
Abstract
Introduction
T
A patient who undergoes some form of AKI has a high risk for developing CKD within 10–15 years. The causes for AKI are variable, as are the causes for CKD. While AKI is typically the result of an acute transient episode of systemic damage, CKD is brought about usually due to genetic predisposition, syndrome, or illness that leads to secondary kidney injury or a previous bout of AKI. Regardless, in every case of impaired kidney function, either acute or chronic, oxidative stress plays a role in renal damage and offers a potential target for therapeutic intervention. In fact, oxidative stress has been linked to the pathogenesis of a variety of diseases, such as cardiovascular disease, atherosclerosis, hypertension, cancer, diabetes, arthritis neurodegenerative diseases (i.e., Alzheimer's and Parkinson's disease), and aging (300).
Oxidative stress is characterized by increases in reactive oxygen species (ROS) and/or reactive nitrogen species (RNS). While many reports exist that indicate ROS and RNS are elevated in both human and animal models of kidney injury (including cardiovascular injury) due to various etiologies, few definitive reports exist that directly define the factors and mechanisms responsible for enhancement of ROS and RNS in the kidney, the signaling mechanisms these oxidative species stimulate to promote damage, and the effect oxidative stress has on endogenous antioxidant systems that protect the kidney from damage and subsequently maintain normal kidney function. Due to the commonality of enhanced oxidative stress in all forms of kidney injury and disease, a broader understanding of these phenomena would be beneficial for the design of effective therapeutic strategies against kidney failure. The lack of in vivo reports is not necessarily due to an absence in effort, but rather the challenges in studying ROS and RNS directly in whole animal and human models due to the specific cellular localizations and active metabolism or instability of ROS and RNS.
While much is left to investigate regarding oxidative stress during AKI and CKD, this review will attempt to summarize what is currently known about ROS and RNS in the kidney by drawing from in vitro and in vivo (animal and human models) research. The beginning of the review will briefly describe the specific ROS and RNS that are responsible for oxidative stress. This section is kept brief since numerous reviews have been written on the subject. Next, we describe the sources of oxidative stress in the kidney and the antioxidant systems that are responsible for the organ's protection against pathological generation of ROS and RNS. Oxidative stress both directly and indirectly affects all facets of the kidney, including vascular reactivity and renal hemodynamics, glomerular filtration, and tubular reabsorption and secretion in all nephron segments. During injury or disease, oxidative stress signaling alters all of these processes and promotes prodamage pathways that lead to cellular apoptosis, necrosis, altered gene expression, progression of tissue damage, promotion of fibrosis, and abnormal kidney function. The review will summarize what is currently known with regard to these aspects in the kidney during both AKI and CKD. We conclude by describing some of the current antioxidative therapeutic treatments that have been tested in animals and humans and potential therapeutic treatments that may hold promise for future exploration.
The Players of Oxidative Stress
The one-electron reduction of molecular oxygen results in the generation of superoxide anion (O2−) and this leads to a variety of ROS (206) associated with processes described in Table 1. Superoxide is generated by several oxidase enzymes and by components of the mitochondrial electron transport chain. Superoxide anion has rather selective reactive properties and poorly diffuses across biological membranes due to it primarily being transported through anion channels. Superoxide anion also undergoes dismutation (either spontaneously or by enzymatic catalysis) to form another ROS, the nonradical species—hydrogen peroxide (H2O2). Hydrogen peroxide can also be formed by direct reduction of oxygen (O2) by oxidases. Unlike superoxide, hydrogen peroxide has membrane transport properties similar to water and this enables it to diffuse across membranes and influence regulatory systems in other cellular compartments or nearby cells.
cGMP, cyclic guanosine monophosphate; iNOS, inducible nitric oxide synthase; nNOS, neuronal nitric oxide synthase; RNS, reactive nitrogen species; SOD, superoxide dismutase.
Superoxide releases Fe3+ from iron-sulfur proteins and ferritin, thus promoting generation of additional ROS. For example, peroxides react with various forms of Fe either bound to proteins (e.g., heme) or small molecular weight molecules to generate additional highly reactive oxidant species. One potential species is the hydroxyl radical (•OH), which is readily generated by the Fenton reaction of peroxide with Fe2+. Hydroxyl radicals attack and react with almost all cellular constituents and generate additional reactive free radicals. In addition, reactive species resembling the hydroxyl radical can also be formed from decomposition of RNS such as peroxynitrite, which is generated from the very efficient reaction of superoxide with nitric oxide (•NO). Reactive species resembling hydroxyl react with cellular constituents, such as unsaturated fatty acids in lipids and nucleic acids, and promote lipid peroxidation and damage of mitochondrial and nuclear DNA.
Hypochlorous acid (HOCl) is another ROS, which is lipid soluble and extremely reactive. Hypochlorous acid is formed by phagocyte cell myeloperoxidase when it metabolizes hydrogen peroxide. In the presence of nitrite, myeloperoxidase reacts to generate highly reactive species (such as nitrogen dioxide), which behave similar to peroxynitrite in promoting tyrosine nitration and other reactions. Low levels of ROS have important signaling roles in cells, but as their levels increase, ROS actively participate in pathophysiology processes, including induction of various forms of cell death.
RNS derived from the free radical nitric oxide (including nitrogen dioxide [•NO2], nonradical peroxynitrite [ONOO−], and other RNS) are additional factors in renal pathophysiology. Nitric oxide is formed by the various isoforms of nitric oxide synthase (NOS) that use L-arginine, oxygen, and other cofactors, including tetrahydrobiopterin, to produce nitric oxide. The free radical nitric oxide is very selective in its reactive properties and it can go on to form other RNS, including peroxynitrite, which is formed by reaction of nitric oxide with superoxide. RNS are often observed to interact with cellular components, including thiol groups and unsaturated fatty acids, to promote regulation through processes such as thiol (RSH) or lipid oxidation or other modifications, including nitrosation (RSNO) or nitration (RSNO2). While some of these interactions are part of signaling, as RNS levels increase, they actively contribute to pathological processes in the cardiovascular and renal systems.
Renal Antioxidants
Superoxide dismutase
There are multiple antioxidant systems described in Table 2 that attempt to protect kidney tissues and cells from ROS or RNS-induced oxidative stress. The first intrinsic enzymes that combat oxidative stress are the superoxide dismutase (SOD) isoforms. SOD catalyzes the dismutation of superoxide into oxygen and hydrogen peroxide (178, 179). All three SOD isoforms are normally found in the kidney and are localized to the mitochondria, cytoplasm, and extracellular space where it is anchored by its heparin binding domain (74, 209, 210, 216, 294, 295, 337). Of the SOD isoforms, manganese SOD (SOD2) is present in the mitochondria, with copper/zinc SOD (SOD1 and SOD3) in the cytosol and the extracellular space, respectively (178, 179). The SOD1 isoform accounts for up to 80% of total SOD activity in the mammalian kidney (177), but only approximately one-third of SOD activity in the renal vessels, which prevents interruption of NO signaling (30). Interestingly, despite the imbalance with SOD1 accounting for the bulk of SOD activity in the kidneys, ablation of SOD2 in mice results in a dramatically more severe pathological phenotype compared with SOD1 ablation [reviewed by Schieber and Chandel (271)]. This highlights the importance of where in the cell ROS and RNS are generated and indicates that oxidative stress generated by the mitochondria plays a vital role in the pathogenesis of kidney disease and injury.
GSH, glutathione; HO-1, heme oxygenase 1; ROS, reactive oxygen species; Trx, thioredoxin.
While there has been reported species-dependent variability in the localization of where each of the SOD isoforms is found within the kidney, in general, SOD activity in the kidney is similar in human, pig, sheep, cow, rabbit, and mouse (177). SOD has been reported in the cortical and juxtamedullary proximal tubule, the thick ascending limb, and in vascular smooth muscle cells (VSMCs) of renal blood vessels, with reduced activity in the medulla and glomeruli of rodents and canines (73, 216, 294, 295). However, despite minimal SOD activity in the renal medulla, SOD inhibition results in enhanced ROS in the kidneys, an effect that consequently reduces medullary blood flow, sodium excretion, and glomerular filtration while potentially inducing hypertension in rats (170, 171, 334).
Other antioxidants
After SOD generates hydrogen peroxide from superoxide, peroxidases and catalase then convert hydrogen peroxide into oxygen and/or water. Catalase is present in all aerobic cells with elevated expression in the kidney (56). Catalase plays a critical role in reducing ROS and preventing lipid peroxidation. Catalase deficiency results in accumulation of mitochondrial ROS, an effect that leads to functional impairment of mitochondria (124). Furthermore, lipopolysaccharide (LPS)-induced loss of catalase activity was demonstrated to significantly aggravate kidney damage in a mouse model of endotoxemia (302) [see review by Vasko and Goligorsky (301)].
