
Abstract
Aims:
Nonalcoholic fatty liver (NAFL) is a common liver disease associated with metabolic syndrome, obesity, and diabetes that is rising in prevalence worldwide. Various molecular perturbations of key regulators and enzymes in hepatic lipid metabolism cause NAFL. However, redox regulation through glutathione (GSH) adducts in NAFL remains largely elusive. Glutaredoxin-1 (Glrx) is a small thioltransferase that removes protein GSH adducts without having direct antioxidant properties. The liver contains abundant Glrx but its metabolic function is unknown.
Results:
Here we report that normal diet-fed Glrx-deficient mice (Glrx−/− ) spontaneously develop obesity, hyperlipidemia, and hepatic steatosis by 8 months of age. Adenoviral Glrx repletion in the liver of Glrx−/− mice corrected lipid metabolism. Glrx−/− mice exhibited decreased sirtuin-1 (SirT1) activity that leads to hyperacetylation and activation of SREBP-1 and upregulation of key hepatic enzymes involved in lipid synthesis. We found that GSH adducts inhibited SirT1 activity in Glrx−/− mice. Hepatic expression of nonoxidizable cysteine mutant SirT1 corrected hepatic lipids in Glrx−/− mice. Wild-type mice fed high-fat diet develop metabolic syndrome, diabetes, and NAFL within several months. Glrx deficiency accelerated high-fat-induced NAFL and progression to steatohepatitis, manifested by hepatic damage and inflammation.
Innovation:
These data suggest an essential role of hepatic Glrx in regulating SirT1, which controls protein glutathione adducts in the pathogenesis of hepatic steatosis.
Conclusion:
We provide a novel redox-dependent mechanism for regulation of hepatic lipid metabolism, and propose that upregulation of hepatic Glrx may be a beneficial strategy for NAFL. Antioxid. Redox Signal. 27, 313–327.
Introduction
N
Nonalcoholic fatty liver (NAFL) is a common liver disease associated with oxidative stress. However, the effects of oxidative post-translational modifications including protein glutathione (GSH) adducts on hepatic lipid metabolism are unknown. Ablation of glutaredoxin-1 (Glrx) increased protein GSH adducts, hepatic lipid synthesis, and steatosis in mouse liver. Sirtuin-1, an important metabolic regulator orchestrating hepatic lipid metabolism, was inactivated by GSH adducts and promoted fatty acid synthase expression. Overexpression of Glrx or a nonoxidizable Cys-mutant SirT1 in vivo normalized hepatic lipid synthesis. These data suggest that Glrx deficiency and oxidative inactivation of SirT1 play an important role in the pathogenesis of NAFL.
Hepatic lipid accumulation (hepatic steatosis) is the initial step in the pathogenesis of NAFL, arising from an imbalance of anabolic and catabolic processes in lipid metabolism (21), including lipid uptake, de novo lipogenesis, excretion, and oxidation. These processes are under tight transcriptional control through well-characterized networks of transcription factors such as the sterol regulatory element-binding proteins (SREBPs) regulating de novo fatty acid and cholesterol biosynthesis, and peroxisome proliferator-activated receptor gamma coactivator (PGC) 1α controlling β-oxidation (25, 60, 63, 76).
An emerging paradigm in disease processes is signaling pathways modulated by reactive oxygen and nitrogen species. In the presence of oxidants, reactive cysteines of proteins form reversible modifications that regulate enzyme activity, localization, protein interactions, and stability (17, 32, 33). Owing to abundant intracellular glutathione (GSH), protein GSH adducts are a key modification (referred to as protein S-glutathionylation [Prot-SG]) that is reversed by the enzyme glutaredoxin-1 (Glrx). Although Glrx has reactive thiols, deficient mice exhibited no aggravated oxidative damage upon angiotensin II infusion, ischemia-reperfusion, or hyperoxia (7, 34). Recent studies have demonstrated that GSH adducts, controlled by Glrx, participate in various processes, including cellular growth, apoptosis, cytoskeletal regulation, angiogenesis, and inflammation (1, 3, 4, 51, 68, 74). Glrx is an abundant liver protein that affects numerous proteins, but in the context of hepatic metabolism, only a few are identified and functionally studied.
Sirtuin-1 (SirT1), an NAD+-dependent class III histone deacetylase, modulates key transcription factors orchestrating hepatic lipid metabolism (30, 57, 60). Activation of SirT1 improved NAFL and conversely hepatic SirT1 deficiency led to steatosis (56). Inhibition of SirT1 activity by reversible GSH adducts has recently been described by our group and was confirmed by other investigators (11, 67, 71, 77). Because NAFL (58) is associated with oxidative stress, increased protein GSH adducts may play an important role in the pathogenesis of steatotic livers.
We report here that Glrx knockout mice (Glrx−/− ) fed normal diet (ND) develop spontaneous fatty liver and hyperlipidemia, suggesting mechanistic importance of Glrx in the development of NAFL. Furthermore, we demonstrate that inactivation of SirT1 by GSH adducts may be a major contributor to steatosis induced by Glrx deficiency.
Results
Glrx−/− mice fed normal diet develop metabolic disorders
Glrx−/−
mice fed ND became obese by 8 months of age compared with age-matched wild type (WT) mice. Body weight (BW) and relative fat mass ratio increased significantly by about 20% in Glrx−/−
mice (Fig. 1A and Supplementary Table S1; Supplementary Data are available online at
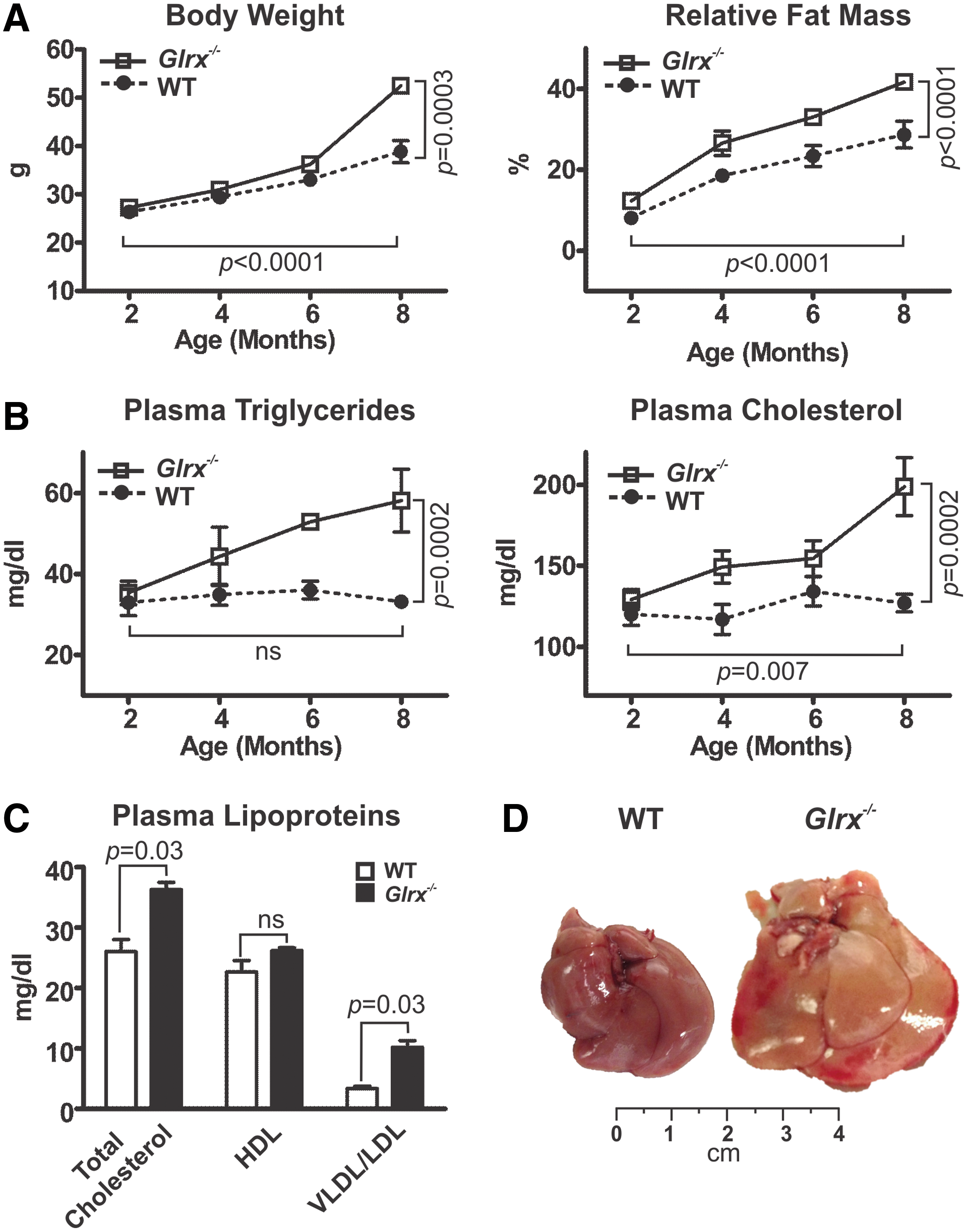
Plasma glucose and insulin levels at 8 months of age were similar between WT and Glrx−/− mice under fasting or fed conditions (Supplementary Table S1). Glrx−/− mice, however, did exhibit mild but significant glucose intolerance and insulin resistance measured by glucose and insulin tolerance tests (Supplementary Fig. S1), consistent with a prediabetic phenotype.
