
Abstract
Introduction
C
Transcription factor NRF2, a master regulator of redox homeostasis, provides additional protection against α-synuclein proteinopathy in Parkinson's disease. The repurposing of dimethyl fumarate (Tecfidera) to target NRF2 in the brain offers a compelling rationale to start clinical trials.
The main mechanism to control NRF2 is at the level of protein stability by the ubiquitin E3 ligase adapter KEAP1 (49). This protein contains several cysteine residues that are capable of undergoing redox modifications and adduct formation with electrophilic compounds. Therefore, NRF2 levels can be modulated pharmacologically to phenocopy this protective NRF2 haplotype. A protective role of NRF2 has been suggested in several cellular and animal models of PD, based on intoxication with mitochondrial complex I inhibitors that induce oxidative stress and ATP depletion (5, 17, 34, 39, 55). Conversely, pharmacological induction of NRF2 protected the brain against these toxins (12, 34, 35, 39, 68). An important example of these studies is the recent observation that activating NRF2 with dimethyl fumarate (DMF) attenuated oxidative stress and was neuroprotective against 6-hydroxydopamine-induced striatal oxidative stress (35). These results, though promising, would be somewhat expected, that is, boosting the cellular antioxidant capacity by targeting NRF2 should provide protection against toxins that induce oxidative stress. Even if this outcome is expected, the usefulness of NRF2, merely considered as antioxidant target, is not clear in humans, because simple antioxidant therapies such as tocopherol or Coenzyme Q10 supplements have provided little benefit or even deleterious effects. One example is the QE3 phase 3 trial for PD, which showed no evidence of clinical benefit (50). Therefore, to provide compelling preclinical evidence that NRF2 is a valid target to slow or prevent PD progression, it is necessary to determine its protective effect in an animal model that better replicates the human pathology, and this requires addressing the proteinopathy associated with human PD, that is the α-synucleinopathy. Attempts to study the Nrf2 signature in transgenic mice expressing human α-SYN have been inconclusive, because either α-SYN pathology was not reproduced in the basal ganglia (24) or there was not a clear loss of nigral dopaminergic neurons and the early expression of Nrf2-regulated genes was followed by loss of induction (4). By contrast, we have been using an α-synucleinopathy model based on stereotaxic delivery of pseudotype 6 recombinant adeno-associated vector (rAAV-6-α-SYN) to the ventral midbrain (VMB) that replicates α-SYN aggregates and dystrophic Lewy neurites (45). With this model, we reported that NRF2 deficiency aggravates protein aggregation, neuronal death, and inflammation in early-stage PD. Those experiments provided a new scenario to validate NRF2 as a new target for PD in a more meaningful preclinical setting.
A wealth of NRF2 activators has been identified that appear to disrupt the KEAP1/NRF2 interaction (28). However, from the point of view of a potential brain therapy, only three compounds have consistently demonstrated sufficient pharmacokinetic and pharmacodynamic properties to act in this tissue: sulforaphane (22), CDDO-methyl ester (also known as bardoxolone methyl) (73), and DMF. The most successful case reported so far in targeting NRF2 is this dimethyl ester derivative of fumaric acid. DMF crosses the gastrointestinal barrier, after which it is converted into monomethyl fumarate (MMF) (58). In theory, the esterase end-product is fumarate but it is not detectable in plasma, suggesting that the TCA cycle is capable of rapidly processing any fumarate that forms from DMF/MMF. Both DMF and MMF (and fumarate if it were present) covalently modify KEAP1, which results in an accumulation of NRF2, leading to up-regulation of the transcriptional NRF2 signature (11). The immune modulatory effect of DMF has been exploited for other autoimmune diseases such as psoriasis (2), lupus erythematosus (69), asthma, and arthritis (63). Very importantly, DMF is an approved drug for relapsing–remitting multiple sclerosis as an oral formulation termed BG-12. After the success with BG-12 in two clinical trials, CONFIRM and DEFINE, this formulation has been commercialized with the name of Tecfidera by Biogen (6), becoming one of the most successful drugs ever developed by the biopharmaceutical industry.
In this study, we aimed at analyzing whether DMF could be used for PD therapy in a murine model of α-synucleinopathy and the mechanisms of action. Our results indicate that DMF targets NRF2 at the basal ganglia and exerts a very significant brain protective effect against α-SYN toxicity by both activating autophagy and modulating neuroinflammation.
Results
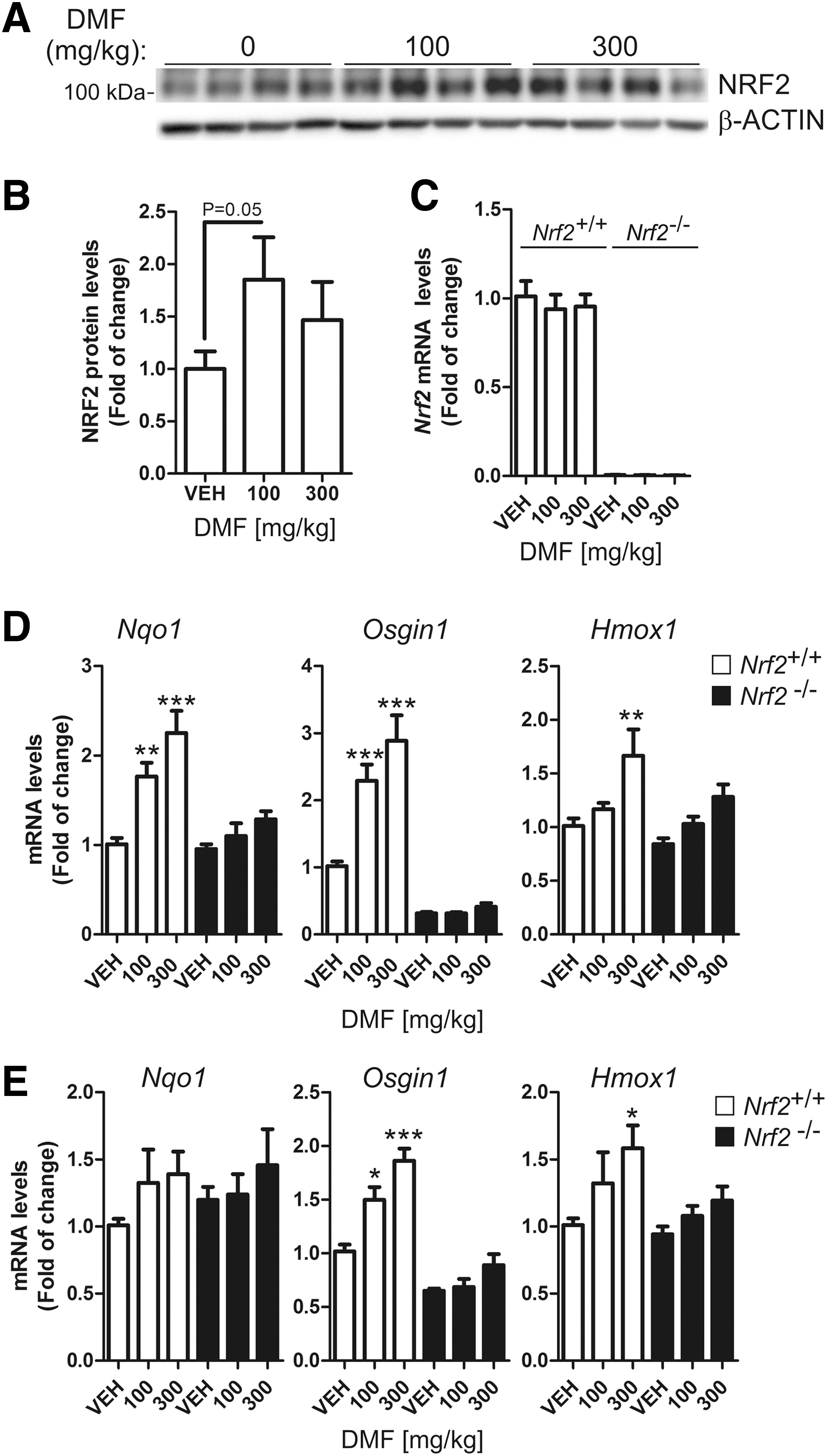
Oral administration DMF induces the NRF2 signature in the brain
We performed a small pharmacodynamic assay to confirm that DMF crosses the blood–brain barrier and activates NRF2 in the brain. As a control, we used kidneys from the same mice (Supplementary Fig. S1; Supplementary Data are available online at
Induction of NRF2 signature in the brain by DMF evokes neuroprotective effects in a PD mouse model
We used an in vivo multifactorial model of PD, based on stereotaxic delivery of an rAAV6-α-SYN viral vector (67) at the VMB of Nrf2 +/+ and Nrf2 −/− mice (45). A control rAAV6 vector expressing GFP did not have any significant effect on neuron viability, gliosis, or inflammation (45). DMF (100 mg/kg) was administered daily by oral gavage for 1 and 3 weeks, and every other day for 8 weeks (Fig. 2A) after stereotaxic injection. One day before sacrifice, animals were evaluated with a double-blind protocol for motor asymmetry based on the elevated body swing test (Fig. 2B). During the whole time course, vehicle-treated Nrf2 −/− animals presented increased contralateral body torsion compared with Nrf2 +/+ mice. Very importantly, DMF prevented this behavioral alteration in Nrf2 +/+ but had a minor nonstatistically significant effect in Nrf2 −/− mice. The alteration in motor performance predicted two important findings that would be confirmed later by immunohistochemistry: (i) The extent of the dopaminergic lesion would be large enough to manifest motor alterations; (ii) DMF might be preventing, at least in part, the motor deficit in Nrf2 +/+ mice but not in Nrf2 −/− mice.