Other vitally important defenses include the glutathione and the thioredoxin peroxidase systems. The enzymes of the glutathione and thioredoxin systems are distributed throughout the cell, including in the mitochondria, cytoplasm, and nucleus (66, 211). The glutathione and thioredoxin systems have been implicated in both the regulation of cellular redox and in many signaling mechanisms, controlling processes such as DNA synthesis, cell proliferation, and apoptosis (80). It has been reported that in addition to reduced SOD and catalase levels, glutathione is also diminished in a rat model of diabetic nephropathy (35) and in CKD patients (32, 215, 282). Conversely, enhancement of catalase and glutathione has been shown to provide resistance against hydrogen peroxide in renal tubular epithelial cells (95). Some of the other factors that act as antioxidants influencing renal function include ascorbic acid (vitamin C), tocopherol (vitamin E), cysteine (55, 132), and the most sensitive indicator of cellular stress, heme oxygenase-1 (HO-1) (239).
While antioxidants play a critical role in maintaining cellular and tissue physiological and pathophysiological homeostasis associated with preventing accumulation of ROS and RNS, when their neutralizing ability is exceeded by oxidative stress then consequently pathological tissue damage ensues. For instance, dysregulation of antioxidants, particularly catalase and peroxidases, results in the accumulation of hydrogen peroxide, which can react with transition metals to subsequently form highly reactive and damaging species with reactivities, such as the hydroxyl radical. Furthermore, studies indicate the overproduction of superoxide, primarily by NADPH oxidase and mitochondria, and reduced superoxide metabolism by SOD and other antioxidants, can initiate or potentiate the development of hypertension (8). For example, it has been shown that during the onset of hypertension, patients have 7% lower antioxidant capacity, including decreased erythrocyte catalase content and enhanced oxidation of low-density lipoproteins (LDLs) (45, 266). As hypertension progresses, the antioxidant systems in these patients continue to falter due to reduced levels of SOD, glutathione peroxidase, and catalase (249, 263, 285), resulting in the development of renal damage. At this point, NADPH oxidase-induced superoxide and ROS production is elevated in VSMCs of renal resistance arteries and in infiltrating leukocytes (182, 314, 331), causing increased plasma asymmetric dimethylarginine and lipid peroxidation (314). As the effects continue to mount, kidney damage progresses and kidney function wanes.
Renal Sources of Oxidative Stress
The role of ROS and RNS is considered a critical factor involved in initiation and progression of various mammalian kidney diseases and injuries, including diabetic nephropathy, hypertension-related kidney injury, ischemia–reperfusion injury (IRI), toxic-induced nephropathy, and several forms of inflammatory syndromes (29, 75, 148, 278, 297). There is generation of ROS and RNS in both renal tubular and vascular cells during numerous tissue and cellular stresses. In the kidney and vascular systems, there are many sources of ROS and RNS that occur during both normal and pathological states, but the major sources of oxidative stress in the kidney come from NADPH oxidase and mitochondria generation (12, 27, 213, 244). It is important to point out that low levels of ROS and RNS are important for normal redox signaling that promotes cell survival, proliferation and growth, and vasoreactivity in the kidney, while also providing cells with a sensor for hypoxia. However, as cells and tissues are stressed, the balance between ROS and RNS generation and elimination is lost due to upregulated ROS/RNS formation and/or reduced antioxidant activity, resulting in the accumulation of these molecules. As ROS and RNS levels are increased, cell damage occurs and tissue function is impaired.
NADPH oxidase ROS
NADPH oxidases, along with the mitochondria, are the major producers of superoxide and are the principal sources of abnormal cellular signaling (205, 236, 262). As NADPH oxidase transfers electrons from NADPH, they react with oxygen to form superoxide, which is usually rapidly converted to hydrogen peroxide. All of the components necessary for NADPH oxidase to function are present in the kidney, including the presence of four different isoforms of this oxidase—NOX1, NOX2, NOX4, and NOX5 (8, 33, 112). The expression of these different NADPH oxidase isoform components has been observed throughout the cortex and medulla of the mammalian (including human) kidney, including in the mesangium (97, 136, 184, 190), proximal convoluted tubules, distal convoluted tubules, collecting duct, macula densa (17, 77, 139, 217, 281, 309, 322), endothelium, and VSMCs (8).
NADPH oxidase is localized in the plasma membrane and generates superoxide for redox signaling for processes such as cell proliferation and interaction with the immune system that includes directing phagocytotic killing of invading microbes (168). Of the NADPH oxidase isoforms in the kidney, NOX4 is expressed the highest (90), producing large amounts of hydrogen peroxide constitutively (296). It has been suggested that the NOX4 isoform of NADPH oxidase is also localized in the mitochondria of the rat kidney cortex. This location would allow superoxide generated by NADPH oxidase to directly trigger mitochondrial generation of ROS (21). NADPH oxidase activity (including induction of NOX2 expression) can be stimulated by angiotensin II (Ang II) (106) and proinflammatory cytokines such as interferon gamma (IFNγ) (201). Recent reports indicate the NOX5 isoform may also influence tubular physiology and contribute to glomerulopathies (112).
Mitochondrial ROS
Mitochondria in renal cells normally produce low levels of ROS, which are thought to play a role in cell signaling processes such as sensing hypoxia. However, the production of superoxide in the mitochondria is markedly enhanced by a variety of pathophysiological stimuli, including Ang II, tumor necrosis factor alpha (TNFα), integrin ligation, diabetes and high glucose, oxidized LDL, and superoxide generated by NADPH oxidase (27, 298). These stimuli increase mitochondrial superoxide generation by affecting intracellular regulatory systems that increase intracellular calcium, open mitochondrial KATP channels, and increase membrane potential. As a result, superoxide production occurs in complex I and complex III of the mitochondrial electron transport chain. In addition, hydrogen peroxide and lipid oxidation products can also enhance mitochondrial ROS generation in renal cells. A few well-understood mechanisms of mitochondrial ROS generation include the consequences of disruption of Fe-S clusters, inhibitory interactions with cytochrome c (cyt-c) oxidase, and relative changes in expression of electron transport chain components. It was also demonstrated that endothelial cells contain a mitochondrial NOS isoform that is capable of generating superoxide during stress (27).
Superoxide levels are normally kept at low levels in the mitochondrial matrix due to its conversion to hydrogen peroxide by mitochondrial matrix Mn-SOD (SOD2). The conversion of superoxide to hydrogen peroxide and its secretion from mitochondria appears to be important in renal signaling processes. However, it is critical for excessive mitochondrial superoxide to be detoxified to prevent damage to the mitochondria. If superoxide is not detoxified, it causes mitochondrial oxidative stress by disrupting iron-sulfur centers, releasing iron, and promoting processes that impair mitochondrial function. If superoxide and ROS generation is excessive in the mitochondria, mitochondrial membrane channels (including anion channels in the inner membrane) open (158), resulting in collapse of the mitochondria membrane potential and further generation of ROS by the electron transport chain (315).
Oxidative stress is worsened by reduced SOD2 levels. For instance, in CKD patients, SOD2 is downregulated in neutrophils during stress, causing their enhanced generation of ROS owing to the neutrophil dysfunction these patients have (214). In animal studies, ablation of SOD2 in mice results in accelerated renal cellular senescence and enhanced tubular damage, glomerular sclerosis, and renal interstitial inflammation upon aging (257). As superoxide and ROS generation in the mitochondria continues, it induces accumulation of uncoupling protein-2 (UCP2) (253). Increased UCP2 leads to the inward proton leak that interferes with the function of ATP synthase and reduces ATP synthesis (206), an effect that would impair ATP-dependent reabsorption processes in renal tubular epithelial cells. If these effects are severe enough, autophagy, apoptosis, or necrosis of these cells will occur (241).
Leakage of electrons from complex of I and II has been shown to mediate ROS-induced autophagy (40) and apoptosis. As ROS increases, oxidation of the acyl chains of cardiolipin reduces its binding affinity for cyt-c and liberates it from the inner mitochondrial membrane. Oxidized cardiolipin, along with permeabilization of the mitochondrial outer membrane by proapoptotic proteins of the B-cell lymphoma-2 (Bcl-2) family, causes release of cyt-c from the mitochondrial membrane into the cytosol, followed by formation of apoptosome, caspase activation, and consequently apoptosis (222).
