
Abstract
Introduction
I
Peroxisomes originally designated as fossil organelles have attracted growing attention and are nowadays recognized as essential players under physiological conditions and in disease.
Mammalian peroxisomes are 0.1–0.5-μm-sized single-membrane organelles present in the majority of eukaryotic cells with the highest abundance in the liver and the kidney. The evolutionary origin of peroxisomes is still a matter of discussion (30). One of the recent theories postulates that the evolution of peroxisomes was a consequence of raising oxygen concentration in the archaic atmosphere (16). In contrast to mitochondria, peroxisomes are not remnants of endosymbiotic organisms, but rather have developed from an endomembrane compartment, most likely from specialized parts of the endoplasmic reticulum (91).
Similar to the evolutionary concept, the origin of peroxisomes (biogenesis) at the subcellular level is not completely resolved. The classical view of peroxisome biogenesis emphasizes the formation of new organelles from pre-existing peroxisomes through growth and division (57). However, several recent studies have demonstrated that peroxisomes can also arise de novo in a maturation process employing preperoxisomal vesicles budding from the endoplasmic reticulum (75).
Peroxisomes have a protein-rich fluid matrix that contains numerous enzymes. Peroxisomes possess specific proteins called peroxins that are encoded by corresponding PEX genes (82). More than 30 different peroxins have been described so far. These proteins regulate peroxisomal proliferation, division, movement, import, and participate in degradation of aged or damaged organelles. Peroxisomes do not have DNA and must import proteins from the cytoplasm. Newly synthesized proteins are transported to peroxisomes by specific transport peroxins. These cytosolic protein transporters recognize a distinct amino acid sequence present on proteins predestined for peroxisomes, the so-called peroxisomal targeting signal (PTS) (11). Peroxisomal targeting signal 1 (PTS1) is a carboxy-terminal serine-lysine-leucine (SKL) tripeptide sequence recognized by PEX5 and represents the main PTS for peroxisomal matrix proteins. Peroxisomal targeting signal 2 (PTS2) is an amino-terminal 11 amino acid sequence present on a small number of peroxisomal matrix proteins. Peroxisomal membrane proteins are tagged by a membrane-targeting signal (mPTS) that has not been explored in such detail as PTS for the matrix proteins.
The exact steps of recognition, binding, and import of peroxisomal proteins from cytosol to peroxisomes are far too complex to describe in its entirety here. For more information, please refer to an excellent review by Rucktäschel et al. (88). This review will briefly describe the relevant molecular mechanisms involved in protein import into peroxisomes. The import of peroxisomal matrix proteins can be divided into four consecutive steps (49). (i) The cytosolic receptors (peroxins) recognize peroxisomal matrix proteins located after synthesis in the cytoplasm via PTS. Peroxin 5 (PEX5) recognizes and binds PTS1-containing proteins and peroxin 7 (PEX7) PTS2-containing proteins in the cytosol. The protein–receptor complexes are transported to the peroxisomal membrane (ii) where they dock to peroxin 14 (PEX14). Then, the protein–receptor complex is integrated into (iii) and subsequently translocated across the peroxisomal membrane. (iv) After the cargo release within the peroxisomal matrix, PEX5 or PEX7 is detached and exported back to cytosol (Fig. 1) (78). The exact mechanisms of protein translocation across the peroxisomal membrane are not clear. One of the current theories assumes the formation of a transient membrane pore through the interaction of PTS receptor PEX5 with the docking peroxin PEX14 (68).

Peroxisomal membrane proteins are recognized and bound by cytosolic peroxin 19 (PEX19) via a specific mPTS. PEX19 interacts with peroxisomal membrane-anchored PEX3, which is followed by release and incorporation of the transported proteins into the membrane. Then, PEX19 is exported back to the cytoplasm (87) (Fig. 1). A small group of membrane proteins lacks the mPTS sequence recognized by PEX19. These proteins are transported to peroxisomes by a vesicle-mediated mechanism via the endoplasmic reticulum (67).
Peroxisomes are surprisingly versatile organelles that accomplish a multitude of biological tasks. Their comprehensive metabolic apparatus encompasses more than 50 enzymes. The numerous metabolic tasks require reliable substrate and cofactor transport systems, in particular for the two most important peroxisomal tasks, the β-oxidation of fatty acids (FAO) and H2O2 metabolism (77).
The peroxisomal membrane is permeable to small metabolites up to 500 Da in size. The internalization of larger molecules, such as fatty acids, adenosine triphosphate (ATP), or coenzyme A, requires specific receptors. Several membrane-incorporated ATP-binding cassette (ABC) transporters facilitate the import of fatty acids processed by peroxisomal FAO (104). Three ABC transporters belonging to subfamily D have been identified in mammalian peroxisomes (73). ABCD3, also known as peroxisomal membrane protein 70 (PMP70), is experimentally used as a structural marker of peroxisomes (86).
Contribution of peroxisomes to the pathogenesis of human diseases has been widely accepted since the discovery of disorders caused by defective peroxisomal biogenesis and/or dysfunction of specific peroxisomal enzymes. Accumulating evidence indicates that peroxisomes are actively involved in apoptosis and inflammation (112), innate immunity (21), aging, and in the pathogenesis of age-related diseases, such as diabetes mellitus and cancer (27).
Peroxisomal Fatty Acid Oxidation
Renal cortex and particularly proximal tubules are heavily dependent on FAO as the major source of energy for numerous transport systems localized in this part of the nephron (58). FAO is exclusive to peroxisomes in prokaryotic organisms. In eukaryotes and particularly in mammals, peroxisomes cooperate closely with mitochondria and form a strategic partnership in cell energy metabolism (6). Mitochondria preferentially oxidize short and medium-chain fatty acids, whereas peroxisomes metabolize very long-chain fatty acids (VLCFA) with more than 22 carbons (69). Peroxisomal enzymes shorten the long chain of VLCFA, which are subsequently transported to mitochondria and oxidized to acetyl-CoA (83). Since peroxisomes do not contain respiratory chain enzymes, peroxisomal FAO is not directly coupled to generation of ATP (indirectly through anaplerotic reactions for mitochondria) and most of the energy is released as heat. Peroxisomal FAO is unique due to the generation of H2O2, a by-product of the oxidative reactions. Despite the functional interconnection between peroxisomes and mitochondria, FAO enzymes differ between these two organelles. The first step in peroxisomal FAO is catalyzed by acyl-CoA oxidase (ACOX), followed by reactions catalyzed by bifunctional enzyme and 3-ketoacyl-CoA thiolase (37, 43). Dysfunction of peroxisomal enzymes, in particular of the rate-limiting ACOX, results in accumulation of unmetabolized fatty acids with potentially harmful consequences for the cell. Peroxisomes can perform different metabolic functions, depending on cell type and environment. A short overview of peroxisomal metabolic tasks is presented in Table 1 [adapted from Smith and Aitchison (94)]. In addition to FAO, peroxisomes can α- and β-oxidize a wide range of natural and xenobiotic compounds containing the fatty acyl chain. Peroxisomes play a crucial role in the biosynthesis of ether phospholipids (plasmalogens) that are abundant in the nervous system. Peroxisomal participation in the metabolism of arachidonic acid advocates in favor of their role in cell signaling and apoptotic pathways (70).
