
Abstract
Introduction
R
Different pharmacological strategies have been pursued to prevent or restore such supposedly systemic redox imbalances and to improve disease outcomes, typically with antioxidant drugs or vitamins. However, intervention trials with small molecules, especially antioxidants, have been mostly ineffective (78) or even harmful (122). Hence, no direct antioxidant approach is currently part of any evidence guideline and the oxidative stress hypothesis still awaits validation in humans.
One crucial reason for these failures may reside in the dichotomy between disease-triggering and beneficial ROS and differences in this between humans and animal models of disease (132). To target redox-dependent diseases safely and effectively, physiological ROS sources that are relevant for signaling need to remain untouched, while disease-triggering ROS should be effectively reduced.
Instead of antioxidants, a more recent and innovative approach uses pharmacological agents that selectively suppress the activity of ROS-forming or toxifying enzymes (2, 3) whose activity or expression is increased under pathological conditions. These include the ROS generators, such as nitric oxide synthase (NOS), monoamine oxidase (MAO), xanthine oxidase (XO), and NADPH oxidase (NOX), or the ROS toxifier, myeloperoxidase (MPO) (Fig. 1). In addition, we will review a third and possibly synergistic strategy to functionally repair proteins that have been damaged by ROS (see related review by Dao et al. in this Forum on the New ROS Pharmacology). It is also possible to reinforce redox homeostasis by targeting the transcription factor Nrf2, a master regulator of the antioxidant control (122). We jointly review these ROS-based interventions of high clinical potential and place them into context. However, all targets included in this review are described at different levels based on their clinical relevance and maturity in drug development.
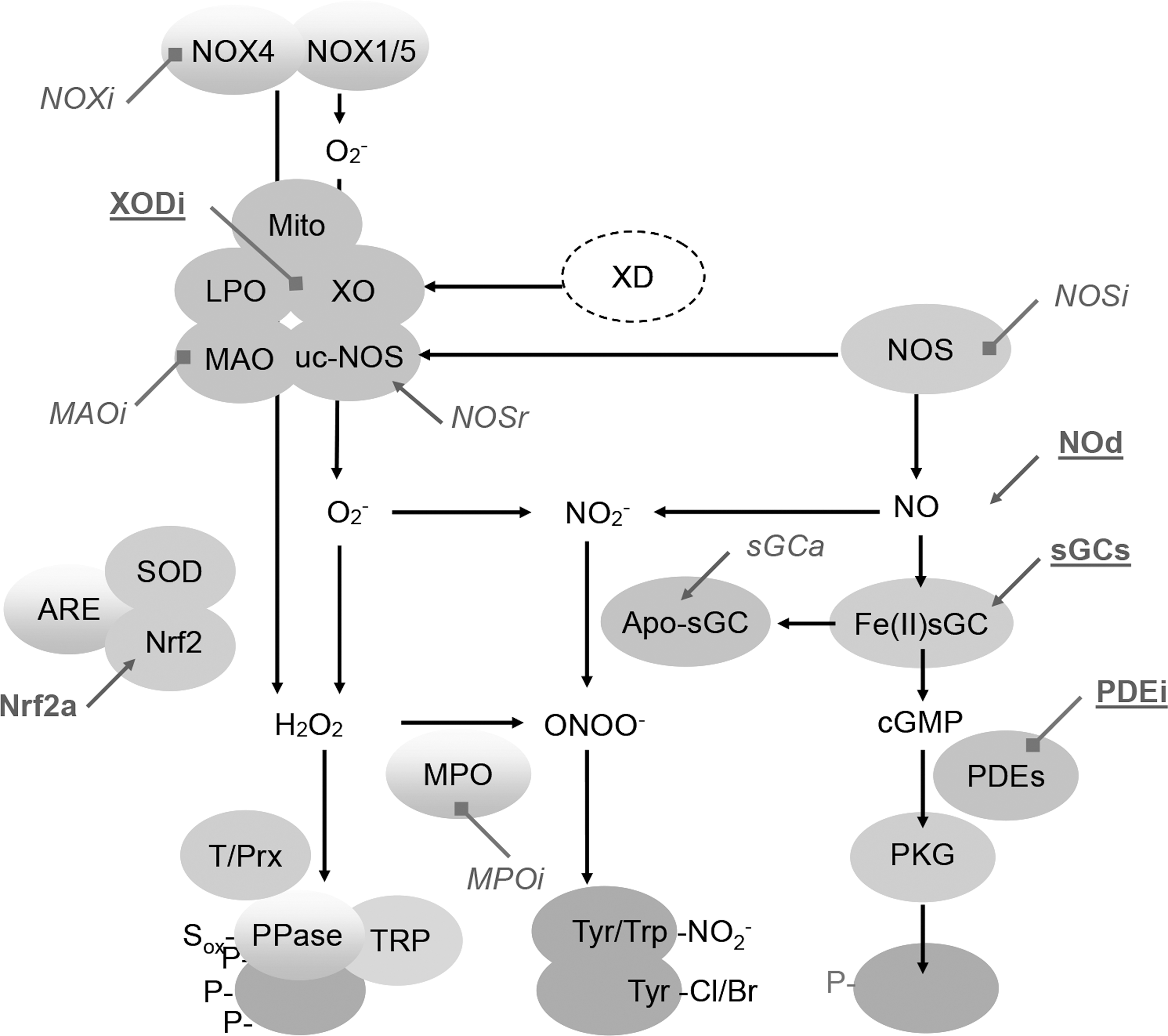
Comparison between pharmacological inhibition of enzymes and changes observed in knockout (KO) animals is detailed in the review. In most cases, a KO represents a de novo deficiency possibly leading to adaptive responses. However, most drug interventions are initiated after onset of the disease (see related review by Dao et al. in this Forum on the New ROS Pharmacology). Therefore, although both approaches are not always comparable, they are needed for targeting validation.
Physiology of ROS