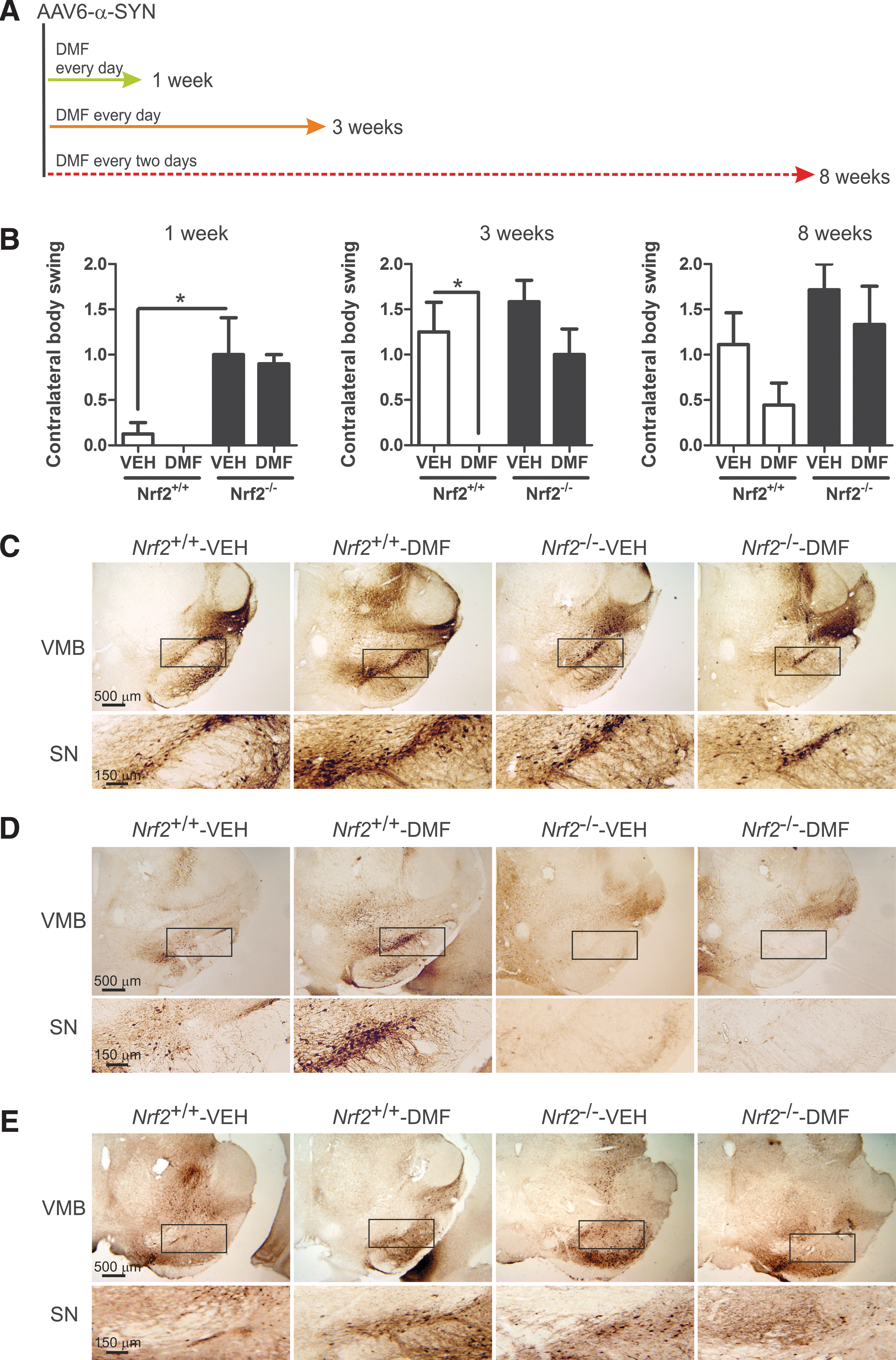
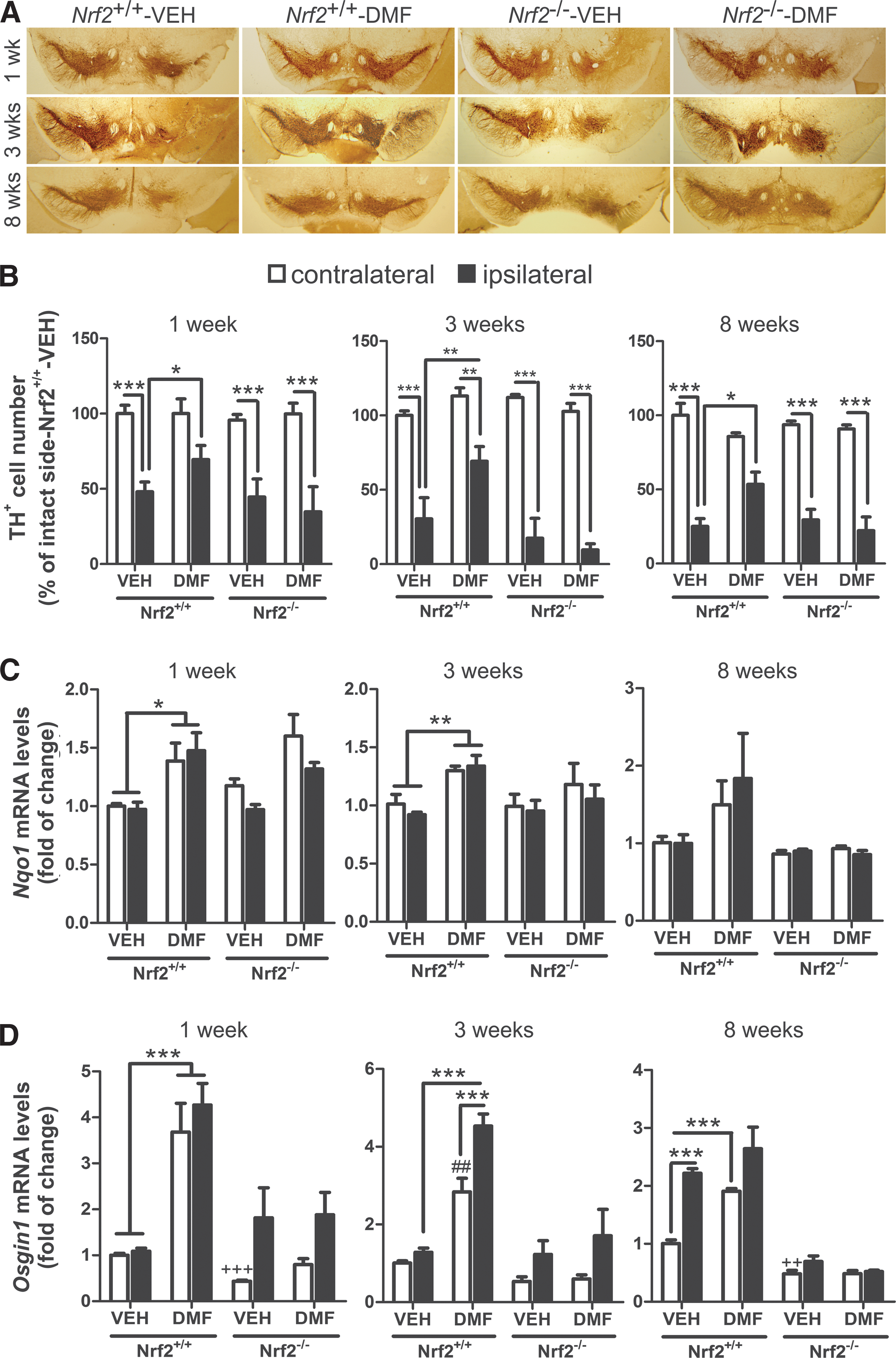
To confirm proper stereotaxic delivery and expression of the rAAV6-α-SYN vector to the basal ganglia, 30 μm-thick sections from STR and VMB were stained with an antibody that was specific for human α-SYN (Fig. 2C–E). In most animals, the needle tract was observed in the precise position, reaching the substantia nigra (SN), and some scattered neurons were found to express α-SYN along the needle tract or close to it, indicating proper delivery of the viral vector. Animals with mislocated injections were discarded. After 1 week from infection, the SN of all animals expressed human α-SYN, and the expression in DMF-treated Nrf2 +/+ mice was the strongest. However, at 3 and 8 weeks, only DMF-treated Nrf2 +/+ mice still supported a significant number of neurons expressing α-SYN, therefore suggesting that they could tolerate α-SYN toxicity better than the other experimental settings.