Peroxisome proliferator-activated receptor alpha (PPARα) is the principal regulator of activation and proliferation of peroxisomes. PPARα is stimulated by hypolipidemic drugs (fibrates) and by naturally occurring ligands of the arachidonic cascade, such as leukotrienes. Upon activation, PPARα translocates into the nucleus and binds to a specific DNA sequence called peroxisome proliferator-activated receptor (PPAR) response element located in the promoter region of genes encoding peroxisomal (and mitochondrial) FAO enzymes (92). PPARα knockout mice have a normal number of peroxisomes, but an abolished proliferation after stimulation with PPARα ligands (59). In recent years, several PPARα-independent activators of peroxisomes, such as PPAR-gamma coactivator (PGC-1α) or sirtuin 1 (SIRT1), have been reported in the literature (3, 34).
Peroxisomal Metabolism of Reactive Oxygen Species
Peroxisomal oxidases generate excessive amount of H2O2 and require reliable scavenger mechanisms. This is exemplified by the highest cellular concentration of catalase in peroxisomes. The dual function of peroxisomes in metabolism of H2O2 predestines them for a decisive role in the precisely regulated redox homeostasis. Dysfunction of peroxisomal antioxidant enzymes may result in H2O2 leakage into the cytoplasm with harmful consequences for the cell. There is increasing evidence on the close connection between peroxisomal, mitochondrial, and cellular redox homeostasis. The inactivation of peroxisomal catalase causes alterations of mitochondrial membrane potential and stimulates generation of mitochondrial reactive oxygen species (ROS) (51). Koepke et al. demonstrated progeric effects of catalase dysfunction (56). Cellular aging is associated with peroxisome senescence and decreased import of catalase. This study also demonstrated that restoration of peroxisomal catalase in aging cells improves the overall redox balance and reduces cellular senescence.
It is important to mention that low (hormetic) concentrations of ROS function as mediators of cell signaling. Low hormetic levels of H2O2 stimulate antiaging pathways (e.g., activation of autophagy responsible for cell surveillance and removal of aged organelles). High concentrations of H2O2 exacerbate organellar and cellular aging (90). Recalling the close link between peroxisomal and mitochondrial metabolism, it becomes evident that the peroxisome–mitochondria partnership must have a major role in cellular metabolism and functions. The previously held rigid view of mitochondria as the exclusive generator of ROS has been challenged by new findings coming from studies on peroxisomes. Further support for their communication and cooperation is furnished by the evidence that both organelles share the components of their division machinery (92). Taken together, peroxisomal disturbances may result in significant, and under certain circumstances, fatal consequences for individual cells, organs, or even the whole organism, as demonstrated in primary peroxisomal disorders.
Peroxisomal Disorders and Their Animal Models
Hereditary peroxisomal disorders cause devastating human diseases. In general, inborn peroxisomal defects can be categorized in (i) peroxisomal biogenesis disorders and (ii) peroxisomal disorders caused by defects of peroxisomal enzymes or transporters. The common metabolic feature observed in peroxisomal diseases is the accumulation of VLCFA normally oxidized in peroxisomes. (i) Zellweger syndrome (cerebrohepatorenal syndrome) is the prototypical peroxisomal biogenesis disease caused by mutations in PEX genes encoding specific peroxins. Depending on the mutation, peroxisomes may be completely absent or present as empty ghost peroxisomes with severely impaired import functions. (ii) Single enzyme deficiencies or transporter defects cause peroxisomal disorders characterized by accumulation of specific metabolic substrate (e.g., phytanic acid in Refsum disease). Patients with peroxisomal enzyme or transporter disorders have structurally intact, but functionally impaired, organelles. A recent review by Braverman provides more detailed and updated information on primary peroxisomal disorders (9).
There is increasing evidence that minor and/or temporary perturbations of peroxisomal functions may be involved in the pathogenesis of common human diseases. Peroxisomal dysfunction has been reported in the molecular pathogenesis of Parkinson's and Alzheimer's diseases (48, 108). Peroxisomes play an important role in human aging and in the pathogenesis of age-related diseases (27). The emerging evidence that human immunodeficiency virus and influenza virus are exploiting peroxisomes for replicative purposes may provide novel targets for therapeutic intervention against viral infections (76).
The majority of the data on peroxisomes originated in studies of peroxisomal mutants generated in different yeasts. There is no naturally occurring animal model of a peroxisomal disorder. Baes created the first experimental mouse model for PEX5 in 1997 (2). At present, animal models with deficiency of PEX5, PEX7, PEX11, and PEX13 are available. The common problems observed in these animals are severe neurodevelopmental disturbances caused by widespread tissue accumulation of VLCFA. The high perinatal and postnatal mortality in peroxin knockout models precludes their use for investigating the pathogenesis of temporary and minor alterations of peroxisomal functions. The implementation of conditional knockouts with elimination of peroxisomes in specific tissues and at a specific developmental stage may overcome these hurdles in the future (100). The lack of natural models for specific peroxisomal defects also implicates rather subtle and/or temporary alterations of peroxisomes in the pathogenesis of common disorders.
Peroxisomal Homeostasis and Autophagy
Cells regulate the number of organelles through continual interplay between biogenesis and degradation. This constant turnover is essential for preservation of cellular and organellar competence. Experimental studies demonstrated that peroxisomes have a half-life of circa 48 h under basal conditions and are primarily degraded by autophagy (44). Autophagy is a highly conserved lysosome-dependent pathway that maintains the quality of cellular components through continual removal of redundant or damaged proteins or entire organelles (72). The molecular basis of autophagy-mediated degradation of peroxisomes called pexophagy has been extensively studied in yeast and reviewed in (97). Contrary to yeast, the molecular details of mammalian pexophagy are not completely understood.
There are three independent systems for clearance of peroxisomes: (i) Lon protease, (ii) 15-lipooxygenase-mediated lysis, and (iii) pexophagy (autophagy of peroxisomes) (110). Lon protease is an ATP-dependent enzyme that catalyzes proteolytic digestion of peroxisomal proteins and accounts for removal of about 20% of peroxisomes. Activation of 15-lipooxygenase leads to disruption of the peroxisomal membrane; however, this mechanism eliminates only a small fraction of peroxisomes (111). The most important pathway for removal of peroxisomes is autophagy. Activation of autophagy in yeast is primarily a metabolic adaptation to changed environment to maintain nutritive balance under starvation. In mammalian cells, autophagy regulates and maintains homeostasis by eliminating aged, damaged, and dysfunctional cellular constituents. Three main types of autophagy have been described: macroautophagy, microautophagy, and chaperone-mediated autophagy (98).
