
Abstract
Aims:
The use of doxorubicin, a potent chemotherapeutic agent, is limited by cardiotoxicity. We tested the hypothesis that decreased soluble guanylate cyclase (sGC) enzyme activity contributes to the development of doxorubicin-induced cardiotoxicity.
Results:
Doxorubicin administration (20 mg/kg, intraperitoneally [IP]) reduced cardiac sGC activity in wild-type (WT) mice. To investigate whether decreased sGC activity contributes to doxorubicin-induced cardiotoxicity, we studied mice with cardiomyocyte-specific deficiency of the sGC α1-subunit (mice with cardiomyocyte-specific deletion of exon 6 of the sGCα1 allele [sGCα1−/−CM]). After 12 weeks of doxorubicin administration (2 mg/kg/week IP), left ventricular (LV) systolic dysfunction was greater in sGCα1−/−CM than WT mice. To further assess whether reduced sGC activity plays a pathogenic role in doxorubicin-induced cardiotoxicity, we studied a mouse model in which decreased cardiac sGC activity was induced by cardiomyocyte-specific expression of a dominant negative sGCα1 mutant (DNsGCα1) upon doxycycline removal (Tet-off). After 8 weeks of doxorubicin administration, DNsGCα1tg/+, but not WT, mice displayed LV systolic dysfunction and dilatation. The difference in cardiac function and remodeling between DNsGCα1tg/+ and WT mice was even more pronounced after 12 weeks of treatment. Further impairment of cardiac function was attenuated when DNsGCα1 gene expression was inhibited (beginning at 8 weeks of doxorubicin treatment) by administering doxycycline. Furthermore, doxorubicin-associated reactive oxygen species generation was higher in sGCα1-deficient than WT hearts.
Innovation and Conclusion:
These data demonstrate that a reduction in cardiac sGC activity worsens doxorubicin-induced cardiotoxicity in mice and identify sGC as a potential therapeutic target. Various pharmacological sGC agonists are in clinical development or use and may represent a promising approach to limit doxorubicin-associated cardiotoxicity. Antioxid. Redox Signal. 26, 153–164.
Introduction
S
Adverse effects observed in chemotherapy patients receiving dexrazoxane, the only drug approved to attenuate doxorubicin-induced cardiotoxicity in patients, have restricted its use. This study illustrates that doxorubicin reduces cardiac soluble guanylate cyclase (sGC) activity and that reduced sGC activity exacerbates doxorubicin-induced cardiotoxicity. Increasing interest in sGC as a therapeutic target is driving clinical development of pharmacological sGC agonists, and the first drug in this class was recently approved to treat pulmonary hypertension. Because our data suggest that sGC protects against doxorubicin-induced cardiotoxicity, pharmacological sGC agonists could represent a novel therapeutic strategy to treat cardiotoxicity in patients undergoing chemotherapy.
Although various pathogenic mechanisms have been suggested to underlie doxorubicin-induced cardiotoxicity, many studies support a pivotal role of oxidative stress (11, 22, 30, 32, 50, 56). Cardiomyocytes are particularly prone to damage by free radicals because of the high oxidative metabolism, low concentrations of antioxidant enzymes, and abundance of mitochondria (14, 15).
With a univalent redox potential of approximately −320 mV, doxorubicin is a good substrate for intracellular oxidoreductases such as xanthine oxidase, NADPH cytochrome P450 reductase, and NADH dehydrogenase (12 –14, 49). Reduction of the quinone moiety of doxorubicin results in an unstable semiquinone radical that donates an electron to molecular oxygen, thereby forming superoxide radicals. If free iron is present, the semiquinone radical can also form a complex with iron, resulting in an anthracycline–iron free radical complex that in turn reduces molecular oxygen (31).
Dexrazoxane (ICRF-187), the only drug currently in clinical use to protect against doxorubicin-induced cardiotoxicity, is thought to confer cardioprotection by chelating iron and subsequently reducing anthracycline–iron free radical complex formation and superoxide radicals (21). Because of reports of lower tumor responsiveness or enhanced myelosuppression in chemotherapy-receiving patients treated with dexrazoxane, the US Food and Drug Administration and the European Medicines Agency restricted the use of dexrazoxane (38, 47). Although a growing body of literature suggests that dexrazoxane does not adversely affect therapeutic response (28, 40, 41, 53), additional mechanism-based therapies for doxorubicin-induced cardiotoxicity are needed.
The second messenger cyclic guanosine 3′,5′-monophosphate (cGMP) regulates a vast array of physiological processes vital to endothelial, vascular smooth muscle, and cardiomyocyte function. cGMP is generated by particulate guanylate cyclase in response to natriuretic peptides and by soluble guanylate cyclase (sGC) upon activation by nitric oxide (NO) and, less potently, carbon monoxide (CO). Disruption of the cGMP signaling pathway has been implicated in adverse cardiac remodeling and heart failure. Interestingly, doxorubicin administration reduced cardiac cGMP levels, which could be prevented by inhalation of CO (45).
In this study, we investigated the role of sGC in doxorubicin-induced cardiotoxicity. We observed that doxorubicin administration decreased cardiac sGC activity and that reduced sGC activity exacerbated reactive oxygen species (ROS) formation in the heart and development of doxorubicin-induced cardiac dysfunction.
Results
Doxorubicin administration reduces cardiac sGC activity
To examine the effect of doxorubicin treatment on sGC enzymatic activity in the heart, the increase in cGMP production induced by the NO donor diethylenetriamine NONOate (DETA-NO) was measured in cardiac extracts from wild-type (WT) mice 24 h after doxorubicin (20 mg/kg, intraperitoneally [IP]) or saline administration. Doxorubicin treatment reduced cardiac NO-stimulated sGC activity by ∼20% (Fig. 1A).