To correlate putative effects of α-SYN toxicity with dopaminergic neuron death, parallel series of 30 μm-thick coronal sections were processed with anti-tyrosine hydroxylase (TH) antibody to assess loss of dopaminergic neuron bodies in SN (Fig. 3A) and fibers in STR (Supplementary Fig. S2). Stereological counting indicated that TH+ neurons of Nrf2 +/+-VEH, Nrf2 −/− VEH, and Nrf2 −/− DMF groups were partially lost at postinfection week 1 (∼50%) and progressed through weeks 3 and 8 (∼75–80%) (Fig. 3B). Similarly, α-SYN overexpression induced a depletion of TH+ terminals at the STR, in a time-dependent fashion in vehicle- and DMF-treated Nrf2 −/− mice (Supplementary Fig. S2A, B). More importantly, the dopaminergic neuron loss was greatly attenuated in Nrf2 +/+ mice treated with DMF (about 75% of neurons preserved). In the α-SYN-expressing side of the STR (ipsilateral side), the dopamine (DA) and 3,4-dihydroxyphenylacetic acid (DOPAC) levels were significantly decreased at 3 weeks in vehicle-treated Nrf2 +/+ mice but DMF attenuated these neurotransmitter changes (Supplementary Fig. S2C). Consistently, DMF increased Nqo1 and Osgin1 mRNA expression at the VMB of Nrf2 +/+ but not Nrf2 −/− mice (Fig. 3C, D, respectively). Osgin1 mRNA levels were also increased by α-SYN expression even in the absence of DMF, suggesting additional mechanisms of regulation for this gene. Taken together, the results shown in Figures 2 and 3 indicate that DMF elicits a protective defense against α-SYN overexpression and that this effect is mediated through NRF2.
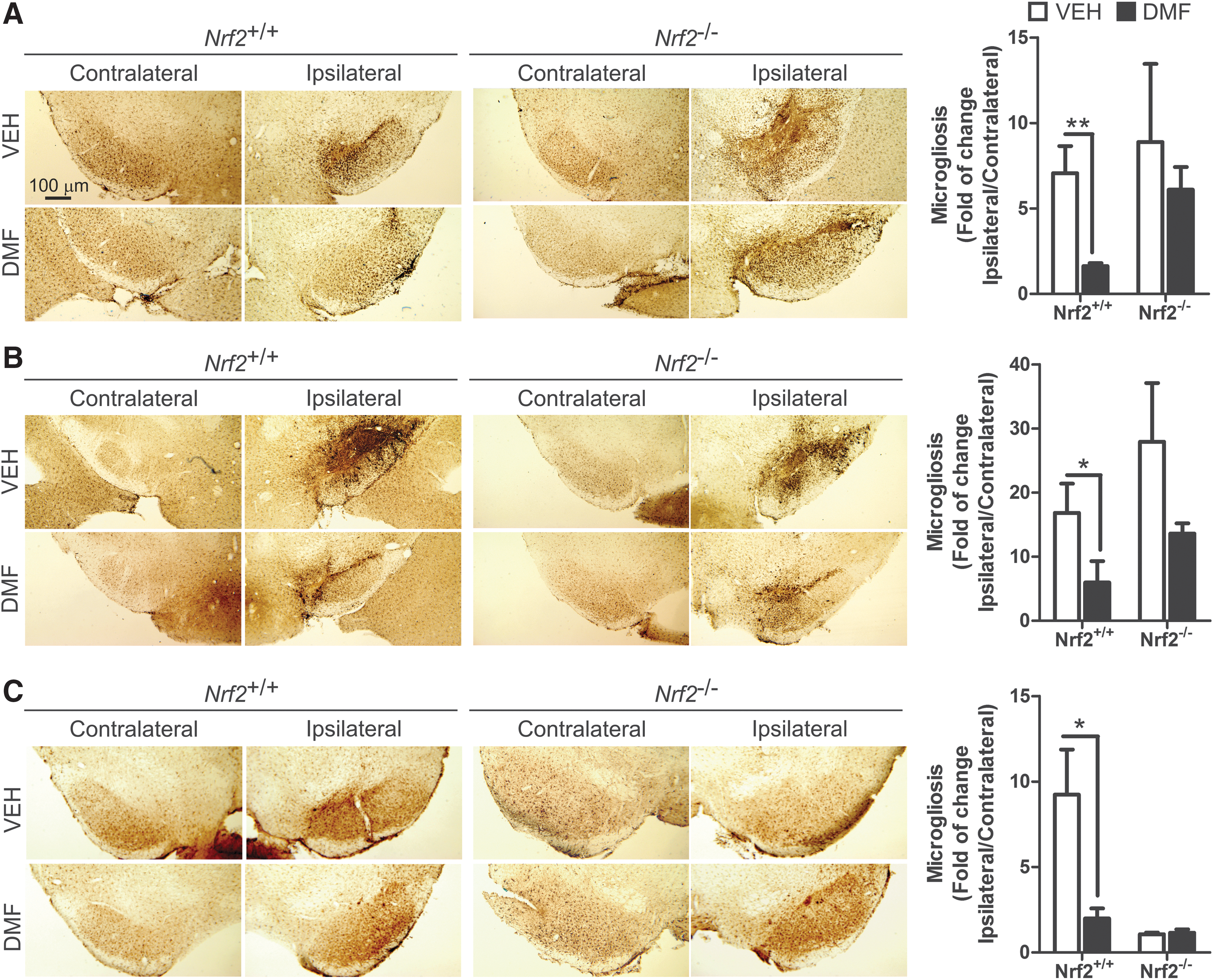
DMF treatment decreased α-SYN-induced astrocytosis and microgliosis
A distinctive hallmark of PD is the presence of low-grade chronic inflammation that is characterized by microgliosis and astrocytosis at the basal ganglia. Therefore, our next step was to evaluate whether DMF could modulate the gliosis triggered by α-SYN expression. Regarding astrocytosis, α-SYN toxicity correlated with a very significant increase in GFAP+ astrocytes at the ipsilateral VMB side of Nrf2 +/+mice, with a maximum at 3 weeks after infection, and an astrocyte scar remained visible at 3 and 8 weeks (Fig. 4). Importantly, DMF reduced astrogliosis in Nrf2 +/+ mice, with a slight effect in Nrf2 −/− mice. Similarly, the astrogliosis observed in the ipsilateral STR for vehicle-treated Nrf2 +/+ was abolished in the DMF-treated Nrf2 +/+, without any effect in Nrf2 −/− mice (Supplementary Fig. S3). Considering the crucial role of astrocytes in filling gaps left by dead neurons, these results further complement the observations of Figures 2 and 3, indicating that DMF attenuates neuronal death caused by α-SYN toxicity.