Oxidative Stress in the Renal Vasculature and Glomerulus
Renal vasculature
The effects of ROS and RNS within the kidney during disease and/or injury appear to be spatially and temporally coordinated with potential multiple factors. All the cells associated with the renal vasculature, including endothelial cells, VSMCs, adventitial fibroblast, and both resident and infiltrating inflammatory cells, appear to be able to produce levels of ROS and/or RNS that promote pathophysiology. While ROS generation and signaling influence reabsorption of Na+ in tubular cells throughout the nephron, it has direct effects on vasoreactivity in renal vessels with increased ROS associated with enhanced renal vascular resistance (RVR) (317). For instance, superoxide, primarily generated by NADPH oxidase in VSMCs, causes vasoconstriction and enhances basal tone in the medullary circulation of mice (340). Vascular oxidative stress plays a critical role in progression of kidney damage during disease. The endothelium is particularly vulnerable to oxidative stress and undergoes a change in phenotype as ROS levels in and around the blood vessels become moderate to severe. This adverse effect on the endothelium promotes inflammation, including enhanced cytokine production and expression of surface adhesion molecules (160) effectively promoting vascular remodeling.
Nitric oxide also plays a critical role in the endothelium during renal disease and oxidative stress. Low levels of nitric oxide (generated by endothelial nitric oxide synthase [eNOS]) induce expression of antioxidative genes, protect renal endothelial and mesangial cells from apoptosis and fibrosis (15, 62, 144, 237, 270, 313), and promote normal renal hemodynamics. In endothelial cells, in addition to being tethered to the cell membrane, eNOS can also present on the cytosolic face of the outer mitochondrial membrane (84). Physiological concentrations of nitric oxide in cells inhibit cyt-c oxidase (28), potentially altering mitochondrial ROS generation.
Moderately increased mitochondria ROS results in stabilization of hypoxia-inducible factor (HIF) in endothelial cells (99) and in stimulating nuclear factor E2-related factor (NRF-2) (235), effects that are both protective against oxidative stress and renoprotective (107, 196). The transcription factor NRF-2 is usually deactivated in an inhibitory complex with Kelch-like ECH-associated protein-1 (Keap-1). Once oxidative stress increases and Keap-1 is oxidized, NRF-2 is released and binds to the promotor region of nuclear respiratory factor-1 (NRF-1). NRF-2 binding promotes expression of numerous antioxidative molecules, including various enzymes of the glutathione and thioredoxin systems, SOD, catalase, HO-1, and other antioxidant molecules. In addition, ROS-induced stimulation of NRF-1 promotes activation of mitochondrial transcription factor A (Tfam) and stimulation of mitochondrial DNA replication/transcription and biogenesis (93, 326), effects aimed at preventing tissue damage. Reduced activity of NRF-2 has been implicated in the progression of renal failure in a mouse model of CKD (147).
During hypertension and CKD, there is enhanced production of ROS in the kidney due to elevated NADPH oxidase activity, enhanced mitochondria respiration, and decreased efficiency in oxygen usage for Na+ transport (8, 317). The increased level of ROS diminishes functional endothelium-derived nitric oxide by impairing and/or uncoupling NOS enzymes. This reduction in endothelial-derived nitric oxide is paramount in the progression of renal hemodynamic pathogenesis due to the loss ability of nitric oxide to induce vasodilation and counterbalance vasoconstrictors such Ang II, endothelin-1 (ET-1), and sympathetic nervous system outflow. As nitric oxide bioavailability continues to decrease, due to its reaction with superoxide and subsequent formation of peroxynitrite, RVR increases and nitric oxide-mediated pressure natriuresis is attenuated (306, 319). If the reduction in nitric oxide persists, endothelial dysfunction ensues and is potentially followed by the development of cardiovascular and renal diseases (71, 101, 104, 133).
Glomeruli
During the initial period of inflammation, the glomerular mesangium reacts by generating superoxide and hydrogen peroxide, mainly by NADPH oxidase (230). During certain disease states when there are elevated circulating levels of glucose, Ang II, and aldosterone (280), these factors further induce generation of ROS in mesangial cells (156, 262). In the case of Ang II and aldosterone, NADPH oxidase (NOX1) activity and superoxide production are induced via stimulation of the angiotensin II type 1 (AT1) receptor and mineralocorticoid receptors, respectively (121). The increased levels of ROS induce glomerular podocytes and mesangial cells to generate Ang II and reinforce feedback that promotes further ROS generation (19, 165, 328).
As ROS generation endures, mitochondrial oxidative stress heightens (19, 165, 328) and various kinase pathways are stimulated in mesangial cells, including protein kinase C (PKC) (154), protein kinase B (PKB)/Akt (97), PDK-1 (20), and c-Jun N-terminus kinase (JNK) (60). The stimulated kinase activity alters gene expression in mesangial cells. The perpetuating generation of ROS and resulting signaling pathways induces ROS modification of podocyte and glomerular proteins, as demonstrated in patients with CKD (48). A recent report demonstrated that the ROS can also directly influence the permeability of the glomerular basement membrane and alter glomerular filtration (292). If ROS generation continues to worsen, oxidative stress promotes autophagy and apoptosis in podocytes (59, 134, 251) and mesangial cells (24, 189), promotion of profibrotic pathways and glomerulosclerosis, and interruption of the glomerular filtration barrier leading to proteinuria (292). The effects of ROS and RNS on cells of the renal glomerulus are illustrated in Figure 1.
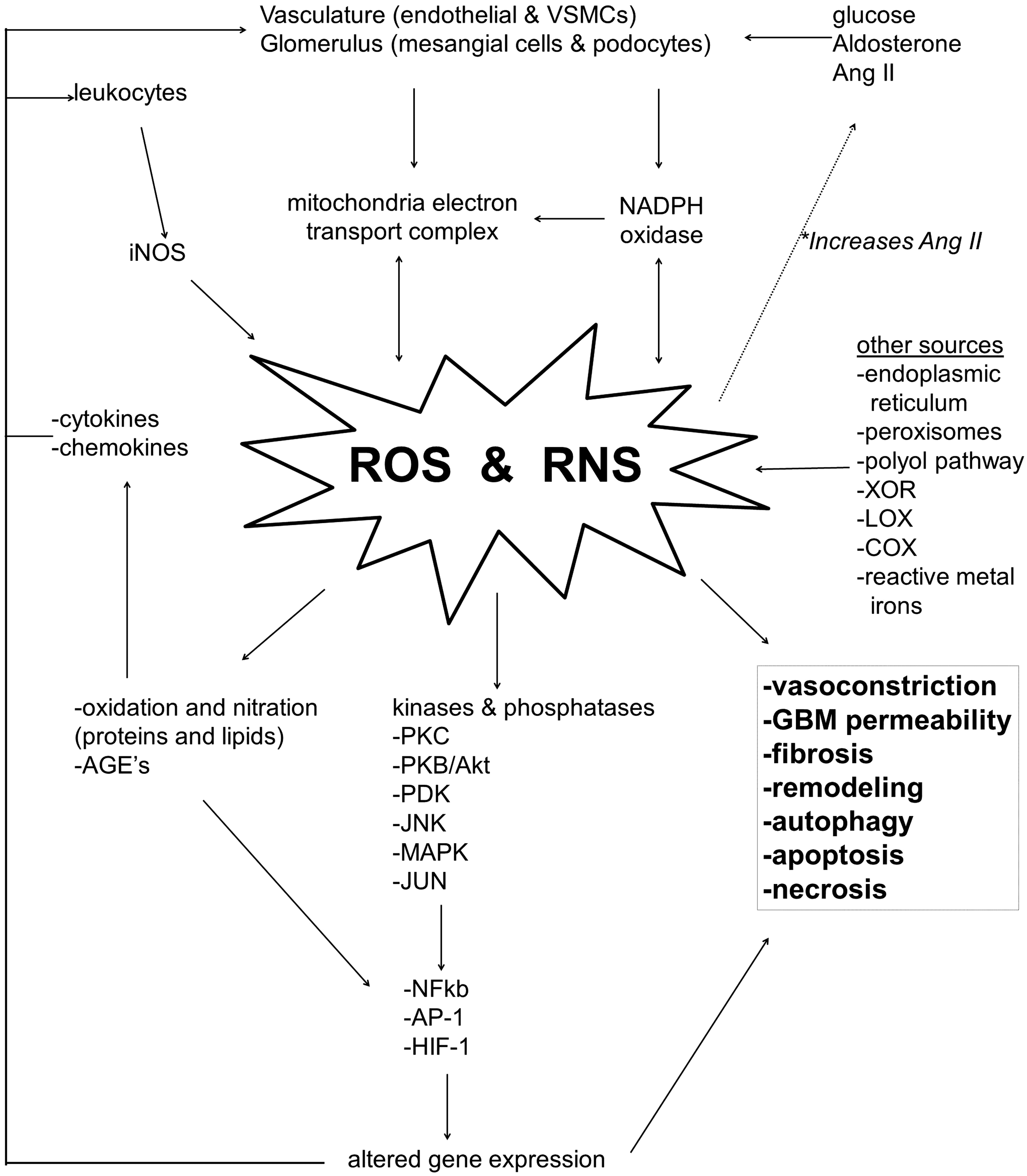
Infiltrating leukocytes
As inflammation proceeds in the murine kidney, stressed mesangial and endothelial cells help recruit leukocytes to the glomerulus and perivascular regions by releasing ROS and proinflammatory cyto-/chemokines, including monocyte chemoattractant protein 1 (MCP-1), monocyte colony-stimulating factor 1 (CSF-1), RANTES (regulated upon activation, normal T cell expressed and secreted), and intracellular adhesion molecule-1 (ICAM) (269). Consequently, leukocytes infiltrate the kidney and contribute significantly to the prodamage oxidative milieu. Despite the contribution of ROS by endothelial and mesangial cells, the main source of oxidative and nitrosative stresses in the glomerulus and perivasculature during kidney injury, such as that associated with sepsis, is derived from invading immune cells. Infiltrating leukocytes (including neutrophils and macrophages) invade the glomerulus and perivascular segments, release large amounts of proinflammatory cytokines such as interleukin-1beta (IL-1β) and TNFα, and undergo a respiratory burst, which includes myeloperoxidase-enhanced release of superoxide (primarily by NADPH oxidase), hydroxyl radical-like species, hypochlorous acid, hydrogen peroxide, nitric oxide (via inducible nitric oxide synthase [iNOS]), and nitrogen dioxide (260, 329). The myeloid cells and macrophages that enter the kidney from the circulation also release proinflammatory cytokines due to induced ROS-dependent activation of inflammasomes (6, 51).