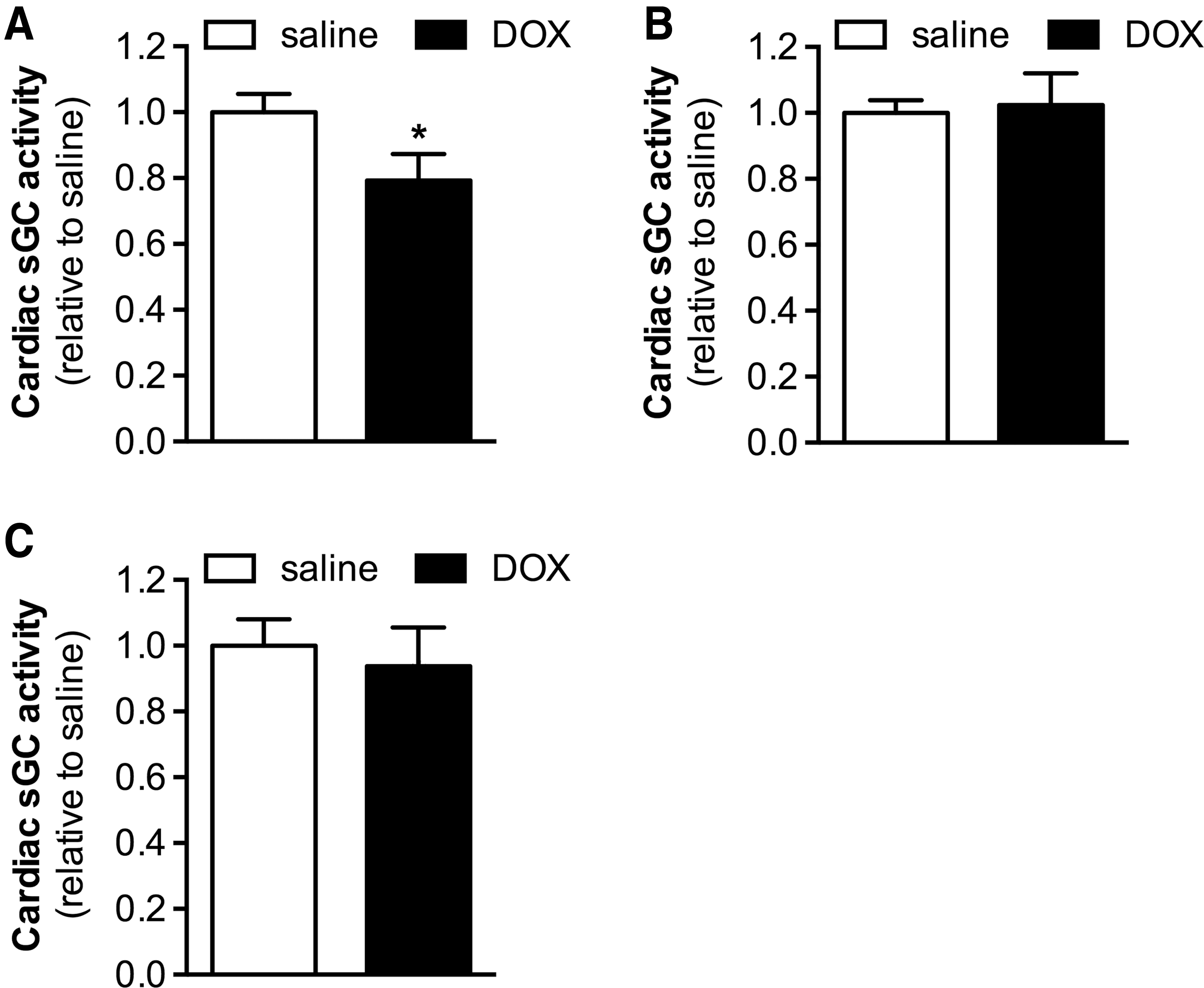
sGC is a heterodimeric enzyme comprising a β1 subunit and either an α1 or α2 subunit, with the sGCα1β1 isoform being the predominant isoform in the heart (6). Immunoblot analysis did not reveal changes in sGCα1 or sGCβ1 protein expression in hearts of doxorubicin-treated mice (Supplementary Fig. S1; Supplementary Data are available online at
It is conceivable that the oxidative stress associated with doxorubicin administration results in direct oxidative modification of sGC, potentially leading to loss of its prosthetic heme moiety (2, 4, 26, 42, 48) and decreased enzyme activity. To explore this possibility, we administered the radical scavenger tempol to WT mice before treatment with doxorubicin (20 mg/kg, IP) or saline and evaluated cardiac sGC activity in the presence of DETA-NO. Tempol administration prevented the doxorubicin-induced decrease in cardiac NO-stimulated sGC activity (Fig. 1B), suggesting that this decrease in activity is due to oxidative modification of sGC.
Our hypothesis that doxorubicin treatment results in oxidation of sGC was further corroborated by the observation that BAY 58-2667 (cinaciguat), an sGC agonist specifically targeting oxidized and heme-free sGC (42), increased sGC activity in cardiac extracts obtained from doxorubicin- and saline-treated WT mice to the same extent (Fig. 1C). Thus, whereas in hearts of doxorubicin-treated mice, activation of sGC by NO—requiring the heme group of sGC to be in a reduced state—was impaired, sGC activation by BAY 58-2667 was not.
Constitutive myocardial sGCα1 deficiency exacerbates doxorubicin-induced cardiac dysfunction
To evaluate whether the doxorubicin-induced decrease in sGC activity contributes to the development of cardiotoxicity, we studied mice with cardiomyocyte-specific reduction of sGC activity (sGCα1−/−CM). Cre-mediated deletion of exon 6 of sGCα1 in cardiomyocytes attenuated the ability of DETA-NO to activate sGC in the heart: the relative fold increase in cGMP synthesis was 0.64 ± 0.09 versus 1.00 ± 0.18 in sGCα1−/−CM and WT mice, respectively (n = 10 and 8, p < 0.05). Restriction of sGCα1 deficiency to the heart in sGCα1−/−CM mice was confirmed by polymerase chain reaction (PCR) analysis (Supplementary Fig. S2). Baseline echocardiographic parameters, including left ventricular (LV) end-systolic and end-diastolic internal diameters and fractional shortening (FS), were similar in sGCα1−/−CM mice and WT littermates (Supplementary Table S1).
Cardiotoxicity was induced in sGCα1−/−CM and WT mice (n = 14 for both) by administering doxorubicin (2 mg/kg IP, once weekly) for 12 weeks, as previously reported (51). Following 8 weeks of doxorubicin administration, echocardiography revealed that end-systolic internal diameter (LVIDES) was increased (Fig. 2A) and FS decreased (Fig. 2B) in both sGCα1−/−CM and WT mice compared with baseline measurements obtained before the first doxorubicin injection.