Glrx−/− mice fed ND develop NAFL disease
The hepatic lipid content of Glrx−/− mice at 8 months of age was significantly increased compared with WT littermate controls, as measured by Oil Red O and hematoxylin and eosin (H&E) staining of liver sections (Fig. 2A upper and middle rows) and quantification of extracted liver triglycerides and cholesterol (Fig. 2B). These data indicate a typical pathology of liver steatosis. Besides a mildly increased plasma alanine aminotransferase (ALT) activity, Glrx−/− mice showed no other signs of liver damage or inflammation, including changes in plasma aspartate aminotransferase (AST) activity and inflammatory cytokines (Fig. 2C, D and Supplementary Table S1). Liver proteins of Glrx−/− mice had significantly more GSH adducts (Fig. 2A lower rows, E and Supplementary Fig. S14) and reversible oxidation (Supplementary Fig. S2). Hepatic oxidized glutathione (GSSG) was below the detection limit by HPLC, and GSH levels in livers of Glrx−/− mice were comparable with those of WT (Supplementary Fig. S3). Taken together, these data suggest that increased reversible oxidative modifications of liver proteins because of the lack of Glrx may promote hepatic lipid accumulation and contribute to the pathogenesis of NAFL.
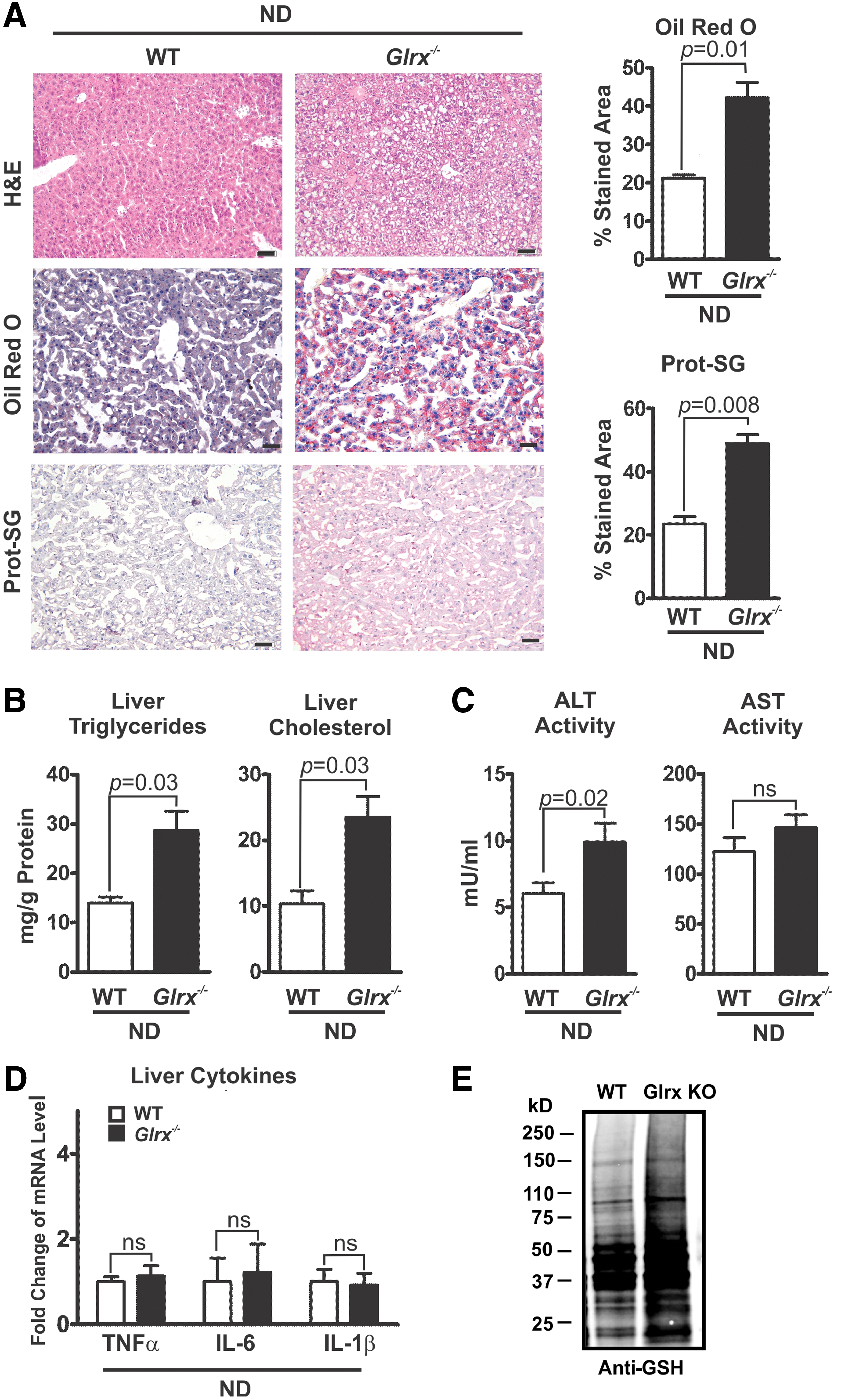
Hepatic Glrx regulates lipid metabolism and controls plasma lipid levels
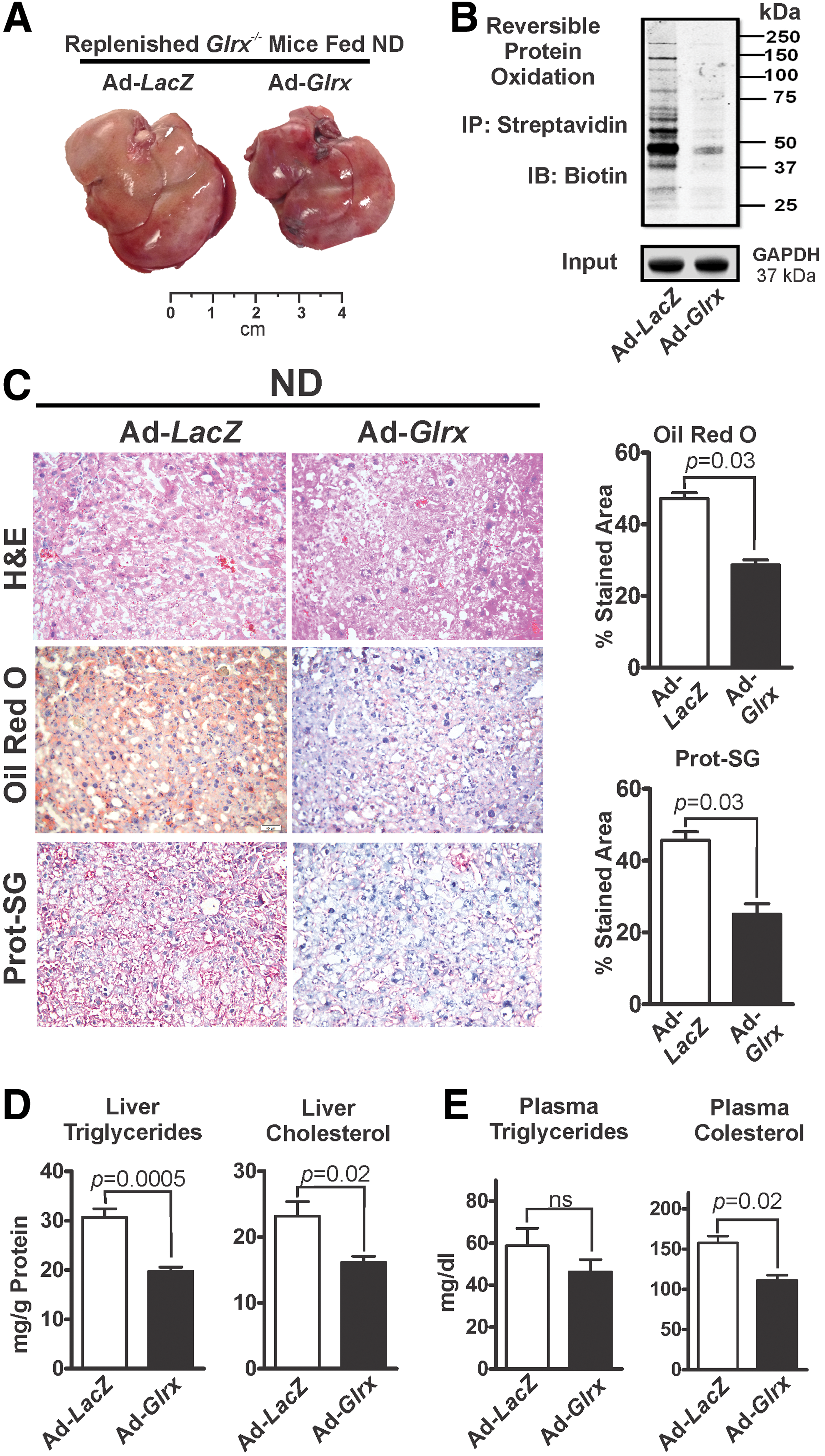
To evaluate the ability of hepatic Glrx to maintain lipid homeostasis, adenovirus-mediated gene repletion (8, 43) of Glrx or LacZ (control) was employed in Glrx−/− mice with hepatic steatosis at 8 months of age. Ten days postadenovirus injection, Western blot analysis confirmed repletion of the liver with Glrx (Supplementary Fig. S4). Glrx expression in other tissues was unaffected by the adenovirus (Supplementary Fig. S4).