The dominant form of organellar autophagy in mammalian cells is macroautophagy. Macroautophagy begins with formation of a double-membrane structure around a cytoplasmic component called autophagosome. The autophagosome with engulfed proteins or organelles moves through the cytoplasm and fuses with the lysosome. The selective autophagy of peroxisomes requires the presence of specific cargo receptors (97). Among them, p62/SQSTM1 has been directly associated with mammalian pexophagy. Autophagy recognizes monoubiquitinated proteins, and monoubiquitination of peroxisomal proteins leads to activation of pexophagy (55). P62/SQSTM1 possesses an ubiquitin-binding domain (UBD) and an LC3-interacting region (LIR) (4). During autophagy, the cytosolic form of LC3 (LC3-I, microtubule-associated protein light chain 3) is conjugated with phosphatidylethanolamine (PE) to generate LC3-II, which is recruited to the autophagosomal membrane (highly lipophilic PE promotes the integration of LC3-II into the lipid membrane). P62/SQSTM1 binds ubiquitinated proteins of the peroxisomal membrane and LC3-II on the autophagosome via UBD and LIR, respectively, and docks peroxisomes to the autophagosome. The subsequent fusion of autophagosome with lysosome creates an autolysosome (autophagy lysosome), where the final stage of digestion takes place (Fig. 2). Since p62/SQSTM1 is degraded by autophagy at the same time as its ubiquitinated cargo, this protein has been used as a marker of the autophagy/pexophagy flux. In other words, the inhibition of autophagy leads to accumulation of p62/SQSTM1 and vice versa. The accumulation of peroxisomes in p62/SQSTM1 knockout models supports the role of p62/SQSTM1 in pexophagy. The preserved motility of peroxisomes is an important prerequisite for accurate functioning of pexophagy. PEX14 has been shown to interact with tubulin, which may affect the motility of peroxisomes. This assumption is corroborated by experimental findings demonstrating that PEX14-deficient peroxisomes lose their ability to move along microtubules (5).

Peroxisomes and Kidney
Peroxisomes are most abundant in the liver and the kidney where they were originally described by Rhodin (85). They are particularly dense in proximal tubules with negligible presence in glomeruli, distal tubules, and collecting ducts (64, 65). Immunohistochemical studies demonstrated the presence of peroxisomal FAO enzymes (ACOX, bifunctional protein, and 3-ketoacyl-CoA thiolase) and catalase in tissue samples from human renal biopsies (64).
Most of the previous studies focused on mitochondria and by and large neglected peroxisomes. Studies examining the contribution of peroxisomes to the pathogenesis of renal disorders are quite scarce.
All parts of the nephron are able to utilize fatty acids as a source of energy. Mitochondrial FAO is equally distributed in the proximal and the distal parts of the nephron. The proximal tubule is the site of numerous energy-consuming transport processes. Proximal tubules exhibit a low rate of glycolysis and critically depend on a functional FAO. Therefore, in addition to mitochondrial FAO, proximal tubular cells possess peroxisomal FAO with a capacity comparable with that of the liver cells (58). The distribution of ACOX, the key peroxisomal FAO enzyme is restricted to the proximal tubule. Disturbances in mitochondrial and/or peroxisomal FAO lead to an increase of unmetabolized fatty acids (backward failure) and simultaneously to decreased energy production (forward failure). High tissue levels of free fatty acids (FFAs) inhibit proximal tubule Na+K+-ATPase, destabilize mitochondrial membrane potential, and stimulate inflammatory response (25, 79).
Most of the pioneer data on peroxisomes in the pathogenesis of kidney injury originate from studies using ischemia–reperfusion models of acute kidney injury (AKI). During tubular regeneration after AKI, mitochondria and endoplasmic reticulum appear before peroxisomes. Peroxisomes regenerate later at the same time as lysosomes (8). In mercuric chloride-induced acute tubular necrosis, peroxisomes appear on the fourth day of regeneration and their number reaches the normal distribution in the fourth week (8).
Ischemic AKI
Proximal tubular cells are the major targets of ischemic injury during acute renal failure (63). Increased parenchymal levels of FFAs and decreased ATP levels are consistent findings in the ischemic kidney.
Mitochondria have long been considered to play a central role in cell dysfunction during ischemia–reperfusion and hypoxia–reoxygenation kidney injury (105). Ruidera et al. showed that renal ischemia impairs FAO in both organelles, mitochondria and peroxisomes. The study demonstrated a comparable loss of FAO activity for short-, long-, and very long-chain fatty acids and an equal degree of mitochondrial and peroxisomal FAO dysfunction (89). Sixty minutes of renal ischemia reduces the activity of FAO almost by 70 percent. Phospholipase A2 activated in the ischemic kidney degrades phospholipid-rich membranes and additionally increases the levels of unsaturated fatty acids elevated due to the dysfunctional FAO. Excessive parenchymal accumulation of FFAs stimulates their oxidation to lipid peroxides. Tissue levels of lipid peroxides in kidneys explanted from rats subjected to renal ischemia are about 2.5-fold higher than in control animals (89).
ROS are the main etiological agents in the pathophysiology of ischemia–reperfusion injury. Reperfusion of ischemic kidney leads to excessive generation of ROS and further aggravates the ischemic injury. Peroxisomal antioxidant mechanisms, including superoxide dismutase, glutathione peroxidase, and the key enzyme catalase, participate in the detoxification of peroxisomal (and cellular) ROS.
The harmful effects of ischemia–reperfusion injury on organelles other than peroxisomes, in particular on mitochondria, have been reported by many studies. Gulati et al. showed for the first time that renal ischemia–reperfusion is accompanied by functional and structural damage of peroxisomes (33). Almost 90% of cellular catalase activity is localized to peroxisomes. The loss of catalase activity during renal ischemia is largely caused by the inactivation of the enzyme. Ischemia-induced generation of H2O2, together with intracellular acidosis, predisposes catalase to form an enzymatically inactive complex. In contrast to ischemia, proteolysis and decreased synthesis of catalase protein are responsible for the suppression of catalase activity during reperfusion. The rate of peroxisomal FAO assessed by the oxidation of lignoceric acid (24 carbon VLCFA metabolized almost exclusively by peroxisomes) decreases with the duration of renal ischemia (53% of control after 60 min and 43% of control after 90 min of ischemia). During reperfusion following ischemia, the kidney is able to restore FAO and catalase activity unless the duration of the preceding ischemia exceeds 60 min. After a longer period of ischemia, subsequent reperfusion causes structural alterations of peroxisomes with cytosolic release of their enzymes and finally a global deterioration of the organelle (33).