The expression of iNOS is upregulated in rat mesangial cells during stress by various cytokines (including IL-1β and IFNγ) (64, 65, 202, 327), endotoxins (LPS), basic fibroblast growth factor (152), and by generators of ROS, including the hypoxanthine/xanthine oxidase (XO) system (14). High levels of iNOS-generated nitric oxide mediate proinflammatory effects by activating nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) and activator protein-1 (AP-1) regulated transcription and further promoting production of iNOS and macrophage inflammatory protein-2 (MIP-2) (191, 313). The activity of iNOS continues to elevate RNS-induced toxic events in the kidney by nitrating proteins and lipids (231), an effect that further progresses renal damage in the rat (94, 96, 207, 208, 228, 304).
Controversy currently exists regarding the role of iNOS-generated nitric oxide in the glomerulus. Various laboratories using murine models have reported that nitric oxide and RNS participate in apoptotic and necrotic death in mesangial cells (24, 118, 189, 192, 265), resulting in loss of mesangial cells, matrix accumulation, and progression of glomerulonephritis (197, 316). In contrast, other reports have indicated that peroxynitrite and nitrosative stresses exert only minimal toxic effects on mesangial cells due to potential intrinsic defense mechanisms against RNS in these cells (265) and that inhibition of iNOS instead causes enhanced fibronectin deposition, hypercellularity, and proteinuria (268, 318). The discrepancy of the effects of nitric oxide appears to be related to its concentration. For instance, nitric oxide mediates renoprotective effects at a specific concentration range when its levels are low, but as iNOS-induced production of nitric oxide increases to pathological concentrations, its protective effects are lost and damage ensues. In comparison, vascular endothelial cells have demonstrated particular susceptibility to the toxic effects of peroxynitrite. Unlike their mesangial counterparts, endothelial cells in the glomerulus and perivascular structures do not appear to have a comparable protective system against nitrosative stress (230). Despite the conflicting reports, it appears, the generation of nitric oxide and ROS continues to play a part in their feedback loop effectively enhancing the generation and release of superoxide and hydrogen peroxide in leukocytes and glomerular mesangial cells. As tissue injury and inflammation progress, circulating and local tissue levels of cytokines such as IL-1β and TNFα (245) continue to increase, NADPH oxidase in renal cells is further stimulated, and ROS production becomes even more pronounced. These processes effectively amplify proinflammatory processes and tissue damage, causing cytoskeletal and vascular remodeling, fibrosis (25), and potential kidney failure.
After prolonged oxidative stress, an eventual shift in the balance of nitric oxide produced compared with ROS and RNS occurs in the renal glomerulus. During the shift, ROS and RNS generation is replaced by nitric oxide-dominated production in mesangial cells (231). It is believed that the shift results from the nitric oxide and ROS-induced amplification of cytokine-induced iNOS expression and subsequent upregulated nitric oxide generation in mesangial cells (14, 191, 230). As a result, ROS generation by NADPH oxidase is repressed by the presence of high amounts of generated nitric oxide (237), which effectively controls expression of the NOX1 homolog of NADPH oxidase via cyclic guanosine monophosphate (cGMP)-dependent mechanisms (237). Afterward, nitric oxide production eventually slows due to exhaustion of NOS cofactors and/or nitric oxide-mediated inhibition of iNOS (230).
Other renal ROS sources and their targets
There are various other sources of ROS in the kidney. The uncoupling of eNOS results in superoxide generation during pathological conditions, such as diabetes and inflammation, when the availability of biopterin is reduced due to its oxidation (50). The endoplasmic reticulum generates ROS as a by-product of protein folding and oxidation (173). Fatty acid breakdown by peroxisomes accounts for superoxide generation [reviewed by Vasko and Goligorsky (301)]. Defects in the polyol pathway produce ROS while reducing glutathione synthesis. The enzymes, XO, lipoxygenase (LOX), and cyclooxygenase (COX), also contribute to ROS generation in the kidney. It was demonstrated that COX-2 can enhance production of ROS and subsequently enhance apoptosis in cultured mesangial and proximal tubular cells (PTCs). In addition, COX-2 inhibition with nonsteroidal anti-inflammatories (NSAIDs) reduces 25% of total kidney ROS production in the rodent kidney (146).
Other contributors to oxidative stress in the kidney include metal ions such as Fe3+ and Cu2+. In a rat model of cisplatin nephrotoxicity, iron release was blamed for renal damage due to its redox activity. Redox-active iron catalyzes (via the Haber–Weiss reaction) oxidative damage to proteins and lipids (lipid peroxidation), events that are strongly associated with renal disease and its severity (187). In fact, the level of lipid peroxidation in the kidney is typically used as a marker for severity of kidney damage and can assist physicians in screening kidneys that are suitable for transplant. Increased lipid peroxidation and the subsequent formation and renal accumulation of advanced lipoxidation end products trigger proinflammatory pathways that include activation of the receptor for advanced glycation end products (125).
Amino acids that contain sulfhydryl groups, such as cysteine and methionine, are the major targets of oxidation due to the susceptibility of their thiol groups to become oxidized. The sulfhydryl groups in these amino acids can easily be oxidized by hypochlorous acid, peroxides, and peroxynitrite (187). Tyrosine, tryptophan, arginine, proline, lysine, and histidine are also susceptible to oxidation (168), while lysine is particularly susceptible to modification due to lipid peroxidation (103). The post-translational oxidative modification of proteins at these amino acids can alter the protein's function. For instance, the prodamage alarmin HMGB1 is rapidly released into the circulation from renal endothelial and epithelial cells after injury, as demonstrated in numerous rodent studies (38, 46, 159, 166, 242, 324). Three key cysteine motifs in HMGB1 dictate the protein's function. If these cysteine residues are reduced thiols, then circulating HMGB1 induces repair of the injured tissues (272, 310). However, as these cysteine residues become oxidized to disulfide bonds, the reparative effects of the protein are lost and instead HMGB1 promotes inflammation and damage (307, 330). Other examples include the regulation of kinases, phosphatases, and even histone deacetylases (HDAC) (246). The immediate cellular redox microenvironment dictates the thiol–disulfide redox state of cysteine residues within these enzymes, an effect that subsequently either activates or inhibits the enzyme's activity.