Glrx-replenished Glrx−/− mice had significantly decreased levels of GSH adducts (Fig. 3C lower rows) and reversibly oxidized proteins (Fig. 3B and Supplementary Fig. S14), consistent with reacquired Glrx function. Strikingly, Glrx repletion for 10 days markedly diminished liver mass (Fig. 3A and Supplementary Fig. S5) and alleviated steatosis (Fig. 3C upper and middle panels, D) compared with LacZ-transduced mice. BW, however, remained unchanged (Supplementary Fig. S5). In addition, decreased plasma cholesterol level reflected the improved liver function in Glrx-replenished Glrx−/− mice (Fig. 3E). In summary, these data strongly support a critical role for hepatic Glrx in controlling liver and plasma lipids.
Glrx deficiency increases lipogenesis and cholesterol synthesis
Hepatic lipid metabolism is regulated by a delicate balance of anabolic and catabolic processes (9, 14, 21, 39, 40, 52, 70). Hence, we analyzed the expression of hepatic genes involved in lipid synthesis, uptake, degradation, and transport in WT and Glrx−/− mice fed ND at 8 months of age. Glrx−/− mice expressed significantly higher levels of fatty acid metabolism genes, including sterol regulatory element-binding transcription factor 1 (Srebf1), fatty acid synthase (Fasn), stearoyl-CoA desaturase (Scd1), and fatty acid translocase/CD36 (Cd36) (Fig. 4A). Although expression of acetyl-CoA carboxylase (ACC)—the rate-limiting enzyme in fatty acid synthesis producing the precursor malonyl CoA—was unchanged, Glrx−/− mice had lower levels of active dephosphorylated ACC. Active phosphorylated AMPK, which is upstream of ACC and controls its phosphorylation, was downregulated in Glrx −/− mice consistent with increased fatty acid synthesis (Supplementary Fig. S6).
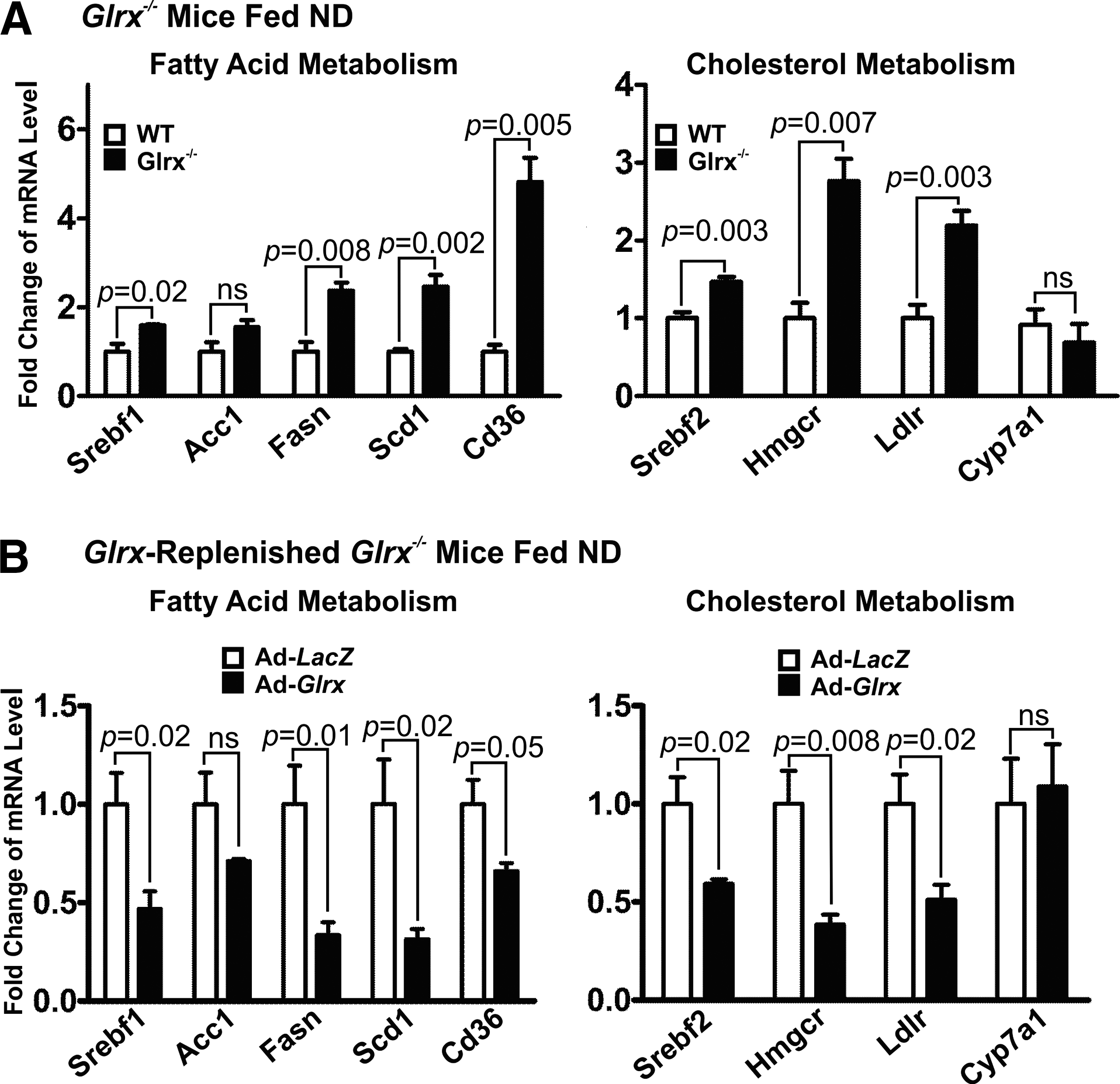
Glrx−/− mice also exhibited higher expression levels of genes related to cholesterol metabolism, including HMG-CoA reductase (Hmgcr), LDL-receptor (Ldlr), and sterol regulatory element-binding transcription factor 2 (Srebf2) (Fig. 4A). The Cytochrome P-450 7A1 (Cyp7a1), which lowers cholesterol by catalysis of the rate-limiting step in bile acid biosynthesis, remained unchanged in Glrx−/− mice (Fig. 4A).