Peroxisomal FAO includes several steps catalyzed by different enzymes. The initial and rate-limiting step is catalyzed by ACOX, whereby fatty acyl-CoA is reduced to trans-2-enoyl-CoA accompanied by consumption of oxygen and generation of H2O2. A multifunctional protein possessing the activities of enoyl-CoA hydratase and 3-hydroxyacyl-CoA dehydrogenase catalyzes the following two reactions. In the final step, 3-ketoacyl-CoA is cleaved by 3-ketoacyl-CoA thiolase into acetyl-CoA and a saturated acyl-CoA having two carbons less than the original molecules, whereas peroxisomal FAO seems to be mostly inducible; mitochondrial FAO has a rather constitutive nature. Peroxisomal FAO enzyme activities are regulated mainly by the rate of their protein synthesis (36).
Peroxisomal FAO, the main source of cellular H2O2, is sensitive to excess H2O2. The exposure of peroxisomes, isolated from a healthy rat kidney, to high H2O2 results in inhibition of lignoceric acid oxidation. Similarly, the inhibition of catalase activity by aminotriazole (catalase inhibitor) reduces the oxidation of lignoceric acid in peroxisomes to about 55% of control (32). Among peroxisomal FAO enzymes, the rate-limiting ACOX is more sensitive to ischemic stress and largely contributes to the loss of FAO capacity in ischemia–reperfusion kidney injury.
Renal ischemia decreases FAO and catalase activity. The reperfusion phase following ischemia generates significant oxidative stress. The accumulation of unmetabolized fatty acids augmented by inactivated and/or degraded catalase promotes lipid peroxidation and cellular damage. Interestingly, renal ischemia–reperfusion injury exhibits negative effects also on distant organs. Fifty minutes of renal ischemia and subsequent reperfusion induce oxidative cardiac injury characterized by increased myocardial levels of lipid peroxides and decreased activities of myocardial antioxidant enzymes (46).
Septic AKI
Sepsis is the most common cause of AKI in critically ill patients. Magnetic resonance imaging shows alterations in renal parenchyma already 6 h after septic insult and underlines the importance of early subclinical alterations in septic kidney (18, 23).
Endotoxin-mediated injury is a very good model of cellular stress that extensively stimulates metabolic and oxidative pathways, two processes related to peroxisomes. The proximal tubule can be divided into three segments that differ anatomically and functionally. The S1 segment corresponds to the initial two-thirds of the pars convoluta, the S2 segment extends to the initial part of the pars recta, and the S3 segment includes the rest of the pars recta to its termination in the outer medulla (13). Systemically administered endotoxin is freely filtered through the glomerulus and almost exclusively reabsorbed in the S1 segment through Toll-like receptor 4-dependent endocytosis. The interaction with endotoxin activates the S1 segment to secrete inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) (52). In contrast to severe oxidative injury in the adjacent S2 and S3 segments, the S1 segment exhibits practically no oxidative stress, despite the heavy uptake of endotoxin. The S1 segment possesses several defense mechanisms, among them antioxidant/cytoprotective enzymes, heme oxygenase-1 and SIRT1 (34, 52). Interestingly, whereas S2 and S3 segments have the highest density of peroxisomes, the S1 segment lacks these organelles. Moreover, S2 and S3 segments (but not S1) express TNF-α receptor-1 that makes this part of the nephron more susceptible to TNF-α-mediated injury. Both, S2 and S3 segments exhibit severe oxidative peroxisome damage already 4 h after administration of endotoxin, even before the mitochondrial injury occurs (24).
