
Abstract
Aims:
Cardiac-specific metallothionein (MT) overexpression extends lifespan, but the mechanism underlying the effect of MT protection against age-associated cardiovascular diseases (CVD) remains elusive. To elucidate this, male wild-type and two lines of MT-transgenic (MT-TG) mice, MM and MT-1 (cardiac-specific overexpressing MT about 10- and 80-fold, respectively) at three representative ages (2–3, 9–10, and 18–20 months), were utilized. A stable human MT2A overexpressing cardiomyocytes (H9c2MT7) was also introduced.
Results:
Histomorphology and echocardiographic analysis revealed that age-associated cardiac hypertrophy, remodeling, and dysfunction were ameliorated in MT-TG mice. Also, aging-accompanied NF-κB activation, characterized by increased nuclear p65 translocation, elevated DNA-binding activity, and upregulation of inflammatory cytokines, was largely attenuated by MT overexpression. Treatment of H9c2 cardiomyocytes with tumor necrosis factor-α (TNF-α), which mimicked an inflammatory environment, significantly increased NF-κB activity, and some age-related phenotypes appeared. The NF-κB activation was further proved to be pivotal for both age-associated and TNF-α-induced nitrative damage to cardiac 2-oxoglutarate dehydrogenase (2-OGD) by virtue of NF-κB p65 gene silencing. MT inhibited NF-κB activation and associated nitrative damage to cardiac 2-OGD in both old MT-TG hearts and TNF-α-treated H9c2MT7 cardiomyocytes; these protective effects were abolished in H9c2MT7 cardiomyocytes by MT-specific gene silencing.
Innovation and Conclusion:
Together, these findings indicate that the protective effects of MT against age-associated CVD can be attributed mainly to its role in NF-κB inhibition and resultant alleviation of nitrative damage to 2-OGD. Antioxid. Redox Signal. 25, 936–952.
Introduction
A
Over the course of aging, cardiomyocytes become more susceptible to oxidative stress caused by the imbalance of redox system, resulting in the increasing rate of cardiomyocyte death (8). When cardiomyocyte necrosis occurs, a release of cellular components is accompanied, which affects the viability of cells in the vicinity. This process also promotes the development of proinflammatory environments (32), which could further enhance oxidative stress and activate the reactive oxygen species (ROS)-dependent NF-κB pathway (15).
We observed for the first time that 2-oxoglutarate dehydrogenase complex (2-OGDHC) nitration and its subsequent dysfunction are involved in aging, providing a link between inflamm-aging and age-associated cardiovascular diseases (CVD). Moreover, we showed that the functional importance of metallothionein (MT) is not limited to its role as a defender against oxidative stress. Instead, its novel role in inhibiting NF-κB activation and the resultant nitrative damage to 2-OGD ensure that cardiac cells have an adequate energy supply. Thus, MT plays a crucial role in preventing aging-associated CVD.
Of note, the highly oxidative inflammatory environment induces oxidative modifications to kinds of biomacromolecules (7), of which the protein oxidation is especially hazardous as the consequent damage and conformational alterations in structures can make target proteins inactive, resulting in functional abnormalities (2, 7). Consistent with this view, a number of important enzymes that are involved in glycolysis, gluconeogenesis, and the citrate cycle have been demonstrated to be modified during aging (3, 6). These results suggest a potential link between protein oxidation and the disturbance of cellular energy metabolism.
The aging process is linked with a subclinical systemic inflammatory response, referred to as “inflamm-aging,” which was first proposed and explained by Franceschi et al. (20). Inflammatory cytokines such as tumor necrosis factor-α (TNF-α) induce the production of nitric oxide (NO) by activating inducible nitric oxide synthase (iNOS) (36). The dual activation of superoxide- and NO-generating systems renders the intracellular environment favorable for the formation of peroxynitrite (ONOO−) and results in the nitration of tyrosine-containing proteins (42), and 3-nitrotyrosine (3-NT) is, thus, formed. More importantly, studies in animals and cultured cells demonstrated that 3-NT is a crucial pathogenic agent of CVD during oxidative stress (18).
Metallothionein (MT) is a family of metal-binding proteins harboring high numbers of cysteine residues, which makes it an important component for defending against oxidative stress. Downregulation of oxidative stress-mediated apoptosis has been demonstrated (17). Moreover, MT prevents the inflammatory cascades from initiating after tissue injury (24), and it also modulates metabolism (46). Over the past few years, extensive studies have drawn much attention to the role of MT in modulating lifespan, highlighting it as an attractive target for aging research (44). To date, however, the molecular mechanism underlying the protective effect of MT on age-associated CVD remains unclear.
This study aimed at investigating how the aging-accompanied inflammatory response was mediated by MT overexpression, and its related effects in old mice hearts. Additionally, MT-mediated alleviation of both age-associated and TNF-α-induced nitrative damage to certain proteins was also analyzed for further elaborating the protective role of MT in age-associated CVD.
Results
Cardiac function and remodeling were partially preserved in old MT-transgenic mice
Hematoxylin-eosin and Sirius red staining revealed that aging is accompanied by myocardial structure disturbance, myofibrillar discontinuation, and enhanced fibrosis (Fig. 1A). The extent of myocardial remolding clearly increased with aging in the hearts of wild-type (WT) mice, but to a significantly lesser extent in MT-transgenic (TG) mice.
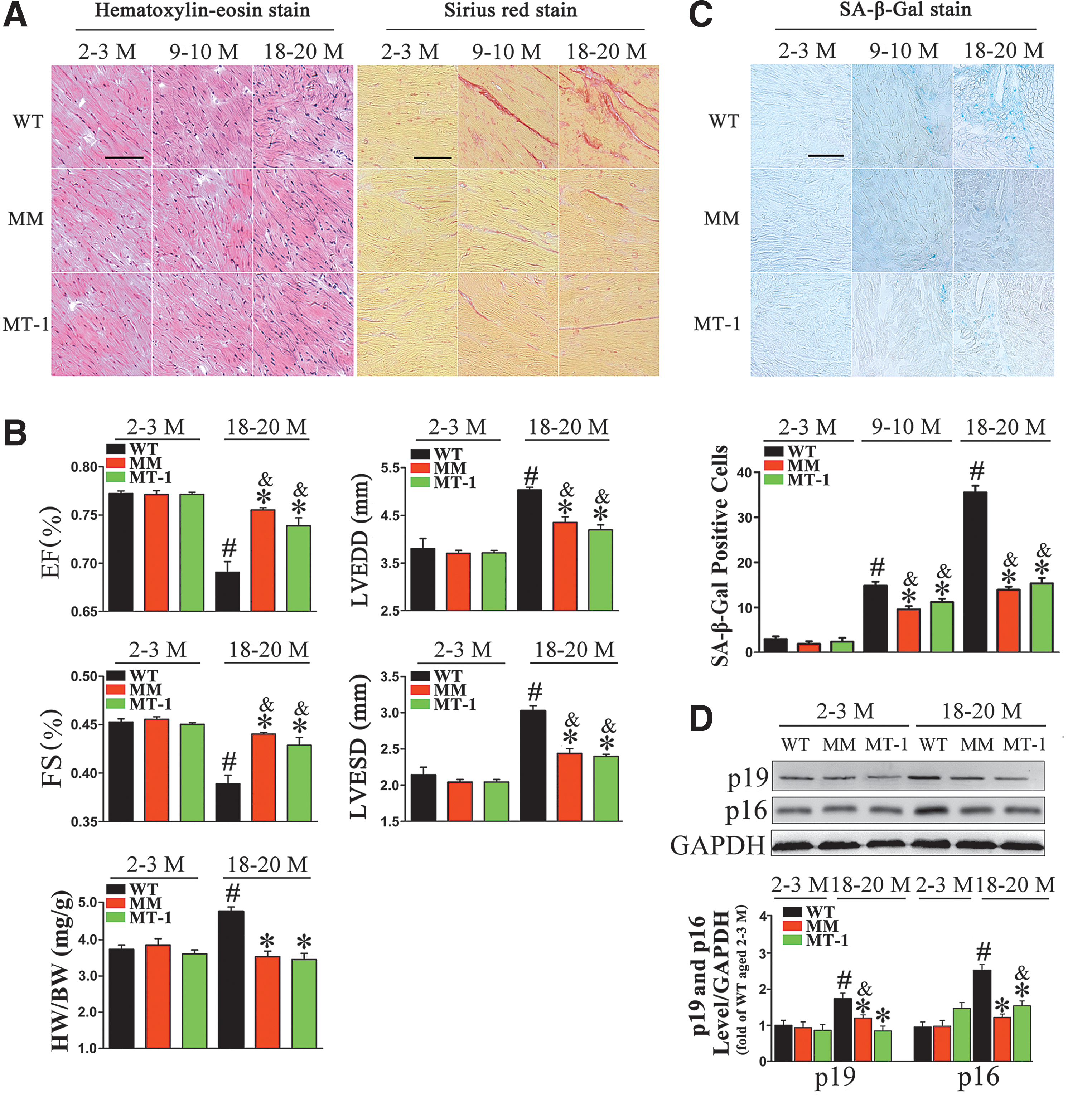
Echocardiographic assessment of in vivo left ventricular (LV) function was performed by using the two lines of MT-TG mice at ages of 2–3 and 18–20 months (hereafter, “young” and “old,” respectively). LV end-systolic dimensions (LVESD), LV end-diastolic dimensions (LVEDD), fractional shortening (FS%), and ejection fraction (EF%) were similar between the young MT-TG and WT mice (Fig. 1B).
