
Abstract
Introduction
D

ROS-Mediated Renal Inflammation in DKD
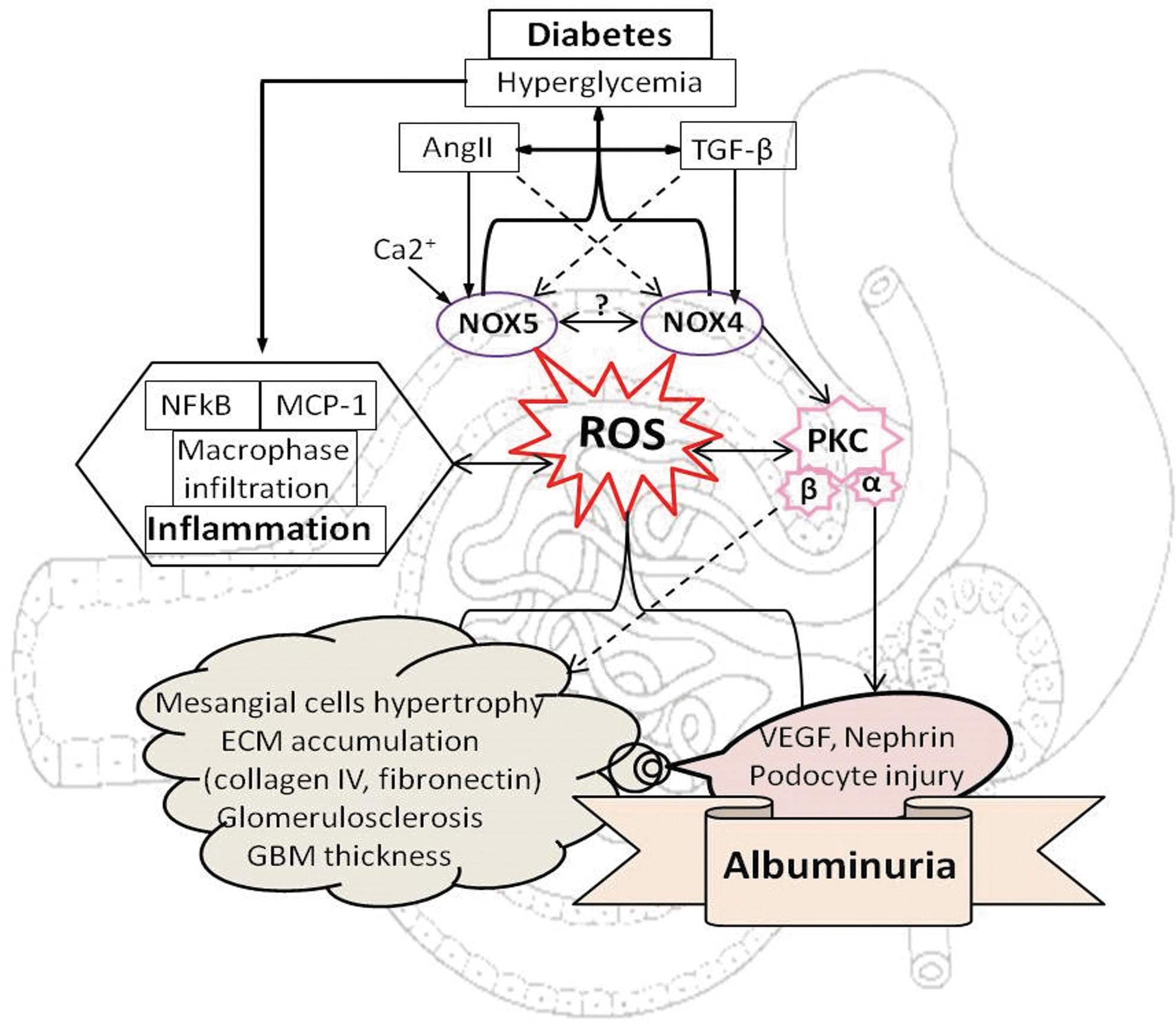
Under physiological conditions, ROS plays an important role in cell signaling implicated in proliferation, differentiation, apoptosis, and immune defense in various cell lineages, including renal cells (197). However, under pathological situations, including in diabetes, the overproduction of ROS in the kidney is implicated in renal inflammation, affecting renal structure and function and subsequently leading to ESRD. Hyperglycemia-induced ROS production stimulates the recruitment of numerous inflammatory cells and production of inflammatory cytokines, growth factors, and transcription factors implicated in the pathological processes of DN (Fig. 2). Excessive infiltration of macrophages and T cells plays a pivotal role in initiating renal damage in DN (46, 47). Activity and recruitment of these immune cells are often regulated by monocyte chemotactic protein-1 (MCP-1) (174). MCP-1 is predominantly expressed in renal monocytes, endothelial cells, and mesangial cells and is highly regulated by tumor necrosis factor-alpha (TNF-α) and interleukin (IL)-1 (174, 191). In addition, MCP-1 was found to be upregulated in the glomerulus (124) and tubulointerstitium (273) in experimental models of type 1 diabetes. Increased oxidative stress has been demonstrated to significantly induce macrophage recruitment and MCP-1 levels (273). Moreover, urinary excretion of MCP-1 and MCP-1 levels in renal biopsies were significantly elevated in diabetic patients compared with healthy control individuals (250). Of importance, a direct correlation exists between MCP-1 excretion and the level of albuminuria (261). Additionally, the levels of IL-6 and TNF-α were found to be positively correlated with the progression of renal disease. IL-6 plays a key role in promoting mesangial cell proliferation, ECM expansion, and increasing endothelial cell permeability (186, 205), while TNF-α has been shown to exert a positive feedback loop on ROS production (149, 275). Furthermore, numerous studies have implicated nuclear factor-kappa B (NF-κB) as the main transcription factor in the initiation of inflammatory responses in the diabetes milieu (182). NF-κB expression is increased in the kidneys in diabetic experimental models and activates mesangial cells to cause renal injury (106, 168). Key downstream effects of NF-κB include stimulation of adhesion molecules and expression of proinflammatory genes, including MCP-1, TNF-α, and IL-6 (168, 186, 261, 275), which are all implicated in the development of DN.
ROS-Mediated Renal Fibrosis in DKD
Renal fibrosis is an integral pathological process in chronic kidney disease, including DKD. Chronic exposure of hyperglycemia drives the formation and accumulation of ECM proteins (collagen I, IV, and fibronectin) and contributes to the pathology and dysfunction of the kidney (42) (Fig. 2). Increased ROS production, along with the activation of profibrotic growth factors such as transforming growth factor-beta (TGF-β) and connective tissue growth factor (CTGF), leads to the recruitment of ECM-producing cells, which drives the progression of renal fibrosis and sclerosis (16). Enhanced production of vasoactive agents, such as angiotensin II (AngII), endothelin, and urotensin, has been shown to increase expression of TGF-β in cultured renal cells and experimental animal models of DN (233, 272, 293). Furthermore, TGF-β upregulates plasminogen activator inhibitor-1, which decreases ECM degradation (15) and CTGF, an important downstream prosclerotic cytokine of TGF-β (218). In vitro studies have demonstrated that CTGF mediates TGF-β-induced elevation in the levels of fibronectin and collagen IV in renal cells (267). Although the kidney consists of at least 20 cell types, fibroblasts are increasingly recognized as the prominent matrix-producing cells leading to ECM accumulation (16, 240). Several in vitro studies have emphasized the importance of fibroblast activation, but it should be kept in mind that no single type of cell in isolation is capable of initiating and sustaining the full scale of renal fibrosis. Renal fibrogenesis clearly necessitates the participation and interaction of many types of resident kidney cells, as well as infiltrating cells. The underlying cellular events leading to fibrogenesis are complex, which involves the activation of mesangial cells (55), dedifferentiation of epithelial and endothelial cells (16), and infiltration of bone marrow-derived fibrocytes (27, 260). ROS play a critical role in profibrotic pathways, including stimulation of many growth factors and cytokines such as TGF-β1, CTGF, platelet-derived growth factor, and vascular endothelial growth factor (VEGF). Increased expression of these profibrotic factors in association with ROS results in an excessive buildup of ECM, which further exacerbates kidney injury (44).
Physiological Oxidants and Antioxidants
In the biological system, molecular oxygen undergoes a series of reductive biosynthetic steps forming several reactive oxygen intermediates commonly known as ROS, which are oxygen-derived, unstable, highly reactive, energized small molecules. ROS are either free radical molecules, including superoxide (O2 •−), hydroxyl (•OH), peroxyl (ROO•), and alkoxyl (RO•), or nonradicals such as hypochlorous acid (HOCl), ozone (O3), singlet oxygen (1O2), and hydrogen peroxide (H2O2). These nonradical ROS are oxidizing agents that are easily converted into free radicals (Fig. 3). Under physiological conditions, both endogenous and exogenous antioxidants interact with these oxidants to counteract the oxidative damage to cells (82). The antioxidant defense mechanisms include superoxide dismutase (SOD): manganese SOD and copper/zinc SOD; glutathione system: glutathione peroxidase and glutathione reductase; catalase; and coenzyme Q (Fig. 2). Antioxidant enzymes mainly convert ROS into nonreactive oxygen molecules, ultimately forming water. The entire antioxidant redox system mainly utilizes NADPH as a chemical reductant, which is mostly produced by glucose-6-phosphate dehydrogenase (61). During the process of sequential generation of ROS, superoxide is the primary reactive oxygen intermediate, which is rapidly converted to H2O2 by spontaneous dismutation as well as by SOD-catalyzed dismutation. The intermediate H2O2 is then transformed to water either by catalase or by glutathione peroxidase, which utilizes reduced glutathione produced by glutathione reductase. On the other hand, myeloperoxidase converts H2O2 to the very toxic hypochlorous acid and hydroxyl radical is formed when superoxide and H2O2 interact with iron by Fenton reaction (142, 253). In addition, superoxide reacts with the nitrogen-containing oxidant, nitric oxide (NO), to form highly reactive nitrogen species, peroxynitrite (OONO−), which reduces NO bioavailability causing NO toxicity to the cells (79) (Fig. 3).