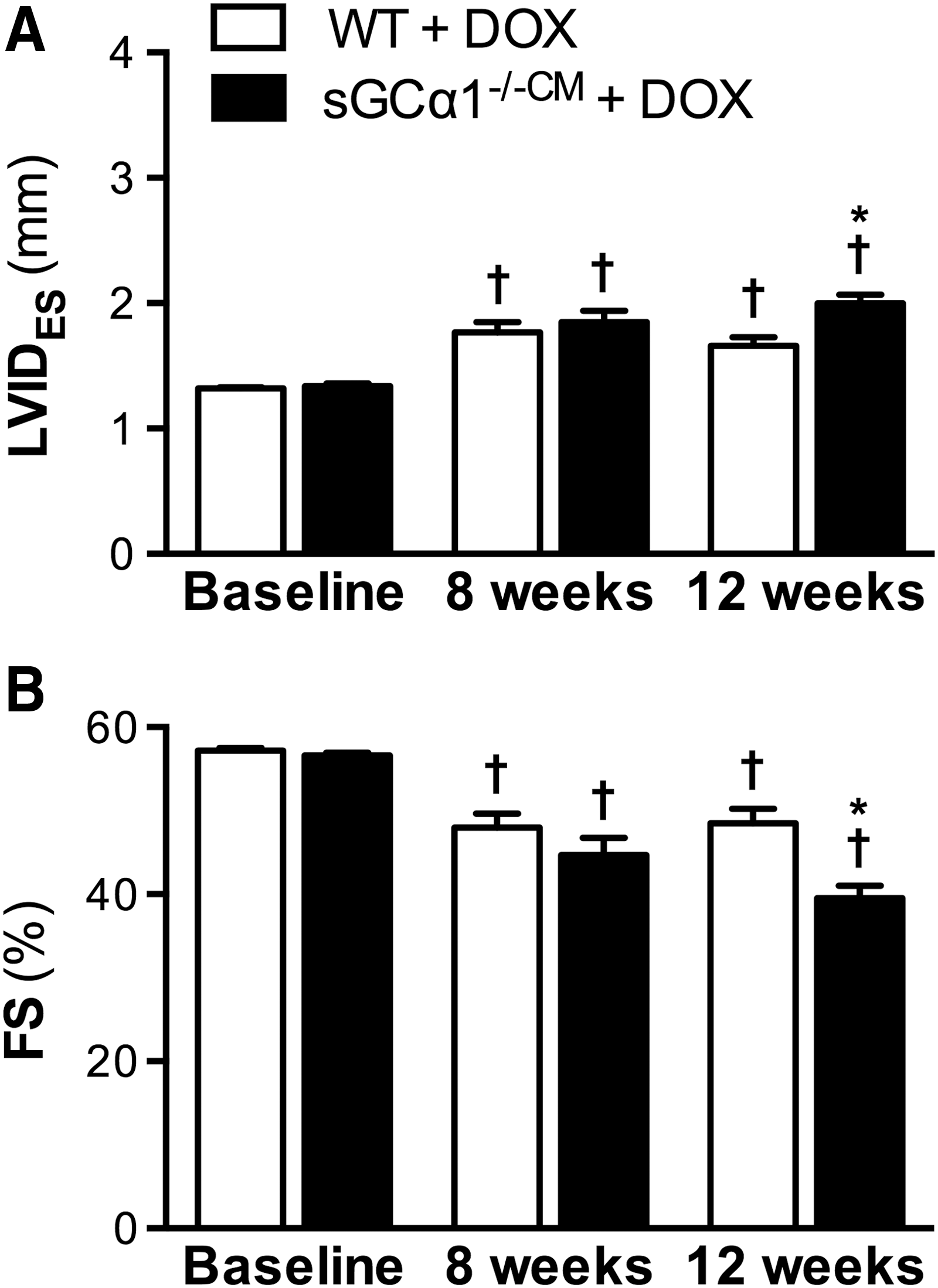
However, after 12 weeks, doxorubicin-induced systolic dysfunction was significantly greater in sGCα1−/−CM mice than in WT littermates: LVIDES was greater (Fig. 2A) and FS lower (Fig. 2B) in sGCα1−/−CM than in WT mice. In addition, compared with baseline, LV end-diastolic internal diameter (LVIDED) was modestly increased after 12 weeks of doxorubicin treatment in sGCα1−/−CM mice (3.3 ± 0.04 vs. 3.1 ± 0.03 mm, respectively, p < 0.05), but not in WT mice (3.2 ± 0.05 vs. 3.1 ± 0.03 mm, p > 0.05). Invasive LV pressure–volume measurements confirmed more pronounced systolic dysfunction, as evidenced by the reduced ejection fraction (EF), in doxorubicin-treated sGCα1−/−CM versus WT mice (Table 1).
Invasive hemodynamic measurements revealed increased systolic dysfunction in sGCα1−/−CM compared with WT mice after 12 weeks of DOX administration.
Multiplicity-adjusted p-values for comparison between WT + DOX and sGCα1−/−CM + DOX are reported. † p < 0.05 versus saline and * p < 0.05 versus WT + doxorubicin.
bpm, beats per minute; DOX, doxorubicin; dP/dtmax, maximum first derivative of developed LV pressure; dP/dtmin, minimum first derivative of developed LV pressure; Ea, arterial elastance; EDPVR, end-diastolic pressure volume relationship; EDV, end-diastolic volume; EES, end-systolic elastance; EF, ejection fraction; ESV, end-systolic volume; HR, heart rate; LV, left ventricular; MAP, mean arterial pressure; PED, end-diastolic pressure; PES, end-systolic pressure; PRSW, preload recruitable stroke work; sGC, soluble guanylate cyclase; sGCα1−/−CM, mice with cardiomyocyte-specific deletion of exon 6 of the sGCα1 allele; Tau, time constant for isovolumic relaxation; WT, wild-type.
Similar to previous reports (10, 35, 36), doxorubicin administration in mice was associated with a decrease in heart rate (HR), assessed via invasive hemodynamic measurements (Table 1) and echocardiography (sGCα1−/−CM + doxorubicin vs. sGCα1−/−CM + saline; 426 ± 18 vs. 505 ± 18 beats per minute [bpm] and WT + doxorubicin vs. WT + saline; 428 ± 17 vs. 531 ± 13 bpm, p < 0.05 for doxorubicin vs. saline in both genotypes). HR did not differ between doxorubicin-treated sGCα1−/−CM and WT mice (p > 0.05).
Furthermore, doxorubicin administration reduced survival rates to a similar extent in sGCα1−/−CM and WT mice (71% vs. 86%, respectively, n = 14 for both, p > 0.05), as well as body weights (−23 ± 3% vs. −19 ± 2%, respectively, n = 10 and 12, p > 0.05).
Doxorubicin-associated cardiotoxicity is increased in mice with inducible cardiomyocyte-specific expression of a dominant negative sGCα1 mutant
To further assess whether reduced sGC activity plays a pathogenic role in doxorubicin-induced cardiotoxicity, a second mouse model with inducible cardiomyocyte-specific sGCα1 deficiency was generated and studied. Upon withdrawal of doxycycline from the diet, a dominant negative sGCα1 mutant (DNsGCα1, Tet-Off system) is expressed that competes with sGCα1 and sGCα2 for binding to sGCβ1, thereby inhibiting the formation of the two catalytically active sGC heterodimers in the heart.
Four weeks after doxycycline removal from the diet, the ability of DETA-NO to activate sGC was attenuated in the hearts of mice with cardiomyocyte-specific expression of a dominant negative mutation of sGCα1 (DNsGCα1tg/+) mice: the relative fold increase in cGMP synthesis was 0.59 ± 0.07 versus 1.00 ± 0.11 in DNsGCα1tg/+ and WT mice, respectively (n = 8 each, p < 0.05). Baseline echocardiographic parameters were similar in DNsGCα1tg/+ mice and WT littermates (Supplementary Table S2).
After 8 weeks of doxorubicin treatment (2 mg/kg IP, once weekly), systolic dysfunction and dilatation, characterized by increased LVIDES and LVIDED (Fig. 3A, B) and decreased FS (Fig. 3C), were apparent in DNsGCα1tg/+ mice, but not WT mice. After 12 weeks of doxorubicin treatment, LV functional impairment and dilatation had further progressed in DNsGCα1tg/+ mice. Overall, these data are consistent with the greater doxorubicin-induced systolic dysfunction observed in sGCα1−/−CM mice and suggest that sGC may protect against cardiac dysfunction associated with chronic doxorubicin treatment.