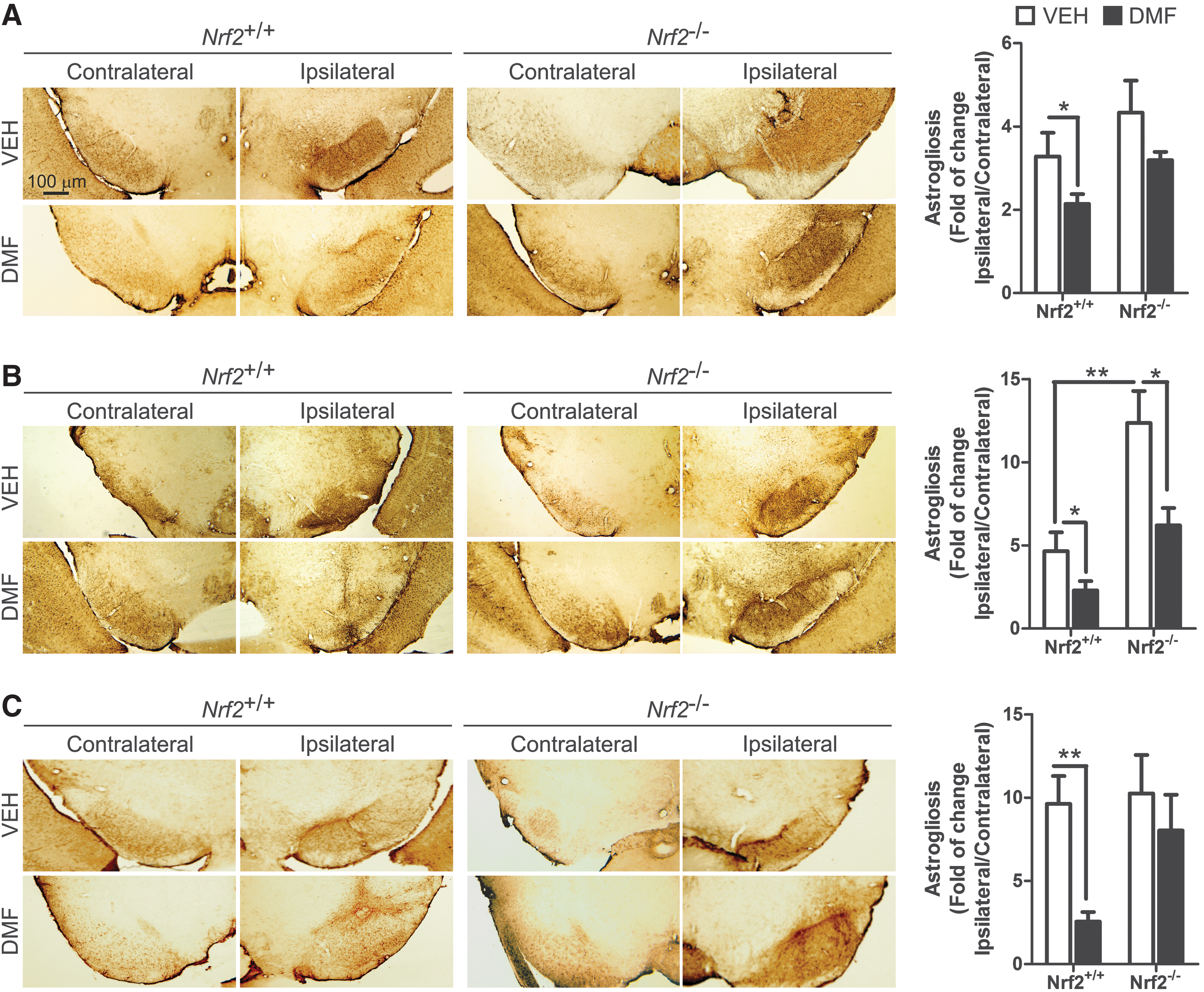
Regarding microglia, Iba1 immunohistochemistry showed that α-SYN expression induced microgliosis at the ipsilateral side of vehicle-treated Nrf2 +/+ and Nrf2 −/− mice after 1 week, reached a maximum after 3 weeks, and returned to close to basal levels at 8 weeks (Fig. 5), indicating that events related to microglial activation took place several weeks before this later time point. However, DMF greatly ameliorated the microgliosis of Nrf2 +/+ and had a minor effect on Nrf2 −/− mice, suggesting that this cell type is very sensitive to NRF2 activation. In the STR, we did not observe microgliosis in the ipsilateral side at any time after α-SYN injection (Supplementary Fig. S4).
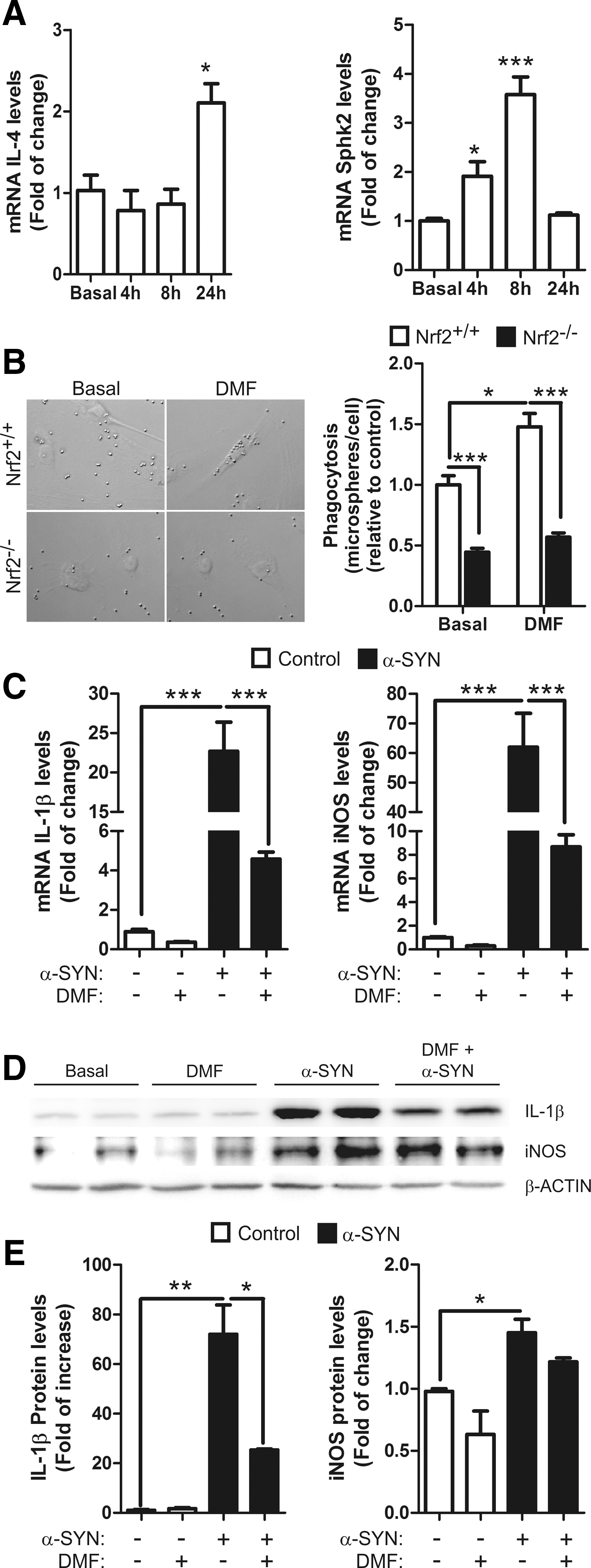
Modulation of classic versus alternative microglial phenotype by DMF
According to their activation state, microglia express specific classical pro-inflammatory (iNOS; IL-1β), anti-inflammatory (IL-4), or type II-deactivating (SPHK1/2) phenotypic markers (18). BV2 microglial cells treated with DMF (20 μM) expressed increased mRNA levels of IL-4 and sphingosine-kinase 2 (SPHK2) in a time-dependent manner (Fig. 6A), whereas SPHK1 levels were undetectable (data not shown). Moreover, Nrf2 −/− microglia showed impaired phagocytosis in comparison to wild-type microglia, as previously reported (45), and DMF increased the phagocytic activity of Nrf2 +/+ but not in Nrf2 −/− microglia (Fig. 6B). To examine the anti-inflammatory effect of DMF against α-SYN, BV2 cells were pretreated with this drug (20 μM) and after 2 h, the cells were shifted to normal medium or to medium supplemented with recombinant α-SYN (1 μM) and incubated for 6 h. As shown in Figure 6C–E, α-SYN induced mRNA and protein levels of the classical pro-inflammatory markers IL-1β and iNOS, and the pretreatment with DMF attenuated this effect. These results suggest that DMF reduces the ratio of microglial classical versus alternative phenotypes.