Sources of ROS in the Kidney
Multiple intracellular mechanisms are involved in ROS production in the kidney, including xanthine oxidase, cytochrome P450 systems, uncoupled NO synthase (NOS), mitochondrial respiratory chain, and NOXs (92, 152) (Fig. 3). Under physiological conditions, xanthine oxidase produces an undetectable amount of ROS in the kidney via the purine metabolism pathway. Xanthine oxidase-derived ROS is found to be associated with endothelial dysfunction in patients with coronary disease and contractile dysfunction in heart failure (228). However, its contribution to DKD remains poorly understood. On the other hand, the cytochrome P450 system has an indirect role in ROS production in kidney. A study by Eid et al. demonstrated that cytochrome P450, particularly CYP4A, produces ROS via activation of NOX, causing renal cell injury and death in diabetic mice (74). Uncoupled NOS contributes to ROS generation, which leads to low levels of endothelial NO bioavailability, resulting in vascular endothelial dysfunction in DKD (105, 227). The most important source of these ROS in the kidney remains controversial. In the context of DKD, it has been suggested that mitochondrial generation of superoxide and NOX-derived ROS play a significant role in the kidney (84, 96, 128, 156). NOXs are particularly important as they have been identified to produce ROS not as a by-product, but as their sole biological function (18). In this section of review, we will discuss the involvement of ROS in the pathogenesis of DKD mainly derived from NOX and mitochondrial dysfunction.
NADPH Oxidases
Over the past several years, significant progress has been made to better understand the role of NOXs in renal ROS biology and the contribution of individual NOX isoforms in the pathogenesis of DKD. Under physiological conditions, most NOXs have very low or no constitutive activity, but their expression at the transcription or translational level may be increased, or the enzyme could be activated in disease states such as hypertension and diabetes. In these situations, the increased NOX-generated O2 − surpasses the handling capacity of the endogenous antioxidant system, thus leading to increased oxidative stress and ultimately tissue injury. In the second half of 20th century, it was demonstrated that NOX-derived superoxide is responsible for the oxidative burst in the phagocytes (11, 221). Later in 1986, gp91phox, commonly known as NOX2, was cloned as the first catalytic subunit of phagocyte NOXs (222, 249). After the identification of NOX2, several ROS-producing enzymes have been identified in the nonphagocytic cells, which differ at the molecular level, but share similar structural homology with phagocyte NOX, NOX2. These enzymes together are now considered as members of the NOX family. To date, seven known isoforms of NOXs—NOX2 (prototype NOX, formerly termed gp91phox), NOX1, NOX3, NOX4 (formerly termed Renox), NOX5, and dual oxidases (DUOX1 and DUOX2)—have been identified in various tissues. These NOX enzymes share common, conserved structural properties, which include the NADPH binding site at the very C-terminus, the flavin adenine dinucleotide (FAD) binding region in proximity of the C-terminal transmembrane domain, six conserved transmembrane domains, and four conserved heme-binding histidines (18). All NOX isoforms are transmembrane proteins that transfer electrons across the biological membrane from NADPH to reduce molecular oxygen to superoxide (O2 −) (10), as illustrated in Figure 3.

NOX Catalytic Isoforms and Regulatory Subunits
An assembly of individual catalytic isoforms of NOX and their respective regulatory subunits is required for the activation and thereby generation of superoxide in the tissue. The classic prototype NOX (NOX2) was first characterized in phagocytes and is a multicomponent enzyme consisting of the highly glycosylated membrane-bound catalytic subunits, gp91phox and p22phox, which associate with regulatory cytosolic subunits, p47phox, p67phox, p40phox, Rac GTPase, and Rap 1A, during the process of enzyme activation (Fig. 3) (23, 242). Following stimulation, the cytosolic subunits translocate to the membrane, where they interact with the flavocytochrome to form the activated oxidase, leading to transfer of electrons from NADPH to molecular oxygen, thereby generating large amounts of extracellular superoxide (242) that plays a pivotal role in host defense against microbial infections (2). Two Nox2 splice variants, named Nox2S and Nox2β, have been identified with Nox2β being suggested to have a functional role in the regulation of NOX activity in macrophages (109). Other NOX isoforms variably require the binding of distinct regulatory subunits for their activity. For instance, NOX1 binds to p22phox, Rac1, and to homologs of p47phox and p67phox called NOX organizer 1 (NOXO1) and NOX activator 1 (NOXA1). NOX1 shares 60% homology with NOX2 and was identified as the first homolog of NOX2 (13, 241). Two Nox1 splice variants, Nox1-L and Nox1-S, have been identified and are considered to be functionally distinct (108). NOX3 was first identified by Kikuchi et al. in 2000 and shares 56% homology with NOX2 (136). NOX3 can function as a heterodimer with p22phox, but its activity is enhanced by interaction with p47phox and p67phox, although it does not seem to require Rac GTPase (2). NOX4 was identified by Geiszt et al. in 2000 and initially characterized as a renal gp91phox homolog (91). However, it was found that NOX4 is relatively distinct in structure from other NOX homologs. NOX4 shares only 39% homology with NOX2 and 35% homology with NOX1 (91). The activation of NOX4 also requires interaction with p22phox, but differs from the other NOX isoforms, in that it does not require any other cytosolic regulatory subunit for ROS production. In addition, the interaction of NOX4 with Rac GTPase remains unclear (99). NOX4 appears to be constitutionally active, with regulation thought to occur mainly at the transcriptional level (167). Furthermore, interaction of NOX4/p22phox with Poldip2, a polymerase (DNA-directed) delta-interacting protein 2, has also been identified as a positive regulator in vascular smooth muscle cells (163). Interestingly, recent reports suggest that NOX4 may have a propensity for predominant H2O2 rather than O2 •− production, in marked contrast to NOX2 (64, 167). The biochemistry behind this is controversial and one hypothesis is that the superoxide produced by NOX4 is rapidly dismutated to H2O2, which makes it practically undetectable from superoxide released from NOX4 (16, 153). In addition, NOX4, unlike the other NOX isoforms, has an extended E-loop whose alteration can switch it from an H2O2-generating system into an O2 −-generating system. Depending on the cellular context, minor structural changes in the E-loop may influence the mechanism of ROS production by these NOX enzymes (247). In human lung tissue, four Nox4 splice variants, Nox4B, Nox4C, Nox4D, and Nox4E, have been identified, but it remains unclear which of these splice variants is the most important in the kidney (100). NOX5 is the most recently identified member of the NOX family and is structurally distinct. In humans, the Nox5 gene is found on chromosome 15, and five known splice variants of Nox5-α, -β, -δ, -γ, and a truncated variant (Nox5-S) (88) have been described, with the Nox5-α and -β splice variants producing functional ROS-generating proteins (19). Despite sharing significant homology to NOX1 and NOX2, NOX5 possesses an amino-terminal calmodulin-like domain with 4 binding sites for Ca2+ and does not require other subunits for its activation. The increase in intracellular calcium and the consequent binding of Ca2+ to NOX5 causes an increase in superoxide generation (180). On the other hand, the dual oxidases, DUOX1 and DUOX2, were first identified in the thyroid gland in 1999 and 2000, respectively, by Dupuy et al. (71) and by De Deken et al. (58). These enzymes share 50% homology with NOX2 and have a similar membrane domain to that of NOX1-4, an EF-hand region such as NOX5, and a seventh transmembrane domain at the N-terminus (49). In addition, two maturation factors, DUOXA1 and DUOXA2, have been identified in 2006 by Grasberger et al. (102). Unlike, other NOX isoforms, interaction of DUOX proteins with p22phox is unclear and these enzymes can be activated through Ca2+ without requiring any cytosolic regulators (57, 262).
Localization and Expression of NOX Isoforms in the Kidney
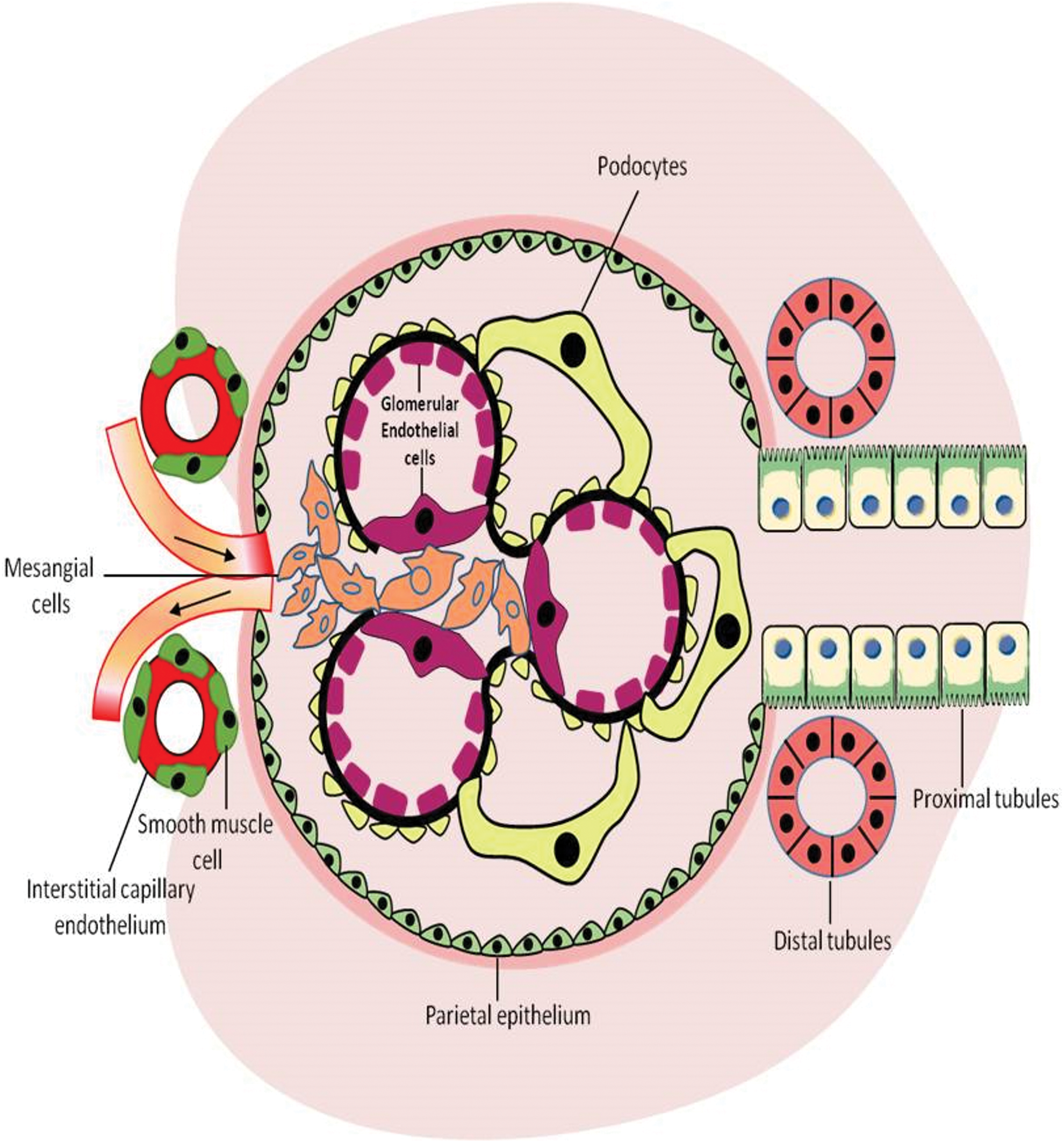