In addition to protein modification, ROS and RNS can oxidize carbohydrates (producing advanced glycated end products [AGEs]) and damage DNA. A strong correlation exists in human patients with severity of kidney disease and elevated levels of AGEs (16, 172, 185, 188, 226, 305). Circulating and cellular AGEs induce lipid peroxidation products in the kidney and/or bind to receptors, activate intracellular signaling pathways, including NFkB, and subsequently activate cytokine and ROS production (227, 273). These proinflammatory effects are worsened as AGE suppresses intracellular antioxidant systems, including the glutathione system, an effect which further promotes renal damage.
ROS-induced gene expression and fibrosis
The effects of ROS and RNS also induce kidney damage by influencing gene expression at the transcriptional level and altering protein function via post-translational modifications (13, 23, 232, 233, 276). Altered gene expression results from the induction of several redox-sensitive transcription factors (including NFkB, AP-1, and HIF-1) and the oxidation, nitration, and nitrosation of the potential 500 kinases and 200 phosphatases contained in the cells of the human kidney (23, 91, 176, 181, 233, 234). Oxidative stress downregulates protein tyrosine phosphatases, while upregulating protein tyrosine kinases, including JNK and p42/p44 (ERK-1/ERK-2) mitogen-activated protein kinase (MAPK) pathways (23, 123, 130, 151, 290), although nitric oxide-induced activation of soluble guanylate cyclase and generation of cGMP triggers phosphorylation events that may counteract some of these effects.
High-glucose levels, as occurs during diabetic nephropathy, lead to increased ROS in mesangial cells via activation of PKC (ROS can also directly enhance PKC activity constituting a positive feedback loop) (100). ROS-stimulated PKC signaling then subsequently activates MAP kinases in mesangial and endothelial cells, followed by activation of the transcription factors, NFkB and AP-1, altered expression of MCP-1 and metalloprotease 9 (63, 89, 230), and stimulation of the proinflammatory effects of heat shock proteins (HSPs) (238) and osteopontin (97).
ROS upregulation of transforming growth factor beta (TGF-β) in mesangial and endothelial cells stimulates SMAD signaling and subsequently enhances expression of collagen 1, 3, and 4, fibronectin, and plasminogen activator inhibitor-1, while attenuating activity of extracellular matrix (ECM) degradation factors (11, 126, 221, 230). It is well documented that activation of these signaling pathways leads to enhanced mesangial cell proliferation and hypercellularity, increased mesangial matrix formation, and renal fibrosis in mammals. Glomerulosclerosis also results from ROS generated as a result of aldosterone and Ang II signaling, which activates p38 MAPK and promotes smooth muscle and mesangial cell proliferation, production of fibronectin and collagen, and subsequent fibrotic expansion of the ECM (78, 97, 165, 184).
Oxidative Stress in the Renal Tubules
Proximal tubule
Within the nephron, the proximal tubule is a major site of ROS generation due to its immense production of ATP by oxygen consumption to support the massive active transport that takes place in these epithelial cells. The PTC requires ATP for critical Na+/K+ ATPase, which maintains the intracellular–extracellular electrochemical gradient that drives the transport processes of several factors (175). The PTCs produce the required ATP by highly oxidative glucose metabolism, which renders these cells sensitive to oxidative stress due to a highly active mitochondrial electron transport chain. To make matters worse, studies on rats have indicated that unlike other segments of the nephron, proximal tubules cannot synthesize glutathione, but instead depend on circulating glutathione for antioxidant protection against mitochondrial-generated ROS (311). The enhanced vulnerability of PTCs to oxidative damage plays a central role in the renal pathogenesis of conditions such as diabetes.
During diabetes and hyperglycemia, excessive glucose is filtered and reabsorbed by PTCs by sodium-dependent glucose transporters (175). The excessive availability of glucose enhances glycolysis, which leads to a high mitochondrial proton gradient in PTCs, resulting in the elevated production and release of mitochondrial superoxide and ROS (203). Increased glucose uptake also stimulates PKC-dependent activation of NADPH oxidase (which is highly expressed in PTCs, particularly NOX4), resulting in excessive production of superoxide by NADPH oxidase (21, 262). In addition to high glucose, elevated levels of Ang II (acting on the AT1 receptor) also cause a rise in NADPH oxidase-derived superoxide generation in PTCs (primarily by NOX4) (224).
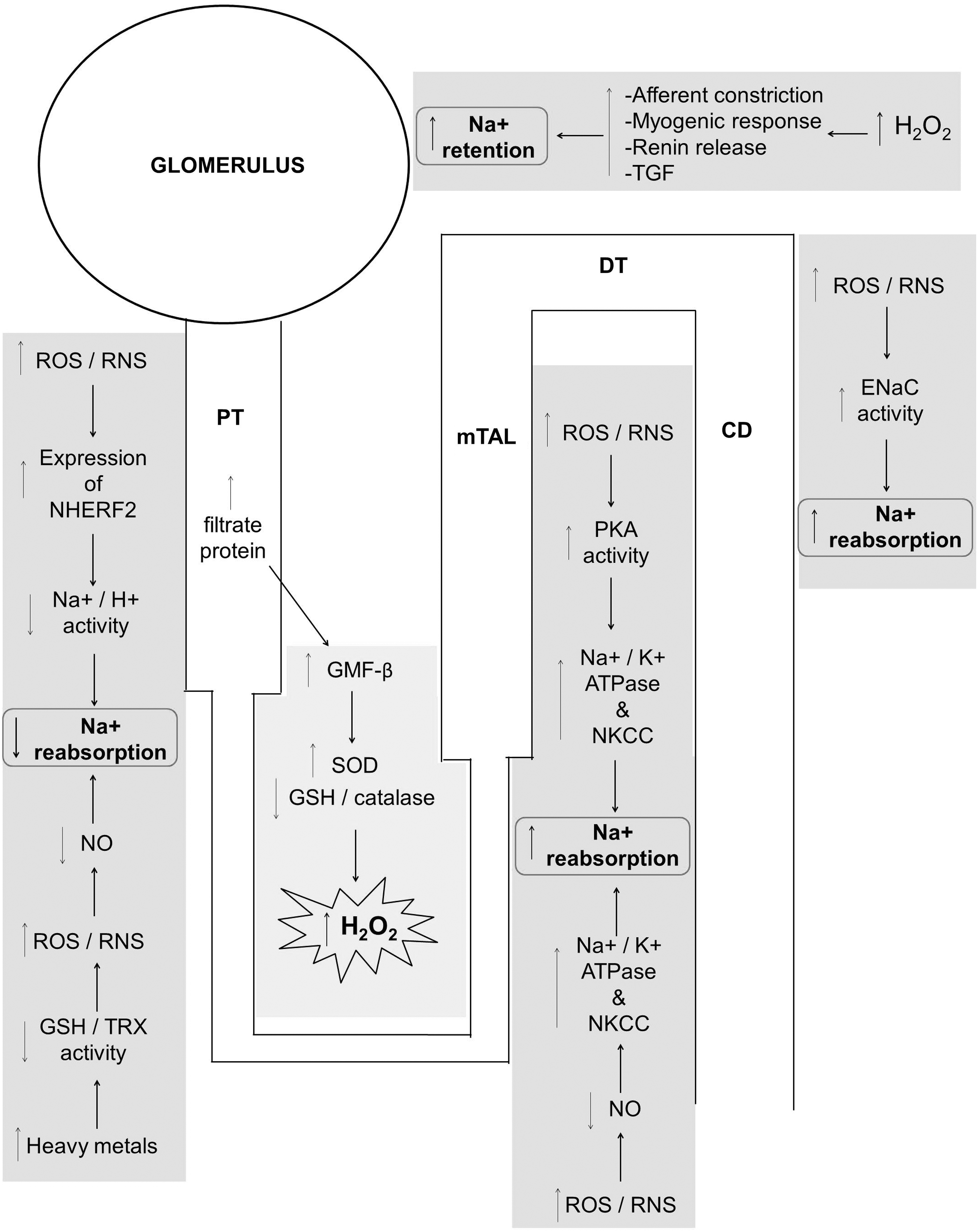
Enhanced generation of superoxide and ROS reduces Na+ reabsorption in the proximal tubule of rats by increasing the expression of the Na+/H+ exchanger regulatory factor (NHERF2), which inhibits Na+/H+ exchanger (NHE3) activity (224). Nitric oxide typically enhances Na+ and fluid reabsorption in the proximal tubule, thus the observed reduction of Na+ reabsorption that occurs in the proximal tubule appears to also be a result of reduced availability of nitric oxide, possibly due to NOS uncoupling and/or nitric oxide reaction with increased superoxide. Thus, various signaling factors converge synergistically on the proximal tubule to blunt Na+ reabsorption. However, Na+ reabsorption is compensated for in other segments of the nephron downstream from the proximal tubule as superoxide enhances Na+ reabsorption in the medullary thick ascending limb (mTAL) and distal nephron, in contrast to the effects of ROS in the proximal tubule (224).