Our group demonstrated that systemic injection of a sublethal dose of endotoxin induces a biphasic response starting with a transient decrease followed by an increase of peroxisome numbers in the kidney of normal mice (102). Experimental studies revealed that peroxisomes have a half-life of 48 h and are mainly degraded by autophagy under basal conditions (44). The initial reduction of peroxisomes after the application of endotoxin results from increased degradation by activated pexophagy. Endotoxin-induced pexophagy eliminates damaged and dysfunctional organelles and helps to rejuvenate the kidney peroxisomal pool (Fig. 3). The activated pexophagy eliminates a substantial number of peroxisomes already 3 h after exposure to endotoxin. Pexophagy removes peroxisomes in toto and consequently reduces both generation and decomposition of H2O2 by FAO and catalase, respectively. Pexophagy preserves in this way the peroxisomal redox homeostasis (Fig. 4). Defective peroxisome removal/recycling due to dysfunctional pexophagy consequently alters cellular redox homeostasis even under normal conditions. Cells with dysfunctional peroxisomes exhibit increased levels of oxidative stress and show a protracted elevation of H2O2 levels after exposure to endotoxin. Interestingly, mice with impaired pexophagy accumulate dysfunctional peroxisomes with active FAO and inactivated catalase. The animals suffer from chronically increased oxidative stress and show an exaggerated oxidative stress response to endotoxin.
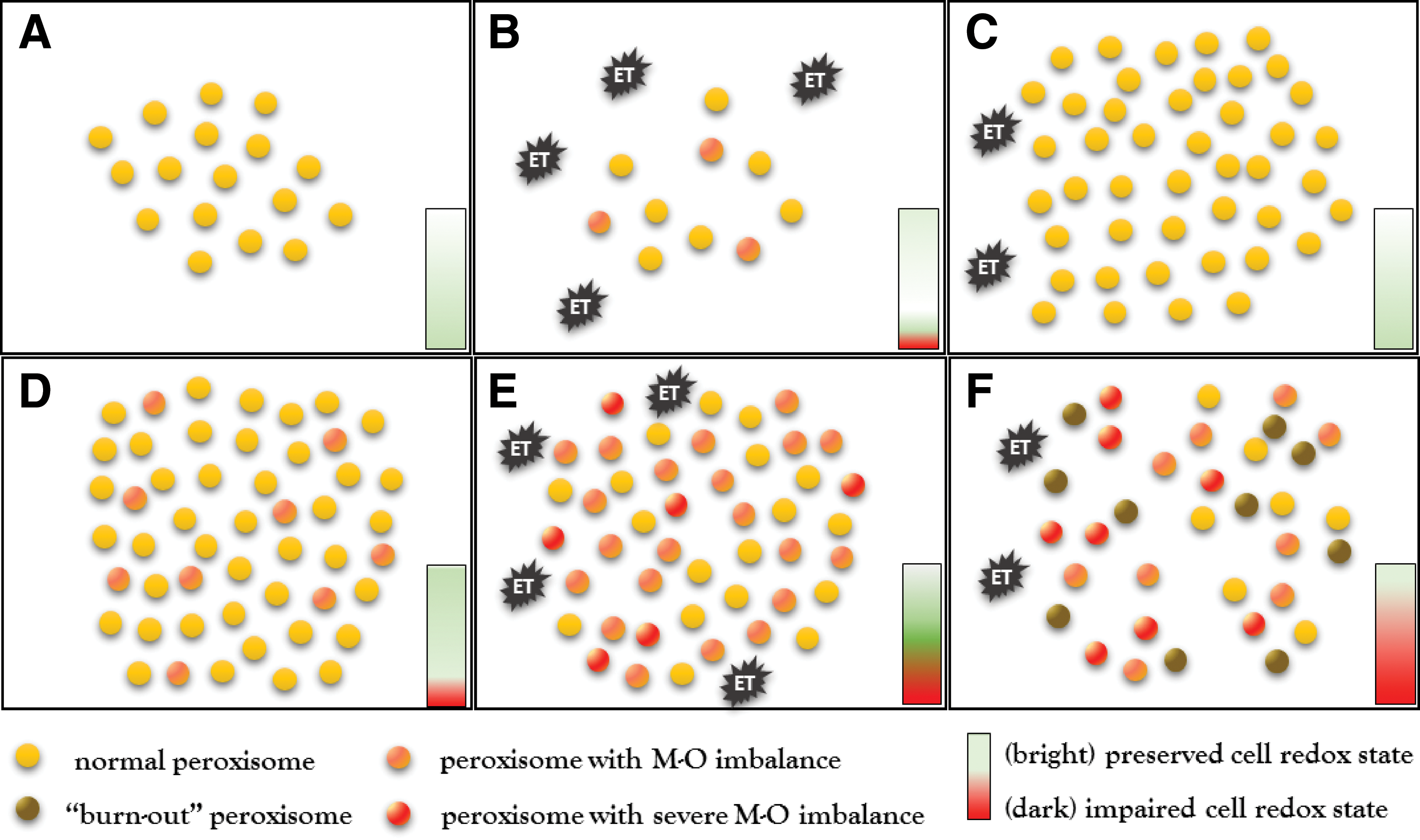
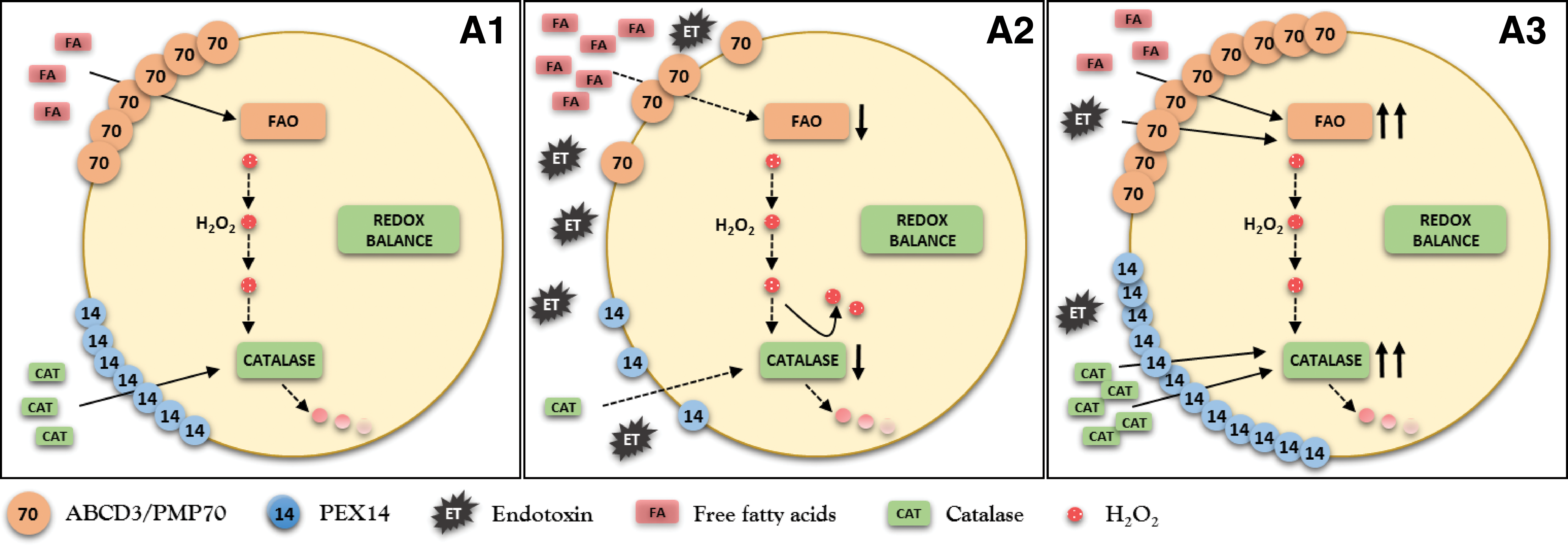
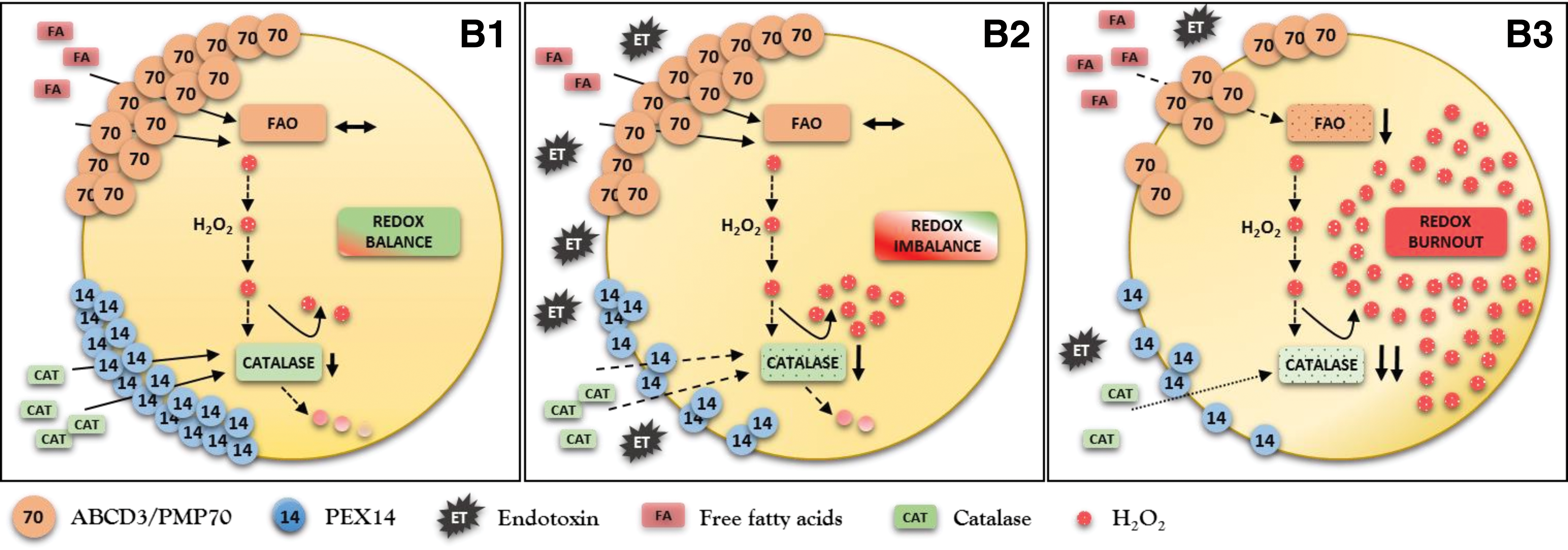
Dysfunctional pexophagy impairs regeneration of peroxisomes and induces intraorganellar redox imbalance (Fig. 5). Peroxisomes import fatty acids via specific ABC transporters (ABCD) residing in the peroxisomal membrane. ABCD3/PMP70 functions as a transporter for long-chain fatty acids that are preferentially oxidized in peroxisomes (47). PEX14 is a small membrane-attached peroxin with an important role in catalase import into peroxisomes (Fig. 6). Endotoxin decreases the expression of PEX14 more than of ABCD3/PMP70 and impairs the import of catalase more substantially compared with fatty acids. Peroxisomes exposed to endotoxin upregulate nitric oxide synthase and produce increased amounts of nitric oxide (95). Nitric oxide interacts with heme groups of catalase and inactivates the enzyme without affecting the protein levels (93). The activity of peroxisomal ACOX during endotoxic stress is initially increased in cells with impaired pexophagy, perhaps as a consequence of an aberrant activation of PPARα. This produces a functional uncoupling between active ACOX and inactive catalase. The accumulation of oxidative stress further perturbs peroxisomal redox state and gradually decreases the initially preserved FAO (ACOX). This may result in global deterioration of peroxisomal homeostasis—peroxisome burnout. Hence, endotoxin-activated pexophagy not only rejuvenates the peroxisomal pool but also prevents the development of intraperoxisomal redox imbalance. Endotoxic AKI is an extreme model of peroxisomal dysfunction. A chronic low-grade imbalance between pro- and antioxidant peroxisomal pathways has been previously reported in hereditary peroxisomal disorders (26). The above data demonstrate that such metabolic-oxidative imbalance patterns can arise in endotoxin-stressed renal peroxisomes. There is emerging evidence that very low concentrations of circulating endotoxin released by bacterial flora of the digestive system are associated with increased cardiovascular burden in patients with chronic kidney disease (71). Szeto demonstrated a positive association between the levels of circulating endotoxin and the degree of renal impairment (96). Keeping in mind the interaction of endotoxin with peroxisomes, these organelles should be considered as potential culprits in the progression of chronic kidney disease.