The NOXs are expressed in many tissues and mediate diverse biological functions, including regulating intracellular signaling. Initially, NOX1 was thought to be found in colon epithelium and to play a role in host defense and cell growth. However, recent studies demonstrate expression of NOX1 in variety of other cell types, including vascular smooth muscle cells, endothelial cells, uterus and placenta, and kidney (18, 124). NOX2 is the catalytic subunit of the respiratory burst oxidase in phagocytes and plays a role in innate immunity, but it is also expressed in nonphagocytic cells such as vascular, cardiac, renal, and neural cells (242); NOX3 is found in fetal tissue and the adult inner ear and is involved in vestibular function (12, 43). Current evidence indicates that NOX3 has a developmental role in otolith biosynthesis. Its absence causes a failure in this process, leading (in the adult animal) to vestibular dysfunction. Thus, the linkage between NOX3 and vestibular function is quite an indirect one (199). NOX4 was originally termed Renox (renal oxidase) because of its abundance in the kidney and thought to be specific to the kidney, but recently it has been also found in nonrenal cells, including osteoclasts, neurons, fibroblasts, vascular smooth muscle cells, cardiomyocytes, and endothelial cells (48, 63, 258, 269, 277). The subcellular localization of NOX4 remains controversial; this is partly because of the unavailability of reliable anti-NOX4 antibodies. Nevertheless, NOX4 has been shown to be associated with intracellular membranes of compartments or cell organelles such as focal adhesions, endoplasmic reticulum, plasma membrane, mitochondria, and nucleus (3, 18, 22, 113, 153, 167, 206). The human NOX isoform, NOX5, is a Ca2+-dependent homolog found in testis and lymphoid tissue, as well as in vascular and renal cells (14). While most NOX enzymes are present in rodents and humans, the mouse and rat genomes do not contain the Nox5 gene (14). Among these isoforms, the expression of NOX1, NOX2, NOX4, and NOX5 is identified in the target organs of diabetic complications, including blood vessels, retina, kidney, and peripheral nerve (18). Currently, there is no evidence of involvement of NOX3, DUOX1, and DUOX2 in diabetes and its complications. Although there has been enormous progress in the biochemical, chemical, and structural characterization of NOX isoforms, the regulation and function of each NOX remain unclear, but it is evident that NOX enzymes are critical for normal biological responses and that they contribute to end-organ injury in diabetes, cardiovascular and renal disease, hypertension, and atherosclerosis. Moreover, the relative importance of NOX1, NOX2, NOX4, and NOX5 isoforms in DN remains unclear. Experimental evidence shows that within the kidney, NOX1, NOX2, NOX4, and NOX5 isoforms are expressed in glomerular cells, including mesangial cells, vascular smooth muscle cells, endothelial cells, podocytes, tubular epithelial cells (proximal and distal tubular cells), and interstitial fibroblasts (92, 95). Localization and expression of NOX catalytic isoforms and its regulatory subunits in the renal tissues and cells are summarized in Table 1.
CCD, cortical collecting duct; MCD, medullary collecting duct; TAL, thick ascending limb of the loop of Henle.
NOX-Derived ROS in DKD
At basal levels, ROS may function as second messengers to influence redox-sensitive signal transduction pathways (237). However, in pathological conditions, including DKD, NOX-dependent overproduction of ROS leads to an imbalance of redox homeostasis in the kidney. Chronic hyperglycemia along with various diabetes-associated stimuli, including AGEs, AngII, and TGF-β, participates in increasing expression and the activity of various NOX isoforms, including NOX1, NOX2, NOX4, and NOX5, and thereby generating unwanted amounts of ROS, causing oxidative damage to the renal tissues (18, 21, 22, 74, 92, 96, 114, 124, 251, 252, 269). Thus, NOX isoforms may represent new therapeutic targets for renoprotection in DKD. In this section of the review, we will discuss the experimental evidences supporting the involvement and contribution of specific NOX isoform-derived ROS implicated in glomerular injury and tubulointerstitial fibrosis, both important features of DKD (Figs. 4 –6).


NOX-Mediated Glomerular Injury in DKD
All NOX isoforms have been found to be upregulated in glomerular cells (mesangial cells, endothelial cells, and podocytes) in response to high glucose in vitro as well as in experimental models of diabetes (1, 18, 21, 22, 74, 96, 97, 99, 114, 124, 164, 251, 252, 269, 294). Indeed, ROS (superoxide and H2O2) have been recognized to be important mediators of early mesangial cell hypertrophy, mesangial expansion, accumulation of ECM proteins, glomerulosclerosis, endothelial dysfunction, podocyte apoptosis, and subsequent development of albuminuria, as well as disruption of glomerular hemodynamics in DKD (1, 18, 21, 22, 74, 96, 97, 99, 114, 124, 164, 239, 251, 252, 269, 294) (Fig. 4). Among NOX isoforms, NOX4 and more recently also NOX5-derived ROS have been suggested to mediate glomerular injury and podocytopathy, leading to albuminuria, which is considered a key feature of DKD (21, 22, 96, 97, 99, 114, 124, 251) (Fig. 5).
NOX-Mediated Mesangial Hypertrophy, ECM Accumulation, and Glomerulosclerosis
NOX4
NOX4 is the most extensively studied isoform of NOX in the context of DKD and this is mainly because NOX4 is highly expressed in the kidney and is upregulated in diabetes. The contribution of NOX4-derived ROS in DKD perpetuating glomerular hypertrophy and mesangial expansion is supported by numerous experimental studies (95, 124, 164). Enhanced ROS level in response to high glucose was found to be associated with increased expression of Nox4 in the mesangial cells (22, 75, 203, 231). NOX4-derived ROS drives uncoupling of endothelial NOS (eNOS), and a decrease in NO bioavailability (eNOS dysfunction) in diabetes can initiate fibrotic injury to mesangial cells, suggesting that NOX4 functions upstream of eNOS (75). In addition, blockade of NOX4-dependent eNOS dysfunction by the antioxidant sestrin 2-dependent AMP-activated protein kinase showed an antifibrotic effect in mesangial cells exposed to high glucose (75). Another study has shown that high-glucose-induced ROS production by NOX4 in mesangial cells is mediated by thioredoxin-interacting protein (TxNIP) (231).
With regard to the in vivo studies, it is postulated that during the early stages of diabetes, increased expression of Nox4 and the subsequent ROS production in glomeruli of streptozotocin (STZ)-induced diabetic rats mediate oxidative damage to the glomeruli, leading to glomerular hypertrophy and increased fibronectin expression (96). In addition, blocking of ROS production in the renal cortex by treating with antisense oligonucleotides to Nox4 was reported to reduce fibronectin expression and ECM accumulation associated with reduced renal hypertrophy in diabetic rats (96). This finding was supported by another study from the same group in establishing a link between NOX4 and ECM production and has identified that the matrix metalloproteinase ADAM17 can modulate Nox4 expression in the kidney (85). Indeed, we have recently shown that global genetic deletion of Nox4 significantly attenuated the diabetes-induced increased mesangial expansion, glomerulosclerosis, and accumulation of ECM proteins (collagen IV and fibronectin) via a reduction in ROS production in a mouse model of type 1 diabetes (124, 251). In addition, deletion of Nox4 in diabetic mice was also associated with reduction in glomerular macrophage infiltration and downregulation of MCP-1 and NF-κB, important mediators of inflammation in diabetes (124). Furthermore, of clinical relevance, the administration of a novel NOX1/4 inhibitor, GKT137831 (
Involvement of AngII or TGF-β alone or in association with high glucose in the regulation of NOX4-derived ROS-induced signaling in renal cells, including mesangial cells, has also been reported by several studies (74, 117, 124, 166). Indeed, recently, TGF-β was found to induce both the activity of NOX and expression of Nox2 and Nox4, indicating that this growth factor induces production of ROS in kidney myofibroblasts (24). In addition, silencing of Nox4 markedly inhibited TGF-β-induced stimulation of NOX activity, and inhibition of transforming growth factor receptor 1 (TGF-βR1) blocked Smad3 phosphorylation, reduced TGF-β-enhanced NOX activity, and decreased expression of Nox4 (24). It is postulated that inactivation of 5′ AMP-activated protein kinase (AMPK) is implicated in driving the effect of TGF-β/NOX4 signaling on ECM accumulation (203). AMPK is a stress-induced protein critical in suppression of ROS and thus protects against oxidative stress in diabetes (33).
Renal tissue AngII activates NOX in mesangial cells and angiotensin-converting enzyme (ACE) inhibitors were shown to reduce renal and urine ROS production in STZ-induced diabetic rats (196). Another study has found a link between NOX4-derived ROS and AngII-mediated activation of Akt/protein kinase B (PKB) in mesangial cells (99). Recently, NOX4 was identified as a mediator of peroxynitrite-dependent eNOS uncoupling in response to AngII, suggesting the involvement of AngII/Nox4 in mesangial cells and in eNOS dysfunction, thus imposing fibrotic injury to mesangial cells (154). Involvement of AngII-induced generation of ROS acts through Nox4 as upstream activators of extracellular signal-related kinases 1 and 2 (ERK1/2), PDK-1, and Akt/PKB that lead to cell hypertrophy and increased fibronectin expression in the kidney (16, 21, 95, 96, 99, 154).
Furthermore, there is a link between NOX-derived ROS and certain PKC isoforms, including PKC-α and PKC-β, in the pathogenesis of DKD (93, 251). Indeed, overactivation of PKC-β was found to upregulate expression of TGF-β and to increase ECM protein components in the glomeruli of diabetic rats via NOX4-dependent ROS formation (147). Recently, we have shown that NOX4-derived ROS can activate PKC-α within the kidney, thereby promoting glomerular injury in experimental diabetes (125, 251). Another study has also shown a link between NOX-derived ROS, PKC-α, and advanced oxidation protein product (AOPP) in renal fibrosis. Exposure of AOPPs to mesangial cells resulted in increased expression of collagen IV, fibronectin, TGF-β, p47phox, p22phox, and Nox4, as well as increased activity of PKC-α, suggesting involvement of the PKC/NOX-dependent pathway in glomerular injury (266). However, it is still a matter of debate if NOX4 is upstream or downstream of PKC-α. Nevertheless, recently, we have shown that deficiency of Nox4 in podocytes was associated with a decrease in protein expression of PKC-α in diabetic mice with subsequent reduction in glomerular injury (125) (Figs. 4 and 5).