In addition to diabetic nephropathy, the proximal tubule is severely damaged during various toxin-induced nephropathies. For example, nephrotoxic substances, including antibiotics (e.g., gentamicin), antivirals (e.g., tenofovir), immunoglobins, radio-contrast media, heavy metals (e.g., cadium), and chemotherapy reagents such as cisplatin (293), directly injure the proximal tubule. Many of these toxins induce ROS and RNS in the proximal tubule (43, 297) and inhibit the mitochondrial electron transport chain, prompting mitochondrial damage and consequently disrupt tubular function. Heavy metals can react with antioxidants and/or their cofactors and effectively inhibit their activity. Heavy metals have been shown to deplete antioxidant systems, including the glutathione and thioredoxin systems, thus allowing for accumulation of ROS. Through the Fenton reaction, metals can also induce the formation of highly reactive prodamage hydroxyl-like radicals. As a result, the enhanced oxidative stress causes DNA damage, protein oxidation, and lipid peroxidation, factors that lead to cell dysfunction and/or cell death by either apoptosis or necrosis (293).
The effects of oxidative stress on apoptosis have been closely examined; however, the mechanisms by which oxidative stress induces necrosis are not yet completely understood. As oxidative stress becomes more severe and prolonged in the kidney, cells undergo necrosis along with apoptosis (Fig. 2). The specific redox signaling that determines if a cell will undergo apoptosis or necrosis is still not clearly defined. However, it is known that ROS and RNS promote apoptosis by directly activating caspase 3 and 8 (via cysteine oxidization) (26) and inducing death receptor clustering on the plasma membrane (70, 261). As the mitochondrial permeability transition complex fails, there is outer membrane permeabilization and cyt-c translocation. These events trigger caspase signaling, apoptosome formation, and apoptosis (70, 261). As oxidative stress continues, there is mitochondrial swelling, dysfunction, and failed ATP production, events that promote necrosis (44). ROS activation of JNK results in stimulation of both the proapoptotic apoptosis signaling kinase 1 (ASK1) (via JNK) (57, 164) and the necrotic death promoter poly(ADP-ribosyl) polyperase (PARP-1) (44). Necrosis also results from the enhanced release of proinflammatory factors, increased lipid peroxidation, and loss of membrane integrity, all factors directly induced by ROS and/or RNS (105, 162).
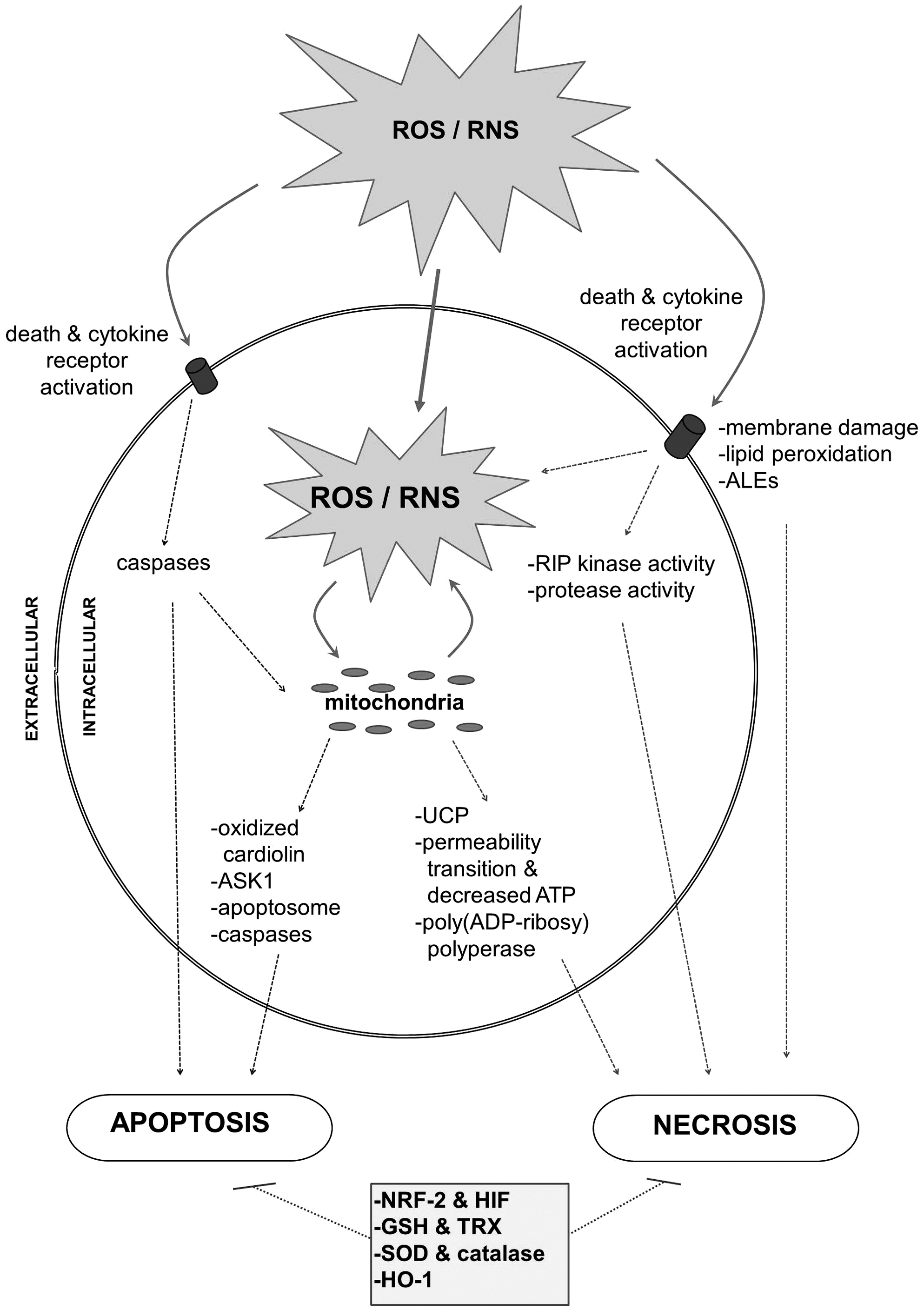
Similar to mesangial cells in glomeruli, ROS in PTCs stimulate the PKB/Akt, MAP, and Janus kinase phosphorylation cascade, which stimulates the activation of various transcription factors (including NFkB, AP-1, SP-1, and STAT) and upregulates expression of TGF-β (120). As discussed previously, the cytokine, TGF-β, plays a critical role in the development of renal fibrosis in many pathological states. Signaling of TGF-β via the SMAD pathway induces endothelial- and epithelial–mesenchymal transition, in which differentiated kidney cells lose their functional phenotype and instead become myofibroblasts. Once these cells transition into myofibroblasts, they begin producing excessive extracellular matrix proteins, including various collagens leading to tubulointerstitial fibrosis and impairment of kidney function (289, 338). Ultimately, the severity of tubulointerstitial fibrosis is closely related to both TGF-β levels and the progression of kidney disease (67).
Genetic tubular diseases
Oxidative stress in the proximal tubule also plays a role in the progression of human genetic renal diseases. For example, enhanced ROS formation has been linked to cystinosis and development of the proximal tubular disease Fanconi syndrome (81). More specifically, cystinosis is a disease that results in renal proximal tubular dysfunction and defective transport of cystine across the lysosomal membrane (293). The accumulation of intracellular cystine induces oxidative stress and reduces mitochondrial ATP generation, leading to PTC damage and apoptosis (248). The loss of renal PTCs leads to Fanconi syndrome, decreased activity of Na+/K+ ATPase activity in the proximal tubule, and ensuing attenuation of tubular transport of glucose, amino acids, electrolytes, and water (18, 76). Dent's disease, which is an X-linked recessive inherited condition and another cause of Fanconi syndrome, has also recently been suggested to be caused, in part, by enhanced oxidative stress (97).
Medulla
As the proximal tubule dives deeper into the cortex and into the medullary region of the kidney, levels of hydrogen peroxide are exacerbated by twofold, as was demonstrated in a rat model (42). The enhancement of hydrogen peroxide in the renal medulla is further exaggerated during hypertension (42) and proteinuria. In the case of proteinuria, proteins that leak into the filtrate consequently activate glia maturation factor-β (GMF-β) in the proximal tubule (140). Activated GMF-β increases the generation of hydrogen peroxide in and around the proximal tubules by upregulating hydrogen peroxide-producing enzymes (CuZn-SOD) and concomitantly attenuating hydrogen peroxide-reducing enzymes (glutathione peroxidase, glutathione, and catalase) (140, 293).
