
Abstract
Thrombus Formation
T
Overview of Thiol Isomerases
There are 20 known members of the thiol isomerase family (Fig. 1), PDI being the best known since the pioneering work of Anfinsen and colleagues (19, 34). PDI family proteins share at least one thioredoxin-like fold, a molecular scaffold consisting of a four-stranded antiparallel β sheet surrounded by three α helices (82). Eleven of these proteins contain at least one and as many as four active site Cysteine-X-X-Cysteine (CXXC) motifs that can alternate between the disulfide and dithiol states to transfer electrons in redox processes. Eight of these thioredoxin domain-containing proteins are redox inactive, either lacking any functional CXXC motif (ERp27, ERp29,
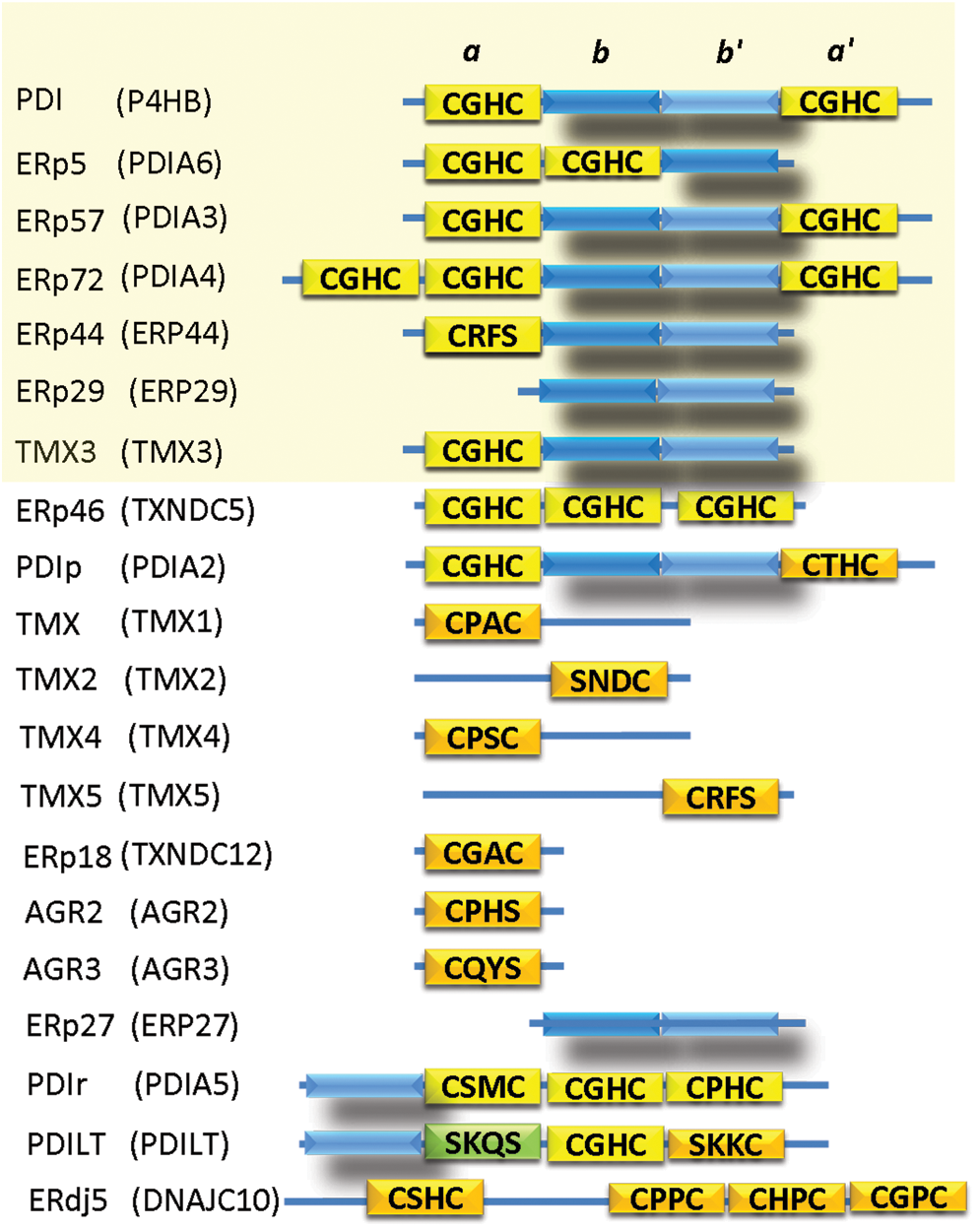
Protein Disulfide Isomerase
PDI is the best characterized and most abundant thioredoxin-like protein in the ER. A polypeptide with a molecular weight of 57,000 encoded by the
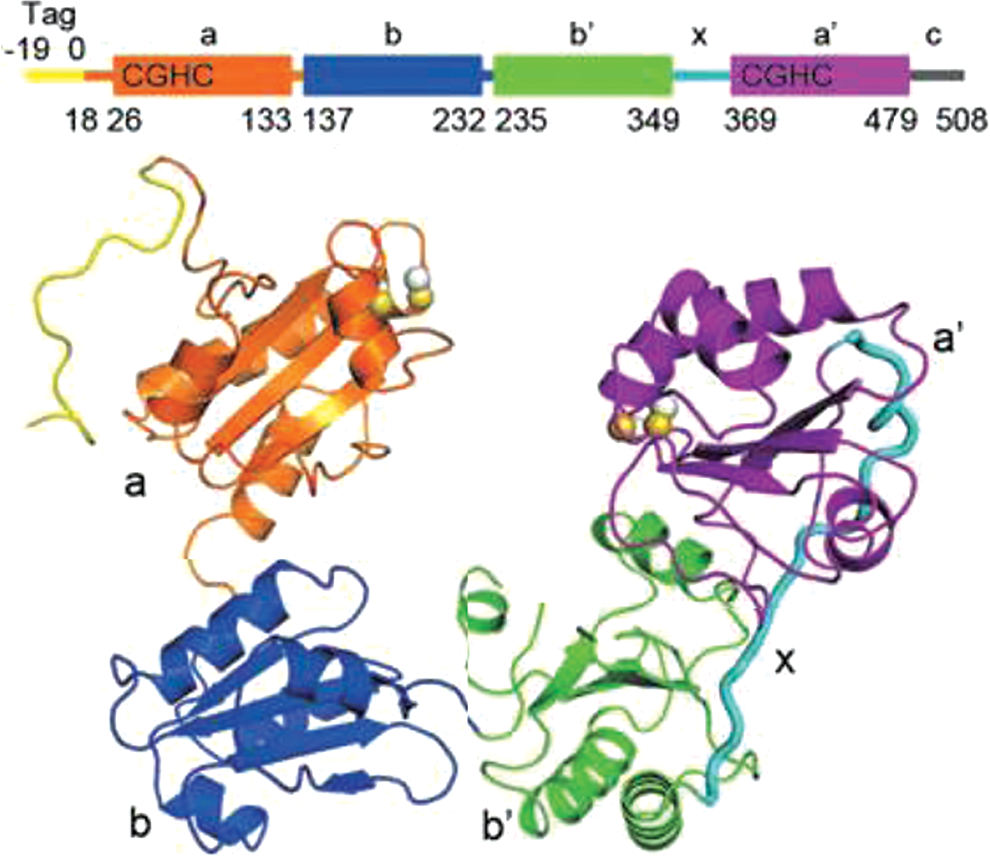
PDI was originally identified as the microsomal disulfide interchange activity capable of accelerating the oxidative folding of ribonuclease A, a protein that contains four disulfide bonds (34, 108). A number of specific oxidative folding substrates have been described for PDI in the ER, including procollagen (27), thyroglobulin (20), and γ-gliadin (9). Beyond its role in disulfide oxidation, reduction, and isomerization reactions, PDI also acts as a noncatalytic chaperone in complex with a variety of enzymes to facilitate, for example, the hydroxylation of proline (78) and membrane transport of triglycerides (117). The entrance of PDI into the ER is specified by a cleavable N-terminal signal sequence containing the C-terminal KDEL ER retrieval motif (107). The a and a′ domain CXXC motifs of PDI appear to be predominantly reduced
