EC. Furthermore, the present data extend our earlier observations that therapeutically applied MSCs are well suited to protect lung EC from vascular dysfunction at early time points post-WTI and to limit associated immune cell infiltration in the previously irradiated lungs at the onset of the pneumonitic phase (44).
In more detail, we showed for the first time that adoptive transfer of MSC within the early phase after irradiation has the potential to provide a long-term protection of pulmonary EC from radiation-induced damage. It is generally accepted that blood vessels are critical components of the radiation response and that vascular damage upon irradiation is particularly prominent in the radiation response of normal tissues (9, 25, 31, 44, 84). We and others showed that thorax irradiation results in impairment of various pulmonary vascular parameters such as structural changes in pulmonary blood vessels, vascular leakage, increased extravasation of circulating immune and tumor cells, as well as stenosis and lumen occlusion promoting increased pulmonary vascular resistance (25, 29, 34, 44).
In general, the response of the vasculature to radiation is classified in acute and late effects, both of which contribute to the initiation, progression, and maintenance of tissue damage (15). High doses of radiation (>10 Gy) can cause a rapid wave of EC apoptosis, whereas surviving cells develop a dysfunctional vascular phenotype (57). Long-term effects include microvessel collapse, thickening of the basement membrane, and persistence of an activated, procoagulant endothelial phenotype (15, 29). As a consequence, the irradiated tissue is converted into a hypoxic proinflammatory environment causing further damage to other normal cells by inducing ischemia, necrosis, and fibrosis (15, 74).
The special importance of the primary injury to the EC for radiation-induced late toxicity in the lungs is emphasized by studies on pulmonary arterial hypertension (PAH), where a functional impairment or even partial loss of the EC could be detected immediately after a selective lung irradiation and long before the manifestation of the first clinical symptoms (33). In this study, we show now that adoptive transfer of MSCs within the early phase after irradiation also provides long-term protection from radiation-induced EC damage, EC loss, and immune cell infiltration that translates in protection from fibrosis development. Our findings support the assumption that adoptive transfer of adult MSCs may be a valuable therapeutic option for the prevention of lung diseases, the regeneration of diseased lung tissue or both because of their relatively easy availability, multipotent differentiation capacities, and immunomodulatory effects (4, 77).
Importantly, in the present study, aorta-derived MSCs were more potent than classically derived BM-MSC to protect lung EC from the adverse late effects of RT. These findings support our assumption that the high activity of the AoMSCs for EC protection might be due to the fact that tissue-specific stem cells mainly support the tissue type from which they originate (26, 42, 44). Within all our experiments concerning the EC (e.g., Western blot analysis, qRT-PCR, and IHC for VE-Cad expression, CD31 FACS analysis, as well as determination of Tagln-positive vessels), BM-MSCs did not appear to produce a robust improvement of the radiation-induced vascular damage, although BM-MSCs were able to limit fibrosis progression, even though not as effective as AoMSCs. Our data might suggest that BM-MSCs have a weaker response overall and in particular for the radioprotection of vascular EC. Affirmative, earlier reports already suggested that BM-MSCs were less effective for MSC therapy compared to other stem cell sources, for example, compared to adipose tissue-derived or fetal MSCs (64, 66, 86).
Mechanistically, MSC-mediated protection was accompanied by restoration of normal expression levels of the antioxidant SOD1 in WTI lungs. Importantly, we identified SOD1 by an MS approach as a paracrine factor that is secreted by aortic and BM-MSCs, but in particularly high amounts by AoMSCs. This corroborates the general view that engraftment in the lung is currently not considered the main mechanism by which MSCs repair a diseased lung tissue (36, 78). Consistently we detected only a few, preferably single donor cells in WTI lung sections while circulating enhanced green fluorescent protein [EGFP(+)] MSCs could be detected in peripheral blood even 25 weeks after transplantation, supporting the idea that tissue protection is due to paracrine signaling (44). Today it is widely accepted that the positive outcome of MSC therapy is not due to a direct engraftment of these cells into the lung, for example, as endothelial and epithelial cells, instead paracrine factors are now considered to be the main mechanism through which stem and progenitor cells exert their therapeutic effect (13). Herein, it was suggested that MSCs may mediate their function through a “hit and run” mechanism, where MSCs, once temporarily localized to the lung, may provide a local source of trophic factors in the pulmonary environment (8, 13).