In old WT mice, the LV was significantly dilated (higher LV long-axis diameter at end-diastole and end-systole stages). Mild LV systolic dysfunction (reduced FS% and EF%) was also observed in old WT mice. By contrast, these phenotypes were alleviated in the same-age MT-TG mice. Similarly, a remarkable increase in heart weight normalized to body weight (HW/BW) was observed in old WT mice, but not in old MT-TG mice (Fig. 1B and Supplementary Table S1; Supplementary Data are available online at
Senescence-associated phenotypes were partially alleviated in old MT-TG mice
Cellular senescence, which is considered the hallmark of aging, is characterized by phenotypes, including elevated senescence-associated beta-galactosidase (SA-β-gal) activity and increased expression of cell-cycle inhibitors (34). As shown in Figure 1C, SA-β-gal activity was markedly elevated in old WT mice, as reflected by the extended SA-β-gal-positive area, but it was significantly lower in old MT-TG mice.
In many organs of rodents, expression of INK4/ARF family proteins increases with age (26). We confirmed that levels of p16INK4a and p19ARF increased with age in WT mice, but they remained low in old MT-TG mice (Fig. 1D). These results demonstrated that cellular senescence, to some extent, could be prevented by MT overexpression.
MT prevents cardiac NF-κB activation and the subsequent inflammatory response in old mice hearts
Inflammation is a crucial etiological factor in the progression of several age-associated diseases, including age-associated CVD. Accordingly, we analyzed inflammatory cell infiltration during the aging process. As shown in Figure 2A, the number of myeloperoxidase (MPO)-peroxidase+ neutrophils increased significantly in old WT hearts. Fewer MPO+ cells were present in old MT-TG hearts, indicating that MT overexpression effectively attenuated this increase.
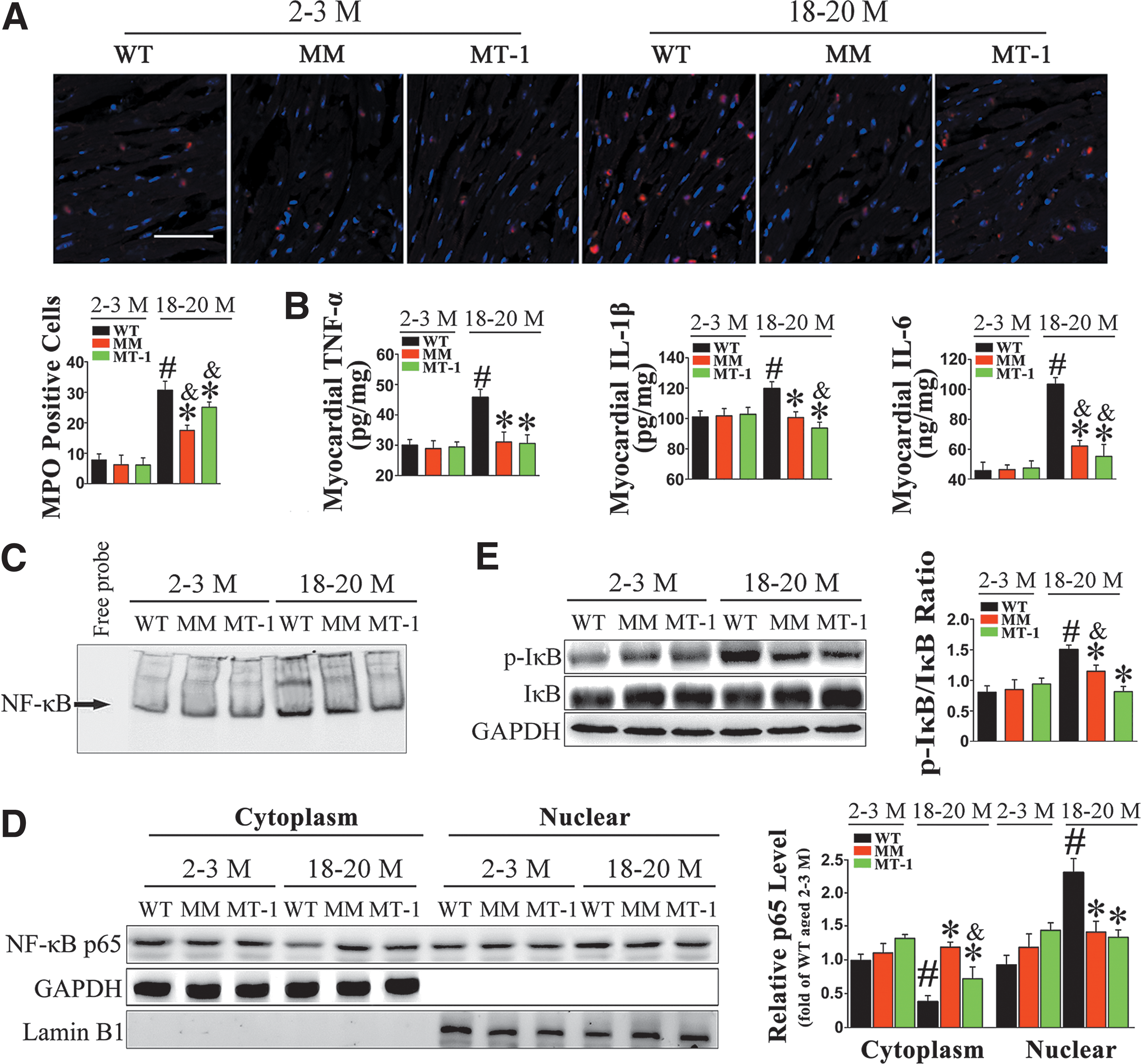
Next, we compared the levels of proinflammatory cytokines in myocardial tissue (Fig. 2B and Supplementary Fig. S1A) and plasma (Supplementary Fig. S1B) of young and old mice. The protein levels of TNF-α, interleukin-1β (IL-1β), and IL-6 were elevated in both myocardial tissue and plasma of old mice. However, cardiac-specific MT overexpression decreased the levels of proinflammatory cytokines in myocardial tissue from old hearts without altering the corresponding plasma levels.
In light of the effect on cytokine production, we explored NF-κB activity. Nuclear extracts from young and old WT and MT-TG hearts were subjected to electrophoretic mobility shift assay (EMSA) for NF-κB DNA-binding activity assessment (Fig. 2C). DNA-binding activity of NF-κB was elevated in old WT hearts, but it remained low in old MT-TG hearts. To investigate the underlying molecular mechanism, we performed immunoblots of p65 (Fig. 2D). In the WT, the nuclear translocation of p65 was obviously higher in old hearts than in young hearts. Consistently, MT suppressed age-associated nuclear translocation of p65. Furthermore, MT inhibited IκB phosphorylation and its subsequent degradation, which is the last step for NF-κB activation in the cytoplasm (40) (Fig. 2E).
To further explore the inhibitory effects of MT during the inflammatory process, we investigated the effect of MT on the cardiac response to lipopolysaccharide (LPS) (Supplementary Fig. S2). Results showed that MT overexpression significantly suppressed LPS-induced nuclear translocation of p65, along with an attenuated DNA-binding activity of NF-κB in MT-TG hearts (Supplementary Fig. S2A, B). The elevation of cardiac transcription levels of TNF-α, IL-1β, and IL-6 after LPS injection was effectively alleviated in MT-TG hearts (Supplementary Fig. S2C).
Furthermore, LPS-induced cardiac apoptosis was also suppressed by the overexpression of MT (Supplementary Fig. S2D). Therefore, these results confirmed the role of MT in protecting against the activation of NF-κB.
MT prevents cardiac apoptosis in old mice
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) analysis revealed that the cardiac apoptosis rate was higher in old WT hearts than in young WT hearts. This effect was significantly alleviated in old MT-TG hearts (Fig. 3A).