Cecal ligation and puncture (CLP) is a model of severe polymicrobial sepsis that simulates human septic peritonitis (40). During activation of autophagy, cytosolic LC3-I is conjugated to PE to form LC3-II that aggregates on the autophagosomal membrane. Increased amounts of LC3 aggregates (autophagosomal membrane-bound) present in renal parenchyma 3 h after CLP indicate activation of autophagy. The predominant occurrence of the cytosolic LC3-I form suggests suppression of autophagy 9 and 18 h after CLP. At this later time point, the animals show increased serum creatinine levels and severe renal injury. Different from the sublethal model of endotoxic AKI, the decline of autophagy in severe polymicrobial sepsis appears to aggravate tubular injury (40). The initially upregulated and later downregulated autophagy/pexophagy (coordinated biphasic response) seem to be protective in endotoxic AKI. Hence, accurately dosed and precisely coordinated activation and deactivation of autophagy/pexophagy play a critical role in organellar and cellular homeostasis during septic renal injury.
Peroxisomal alterations reported in various models of AKI may represent mechanisms involved in more common kidney disorders, however, at a lower intensity and with long-term implications.
Diabetic nephropathy
Diabetic nephropathy (DN) is the leading cause of end-stage renal disease in developed countries. Chronic hyperglycemia stimulates the production of ROS and angiotensin, which in turn accelerates the progression of tubulointerstitial fibrosis (41, 101). Experimental data indicate an important role for ROS in the nephropathy of diabetes. Hence, most of the studies analyzing peroxisomes in DN focused on the oxidative stress and the role of peroxisomal catalase.
Mammalian catalase is a highly conserved heme-containing tetrameric protein with a molecular mass of 240 kDa (84). The overexpression of catalase reduces oxidative stress and mitigates oxidative injury in vascular smooth muscle cells and ischemic myocardium (12, 54). Transgenic diabetic mice overexpressing catalase in proximal tubules exhibit decreased levels of ROS, angiotensin, plasminogen activator inhibitor type 1 (PAI-1), and proapoptotic p53 (10).
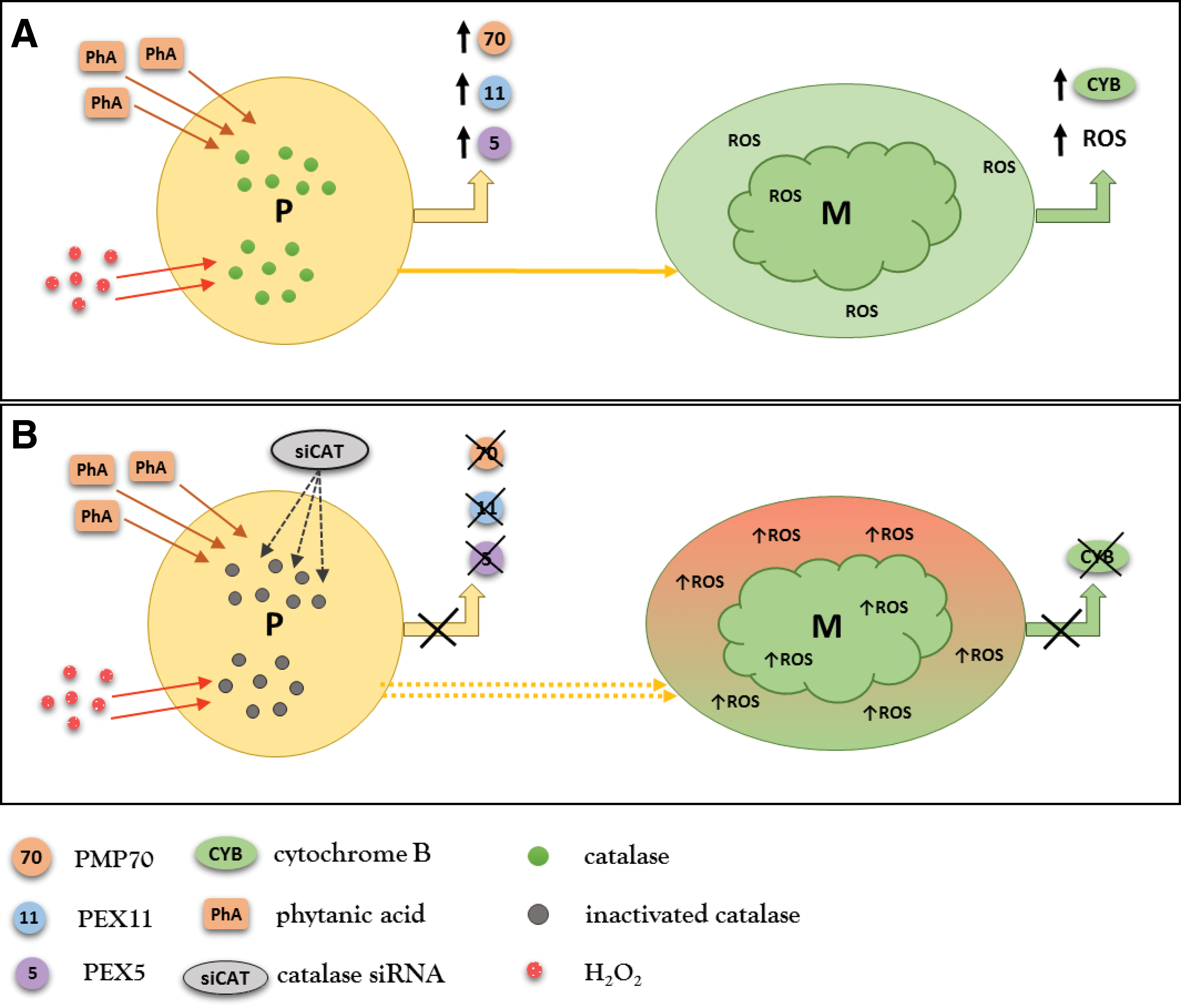
Studies scrutinizing mechanisms induced by catalase deficiency are helpful in defining the potential contribution of peroxisomes to diabetic kidney disease. Diabetic catalase knockout (CKO) mice have higher serum creatinine and FFA levels. They develop higher level proteinuria compared with diabetic wild-type mice, despite the blood glucose levels being similar (45). CKO mice exhibit increased renal expression of profibrotic-transforming growth factor β1 and decreased expression of antifibrotic bone morphogenetic protein 7. CKO mice and cultured catalase knockdown mesangial cells have impaired biogenesis of peroxisomes resulting from increased levels of H2O2. Mesangial cells typically increase peroxisomal biogenesis in response to physiological concentrations of H2O2 and this effect disappears when the H2O2 concentration exceeds a certain level (45). The nontoxic levels of H2O2 generated by FAO stimulate peroxisomal biogenesis to meet the metabolic demand of the cell. However, H2O2 concentrations above a certain level (modified by the balance between FAO and catalase) may cause peroxisomal injury. Catalase knockdown mesangial cells stimulated with low concentrations of H2O2 or phytanic acid (physiological substrate of peroxisomes) do not respond with activation of peroxisomal biogenesis and, even more, exhibit defective biogenesis of mitochondria (45) (Fig. 7). Moreover, the basal levels of mitochondrial ROS are significantly increased in catalase knockdown mesangial cells, supporting the idea of close interactions between mitochondrial and peroxisomal pathways. The above data provide evidence that endogenous catalase fulfills an important role in protecting the kidney from diabetic stress by maintaining peroxisomal and mitochondrial fitness.
In the streptozotocin-induced model of DN, a mismatch between increased FAO (ACOX) and decreased catalase activity results in metabolic-oxidative imbalance with overproduction of H2O2 and accelerated progression of the diabetic kidney disease (19).