However, Glrx−/− mice exhibited no expression changes of genes involved in hepatic fatty acid oxidation, including the mitochondrial fatty acid transporter carnitine plamitoyl-transferase (Cpt) 1a, peroxisome proliferator-activated receptor alpha (Ppara), acyl-coenzyme A dehydrogenase for mitochondrial β-oxidation (Acadm), and cytochrome P450 (Cyp) 4a10 for peroxisomal β-oxidation (Supplementary Fig. S7). Thus, hepatic lipid accumulation in Glrx−/− mice is unlikely increased to result from decreased lipid oxidation, but rather from de novo biosynthesis.
Expression of Glrx2—the mitochondrial Glrx isoform—and members of the thioredoxin system including thioredoxin (Trx) 1, 2 and thioredoxin-interacting protein (Txnip) (20) also remained unaltered in Glrx−/− mice (Supplementary Fig. S8) and likely did not contribute to the metabolic phenotype.
Consistent with our hypothesis that Glrx directly influences lipid metabolism, Glrx repletion consistently decreased the expression of all genes that were induced in the Glrx−/− mice livers (Fig. 4B), indicating a pivotal regulatory role of liver Glrx in lipid homeostasis.
Glrx deficiency induces hepatic steatosis through inhibition of SirT1 by reversible cysteine oxidation
SirT1, an NAD+-dependent class III histone deacetylase, has emerged as a central regulator of hepatic lipid metabolism (46, 60, 63, 67, 75), and we have recently described it can be modified by GSH adducts (67).
Glrx deficiency in mice markedly increased GSH adducts on hepatic SirT1 (Fig. 5A left panel and Supplementary Figs. 9A and S15), impairing the enzymes deacetylase activity (Fig. 5B left panel and Supplementary Fig. S16). Acetylation of p53 at lysine-379, a deacetylase substrate of SirT1 (30, 42, 48, 60, 72), was increased in livers of Glrx−/− mice, indicating the inhibition of SirT1 activity (Fig. 5C left panel and Supplementary Fig. S16). Consistent with this finding, Glrx deficiency in mice also increased acetylation of lysine-289 and 309 of SREBP1C, another SirT1 substrate that regulates transcription of fatty acid metabolism genes (60) (Supplementary Fig. S10A). Expression level of fatty acid synthase (FAS), which is a key enzyme in de novo lipogenesis and that is downregulated by active SirT1 via SREBP1C (60), was induced in livers of Glrx−/− mice (Fig. 5D left panel and Supplementary Fig. S17). Conversely, Glrx-replenished Glrx−/− mice showed decreased SirT1 GSH adducts (Fig. 5A right panel and Supplementary Figs. S9B and S15), increased SirT1 deacetylase activity (Fig. 5B right panel), decreased acetylated p53 (Fig. 5C right panel and Supplementary Fig. S16) and SREBP1C (Supplementary Fig. S10B), and diminished expression levels of FAS (Fig. 5D right panel and Supplementary Fig. S17). Of importance, neither Glrx gene deletion nor repletion altered hepatic SirT1 protein expression (Fig. 5B and Supplementary Fig. S15). Collectively, these data suggest that SirT1 is an important redox target of Glrx, and may mediate the effect of Glrx on lipid metabolism.
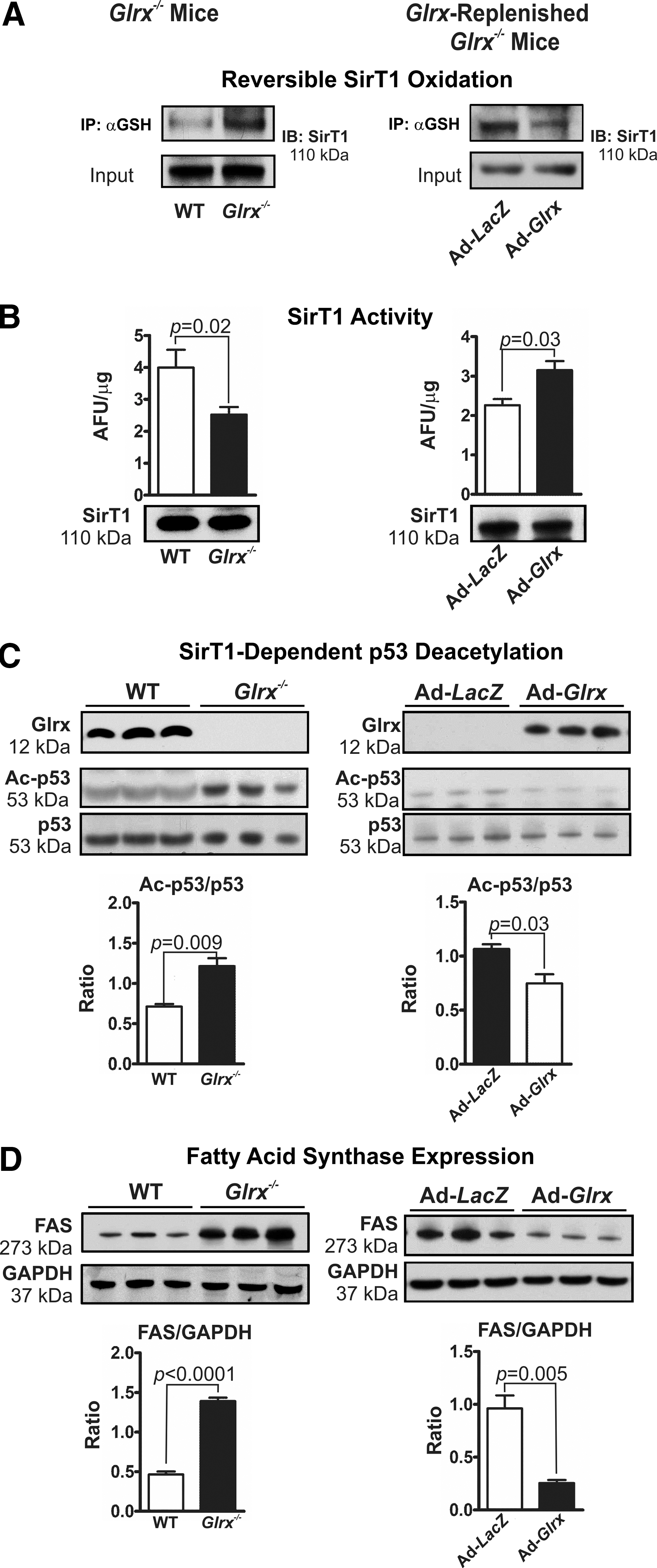
Glrx deficiency accumulates lipids through increased reversible oxidation of SirT1 cysteines
Employing HepG2 cells, a well-established human hepatocellular in vitro model, we further investigated molecular mechanisms by which Glrx regulates hepatic lipid metabolism. High-palmitate and high-glucose (HPHG) treatment increases intracellular oxidants (67) and subsequent protein cysteine oxidation (Fig. 6A and Supplementary Figs. S18 and S19). siRNA-mediated Glrx ablation exacerbated this effect. Consistent with the high-fat diet (HFD)-fed mouse model, Glrx depletion and HPHG treatment further increased reversible cysteine oxidation of SirT1 and acetylated-p53 (Fig. 6B and Supplementary Figs. S18 and S19), indicative of SirT1 inhibition. Transcriptional regulation of FAS, which is a major regulator of lipogenesis and is suppressed by active SirT1 (60), was measured with the FAS-promoter luciferase reporter assay. FAS-promoter activity increased in response to HPHG treatment and Glrx ablation (Fig. 6C left panel), leading to higher FAS protein expression (Fig. 6C right panel and Supplementary Figs. S18 and S19) and accumulation of lipids—triglycerides and cholesterol—in HepG2 cells (Fig. 6E).