Effect of DMF in autophagy
As shown in Figure 7A, α-SYN accumulated in neuron bodies and neurites in Nrf2 +/+ and Nrf2 −/− mice, suggesting a limiting capacity of the proteolytic clearance systems. Degradation of α-SYN may be achieved through proteasome and autophagy pathways (76). The mRNA levels of several proteasome subunits did not change significantly after DMF treatment in MN9D, BV2, and IMA2.1 cells (Supplementary Fig. S5), although we have previously reported that NRF2-deficiency is implicated in regulating proteasome activity (45). This discrepancy may be due to the slow turnover of proteasome subunits that cannot be substantially increased by DMF. For the purpose of this study, we focused on autophagy. We analyzed the levels of autophagosome cargo protein p62, because its expression is regulated by NRF2 (32, 41) and it has been reported to participate in autophagy of α-SYN (66, 74). We also analyzed LC3, a protein specifically associated with autophagic vesicles whose lipidized levels (LC3-II) correlate with autophagosome abundance.
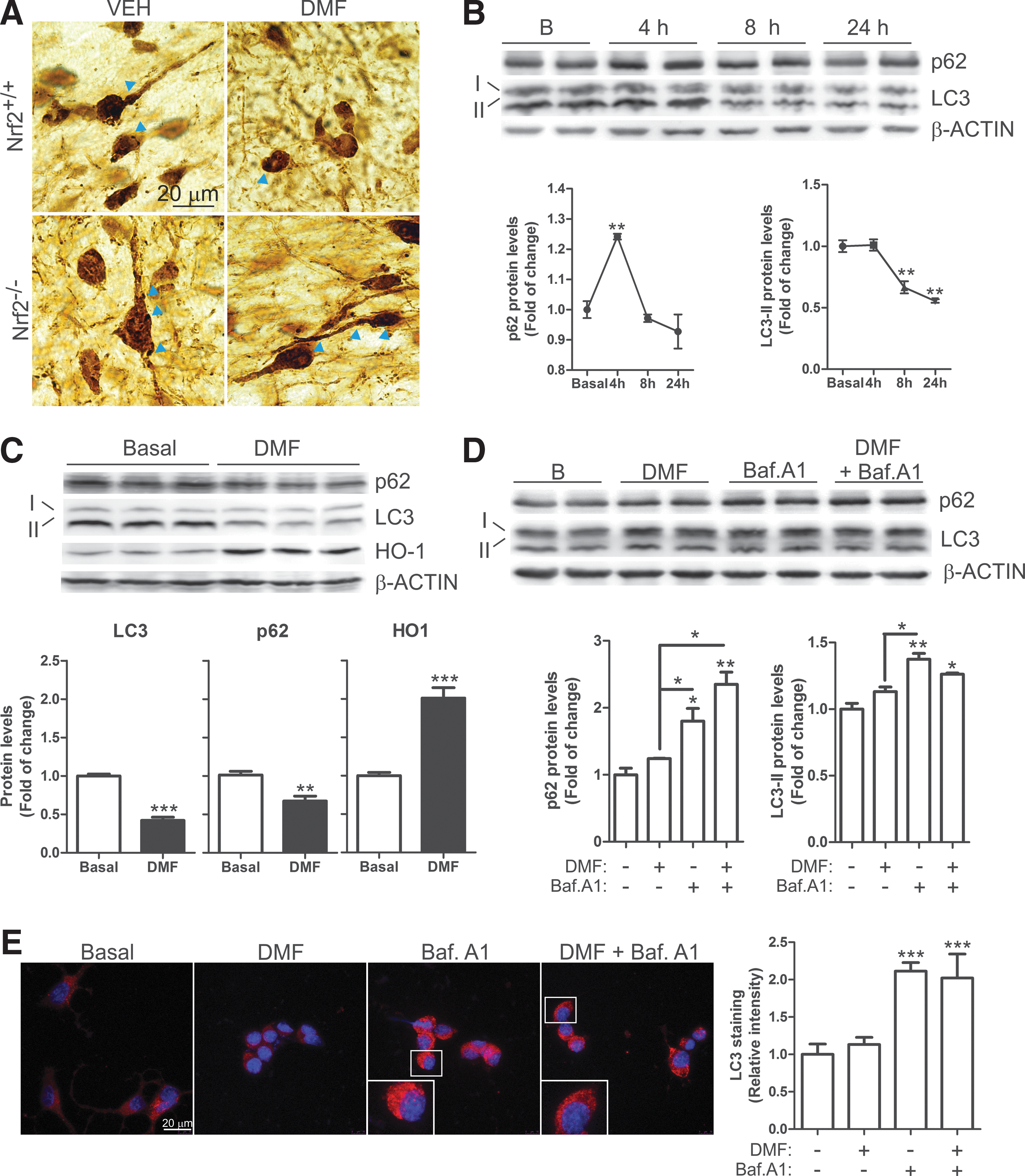
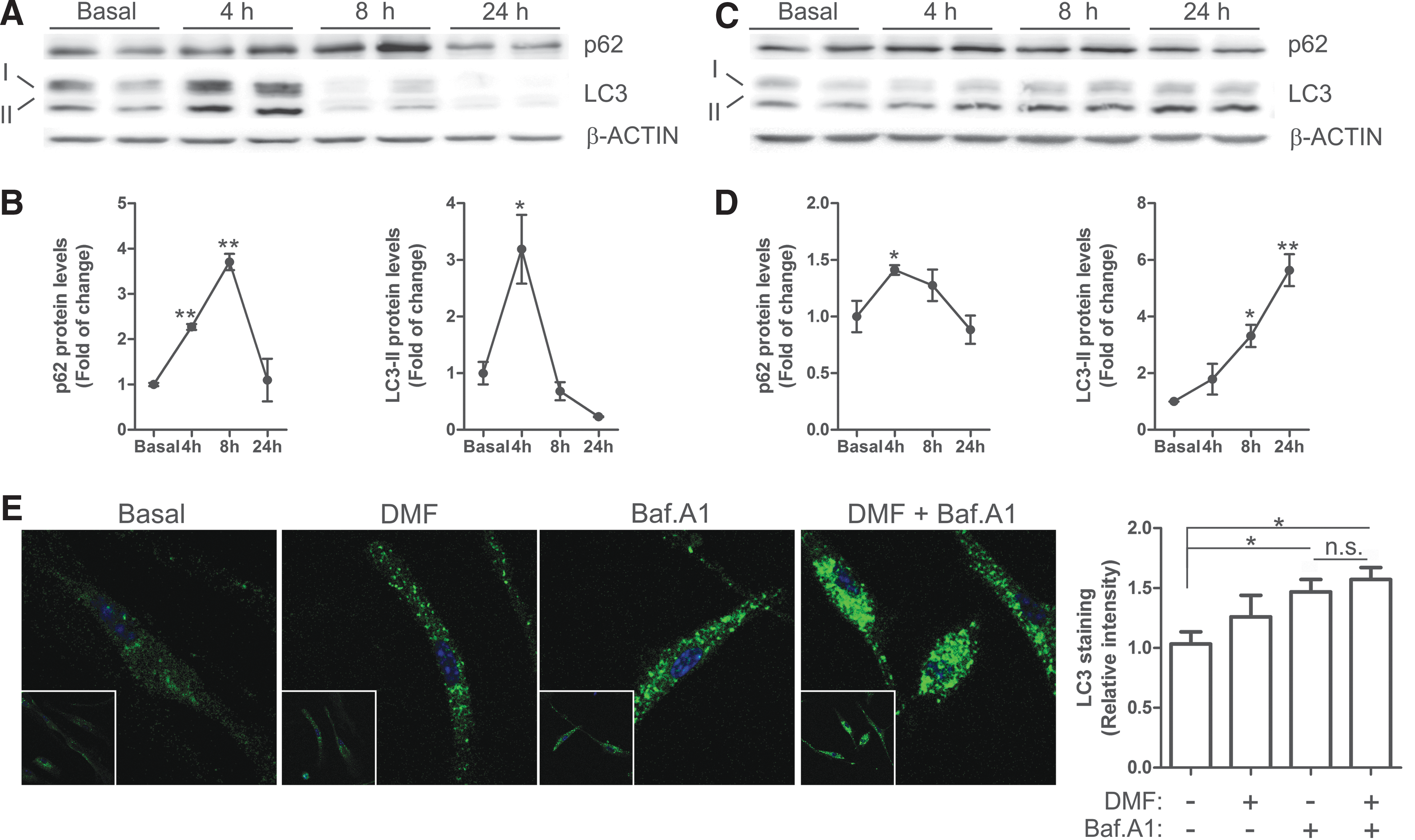
In neuron-derived dopaminergic MN9D cells, DMF increased the levels of p62 with a maximum at 4 h, which then decreased even below basal levels by 24 h (Fig. 7B, C). LC3-II remained stable during the first 4 h but decreased after 8–24 h. To further determine whether these changes might be due to a modification in the autophagic flux, we used the autophagy inhibitor bafilomycin A1 (Baf.A1). MN9D cells were serum-deprived for 16 h, then pretreated with Baf.A1 (400 nM, 1 h) before further addition of DMF or vehicle (20 μM, 4 h) (Fig. 7D). As expected, the blockage of autophagy flux by Baf.A1 led to the accumulation of p62 and LC3-II. The combined Baf.A1/DMF treatment resulted in further accumulation of p62 but not of LC3. Moreover, immunocytochemical analysis of LC3 (Fig. 7E) showed the accumulation of autophagic vacuoles in the presence of Baf.A1 but DMF co-treated cells did not show a further increase. These results suggest that DMF favors autophagy by providing more cargo protein p62 but has a minor role in the autophagic flux since the levels of LC3-II (Fig. 7D) and LC3-positive vacuoles (Fig. 7E) were not significantly altered by this drug. Then, we analyzed these autophagy makers in microglia and astrocytes. In microglial BV2 cells, DMF induced a time-dependent variation of the levels of p62 with a maximum at 8 h and a return to baseline by 24 h (Fig. 8A, B). LC3-II increased at 4 h but decreased even below baseline by 8 h (Fig. 8A, B). In murine IMA2.1 astrocytes, DMF induced a slight accumulation of p62, maximal at 4 h, but contrary to BV2 cells, LC3-II accumulated gradually at least during the first 24 h of treatment (Fig. 8C, D). We further analyzed the autophagy flux in primary microglia submitted to Baf.A1 or DMF (Fig. 8E). Like in MN9D cells, the amount of LC3-positive vacuoles was augmented after Baf.A1 treatment but was not significantly increased by DMF. Taken together, these results suggest that DMF modulates p62-dependent autophagy but different nerve cells may have slightly different responses.