Our observation on the importance of upregulation of the antioxidant enzyme SOD1 for MSC-mediated protection supports the current hypothesis that late pulmonary fibrosis develops from early damage to resident lung cells and that this early damage involves radiation-induced oxidative stress (29, 48, 80, 87). Oxidative stress as a consequence of chemotherapy or radiotherapy or tissue inflammation promotes tissue damage, including damage to the vasculature by activating a series of signaling pathways (47, 65). Moreover, accumulation of reactive oxygen species (ROS) causes cell apoptosis and necrosis and can further contribute to myofibroblast activation (10, 72).
Consequently, disease-promoting processes that are at least in part caused by oxidative damage can be targeted by increasing the antioxidant defense, for example, by the intake of, or supplementation with, antioxidants (19, 28, 59). Consistently, antioxidant treatment with the SOD mimetic EUK207 during the first month after thorax irradiation was able to protect against excessive collagen synthesis during the fibrotic stage (25–30 weeks) (29). The protective effect of EUK207 applied during the early phase after irradiation was associated with a partial mitigation of the radiation-induced vascular injury and reduction in blood vessel density observed as a late effect of TBI of mice with 11 Gy (29), highlighting a role of oxidative stress for radiation-induced vascular injury. In line with these findings, continuous administration of EUK207 via implanted subcutaneous infusion pumps from 1 h until 14 weeks after irradiation also mitigated radiation-induced lung damage in rats (49). Even, when given after irradiation, a single injection of the SOD mimetic EUK189 had a small but mitigating effect in rodents, especially in the lung (48).
For some normal tissue radiation mitigation indications, a topical mean of delivering salen Mn complexes turned already been out to be therapeutically practical (68). Herein, EUK134 was reported as an effective combination catalase/SOD mimetic. In particular, topical treatment with EUK134 shortly before ultraviolet A (UVA) exposure resulted in reduced levels of lipid peroxides at the surface of UVA-exposed skin, but also baseline peroxide levels on nonirradiated skin were reduced in a dose-dependent manner (20). As inhibition of H2O2 signaling using an EUK134-like compound has already been shown to be a promising approach, for example, to breast cancer therapy it might also be possible that the protective effect of SOD1 and/or EUK134 on counteracting radiation-induced EC loss could be mediated by H2O2 decomposition (71).
Our novel findings demonstrate for the first time that adoptive transfer of MSCs early after irradiation is also suited to enhance the antioxidant defense and to prevent radiation-induced late effects in irradiated WTI lungs by restoring expression levels of SOD1. MSC-mediated protection limited fibrosis progression presumably through secretion of SOD1. Notably, this protective effect of MSC therapy could be mimicked by the application of the SOD mimetic EUK134: application of EUK134 within the early phase after radiation counteracted EC loss and slightly but significantly reduced fibrosis development as long-term adverse effects of WTI. Furthermore, adoptive transfer of MSC also efficiently counteracted infiltration of immune cells at early (44) and at late stages as shown here. This is particularly important since oxidative stress generated by radiotherapy or chemotherapy is known to initiate a cascade of acute and chronic inflammatory reactions that may further increase oxidative stress in the inflamed tissue (3, 30). Thus, by addressing both, oxidative stress and tissue inflammation, MSC therapy may serve as an ideal mitigator of adverse late effects of radiotherapy. Herein, mitigation refers to therapies that are started after irradiation, but before there is overt evidence of clinical disease (29, 58). Interestingly, MSC treatment particularly reduced the infiltration of profibrotic myeloid cells that had been shown to facilitate the progression of pulmonary fibrosis in the bleomycin-induced fibrosis model (35, 63). The reduced infiltration of myeloid cells with an assumed profibrotic phenotype here after MSC therapy highlighted the importance of the proper function of the vascular system to avoid fibrosis development.