While the existence of endogenous antioxidant enzymes (i.e., superoxide dismutase, glutathione peroxidase, catalase) suggests that the capacity to eliminate ROS is of evolutionary benefit (121), evidence that ROS fulfill equally essential physiological functions stems from the existence of the NOX enzyme family that has no other known function than to produce ROS (72). Table 1 lists the major enzymatic sources of ROS, ROS toxifiers, and their biological effects. Importantly, these physiological effects, which can be ascribed to a specific ROS source or toxifying enzyme, need to be kept in mind as potential side effects of enzyme inhibitors during chronic therapy.
Potential side effects of drugs targeting these enzymes.
CGD, chronic granulomatous disease; DUOX, dual oxidase; GI, gastrointestinal; MAO, monoamine oxidase; MPO, myeloperoxidase; NOX, nicotinamide adenine dinucleotide phosphate oxidase; XO, xanthine oxidase.
From chemical antioxidants to defined enzyme targets
Antioxidants may have a benefit in acute parenteral treatment, but evidence in chronic therapy is lacking (132). One conceptual problem with the antioxidant approach is the fact that it overlooks that ROS also may have beneficial effects. Thus, scavenging ROS systemically may interfere with physiological as well as with pathological processes. However, three alternative approaches to ROS scavenging have been described, including the targeting of the relevant sources of ROS, ROS toxifiers, or repairing previously oxidized proteins (e.g., oxidized soluble guanylate cyclase [sGC] or endothelial nitric oxide synthase [eNOS]). In fact, targeting disease-relevant enzymatic sources of ROS is one of the most promising options. In this strategy, NOXs are a major target. All other enzymes generate ROS together with other products (e.g., MAO) or start to form ROS as a result of a biochemical accident induced by their proteolytic or oxidative modifications. (125a) The latter include XO (57), uncoupled endothelial NOS (uc-eNOS from eNOS) (95), and several mitochondrial enzymes, particularly respiratory chain complexes. Another important category of enzyme targets includes ROS toxifiers (70). We define them as enzymes that convert relatively nontoxic ROS such as hydrogen peroxide (H2O2) to more reactive species. A typical example is MPO, which converts H2O2 into hypochlorous acid (HOCl) (150). In a particular disease condition, specific inhibition of either a source of ROS or an ROS toxifier may become an effective and safe intervention. Recently, proof of principle has also been shown for a surprising third alternative, that is, the functional repair of oxidatively damaged proteins (see related review by Dao et al. in this Forum on the New ROS Pharmacology).