Farnesyl transferase catalyzes farnesylation of small GTPase proteins such as Ras and Rho and plays therefore an important signaling role in the regulation of cell proliferation. Treatment of diabetic rats with farnesyl protein transferase inhibitor, FPTIII, has been shown to diminish diabetes-induced inhibition of catalase. This study indicates that inhibition of farnesyl transferase might prove beneficial in DN and that modulation of peroxisomal functions through Ras-/Rho-GTPase signaling pathways may play a role (20).
Renal cancer
Resistance to chemotherapy and radiotherapy is a common feature of renal cancer. Because of defective mitochondrial oxidation, renal cancer cells obtain their energy primarily from glycolysis rather than oxidative phosphorylation (38). Interestingly, whereas proximal tubules of normal human kidney contain large numbers of peroxisomes, these organelles seem to be entirely absent in renal cancer cells. The absence of peroxisomes and mitochondrial disturbances in renal cancer cells result in alterations of fatty acid metabolism that may serve as potential targets for future therapeutic interventions (28, 29).
PPAR alpha
The results of numerous studies indicated that inhibited or dysfunctional FAO exhibits negative effects on renal injury. Functional FAO prevents accumulation of fatty acids, their peroxidation, and formation of lipid aldehydes that can further aggravate renal injury (66). For that reason, many of the follow-up studies focused on the potential protective effects of FAO stimulation in AKI.
PPARs belong to the nuclear receptor family and function as ligand-activated transcription factors. The name originated from early observations that certain compounds such as fibrates stimulate the growth and proliferation of peroxisomes (99). The receptor responsible for this effect was later identified and named PPARα (39, 50). PPARα is the principal activator/stimulator of peroxisomes. Renal PPARα resides predominantly in the proximal tubule and corresponds to the highest density of peroxisomes in this part of the nephron (42).
In the proximal tubule, PPARα modulates fatty acid metabolism, oxidative stress, inflammatory response, and apoptosis. Normally, FFAs bound to plasma albumin are filtered through glomeruli, reabsorbed, and metabolized in proximal tubular cells. In the protein overload nephropathy model of AKI, repetitive injections of high amounts of albumin-bound fatty acids induce severe proximal tubular injury in PPARα knockout mice, but not in wild-type mice (53). Tubular damage in PPARα knockout mice is associated with accumulation of fatty acids, increased oxidative stress, and apoptosis. Even though the study did not specifically focus on peroxisomes, the authors described an obvious damage of mitochondria characterized by swelling and rupture of the organelle (53).
Peroxin 11 (PEX11) is a small peroxin implicated in elongation and division of peroxisomes. PEX11 knockout mice have significantly lower numbers of functional peroxisomes. In the protein overload nephropathy model of AKI, PEX11 knockout mice show marked lipid infiltration of renal parenchyma and more severe tubulointerstitial injury compared with wild-type mice (106). PEX11 is a crucial regulator of peroxisomal proliferation and its deficiency impairs the biogenesis of peroxisomes. Similar to dysfunctional pexophagy, PEX11 deficiency negatively affects the regeneration of peroxisomes and aggravates renal injury.
PPARα knockout mice subjected to kidney ischemia–reperfusion injury develop more extensive necrosis of the corticomedullary area and severe deterioration of renal function compared with wild-type littermates (81). Renal ischemia–reperfusion injury causes repression of PPARα target genes, among them peroxisomal rate-limiting ACOX. Treatment of normal rats with PPARα ligand, clofibrate, before the induction of renal ischemia reverses the inhibitory effect on FAO and significantly ameliorates acute tubular necrosis (81).