Interestingly, a normal function of resident MSCs in adult lungs is crucial for pulmonary tissue homeostasis as they contribute to the maintenance of tissue integrity by various mechanisms (39, 53, 55). As an example, endogenous lung MSCs normally exert anti-inflammatory properties; these are, however, negatively affected by bleomycin treatment, thereby contributing to fibrosis development in a murine model of bleomycin-induced fibrosis (39). In line with these findings, exogenous administration of untreated lung MSCs protected lung integrity from bleomycin-induced lung injury and associated oxidative stress (39). Thus, in certain situations, external stimulation, for example, exogenously applied MSCs, may be required to catalyze the repair, suggesting an important role of transplanted cells to act as an initiator to trigger endogenous stem cell-based tissue repair (23).
In contrast, endogenous lung-resident MSCs were also shown to promote fibrotic remodeling by acquisition of a profibrotic myofibroblast phenotype, for example, upon stimulation with the profibrotic cytokine TGFb (52, 73). In this study, we used cultured AoMSC as an in vitro model for endogenous lung MSCs and observed that radiation induced the acquisition of a fibroblast-like phenotype that was associated with the downregulation of SOD1 expression and secretion. We are well aware of the fact that tissue-resident MSCs are heterogeneous and that different MSC subsets exist. The characterization and/or isolation of the stem cell subpopulations represent a major challenge to improve the efficacy of transplantation protocols used in regenerative medicine and applied to lung disorders (13). Primary lung MSCs that were enriched in the CD90/CD105 mononuclear cell fraction were shown to be located perivascularly (67). Therefore, we think that vascular wall-derived MSCs used in our studies were a suitable model. We further used NestGFP transgenic mice in which endogenous Nestin-expressing MSCs were GFP tagged to show that these lung-resident MSCs contribute to remodeling of lung tissue upon fibrosis development after WTI. Using BM chimeras we further confirmed that indeed tissue-resident and not BM-derived MSCs were predominantly involved in fibrosis development. Interestingly, increasing amounts of GFP were detected in fibrotic lungs of NestGFP mice after WTI and in Nest wtBM chimeras after TBI, although an induction of resident MSCs differentiation would firstly suggest a downregulation of MSC marker proteins (namely Nestin) and thus of GFP. In line with our findings, immunofluorescence microscopy already revealed that Nestin is expressed in small proportions of fibroblasts (70). Furthermore, a concomitant appearance of Nestin- and CD34-positive myofibroblasts was observed under fibrosing conditions, suggesting that the differential expression of Nestin may not only indicate phenotypic and functional heterogeneity but may also indicate that Nestin-positive myofibroblast may represent a relatively immature subpopulation of cells with multipotentiality (41).
These findings demonstrate that damage to tissue-resident lung MSCs may contribute to progression of radiation-induced fibrosis. However, we show for the first time that a reduced antioxidant capacity of MSC, for example, induced by the loss of SOD1, may be critical for development of a profibrotic MSC phenotype. In line with this assumption, Chow et al. previously demonstrated that depletion of extracellular SOD in resident lung MSCs resulted in microvascular remodeling and increasing muscularization in a murine model of PAH (11). Moreover, loss of SOD in cultured lung MSCs resulted in accelerated proliferation as well as increased multilineage differentiation potential and promoted the transition to a contractile phenotype (11) while suppression or elimination of excessive ROS turned out to be an effective strategy for improving the survival rate of transplanted MSC (85). Altogether, these findings demonstrate that adoptive transfer of functional MSC has the potential to protect, besides EC also the endogenous lung-resident MSCs from radiation-induced activation and subsequent differentiation into fibrosis-promoting fibroblast-like cells.