These three approaches hold great therapeutic promise for chronic therapy as long as they are optimally targeted, dosed, and leave physiological ROS formation intact. All are currently in clinical development with the aim to (i) prevent exacerbated ROS production by enzyme inhibition (NOX and XO); (ii) prevent the toxification of ROS such as H2O2 to secondary reactive products (e.g., by MPO); and (iii) promote functional repair of proteins that were damaged by ROS. However, these three approaches require thorough knowledge about the target proteins in addition to possible pharmacological inhibition (see related review by Dao et al. in this Forum on the New ROS Pharmacology). In this review, we focus on these target enzymes and the possible future clinical indications for drugs targeting them.
Disease-Relevant Enzymatic Sources of ROS
NADPH oxidases
NOXs are multiprotein complexes, which contain six or seven transmembrane-spanning domains (72). The NOX enzyme family contains seven members, NOX1-5 and dual oxidase (DUOX)1-2 (also termed NOX6-7). Each isoform has a particular pattern of activity regulation, tissue expression, type of ROS produced, and function (Table 2) (12, 81). The catalytic core of all NOXs contains one multimodular NADPH binding site at the C-terminus and a bimodular flavin adenine dinucleotide (FAD) binding site, as well as four conserved histidine residues involved in the binding of two heme moieties in the membrane. NOXs use NADPH as an electron donor and proximal or extracellular oxygen as an electron acceptor (27). Most NOX family members have similar redox centers (68) as well as the mechanism to generate O2 − as the main product. However, NOX4 and DUOX produce H2O2 as their primary product (86, 126). Other membrane, cytosolic, and regulating domains are involved in NOX activity. While NOX1-3 needs docking of cytosolic factors for complete activation, NOX4 seems to produce ROS constitutively. Yet, NOX5 and DUOX are activated by elevated cellular Ca2+ concentrations via N-terminal EF-hand domains (12).
DUOX, dual oxidase; O2 −, superoxide anion radical; H2O2, hydrogen peroxide; KO, knockout; NOX, nicotinamide adenine dinucleotide phosphate oxidase; ROS, reactive oxygen species.
Because NOX is the only known enzyme family with the sole function to produce ROS (unlike XO, uc-eNOS, and mitochondria), it may represent the primary disease mechanism and thus targets for mechanism-based prevention of oxidative damage (12, 110). Moreover, some NOX isoforms are critically regulated by Ser/Thr kinases (e.g., PKC) (15), although PKC itself is upregulated by ROS (74). Of the seven isoforms, three are best studied: NOX1, NOX2, and NOX4. NOX3 appears to have a very limited organ-specific role both in physiology and pathophysiology. It is mostly expressed in the vestibular system of the inner ear where it controls the formation of otoconia, small biomineral particles (103). Mutations affecting NOX3 activity have not been described so far in humans, but its loss of function leads to severe imbalance in the head tilt mouse (69). NOX5 is not expressed in mice and rats and is thus understudied and remains the big unknown when it comes to translating animal data toward human pathology.
An involvement of NOX2 has been suggested in many disease states (76). The complete loss of function of NOX2 results in chronic granulomatous disease (CGD), which is characterized by susceptibility to certain fungal and bacterial infections (120). Foremost, CGD carriers are prone to developing autoimmune diseases, such as polyarthritis and lupus erythematosus (59, 120). Whether it may therefore become a safety risk for pharmacological inhibition of NOX2 and thereby compromise the innate immune response remains to be tested.
With respect to NOX1, it seems to be implicated in systemic hypertension (148). NOX1-deficient mice show a decreased angiotensin II-induced hypertensive response (42, 87). One study also found a significant effect of NOX1 deletion on basal blood pressure (43). Similarly, NOX1 overexpression potentiates angiotensin II-induced hypertension (31). NOX1-deficient mice were also protected from angiotensin II-induced aortic aneurysms (43) and diabetic vasculopathies (Fig. 2), both in the retina (147) and in large vessel atherosclerosis (48). Taken together, NOX1 may be involved in a whole range of vascular diseases and remodeling of the vascular wall.