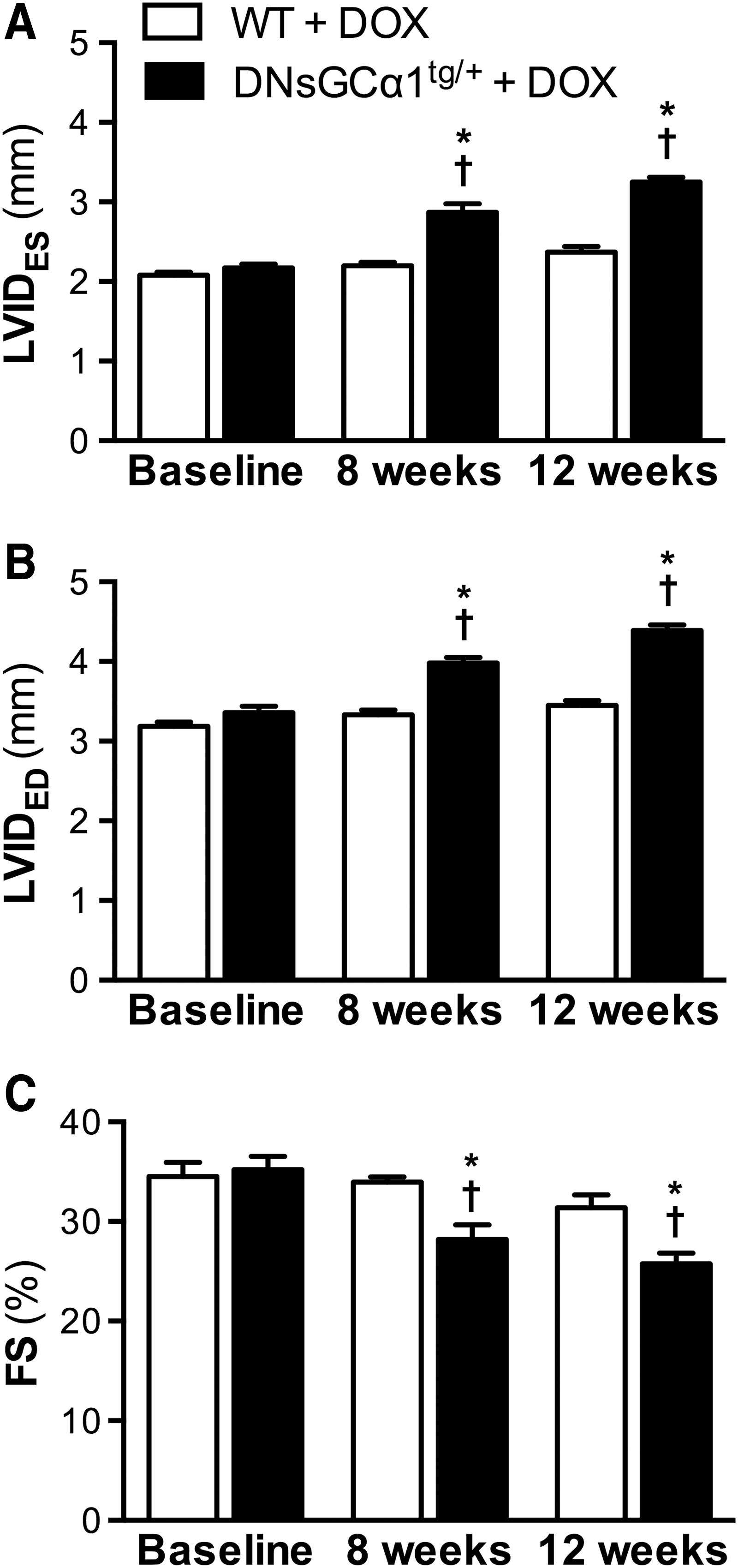
Of note, treating mice with doxorubicin for 12 weeks decreased survival rates to a similar extent in DNsGCα1tg/+ and WT mice (67% vs. 56%, respectively, n = 30 and 34, p > 0.05), as well as body weights (−12 ± 0% vs. −15 ± 1%, respectively, n = 18 and 17, p > 0.05).
Doxorubicin-induced cardiotoxicity in mice with induced expression of mutated sGCα1 is attenuated by reversal of sGCα1 mutant expression
The observation that systolic dysfunction occurs within 8 weeks of doxorubicin treatment in DNsGCα1tg/+, but not WT, mice provided a model to test the therapeutic potential of removing sGC activity inhibition by readministering doxycycline. DNsGCα1tg/+ and WT mice were exposed to doxorubicin for 12 weeks (2 mg/kg IP, once weekly), and after 8 weeks of doxorubicin treatment, doxycycline was added to the diet of both DNsGCα1tg/+ and WT mice for an additional 8 weeks, restoring cardiac sGC activity in DNsGCα1tg/+ mice (Fig. 4A).
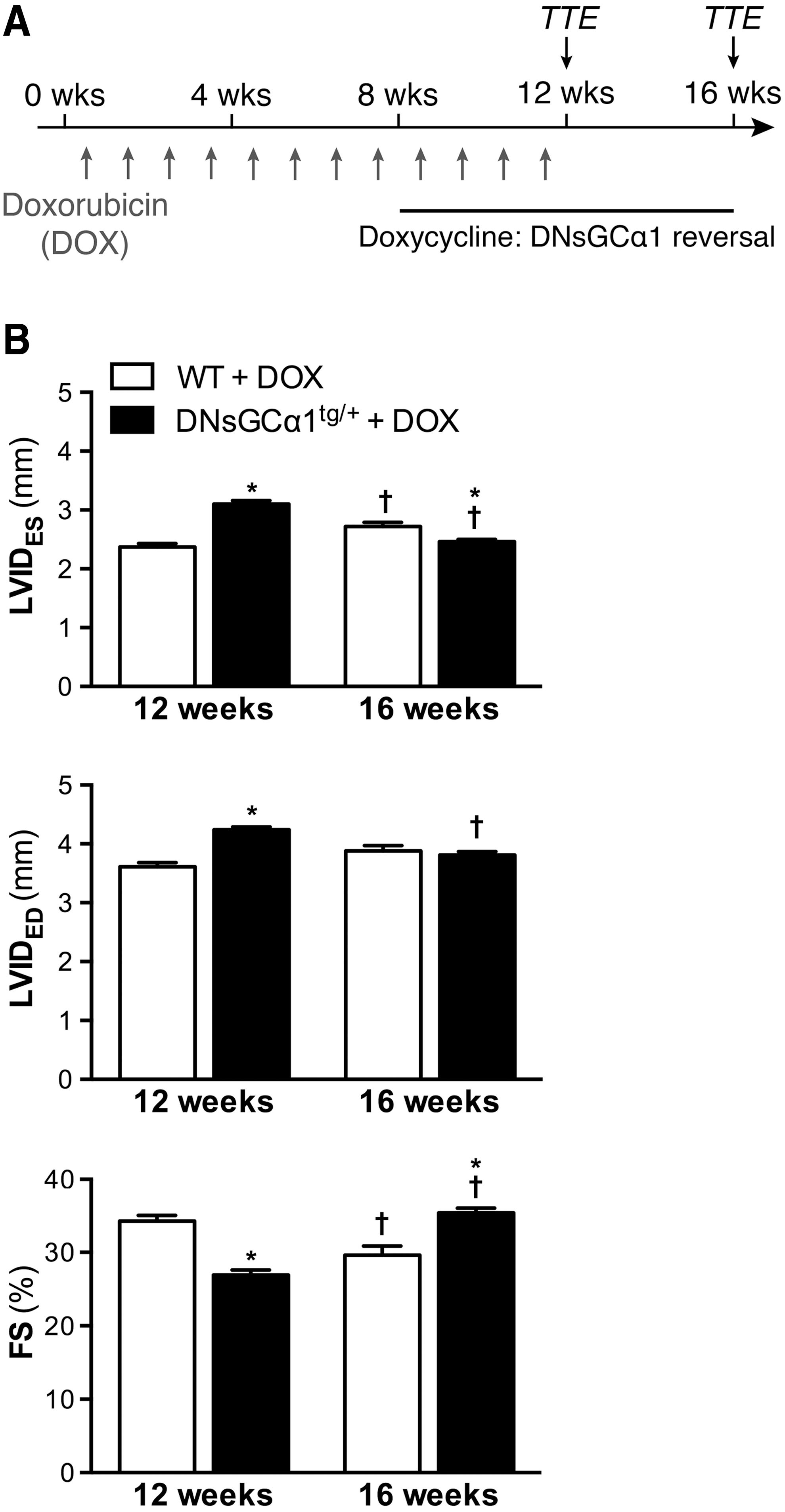
Cardiac function was assessed by echocardiography after 12 weeks of doxorubicin administration (T12 weeks) and 4 weeks later (T16 weeks). After 12 weeks of doxorubicin treatment, including 4 weeks of doxycycline administration, cardiac dysfunction and dilatation were still more pronounced in DNsGCα1tg/+ than in WT mice (Fig. 4B). However, 4 weeks later, functional deterioration and adverse remodeling had progressed further in WT mice, whereas cardiac dysfunction and dilatation were reduced in DNsGCα1tg/+ mice (Fig. 4B). These findings suggest that restoring sGC activity can attenuate doxorubicin-induced LV dysfunction in mice.