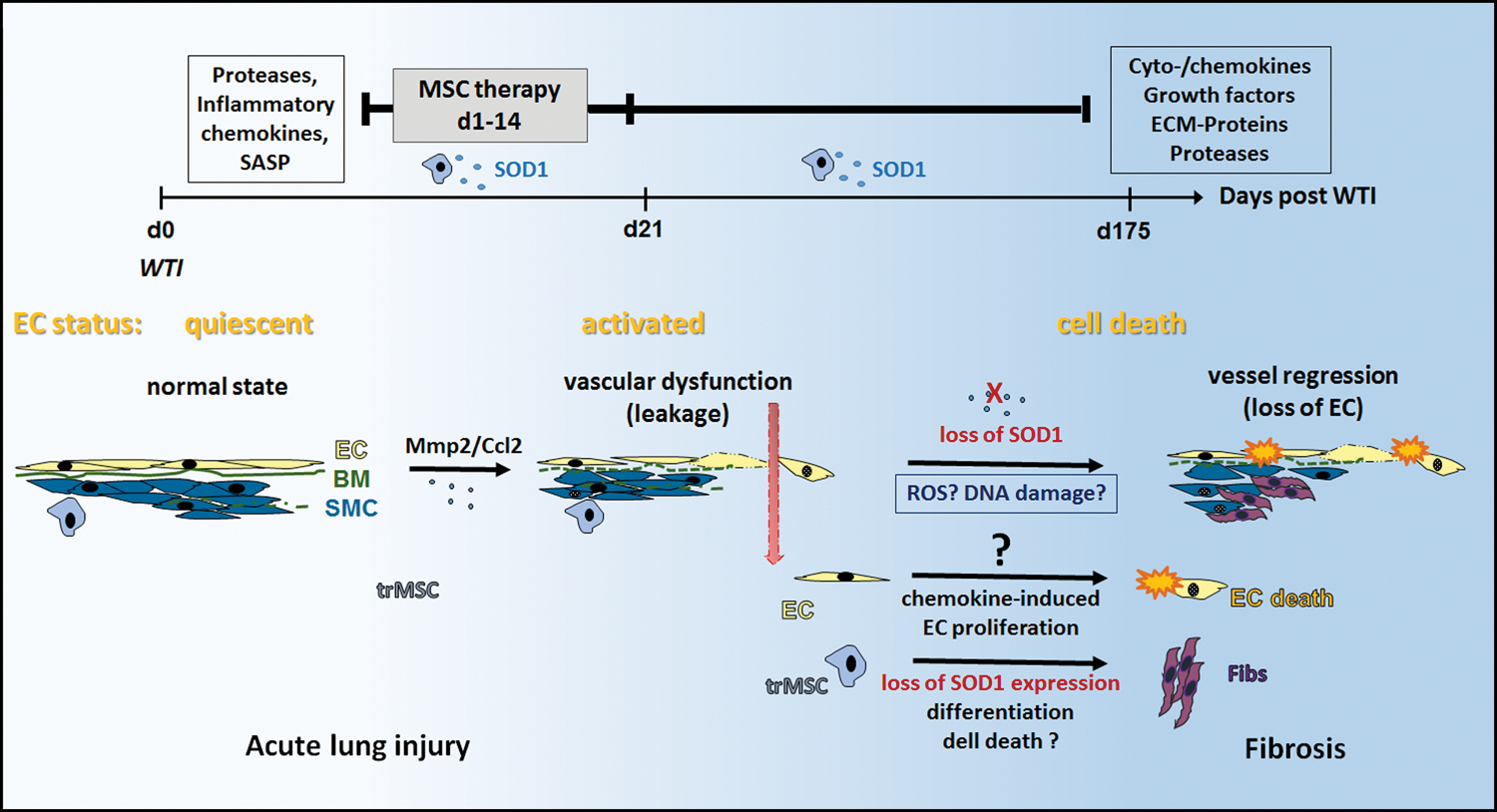
Conclusively, our findings highlight the importance of radiation-induced damage to the vascular system within the processes of fibrosis progression (Fig. 9). While the exact mechanisms by which therapeutically applied MSCs as well as the SOD-mimetic EU134 prevent vascular cell loss are not known, their ability to reduce radiation-induced apoptosis in capillary EC cultures may be relevant as they result in improved clonogenic EC survival as shown here and by others (21, 79). In addition to a direct protective effect on the lung endothelium, proper vascular function and restoration of SOD1 expression may contribute to a microenvironment, for example, through normalization of immune cell infiltration, which favors both prevention of and recovery from radiation injury to vascular and other resident lung cells. In support of this hypothesis, we show here that elevated SOD1 expression levels after treatment protect resident MSCs presumably by inhibiting differentiation into a fibroblast-like phenotype. However, further studies are needed to clarify the mechanistic link between effects on ECs and lung-resident MSCs and damage of other lung parenchymal cells, for example, alveolar or bronchial epithelial cells for fibrosis development. MSC treatment could also antagonize radiation-induced senescence of bronchial–alveolar epithelial cells and thus prevent upregulation of the senescence-associated secretory phenotype (SASP) factors and subsequent recruitment of inflammatory myeloid cells as late adverse effects that were already shown for to be blocked early after radiation (44). Further investigations will also be performed in the future to corroborate our findings in more clinical relevant settings such as fractionated low-dose irradiation. Understanding the mechanisms and, in particular, the advantage of AoMSC-mediated protection of EC from the adverse effects of IR is required to if we aim to develop MSC-based protective treatment strategies.