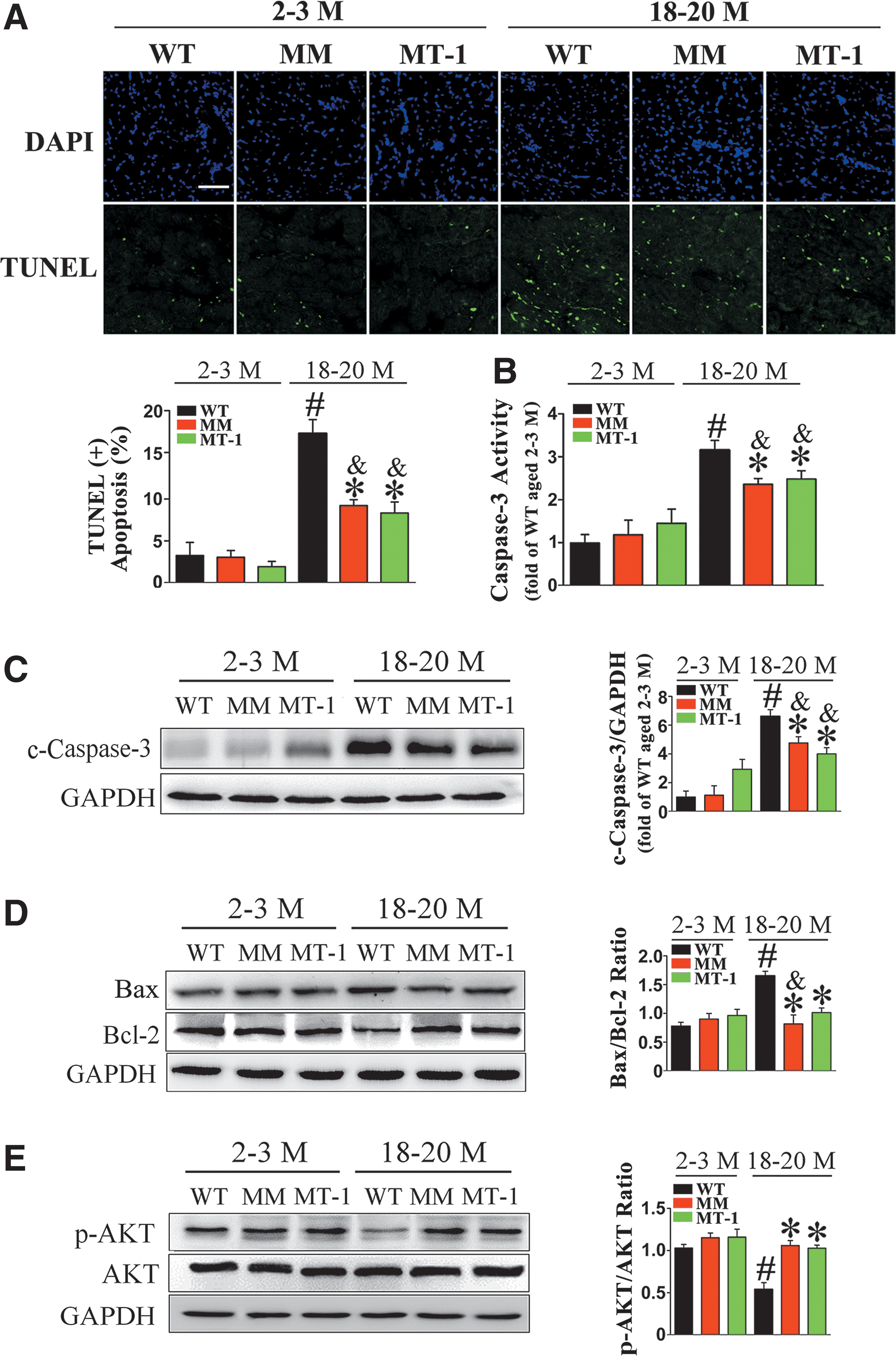
We further tested the protective effect of MT on aging-associated cardiac apoptosis by examining caspase-3 activity, cleaved caspase-3 expression, and Bax/Bcl-2 ratio; consistent results were obtained in all cases (Fig. 3B–D). In addition, MT promoted cellular survival in old hearts, as demonstrated by the elevated ratio of phospho-AKT to total AKT (Fig. 3E).
3-NT modification of 2-oxoglutarate dehydrogenase is involved in cardiac aging, whereas MT inhibits age-associated nitrative damage, thereby preserving enzyme activity and energy equilibrium in the old heart
Oxidative stress plays a key role in inflamm-aging and associated diseases (23). We observed more intense fluorescence-labeled dihydroethidium (DHE) staining in the hearts of old WT mice in comparison with young animals, suggesting elevated accumulation of superoxide with aging. By contrast, old MT-TG mice had lower DHE fluorescent intensity (Fig. 4A).
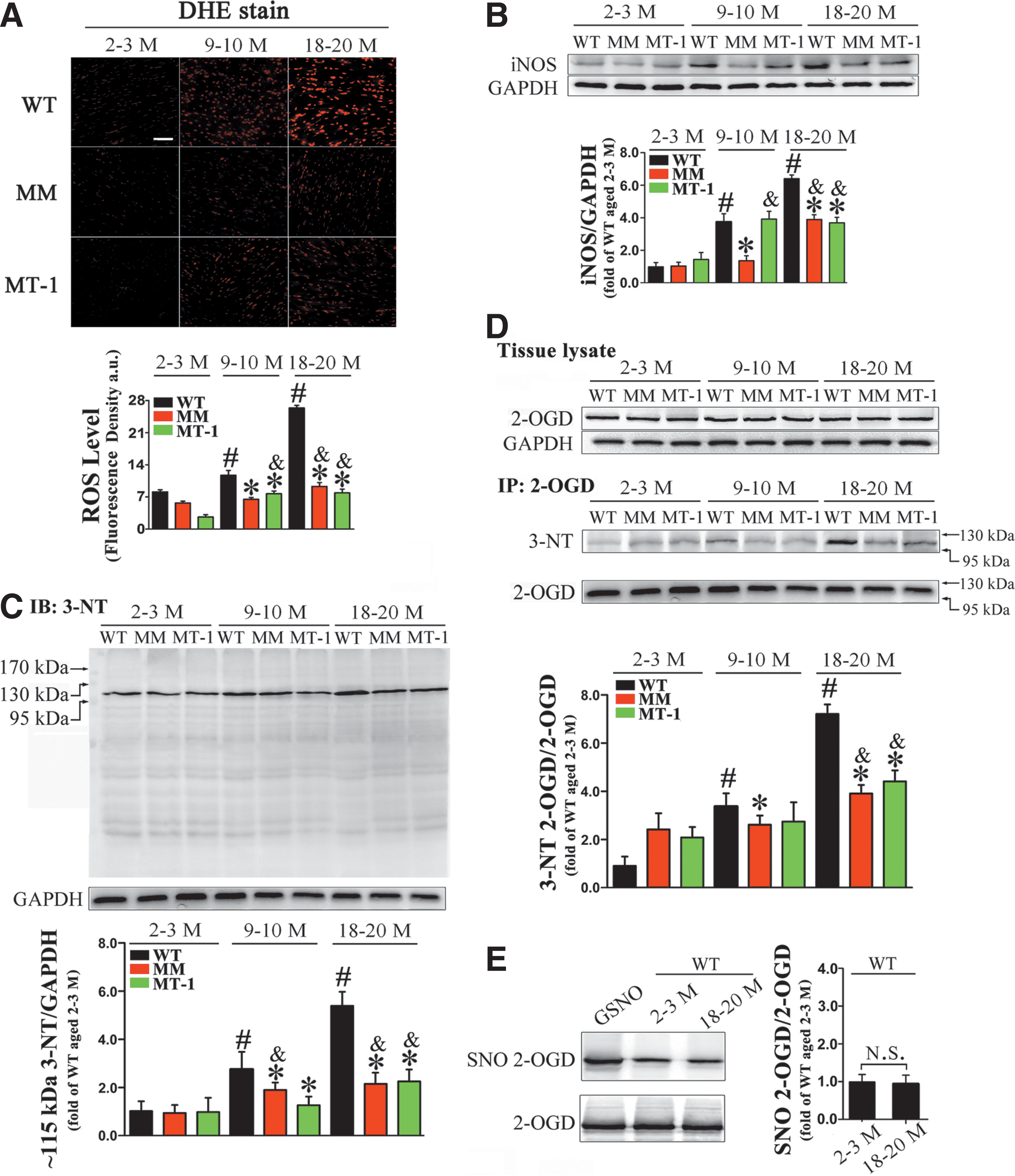
In addition, the level of iNOS in heart homogenates increased dramatically (Fig. 4B), concomitant with elevated nitrite production, during the aging process (Supplementary Fig. S3A). Hyperproduction of NO due to induction of iNOS leads to the formation of ONOO−, which can react with particular residues in proteins, thereby affecting their biological function (19). Therefore, we assessed the possible involvement of nitrative damage in age-associated CVD and the effect of MT on 3-NT formation in WT and MT-TG mice hearts with mean ages of 2–3, 9–10 (hereafter, “middle-aged”), and 18–20 months.
In WT mice, quantitative analysis by immunoblotting revealed that 3-NT accumulation was much higher in old mice than in young mice. 3-NT was predominantly present in proteins of ∼115 kDa (Fig. 4C), where 2-oxoglutarate dehydrogenase (2-OGD) are located (Supplementary Table S2). In contrast to WT mice, old MT-TG mice exhibited less 3-NT modification in proteins of ∼115 kDa (Fig. 4C). In addition, we also identified other less nitrated proteins in old hearts (Supplementary Fig. S4 and Supplementary Table S3).