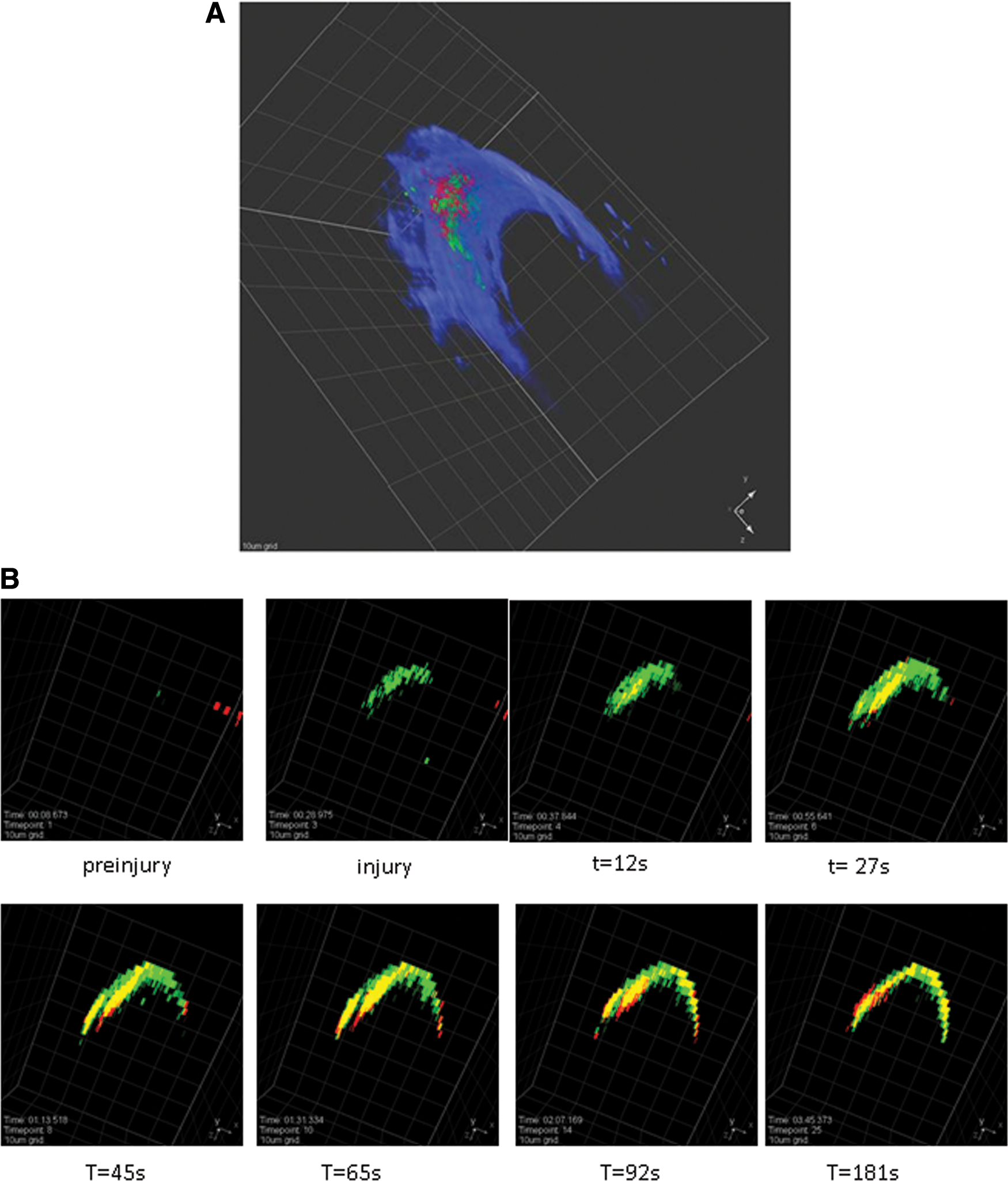
PDI is abundant in the ER. However, it is also located in small secretory granules, including the GRO-α granule of the endothelial cell and the T-granule of the platelet (45, 105). Following laser injury in a live mouse, PDI appears rapidly first on the surface of the endothelium and subsequently on the surface of bound platelets (16, 45). The binding of secreted PDI to the developing thrombus involves the interaction of PDI with the β3 integrins, αvβ3 on the endothelium, and αIIbβ3 on bound platelets (17). Tissue factor and PDI are colocalized on the endothelium surface following cell activation by the laser injury (Fig. 3A). The expression of PDI on the lumenal wall of an arteriole immediately follows laser injury and is rapidly followed by the expression of tissue factor (Fig. 3B). The endothelial surface is also shown. These events occur following calcium mobilization (45). Similarly, PDI has been known for some time to be secreted rapidly from platelets
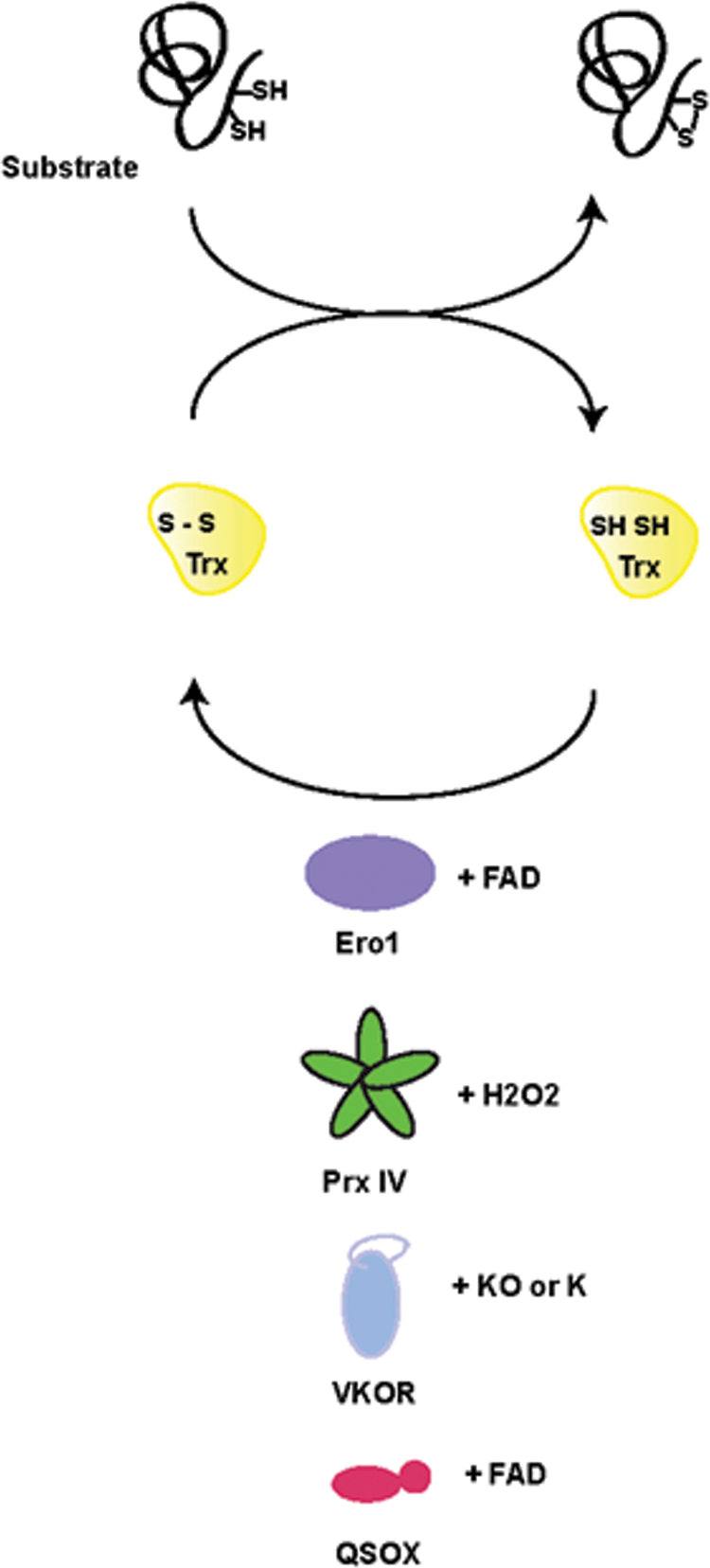
Oxidoreductase activity of PDI
The CGHC motif active sites of PDI enable it to alternate between the reduced dithiol and disulfide-bonded states and, thus, participate in biochemical oxidation and reduction reactions. While PDI does directly oxidize disulfide bridge formation in a number of proteins, it is itself unable to generate a disulfide
A general molecular logic unites these pathways (Fig. 4). A sulfhydryl oxidase couples the oxidation of a CXXC motif in PDI to the reduction of a small molecule cofactor; this cofactor may be a flavin adenine dinucleotide (FAD) molecule for the Ero1 and QSOX proteins or a quinone such as vitamin K in the case of the VKORs. The reaction proceeds through a mixed disulfide intermediate between the sulfhydryl oxidase and a catalytic thioredoxin domain of the PDI family protein that can be trapped