MicroRNAs are also important modulators of renal function and disease. Indeed, recent studies also show the association of NOX4 with expression of certain microRNAs, particularly microRNA-25 (mir-25) in diabetes-associated renal pathology (86). Downregulation of mir-25 in mesangial cells in response to high glucose or in the diabetic kidney was found to be associated with increased expression of Nox4 and subsequent increase in ROS production in the kidney (86). In addition, microRNA-146a attenuated high-glucose/thrombin-induced endothelial inflammation in human aortic endothelial cells by inhibiting Nox4 expression (264) (Fig. 4).
While the above evidence supports the fact that NOX4 plays a significant role in mediating glomerular injury in diabetes, its role remains to be more clearly defined. Babelova et al. (9) did not find an upregulation of Nox4 in the kidneys of C57/BL6 diabetic mice and Nox4 deficiency did not attenuate nephropathy. This may be due to the fact that C57/BL6 mice are relatively resistant to the development of the typical morphologic features of DN (28). Nevertheless, the majority of studies have indicated that contribution of NOX4 is critical in ROS-mediated renal glomerulopathy in diabetes.
NOX5
Few studies have investigated the involvement of NOX5-derived ROS in development of glomerular injury in diabetes. This is partly because of the lack of the Nox5 gene in mice or rats, making it difficult for experimental studies. However, recently, Holterman et al. (114) have shown increased glomerular injury in Nox5 transgenic mice with selective expression of Nox5 in the podocytes in the presence of diabetes and hypertension. However, the translation relevance of such transgenic approaches remains to be fully defined.
NOX2
NOX2-derived superoxide production from macrophages as well as from resident renal cells may also contribute to renal injury. Indeed, we have previously shown increased macrophage infiltration and expression of MCP-1 and vascular cell adhesion protein-1 (VCAM-1) in kidneys of diabetic ApoE KO mice in association with increased nitrotyrosine expression, a marker of oxidative stress (238). The expression of Nox2 and enhanced membrane translocation of p47phox were reported in the kidneys of diabetic mice (7). Supporting this statement, studies by Shi et al. reportedly demonstrated that AOPPs promote inflammatory responses in the diabetic rat kidney through the upregulation of membrane expression of Nox2 and p47phox (235). Moreover, a recent study has shown that deletion of Nox2 did not attenuate glomerulosclerosis in diabetic mice (281). However, our group has recently reported that global deletion of Nox2 in diabetic mice was associated with increased susceptibility to infections and 100% mortality at week 20 of diabetes (103) with animals surviving only when placed on prophylactic antibiotic treatment. Nevertheless, one cannot exclude a potential role for partial Nox2 inhibition, but the lethality of severe Nox2 deficiency in hyperglycemic states is likely to narrow the therapeutic window of such an approach.
NOX1
On the other hand, ROS derived from NOX1 has been implicated in the development of atherosclerosis (103, 234) and hypertension (171), but very little is known about the potential role of NOX1 in DKD. It was reported that AngII infusion promotes oxidative stress in the renal cortex of mice via increased mRNA expression of Nox1 and p22phox (35). A recent study has suggested that NOX1 plays a key role in the modulation of systemic and renal oxidative stress and redox-dependent signaling, but not in the elevation of blood pressure in a model of chronic AngII-dependent hypertension (279). In Dahl salt-sensitive hypertensive rats, which exhibit proteinuria and increased cortical collagen content, enhanced renal cortical expression of Nox1 and p22phox in association with activation of the ERK1/2 pathway was demonstrated (189). In addition, NOX1 was suggested to play a role in the modulation of renal oxidative stress and redox-dependent signaling of c-Src, p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), and focal adhesion kinase (279). In addition, a study by Gao et al. showed that silencing of Nox1, but not Nox2 or Nox4, suppressed the levels of superoxide and peroxynitrite, as well as reduced expression of TGF-β and fibronectin in human mesangial cells (90). However, recently, we have shown that deletion of NOX1 in diabetic ApoE KO mice did not provide protection against glomerular injury (124). These authors reported that NOX1-derived ROS-mediated fibrogenesis acts via activation of inducible NOS and the latter is involved in mediating AGE-driven ECM remodeling via peroxynitrite formation (39). Thus, the role of NOX1 warrants further elucidation.
Another important feature of DN is impaired contractility of mesangial cells in diabetes, thus altering normal glomerular hemodynamics and contributing to diabetic hyperfiltration. TRPC6 is a member of the TRPC family of Ca2+-permeable cation channels, which regulates Ca2+ signaling and participates in mesangial cell contractile function. In this study, Graham et al. found that NOX4-derived ROS in the hyperglycemic setting downregulates TRPC6 expression via activation of PKC-α, impairing the contractile function of mesangial cells and thus contributes to diabetic hyperfiltration (101). However, the role of TRPC6 in glomerular pathology was found to be cell specific. In particular, elevated TRPC6 channel activity mediated by increased ROS may account for podocyte apoptosis and diabetic glomerulosclerosis (161). With the knowledge that NOX5 is regulated by Ca2+ levels, the role of NOX5 in the contractile function of mesangial cells in diabetes warrants further investigation.
NOX-Mediated Glomerular Endothelial Dysfunction
Modification of glomerular capillaries and endothelial dysfunction are key contributors to epithelial cell injury in the progression of DKD. The hemodynamic alterations observed in early diabetes such as increased glomerular intercapillary pressure are partly attributed to endothelial dysfunction. With the glomerular endothelial cell strategic location at the interface between the blood compartment and the glomerular mesangium, it serves an important role in regulating glomerular microcirculation. In diabetic conditions, superoxide anions interact with NO, which is produced physiologically by constitutive sources such as the eNOS. This process leads to the formation of peroxynitrite (ONOO−), which binds to tyrosine and other protein residues yielding cytotoxic compounds such as nitrotyrosine (198). While diabetes is associated with reduced NO bioavailability, it is observed that in early stages of DN, eNOS expression in the kidney is increased (60). There is paucity of information on the involvement of NOXs specifically in glomerular endothelial dysfunction. Gene expression of the catalytic subunits, Nox1, Nox2, Nox4, and Nox5, and regulatory subunits, p22phox, p67phox, and p47phox, has been demonstrated in cultured endothelial cells (1, 127, 294).
Numerous studies have demonstrated that in the diabetic endothelium, eNOS represents the predominant source of superoxide, following activation of the Ang-II-dependent NOX pathway (38, 121, 193). Indeed, a study by Jaimes et al. (120) found that exposure of human renal glomerular endothelial cells (HRGECs) to either AngII or high glucose resulted in increased expression of cyclooxygenase 2 (COX2) and uncoupled eNOS, and this effect was prevented by the use of an NOX nonspecific inhibitor, diphenylene iodonium (DPI). eNOS dysfunction in the kidney has been reported in experimental models of type 1 diabetes. Indeed, eNOS KO diabetic mice appeared to have increased albuminuria and mesangial matrix expansion, as well as advanced renal lesions such as arteriolar hyalinosis and Kimmelstiel–Wilson nodules (184). Recently, NOX4-derived ROS was identified as a critical mediator in eNOS uncoupling in mesangial cells (75). This is intriguing given the current findings that NOX4 generates predominantly H2O2 rather than superoxide. This property of NOX4 has important implications in NO signaling since H2O2, unlike superoxide, does not react with NO to form peroxynitrite and may even stimulate eNOS activity. However, one explanation is that even if NOX4 generates relatively less superoxide than H2O2, the release of superoxide is still capable of reacting with NO to form peroxynitrite, leading to endothelial dysfunction. One cannot exclude the fact that effects of peroxynitrite on individual cells could occur not only as a result of production by the cells themselves but also by adjacent cells of a different lineage. For example, the effect of peroxynitrite in endothelial cells could have occurred as a result of peroxynitrite generated by adjacent cells, including podocytes and mesangial cells. Another possible explanation is that mesangial cells express not only Nox4 but also Nox1, Nox2, and Nox5. H2O2 may stimulate Nox1 or Nox2 or Nox5, which predominantly produces superoxide, thus ultimately causing endothelial dysfunction. Indeed, El Jamali et al. demonstrated a regulation of Nox2 and Nox5 by H2O2 through a Ca (2+)/c-Abl signaling pathway (77, 78).
Endothelial cells have also been shown to participate in glomerular inflammation. Glomerular endothelial cells in response to AngII was found to be associated with increase in ROS production and p47phox protein levels, as well as inflammatory parameters such as NF-κB and MCP-1 (201a). The link between inflammation and a specific NOX isoform, Nox4, has been previously suggested. Specifically, Nox4 downregulation using an siRNA approach attenuated lipopolysaccharide (LPS)-induced proinflammatory responses in human endothelial cells (204). Furthermore, uncoupling of eNOS was found to be intact in diabetic Nox2 KO mice (283). PKC-dependent activation of NOX4-derived ROS in endothelial cells in response to high glucose was also reported (66, 118, 278). A novel mechanism linking calcium to NOX5-drived ROS and ERK1/2 signaling, which is crucial for regulation of vascular function, has been identified in endothelial cells (180). Dysregulation of these processes could contribute to endothelial dysfunction and renal disease. However, the specific role for the recently described human isoform, NOX5, in endothelial cells in the context of diabetes needs further study.
Limitations on studies with regard to endothelial cells in the context of DN include lack of specific isolation of glomerular endothelial cells. Most studies relied largely on surrogate cell lines such as human umbilical vein endothelial cells (HUVECs) (127) and aortic endothelial cells (118). Endothelial cells are known to exhibit functional heterogeneity. Hyperglycemia-induced endothelial dysfunction is differentially regulated in endothelial cells from different parts of the vascular system. For instance, Karbach et al. demonstrated that HUVECs and EA.hy926 cells both represent cellular models for diabetic vascular complications such as increase in oxidative stress and dysfunctional NO signaling. However, these cell lines differ in their viability and superoxide production in hyperglycemic conditions (132). Wang et al. showed a disparity between the expression of growth factors and impaired angiogenesis between microvascular and aorta-specific endothelial cells (265). Furthermore, HUVECs and HRGECs have distinct biological properties and specific gene expression features in response to IL-1β. HRGECs demonstrated higher permeability and reduced expression of VE-cadherin. Moreover, glomerular endothelial cells remain to be fully characterized in diabetic and nondiabetic conditions. Thus, studies on endothelial dysfunction in diabetic renal disease perspective require more specific cellular or animal models.