To further confirm the nitrative damage of 2-OGD, we examined the protein level of 2-OGD in young, middle-aged, and old MT-TG and WT mice hearts. We observed no obvious up/downregulation of 2-OGD expression level in any age group (Fig. 4D). Heart tissue samples were then immunoprecipitated with anti-2-OGD antibody and blotted with anti-3-NT antibody to examine the expression level of 3-NT-modified 2-OGD (Fig. 4D). The results confirmed an age-dependent increase in the modification of 3-NT to 2-OGD in WT mice, which was markedly suppressed in MT-TG mice. The age-associated nitration of 2-OGD was not detected in other tissues (kidney, brain, and liver) from old WT mice, suggesting a unique role for 2-OGD nitration in old hearts (Supplementary Fig. S5A–D).
Considering that the thiol-containing cysteine in redox-sensitive proteins is particularly sensitive to alterations in the redox environment (9, 16), the possible role of cysteine oxidation in 2-OGD during cardiac aging was assayed. Accordingly, we used the biotin-switch method to examine S-nitrosation, a reversible NO-induced modification, on the cysteine in 2-OGD (Fig. 4E). We observed no apparent difference of S-nitrosation on 2-OGD between young and old WT hearts. Furthermore, the irreversible cysteine oxidation in 2-OGD (sulfinic and sulfonic acid formation) was also assessed by MS analysis, but only tyrosine nitration in 2-OGD was detected in old WT hearts (Supplementary Table S2).
To determine whether the elevated rate of nitration adduction affected the function of 2-oxoglutarate dehydrogenase complex (2-OGDHC), we monitored 2-OGDHC activity in isolated cardiac mitochondrial extracts. As shown in Supplementary Figure S6A, 2-OGDHC activity declined with aging. In WT mice, enzyme activity did not differ significantly between middle-aged and young animals; whereas in old mice, cardiac 2-OGDHC activity was reduced by ∼30%.
Moreover, kinetics analysis of 2-OGDHC revealed that K m for substrate a-ketoglutarate and V max varied among age groups (Supplementary Fig. S7 and Supplementary Table S4). In contrast with that in young mice, the K m in middle-aged and old mice was elevated by ∼18% and ∼34%, and V max was reduced by ∼18% and ∼26%, respectively. Together, these observations indicate a reduced affinity for the substrate and slower catalysis in older animals. On the other hand, in MT-TG mice, enzyme kinetic characteristics were well preserved with advancing age. These results suggest a possible link between 2-OGD nitration and 2-OGDHC dysfunction.
In view of the fact that 2-OGDHC is a rate-limiting enzyme of the tricarboxylic acid (TCA) cycle, which is functionally connected to NADH-linked respiration and energy production, we examined the total ATP level in hearts and NADH content in isolated mitochondria (Supplementary Fig. S6B, C). In WT hearts, we observed an obvious decrease in NADH production over the course of aging, especially in old mice. In addition, we observed a similar trend in total ATP content, as determined by luciferin/luciferase assay. By contrast, heart-specific MT overexpression could partially improve these age-dependent conditions.
To further monitor the influence of 2-OGD nitration on overall energy metabolism, we used ion-pairing LC-MS to analyze the energy charge in each age group. The results revealed that energy charge declined mildly (∼6%) in middle-aged WT mice, whereas a ∼16% decline in old WT mice was noted with significant difference. In MT-TG mice, energy charge was well preserved, even in old mice (Supplementary Fig. S6D).
NF-κB inhibition acts as a major role in the attenuation of nitrative damage to 2-OGD
Our in vivo results indicated that the protective effects of MT against aging-associated CVD might be attributed to its role in inhibiting the NF-κB pathway. However, we lacked direct proof of the link between NF-κB activity and subsequent nitrative damage during aging.
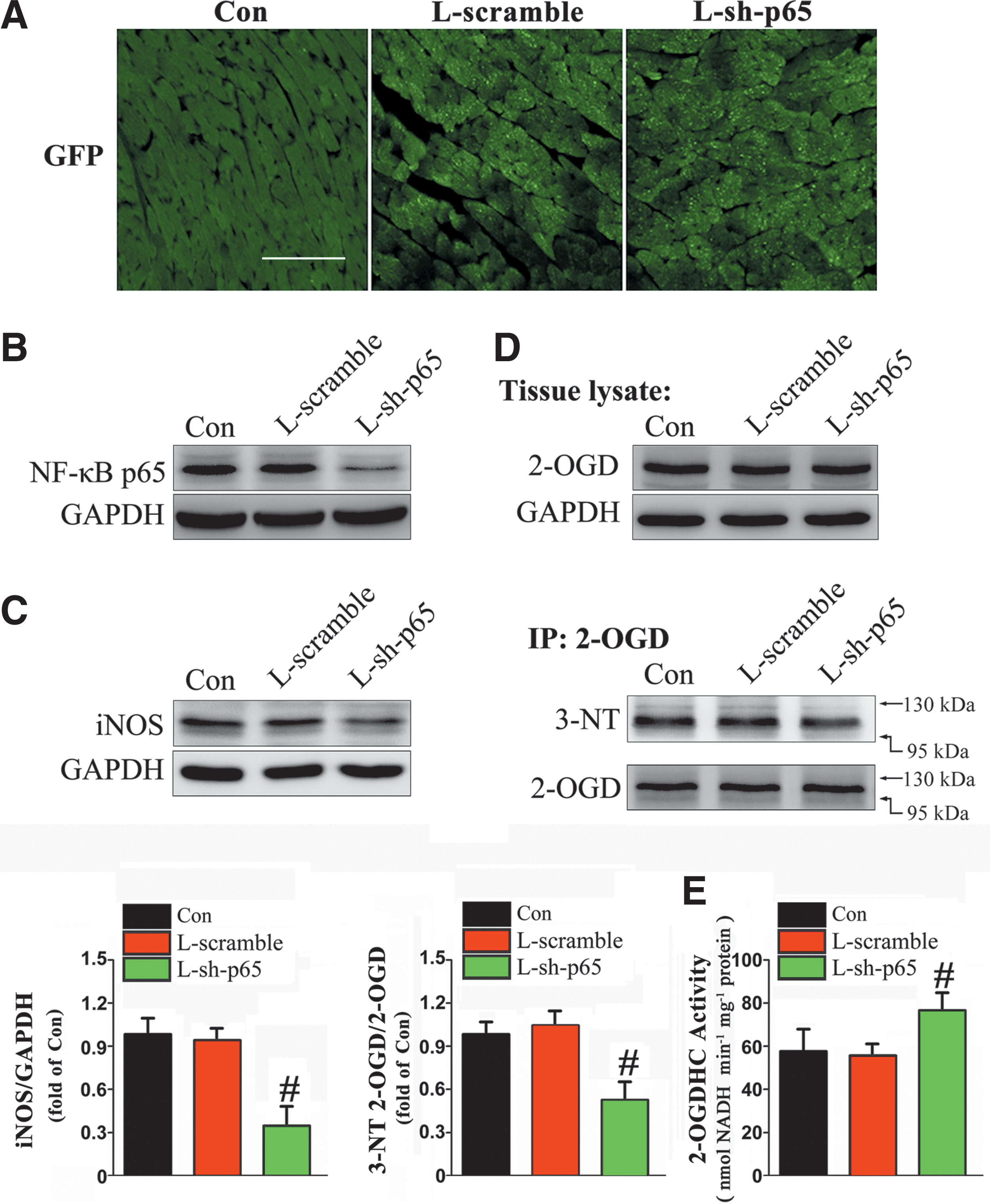
To obtain this, we directly injected lentiviral plasmids containing sh-p65 (LRV-U6-p65-CMV-EGFP) and a scrambled sequence (LRV-U6-scramble-CMV-EGFP) into the myocardium of 15-month-old WT mice over a period of 12 weeks, and the transfection was confirmed by the presence of EGFP in the myocardium (Fig. 5A). L-sh-p65 injection dramatically suppressed NF-κB p65 expression in WT hearts (Fig. 5B). As expected, iNOS protein was downregulated after L-sh-p65 treatment (Fig. 5C). L-sh-p65-treated WT mice also exhibited significant attenuation of aging-induced nitrative damage to 2-OGD in comparison with the scramble-treated or non-treated group (Fig. 5D). Meanwhile, 2-OGDHC activity was efficiently restored in WT hearts due to p65 knockdown by L-sh-p65 (Fig. 5E).