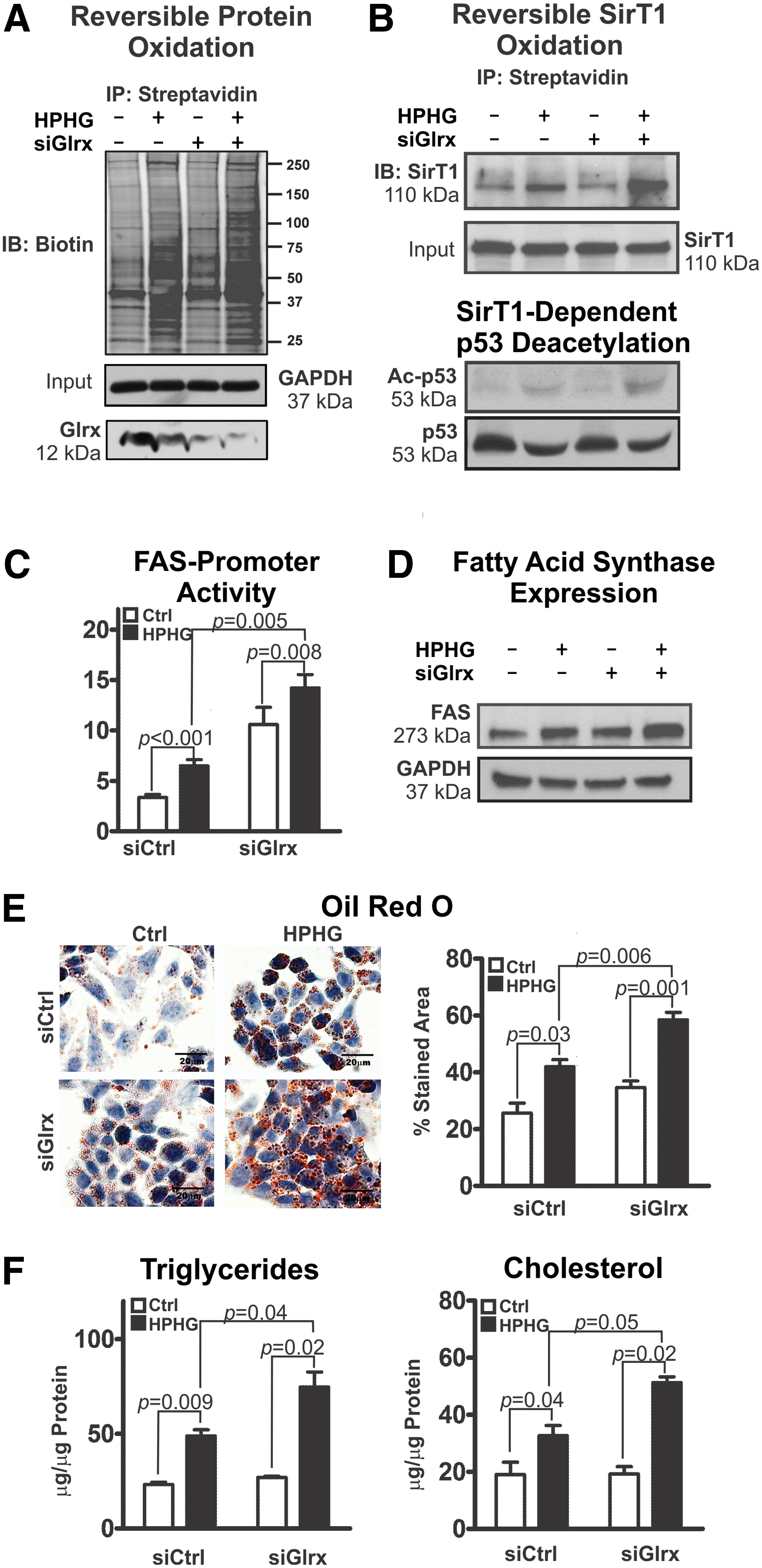
In previous work, we have created and characterized a nonoxidizable mutant SirT1 (Mut SirT1) (C61S+C318S+C613S), in which we replaced three essential cysteine residues by serine. Under oxidative and metabolic stress, mutant SirT1 maintains full activity and exhibits no reversible oxidative modifications (67). Overexpression of the mutant SirT1, as compared with WT, markedly attenuated lipid accumulation—triglycerides and cholesterol—in HepG2 cells under HPHG treatment and Glrx ablation (Fig. 6F). Adenoviral gene transfer of WT (Ad-SirT1), mutant SirT1 (Ad-Mut SirT1), or control (Ad-LacZ) into Glrx−/− mice at around 10 months of age (Supplementary Fig. S11D) was performed to compare the effects on lipid accumulation in vivo. Liver lipids (Supplementary Fig. S11A, B) and plasma cholesterol (Supplementary Fig. S11C) were significantly decreased in mutant SirT1-injected mice. Lipids in WT SirT1-injected mice also improved, but to a lesser degree, consistent with partial oxidative inhibition of WT SirT1. These data together indicate that Glrx through SirT1 also regulate liver lipid metabolism in vivo.
Glrx deficiency accelerates diet-induced NAFL
Our previous study (67) showed that HFD induced metabolic syndrome in mice and increased reversible cysteine oxidation of proteins in the liver. To determine whether elevated oxidative cysteine modifications caused by HFD can accelerate the pathogenesis of NAFL in Glrx−/− mice, 2 months old WT and Glrx−/− mice were fed HFD for 12 weeks (Supplementary Table S2). As expected, hepatic and plasma lipids (Fig. 7A–C) and reversible cysteine oxidation of hepatic proteins (Fig. 7D and Supplementary Figs. S18 and S19) were significantly increased in HFD-fed Glrx−/− mice.