Chaperone activity of PDI
Chaperones share the ability to recognize and bind nonnative proteins under biophysical conditions that can promote proper folding, as occurs in the ER (8). PDI has molecular chaperone activity (115). The complete deletion of PDI in yeast is lethal, yet cells with mutant PDI that possess only inactivated catalytic sites (AXXA) are viable (62). Consistent with these observations, no viable strains of PDI knockout mice have been described (37). These findings suggest that the essential function of PDI in cell growth and survival may lie not in its catalytic activity but rather in its role as a chaperone protein. The structural determinants of PDI chaperone activity remain unclear. A mutant PDI with the 51 C-terminal amino acid residues deleted (abb′xa′) lost its chaperone activity but retained its isomerase and oxidoreductase activities (18). While this result seems to indicate that the C-terminus of PDI may be necessary for its chaperone activity, the abb′xa′ mutant may also result in a loss of substrate binding by the b′ domain due to destabilization of the a′ domain (37). If true, this would, in turn, suggest that the main effect being observed is not the importance for chaperone activity of the c-extension but rather the critical substrate binding
We have recently demonstrated that a specific inhibitor of PDI reductase activity identified from a high-throughput screen, quercetin-3-rutinoside, binds directly to the PDI b′ domain (65). Quercetin-3-rutinoside binds to the b′x domain and to the bb′ domains and inhibits PDI reductase activity. Reversal of quercetin-3-rutinoside inhibition of thrombus formation in a mouse model is observed with infusion of the PDI b′x domain (65).