MT inhibits TNF-α-induced NF-κB activation and aging phenotypes in vitro
To further elucidate the relationship between inflammation and aging, we administered exogenous TNF-α to H9c2 and H9c2MT7 cardiomyocytes for 24 h to mimic an inflammatory environment. To measure ROS production in the cytosol, we transfected both cell lines with the cytosolic-localized redox-sensitive GFP mutant (Cyto-RoGFP) (Fig. 6A). In H9c2 cardiomyocytes, TNF-α treatment significantly increased the fluorescence ratio (400 nm/484 nm), suggesting that the ROS level in the cytosol was elevated. In addition, NF-κB DNA-binding activity, nuclear p65 translocation, and IκB degradation were also increased by TNF-α treatment in H9c2 cardiomyocytes (Fig. 6B–D), resulting in higher levels of proinflammatory cytokines in both lysate and culture medium (Supplementary Fig. S8A, B).
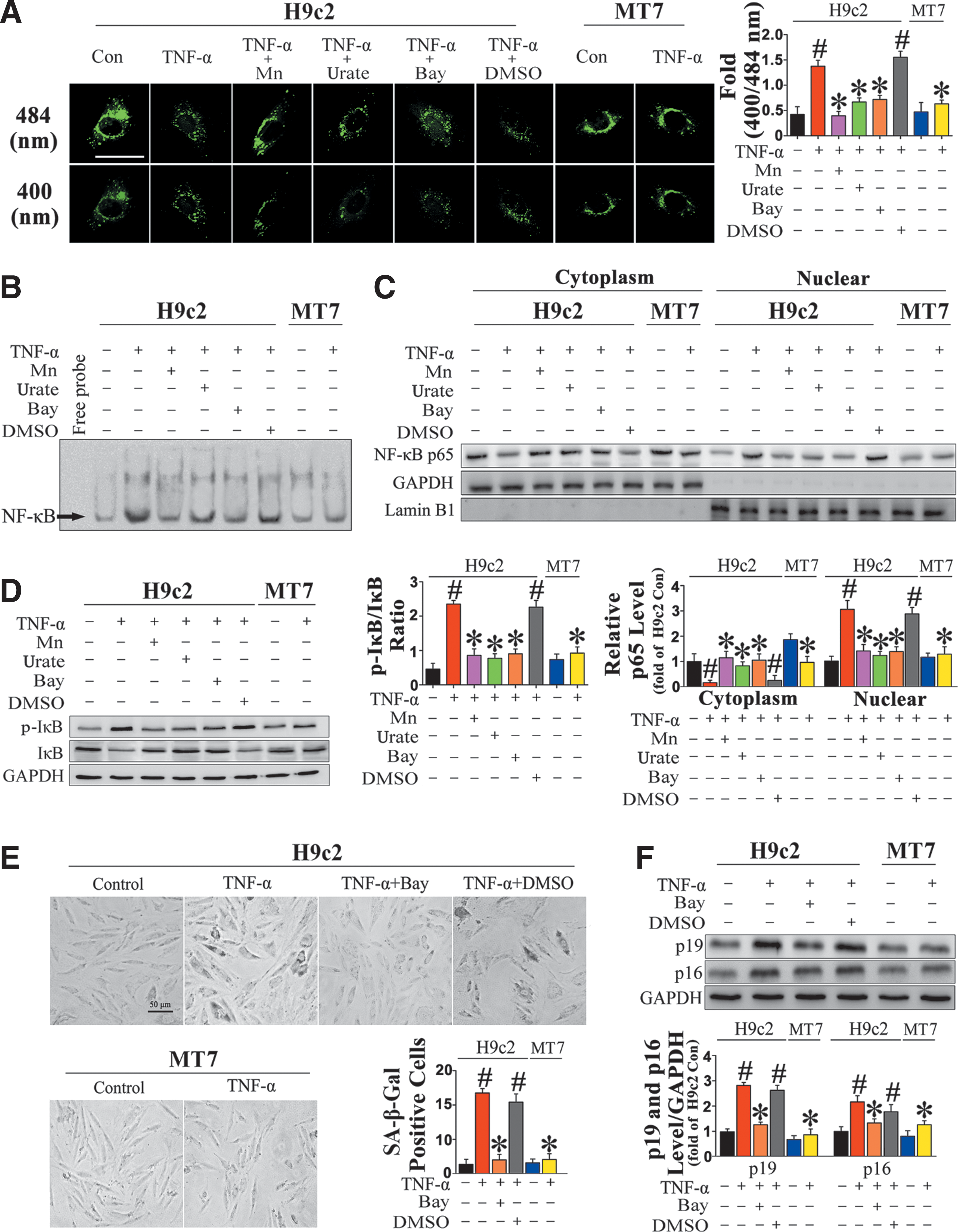
By contrast, when H9c2 cardiomyocytes were treated for 24 h with TNF-α in the presence of 100 μM urate, an ONOO− scavenger, or 100 μM Mn(III) tetrakis 1-methyl 4-pyridyl porphyrin pentachloride (MnTMPyP), an SOD mimic, the cytosolic ROS level was markedly reduced (Fig. 6A). Importantly, NF-κB DNA-binding activity, nuclear p65 translocation, and IκB degradation were subsequently inhibited (Fig. 6B–D), suggesting that NF-κB activation was ROS dependent.
In addition, some age-related phenotypes, such as an increase in SA-β-gal-positive cardiomyocytes (Fig. 6E) and upregulation of the senescence markers p16INK4a and p19ARF (Fig. 6F), were observed in TNF-α-treated H9c2 cardiomyocytes. When the cells were pretreated with Bay-117082, an NF-κB pathway inhibitor, before TNF-α administration, not only was expression of proinflammatory cytokines attenuated, but also SA-β-gal activity and expression of senescence markers were reduced (Fig. 6B–F). These results suggest a plausible link between NF-κB activation and aging. Likewise, in H9c2MT7 cardiomyocytes, we found that TNF-α-induced ROS production, NF-κB activation, proinflammatory cytokine expression, and age-associated phenomena were effectively decreased by MT overexpression (Fig. 6A–F).
Furthermore, TNF-α treatment induced high levels of apoptosis in H9c2 cardiomyocytes, as demonstrated by increases in the proportion of TUNEL-positive cells (Supplementary Fig. S9A) and caspase-3 activity (Supplementary Fig. S9B). According to both metrics, apoptosis was effectively inhibited by co-treatment with MnTMPyP, urate, or Bay-117082, similar to the effect of MT overexpression in H9c2MT7 cardiomyocytes (Supplementary Fig. S9A, B). Next, we investigated the mechanism underlying TNF-α-induced apoptosis and the inhibitory effect of MT.
TNFR1 protein level was not altered, but TNF-α-induced apoptosis was shown to be closely linked to the induction of a receptor-mediated downstream signaling cascade, demonstrated by immunoprecipitation of TNFR1-associated death domain (TRADD) with TNFR1 protein (Supplementary Fig. S9C), as well as higher levels of active caspase-8 in H9c2 cardiomyocytes (Supplementary Fig. S9D). Moreover, the levels of Bax/Bcl-2, mitochondrial Cytochrome C, and active caspase-9 were significantly elevated after TNF-α treatment of H9c2 cardiomyocytes, indicating that the mitochondrial apoptotic cascade also plays a role in TNF-α-induced cardiac apoptosis (Supplementary Fig. S9D). On the other hand, the overexpressed MT comprehensively inhibited TNF-α-induced apoptosis via both the receptor-mediated and mitochondrial pathways (Supplementary Fig. S9C, D).
MT prevents NF-κB activation-mediated nitrative damage to 2-OGD, thus preserving the enzyme activity and energy equilibrium in vitro
As NF-κB activation is connected with age-associated symptoms in cardiomyocytes, the possibility of an NF-κB-mediated increase in the 2-OGD nitration level in TNF-α-treated H9c2 cardiomyocytes was investigated. To this end, we first monitored iNOS protein expression (Fig. 7A) and nitrite production in H9c2 cardiomyocytes (Supplementary Fig. S3B) after TNF-α treatment. Next, we assessed 3-NT levels in H9c2 cardiomyocytes after TNF-α administration for 24 h. Coincidentally, quantitative analysis by immunoblotting revealed an obvious enhancement of 3-NT modification in treated cells, mainly in a protein band at ∼115 kDa (Supplementary Fig. S10).The immunoprecipitation verified an increase in 2-OGD nitration level induced by TNF-α, without a change in 2-OGD expression (Fig. 7B).