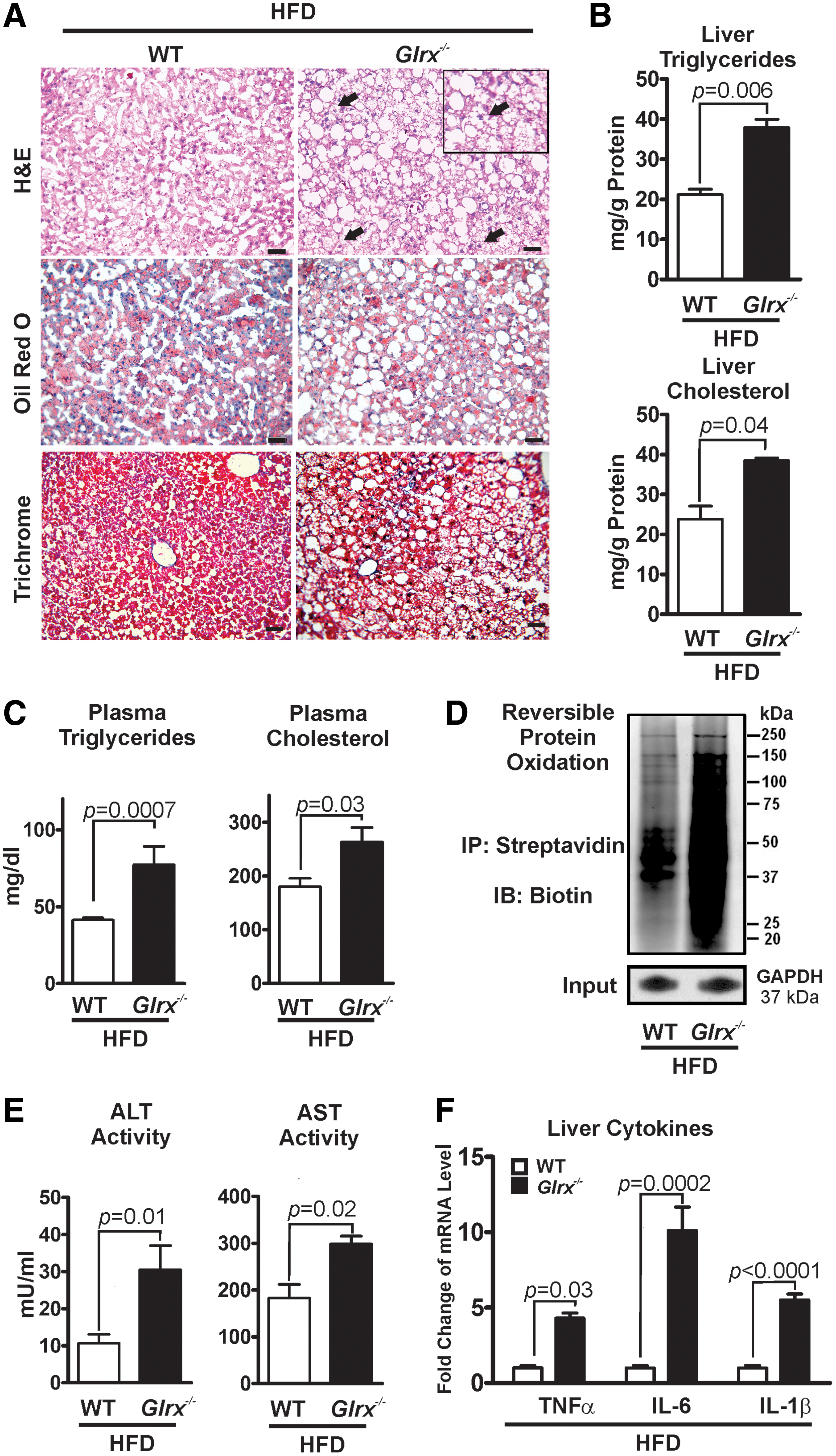
In contrast to the Glrx−/− mice fed ND (Fig. 2), the plasma levels of ALT and AST (Fig. 7E and Supplementary Table S2) as well as inflammatory cytokines were markedly induced (Fig. 7F). In addition, hepatocellular ballooning was observed (Fig. 7A right upper row), demonstrating an accelerated NAFL to NASH progression in HFD-fed Glrx−/− mice. To further investigate the effect of Glrx deficiency on fibrosis, liver sections were stained with Masson's trichrome, a marker of collagen deposition (Fig. 7A left lower row). Very mild hepatic fibrosis was detected with no significant difference between WT and Glrx−/− mice.
To investigate whether GSH adducts and Glrx level are related to NAFL, GSH adducts and Glrx expression levels were measured in liver biopsy sections. Patients diagnosed with hepatic steatosis (Supplementary Fig. S12) showed diminished Glrx protein expression and increased protein GSH adducts, although we need more samples to conclude significance in human liver.
Collectively, these data suggest that decreased Glrx level and accumulation of GSH adducts of hepatic proteins may contribute to the pathogenesis of NAFL, providing a rationale to increase Glrx expression in treating NAFL.
Discussion
Our results define a novel role of Glrx and protein GSH adducts in regulating hepatic lipid homeostasis.
Molecular mechanisms of NAFL
Various molecular mechanisms causing NAFL have been previously described (5). We investigated the pathogenesis-causing steatosis in Glrx−/− mice. Hepatic lipid content, including cholesterol and fatty acids, is controlled by a delicate balance between lipid uptake, synthesis, degradation, and excretion (18, 38, 41). In many cases, hepatic lipogenesis and cholesterol synthesis greatly contribute to liver steatosis (61). However, diminished mitochondrial lipid uptake by inhibition of the long chain fatty acid transporter Cpt1a (6) or attenuated hydrolysis of tryglycerides by adipose triglyceride lipase also causes hepatic steatosis (28, 29).
Perturbations in hepatic lipid metabolism, as demonstrated for Glrx−/− mice, can severely affect plasma lipids and is a risk factor for atherosclerosis and cardiovascular disease (2, 53, 69).
Glrx−/− mice increased and liver-specific Glrx gene repletion-corrected mRNA expression levels of all three rate-limiting enzymes: fatty acid synthase, acyl-CoA desaturase (monounsaturated fatty acids), and hydroxy-methylglutaryl-CoA reductase (cholesterol) (Fig. 4A, B). The transcription factors, SREBP 1c for fatty acids and SREBP 2 for cholesterol, respectively, regulate the expression of these enzymes. Both transcription factors are associated with NAFL (12, 18, 23, 50) and were upregulated in Glrx−/− mice. Importantly, SirT1 also regulates SREBPs (56, 63, 75). Activation or overexpression of SirT1 can alleviate diet-induced NAFL (35, 46, 56) through downregulation of SREBP1 (56). Conversely, hepatocyte-specific deletion of SirT1 upregulated SREBPs, hydroxy-methylglutaryl-CoA reductase, fatty acid synthase, acyl-CoA desaturase, and induced weight gain and hepatic steatosis in mice (46, 73).
We have previously shown that metabolic or nitrosative stress increases SirT1 reversible oxidative modification, which inhibits its enzyme activity and promotes hepatocyte apoptosis (67). Consistent with these findings, here we demonstrated that Glrx deficiency decreases SirT1 activity by GSH adducts and consequently increases acetylation of Srebp1 and expression of lipid synthesis genes, including Fasn and Scd1 in the liver (Fig. 4A). Importantly, Glrx gene repletion restored liver SirT1 activity, decreased Srebp1 acetylation, suppressed the downstream genes, and ameliorated the fatty liver phenotype (Figs. 3 and 4B). Meanwhile, metabolic pathways for degrading hepatic lipids including fatty acid β-oxidation (Supplementary Fig. S7) were unaltered at mRNA levels in Glrx−/− mice.
Because plasma cholesterol was elevated in Glrx −/− mice, we measured hepatic-free cholesterol (12, 49, 62) and expression of enzymes involved in the classic (cytoplasmic) or “alternative” (acidic) mitochondrial pathway of bile acid formation (27, 55). Expression of microsomal cholesterol 7α-hydroxylase (CYP7A1) (27), the key enzyme of classic bile acid formation, was unchanged (Fig. 4). The alternative bile acid synthesis pathway in hepatocytes utilizes the mitochondrial transporter “steroidogenic acute regulatory protein” to transport cholesterol into mitochondria, which is then metabolized by cytochrome p450 sterol 27-hydroxylase (CYP27). Expression of both enzymes was unaltered. Thus, lipid uptake as shown by increased CD36 and LDL receptor expression and de novo cholesterol synthesis are likely to play a major role in Glrx −/− mice.
The glutaredoxin system may affect the thioredoxin system as demonstrated in Escherichia coli. Ablation of Txnip or Trx reductase-1 (TrxR1) in mice caused altered lipid metabolism (20, 36). However, gene expression of the thioredoxin system was unaltered in Glrx −/− mice. Furthermore, the mitochondrial isoform glutaredoxin-2, which is exclusively mitochondrial matrix localized (54) and controls iron–sulfur clusters (24) as well as mitochondrial protein GSH adducts (19), showed no changes in gene expression.
Upregulation of SirT1 deacetylase activity could mediate the beneficial metabolic effects of caloric restriction through rising cosubstrate NAD+ levels (47). Conversely, decreased SirT1 activity may not necessarily reflect changes of cellular NAD+ concentration (22, 26). We also found that hepatic NAD+ concentrations of Glrx−/− mice were similar to those of WT mice (Supplementary Fig. S13).