Depending on the vascular bed, hydrogen peroxide can elicit vasodilation, vasoconstriction, or a biphasic effect (41, 52, 85). In the rodent kidney, hydrogen peroxide reduces blood flow in the renal medulla and enhances contraction of the afferent arteriole to Ang II and ET-1 (250). These effects by hydrogen peroxide alter renal autoregulatory mechanisms by enhancing the myogenic and macula densa tubuloglomerular feedback responses, modify the expression and release of renin by juxtaglomerular cells (82, 83, 131, 250), and subsequently reduce sodium excretion (in contrast to the increased Na+ excretion that occurs in response to elevated ROS in the cortical proximal tubule) (247). The hydrogen peroxide-induced sodium retention results in salt-sensitive augmentations to blood pressure in the rat (321). This is further compounded by the ability of ROS to impair dopamine receptors in the tubules and decrease natriuresis (10, 288, 335). Furthermore, hydrogen peroxide directly affects apoptosis and cell survival by inducing Ask1 and activation of phospholipase A2 and the p38 MAPK pathway in PTCs (3, 53, 95, 194). These affects at least contribute, if not mediate, hydrogen peroxide promotion of cardiovascular and renal dysfunction (42, 286, 287).
Albumin
Serum albumin has been examined for its role in contributing or preventing oxidative stress in the proximal tubules. It appears that albumin has antioxidant capabilities at lower concentrations, but in contrast, it induces generation of ROS at higher levels. Albumin is the major protein constituent in plasma and undergoes small-scale filtration in the glomerulus. At physiological low concentrations, albumin protects renal PTCs by activating PKB, which leads to phosphorylation of the Bad protein and inhibition of apoptosis (31, 293). At low concentrations, albumin also effectively functions as an antioxidant by scavenging ROS and preventing formation of hydroxyl radical-like species via the Fenton reaction due to its binding to highly oxidative transition metals such Fe3+ and Cu2+ (102, 293). In addition to scavenging oxidants extracellularly, filtered albumin also protects PTCs upon endocytosis. For example, studies have shown that when albumin uptake by the PTC is reduced, the antioxidative effect of albumin is removed and subsequent oxidative stress ensues, followed by renal disease progression and tubulointerstitial fibrosis (81, 167, 254).
In contrast, at high concentrations, albumin promotes deleterious effects. During periods of glomeruli injury or damage to the glomerular filtration barrier, which results in augmented protein load in the tubular filtrate, there is excessive albumin endocytosis by PTCs. The high albumin load within PTCs activates PKC activity, followed by activation of (NOX4) NADPH oxidase and generation of superoxide. High levels of albumin also enhance its binding to fatty acids, which effectively impairs SOD2 activity in PTCs while promoting mitochondrial ROS production (128). The increase in ROS in the cytosol of PTCs activates STAT, NFkB, and AP-1 intracellular signaling cascades, leading to expression of proinflammatory genes and promotion of interstitial fibrosis in the kidney (293).
Medullary thick ascending limb
Continuing through the nephron, a large fraction of Na+ is reabsorbed from the filtrate in the mTAL (also referred to as the distal straight tubule), particularly when proximal tubule reabsorption is reduced. The apical Na+/K+/2Cl− cotransporter (NKCC) accounts for the bulk of Na+ reabsorption in this portion of the nephron (upward of 80%), with NHE3 handling the rest of Na+ reabsorption in this segment (8). During various episodes of renal stress and/or disease, this region of the nephron is subject to the adverse effects of oxidative stress.
In the mTAL, various sources contribute to the generation of superoxide, including XO-reductase, COX, and CYP450 enzymes, although the major producer of superoxide in this segment is NADPH oxidase (8, 88). NADPH oxidase is also the main source of superoxide generation in the mTAL in response to Ang II and increased luminal flow (8, 114, 274, 284). Superoxide generated in the mTAL of the rat kidney activates the PKA signaling pathway and stimulates Na+/K+ ATPase and NKCC activity, which subsequently enhances Na+ transport and reabsorption (218, 284). However, in contrast to its action in the proximal tubule, nitric oxide inhibits NKCC and NHE3 in the mTAL, thus effectively reducing Na+ reabsorption and potentially counteracting the superoxide-induced enhancement of Na+ reabsorption (87, 219, 220). It is expected then that since Na+ reabsorption in the mTAL is enhanced during periods of oxidative stress, nitric oxide availability is decreased, possibly due to NOS uncoupling or nitric oxide reaction with ROS to form RNS (86, 87). It also appears that the arachidonic acid metabolite, 20-hydroxyeicosatetraenoic acid (20-HETE), plays a role in ROS-dependent enhanced Na+ reabsorption in the mTAL. Increased ROS causes 20-HETE levels to decline, which in turn enhances mTAL Na+ reabsorption in rodent models by unclear mechanisms (110, 320).
Distal tubule and collecting duct
In the distal tubule and collecting duct, the vast majority of Na+ reabsorption occurs via the epithelial Na+ channel (ENaC). Ang II (acting on AT1 receptors) and aldosterone have been shown to increase ENaC activity in the collecting duct by NADPH oxidase-dependent generation of ROS (291, 336). High intake of NaCl has also been shown to increase ENaC activity by promoting hydrogen peroxide formation (291). Thus, ROS increases Na+ reabsorption in the distal tubule and collecting duct by stimulating ENaC.
Despite the decrease in Na+ reabsorption that occurs in the proximal tubule, oxidative stress overall results in sodium retention due to enhanced RVR, altered tubuloglomerular feedback, reduced GFR, and increased sodium transporter activity in the later portion of the nephron (Fig. 3).
Therapeutic Antioxidant Treatment
Despite reports that have demonstrated that oxidative stress plays a significant role in AKI and CKD along with cardiovascular disease, application of antioxidants clinically to patients with a variety of kidney injuries and diseases has been met with limited success. A major hurdle that poses a significant problem for any therapeutic treatment is the complexity of the multifaceted sources of oxidative stress and their dynamic interconnection. While resident renal cells, such as endothelial, mesangial, and tubular cells, generate ROS during stress, infiltrating leukocytes also produce damaging ROS and RNS as well. This makes cellular targeting of therapeutic antioxidants difficult due to the widespread nature of the cells involved in the process. This is further complexed by the generation of ROS and RNS in multiple compartments within the cell and in the extracellular environment. This means therapeutic treatments must specifically target both the source of oxidative stress and its specific cellular location.
To make matters more complex, low-level ROS signaling is required to maintain normal physiological function and for preservation of cellular homeostasis. Thus, complete inhibition of oxidative stress generation would impair the normal function of the cells by suppression of prosurvival and reparative signaling. Such is the case in regard to hydrogen peroxide. Low levels of hydrogen peroxide play a role in maintaining basal tone in vascular beds throughout the body, including the kidneys, thus its removal would impair normal hemodynamic function. Another example is nitric oxide, which also maintains normal vasoreactivity in vascular beds in addition to suppressing cellular apoptosis and leukocyte adhesion in vessels.
Regardless of the hurdles associated with developing effective antioxidant therapeutic strategies, the administration of various antioxidants in in vitro experiments and in animal models of both acute and chronic kidney injuries has shown therapeutic potential. However, when the same antioxidant molecules are tested in humans, the potential is not realized, and in some cases, these antioxidants worsened patient renal impairment and outcome. The conflicting results may, in some regard, be due to administration of antioxidants at doses that are not optimal. Table 3 lists some of the current and significant antioxidants that are being examined for their renoprotective effects.
AKI, acute kidney injury; Ang II, angiotensin II; AT1, angiotensin II type 1 receptor; CKD, chronic kidney disease; CoQ10, coenzyme Q10; ESRD, end-stage renal disease; NAC, N-acetylcysteine; NRF-2, nuclear factor E2-related factor.
N-acetylcysteine
While clinical trials to date have not been encouraging, there are a few antioxidant molecules that have demonstrated limited therapeutic efficacy in humans. The antioxidant compound that has undergone the most elaborate clinical testing is N-acetylcysteine (NAC). NAC elicits its potential therapeutic effects by replenishing intracellular levels of glutathione and also by direct scavenging of free radicals. While NAC has been shown to reduce lipid peroxidation, maintain mitochondrial membrane potential, and prevent apoptosis in various animal models (rats, pigs, and canines) (4, 135, 163) and cultured human PTCs (332), its use for the treatment of patients with CKD has been disappointing (186, 252). However, in patients with end-stage renal disease, NAC did reduce inflammatory cytokines (119, 198) while improving early outcomes in kidney transplant recipients (54).