A role for the chaperone function of PDI in thrombosis has been suggested. Versteeg used S-methyl methanethiosulfonate (MMTS) to inactivate the catalytic sites of PDI (109). PDI modified covalently with MMTS showed a significant reduction in oxidoreductase activity, but its chaperone activity was unaffected. However, both unmodified and MMTS-treated PDI significantly increased the rate of tissue factor-dependent factor Xa generation in a chromogenic assay (99, 109). Furthermore, the wasp venom-derived peptide mastoparan, which binds in the b′ hydrophobic pocket of PDI necessary for chaperone activity, inhibited the ability of bovine PDI to enhance factor Xa generation. PDI treated with N-ethylmaleimide enhanced factor Xa generation in a manner similar to unmodified PDI (86).
PDI and thrombus formation
Of uncertain physiologic relevance at the time, PDI was identified on the surface of resting platelets (22) and secreted from activated platelets (13, 14). The supernatant of activated platelets contained a bacitracin-sensitive activity capable of refolding disulfide-reduced RNAse to restore its function (14). Membrane impermeable thiol modifiers, anti-PDI Fab fragments, and the nonselective thiol isomerase inhibitor bacitracin impaired platelet aggregation
Thrombospondin-1 was the first cell surface substrate of PDI to be identified (41), with the resulting disulfide arrangement of TSP-1 exposing a cryptic RGD motif to facilitate enhanced integrin receptor binding (42, 100). PDI was also demonstrated to catalyze the formation of thrombin–antithrombin–vitronectin and thrombin–antithrombin–thrombospondin-1 complexes
The relevance of these observations was realized by
The role of PDI in thrombus formation was also studied in a thrombosis model in which vessel injury was initiated with ferric chloride (Fig. 5). PDI antigen, not detected in the intact arteriole of the mesentery, accumulated at the site of injury. PDI antigen expression in the injured arteriole peaked at approximately 5 min and then slowly decreased. Thus, PDI antigen is expressed extracellularly during FeCl3-initated thrombus formation as well as following laser-induced thrombus formation.
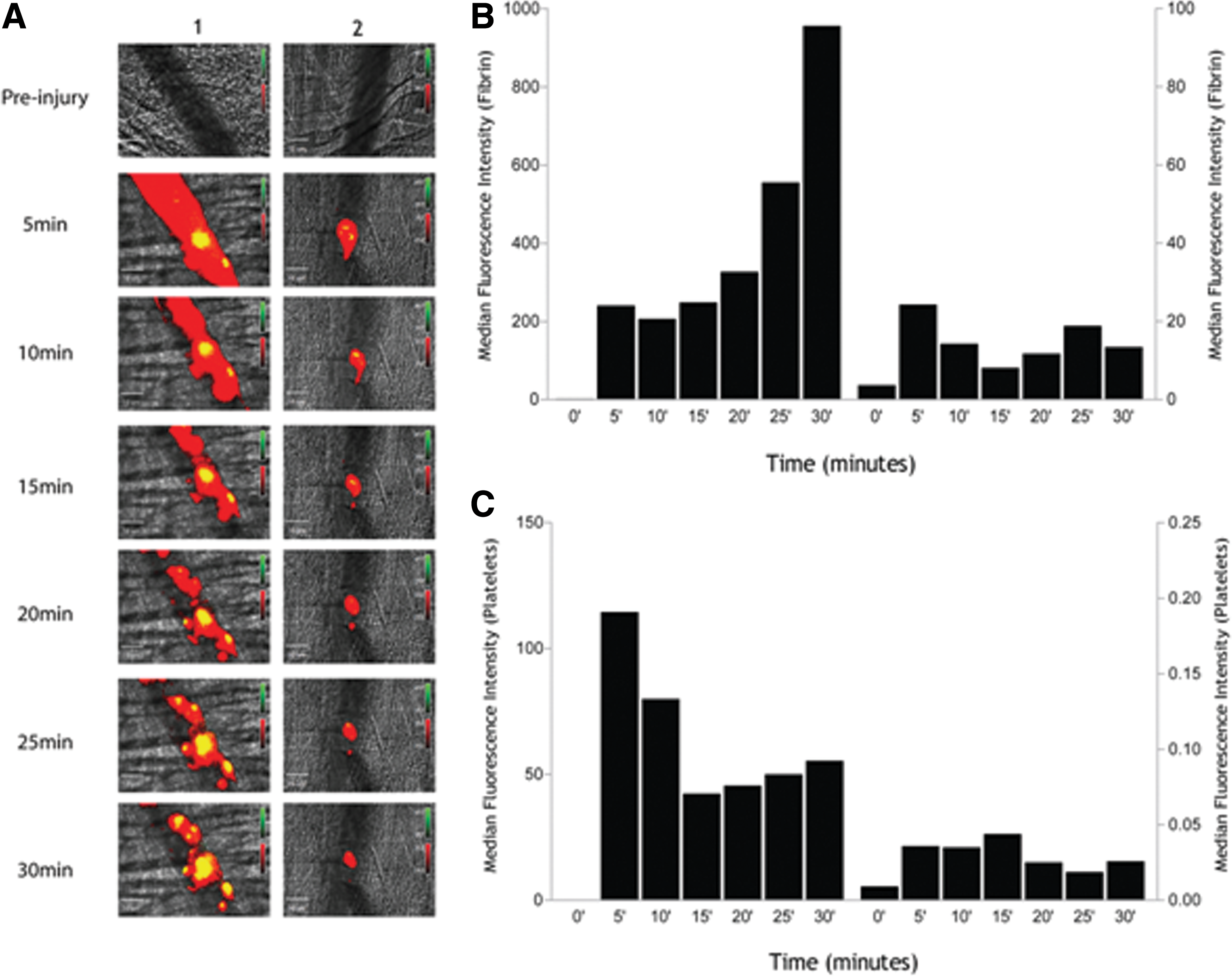
To prove that PDI specifically plays a role in FeCl3-induced platelet thrombus formation, the effect of an inhibitory anti-PDI antibody RL90 [known to cross-react with ERp57 (118)] on platelet deposition after FeCl3-induced laser injury was studied (Fig. 5). Pretreatment of mice with this antibody delayed the appearance of the initial platelet thrombus; no platelet accumulation was observed in about half of the mice treated at higher antibody doses and only about one third of injured vessels occluded within 30 min. Markedly reduced fibrin deposition was detected following administration of this antibody. These results indicate that PDI, or possibly ERp57 as well, is important for both platelet accumulation and fibrin deposition during thrombus formation following FeCl3-induced vessel wall injury. Quercetin-3-rutinoside, which inhibits PDI but not ERp57 activity, also blocks platelet accumulation and fibrin formation following FeCl3 exposure, confirming a role for PDI (46). This extracellular PDI release triggered by cell activation is further supported by the observations that PDI rapidly adheres to sites of tissue injury even in severely thrombocytopenic mice (87), animals treated with the αIIbβ3 inhibitor eptifibatide (45), and mice genetically deficient in platelet-derived PDI (55).
PDI appears to control the initiation of thrombus formation, but the mechanism by which it does so is currently unknown. Specifically, the targets of vascular PDI enzymatic activity have not been identified although some preliminary results are available. PDI has been proposed to modulate the initiation of tissue factor activity, thus initiating the blood coagulation cascade and ultimately thrombin generation and fibrin formation (15). Tissue factor exists in an encrypted inactive form, but the molecular basis of this inactivity has been elusive. Encrypted tissue factor can be detected as a tissue factor antigen on cell surfaces that binds to factor VIIa but its procoagulant activity is diminished or absent (6, 71).