NOX-Mediated Podocyte Injury, Apoptosis, and Albuminuria
While the earliest clinically detectable abnormality in DN is microalbuminuria that eventually progresses to proteinuria, very little is known about the cellular events that precede the onset of clinical albuminuria. The degree of proteinuria correlates with the progression of glomerulosclerosis and tubulointerstitial fibrosis of kidney. While glomerular hypertrophy, matrix accumulation, mesangial expansion, and GBM thickening are classical features of diabetic glomerular lesions, several preclinical and clinical evidences suggest that the onset of albuminuria is closely associated with podocyte injury, which includes foot process effacement, podocyte detachment, and depletion, as well as glomerular endothelial cell dysfunction (122). Podocyte apoptosis is an early glomerular phenotype in DKD that contributes to progressive depletion of podocytes and subsequent excretion of albumin in urine (245). Numerous studies have reported that excess levels of ROS induce podocyte apoptosis in a diabetic animal in vivo or in response to high glucose in vitro (96, 137). Indeed, NOX was found to be involved in podocyte depletion (74) and apoptosis through proapoptotic mediators, including p38 MAPK and caspase 3 in type 1 and 2 diabetic mice (245). In addition to hyperglycemia, other diabetes-related stimuli such as TGF-β and AngII have been found to be associated with podocyte injury and apoptosis via excess production of ROS causing cellular dysfunction leading to increased excretion of albumin in the urine (74, 225, 245). Podocyte injury has also been found to contribute to ECM accumulation and glomerulosclerosis in diabetes (65, 225). An increase in TGF-β can stimulate ECM synthesis by the podocyte itself in an autocrine manner or may reach mesangial cells to induce a sclerosing phenotype in these cells. In addition, AGEs and their receptor RAGE via induction of TGF-β have also been found to induce podocyte injury and apoptosis (271). It was also reported that systemic administration of a nonspecific NOX inhibitor, apocynin, prevented podocyte apoptosis and ameliorated urinary albumin excretion in diabetic mice (7, 245). Furthermore; high glucose stimulates hypertrophy of podocytes through ROS-dependent activation of ERK1/2 and Akt/PKB pathways (96, 137).
In the study by Durvasula (72), exposure of podocytes to high glucose resulted in increase in expression of AngII via elevation in renin activity and AT1R expression. In addition, exposure of podocytes to AngII has been found to upregulate expression of Nox4, Nox2, Rac, and p22phox (21, 190, 268).
NOX4
Numerous experimental studies demonstrate the deleterious effect of NOX4-derived ROS in podocyte injury, leading to albuminuria, foot process effacement, and loss of podocytes in DKD (74, 75, 125, 124, 135) (Fig. 5). We and others have shown enhanced ROS production through upregulation of Nox4 in podocytes in response to high glucose and TGF-β (74, 96, 124, 137, 212, 229). TGF-β/Nox4-derived ROS mediates activation of caspase 3 via the Smad2/3 pathway leading to mouse podocyte apoptosis (56). In another study, high-glucose-induced apoptosis of podocytes was found to be mediated via sequential upregulation of Nox1 and Nox4 by cytochrome P450 of the 4A family (CYP4A) (74). Nox4 was also shown to promote podocyte cell death by activating p53-upregulated modulator of apoptosis (PUMA)-dependent apoptotic pathways (73). Similar to mesangial cells, AMPK inhibits the mTORC1 pathway and was found to reduce NOX4-derived ROS in OVE26 type 1 diabetic mice, as well as in cultured podocytes exposed to high glucose (75). In addition, previously, we have shown that silencing of Nox4 in human podocytes in response to high glucose attenuated a range of inflammatory and fibrotic markers via reduction in ROS formation (124). A correlation between albuminuria and podocyte damage in diabetes has been documented by several studies. Previously, we have shown that global genetic deletion of Nox4 in ApoE KO and C57/BL6 STZ diabetic mice attenuated albuminuria and glomerulosclerosis (90). In addition, You et al. have shown that podocyte-specific induction of Nox4 in mice induces renal pathology, including glomerular hypertrophy, mesangial matrix accumulation, and GBM thickening, as well as albuminuria and loss of podocytes (282). More recently, we have demonstrated that deletion of Nox4 only in podocytes attenuates the diabetes-induced increase in albuminuria by ∼50% in diabetic mice (125). This finding strongly supports the view that NOX4-derived ROS in the podocyte play a significant role in the regulation of albuminuria in diabetes. Decreased expression of nephrin in diabetic mice has been found to be associated with GBM thickening, podocyte foot process effacement, broadening of slit pore, podocyte detachment, and depletion in number of podocytes in correlation with albuminuria (51, 68, 176). Indeed, we have shown that Nox4 deletion in podocytes preserved nephrin expression and protected from ultraglomerular structural damage, including foot process effacement (125).The effect of NOX-derived ROS formation on VEGF expression, a growth factor expressed by renal cells, including podocytes associated with increased vascular permeability to macromolecules and albuminuria in diabetes, has been confirmed by various experimental studies (52, 244, 251). Indeed, both global and podocyte-specific NOX4 deletions were found to be associated with decreased renal VEGF expression in diabetic mice. In addition, we and others have shown increased expression of VEGF in podocytes in response to high glucose and silencing of Nox4-attenuated VEGF expression (124, 244, 251). Furthermore, a recent report suggested that blockade of VEGF signaling by a receptor kinase inhibitor ameliorates diabetic albuminuria in db/db mice (244). These findings strengthen the postulate that NOX4-derived ROS play a crucial role in the modulation and regulation of VEGF expression in podocytes. It has been demonstrated that acute hyperglycemia increases nephrin and endocytosis in a PKC-α-dependent manner (215) and this effect is considered to promote albuminuria. Indeed, we have shown a direct link between diabetes-induced PKC activation and NOX4-derived ROS production in the podocytes in association with albuminuria (125, 251).
There is a close connection between diabetes and renal inflammation in DKD. Indeed, NOX4 deletion has been found to reduce macrophage infiltration and decrease expression of the key proinflammatory transcription factor NF-κB and the chemokine MCP-1 in renal cortex. In addition, high-glucose-induced upregulation of NF-κB and MCP-1 in podocytes was found to be attenuated by Nox4 silencing (124). Furthermore, the diabetes-induced increase in glomerular MCP-1 expression attenuated in podocyte-specific NOX4-deficient diabetic mice. This indicates that targeting NOX4 in podocytes not only prevents podocytopathy but may also play a key role in attenuating intrarenal inflammation. It is apparent from these studies that NOX4-derived ROS mediate podocyte damage, albuminuria, and ultimately other markers of renal injury.
NOX5
While the evidence for a role of NOX4 mediating podocyte injury and apoptosis in association with albuminuria in diabetes is clear, the role of NOX5 in podocyte dysfunction is currently getting more attention. A recent study by Holterman et al. found increased expression of NOX5 protein in human kidney material from diabetic patients (114). They reported that AngII stimulates NOX5-dependent ROS production in human podocytes and in conditionally immortalized mouse podocytes transfected with adenovirus-expressing Nox5 (Fig. 5). De novo human Nox5 expression in mouse podocytes induced actin cytoskeleton rearrangement and Rac1 activation, which led to increased cellular motility. The increased cellular motility was thought to be comparable with podocyte foot process effacement associated with development of albuminuria (175). This study also found that ROS generated by NOX5 is additive when podocytes are costimulated with high glucose combined with AngII. The role of NOX5 in triggering podocyte damage was confirmed in vivo using podocyte-specific Nox5 transgenic mice. Transgenic mice expressing Nox5 in a podocyte-specific manner (Nox5pod+) exhibited renal dysfunction, including early onset of albuminuria and podocyte foot process effacement (114). These findings support our own observations (unpublished data) that exposure of human podocytes to high glucose increases the expression of Nox5 and silencing of Nox5 resulted in attenuation of markers of fibrosis (collagen IV, fibronectin) and inflammation (MCP-1) via reduction in ROS formation. The relative contribution of individual isoforms, NOX4 and NOX5, in podocyte injury needs further investigation.
NOX1
Based on published data, it is unlikely that NOX1 plays an important role in podocyte dysfunction. Instead, our own data suggest that Nox1 deletion in ApoE KO diabetic mice did not attenuate albuminuria (124). This could be because of the low expression of Nox1 in the kidney, specifically in the podocyte. However, mTORC1-derived increased expression of NOX1 protein was reported in cultured podocytes in response to high glucose (75).
NOX2
The involvement of NOX2 in the development of albuminuria remains unclear. It was reported that insulin-deficient diabetic Nox2 KO mice, despite a reduction in macrophage infiltration, did not demonstrate reduced albuminuria as well as glomerular ECM accumulation and tubulointerstitial fibrosis, suggesting that this lack of renoprotection may be due to upregulation of renal Nox4 (284). However, a recent study has shown that administration of antioxidant, probucol, was associated with attenuation of albuminuria, ECM protein collagen IV accumulation, and podocyte damage via inhibition of NOX2 expression. In addition, another recent study examining a mouse model of fibrosis demonstrated that p47phox KO mouse attenuated albuminuria and glomerulosclerosis via reduction in ROS (263).
NOX-Mediated Tubulointerstitial Injury in DKD
Not only glomerular cells but also renal tubular cells are adversely affected by diabetes. Changes in podocytes, which participate in the initiation of glomerulosclerosis and leakage of plasma proteins, and tubulointerstitial fibrosis are other key events in the progression of DKD. It is believed that downstream of the glomeruli, exposure of plasma proteins in association with chronic hyperglycemia across the tubular compartment of the nephron can trigger profibrotic and proinflammatory mechanisms in tubular epithelial cells, thereby inducing the development of tubulointerstitial fibrosis (200). Tubulointerstitial fibrosis is characterized by accumulation of interstitial fibroblasts and excessive ECM deposition in the tubulointerstitial space (119), ultimately leading to disrupted tubular reabsorption. The proximal tubular epithelial cells are considered to be major players in orchestrating renal interstitial fibrosis in DN (172). Similar to glomerular injury, NOX-derived ROS are involved in the process of tubulointerstitial fibrosis in diabetes (Fig. 6). Indeed, inhibition of NOX activity by apocynin was found to result in reduction in renal gluconeogenesis via activation of the ERK1/2 pathway in rat proximal tubules exposed to high glucose as well as in Zucker diabetic fatty rats (270), suggesting the implication of NOX-derived ROS in renal glucose regulation. In addition, NOX-dependent ROS in renal tubular cells in response to high glucose is found to be associated with stimulation of MAPKs and the redox-sensitive transcription factor, NF-κB, leading to upregulation of the proinflammatory gene MCP-1 (276). One of the potential mechanisms for renal fibrosis is epithelial–mesenchymal transition (EMT), in which there is a transdifferentiation of epithelial cells into motile mesenchymal cells (16). Hyperglycemia along with TGF-β, AngII, CTGF, albumin, and AGEs induces EMT in renal tubular cells with upregulation of alpha-smooth muscle actin (α-SMA) and vimentin and downregulation of E-cadherin (34, 40, 154, 275).