Materials and Methods
WTI mouse model
Wild-type C57BL/6 and NestGFP transgenic donor mice (mixed gender) received 15 Gy of WTI in a single dose of a Cobalt 60 source (60Co γ-rays at 0.5 Gy/min) as previously described (43, 44, 81). Single cell suspensions of cultured MSCs (0.5 × 106 cells) were intravenously transplanted into the tail vein of WTI mice 24 h or 14 days after irradiation or in sham-irradiated (0 Gy) control animals as previously described (44). All procedures involving mice were approved by the local institutional Animal Care Committee (Regierungspräsidium Düsseldorf Az84-02.04.2012.A137; 84-02.04.2012.A034). For mimicking MSC action (restoring SOD1 expression), a synthetic superoxide dismutase mimetic (EUK134; Selleckchem, Houston, TX) was used. Within combined treatment, WT mice were exposed to WTI and subsequently treated three times a week (starting within 24 h after WTI) within the first 3 weeks postirradiation by intraperitoneal injection with 100 μl solvent (phosphate-buffered saline [PBS]) or 10 μg/g bodyweight EUK134 (diluted in 100 μl PBS). Mice were sacrificed at 25–30 weeks postirradiation, and lung tissues were collected for further analysis.
TBI mouse model
A mouse TBI model was used as described before (43, 44). In brief, BM cells were harvested aseptically by flushing the tibias and femurs of adult animals and subjected to erythrocyte lysis. C57Bl/6J (both gender) mice were lethally irradiated with a split dose (7 + 3 Gy) of an X-ray source and were intravenously transplanted with 1 × 106 unfractionated murine NestGFP-expressing BM cells from NestGFP transgenic donor mice into the tail vein (wt NestBM) (83). After 25 weeks, animals were sacrificed and lungs were isolated. In addition, a vice versa experiment was performed: NestGFP mice were lethally irradiated and BM cells from C57Bl/6 wild-type donor mice were transplanted (Nest wtBM). Experiments were repeated three times.
Isolation and purification of aortic MSCs and BM MSCs
Vascular wall-resident MSCs were isolated from aortas of C57BL/6-Tg(CAG-EGFP)1Osb/J mice (Jackson Laboratory, Bar Harbor, ME) as previously described (43, 44). In brief, tissue pieces were mechanically minced and dissociated for 15 min at 37°C in OptiMEM I medium containing 0.2% type 2-collagenase (CLS2, 43J14367B; Worthington, Lakewood, WA). Pure MSCs were generated using a Sca-1 antibody (130-092-529) and magnetic activated cell sorting (MACS) technology (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Primary MSCs were cultivated on plastic plates in Dulbecco's modified Eagle's medium (DMEM)/20% fetal calf serum (FCS). Primary cultures were clonally expanded under limiting dilution conditions. BM cells were harvested and cultured using complete DMEM/20% FCS as previously described (44).