Cisplatin-induced nephrotoxicity is partially mediated by repression of ACOX and carnitine palmitoyl transferase (CPT) mRNA levels, the rate-limiting peroxisomal and mitochondrial FAO enzymes. The negative effect of cisplatin on FAO is delivered via inhibition of PPARα. Cisplatin decreases PPARα binding ability to its target genes and does not affect PPARα protein levels (80). Treatment with PPARα ligand, WY14643, prevents the repression of mitochondrial FAO and ameliorates cisplatin-induced AKI. This protective effect of WY14643 is not present in PPARα knockout mice. The authors also demonstrated that intact function of PPARγ-coactivator-1 (PGC-1), a transcriptional coactivator of PPARα and PPARγ, further increases the survival rates of proximal tubular cells during cisplatin-induced AKI (62). Transgenic mice overexpressing PPARα in proximal tubules demonstrate improved renal function and morphology during cisplatin- and ischemia-induced AKI. Histopathological examination reveals a substantial reduction of tubular damage, particularly in the S3 segments typically affected in AKI. The protective effect of PPARα overexpression in the proximal tubule is mediated mainly through improvement of fatty acid metabolism. PPARα-mediated nephroprotection does not require the presence of an exogenous PPARα ligand, which emphasizes the essential role of PPARα per se (61).
Fatty acid-binding proteins (FABPs) are members of a conserved multigene family involved in fatty acid transport from the plasma membrane to intracellular organelles, such as mitochondria and peroxisomes (15). L-FABP is an isoform expressed in the liver and in renal proximal tubules. Treatment with PPARα agonist, fibrate, increases the number of peroxisomes and ameliorates renal damage in cisplatin-induced AKI. L-FABP protein is increased in proximal tubules of fibrate-treated mice and upregulation of L-FABP depends on intact PPARα signaling (74). Treatment of rat hepatocytes with fibrates increases the peroxisomal L-FABP concentration above the total cellular L-FABP levels, which indicates a more efficient translocation of L-FABP into peroxisomes during fibrate treatment. The induction of L-FABP in liver cells goes hand in hand with the induction of peroxisomal FAO (1). In renal ischemia–reperfusion injury, L-FABP binds excessive FFAs and lipid peroxidation products in tubular cells and transports these into the tubular lumen (109). Several studies confirmed the ability of L-FABP to attenuate renal injury and demonstrated the potential of L-FABP for use as a biomarker of kidney damage (107).
Transgenic mice overexpressing PPARα in proximal tubules show decreased expression of inflammatory and profibrotic cytokines in a unilateral ureteral obstruction model of renal injury. The expression of fibrogenic genes, including collagen 1, α-smooth muscle actin, and fibronectin, is significantly reduced. The peritubular fibrosis present in wild-type mice is almost absent in kidneys of PPARα transgenic animals. These results indicate that PPARα overexpression in the proximal tubule is able to reduce the extent of tubulointerstitial fibrosis after kidney injury (60).
Chau et al. investigated changes of microRNA expression in renal fibrosis and showed that microRNA-21 (miR-21) is highly elevated in experimental animal models and in human transplanted kidneys with nephropathy (14). Systemic deletion of miR-21 causes no apparent abnormality in mice. However, miR-21 knockout mice develop significantly less interstitial fibrosis in response to kidney injury. The investigators scrutinized genes silenced in response to kidney injury in miR-21 wild-type mice and compared them with miR-21 wild-type mice treated with anti-miR-21 oligonucleotides. Interestingly, the analysis of genes upregulated in the absence of miR-21 identified genes involved in peroxisomal (ACOX) and mitochondrial (CPT) FAO pathways regulated by PPARα. PPARα deficiency abrogates the antifibrotic effect of anti-miR-21 oligonucleotides and indicates that PPARα-controlled FAO pathways are the major targets of miR-21 in renal disease (14, 22).
These results underline the potential of PPARα-mediated modulation of FAO activity as a potential therapeutic strategy in AKI. One problem that needs to be mentioned is that human cells express significantly less PPARα than rodent cells (31). Hence, the potential of PPAR expression and overexpression in human renal pathogenesis needs further demonstration.
SIRT1 is an NAD-dependent deacetylase that regulates numerous cellular functions and has been associated with longevity in mammals (7). Renal SIRT1 reduces apoptosis and inflammation and improves lipid metabolism, just to name a few. In proximal tubular cells, SIRT1 regulates H2O2 levels by modulating the expression of catalase in either a positive or a negative way, depending on the oxidative status of the cell. Under normal conditions, SIRT1 suppresses catalase and helps to maintain low hormetic levels of ROS, which participate in cell signaling and stimulate peroxisomal biogenesis. SIRT1 upregulation during increased oxidative stress reduces the apoptosis of renal tubular cells by stimulation of catalase synthesis that is mediated through deacetylation of forkhead transcription factor, forkhead box O3 (FOXO3), by SIRT1 (35). Transgenic mice with SIRT1 overexpression in proximal tubules show decreased levels of ROS and diminished tubular cell apoptosis during cisplatin-induced AKI. SIRT1 overexpression rescues proximal tubules from cisplatin-induced injury by maintaining peroxisome number and functions, in particular by upregulation of catalase. This study reveals that peroxisome is an important intracellular target organelle for SIRT1 (34). Protective effects of SIRT1 in acute and chronic renal injury have also been reported by our group (103).
Conclusions
Although the discovery of peroxisomes recently celebrated its 60th anniversary, the contribution of this spectacular organelle to the pathogenesis of common human diseases remains scarcely explored.
Peroxisomes can generate as well as scavenge ROS and are together with mitochondria essential players in tightly regulated redox homeostasis. Many peroxisomal tasks and particularly the FAO are functionally connected to mitochondrial pathways. Peroxisomal oxidases are unique in generating high amounts of H2O2 that is decomposed by the most prominent peroxisomal antioxidant enzyme catalase. Very low hormetic concentrations of H2O2 play a role in cell signaling. Overproduction or impaired degradation of H2O2 results in increase of peroxisomal membrane permeability and leakage of H2O2 and peroxisomal matrix components into the cytoplasm. Peroxisomal dysfunction induces alterations of mitochondrial membrane potential and promotes generation of mitochondrial ROS. Renal tubular cells rely on FAO as an essential source of ATP and alterations of this critical energy-generating pathway may aggravate kidney injury. Peroxisomes increase their metabolic activity and proliferation rates in response to stimuli acting through PPARα. Treatment with PPARα agonists such as fibrates preserves FAO and shows nephroprotective effects in AKI.
The evidence from studies on the physiology and pathophysiology of renal disorders is consistent in showing that neither peroxisomes nor mitochondria alone are involved, but rather the mitochondria–peroxisome partnership plays a major role.
Despite recent progress, further studies are needed to elucidate the molecular mechanisms induced by dysfunctional peroxisomes and the implications of the dysregulated mitochondria–peroxisome axis in the pathogenesis of renal injury.