NOX4
There is experimental evidence to suggest that the expression of Nox4 in the tubular epithelium, particularly in the proximal tubule, is predominantly comparable with other renal cells in diabetes (Fig. 6). Indeed, expression of NOX4 protein and ROS production were found to be increased in the kidneys of diabetic mice and NOX4 expression was largely localized to the tubules (229). Increased protein levels of NOX4, but not of NOX1, NOX2, p22phox, or p47phox, were found in mouse proximal tubules in response to high glucose (80, 229). NOX4-derived ROS formation in mouse proximal tubules exposed to high glucose activates profibrotic processes via NOX4-sensitive p38 MAPK-dependent pathways (80, 229). Excessive deposition of ECM in the tubulointerstitium has a significant impact on the progressive decline in renal function in chronic kidney disease. Indeed, NOX4-dependent ROS production and subsequent Akt phosphorylation resulted in accumulation of fibronectin in proximal tubular epithelial cells in response to high glucose (187). The view that NOX4 is the major source of ROS in the tubular compartment of the diabetic kidney has been supported by our own study demonstrating that genetic deletion of Nox4 in ApoE KO or in C57/BL6 diabetic mice markedly reduced ROS production in renal cortex, which mainly comprises tubular epithelial cells (124, 251). In contrast, this effect was not seen upon Nox1 deletion in the renal cortex of diabetic ApoE KO mice. TGF-β is a known profibrotic growth factor mediating tubular injury via interaction of TGF-β with NOX4. Lee et al. have shown that NOX4-mediated ROS generation in the tubular cells in response to TGF-β was ameliorated by activation of the AMPK pathway (154). In addition, inhibition of NOX4 by plumbagin (a nonspecific NOX4 inhibitor) or silencing of Nox4 attenuated expression of fibronectin and collagen IV in human proximal tubular cells exposed to high glucose and TGF-β (280). Contribution of the matrix metalloprotease (ADAM)-17 in matrix accumulation and renal fibrosis has been suggested in diabetes. Increased expression and activity of ADAM17 in association with upregulation of Nox4 were observed in mouse proximal tubular cells in a high-glucose environment. Interestingly, inhibition of ADAM17 resulted in attenuation of ROS production and fibrosis in diabetic OVE26 mice via downregulation of Nox4, suggesting a role in Nox4-mediated oxidative stress and kidney injury (85).
Another growth factor contributing to ECM protein accumulation in DKD is insulin-like growth factor-I (IGF-I). IGF-I was found to accelerate the fibrotic process via NOX4-derived ROS and activation of Akt/PKB and mTOR/p70S6K signaling pathways in renal tubular epithelial cells (187). Similar to the glomeruli, both PKC-α and PKC-β in association with NOX4 contribute to tubulointerstitial fibrosis in diabetes (194, 251). Furthermore, a recent study has found that chronic exposure of albumin to renal tubular cells induces inflammation through the HSP70-TLR4 axis in mice with early DN (126). Interestingly, in response to glycated albumin, an early stage glycation product, rat tubular epithelial cells demonstrated accelerated fibrosis and apoptosis and silencing of Nox4 ameliorated these changes (214). A recent study by He et al. (110) demonstrated that high-glucose-induced EMT is attenuated by decreasing intracellular ROS levels via downregulation of Nox1 and Nox4, but not of Nox2, and thereby activating the ERK signaling pathway in human tubular epithelial cells. It was also reported that the inhibitory effect of AMPK on TGF-β, AngII, and high-glucose-induced EMT was mediated by suppression of Nox4 expression (158). TGF-β seems to play a critical role in myofibroblast differentiation via activation of Smad2/3, Akt/PKB, and ERK signaling pathways (8, 169). In addition, the TGF-β/Smad2/3 cascade is tightly regulated by the activation of MAPK in mesangial cells and fibroblasts (285). Rhyu et al. found that NOX-dependent ROS is integral in activation of TGF-β/Smad2/3 pathways in proximal tubular epithelial cells (217). Another study using renal interstitial fibroblasts demonstrated that TGF-β/NOX4-induced ROS generation contributes to myofibroblast differentiation to a profibrotic phenotype via activation of Smad 3 and ERK pathways (24). A recent study by Manickam et al. has shown that TGF-β-induced kidney fibroblast differentiation to a myofibroblast is mediated by Rho/Rho-associated protein kinase (ROCK) via activation of NOX4 (165). Furthermore, stimulation of renal tubular epithelial cells with AngII was shown to induce apoptosis and EMT via NOX4-dependent ROS production and resultant Src/caveolin-mediated activation of the epidermal growth factor receptor/ERK signaling pathway (40, 138, 207, 276).
NOX5
Emerging evidence suggests a contribution of NOX5-derived ROS in tubulointerstitial fibrosis. A recent study by Holterman et al. showed early tubulointerstitial fibrosis in podocyte-expressing Nox5 mice even in the absence of diabetes. This was further accelerated in the presence of diabetes (114). In another study, albeit in the hypertensive context, Yu et al. have demonstrated that Nox5 expression is predominantly increased in human proximal tubules obtained from hypertensive subjects and showed increased ROS formation compared with proximal tubules of normotensive individuals. In addition, silencing of Nox5 in hypertensive cells reduces ROS production and fibrosis (284).
NOX2 and NOX1
The importance of NOX2 in mediating tubular injury in diabetes is controversial. Increased expression of Nox2 in the renal cortex was shown in insulin-deficient diabetic rats (96), but the expression of Nox2 and p47phox was not affected in type 2 diabetic mice (229). In addition, You et al. (281) demonstrated that Nox2 KO mice did not attenuate tubulointerstitial injury in insulin-deficient diabetes. The importance of NOX1 is largely unknown in tubulointerstitial fibrosis, including in diabetes. From these findings, it is apparent that NOX4 and more recently NOX5-derived ROS seem to have predominant implications in the molecular mechanisms underlying fibrosis in DN.
NOX-Mediated Tubular Reabsorption Dysfunction in DKD
Diabetes is associated with renal Na+ retention and expanded extracellular fluid volume. Volume expansion is largely responsible for hypertension in diabetes and contributes to the altered renal hemodynamics. The increased urinary albumin excretion is not only due to leakage from the altered glomerular compartment but also due to a decrease in albumin reabsorption by the proximal tubule (200).The precise molecular mechanisms underlying the increase in Na+ retention and proteinuria remain unresolved. However, NOX-derived ROS is proposed to be involved in this pathological process.
NHE3
Reabsorption of salt and water in proximal tubules mainly occurs by the Na+/H+ exchanger isoform 3 (NHE3). In addition, NHE3 also plays a role in receptor-mediated albumin uptake in the proximal tubule. Hyperglycemia and AngII-mediated activation of NOX and subsequent increased ROS production activate NHE3 activity in the proximal tubules in diabetes leading to alteration in tubular Na+ transport (116, 143). This effect was evident by decreased Li+ clearance, a marker of proximal tubular reabsorption after NOX inhibition (210). Importantly, NOX-mediated superoxide was found to modulate the activity of NHE3 in proximal tubules in diabetes (210). These findings suggest that aberrant activity of proximal tubular NHE3 and albumin uptake may confer renal salt/water retention and increased excretion of protein in DKD.
SGLT2
The bulk of glucose filtered by the glomerulus is reabsorbed in the early proximal tubule by the sodium glucose cotransporter, SGLT2, which is expressed mainly on the apical membrane of renal proximal tubules, whereas SGLT1 removes the remaining luminal glucose in the distal proximal tubule (259). Glycosuria can be observed in diabetes; however, glucose reabsorption by SGLT2 is enhanced under diabetic conditions. Oxidative stress regulates the activity of SGLT2, with selective inhibition of SGLT2 that increases glycosuria in diabetes. Interestingly, insulin stimulates SGLT-2-mediated glucose entry into cultured proximal tubular cells via oxidative stress (185). The involvement of NOX-derived ROS in SGLT2-mediated glucose reabsorption remains to be evaluated. Increased levels of ROS formation in proximal tubules in response to high glucose have been shown to be mediated through AngII and subsequent activation of the NOX system; therefore, it is hypothesized that high glucose and AngII regulate renal SGLT2 via NOX, particularly NOX4.
NOX-Derived Markers of Oxidative Stress in Urine
Several preclinical and clinical studies have identified the presence of a range of oxidative stress markers in the urine of diabetic individuals or diabetic animals with nephropathy, including proteinuria and loss of podocytes (137, 179, 251). These urinary oxidative stress markers include 8-oxo-7,8-dihydro-2-deoxyguanosine (8-OHdG), which is a product of oxidative DNA damage and F2 isoprostane 8-iso prostaglandin F2 (8-iso PGF2), which is a widely recognized marker of lipid peroxidation in patients with diabetes. In addition, our own studies have shown that activation of NOX, particularly NOX4-derived ROS, contributes to the generation and subsequent excretion of these oxidative stress markers in the urine of diabetic animals (125, 251).