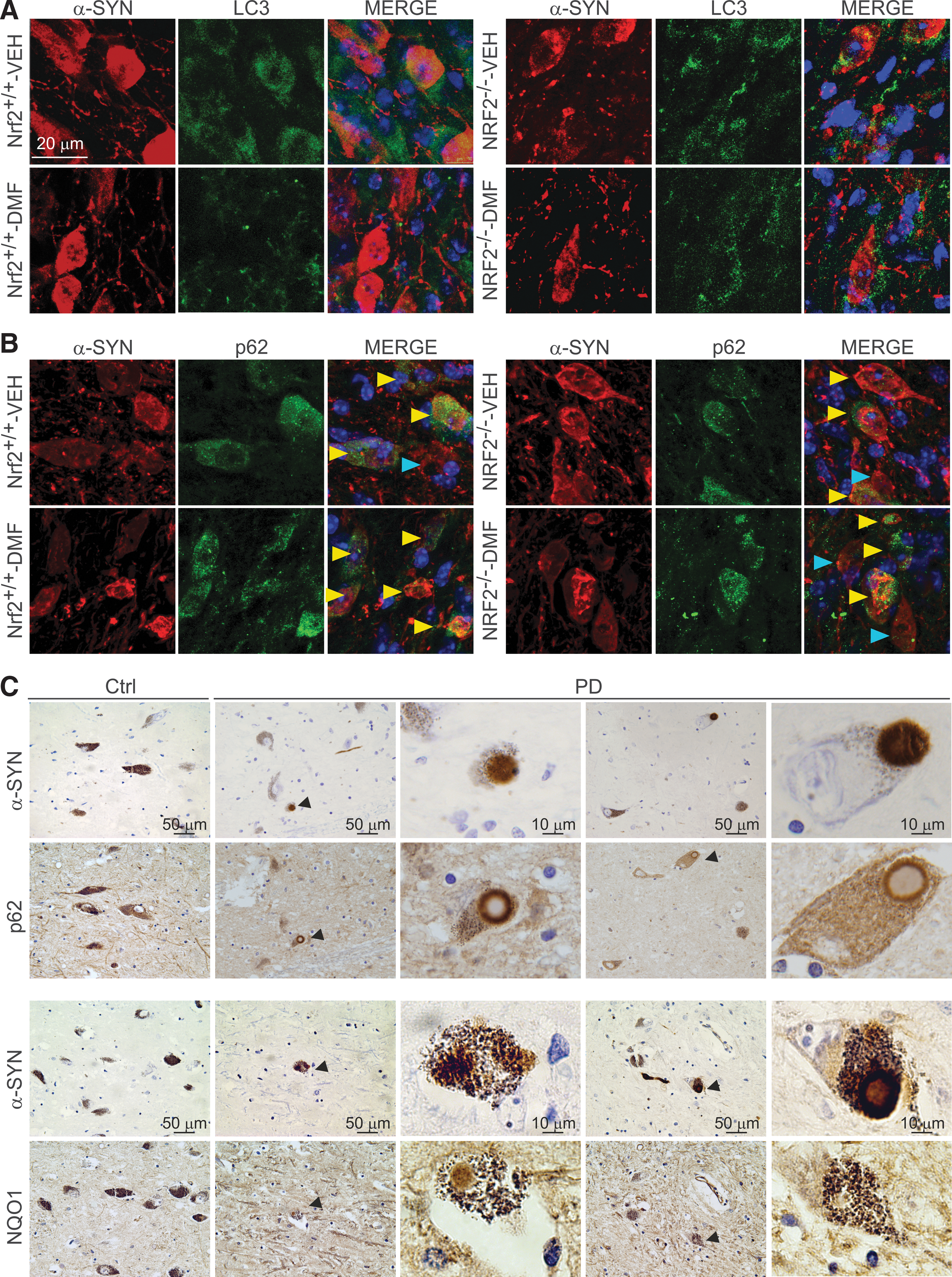
To analyze the effect of synucleinopathy in NRF2-mediated autophagy, we performed double immunofluorescence of human α-SYN versus LC3 or p62 in 30 μm-thick sections of VMB (Fig. 9A). LC3 localization did not clearly correlate with human α-SYN staining but similar to the observations in MN9D cells, DMF decreased the overall level of LC3 in Nrf2 +/+ mice. Regarding p62, we found α-SYN expressing neurons with or without overall expression of p62 but in DMF-treated Nrf2 +/+ mice, there was a tendency to find more cells co-expressing α-SYN and p62 (Fig. 9B). Although to our knowledge there are still no PD patients treated with DMF, we wanted to anticipate a possible correlation between NRF2 activity and this human synucleinopathy. Markers of NRF2 activation were analyzed in postmortem samples of asymptomatic subjects and PD patients. Due to the unspecific staining of available anti-NRF2 antibodies, we used NQO1 and p62 as two combined markers reflecting activation of the NRF2. Also, to circumvent the problem associated with different retrieval requirements for each antigen, we analyzed two adjacent 4 μm-thick sections that, in many cases, showed part of the same cell. As shown in Figure 9C, the SN pars compacta of two patients exhibited scattered dopaminergic neurons loaded with neuromelanin and α-SYN. These neurons were positive for NQO1 staining not only in neuronal bodies but also in Lewy bodies. Similarly, p62 was found not only in the cytoplasm but also decorating the periphery of Lewy bodies of dopaminergic neurons. These results suggest that some dopaminergic neurons up-regulate the NRF2 response, possibly in an attempt to protect from α-SYN toxicity and subsequent oxidative stress but, due to partial sequestration of NRF2-regulated products such as NQO1 and p62 in Lewy bodies, the physiological NRF2 response may be insufficient for mounting an optimal cellular defense and may need a pharmacological reinforcement.
Discussion
Several drugs have been used for proof-of-concept studies that strongly suggest that activation of NRF2 may provide a benefit against the Parkinsonian toxins 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, which generate oxidative stress in the nigrostriatal track of rodents [for a review, see (37)]. From the point of view of clinical significance, these studies present two problems: (i) Most compounds tested lack biopharmaceutical pipelines that could proceed toward a clinical setting; (ii) “curing” oxidative stress-induced parkinsonism in rodents, while interesting and predictable as proof of concept, may have little impact in the much more complex pathology of human PD.