The two hit hypothesis
NAFL encompasses a spectrum of liver pathology that includes hepatic steatosis, inflammatory NASH, and fibrosis. The progression of NAFL is delineated by the “two-hit hypothesis” (5, 15, 16). The “first hit” causes hepatic lipid accumulation and leaves stressed hepatocytes susceptible to injury. The “second hit” advances hepatic steatosis to NASH by induction of inflammation and oxidative stress, causing hepatocyte damage and death (10, 56, 59, 66). NASH can occur with or without fibrosis and then progress to end-stage liver disease, cirrhosis, and hepatocellular carcinoma (13). Damaged hepatocytes release specific enzymes such as ALT and AST into the blood. Therefore, NASH diagnostics require measurement of plasma biomarkers, at times in combination with histological assessment of liver tissue.
In this study, we found that Glrx deficiency increases hepatic lipid content without causing damage to hepatocytes that otherwise would have resulted in inflammation and release of liver enzymes into the blood. Thus, Glrx deficiency results in a “first hit” causing hepatic steatosis and plasma dyslipidemia by SirT1-dependent upregulation of de novo fatty acid and cholesterol synthesis. We tested whether feeding a HFD for 3 months to Glrx−/− mice would aggravate NAFL as a “second hit.” HFD-fed Glrx−/− mice exhibited signs of tissue damage, resulting in elevated plasma levels of ALT and AST, as well as hepatitis associated with increased inflammatory cytokines and hepatocellular ballooning (Fig. 7). Increased reversible oxidative modifications, mainly GSH adducts, coincided with HFD feeding and Glrx deficiency further augmented them. Thus, these experiments suggest that HFD in Glrx−/− mice aggravates NAFL to NASH by causing a greater increase in GSH adducts induced by metabolic stress compared with HFD-fed WT mice (Fig. 7), which may further impair SirT1 function.
Furthermore, increased mitochondrial cholesterol can promote inflammation by mitochondrial GSH depletion and sensitize tumor necrosis factor and FAS signaling (44). Glrx −/− mice fed HFD, in particular, may have increased mitochondrial cholesterol levels and thus show increased inflammation and progression to NASH.
Role of Glrx in liver
Our studies used global Glrx −/− mice, therefore, Glrx ablation in other tissues was a concern. Using an adenovirus transgene coding for Glrx, we selectively replenished the liver of Glrx−/− mice to test whether hepatic Glrx deficiency directly caused steatosis. Surprisingly, replenished Glrx expression nearly normalized liver weight, hepatic lipids, and plasma lipid content after only 10 days. Thus, hepatic Glrx deficiency, and no other systemic metabolic abnormality, causes steatosis. Furthermore, liver biopsies from patients diagnosed with NAFL showed first evidence of decreased Glrx expression and increased protein GSH adducts. This finding is supported by a previous study of Piemonte et al. that observed increased protein GSH adducts in children with NAFL using the same monoclonal antibody (58). Glrx in other tissues such as adipose tissue, however, may also have effects on metabolism and requires further investigation.
Conversely, overexpression of Glrx protected the heart from diabetic complications (44) and preserved function of high-glucose exposed cells (45). Glrx also improves insulin secretion of beta cells (65). Thus, overexpression or activation of liver Glrx could be a strategy to normalize hepatic and plasma lipid metabolism.
In conclusion, our findings indicate that reversible thiol modification in the liver is a major mechanism of lipid metabolism and the development of NAFL. Hepatic Glrx controls lipid homeostasis by regulating protein GSH adducts and specifically those on SirT1, which regulate its activity and downstream lipid regulators. Our results assign a novel role for Glrx and Glrx-mediated regulation of reversible thiol modifications in lipid homeostasis and protection from hepatic steatosis. This provides a new clue into the molecular mechanisms underlying NAFL and opens the possibility for new modes of therapy.
Materials and Methods
Reagents, materials, and antibodies
N-(biotinoyl)-N′-iodoacetyl ethylenediamine (BIAM, B-1591), Zeba™ spin desalting columns (40K MWCO, 87767), Lipofectamine™, and cell culture media were obtained from Life Technologies (Grand Island, NY). Anti-SirT1 mouse monoclonal antibody (ab110304) was from Abcam (Cambridge, MA) 1:5000 dilution for Western blot. Antiacetylated p53 (K382) rabbit polyclonal (#2525), antiacetylated p53 (K379) rabbit polyclonal (#2570), and antibiotin HRP-linked goat antibody (#7075) were from Cell Signaling (Danvers, MA), 1:1000 dilution for Western blot. Antitotal p53 (sc-126) mouse monoclonal, anti-SREBP-1C (k-10 [rabbit polyclonal], 2A4 [mouse monoclonal], and H160 [rabbit polyclonal]) antibodies were from Santa Cruz (Dallas, TX), 1:1000 dilution for Western blot. Anti-Glrx rabbit polyclonal antibody was custom ordered by Bethyl Laboratories (Montgomery, TX), 1:1000 dilution for Western blot. Anti-GSH mouse monoclonal antibody (101-A-100) was from Virogen, 1:200 dilution for immunostaining. For details on antibodies and working dilutions please refer to Supplementary Table S3. The luciferase assay kit was obtained from Promega (Madison, WI). Fluor-de-Lys™ SirT1 activity assay was from Enzo Life Sciences (Farmingdale, NY). Polyvinylidene fluoride membrane, polyacrylamide electrophoresis gels, and other reagents for immunoblotting were obtained from Bio-Rad (Hercules, CA). Western blots were corrected for brightness and contrast. The “Precision Plus Protein Standards—All Blue” were used as molecular mass maker for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (Cat #161-0373; Bio-Rad, Hercules, CA). Western blots were either developed using ECL or the Odyseey infrared scanner (LI-Cor, NE) equipped with two IR channels—700 and 800 nm—as previously published (67). The 700 nm channels visualized the molecular mass marker that was superimposed over the 800 nm channel. Both channels are provided as supplemental information.
Experimental animals
Glrx −/− mice were originally generated by Dr. Y.S. Ho (Wayne State University, Detroit, MI) (18), and backcrossed to C57BL/6NJ background in Dr. Janssen-Heininger's laboratory (University of Vermont). Male mice were used for all experiments. The mouse colony has been maintained in the animal facility at Boston University Medical Campus. For metabolic characterization, Glrx −/− mice and WT littermates were fed ND (4.5% fat, 0.02% cholesterol by weight).
To investigate the effects of metabolic stress, a cohort of 2 months old Glrx−/− mice and WT littermates were fed a HFD (21% fat representing 42% calories, 34% sucrose, and 0.2% cholesterol, TD.88137; Harlan, South Easton, MA) for 3 months. Mice were housed in rooms with 12 h light–dark cycle and in groups of 3–4 whenever possible. The protocol was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine.
Metabolic phenotyping
The mouse body composition including fat mass, lean tissue mass, free water, and total body water was assessed with noninvasive quantitative magnetic resonance in an EchoMRI700 instrument. Values are expressed as a percentage of BW. All studies were performed at the Boston University Metabolic Phenotyping Core.
Homogenization and protein extraction from liver tissue—homogenization and extraction of individual liver pieces were carried out in NP-40 lysis buffer containing 50 mM Tris, 150 mM NaCl, 1 mM EDTA, and protease inhibitor cocktail (Roche Applied Science) at pH 7.4.
Cell culture and treatments
HepG2 cells (ATCC, Manassas, VA) were maintained in DMEM containing 10% FBS and penicillin/streptomycin (Gibco, Grand Island, NY). Transfected cells were treated with control medium containing 5 mM glucose and 0.67% bovine serum albumin (BSA, fatty acid free; Sigma-Aldrich St. Louis, MO) or medium in HPHG (25 mM glucose, 0.4 mM palmitic acid, and 0.67% BSA) for 16 h.
Glrx knockdown in HepG2 cells was achieved using on-target plus siRNA (Dharmacon, Lafayette, CO).