NOX and Therapeutic Perspectives for DKD
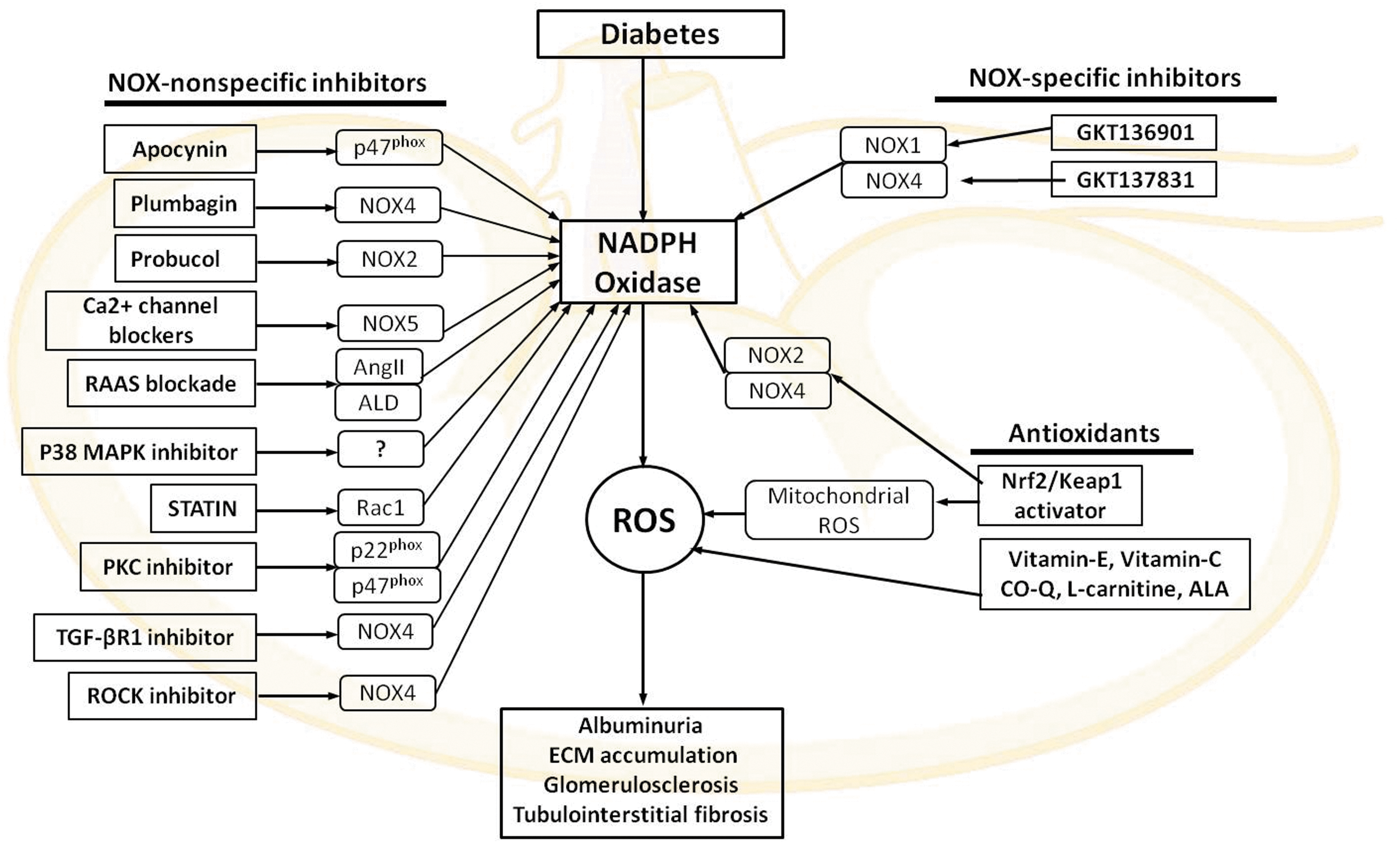
Numerous clinical studies have shown that antioxidant therapies failed to improve the health of patients with cardiovascular and DKD. This is partly because of the lack of information about the specificity and the mechanism of action of these antioxidants. This raises many questions in relation to our current knowledge of the molecular processes involved in ROS formation. The use of dietary antioxidant supplements and selective inhibitors of various enzymatic sources of ROS, including NOX inhibitors, has been employed to combat oxidative damage of tissue (195). NOX has been shown to be a potential target for pharmacological intervention in DKD. In this section, we will update the information about both NOX-specific inhibitors and nonspecific antioxidants that have been tested in diabetes (Fig. 7).
NOX-Specific Inhibitors
GKT
The new advancement in ROS/NOX biology has shown some degree of progress in developing NOX-specific agents. Some of these compounds, considered NOX-specific inhibitors, have shown promising results at the preclinical level. Recently, pyrazolopyridine compounds, NOX-specific agents named GKT136901 and GKT137831, have been developed by Genkotex (
The expression of NOX isoforms varies in different tissues with NOX4 predominantly expressed in the kidney, whereas NOX1 is more widely expressed in the macrovasculature. Treatment of diabetic ApoE−/− mice with GKT137831 attenuated not only renal disease but also atherosclerosis in short- and long-term studies, suggesting that concomitant NOX1 and NOX4 inhibition can provide simultaneous athero- and renoprotection. Considering the presence of NOX5 in patients, the role of NOX5 requires further studies. Isoform-specific NOX inhibitors are currently being developed and it needs to be shown if isoform-specific NOX inhibitors will provide superior protection in individual organs or if combined inhibitors such as GKT137831 are superior because of simultaneous protection of several organs.
NOX Nonspecific Inhibitors
Apocynin
Initially, apocynin was presented as a pharmacological inhibitor of NOX. Administration of apocynin to STZ-induced diabetic rats resulted in a decrease in plasma lipid peroxidation products, renal H2O2 production, and urinary protein excretion via inhibiting the translocation of p47phox from the cytosol to plasma membranes in the kidney (7). In addition, another report suggested that apocynin attenuated albuminuria and prevented podocyte apoptosis, podocyte depletion, and mesangial matrix expansion in db/db diabetic mice (245). However, recently, it was reported that apocynin is not a specific inhibitor of NOXs, but rather a nonspecific antioxidant. In HEK293 cells overexpressing Nox1, Nox2, or Nox4, apocynin failed to inhibit superoxide generation detected by lucigenin chemiluminescence (112). Furthermore, another study has also suggested that apocynin does not inhibit vascular NADPH oxidase-dependent superoxide formation, but rather may have alternative or additional actions, including inhibiting Rho kinase activity (226).
Plumbagin
Plumbagin is a plant-derived compound and has been considered to be an Nox4 inhibitor. Plumbagin has shown antiatherosclerotic and renoprotective effects in diabetic animals (67). Administration of plumbagin attenuated albuminuria, accumulation of ECM proteins, and ROS production via downregulation of Nox4 in a mouse model of DKD (280). However, the mechanism of action and specificity toward NOX4 remain questionable. Further studies are needed to fully characterize the specific action of these drugs on NOX activity in human DN.
GLX351322
Recently, another agent called GLX351322 presented as a specific NOX4 inhibitor has shown to counteract glucose intolerance in high-fat diet-treated C57BL/6 mice (5). However, the compound specificity toward NOX4 remains to be fully evaluated.
Probucol
The antioxidant, probucol (a clinically used lipid-lowering drug), has reported to possess cardioprotective effects in experimental models of heart failure via its antioxidant properties and an enhancement of endogenous antioxidant reserve (76). Recently, it was demonstrated that probucol inhibits NOX2 expression and attenuates podocyte injury and albuminuria in a type 2 diabetes mouse model of nephropathy (291). The calcium-dependent NOX, NOX5, is recognized to be an important source of ROS in atherosclerosis and hypertension, as well as in DKD (107, 114). Administration of calcium channel blockers significantly reduced albuminuria levels in patients with DN (256). Currently, there is no direct evidence suggesting a direct effect of Ca2+ channel blockade on NOX5 inhibition.
RAAS blockade
ACE inhibitors and Ang-II AT1 receptor blockers are currently employed to delay the decline of renal function in DKD. ACE inhibitors and ARB suppress renal ROS production and p47phox expression in association with attenuation of microalbuminuria in STZ-induced diabetic rats (196). The inhibition of NOX downstream of RAS may offer a novel approach to delay the onset of renal dysfunction. In addition, administration of eplerenone, an aldosterone inhibitor, to diabetic animals with obesity, hypercholesterolemia, and hypertension was found to be associated with attenuation of albuminuria and renal ROS production via reducing NOX activity (183).
p38 MAPK inhibitor
It has been reported that p38 MAPK is increased in the kidney of Dahl salt-sensitive hypertensive rats. A specific inhibitor of p38 MAPK, FR-167653, blocked phosphorylation of p38 MAPK and reduced NOX activity and superoxide production in the kidney, resulting in reduction of proteinuria and glomerulosclerosis (255). In experimental animal models and diabetic patients, renal p38 MAPK is increased and has been suggested to enhance early hypertrophy and ECM accumulation (130); therefore, p38 MAPK inhibition may represent an alternative strategy against DN.
Statins
Several reports have demonstrated beneficial effects of inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase, the so-called statins, in DN. Besides their cholesterol-lowering effect, statins suppress membrane translocation of Rac and inhibit NOX stability on the plasma membrane (254). In db/db mice with type 2 diabetes, treatment with pitavastatin ameliorates albuminuria and mesangial expansion by minimizing oxidative stress via downregulation of NOX4 expression in the kidney (87). Thus, it is possible that statins could improve DN via their effects on NOX4. In addition, other drugs used in diabetes and hyperlipidemia, including gliclazide and fibrates, also inhibit NOX and reduce oxidative stress, but further studies are necessary to assess the direct renoprotective effect of these agents.
Inhibitor of PKC
Both PKC-α and PKC-β play an important role in diabetic renal pathology via activation of NOX. A recent study has demonstrated that a conventional PKC (PKC-α and PKC-β) inhibitor, Gö6976, suppresses increased expression of p22phox, p47phox, and collagen IV, along with reduced ROS production in mesangial cells under high-glucose conditions (274). Despite these promising observations, it must be noted that PKC has many roles in the phosphorylation of multiple proteins that control cell permeability, gene transcription, and other cellular responses. Thus, inhibitors of PKC activity are not specific for NOX and could have off-target effects if used systemically.
Inhibitors of TGF-βR1
TGF-β is well recognized to regulate the expression of Nox4 in various renal cells in response to glucose. Silencing of Nox4 in kidney myofibroblasts inhibited TGF-β-induced stimulation of NOX activity, and inhibition of TGF-βR1 blocked Smad3 phosphorylation, reduced enhanced NOX activity, and decreased expression of Nox4 (24). In a recent study, it was demonstrated that administration of soluble human TGF-β type II receptor (sT_RII.Fc) provided renoprotective effects by reducing fibrosis and albuminuria through neutralizing TGF-β (223). However, the systemic administration of TGF-β blockers is not feasible for the treatment of DN because of the immunoprotective effect of TGF-β. On the other hand, blocking of TGF-β may have important hemodynamic effects, which are relevant to diabetic complications (173).
ROCK inhibitor
Recently, it has been reported that treatment with Fasudil, a specific ROCK inhibitor in diabetic rats, significantly reduced albuminuria and decreased levels of urinary 8-hydroxyguanosine, a marker of oxidative stress (94). This finding was supported by another study, in which the administration of an ROCK inhibitor to db/db mice, a model of type 2 diabetes, was associated with decreased albuminuria, mesangial expansion, accumulation of glomerular type IV collagen, and GBM thickening via downregulation of TGF-β and NOX4 (144). However, these findings lack specificity toward Nox4.