Competing hypotheses include the dimerization of tissue factor (7), the absence of negatively charged phospholipid or the presence of neutral or anticoagulant phospholipid (85), sequestration of tissue factor in cholesterol-rich lipid rafts (21, 97), and the reduction–oxidation of the Cys 186-Cys 209 allosteric disulfide bond that corresponds functionally to a molecular switch from inactive to active (15). It has been suggested that the oxidation of tissue factor to generate a disulfide bond between these two residues is the key event in converting the encrypted tissue factor into its active conformation, with PDI playing a crucial role in this process
Using mechanism-based kinetic trapping with active site variants of PDI, we have identified mixed disulfide intermediates between PDI and a number of novel substrates potentially critical for thrombus formation: vitronectin, complement factor 3, complement factor 5, C4b-binding protein, α2-macroglobulin, protein S, histidine-rich glycoprotein, thrombospondin 1, prothrombin, and CD5 antigen-like protein (Bowley, Furie and Furie; unpublished).
ERp57
ERp57 is a single chain protein of 505 amino acids with a molecular weight of 57,000. It is expressed in eukaryotic cells from yeast to man and is widely distributed among mammalian tissues (70). Its domain structure is a-b-b′-a′, and its domains share a high degree of similarity with those of PDI. Both enzymes have four thioredoxin-like domains with the a and a′ domains each characterized by a catalytic active site motif CGHC. A monoclonal antibody, RL90, that was developed against PDI cross reacts with and inhibits the function of ERp57 (118), underscoring the degree of homology between the active domains of these enzymes. In contrast, the b′ domain of ERp57, which is responsible for substrate binding, is distinct from that of PDI. This observation suggests that these two enzymes interact with different substrates (38, 56, 66, 90). That ERp57 is functionally distinct from PDI, despite their structural similarity evidenced by the fact that ERp57 cannot substitute for PDI in rescuing PDI deficiency in yeast (35). Moreover, ERp57 and PDI differ in their capacities for redox modulation by endogenous sulfhydryl oxidases such as Ero1α (48, 57, 72). The gene for ERp57,
ERp57 is localized primarily to the ER of nucleated cells and contains an ER retrieval motif. A subpopulation of ERp57 escapes retention in the ER by a poorly understood mechanism and can be released into the extracellular environment, where it acts on extracellular substrates. As is the case with PDI, it is the extracellular subpopulation of ERp57 that participates in hemostasis and thrombosis. ERp57 is released from platelets upon platelet stimulation (40) and is released from the endothelium upon endothelial stimulation. Its location within platelets has not been determined, but it is present in the membrane fraction of endothelial cells in addition to the ER (45).
The ERp57 b′ domain interacts with the ER lectin chaperones calnexin and calreticulin, while the b′ domain of PDI does not. Thus, intracellular substrates of ERp57 include glycoproteins that enter the calnexin cycle. Both PDI and ERp57 possess similar reduction potentials that are substantially more oxidizing than those of substrate thiols (37). Free thiol groups on the cell surface are important for normal cellular functions. Thiol isomerases on the cell membrane have multiple biological functions, including NO transport
Because ERp57 has many other well-described functions in the ER, including the processing of glycoprotein substrates of the calnexin/calreticulin cycle (76), genetic studies alone would be insufficient to distinguish between a direct extracellular role for the thiol isomerase rather than a bystander effect on another endogenous substrate of ERp57 such as various β3 integrins. Interpreting the genetic (114) and inhibitory antibody (39, 118) studies together, the data favor a nonredundant extracellular role for platelet and endothelial ERp57 in thrombus formation.