This study was aimed at determining whether pharmacological targeting of NRF2 might provide a disease-modifying therapy for the motor symptoms of PD by using a mouse model that replicates the main features of the human synucleinopathy. Our mouse model replicates the presence of α-SYN containing intracellular aggregates and dystrophic neurites, leads to a robust and selective loss of nigral dopaminergic neurons, and correlates with strong astrogliosis and microgliosis. It is generally believed that protofibrils made of conformationally altered α-SYN lead to neuronal death in both idiopathic and, at least, some familiar cases of PD, where the α-SYN coding gene has suffered duplications or triplications. The process of α-SYN aggregation into Lewy bodies has been used to stage PD progression and is discussed in detail in (9). Considering the level of α-SYN aggregation into dystrophic Lewy dendrites and aggregates, this mouse model seems to replicate the early stages of PD where abnormal α-SYN deposition occurs and becomes the driving force in PD pathogenesis (64). A remaining limitation of this model is that it only replicates histological and motor symptoms of PD that are associated with nigrostriatal pathology. However, because DMF is administered systemically and NRF2 appears to provide multiple layers of tissue protection, including oxidative, inflammatory, and proteotoxic stress, it is tempting to speculate that systemic activation of NRF2 will also provide a relief of nonmotor symptoms such as constipation, sleep disturbance, depression, or dementia, among others. Thus, oxidative stress during inflammation causes persistent dysfunction of colonic smooth muscle that leads to motility dysfunction, and it has been reported that the NRF2 activator sulforaphane can attenuate this pathological mark in a model of inflammatory bowel disease (20). Regarding sleep disturbance, NRF2 is regulated by an E-box-mediated circadian rhythm that combats oxidative/fibrotic lung damage, and timed administration of sulforaphane significantly blocks this effect in a murine model of bleomycin-induced lung fibrosis (52). Another co-morbidity associated with PD is depression, and it has been reported that NRF2 activation prevents depressive-like behavior in mice (23, 47). Finally, dementia is becoming one of the most difficult symptoms to palliate in late PD. However, although this issue has not yet been addressed in a preclinical model of PD, there is strong evidence indicating that the NRF2 might protect the cholinergic system in Alzheimer's disease mouse models (40, 62).
In preliminary experiments, we searched for the most suitable dosing of DMF that might provide a modest activation of the NRF2 signature in the brain, yet lack significant toxicity and stay within clinically relevant values. In agreement with former studies in experimental autoimmune encephalomyelitis (10), the 100 mg/kg dose elicited a modest but persistent activation of the NRF2 signature. In humans, the usual dose is 480 mg but the pharmacokinetics between rodents and humans is different. Thus, in a plot of DMF concentration in plasma over time (24 h), we found that the area under the curve in rodents submitted to 100 mg/kg was just two-fold higher than in humans submitted to 480 mg DMF [data not shown (59)]. . Moreover, at the 100 mg/kg dose, we did not observe in mice any significant evidence of toxicity, such as weight loss, hair loss, or other gross alterations. In humans, toxicity of DMF does not appear to be an important concern since some patients with multiple sclerosis have been taking this drug for more than 5 years with little or no evidence of adverse effects.
The elevated body swing test used here to evaluate motor performance was highly predictive of the degree of nigrostriatal injury as previously reported (3, 8). It provided the first evidence of a beneficial effect of DMF that was further confirmed by preservation at 1, 3, and 8 weeks from surgery of TH+ neuron bodies in the SN and fibers in the STR in Nrf2 +/+ but not Nrf2 −/− mice. Other motor tests used, such as the cylinder, apomorphine, and open-field test, were not sensitive enough to predict nigrostriatal damage (data not shown). The denervation of the STR, as a result of retrograde transport of α-SYN from the SN, was not as dramatic as the loss of TH+ neuronal bodies at the SN, which is consistent with the existence of compensatory mechanisms that favor sprouting of remaining dopaminergic fibers (34, 45, 55). But most importantly, the continuous administration of DMF to the Nrf2 +/+ mice strongly attenuated loss of both dopaminergic neuron bodies at the SN and fibers at the STR. This protection was not found in the DMF-treated Nrf2 −/− mice, further indicating that DMF is targeting NRF2 for this protective effect. In fact, only the DMF-treated Nrf2 +/+ mice could stand the expression of human α-SYN after 1, 3, and 8 weeks. It is interesting that DMF also induced a mild reduction of astrogliosis and microgliosis in the Nrf2 −/− mice. This is consistent with the reported inhibitory effect of this drug on NF-κB activity in an NRF2-independent manner (25).
The analysis of the nigrostriatal lesion after 1 week from surgery was very informative about early events leading to synucleinopathy. Several reports suggest that NRF2 up-regulates the expression of proteasome genes and probably autophagy genes (38, 43), and, thus, the Nrf2 −/− mice would be expected to be more sensitive to α-SYN toxicity. In this context, it has been described that p62 interacts directly with KEAP1, demonstrating that p62-mediated NRF2 up-regulation is KEAP1 dependent (46) and NRF2 up-regulates the expression of p62 (32). Consistently, DMF induced p62 protein levels in cultured neural cells but the DMF modulation of autophagy may be more complex than NRF2 targeting, as it has been suggested for sulforaphane, a well-established NRF2 activator, that also promotes autophagy by regulating ERK (36) or AMPK (79). In any case, these observations collectively indicate that DMF regulates autophagy, at least in part, by regulating the NRF2/p62 axis.
DMF led to changes in the levels of the lipidated LC3-II form, although with different kinetics in the three nerve cells analyzed, suggesting a different mechanism of regulation. A puzzling result was that the level of LC-3II and the number of LC-3+ autophagy vesicles was not further increased after treatment with the autophagy flux blocker Baf.A1 in either MN9D cells or microglia. The combination of LC-3 and Baf.A1 has been traditionally used as a method to monitor autophagy flux, and, therefore, it was surprising to find that DMF did not clearly change this flux. Then, the DMF-induced changes in LC-3II levels in the absence of Baf.A1 must be attributed to variations in the composition of autophagosomes rather than to a net change in the autophagy flux. It remains to be determined whether LC3 is modulated by NRF2. The messenger RNA levels of LC-3 are similar in Nrf2 +/+ and Nrf2 −/− cortical neurons (data not shown) and the kinetics of induction of p62 and LC3 by DMF is different (Figs. 7 and 8), therefore suggesting that LC3 is not regulated directly by DMF or NRF2. Taken together, we suggest that DMF regulates the NRF2/p62 axis to provide more cargos such as α-SYN to the autophagosome and has a global but still not fully defined effect on autophagy, as reflected by changes in LC-3II levels.
We and others have reported the relevance of NRF2 in modulation of acute and chronic neuroinflammation (29, 33, 56), and the experimental and clinical evidence suggests that DMF has a very relevant role in targeting inflammation (1, 75). DMF modulated the microglial response to α-SYN, shifting from a more pro-inflammatory phenotype (IL-1 and iNOS) toward a more alterative phenotype (IL-4 and SPHK2) in vitro, and probably during the early stages of damage to the brain parenchyma, because microgliosis was attenuated at 1 and 3 weeks. The fact that we could not see a significant microgliosis at 8 weeks suggests that the initial neuron damage elicited by α-SYN overexpression led to microgliosis that remitted only when most neurons had died. It is interesting to note that DMF induced microglial phagocytosis in an NRF2-dependent manner. This intriguing observation has also been reported for the NRF2 activator sulforaphane (65) and in our previous work (45). In peripheral macrophages, the phagocytosis-stimulating activity of some pro-resolving lipid mediators of inflammation was significantly impaired in Nrf2−/− macrophages, which already had a weakened basal phagocytic function (53). Mechanistically, to our knowledge, there is not yet a conclusive explanation, but we have reported that the expression of some receptors involved in phagocytosis (TAM receptors) is slightly decreased in NRF2-defficient microglia (44, 45). Other evidence suggests that NRF2 modulates the expression of scavenger receptors such as MARCO (macrophage receptor with collagenous structure) (7) and CD36 (30).
Contrary to microglia, astrogliosis was also observed at 8 weeks. This was most likely due to an accumulative effect, since astrocytes will fill the empty spaces left by dead neurons and will remain there as a mark of the neuronal lesion. The protective effect of DMF was very obvious when we analyzed the degree of astrogliosis in mice injected with rAAV-6-α-SYN. Thus, in the STR of vehicle-treated Nrf2 +/+ mice, we observed abundant astrocytes that were largely decreased in the DMF-treated counterparts.