ShRNA lentivirial vector against human SirT1 (RHS4533-EG23411, Dharmacon, Lafayette, CO) was packed into lentiviral particles following manufacturers protocol. In brief, 293T cells were transfected with pLKO-shSirT1 or scrambled control pLKO-pGL2 together with the packaging plasmids encoding Δ8.9 and VSV-G. Supernatants containing lentiviral shRNA against SirT1 were collected 48 h post-transfection. HepG2 cells were incubated with collected medium containing lentiviral particles coding for shSirT1. A stable SirT1 knockdown HepG2 cell line was generated by selection with puromycin (2 μg/ml).
Fasn-promoter luciferase reporter
The luciferase reporter vector containing the promoter region of the human Fasn gene was obtained from Addgene (#8890) (Cambridge, MA). Luciferase activity was measured 24–48 h post-transfection in HepG2 cells according to the manufacturer's protocol using a TECAN Infinite M1000 Pro Microplate Reader (TECAN, San Jose, CA).
SirT1 activity measurement
SirT1 activity was tested by Fluor-de-Lys assay. Then 90 μl of 30 μg of nuclear extraction from mouse liver was incubated with 100 μM acetylated p53 peptide (Arg-His-Lys-Lys[Ac]-AMC) for 30 min at 37°C with 100 μM NAD+ in activity assay buffer (50 mM Tris-HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, pH 8.0). Then 100 μl of 1 mg/ml concentrated trypsin solution was added to release the AMC fluorophore, which allows quantification of the amount of substrate deacetylated by SirT1. The fluorescence intensity was recorded over 60 min using a Fluoroscan Ascent microplate reader (Thermo Fisher, Cambridge, MA) with excitation set to 375 nm and emission to 460 nm.
Biotin-switch assay for labeling of reversibly oxidized cysteines
Labeling with N-(biotinoyl)-N′-iodoacetyl ethylenediamine was used in a biotin-switch assay to detect reversibly oxidized cysteines. Cells were lysed in lysis buffer containing 100 mM maleimide. Excess maleimide was removed by passing the lysates over Zeba spin columns. Lysates were incubated with 5 mM DTT for 1 h and reduced cysteines were labeled with 1 mM N-(biotinoyl)-N′-iodoacetyl ethylenediamine for 1 h. Streptavidin beads were added into the lysates and beads were boiled in 30 μl of 2 × reducing Laemmli buffer and loaded on an SDS Tris-glycine gel. SirT1 was detected by immunoblotting with a total SirT1 antibody (Santa Cruz). Reversible cysteine modifications were detected by antibiotin antibody (Cell signaling).
Liver histology and analysis
For H&E staining, liver tissue was fixed in 4% phosphate-buffered formalin, embedded in paraffin, and cut into 5 μm sections. For Oil Red O staining, livers were embedded in optimal cutting temperature compound, cut into 5 μm cryosections, and stained with Oil Red O. Slides were mounted with aqueous mountant. For Masson's trichrome staining, 5 μm liver sections were stained to assess the hepatic collagen deposition (fibrosis). For immunostaining of Glrx, liver tissue was fixed in 4% phosphate-buffered formalin, embedded in paraffin, and cut into 5 μm sections. GSH adducts staining method of liver sections was also performed as previously described (31, 67).
Tissue and plasma biochemical measurements
Three hundred microliters of liver homogenate was extracted with 5 ml of chloroform–methanol (2:1) and 0.5 ml of 0.1% sulfuric acid (55). An aliquot of the organic phase was collected, dried under nitrogen, and resuspended in 2% Triton X-100. Hepatic triglycerides and cholesterol and plasma triglycerides were measured using the infinity triglycerides and total cholesterol reagent kit (TR13421, TR-22421) (Thermo Fisher). Hepatic lipid contents were normalized for differences in protein concentration. Plasma HDL, LDL/VLDL cholesterol was measured using HDL and LDL/VLDL cholesterol assay kit (ab65390) (Abcam). Plasma alanine (ALT) and AST were detected using ALT and AST activity assay kits (K752, K753) (BioVision, San Francisco, CA). NAD NAD+/NADH ratio in tissues was measured using an assay kit (ab65348) (Abcam) according to the manufacturer's instructions. Tissue GSH and GSSG levels were measured using a modified HPLC-based method as established by Reed et al. (37, 64).
Quantitative reverse transcriptase–polymerase chain reaction
Total RNA was isolated from tissues or cells using TRIzol™ reagent and cDNA generated utilizing High Capacity RNA-to-cDNA kit. Quantitative PCR was conducted using inventory gene-specific TaqMan™ primers (Life Technologies): Fasn (Mm00662319_m1), Acc1 (Mm01304257_m1), Scd1 (Mm00772290_m1), Srebf1 (Mm00550338_m1), Cd36 (Mm01135198_m1), Hmgcr (Mm01282499_m1), Srebf2 (Mm01306292_m1), Ldlr (Mm01177349_m1), Cyp7a1 (Mm00484150_m1), Cpt1a (Mm01231183_m1), Acadm (Mm01323360_g1), Ppara (Mm00440939_m1), Cyp4a10 (Mm01188913_g1), Tnfa (Mm00443258_m1), Il1b (Mm00434228_m1), Il6 (Mm00446190_m1), Glrx (Mm00728386_m1), Trx-1 (Mm00726847_s1), Trx-2 (Mm00444931_m1), Txnip (Mm01265659_g1), Glrx-2 (Mm00469836_m1), and Actb (Mm00607939_s1) (Supplementary Table S4). Expression was obtained and analyzed using comparative Ct (ΔΔCT) with StepOne™ quantitative real-time PCR software (Applied Biosystems, Grand Island, NY), normalized to β-actin.
Liver biopsies
We conducted a pilot investigation aimed at determining reversible oxidative protein modifications in liver biopsies of patients with NAFL disease. The study population included two groups: normal liver histology and nonalcoholic hepatic steatosis, all obtained through the Boston University Biospecimen Archive Research Core (BARC). Each of the two groups consisted of three individual patient samples. A single pathologist with specialized training in liver histology reviewed all samples and confirmed the diagnoses in previously specified groups. The Boston University School of Medicine Institutional Review Board (IRB) reviewed the study protocol as “IRB exempt.” All patient studies were conducted in compliance with the principles of the “Declaration of Helsinki.”
Statistical analysis
Statistical analysis was performed using Prism 6.0 (GraphPad Software). Means were compared between two groups by the Mann–Whitney U test. Mann–Whitney U test with Dunn's post-test, paired-test was used in small number animal experiments. Multiple comparisons were conducted with ANOVA. A p value of <0.05 was considered statistically significant.
Footnotes
Acknowledgments
This work was supported by NIH grants P01 HL068758, R37 HL104017, R01 DK076942, and R01 DK103750, R01 HL133013, R01 HL115955, NIH CTSI award 1UL1TR001430, NHLBI, National Institutes of Health, Department of Health and Human Services, under contract Nos. HHSN268201000031C and N01-HV-00239, American Heart Association “Grant in Aid” 16GRNT27660006, European Cooperation in Science and Technology (COST Action BM1203/EU-ROS), and the Metabolic Clinical Research Collaborative. The article contents are solely the responsibility of the authors and do not necessarily represent the official views of the awarding offices. D.S. was supported by an American Heart Association Scientist Postdoctoral Fellowship Award 15POST21790006. J.H. was supported by an NRSA grant T32 HL70024 through the Whitaker Cardiovascular Institute postdoctoral training grant program, 1UL1TR001430 (BU CTSI), and an American Heart Association Scientist Development Grant 14SDG20140036. This work was supported by a Strategic Alliance with Institut de Recherche Servier. M.M.B. was supported by the Evans Junior Faculty Research Award by the Department of Medicine of Boston University. We thank Drs. M. Zang, M. Kirber, T. Balon, and L. Deng and the Boston University School of Medicine “Analytical Instrumentation,” “Immunohistochemistry,” “Cellular Imaging,” and “Metabolic Phenotyping” Cores for their technical support.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.