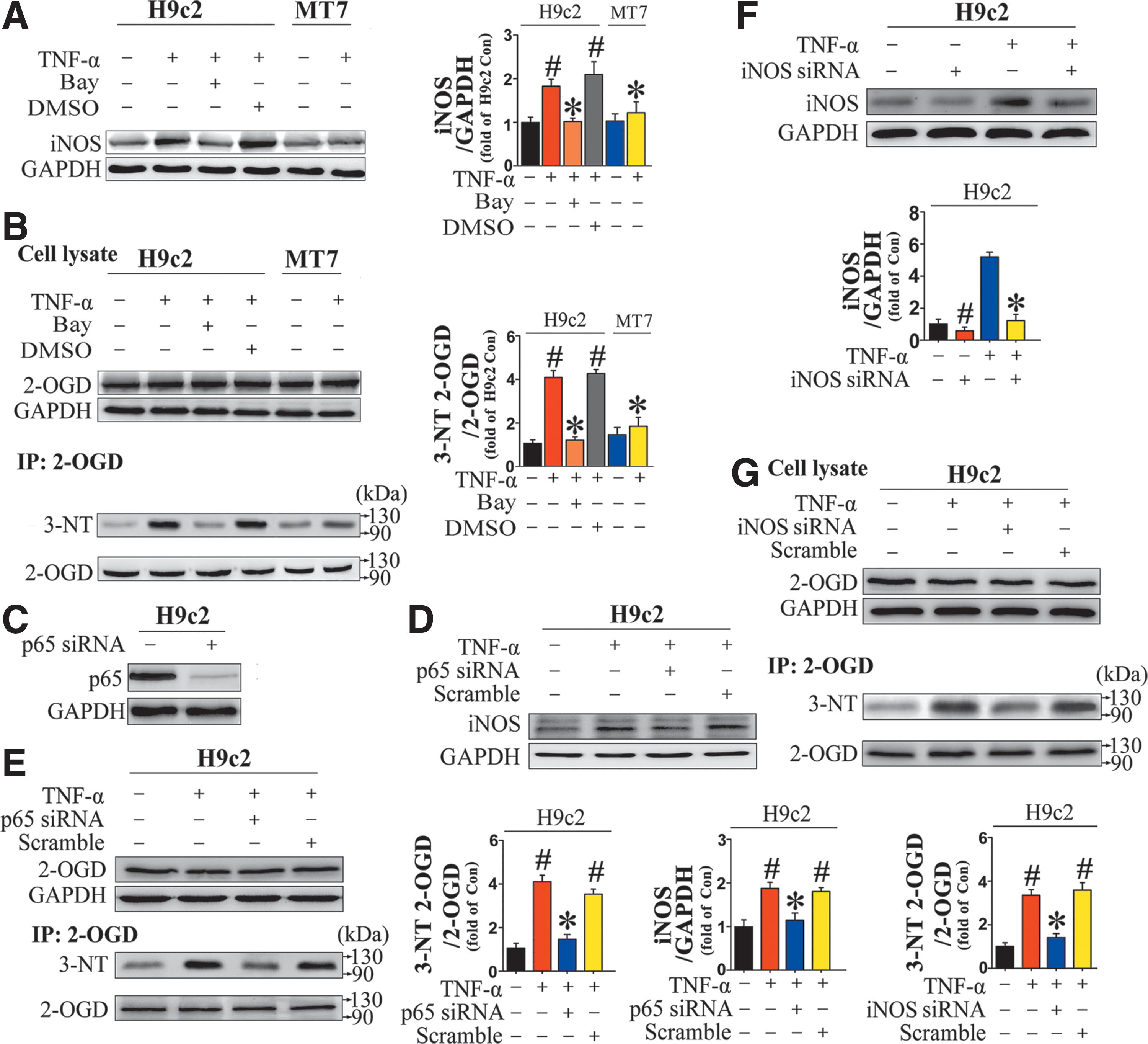
Consistent with results obtained in vivo, TNF-α-induced nitrative damage to 2-OGD also had a significant effect on 2-OGDHC activity, kinetics, and energy charge in H9c2 cardiomyocytes (Supplementary Fig. S11 and Supplementary Table S5). When TNF-α-induced NF-κB activation was restrained in H9c2 cardiomyocytes by pretreatment with Bay-117082, iNOS expression (Fig. 7A) and nitrite production (Supplementary Fig. S3B) were also effectively reduced, concomitant with a reduction in 2-OGD nitration level (Fig. 7B). Subsequently, 2-OGDHC activity, kinetics, and energy charge were well preserved (Supplementary Fig. S11 and Supplementary Table S5).
The roles of TNF-α-induced NF-κB activation and iNOS upregulation in 2-OGD nitrative damage were further confirmed by gene silencing of p65 and iNOS. For these experiments, H9c2 cardiomyocytes were transfected with p65- or iNOS-specific siRNA for 8 h, followed by TNF-α treatment for 24 h. Transfection of p65 siRNA dramatically suppressed NF-κB p65 expression in H9c2 cardiomyocytes (Fig. 7C). In addition, p65 knockdown abolished TNF-α-induced upregulation of proinflammatory cytokines at both the protein and mRNA levels (Supplementary Fig. S12).
The increase of iNOS expression by TNF-α treatment was also effectively inhibited in cells transfected with p65 siRNA (Fig. 7D). Accordingly, TNF-α-induced 2-OGD nitrative damage was well prevented (Fig. 7E and Supplementary S13A). Also, a direct inhibitory effect on TNF-α-induced 2-OGD nitrative damage was achieved by virtue of iNOS siRNA (Fig. 7F, G and Supplementary S13B).
As demonstrated earlier, MT is an effective NF-κB inhibitor in both old mice hearts and TNF-α-treated H9c2 cardiomyocytes. Accordingly, NF-κB-mediated nitrative damage to 2-OGD was less evident in H9c2MT7 cardiomyocytes than in H9c2 cardiomyocytes, which expressed lower levels of iNOS (Fig. 7A), produced less nitrite (Supplementary Fig. S3B), and exhibited less 3-NT modification on 2-OGD on TNF-α treatment (Fig. 7B). In addition, enzyme activity and energy charge were well preserved in H9c2 cardiomyocytes (Supplementary Fig. S11).
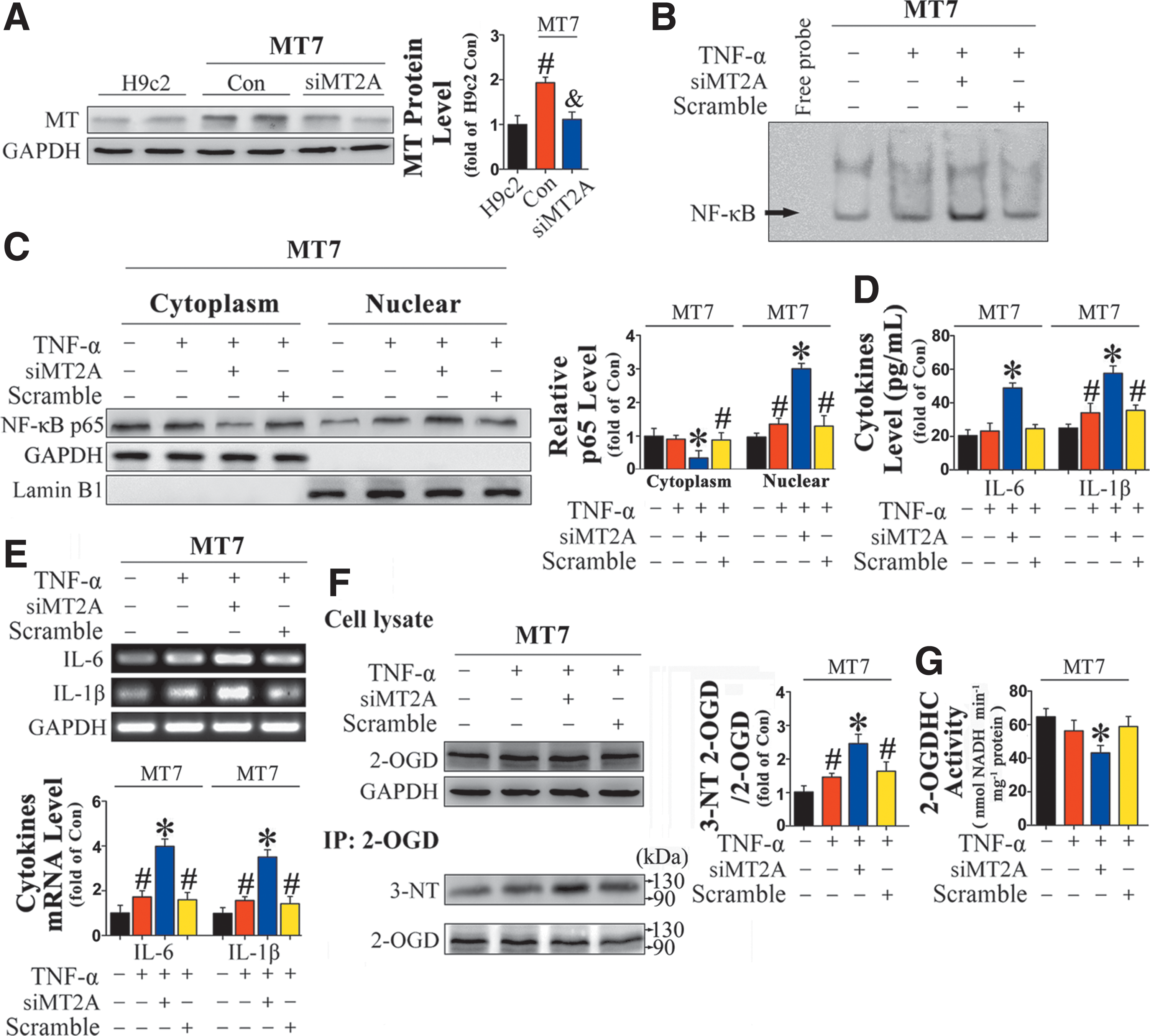
SiRNA-mediated knockdown of MT2A overexpressed in H9c2MT7 cardiomyocytes (Fig. 8A) abolished the protective effects against TNF-α treatment, including MT-mediated inhibition of NF-κB DNA binding (Fig. 8B), reduction in nuclear p65 translocation (Fig. 8C), and downregulation of proinflammatory cytokines (Fig. 8D, E). More importantly, the originally observed alleviation of TNF-α-induced nitrative damage to 2-OGD was abolished by silencing of MT (Fig. 8F, G).