ERp57 in thrombus formation
ERp57 was first detected in platelets in profiling studies designed to identity thiol isomerases in mouse and human platelets and megakaryocytes (40). These experiments showed that stimulated platelets release ERp57 and ERp57-containing microparticles. A second approach used proteomics to identify proteins that are upregulated upon ligation of platelet GPVI showed that ERp57 is released from platelets upon GPVI activation (95). Polyclonal sheep anti-ERp57 antibodies inhibited platelet aggregation and adenosine triphosphate (ATP) secretion from washed platelets stimulated with CRP-XL, whereas PF4 and P-selectin expression was minimally altered (39). These results suggest the involvement of ERp57 in dense granule secretion, but not alpha granule secretion under the conditions used. Furthermore, these antibodies inhibited calcium mobilization, an early event in platelet activation, and the activation of αIIbβ3, as monitored by fibrinogen binding. Using an
A monoclonal antibody to ERp57 inhibits platelet aggregation, whereas ERp57 enhances platelet aggregation and a mutant inactive form of ERp57 perturbs platelet aggregation (118). The activation of αIIbβ3 and the expression of P-selectin are inhibited by the anti-ERp57 monoclonal antibody under the conditions used. Genetically engineered mice lacking platelet-derived ERp57 have prolonged tail bleeding times and occlusion times in a mouse model of ferric chloride-induced carotid injury (116). In mesenteric arterioles, incorporation of ERp57-null platelets into developing thrombi induced with ferric chloride was reduced compared to wild-type platelets and platelet incorporation was dependent upon the presence of β3 integrins. ERp57-null platelets revealed decreased platelet aggregation and decreased activation of αIIbβ3. ERp57 bound to β3 in thrombin-activated platelets and not in platelets lacking β3. These authors propose that ERp57 directly activates αIIbβ3 (116). These experiments were extended to the study of fibrin generation in platelets lacking ERp57 (123). Using the laser-induced thrombosis model, fibrin generation was reduced. In a mouse lacking ERp57 in the endothelium or upon addition of inhibitory antibodies to ERp57 into the mouse, fibrin generation was further reduced. These results parallel those observed for PDI (16, 45).
We have also evaluated the role of platelet-derived and endothelium-derived ERp57 in thrombus formation in a mouse thrombosis model using laser injury of the cremaster arteriole vessel wall. Both the stimulated endothelium and activated platelets release ERp57 at the site of thrombus formation after laser injury. The role of ERp57 in promoting platelet activation was evaluated with knocked down ERp57 in mice using a targeted antisense Vivo-morpholino that acts upstream of the translation start site of ERp57 (Vivo-ERp57). Reduction in expression of ERp57 was induced by injecting mice with 12.5 μg per day per mouse of Vivo-ERp57 or control Vivo-morpholino for 4 consecutive days. To determine the functional importance of ERp57 secreted from activated platelets and endothelium
Infusion of Vivo-ERp57 morpholino resulted in markedly reduced expression of ERp57 levels in platelets as well as the kidney and liver. Platelet aggregation was impaired in platelets from Vivo-ERp57-exposed mice. To determine the functional importance of ERp57 secreted from activated platelet and endothelium