Morphological changes associated with doxorubicin administration were similar in DNsGCα1tg/+ and WT hearts
To assess potential differences in the degree of doxorubicin-induced cardiac atrophy between sGCα1-deficient mice and WT mice, we measured heart weights in both genotypes following 12 weeks of doxorubicin or saline administration. Doxorubicin treatment decreased normalized heart weight by 18% and 24% in sGCα1−/−CM and WT mice, respectively, compared with saline-treated controls (Supplementary Table S3, p > 0.05). Similarly, in doxorubicin-treated DNsGCα1tg/+ and WT mice, heart weight was decreased by 17% and 15%, respectively, compared with saline-treated mice (Supplementary Table S3, p > 0.05).
To further explore the mechanisms underlying the difference in doxorubicin-induced cardiac dysfunction between sGCα1-deficient mice and WT mice, we examined fibrosis, apoptosis, inflammation, and vascular density in hearts of DNsGCα1tg/+ and WT mice following 12 weeks of doxorubicin administration.
To investigate cardiac fibrosis, we measured collagen deposition and expression levels of two key mediators of fibrosis: transforming growth factor (TGF)-β1 and connective tissue growth factor (CTGF). CTGF is the profibrotic mediator through which TGF-β1 triggers connective tissue cell proliferation, adhesion, migration, and the synthesis of extracellular matrix. Analysis of Sirius red-stained tissue sections using circularly polarized light revealed comparable deposition of thick, tightly packed, red birefringent collagen fibers as well as thin, loosely assembled, green birefringent collagen fibers in the two genotypes (Supplementary Table S4). Consistent with these results, TGF-β1 and CTGF transcript levels were similar in doxorubicin-treated DNsGCα1tg/+ and WT mice (Supplementary Table S4).
In addition, the number of apoptotic cardiomyocytes was similar in DNsGCα1tg/+ and WT mice after 12 weeks of doxorubicin treatment. In accordance with these results, cardiac mRNA expression levels of antiapoptotic B cell lymphoma 2 (Bcl-2) and proapoptotic Bcl-2-associated X protein (Bax) did not differ between the two genotypes (Supplementary Table S4).
In addition, the number of infiltrated CD45-positive leukocytes, a measure of cardiac inflammation, was similar in hearts of DNsGCα1tg/+ and WT mice after 12 weeks of doxorubicin administration (relative to the number of cardiomyocyte nuclei; Supplementary Table S4).
Furthermore, vascular density was comparable in DNsGCα1tg/+ and WT hearts after 12 weeks doxorubicin treatment (Supplementary Table S4), suggesting that a lower density of capillaries, supplying cardiomyocytes with oxygen and nutrients, did not contribute to the greater cardiac dysfunction in doxorubicin-treated DNsGCα1tg/+ mice.
Doxorubicin-induced ROS generation is increased in sGCα1-deficient mice
To consider the possibility that doxorubicin induced higher levels of oxidative stress in sGCα1-deficient than in WT mice, we investigated the degree of doxorubicin-induced ROS generation in cardiomyocytes isolated from sGCα1-deficient and WT mice. sGCα1−/− and WT cardiomyocytes were exposed to doxorubicin (50 μM) for 3 h and then incubated with chloromethyl derivate of 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA). This cell-permeable compound is deacetylated by intracellular esterases and then oxidized by hydrogen peroxide (5) and other oxidants (20) to produce fluorescent 2′,7′-dichlorofluorescein (DCF). Three hours after exposure to doxorubicin, greater DCF fluorescence, and thus ROS production, was observed in sGCα1−/− than in WT cardiomyocytes (Fig. 5A).