Considering that the MT-elicited effects are mainly due to its role in iron chelating (38), we next investigated whether similar results could be obtained by other iron chelators. Therefore, a selective iron chelator-desferrioxamine mesylate (DFO) (10) was pretreated in H9c2 cardiomyocytes before TNF-α administration. The results showed that DFO pretreatment could suppress TNF-α-induced NF-κB activation and the following nitrative damage to 2-OGD, to some extent, but the inhibitory effects exerted by DFO were slighter than those of MT overexpression (Supplementary Fig. S14).
Identification and quantitation of nitrated peptides of 2-OGD
Since nitration of 2-OGD is involved in the process of aging, details regarding nitration of this protein need to be explored. As such, LC-MS was introduced for site mapping and quantitation of the nitrated peptides of 2-OGD.
For a full investigation of all nitration sites of 2-OGD, a parallel-reaction monitoring (PRM)-targeted MS/MS method was utilized to conduct analysis in old WT mice hearts, and eight 3-NT-modified peptides of 2-OGD were successfully identified with a high confidence level. The representative mass spectra of identified 3-NT-modified peptides are shown in Supplementary Figure S15.
After PRM, the confirmed 3-NT-modified peptides and their non-nitrated counterparts were defined as targets for further quantitation of the nitration level. We used a selective reaction monitoring (SRM)-based targeted proteomics method to quantitate relative differences in the degree of 2-OGD nitration in WT and MT-TG (MM, MT-1) mice hearts at the three age stages defined earlier (young, middle-aged, and old). The SRM results revealed that Y175, Y460, Y910, Y965, Y966, W984, and Y968 were significantly nitrated in old WT mice, but these modifications were downregulated in old MT-TG mice (Fig. 9A–H i).
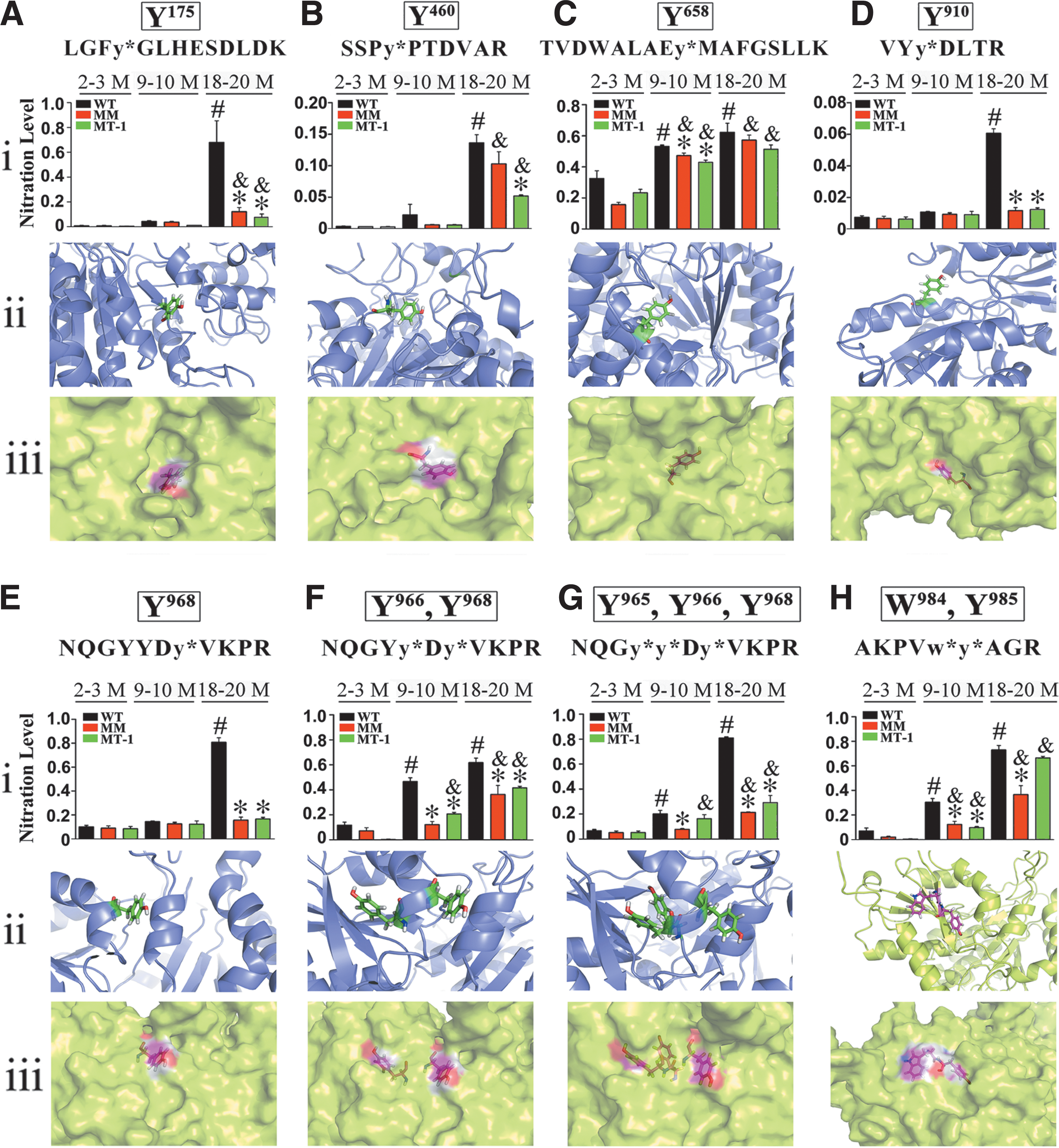
Computational models illustrated that the tyrosine sites identified earlier are located on the surface of 2-OGD, making them more sensitive to nitration (Fig. 9A–H ii, iii). The SRM results of Y658 displayed a relatively high nitration degree in all the three age stages without an obvious age-dependent increase in nitration level. In addition, MT did not significantly protect this residue against nitration (Fig. 9C i). Three-dimensional structural analysis revealed that Y658 is located in the interior of 2-OGD (Fig. 9C ii, iii), which may explain these observations.
To confirm the results obtained in vivo, the same PRM strategy was performed on TNF-α-treated cardiomyocytes. Seven nitration sites were detected with high confidence (Table 1). In addition, a quantitative analysis of the nitration degree for each peptide was also performed (Supplementary Fig. S16). The in vitro results were, to some extent, coincident with the observations in old mice hearts.
2-OGD, 2-oxoglutarate dehydrogenase; PD, Proteome Discoverer; TNF-α, tumor necrosis factor-α.
Discussion
The data here establish that MT overexpression in the heart largely decreases tyrosine nitration levels in this model organism; dramatically alleviates myocardial hypertrophy, dysfunction, and remodeling; and postpones the occurrence of senescence phenotypes. In addition, in the course of our experiments, we found that old mice exhibit elevated NF-κB activity and a resultant chronic inflammation response, which are considered critical in the development of numerous CVD during aging (20). Accordingly, in this study, we observed several senescence-associated phenotypes, such as elevated SA-β-gal activity and upregulation of senescence markers (p16INK4a and p19ARF), in TNF-α-treated H9c2 cardiomyocytes. These phenotypes were abolished when NF-κB activation was blocked, consistent with the notion of a link between inflammation and aging.
NF-κB activation during aging is likely to be responsible for the increased levels of adhesion molecules and iNOS in old mice hearts (14), which is considered the main cause of nitration in tyrosine-containing proteins. Although our results demonstrated that MT suppressed NF-κB activation by the inhibition of IκB phosphorylation and further degradation in the cytoplasm in both old mice hearts and TNF-α-treated H9c2 cardiomyocytes, it remains possible that MT regulates NF-κB activity even in the nucleus.
Consistent with this idea, an EMSA supershift analysis was utilized and it revealed that MT directly combines with NF-κB (1). In addition, MT was demonstrated to bind Zn and regulate cellular Zn homeostasis, which is essential for NF-κB DNA-binding activity (28, 43). The results from this work and other studies suggest that, although by different means, MT acts as a negative regulator of NF-κB activity. Accordingly, NF-κB-dependent upregulation of iNOS, as well as the resultant nitrative damage to certain proteins, could be attenuated by MT overexpression.