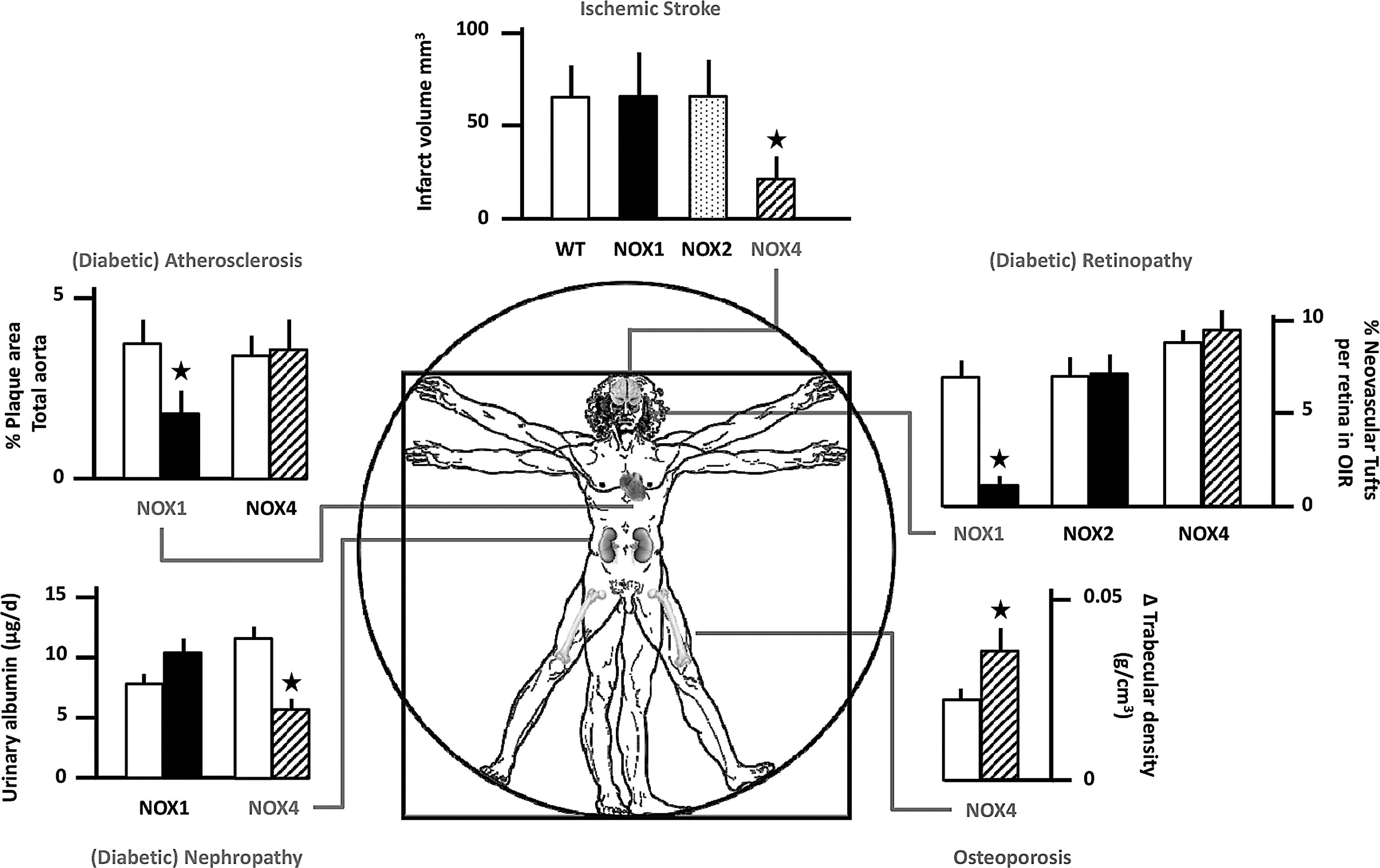
NOX4 is the most widely distributed isoform and can also contribute to diabetic end-organ damage, especially in the kidney (60). NOX4 is also upregulated under hypoxic conditions (94) and can function as an oxygen sensor (99). Pathologically, stroke is one of the best-validated disease indications for NOX4 inhibition (73, 109) (Fig. 2). Moreover, NOX4 expression and activity are strongly increased following TGF-β stimulation of human fibroblasts. This can lead to the transformation of not only normal fibroblasts into myofibroblasts, a key feature of wound healing, but also of chronic fibrotic diseases of the lungs, kidney, or liver. The fact that NOX4 inhibition mitigates myofibroblast transformation in vitro was first shown in cardiac and lung fibroblasts (28, 53) and later in the bleomycin model of pulmonary fibrosis using NOX4-deficient mice (23, 52). Interestingly, the benefit of NOX4 inhibition appears to be tissue specific as NOX4 deletion does not confer protection in urinary obstruction-induced kidney fibrosis (8), a condition where NOX4 inhibition even seems to be deleterious (100). Although the direct connection of NOX4-derived ROS and the tissue-specific fibroblast phenotypic changes are unclear, NOX4 inhibition in idiopathic lung fibrosis represents another promising indication for a pharmacological intervention targeting NOX4.