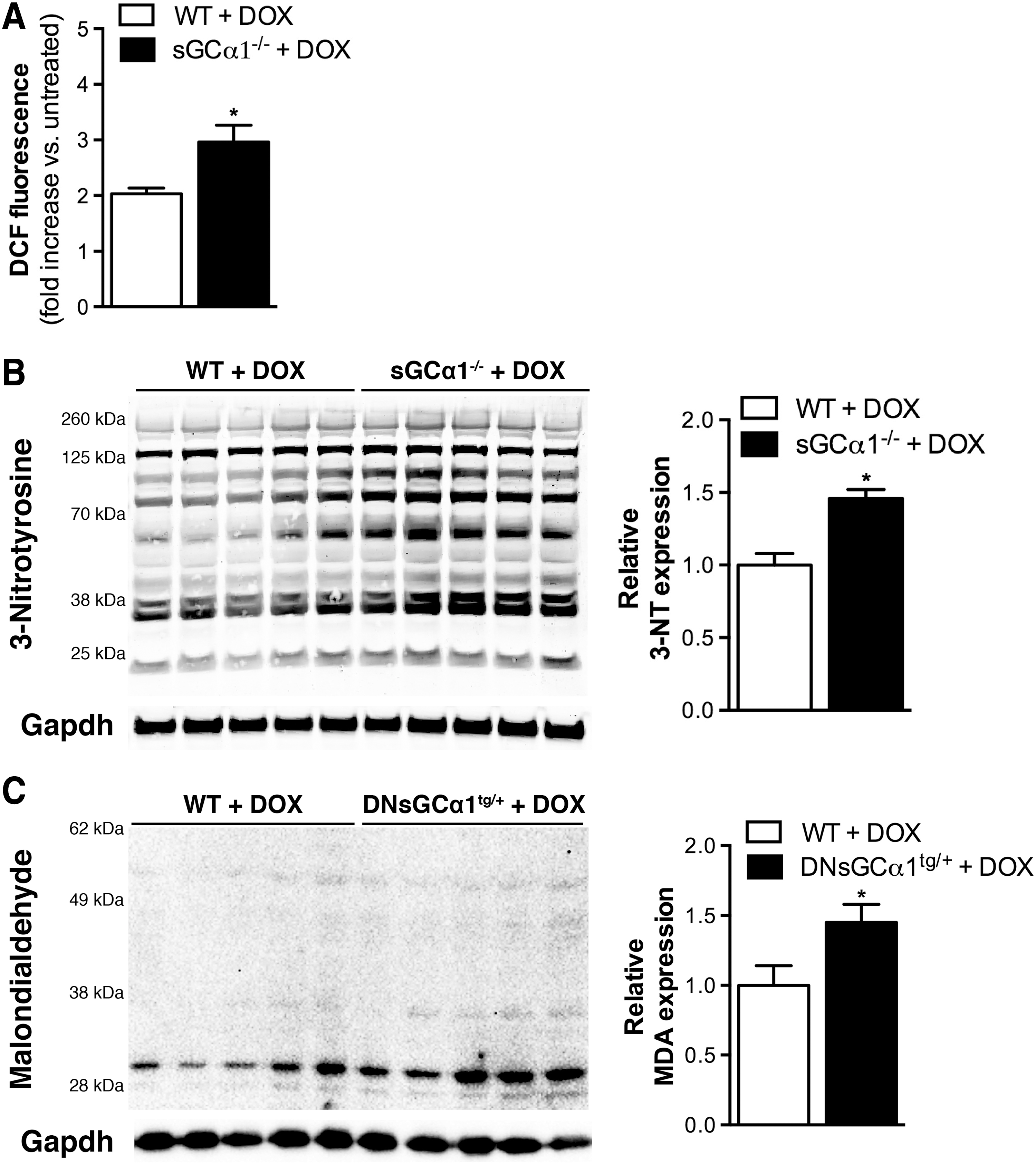
Similarly, reactive nitrogen species-mediated tyrosine nitration was more pronounced in sGCα1−/− than WT hearts 24 h after doxorubicin administration (Fig. 5B). In addition, higher expression of malondialdehyde (MDA) was observed in DNsGCα1tg/+ than WT hearts after 12 weeks of doxorubicin treatment (Fig. 5C). MDA is a product of lipid peroxidation, frequently initiated by hydroxyl and hydroperoxyl radicals (3).
Taken together, these results suggest that doxorubicin-induced oxidative stress is more pronounced when cardiac sGC activity is reduced. This in turn could lead to enhanced sGC oxidation and decrease in activity and subsequent exacerbation of ROS levels via a yet to be identified negative feedback system.
Discussion
The long-term adverse cardiac effects of doxorubicin, which limit the use of this effective chemotherapeutic agent, emphasize the need for novel therapeutic approaches to prevent and treat cardiotoxicity associated with doxorubicin treatment. Our data identify sGC as a potential therapeutic target for doxorubicin-induced cardiotoxicity.
Twenty-four hours after doxorubicin administration, cardiac sGC activity was reduced in mice. Since cardiac sGC expression was not altered by doxorubicin treatment, the observed decrease in activity likely results from post-translational modification of sGC. Oxidative modification of sGC, leading to loss of its prosthetic heme group and generation of NO-insensitive sGC, was previously reported in cardiovascular diseases associated with increased oxidative stress (2, 4, 26, 42, 48). In vivo administration of the radical scavenger, tempol, or targeting oxidized and heme-free sGC by adding the sGC activator, BAY 58-2667, ex vivo rescued the doxorubicin-associated decrease in cardiac sGC activity, suggesting that doxorubicin-induced oxidative modification of sGC represents an underlying mechanism for the reduction in activity.
To determine whether reduced sGC activity contributes to the development of doxorubicin-induced cardiotoxicity, doxorubicin was administered to mice with a cardiomyocyte-specific deficiency in sGCα1. After 12 weeks of doxorubicin treatment, LV systolic dysfunction was more pronounced in sGCα1−/−CM than in WT mice. Similarly, mice with induced expression of a dominant negative mutated sGCα1 (DNsGCα1tg/+) displayed greater LV systolic dysfunction and remodeling than WT mice after 12 weeks of doxorubicin treatment. Together, these data reveal a protective role for sGC against doxorubicin-induced cardiotoxicity.
Furthermore, we wanted to explore whether restoring sGC activity could attenuate established cardiac dysfunction. Since DNsGCα1tg/+ mice displayed LV dysfunction and remodeling after 8 weeks of doxorubicin administration, whereas WT mice did not, the impact of restoring cardiac sGC activity on doxorubicin-induced cardiotoxicity could be evaluated by initiating reversal of sGCα1 mutant expression in DNsGCα1tg/+ mice at this time point. Halting expression of mutated sGCα1 in DNsGCα1tg/+ mice after 8 weeks of doxorubicin administration, resulting in restored sGC activity, eventually reduced LV dysfunction. Four weeks after halting doxorubicin treatment, cardiac function was even less impaired in DNsGCα1tg/+ mice than in WT animals.
It is possible that mechanisms to compensate for reduced cardiac sGC activity are triggered in the DNsGCα1tg/+ mouse model. When expression of mutated sGCα1 is halted in these DNsGCα1tg/+ mice, the resulting increase in sGC activity in combination with potential compensatory mechanisms could lead to enhanced cGMP signaling in comparison with WT mice and may contribute to improvement of cardiac function beyond the level observed in doxorubicin-treated WT mice.
Despite the observation that doxorubicin-induced cardiac dysfunction was greater in sGCα1−/−CM and DNsGCα1tg/+ mice than in WT littermates, heart weights were similar in doxorubicin-treated sGCα1-deficient and WT mice. In addition, cardiac Bcl-2 and Bax mRNA expression and cardiomyocyte apoptosis did not differ between DNsGCα1tg/+ and WT mice following 12 weeks of doxorubicin treatment. These results indicate that the increased cardiac dysfunction in doxorubicin-treated sGCα1-deficient mice did not result from a greater loss of cardiomyocytes. In addition, cardiac fibrosis, inflammation, and vascular density did not differ between sGCα1 mutant and WT mice following 12 weeks of doxorubicin administration.
We cannot exclude that the degree of cardiac fibrosis, apoptosis, inflammation, or vascular density might differ at earlier or later time points. In addition, the difference in cardiac function between doxorubicin-treated sGCα1-deficient and WT mice did not reflect an effect of sGCα1 deficiency on blood pressure, which was similar in doxorubicin-treated sGCα1−/−CM and WT mice.