Endogenous and Dietary Antioxidants
Keap1-Nrf2 pathway
Endogenous antioxidants play an important role in combating oxidative stress in various diseases, including DKD. The transcription factor NFE2-related factor 2 (Nrf2) is considered a regulator of cellular detoxification responses and redox status. Nrf2 in combination with its negative regulator, Kelch-like ECH-associated protein 1 (Keap1), activates the antioxidant response element, leading to increased expression and activity of several antioxidants, including glutathione S-transferase, hemeoxigenase-1, and γ-glutamylcysteine synthetase, and NADPH quinone oxidoreductase (133, 134). Indeed, a recent study has suggested that the Keap1-Nrf2 pathway regulates both mitochondrial and cytosolic ROS production through NOX (146). Nrf2 was found to be upregulated in response to hyperglycemia in the kidney and specifically in certain renal cells (290). Nrf2 activators, such as sulforaphane or cinnamic aldehyde, were shown to attenuate damage and preserve renal function in diabetic mice, indicating that Nrf2 might be a valuable therapeutic target in DN (290). In addition, experimental evidence suggests that Nrf2 KO mice or silencing of Nrf2 in renal cells aggravates accumulation of matrix proteins in diabetes (59). Bardoxolone methyl, an Nrf2 activator has anti-inflammatory effects, and after an initial promising clinical trial study known as the BEAM trial in patients with DN, the subsequent phase 3 trial was prematurely terminated (BEACON; Bardoxolone Methyl Evaluation in Patients With Chronic Kidney Disease and Type 2 Diabetes) because of higher cardiovascular mortality in the treated group (ClinicalTrials.gov NCT01351675) (208, 209). In addition, several trials using dietary antioxidant supplements containing vitamins (e.g., vitamin E, vitamin C, coenzyme Q10, L-carnitine, α-lipoic acid) have been conducted to combat oxidative stress-induced tissue damage in diabetes; however, a meta-analysis on 14 studies on type 2 diabetic individuals showed that many of the trials failed to improve the disease outcome (4) and some had deleterious effects. Hence, one has to be cautious with the use of these dietary antioxidants as their mechanism of action and function remains poorly understood.
Mitochondrial ROS in DKD
Growing number of evidences suggest that mitochondrial respiratory chain-derived ROS are crucial mediators in the progression of DKD (53). It is unknown whether mitochondrial deregulation is a cause or consequence of renal dysfunction, but it appears to be part of a vicious cycle perpetuating ROS generation and end-organ injury. Under normal physiological conditions, mitochondria generate substantial amounts of superoxide caused by electron leakage from the oxidative phosphorylation pathway. However, in diabetes, excess glucose uptake by susceptible cells leads to more glucose-derived pyruvate being oxidized in the citric acid cycle in mitochondria that pushes more electron donors (NADH and FADH2) into the electron transport chain causing electron leakage, resulting in overproduction of superoxide (30, 188). In addition, the hyperglycemic state impairs the activity of enzymes of the mitochondrial respiratory chain complex. As a result, a positive feedback loop of oxidative stress occurs over time leading to mitochondrial dysfunction, cell injury, and apoptosis. Current existing data demonstrate that mitochondrial ROS (mtROS) produced by increased flux through oxidative phosphorylation induce adverse effects in DN, including cell apoptosis, hypertrophy, mesangial matrix expansion, podocyte apoptosis, and inflammation (111, 245), as illustrated in Figure 8. Increased formation of mtROS in the kidney has been observed in experimental diabetes (37, 53, 220, 245). It is postulated that the excess mtROS is mediated by electron leakage at two main sites: complex I (NADH:ubiquinone oxidoreductase) and at the interface of complex III (ubiquionol:cytochrome c oxidoreductase) (20, 145, 211). Indeed, mitochondria from renal cortex of diabetic rats demonstrated diminution of oxidative phosphorylation via a decrease in respiratory complex III activity and increased superoxide production (219). Moreover, opening of the mitochondrial permeability transition pore induced ROS production at the level of the respiratory chain complex I (17). In diabetes, the AGE-RAGE interaction induces H2O2 production, facilitating the induction of mitochondrial permeability transition and promoting complex I deficiency of the mitochondrial respiratory chain (53). Mitochondrial dysfunction is associated with reduced adenosine triphosphate (ATP) production. Indeed, decreased renal mitochondrial ATP production has been observed in the renal cortex of diabetic mice (248). Proximal tubular cells contain a large number of mitochondria owing to their many functional roles, including energetically active reabsorption of glucose from the urinary filtrate. Active transport is key to the reabsorptive processes in the proximal tubule. In diabetes, to maintain the glucose gradient, a greater pool of ATP is demanded. Inability to deliver ATP-driven transport can lead to impairment of proximal tubular epithelial cells in diabetes. Indeed, ATP depletion in proximal tubular cells is associated with impaired solute and ion transport as well as with a disruption of the actin-based cytoskeleton and disturbances in the apical basolateral protein polarization and apoptosis (26).

A central role for mitochondrial dysfunction and oxidative stress in DKD involves fibrosis along with infiltration of inflammatory cells. This is supported by a number of studies demonstrating activation of fibrotic and inflammatory pathways in DKD by mtROS. Indeed, in glucose or AGE-stressed podocytes, mtROS activate the NLrp3 inflammasome, which has been shown to be induced in DKD (232, 292). Under conditions of cellular stress, Nlrp3 induces caspase-1-mediated responses, including secretion of proinflammatory cytokines such as IL-1beta and IL-18 (162). Inhibition of mtROS prevented glomerular inflammasome activation and DN in mice. Moreover, Nlrp3 has been shown to be a crucial element of EMT of tubular epithelial cells, a process associated with tubular atrophy and interstitial fibrosis in chronic kidney disease (162). ROS activates a range of other pathways implicated in renal fibrosis. Inhibition of mitochondrial electron transfer chain complex I, rotenone, significantly blocked high-glucose-induced ROS generation in mesangial cells and high-glucose-driven fibronectin upregulation in tubular epithelial cells (156). Treatment with MitoQ (mitochondria-targeted ubiquinone) has been shown to improve tubular and glomerular function in insulin-deficient Akita mice. MitoQ treatment was associated with a decrease in nuclear accumulation of profibrotic transcription factors, phospho-Smad2/3 and beta-catenin, in Akita mice, indicating reduced TGF-β/Smad signaling in these mice (37).
Whether the decreased mitochondrial function and content contribute to enhanced mtROS production and subsequent renal dysfunction in diabetes is unclear. This issue has been addressed by stimulating mitochondrial biogenesis through the AMPK pathway. AMPK is a ubiquitously expressed protein that plays a key role in energy homeostasis of the cell. It is highly expressed in the renal cells, including podocytes, mesangial cells, glomerular endothelial cells, and particularly in mitochondria-rich cells such as proximal tubular cells (150). In experimental models of diabetes, the activity of AMPK appears to be reduced in the kidney. In energy-depleted conditions, intracellular concentrations of AMP rise, while levels of ATP fall. This leads to activation of AMPK and phosphorylation of substrates that promote catabolic processes to generate ATP. However, in excess energy states such as in diabetes, reduced AMPK activation stimulates protein synthesis and cell growth. In experimental models of diabetes, the activity of AMPK appears to be reduced in the kidney (141). However, two opposing models have been proposed with regard to the role of mtROS in reducing AMPK activation. One model upholds the current prevailing theory that in the diabetic kidney, there is increased mtROS, which leads to reduction of AMPK activity and decreased mitochondrial biogenesis (188).
However, opposite to the prevailing theory that mitochondrial overproduction of superoxide is the basis of DKD, the Dugan et al. study (69) reported reduced superoxide production in STZ diabetes mice, suggesting suppressed mitochondrial activity. In diabetic conditions or in conditions of caloric excess, the mitochondria consume fewer electrons and oxygen and consequently there is a reduction of carbon flow into the mitochondria (69). The authors attributed the reduced mitochondrial function in diabetes to reduced AMPK along with a decrease in pyruvate dehydrogenase activity and PGC1-α expression. This study suggested that increased AMPK activity would increase ROS production by mitochondria.
NOX4-Mediated Mitochondrial Dysfunction
It has been suggested that NOX4-mediated ROS production plays a role in mitochondrial dysfunction in various renal cell lineages, including mesangial cells, cardiac myocytes, and endothelial cells (3, 22, 148). Indeed, a study by Block et al. suggested that NOX4 is localized in the mitochondria and is upregulated in response to high glucose and that silencing of Nox4 is associated with reduced mitochondrial oxidative stress and renal injury (22). Our own data suggested a reduction in ROS formation in both mitochondrial and cytosolic fractions of Nox4-deficient diabetic mice (124). A report by Das et al. (56) suggested that TGF-β induces upregulation of mitochondrial Nox4, which increases the ROS burden, leading to mitochondrial depolarization and podocyte apoptosis. Moreover, AngII-induced mitochondrial NOX4-derived ROS in the mitochondrial fraction of renal tubular cells was found to be associated with mitochondrial dysfunction and apoptosis (138). In addition, Koziel et al. suggested that NOX4 inactivates mitochondrial respiratory chain complex I and thereby induces mitochondrial dysfunction in human endothelial cells (148). However, a recent study by Dugan et al. showed a reduction in mitochondrial superoxide levels in murine models of type 1 diabetes (69). One possibility could be that superoxide rapidly binds to NO to form peroxinitrite and, indeed in the same study, they found an increased level of nitrotyrosine in the kidney.
Conclusion
It has become apparent in recent years that oxidative stress is an important element initiating diabetic microvascular complications, including DKD. Renal oxidative stress in diabetes is the consequence of a series of pathological events causing an imbalance between the pro-oxidant levels and counteracting antioxidant defense mechanisms in response to chronic hyperglycemia. Despite advancement in the field of ROS pathobiology, so far, there are no therapeutic options to reduce directly oxidative stress in the progression of DKD. NOXs, particularly NOX4 and the more recently identified human isoform, NOX5, are important sources of ROS in the kidney. With ongoing technical difficulties in defining, at the protein and enzymatic levels, the status of the different NOX isoforms, it has been a challenge to characterize the relative importance of the contribution of the various NOX isoforms in individual renal cell populations. Currently, the strongest data are related to NOX4, which appears to be widely distributed within the kidney, particularly in tubules and podocytes. However, the role of other NOX isoforms particularly in immune infiltrating cells such as macrophages in the kidney warrants further exploration. The development of NOX4 or NOX5-specific pharmacological inhibitors will provide additional information about the role of ROS in the renal pathology in diabetes. Further studies are required for better understanding of the contribution of mtROS and antioxidant pathways such as the Nrf2-Keap-1 pathway in renal disease in diabetes.
Footnotes
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia and a JDRF Program/Project Grant.