LMEC isolation
LMEC was purified from type 2 collagenase digested cell extracts using the same MACS technology protocol in combination with biotinylated PECAM1 antibody (CD31 MEC 13.3; BD Biosciences, Franklin Lakes, NJ) and Streptavidin Microbeads. Cells were cultured in ECG medium MV (PromoCell, Heidelberg, Germany).
Real-time qRT-PCR
RNA was isolated using RNeasy Mini Kit (74106; Qiagen, Hilden, Germany) according to the manufacturer's instruction and as previously described (44). Expression levels were normalized to the reference gene (beta actin; set as 1) and are shown as relative quantification. Specific primers were designed with the program Primer 3 (
Conditioned media
Aortic MSCs and BM-MSCs were cultured in normal growth media until confluence. Media were replaced, and cells were cultured in the presence of 0.5% fetal bovine serum for 24 h before collection of media. Control media were generated by incubating the same medium (containing 0.5% fetal bovine serum) without cells. Conditioned media were used as 1/1 mixture with NGM. For MS analysis, confluent cells were incubated for 48 h with serum-free media.
Sample preparation for liquid chromatography–mass spectrometry
Proteins were precipitated with acetone (−20°C, overnight) and then resuspended in 50 mM phosphate buffer (pH 7.5). The protein concentration was determined using the Roti Nanoquant (Roth) protein assay. A volume corresponding to 15 μg total proteome was then transferred to a fresh Eppendorf tube and reduced (10 mM DTT, 45 min) and alkylated (20 mM iodoacetamide IAM, 60 min) at 37°C in the presence of 6 M Urea. Afterward, the sample was incubated with LysC (ratio 1:50) at 37°C for 3 h. Next, the urea concentration was reduced to 1.8 M urea by addition of 50 mM ammonium bicarbonate (ABC) buffer. Subsequently, trypsin was added (ratio 1:30), and the samples were incubated overnight at 37°C while vigorously shaking. The digestion reaction was stopped by adding formic acid (FA; final concentration 1%). The tryptic digests were desalted on homemade C18 StageTips. After elution from the StageTips, samples were dried using a vacuum concentrator and the peptides taken up in 15 μl 0.1% FA solution.
Liquid chromatography–MS/MS
Experiments were performed on an Orbitrap Elite instrument (Thermo Fisher Scientific, Waltham, MA) that was coupled to an EASY-nLC 1000 liquid chromatography (LC) system (56). The LC was operated in the two-column mode. The homemade fused silica column equipped with a glass fiber frit was packed with Reprosil-Pur 120 C18-AQ 3 μm resin and connected to the analytical column via an ultra high-pressure liquid chromatography (UHPLC) union (50). The analytical column was a fused silica capillary (75 μm × 25 cm) with integrated PicoFrit emitter packed in-house with Reprosil-Pur 120 C18-AQ 3 μm resin. The analytical column was attached to a nanospray flex ion source (Thermo Fisher Scientific). Peptides were delivered to the precolumn via the integrated autosampler at a flow rate of 2–3 μl/min in 100% solvent A (0.1% FA, in high-pressure liquid chromatography (HPLC) grade water). Peptides were subsequently separated on the analytical column by running a 70-min gradient of solvent A and solvent B (start with 7% B; gradient 7%–35% B [0.1% FA in acetonitrile, ACN] for 60 min; gradient 35%–100% B for 5 min; and 100% B for 5 min) at a flow rate of 300 nl/min.
The mass spectrometer (positive ion mode) was operated using Xcalibur software (version 2.2 SP1.48). Precursor ion scanning was performed in the Orbitrap analyzer Fourier transform-based mass spectrometers (FTMS) in the scan range of m/z 300–1500 and at a resolution of 120,000 with the internal lock mass option turned on (lock mass was 445.120025 m/z, polysiloxane) (60). Product ion spectra were recorded in a data-dependent manner in the ion trap mass spectrometer (ITMS) in a variable scan range and at a rapid scan rate. The ionization potential (spray voltage) was set to 1.6–2.0 kV. Peptides were analyzed using a repeating cycle consisting of a full precursor ion scan (1.0 × 106 ions) followed by 15 product ion scans (1.0 × 104 ions) where peptides are isolated based on their intensity in the full survey scan (threshold of 500 counts) for tandem mass spectrum (MS2) generation that permits peptide sequencing and identification. collision-induced dissociation (CID) collision energy was set to 35% for the generation of MS2 spectra. During MS2 data acquisition, dynamic ion exclusion was set to 120 s with a maximum list of excluded ions consisting of 500 members and a repeat count of 1. Only charge states >1 were considered for fragmentation.