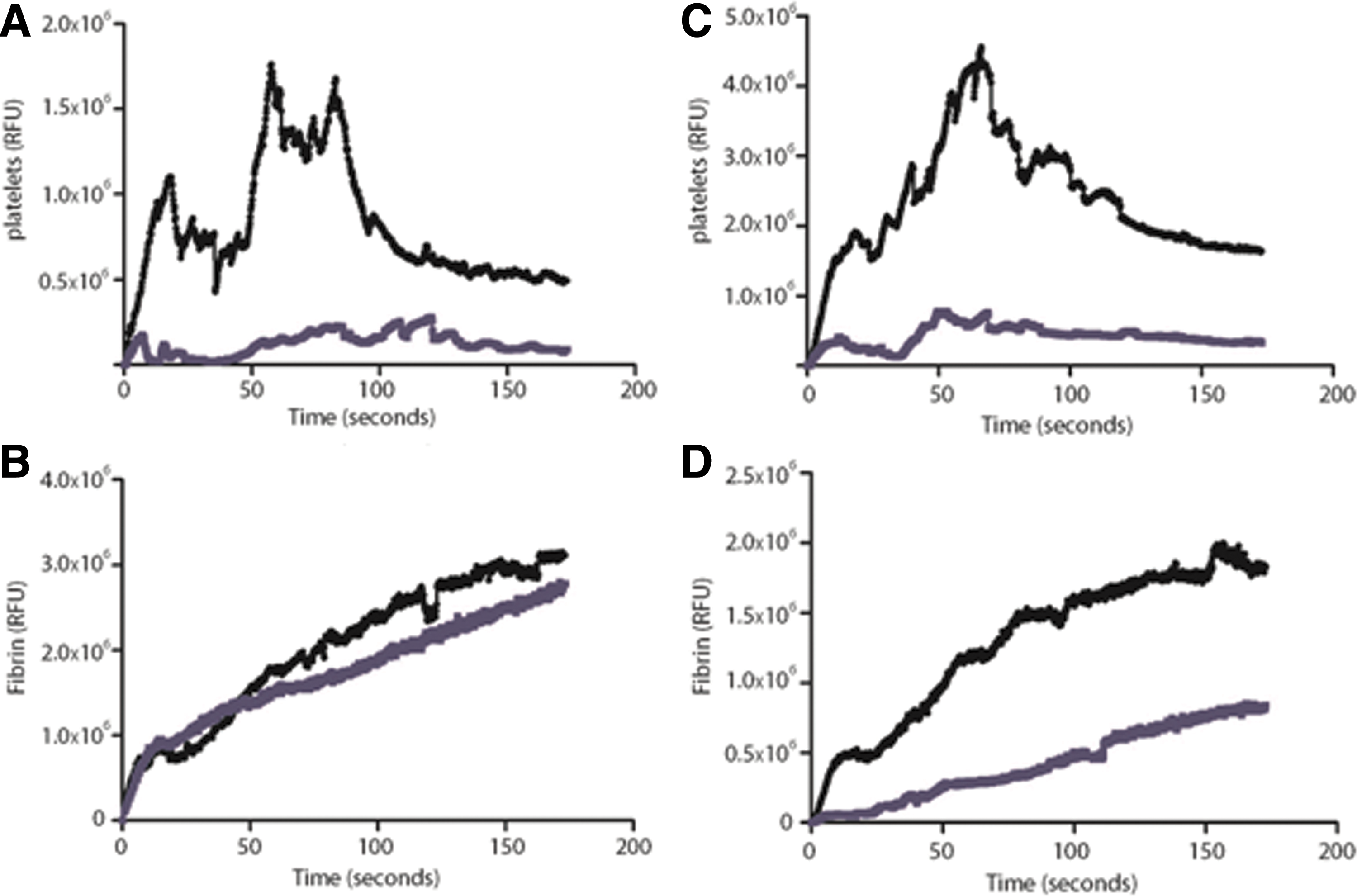
Our group has further explored the role of ERp57 in thrombus formation using the laser injury thrombosis model to determine how isolated deficiency of platelet ERp57 affects thrombus formation
Infusion of specific inhibitory antibodies to ERp57 before laser-induced injury results in diminished platelet thrombus development and fibrin generation. In contrast, platelet-specific ERp57 knockout mice show reduced platelet thrombus formation but normal fibrin accumulation. Our results demonstrate that ERp57 derived from both platelets and the endothelium following vascular injury is important to the thrombotic response. In the laser-injury model, ERp57 secreted from endothelial cells is sufficient to maintain normal fibrin accumulation but cannot restore platelet aggregation in the absence of platelet-derived ERp57.
ERp5
ERp5, encoded by the gene
Specific functions for ERp5 in the ER remain poorly characterized, although this thiol isomerase appears to undergo transcriptional upregulation by spliced XBP-1 as part of the unfolded protein response (63). Despite the presence of a classical C-terminal KDEL ER retrieval motif, many unique roles for ERp5 have been established beyond the cell surface, including thrombus formation (77) as well as in the shedding of tumor-associated ligands required for metastasis (52). For example, the extracellular catalytic activity of ERp5 acts on the tumor ligand, major histocompatibility complex class-I-related ligand MICA, and contributes to tumor immunoevasion (52).
ERp5 in thrombus formation
This enzyme expresses oxidoreductase activity, as demonstrated by the insulin reductase activity and the renaturation of ribonuclease. Like PDI and ERp57, ERp5 is secreted from platelets upon cell activation (51). Inhibition of ERp5 function with an anti-ERp5 antibody prevented fibrinogen binding to activated platelets and platelet aggregation
Interaction of
β
3 Integrins and Thiol Isomerases
The β3 integrins of the vasculature, including αIIbβ3 and αVβ3, have been established as binding partners to extracellular PDI both
β3
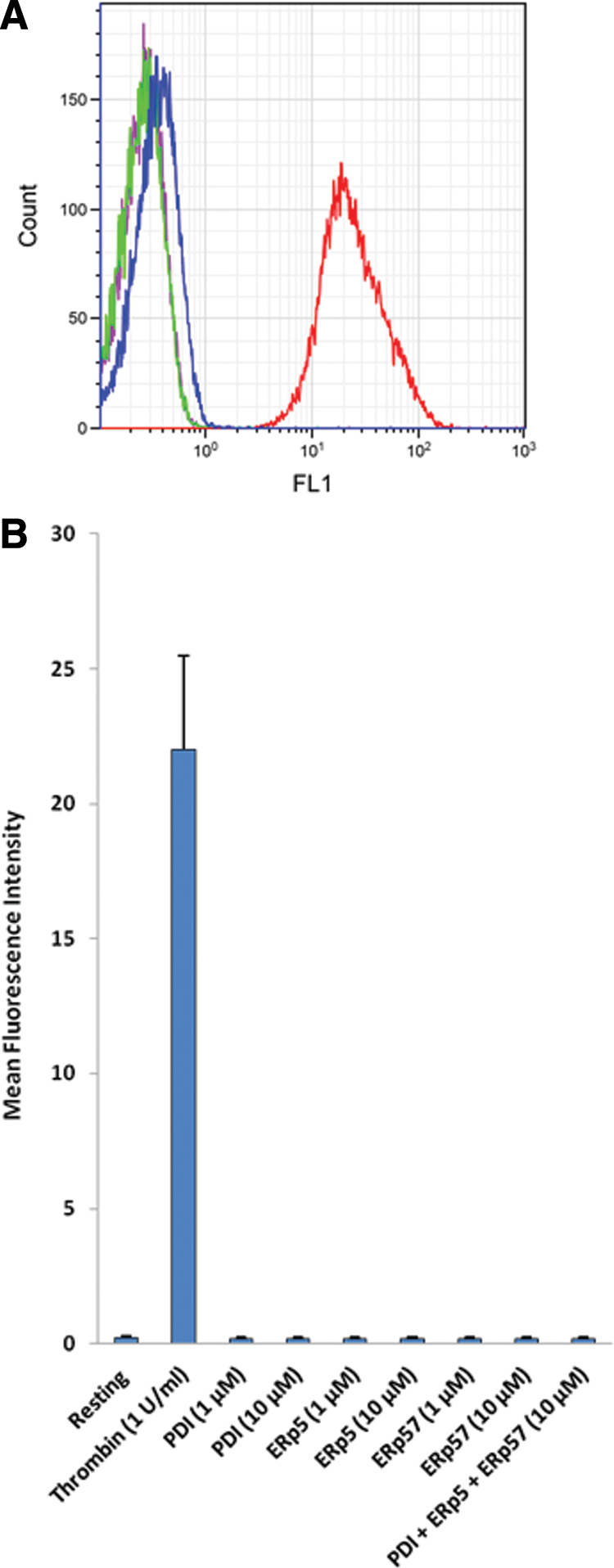
PDI, ERp5, or ERp57 influences β3 integrin function, but the mechanism is unknown. Wu reported the ability of an inhibitory antibody to ERp57 to inhibit platelet aggregation and activation of αIIbβ3, whereas ERp57 potentiates platelet aggregation, and the active site-mutated ERp57 inhibits platelet activation and prolongs the tail bleeding time in mice (118). Exogenous PDI binds to platelet PDI-null platelets but not to β3-null platelets (55). Using the monoclonal antibody, PAC-1, specific for the activated conformer of β3, we examined the expression of this antigen in human platelets treated with PDI, ERp5, and ERp57, alone and in combination by flow cytometry. We did not observe expression of the active conformer upon incubation of PDI, ERp5, ERp57, or a mixture of PDI+ERp5+ERp57 (Fig. 7). Under the conditions studied, β3 does not appear to be a substrate of these thiol isomerases. However, these thiol isomerases all bind to β3. Therefore, it would appear that the pathway to β3 activation involves these thiol isomerases, but they do not directly alter the β3 conformation to express the structure associated with integrin activation.