Oxidative stress has been suggested to play a prominent role in doxorubicin-associated cardiotoxicity. In mice overexpressing endothelial NO synthase (NOS3), doxorubicin-induced cardiotoxicity was greater than in WT mice due to abundant ROS production resulting from NOS3-mediated reduction of doxorubicin (34). We also observed increased doxorubicin-induced ROS production in sGCα1-deficient cardiomyocytes and greater oxidative stress levels in sGCα1-deficient hearts after in vivo exposure to doxorubicin.
It is possible that the more pronounced doxorubicin-induced oxidative stress in sGCα1-deficient cardiomyocytes leads to greater contractile impairment. Oxidative stress can alter expression, phosphorylation, or function of calcium regulatory proteins such as sarcoplasmic reticulum calcium ATPase 2A and ryanodine receptor 2 (1, 25, 54). Oxidative stress also induces structural modifications of sarcomeric proteins and disrupts mitochondrial function in cardiomyocytes (44). Additional studies are required to evaluate whether increased oxidative stress levels in sGCα1-deficient cardiomyocytes result in greater contractile impairment and, if so, which mechanisms are involved.
Further support for a protective role of NO-sGC-cGMP signaling in doxorubicin-induced cardiotoxicity was previously provided by the observation that supplementing dietary nitrate reduced doxorubicin-induced cardiotoxicity (57). This cardioprotection was associated with a decrease in ROS generation, lipid peroxidation, and mitochondrial respiratory chain damage (57).
In addition, pharmacological inhibition of the cGMP-catabolizing enzyme, phosphodiesterase type 5 (PDE5), with sildenafil or tadalafil attenuated doxorubicin-associated cardiotoxicity in mice (17, 24). These PDE5 inhibitors reduced doxorubicin-induced oxidative stress, disruption of the mitochondrial membrane potential, apoptosis, and depletion of prosurvival proteins in the heart (17, 24). Because the ability of PDE5 inhibitors to augment cGMP levels is inherently limited by cGMP synthesis, strategies designed to enhance cGMP synthesis may be more effective in preventing doxorubicin-induced cardiotoxicity. Nitroglycerin and related NO donors are currently used to treat various cardiovascular diseases. However, nitrates produce off-target (cGMP-independent) effects and induce tolerance.
As many beneficial effects of NO are mediated through sGC, pharmacological agents directly targeting sGC could represent a novel mechanism-based approach to limit cardiotoxicity in patients undergoing doxorubicin-based chemotherapy. The therapeutic potential of targeting sGC to prevent or even reverse doxorubicin-induced cardiotoxicity is supported by the observation that doxorubicin-induced LV dysfunction in DNsGCα1tg/+ mice is attenuated by removing inhibition of sGC activity. Future studies exploring the therapeutic potential of sGC agonists for the treatment of cardiotoxicity associated with doxorubicin treatment are warranted.
In conclusion, doxorubicin administration reduces cardiac sGC activity in mice, potentially via oxidative modification of the enzyme. Our studies in two genetically modified mouse models of cardiac sGCα1 deficiency demonstrate that reduced cardiac sGC activity contributes to the development of doxorubicin-induced cardiotoxicity, identifying sGC as a plausible therapeutic target to treat this devastating side effect of anthracycline-based chemotherapy. Carefully designed preclinical studies with pharmacological sGC agonists are required to test whether these drugs may provide an additional tool to cardiologists and oncologists for preventing chronic cardiomyopathy and heart failure in doxorubicin-treated patients.
Materials and Methods
Experimental animals
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Housing and all procedures involving experimental animals were approved by the Institutional Animal Care and Use Committees of Massachusetts General Hospital (Subcommittee on Research Animal Care).
Cardiomyocyte-specific sGCα1−/−CM mice were generated by crossing mice in which exon 6 of the sGCα1 allele is flanked by two LoxP sites [sGCα1fl/fl, generated by our group (7)] with mice expressing Cre recombinase under the control of an α-myosin heavy chain promoter (αMHC-Cre+/−). In cardiomyocytes of sGCα1fl/flαMHC-Cre+/− offspring (referred to as sGCα1−/−CM mice), Cre recombinase induces deletion of exon 6, encoding a portion of the catalytic domain of sGCα1 required for enzyme activity. These mice were maintained on a C57BL/6J background (The Jackson Laboratory, Farmington, CT) and sGCα1fl/flαMHC-Cre−/− littermates served as WT controls.
To establish the DNsGCα1tg/+ mouse model, a first mouse line was generated in which expression of a mutated sGCα1-encoding gene is driven by a doxycycline-responsive promoter element (tetracycline operator [TetO]). The dominant negative point mutation introduced into the sGCα1-encoding gene results in an amino acid change in the catalytic region of sGCα1 (D529A, Asp to Ala) (55). Following implantation of microinjected FVB zygotes into a pseudopregnant Swiss foster mother, offspring were genotyped, and transgenic founders were backcrossed for >6 generations onto a C57BL/6N background (Taconic, Hudson, NY).
This mouse line was crossed with a second mouse line expressing the gene encoding a tetracycline transactivator (tTA) protein under the control of an αMHC promoter, kindly provided by Dr. D.A. Dichek (University of Washington Medical Center, Seattle, WA), and maintained on a C57BL/6N background (Taconic, Hudson, NY).
When doxycycline is removed from the diet (doxycycline-containing chow, 200 mg/kg; Harlan Laboratories, Huntingdon, United Kingdom) of dual heterozygous TetO-DNsGCα1+/−αMHC-tTA+/− offspring (referred to as DNsGCα1tg/+ mice), doxycycline-mediated repression of transcriptional activation is abolished (Tet-Off system), and tTA binds to and activates transcription from the TetO promoter element, inducing expression of the sGCα1 mutant specifically in cardiomyocytes. DNsGCα1tg/+ mice and their WT controls (TetO-DNsGCα1−/−αMHC-tTA−/− littermates) were conceived and raised in the presence of doxycycline. When the mice were 4–6 weeks old, doxycycline was withdrawn, and 4 weeks later, mice were used for experiments.
Doxorubicin treatment regimen
For acute studies, 8-week-old C57BL/6J mice (The Jackson Laboratory) were injected with a single dose of doxorubicin (20 mg/kg IP, using a 2 mg/ml stock of doxorubicin hydrochloride obtained from Pfizer, New York, NY) or saline and sacrificed 24 h later. A subset of these mice were provided with the radical scavenger, tempol (1-oxyl-2,2,6,6-tetramethyl-4-hydroxypiperidine; Thermo Fisher Scientific, Waltham, MA), via drinking water (1 mM) 4 days before doxorubicin or saline treatment and via gavage (30 mg/kg) 1 h before treatment.
For chronic studies, 8–12-week-old male and female DNsGCα1tg/+, sGCα1−/−CM, and WT mice were administered a low dose of doxorubicin for 12 weeks (2 mg/kg IP, once weekly).