Label-free quantification using MaxQuant
RAW spectra were submitted to an Andromeda (18) search in MaxQuant (version 1.5.0.25) using the default settings (17). Label-free quantification and match-between-runs were activated (16). MS2 spectra data were searched against a Uniprot mouse reference database (MOUSE.fasta; 59375 sequences). All searches included a contaminants database (as implemented in MaxQuant, 263 sequences). The contaminants database contains known MS contaminants and was included to estimate the level of contamination. Andromeda searches allowed for oxidation of methionine residues (16 Da) and a static modification on cysteine (57 Da, alkylation with iodoacetamide). Enzyme specificity was set to Trypsin/P. For the Andromeda searches, the default MaxQuant settings were used. Briefly, the precursor peptide tolerance for the first search was 20 ppm and for the main search 4.5 ppm. The ion trap MS/MS match tolerance was 0.5 Da. Label-free quantification and match-between-runs were switched on.
Flow cytometry analyses
Crude cell extracts of freshly isolated lungs were generated, and FACS analysis was performed as previously described (44, 81). Lung cell suspensions were stained with anti-mouse CD45 (30-F11; Cat. 103126 from BioLegend, San Diego, CA) to exclude leukocytes. Lung cells were further fluorochrome labeled with anti-mouse CD31 (390; Cat. 11-0311, from eBioscience Frankfurt, Germany) and anti-mouse CD11b (M1/70, Cat. 101227 from BioLegend). Flow cytometric measurements were performed on a BD LSRII flow cytometer using FACS DIVA software. Analyses of obtained data sets were done using FACS DIVA software (all from BD Biosciences).
Lung sprouting assay
Ex vivo isolated lung pieces were seeded on growth factor-reduced Matrigel in NGM supplemented with or without MSC conditioned medium. Capillary-like outgrowth was quantified by measuring the sprouting distance 4 days postirradiation.
Western blot
Whole cell lysates were generated by scraping cells into ice-cold RIPA-P buffer (150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecylsulfate, 50 mM Tris/HCL pH8, 10 mM sodium fluoride (NaF), 1 mM sodium orthovandanate (Na3VO4) supplemented with complete Protease-Inhibitor-Cocktail (04693159001; Hoffmann-La Roche, Basel, Switzerland) and performing two to three freeze–thaw cycles. Protein samples (50–100 μg total protein) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) electrophoresis, and Western blots were done as previously described using indicated antibodies (18, 60). SOD1 (FL-154, sc22760), p21 (F8, sc271610), Col1A1 (D13, sc-25974), Nestin (10c2, sc-23927), and VE-Cad (C19, sc6458) antibodies were from Santa Cruz (Santa Cruz, CA), FAP (PA5-51057), and GFP (A-11122) antibodies were from Thermo Scientific (Dreieich, Germany), and beta-actin (clone AC-74, A2228) antibody was from Sigma-Aldrich (St. Louis, MO).
IHC and electron microscopy
Paraffin-embedded tissue sections were hydrated using a descending alcohol series, incubated for 10–20 min in target retrieval solution (DAKO, Glostrup, Denmark) and incubated with blocking solution (2% FCS/PBS). After permeabilization, sections were incubated overnight at 4°C with primary antibodies (SOD1, VE Cad, Nestin, GFP each 1/100). Antigen was detected with a horseradish peroxidase-conjugated secondary antibody (1/250) and DAB (3,3′-diaminobenzidine) staining (DAKO). Nuclei were counterstained using hematoxylin. Electron microscopy was done as previously described (44).