Edaravone and ebselen
A variety of antioxidants, including edaravone and ebselen, have demonstrated the ability to improve renal function in animal models of kidney injury/disease, but these effects remain to be validated in human studies. Edaravone is an antioxidant that reduces levels of ROS, including hydroxyl radical-like and peroxyl-free radicals (2, 204, 339). Ebselen has the ability to metabolize peroxides by utilizing glutathione or through direct reduction of thioredoxin reductase and thus has been referred to as a glutathione peroxidase mimetic. While edavarone has not been examined thoroughly in human patients with AKI or CKD, it has been proven to be neuroprotective in stroke patients (117), while protecting renal function in a rat renal IRI model (61). The delivery of the seleno compound ebselen has demonstrated the benefits of scavenging nitrosative species. Ebselen is an antioxidant that scavenges peroxynitrite without effecting endogenous nitric oxide (195), making this antioxidant very attractive for treatment of oxidative stress-related pathological conditions. In various animal models of AKI, ebselen application prevented lipid peroxidation, enhanced endothelial and epithelial cell viability, and improved renal function (35, 39, 58, 94, 150, 207, 223, 228). However, the benefits of ebselen remain to be seen in human subjects.
Mitochondrial antioxidants
Various antioxidants that target and improve mitochondrial oxidative stress and function have been investigated. The molecule coenzyme Q10 (CoQ10), which is a component of cell membranes and plays a role in mitochondrial electron transfer and free radical quenching, has been shown to improve renal function in heminephrectomized rats (127). In humans, CoQ10 appears to prevent mitochondrial dysfunction, but more studies are still required to determine if CoQ10 is effective for treatment of CKD.
Other compounds that target the mitochondria have gained interest in recent years for treatment of kidney injury and disease, including mitoquinone mesylate (a derivative of CoQ10, termed MitoQ) and α-tocopherol-conjugated triphenylphosphonium (otherwise referred to as MitoVit E or mitochondrial targeted vitamin E). Animal studies with MitoQ have shown its ability to improve renal function during diabetic nephropathy, toxin-induced nephropathy, and IRI-induced AKI (34, 58, 150, 223). However, it remains to be seen if MitoQ improves renal function in human CKD patients.
Another mitochondrial targeting agent, the dithiol α-lipoic acid, is a cofactor for mitochondrial bioenergetic enzymes. α-Lipoic acid has been shown to be renoprotective in numerous animal studies that utilized various kidney injury models (47, 180, 275, 333). α-Lipoic acid preserves NOS function by preventing or reversing biopterin oxidation. The effects of α-lipoic acid improve endothelial function in diabetic patients (312) and may prove to be effective in preserving renal function in these patients as well.
Vitamin E
Another approach to combat the oxidative stress associated with kidney injury and disease is to enhance endogenous antioxidative systems by dietary supplementation. Supplementation should contain various metal ion cofactors (such as manganese, chromium, and selenium) and/or vitamins A, C, and E to potentially induce antioxidant benefits. Vitamin E supplementation has received considerable attention for treatment of various oxidative pathological conditions. Vitamin E incorporates into the plasma membrane, scavenges free radicals, and prevents lipid peroxidation (277). After vitamin E is oxidized due to scavenging of ROS (particularly peroxyl radicals), vitamin C reduces and effectively restores the antioxidant capabilities of vitamin E (79, 137, 138), followed by vitamin C restoration by glutathione and lipoic acid (98). Supplementation with vitamin E analogs has demonstrated beneficial effects for treatment of IRI-induced AKI in animals (69, 323). In humans, patients with CKD demonstrate reduced levels of vitamin E. However, clinical trials have reported both beneficial results upon introduction of vitamin E analogs, including reduced risk of CKD (22, 92, 129), and, in contrast, an absence of any beneficial effects (109) and even enhancement of mortality (122).
Naturally occurring antioxidants
While medicinal plants have been used for generations for treatment of various diseases, their therapeutic potential has yet to be fully explored in the clinic. Natural medicines may prove to possess a wealth of effective antioxidant molecules and/or antioxidant-stimulating capabilities that can be used for effective treatment of kidney injury and disease. For instance, the substance calendula has been isolated from the marigold plant and has been successfully used for treatment of cisplatin-induced nephropathy in a rodent model (240, 308), but its therapeutic potential remains to be explored in humans. There are numerous other plants and naturally occurring compounds with antioxidant properties, including carotenoids and phenolic compounds. Flavonoids (such as quercetin, soy isoflavones, and silibinin) represent one group of naturally occurring phenolic compounds that have been studied for their antioxidant capabilities, cardiovascular protection, and anti-inflammatory properties (72). While foods rich in flavonoids are recommended for patients with CKD, it remains to be seen if pharmacological flavonoid treatment offers advantages for patients with CKD.
Other polyphenolic compounds such as curcumin and resveratrol along with the anti-inflammatory compound, ethyl pyruvate, have displayed antioxidant capabilities that may be clinically useful for treatment of AKI and/or CKD. Curcumin improves kidney function in animal models of AKI by effects that include activating NRF-2 and HO-1 (108, 169, 299). Experiments conducted by our laboratory and others demonstrated the renoprotective effects of resveratrol and ethyl pyruvate in various animal models (36, 37, 46, 157, 174, 183, 243, 264). In our experiments using mouse models of sepsis- and IRI-induced AKI, we observed that the beneficial effects of resveratrol and ethyl pyruvate are attributed to their ability to scavenge ROS and prevent release of proinflammatory HMGB1 in the circulation (242). The renoprotective effects of resveratrol and sulforaphane (an isothiocyanate that is found in vegetables) are also mediated by their ability to upregulate NRF-2 signaling and protect mitochondrial function (149, 153, 200, 279). The upregulation of NRF-2 results in downstream stimulation of various antioxidants, including glutathione, thioredoxin, SOD, and catalase. Thus, these compounds are particularly attractive therapeutics due to their ability to scavenge both ROS and RNS while upregulating numerous antioxidant systems via NRF-2 stimulation.
Many of the naturally occurring compounds still require human studies to determine if they confer any relevant clinical effects for treatment of kidney disease. Indeed, in the case of resveratrol, human studies are currently ongoing. Another NRF-2 activator, bardoxolone methyl, was shown to successfully improve kidney function during AKI and CKD in both animals and humans (113, 229, 325). However, a phase 3 study, in which bardoxolone methyl was administered to patients with kidney disease, was recently discontinued due to the high toxicity of the drug and an associated increase in mortality.
Ang II blockers
Other pharmacological agents that have been previously shown to reduce oxidative stress in the kidneys and consequently improve markers of kidney disease are Ang II receptor blockers (141). It is well established that Ang II increases oxidative stress and inflammation, factors that are critically associated with renal pathogenesis, particularly due to hypertension and diabetes (258, 306). While infiltrating and resident kidney cells can produce ROS and RNS as a result of Ang II stimulation (7, 68, 133, 193, 225, 255, 256, 259), infiltrating immune cells are also able to directly increase Ang II levels due to their expression of angiotensinogen, angiotensin-converting enzyme, and renin (111). Angiotensinogen expression is enhanced in these cells by NFkB stimulation, an event that is increased by ROS, effectively creating a self-perpetuating positive feedback loop (5). The AT1 receptor modulates the effects of Ang II by stimulating NADPH oxidase production of superoxide (5) and mitochondrial ROS generation (155), effects that are exaggerated in CKD animals (147, 303). Ang II was also shown to inhibit NRF-2 signaling and reduce the glutathione system in renal epithelial cells (142).
Various AT1 receptor blockers already in use in the clinic for treatment of certain kidney diseases and injuries include losartan, telmisartan, irbesartan, and candesartan. Despite some adverse side effects that sometimes occur with the application of these agents to CKD patients, they are overall effective in improving kidney function and proteinuria. All of these AT1 blockers have been shown to reduce oxidative stress in the kidneys during disease and injury (145, 212, 267), with losartan downregulating expression of NADPH oxidase (9).
Footnotes
Acknowledgments
Support was received from AHA grant 12SDG9080006, ASN grant 010973–101, The New York Community Trust Renal Clinical Fund (B.B.R.), and NIH grant R01HL115124 (M.S.W.).