Mass spectrometry analysis revealed that the most significant nitrated protein identified in the old mice hearts is 2-OGD, which is the E1 subunit of 2-OGDHC. It is reported that 2-OGDHC is a crucial intracellular target for ROS and plays an important role in the bioenergetic shortage on account of oxidative stress. On the other hand, recent evidence has shown that the enzyme itself is able to generate ROS, and, therefore, it could further enhance the extent of intracellular oxidative stress (41). Considering the susceptibility of 2-OGDHC to oxidative stress, it can be presumed that when the radicals are produced by the enzyme near the ROS-sensitive components, the enzyme itself may also be inactivated. Therefore, the dual role of the 2-OGDHC as both a target and a generator of ROS may result in its preferential nitration during the aging process.
To date, the majority of studies regarding 2-OGDHC concentrated on revealing the relationship between its inactivation and age-related neurodegenerative diseases (35, 22). Nevertheless, the phenomenon and influence of 2-OGDHC inactivation by ONOO¬ in age-dependent CVD has been rarely discussed.
In this study, a PRM method was used to identify the actual nitrated peptides of 2-OGD. Eight nitrated peptides of 2-OGD were successfully identified with high confidence, involving eight nitration sites: Y175, Y460, Y658, Y910, Y965, Y966, W984, and Y968. Among these, Y175 is located in the N-terminus of 2-OGD. A previous report demonstrated that 2-OGD, the E1 component of the 2-OGDHC, interacts with the E2 core through the N-terminal domain, which consists of a columnar arrangement of four helices formed by residues 85–190 (21). Therefore, we speculate that the nitration of Y175 may directly influence the binding efficiency of 2-OGD to the E2 component. Y460 is located in a highly conserved region shared by many dehydrogenase enzymes (Pfam), and Y658 resides in the transketolase-like, pyrimidine-binding domain formed by residues 649–866.
Therefore, we hypothesize that nitration of these sites exerts significant influence on the function of the enzyme. In parallel, the same PRM strategy was used to analyze the nitration sites of 2-OGD in TNF-α-treated cardiomyocytes. The results revealed that NF-κB activation-mediated 2-OGD nitration at Y175, Y460, Y910, Y965, Y966, W984, and Y968 is highly concordant with the nitration sites observed in old mice hearts. Therefore, we speculate that the physiological processes of aging and inflammation may share some commonalities, resulting in similar nitrative damage to the enzyme.
In addition to nitration on tyrosine, a reversible modification on the cysteine of 2-OGD has also been reported to have influence on the catalytic activity of 2-OGDHC (33). Considering that the thiol-containing cysteine in redox-sensitive proteins is particularly sensitive to alterations in the redox environment (9, 16), the possible role of cysteine oxidation in 2-OGD during cardiac aging was assayed. S-nitrosation, a representative reversible modification on the cysteine caused by NO hyperproduction (45), was detected by the biotin-switch method. The results revealed no apparent distinction of S-nitrosation in 2-OGD between young and old WT mice hearts. Besides, the irreversible cysteine oxidation in 2-OGD (sulfinic and sulfonic acid formation) was also assessed by MS analysis. However, only tyrosine nitration in 2-OGD was detected in the old WT mice hearts.
The present study revealed that the activity of 2-OGDHC has suffered ∼30% diminishment in both old mice hearts and TNF-α-treated cardiomyocytes. In addition, alterations in KGDHC kinetic characteristics (elevated K m and decreased V max) indicated a reduced affinity for the substrate and slower catalysis. Such a dysfunction of 2-OGDHC has major functional implications considering that the 2-OGDHC-catalyzed reaction is significant for energy production as well as for the metabolic interaction between mitochondria and cytoplasm.
Perturbation of these functions might link 2-OGDHC inactivation and cardiac degenerative diseases, mainly through (i) decreased energy production, especially in energy-consuming organs, such as the heart. Decreased ATP production resulting from 2-OGDHC inactivation in the heart was associated with an exacerbation of cardiac function, as shown by a reduction in FS% and LV hypertrophy (30), and (ii) changes in the normal flow of reducing equivalents and metabolites among cellular compartments (11).
These hypotheses are, indeed, supported by observing a reduction of the total ATP level and NADH content in isolated mitochondria from both old mouse hearts and TNF-α-treated cardiomyocytes. Moreover, our detailed analysis of variation in adenosine nucleotides via ion-pairing LC-MS revealed a decrease of energy charge in both old mice hearts and TNF-α-treated cardiomyocytes (∼16% and ∼28%, respectively). Taken together, the role of 2-OGDHC in overall metabolism renders this enzyme a promising target for metabolic regulation in various kinds of age-related degenerative diseases.
In summary, our findings demonstrate that the protective effects of MT on age-associated CVD can be attributed mainly to its role in NF-κB inhibition and the resultant alleviation of nitrative damage. Moreover, we revealed that 2-OGD, as the E1 component of 2-OGDHC, was preferentially nitrated during inflamm-aging. The nitration of 2-OGD was shown to correlate with the diminution of 2-OGDHC catalytic function and the reduction of energy charge. This novel finding provides a possible insight into the mechanisms by which it might act through alleviating the nitration of the enzyme involved in intermediate metabolism during the aging process, MT preserves the normal function of the enzyme, thus protecting the heart against age-associated cardiomyopathy.
To our knowledge, this is the first study to report these nitration sites on 2-OGD from the standpoint of aging. Further studies are needed to elaborate a causative role of 2-OGD nitration in age-associated CVD by virtue of in vitro expression systems and site-directed mutagenesis.
Materials and Methods
Animals
The MT-TG mice were first generated by Kang et al. (25). MT-1, cardiac-specific overexpressing MT about 80-fold that of normal mice; MM, cardiac-specific overexpressing MT about 10-fold that of normal mice, as shown in Supplementary Figure S17. These two lines of MT-TG mice were gifted by Dr. Lu Cai (Department of Pediatrics, University of Louisville). Briefly, MT-TG mice were reproduced from FVB mice, and they have been well characterized in the literature (12, 13, 37). Both MT-TG-positive mice (heterozygotes) and negative littermates (WT) were kept in the same cage for 4 weeks from birth and then kept in separate, but identical cages.
Based on the standardized protocol, all mice were housed at a stable room temperature with a 12 h light/dark cycle and offered ad libitum access to food and water. MT-TG and WT mice littermates at three representative age stages (2–3, 9–10, and 18–20 months) were euthanized without fasting and analyzed. All animal experiments and methods were approved by the Animal Policy and Welfare Committee of Wenzhou Medical University, Wenzhou, Zhejiang Province, China.
Cell cultures
H9c2 and H9c2MT7 (stable overexpression of human MT-2A) rat cardiomyocytes were purchased from American Type Cell Collection (ATCC) and cultured using Dulbecco's modified Eagle's medium (DMEM) (Mediatech, Inc.) that was supplemented with 10% fetal bovine serum (Invitrogen). H9c2 and H9c2MT7 cardiomyocytes in all the experiments were in passages between 3 and 15 (39). The H9c2 and H9c2MT7 cardiomyocytes were incubated using different concentrations of TNF-α (PreproTech, Rocky Hill, NJ), and a final concentration of 100 ng/ml of TNF-α was added for 24 h of incubation. This concentration was selected because of its explicit effect on inducing iNOS in cardiomyocytes (4, 36) compared with other concentrations (31).
In some experiments, H9c2 cardiomyocytes were also co-incubated with either 100 μM of ONOO¬ scavenger urate (Sigma) or 50 μM MnTMPyP (Calbiochem) plus TNF-α. The influence of TNF-α-induced NF-κB activation on cardiomyocytes was defined by pretreating Bay-117082, a representative NF-κB pathway inhibitor, for 10 min in a concentration of 1.5 μM before TNF-α administration.
For determining the influence of iron chelators on TNF-α-induced effects, H9c2 cardiomyocytes were pretreated for 30 min with or without 10 mM DFO (5) before 100 ng/ml TNF-α administration.
Statistical analysis
Prism 5 software (GraphPad) was utilized for statistical analysis. All data were presented as mean ± standard deviation. Comparison between two groups was analyzed by independent t-tests. Comparisons among multiple groups were analyzed by one-way analysis of variance (ANOVA) or two-way ANOVA, as appropriate. A two-tailed p-value less than 0.05 was considered statistically significant.
More methods and materials were provided in Supplementary Data.
Footnotes
Acknowledgments
This work was supported by grants from the National Nature Science Foundation of China (81371753, 81300171, 81570368, 81500295, and 81470061), the Zhejiang Provincial Natural Science Foundation (LQ14H070002), the Technology Program of Wenzhou (Y20150010), the Public Projects of Zhejiang Province (2015C37064), and the College Students’ Technological Innovation Activities of Zhejiang Province (2014R413080).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.