Future Directions
Despite a firm and growing body of evidence supporting a critical role for PDI family thiol isomerases in thrombus formation and fibrin generation, a number of major questions invite future investigation.
Thiol isomerase substrates involved in thrombus formation
Although thiol isomerases are important for thrombus formation, the mechanism by which they support thrombus formation remains to be elucidated. Specifically, the identity of the substrates of each thiol isomerase needs to be defined as do the changes in structure and function that each substrate undergoes
Redox state
Retaining one or more CXXC motifs, the PDI family thiol isomerases are able to catalyze a variety of chemical reactions, including reduction, oxidation, isomerization, and nitrosylation/denitrosylation of substrate thiols. The reduction potential and resulting redox state of various ER-resident PDI family proteins are not identical [for review, see Hatahet and Ruddock (37)]. However, the redox state of a thioredoxin-like protein dictates the types of reactions in which it may participate. While the active site of platelet surface PDI exists in both the dithiol and disulfide states, the fraction of PDI that is reduced increased substantially upon platelet activation (10). However, it is unclear whether PDI emerged in this state or had already undergone a redox process at the time of assay. The redox state of secreted thiol isomerases remains largely unknown. Similarly, as the key substrates of secreted thiol isomerases remain to be identified, the net vector of electron flow required to support hemostasis at sites of tissue injury must also be clarified.
A limitation of much recent work on PDI family proteins in thrombus formation has been the inability to determine the redox state of the released proteins, particularly
A major question surrounds whether the redox state of these thiol isomerases is actively regulated or modified, either immediately before secretion or beyond the plasma membrane. There is clear precedent for this within the cell, where enzymes such as thioredoxin reductase help maintain a reducing cytosol
By contrast, the QSOX enzymes have an established role in the late secretory pathway (12) and are present in a variety of extracellular fluids, including plasma (44). The single transmembrane domain of QSOX1a is proteolytically processed in the Golgi to enable its cellular egress
Redundancy or specificity
Why are so many thioredoxin-like proteins required beyond the plasma membrane? Are there parallel and redundant pathways present to maximize redox capacity and ensure fidelity of extracellular redox reactions? Or, alternatively, are there separate and discriminatory pathways present to enable selective or regulated activation of specific substrates? As the redox potentials of the vascular thiol isomerases are not identical, one could even hypothesize that different thiol isomerases are secreted in distinct redox states, perhaps regulated by cooperation with specific thiol oxidases or reductases. ERp57 and PDI share a high degree of sequence homology, and the redox potentials for ERp57 (domain a, −0.167V; domain a′ −0.156v) and PDI (−0.175V) are quite similar (29, 68). For these reasons, the question arises whether the roles of these two enzymes in thrombus formation are redundant or whether they have unique functions in this process. Several lines of evidence suggest the latter. Function blocking anti-ERp57 antibodies further decrease thrombin-induced aggregation, ATP secretion, and αIIbβ3 activation of PDI-null mouse platelets already impaired in these functions (55). A mechanism for the participation of both of these thiol isomerases in αIIbβ3 activation is suggested by an elegant study of polyomavirus infection demonstrating that three thiol isomerases coordinately catalyze the unfolding of the C-terminal arm of VP1, the major coat protein of the virus, to facilitate infection (111). Alternatively or additionally, each of these thiol isomerases may interact with a unique set of substrates based on binding to their poorly conserved noncatalytic domains. The specialized role of ERp57 as a glycoprotein oxidoreductase suggests that it may be important in regulating platelet and endothelial cell surface glycoprotein function (47).
Mice specifically lacking platelet PDI suggested a defect in platelet aggregation
Beyond the ER
Another unexplained question is how PDI and related thiol isomerases are secreted by platelets and endothelium? The vascular thiol isomerases all contain a classical C-terminal ER retrieval motif: KDEL in PDI and ERp5; QEDL in ERp57. These short peptide tags interact with a family of KDEL receptors in the early Golgi that should prevent escape to the cell surface and instead specify return of these cargos to the ER
The cell biology of the endothelial cell and the platelet likely plays a critical role in PDI secretion. A KDEL receptor-dependent src kinase signaling pathway is activated by Golgi traffic (81) and has been implicated in cell surface expression of PDI in endothelial cells (112). Platelet PDI is stored in a newly described T-granule compartment that is secreted upon activation (105).
Conclusion
Thiol isomerases were originally described as intracellular actors that primarily functioned to assist in the process of protein folding. However, over the recent past, our understanding of these enzymes has expanded to include their role as extracellular regulators of thrombus formation