Transthoracic echocardiography
To measure LV systolic function and dimensions, sGCα1−/−CM mice were lightly sedated (ketamine, 100 mg/kg, IP) and echocardiography was performed using a 13 MHz ultrasound probe (Vivid 7; GE Healthcare, Wilmington, MA). DNsGCα1tg/+ mice were sedated with 1.5% isoflurane and echocardiograms were obtained using an MS400 transducer on a Vevo 2100 scanner (VisualSonics, Inc., Toronto, Canada). LVIDED and LVIDES, interventricular septal thickness at end-diastole, LV posterior wall thickness at end-diastole, and HR were measured and FS calculated, as previously described (39).
Invasive hemodynamic measurements
Mice were anesthetized by IP injection with ketamine (120 mg/kg), fentanyl (90 μg/kg), and rocuronium (10 mg/kg), intubated, and mechanically ventilated (FiO2 = 1, 10 μl/g, 120 breaths per minute). A fluid-filled catheter was introduced into the carotid artery to record arterial blood pressure and HR. After thoracotomy, a pressure–volume conductance catheter (PVR-1030; Millar Instruments, Houston, TX) was introduced through the apex into the LV, as described previously (19).
LV end-systolic and end-diastolic volumes and LV pressures were measured, and EF, arterial elastance, the maximum and minimum first derivative of developed LV pressure (dP/dtmax and dP/dtmin), and the time constant for isovolumic relaxation (Tau) were calculated. The preload recruitable stroke work, end-systolic elastance, and end-diastolic pressure–volume relationship were obtained by transiently occluding the inferior vena cava. All parameters were measured using LabChart (ADInstruments, Colorado Springs, CO).
sGC activity measurements
sGC enzyme activity was measured as described previously (7). Cardiac tissues were homogenized and supernatants (containing 50 μg of protein) were incubated for 10 min at 37°C in a reaction mixture with DETA-NO (1 mmol/l) or BAY 58-2667 (100 μmol/l). cGMP in the reaction mixture was measured using a commercial radioimmunoassay (Cayman Chemical, Ann Arbor, MI). sGC enzyme activity was expressed as picomoles of cGMP produced per minute per milligram of protein in cardiac extract supernatant, normalized to the contemporaneous control group (saline-treated mice when sGC activity was measured in doxorubicin-treated mice and WT mice when sGC activity was measured in sGCα1-deficient mice).
Histological and immunoblot analysis
Immunohistochemistry was performed as described previously (52). Images were obtained using an Axiovert 200M imaging microscope (Zeiss, Oberkochen, Germany). To assess the degree of cardiac fibrosis, the area of collagen deposition was traced on Sirius red-stained tissue sections using circularly polarized light, allowing evaluation of tightly packed, red birefringent collagen and thin, loosely assembled, green birefringent collagen. The degree of fibrosis was expressed as the area of red or green birefringent collagen relative to the area of the examined LV tissue area.
Apoptotic cardiomyocytes were detected by labeling DNA strand breaks using TUNEL (ApopTag; Millipore, Billerica, MA), according to the manufacturer's instructions. The index of apoptosis was determined by dividing the number of immunolabeled cardiomyocyte nuclei by the area of the examined LV tissue area.
Vascular density was analyzed on BS1-lectin-stained tissue sections (Sigma-Aldrich, St.-Louis, MO) by dividing the number of positively stained small blood vessels by the number of cardiomyocyte nuclei.
Mouse cardiac tissue was homogenized in RIPA buffer containing Halt protease and phosphatase inhibitors (Thermo Fisher Scientific). Homogenates were transferred to Eppendorf tubes, incubated on ice for 20 min, centrifuged at 14,000 g for 10 min, and supernatants collected. Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to Immobilon-FL polyvinylidene difluoride membranes (Millipore) by semidry electroblotting, blocked for 1 h in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE), and probed overnight with antibodies specific for sGCα1 (Abcam, Cambridge, MA), sGCβ1 (Sigma-Aldrich), 3-nitrotyrosine (3-NT; Millipore), or glyceraldehyde-3-phosphate dehydrogenase (Cell Signaling Technology, Danvers, MA). Bound fluorophore-coupled secondary antibodies were visualized using the Odyssey system (LI-COR Biosciences).
For MDA immunoblotting, proteins were transferred to a nitrocellulose membrane, which was blocked for 1 h in 5% nonfat milk. Bound primary antibody (Millipore) was detected using a horseradish peroxidase-conjugated secondary antibody (Dako, Heverlee, Belgium) and visualized using ECL Plus Western blotting detection reagents (GE Healthcare, Buckinghamshire, United Kingdom).
For all immunoblots, densitometric analysis was performed using ImageJ software (NIH, whole lane densitometry for 3-NT and MDA immunoblots).
Quantitative real-time polymerase chain reaction
Cardiac expression of genes involved in fibrosis and apoptosis was measured using quantitative real-time polymerase chain reaction (TaqMan or SYBR green PCR master mix; Life Technologies, Carlsbad, CA). Relative mRNA levels were analyzed using the Livak method (29).
Measurements of DCF fluorescence
Cardiomyocytes were isolated from global sGCα1−/− mice and WT littermates by Langendorff perfusion, as previously described (27). After plating cardiomyocytes onto laminin-coated plates, the cells were incubated with 50 μmol/l doxorubicin (Sigma-Aldrich) for 3 h, washed with phosphate-buffered saline, and lysed by adding 100 μl T-PER (Thermo Fisher Scientific). Next, 140 μl of 50 μM CM-H2DCFDA (Life Technologies) was added to 10 μl cell lysate (96-well plate). Following incubation at 25°C for 10 min in the dark, DCF fluorescence was measured using a spectrofluorometer (BMG Labtech, Ortenberg, Germany), as previously described (16). Fluorescence intensity was normalized to protein concentration and expressed relative to levels in untreated cardiomyocytes (i.e., without doxorubicin).
Statistical analysis
Values are presented as mean ± standard error of the mean. When comparing two groups, normal distribution of the data was assessed using the Shapiro–Wilk test, and an unpaired two-tailed t-test (parametric) or a Mann–Whitney test (nonparametric) was used. Serial echocardiographic data were analyzed by two-way analysis of variance (ANOVA) and Sidak's multiple comparisons test. For these experiments, the required samples sizes were calculated to yield statistical power of ≥0.80 (α = 0.05). Analysis of pressure–volume measurements was performed via one-way ANOVA and Sidak's multiple comparisons test (when passed the Shapiro–Wilk normality test) or the Kruskal–Wallis test and Dunn's multiple comparisons test (when failed the Shapiro–Wilk normality test). Survival was analyzed via the Kaplan–Meier method.
Footnotes
Acknowledgments
The authors would like to thank Dr. Cornelius Busch and Dr. Pieter Vermeersch for their help in generating the DNsGCα1tg/+ mice and Dr. David Dichek for providing the αMHC-tTA mice. This work was supported by a Postdoctoral Fellowship from the Belgian American Education Foundation (to S.V.), Scientist Development Grant 10SDG2610313 from the American Heart Association (to E.S.B.), Research Grant from KU Leuven (PF10/014, to S.P.J.), NIH grant R01EY022746 (to E.S.B.), grants from the FWO-Vlaanderen and the UGent-GOA programs (to P.B.), NIH Grant R01DK082971 (to K.D.B., D.B.B.), and the Leducq Foundation (to K.D.B., D.B.B.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.