The global results of this study are presented in an idealized graph in Supplementary Figure S6. It is predicted that overexpression of human α-SYN leads to a rapid, less than 3-week, intoxication of nigrostriatal dopaminergic neurons of Nrf2 +/+ and Nrf2 −/− mice. This injury is slightly higher in the Nrf2 −/− mice (Fig. 3). In parallel to neuron intoxication, we find microglial activation that will elicit an inflammatory response and remove neuronal debris but will cease once α-SYN intoxicated neurons have disappeared. Microglial activation will be lower in DMF-treated Nrf2 +/+ mice, because they exhibit less neuron damage (Figs. 5 and 3, respectively). Astrocytes are activated in parallel to neuronal intoxication but contrary to the microglia, they remain detectable after the phase of injury, creating a scar in the damaged tissue (Fig. 4). The astroglial scar is smaller in the DMF-treated mice, because the death of dopaminergic neurons was attenuated by this drug. Further work may be required for obtaining a fine analysis of the participation of DMF and NRF2 in prevention of proteinopathy, but from a clinical perspective, DMF is now ready for clinical analysis for the treatment of PD.
Materials and Methods
Cell culture
MN9D is an immortalized dopaminergic neuronal cell line derived from mouse mesencephalon (19). IMA 2.1 astroglial cells were provided by Dr. Stefan Schildknecht (University of Konstanz, Konstanz, Germany). MN9D and IMA2.1 were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 80 μg/ml gentamicin. BV2 microglial cells were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum and 80 μg/ml gentamicin. Primary microglia were prepared from neonatal (P0-P2) mouse cortex from Nrf2 +/+ and Nrf2−/− and were isolated and grown as described in (45). The medium was changed to serum-free DMEM without antibiotics at 16 h before treatment.
Immunoblotting
Whole brain lysates were prepared as previously described (57). Immunoblots were performed as described in (15). The primary antibodies used are described in Supplementary Table S1.
Analysis of mRNA levels by quantitative real-time polymerase chain reaction
Total RNA extraction, reverse transcription, and quantitative polymerase chain reaction (PCR) were done as detailed elsewhere (44). Primer sequences are shown in Supplementary Table S2. Data analysis was based on the ΔΔCT method with normalization of the raw data to housekeeping genes (Applied Biosystems). All PCRs were performed in triplicate.
Phagocytosis assay
About 150,000 cells of primary microglia from Nrf2 +/+ and Nrf2 −/− mice were plated on coverslips for 16 h. Then, the medium was replaced with serum-free DMEM without antibiotics for 24 h before adding 150 microspheres per cell (FluoSpheres polystyrene microspheres; Invitrogen) and DMF (20 μM) and incubating for 2 h. Then, the cells were washed with PBS, fixed with 4% paraformaldehyde, and stained with 4',6-diamidino-2-phenylindole (DAPI) (Molecular Probes). The images were captured by using a 90i Nikon microscope (Nikon) with a 40× objective.
Immunocytochemistry
Primary microglia from Nrf2 +/+ and MN9D cells were seeded in 24-well plates. After 24 h, the cells were shifted to serum-free DMEM without antibiotics. After treatments, the cells were washed with cold PBS and fixed with 4% paraformaldehyde for 10 min. The cells were permeabilized with 0.25% Nonidet NP-40 (Sigma-Aldrich) for 10 min and incubated with primary antibodies for 1.5 h at 37°C in a humidified box. Secondary antibodies were incubated for 45 min at 37°C under the same conditions. Cells were counterstained with DAPI. Fluorescent images were captured by using appropriate filters in a Leica DMIRE2TCS SP2 confocal microscope (Leica).
Animals and treatments
Colonies of Nrf2 −/− mice and Nrf2 +/+ littermates were established from funders kindly provided by Prof. Masayuki Yamamoto (Tohoku University Graduate School of Medicine, Sendai, Japan) (31). Each experimental group comprised 5–8 animals. An adeno-associated pseudotype 6 (rAAV6) viral vector was used to express human α-SYN under the neuron-specific human Synapsin 1 promoter (42). Vector batches yielded 3.5 × 108 TU/μl titers, and stock preparations were diluted two-fold for in vivo injections. Surgical procedures and a unilateral intracerebral injection of viral particles into the right hemisphere were performed as described (45) with several modifications. Briefly, animals received a single 2 μl injection into the right SN at the following coordinates: −2.8 mm posterior and −1.4 mm lateral to bregma, and −4.5 mm ventral relative to dura, calculated according to the mouse atlas of (51). All experiments were performed by certified researchers according to regional, national, and European regulations concerning animal welfare and animal experimentation, and they were authorized by the Ethics Committee for Research of the Autonomous University of Madrid. DMF (100 mg/kg or 300 mg/kg; Biogen) was suspended in 0.8% methocel and given by oral gavage. We did not detect significant weight loss, hair loss, or other gross alterations in the DMF-treated mice either in the 1- and 3-week administration every day or in the 8-week administration every 2 days.
Behavioral test
The elevated body swing test is a simple and easy behavioral test that only requires handling the animal by its tail and recording the direction of swings made by the animal for a certain period (3, 8). Briefly, the animal was held in the vertical axis and a swing was recorded whenever the animal moved its head out of the vertical axis to either side. Before attempting another swing, the animal must return to the vertical position for the next swing to be counted. Each experimental group comprised 5–8 animals.
Immunohistochemistry in mouse and human tissues
Immunohistochemistry in mice was performed on 30 μm-thick coronal brain sections with a standard avidin-biotin immunohistochemical protocol (45). Primary antibodies are described in Supplementary Table S1. Human tissues were obtained from two controls (age 78 and 88 years) and three PD patients (aged 62, 65, and 84 years) within a 12 h-postmortem interval, according to standard procedures of Banco de Tejidos de la Fundación CIEN. The control subjects had no background of neuropsychiatric disease, and a full neuropathological examination on paraffin-embedded tissue excluded relevant brain pathology. In PD patients, clinical diagnosis was confirmed by hematoxylin and eosin and α-SYN staining on paraffin-embedded tissue sections. PD patients were categorized as Braak stage 6 for Lewy body pathology (9). Immunohistochemistry in human SN was done on 4 μ-thick paraffin-embedded samples. Primary antibodies were incubated with Dako REAL antibody diluents. After washing, the sections were incubated with the secondary biotinylated antiserum (Vector) and then with the ABC kit system, and they were developed by using 3,3′-diaminobenzidine.
Stereological analysis
Cell counts were performed every four sections (30 μm-thick) through the SN by using Stereo Investigator Software (MicroBrightfield) that was attached to an E800 Nikon microscope (Nikon). SN, excluding ventral tegmental area, was delineated in low magnification (5× objective), and a point grid was overlaid onto each section. Cell counting was performed as previously described by (21). The error coefficient attributable to the sampling was calculated according to Gundersen and Jensen (26), and values ≤0.10 were accepted.
Determination of monoamines and their metabolites
Hemi-brains were frozen on dry ice until use. DA and its metabolite, DOPAC, were measured by HPLC with an ESA coulochem detector. Briefly, the tissue was sonicated in 6 vol. (weight/volume) of 0.4 N perchloric acid (PCA) with 0.5 mM Na2S2O5 and 2% EDTA and then centrifuged at 10,000 g at 4°C for 20 min. Monoamine levels were determined from 20 μl of the resulting supernatant. The chromatographic conditions were as follows: A column (Nucleosil 5C18); the mobile phase, a citrate/acetate buffer 0.1 M, pH 3.9 with 10% methanol, 1 mM EDTA, and 1.2 mM heptane sulfonic acid; and the detector voltage conditions: D1 (+0.05), D2 (−0.39), and the guard cell (+0.40) have been previously described (48).
Immunofluorescence on mouse tissues
The protocol was previously described (55). Primary antibodies are described in Supplementary Table S1. Secondary antibodies were as follows: Alexa Fluor 546 goat anti-mouse, Alexa 546 goat anti-rabbit, and Alexa Fluor 488 goat anti-mouse (1:500; Life technologies). Control sections were treated by following identical protocols but omitting the primary antibody.
Statistical analyses
Data are presented as mean ± SEM. To determine the statistical test to be used, we employed GraphPad Instat 3, which includes the analysis of the data to normal distribution via Kolmogorov–Smirnov test. In addition, statistical assessments of differences between groups were analyzed (GraphPad Prism 5) by unpaired Student's t-tests when normal distribution and equal variances were fulfilled, or by the nonparametric Mann–Whitney test. One- and two-way ANOVA with post hoc Newman–Keuls test or Bonferroni's test were used, as appropriate.
Footnotes
Acknowledgments
This work was supported by grants from Biogen, SAF2013-143271-R and “Fundación Salud 2000.” ILB was recipient of a Ramón y Cajal contract (MICINN-RYC). The authors thank Johanna Troya-Balseca for her technical support.
Author Disclosure Statement
At the time this work was conducted, R.S. was an employee of and held stock/stock options in Biogen. All other authors declare no competing interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.