Finally, the NOX5 isoform, which is not expressed in mice or rats (11), is unique as it is directly activated by calcium (10) and may thus directly link cellular calcium overload to oxidative stress. In adults, NOX5 is found mostly in the spleen, lymph node, and the reproductive and vascular systems (39); in disease, it may play a role in coronary artery disease (49).
DUOX enzymes are expressed at high levels in the thyroid glands and generate H2O2 at the apical membrane. Mutations in DUOX2 and DUOXA2 lead to defects in thyroidal H2O2 generation, congenital hypothyroidism, and euthyroid goiter (61).
Despite the rich genetic evidence for distinct roles of different NOX isoforms (3), the development of isoform-specific inhibitors is lagging behind (see related review by Dao et al. in this Forum on the New ROS Pharmacology). The first generation of inhibitors was highly unspecific, that is, not even specific for NOX (56, 149), while, more recently, the achieved differences in IC50 of the second-generation NOX-specific compounds are hardly relevant in vivo (4). However, with the increasing interest in this target both in pharmaceutical and biotech industries, a third generation of inhibitors, specific and isoform selective, is on the horizon (see related review by Dao et al. in this Forum on the New ROS Pharmacology).
NOS
In this review, we consider nitric oxide (NO) a member of the ROS family. NO is generated from
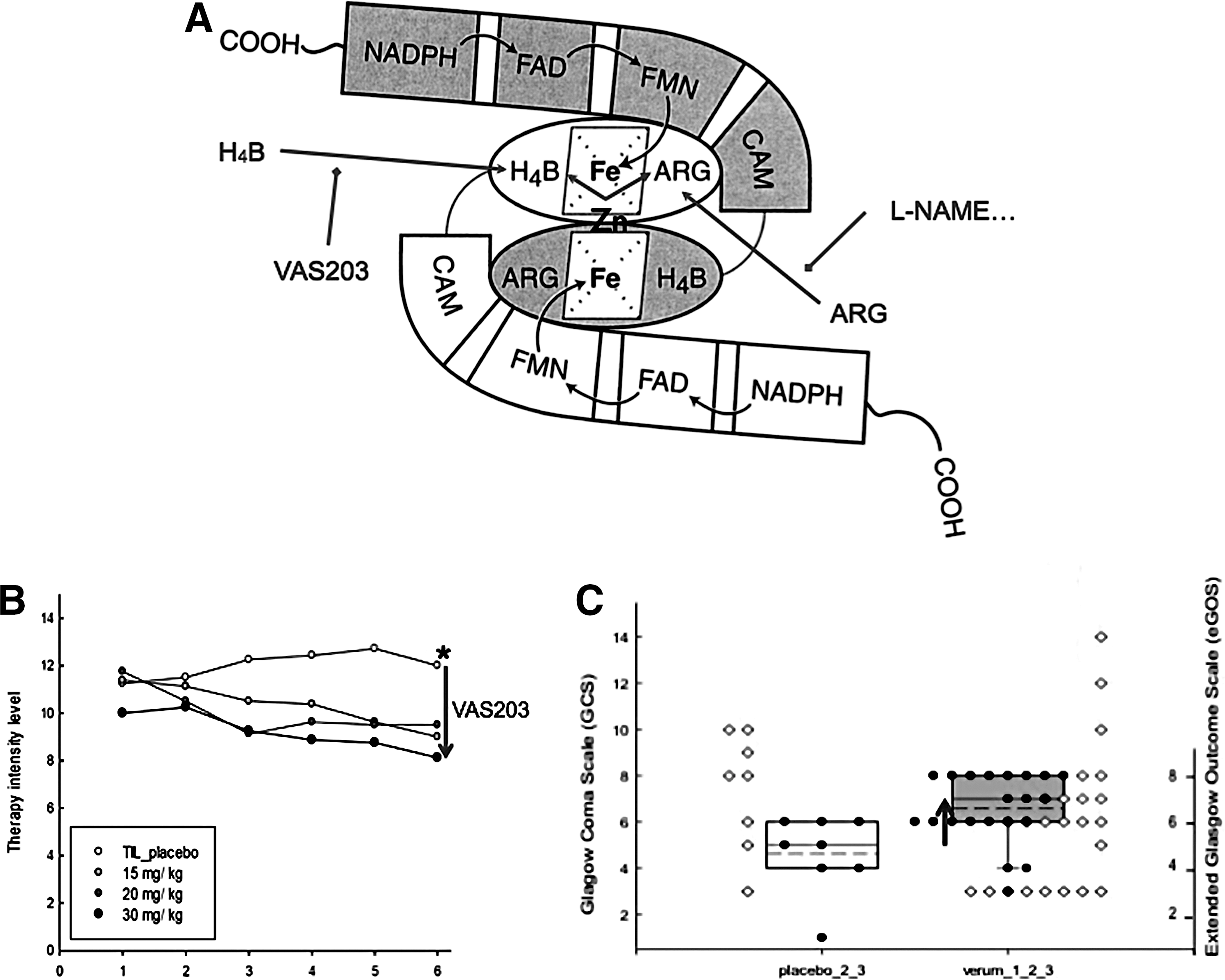