Lung histopathology
For lung histology, mice were narcotized using isoflurane [2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane] and killed by transcardial perfusion with PBS. Whole inflation fixed lungs were taken out, and lung tissue was fixed in 4% formalin and embedded in paraffin. Three to four 5-μm paraffin longitudinal cross sections were taken per mouse lung at the midpoint through the lung block depth. Sections were stained with hematoxylin and eosin or Masson's Goldner Trichrome (MT; Carl Roth Karlsruhe, Germany) for histological evaluation. Samples were then analyzed microscopically with a 20 × objective. Sections were scored blinded to the genotype and treatment group. In whole sections of lung parenchyma, lung fibrosis from each specimen was scored using a 0–8 point Ashcroft scale (2, 82). The mean scores (five per section) were averaged to yield the final score for each specimen. Depicted data represent the mean values of all mice per group (mean of single average number for each mouse/mouse number) as indicated.
Irradiation of cell cultures
Radiation with indicated doses was performed using the Isovolt-320-X-ray machine (Seifert–Pantak, East Haven, CT) at 320 kV, 10 mA with a 1.65-mm aluminum filter, and a distance of about 500 mm to the object being irradiated. The effective photon energy was about 90 kV, and the dose rate about 3 Gy/min.
Colony formation assay
For this long-term assay, 200–1600 cells/well were plated in six-well plates as previously described (45). After radiation with indicated doses, plates were incubated for a total of 10 days to allow growth of single colonies. Cells were then fixed in 3.7% formaldehyde and 70% ethanol and subsequently stained with 0.05% Coomassie Brilliant Blue. Colonies (≥50 cells/colony) were counted under the microscope at fivefold magnification. The survival curves were established by plotting the log of the surviving fraction against the treatment dose.
Cell proliferation
At indicated time points cells were fixed with methanol for 10 min and stained with 0.5% crystal violet dye (in methanol: deionized water, 1:5) for 10 min. Excess crystal violet dye was removed by five washes of deionized water on a shaker (10 min for each wash), and the culture plates were dried overnight. The crystal violet dye was released from cells by incubation with 1% SDS for 1–2 h before optical density (OD 595 nm) measurement. The cell proliferation reagent WST-1 was used as a ready-to-use colorimetric assay for the nonradioactive quantification of cellular viability and cytotoxicity according to the manufacturer's instructions. OD (450 nm) measurements were performed 60–90 min after incubation.
Migration assay
Migration of the cells was investigated via time lapse microscopy for 8 h after IR. Therefore, cells were grown to confluence, irradiated, and a thin wound was introduced by scratching with a 10 μl pipette tip. Wound closure was determined for the different treatments by measuring the migration distance using ImageJ 1.47t (Wayne Rasband, National Institutes of Health, US states).
Statistical analysis
If not otherwise indicated, data were obtained from three independent experiments with at least three mice each. Mean values were calculated and used for analysis of standard error of the mean as indicated by error bars. Statistical significance was evaluated by one-way analysis of variance followed by the Tukey's or Bonferroni multiple comparisons posttest. Statistical significance was set at the level of p ≤ 0.05. Data analysis was performed with Prism 5.0 software (GraphPad, La Jolla, CA).
Footnotes
Acknowledgments
The authors thank L. Lüdemann and M. Groneberg for support with the irradiation of mice, Holger Jastrow for the help in ultrastructural analysis, Mohamed Benchellal, Inge Spratte, and Eva Gau for their excellent technical assistance. The work was supported by grants of the DFG (GRK1739/1; JE275/1; INST 20876/127-1 FUGG M.K.), the BMBF (ZISS 02NUK024-D), an ERC starting grant (M.K. 258413), and the UK Essen/IFORES (D/107-81040).
Authors' Contributions
D.K., J.S., A.W., K.R., F.S., F.W., and F.K. performed experiments, D.K. supervised and analysed results and made the figures; M.K., J.W.F., M.Y., and M.S. provided materials; D.K. and V.J. designed research and wrote the manuscript. All authors read and approved the article.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.