Mice with genetic deficiencies in one of the NOS isoforms are viable. Nos1−/− mice show impaired cognitive performance (145), dramatic enlargement of the stomach, and significantly reduced brain damage after cerebral ischemia (84). Nos2−/− mice suffer from impaired host defense against pathogens and are prone to severe infections. However, they are protected from life-threatening hypotension in septic shock (84). Nos3−/− mice display impaired vasodilation, elevated blood pressure, diminished cardiac contractility (84), and, under stress conditions, impaired adaptation, for example, increased atherogenesis under high-fat diet and accelerated development of diabetic complications (18). Double knockout mice that lack both NOS1 and NOS3 display abnormalities in hippocampal long-term potentiation, a model for learning and memory (7). Triple knockout mice are severely insulin resistant (nephrogenic diabetes insipidus), display a number of cardiovascular risk factors, including hypertension and hypertriglyceridemia, and develop spontaneous myocardial infarction, supporting a critical role of NO in maintaining cardiovascular homeostasis (7, 138). Clinically, many reports suggested therapeutic benefit from inhibiting NOS1 or NOS2, for example, asthma (51), migraine (139), or cardiovascular diseases (CVDs) (2). Currently, the clinically most advanced therapeutic approach for NOS inhibition is in traumatic brain injury (133) (Fig. 3).
XO
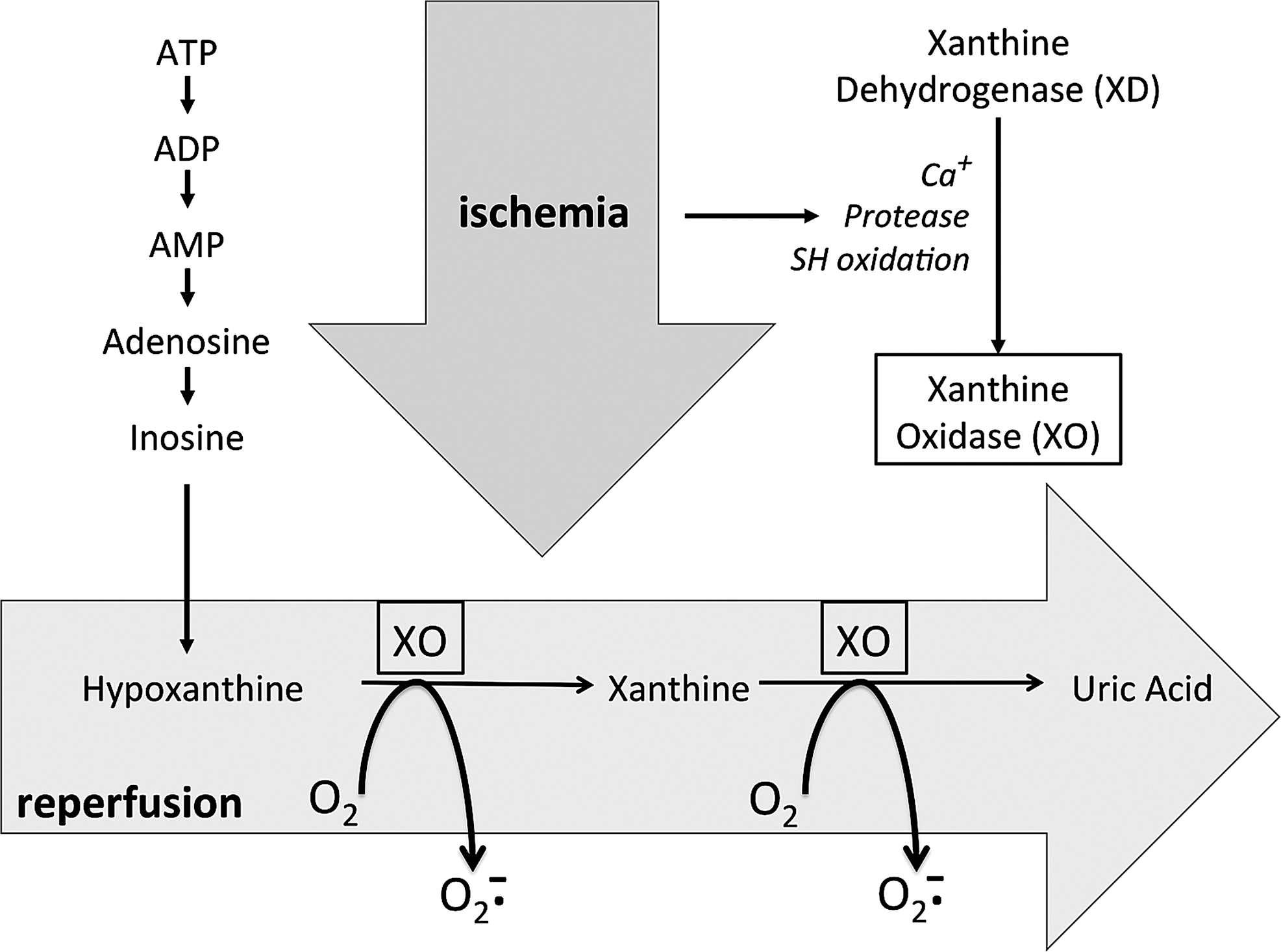
XO is defined as an enzyme activity; it utilizes oxygen as the electron acceptor to form reduced ROS (89) according to the following:
XO is derived from xanthine dehydrogenase (XDH, encoded by Xdh) by reversible sulfhydryl oxidation or by irreversible proteolytic modification (57, 98). As the terminal enzyme in the catabolism of purines, XDH activity utilizes NAD+ as the electron acceptor to convert hypoxanthine to xanthine and the latter to uric acid, according to the following:
In some mammals, such as mice, uric acid is metabolized further by uricase to form allantoin. As XO may arise from XDH by sulfhydryl oxidation, XO activity can be a direct consequence of increased oxidative stress that further contributes to the pathogenesis of various diseases as a feed-forward mechanism of ROS-induced ROS.
Homozygous Xdh −/− mice show early neonatal lethality and display renal dysplasia (106), while heterozygous Xdh+ /− mice have disrupted formation of the milk fat globule, underlining the importance of XDH to lactation (38). Reduced expression of Xdh in mice augments lipid accumulation in adipocytes, accompanied by an increase in oxidative stress, and induces obesity with insulin resistance in older age groups (96).
Inhibition of XO has been clinically applied for decades for the treatment of hyperuricemia and gout (35). In addition, xanthine oxidase inhibitors (XOi) has recently been explored for cardiovascular therapy (Fig. 4) based on animal (124, 135) and clinical studies in patients with type 2 diabetes and idiopathic dilated cardiomyopathy (21, 22). While several studies show clinical efficacy of XOi in CVD, others do not, such as in the case of heart failure patients with hyperuricemia (46). A possible explanation for these contradicting results is that XO inhibition might be a double-edged sword. Thus, while XO generates O2 −/H2O2 as by-products, its final metabolite, uric acid, is also an antioxidant. Depending on the disease condition, one of these opposite effects of XO (ROS or antioxidant production) may prevail. This notion is consistent with the observation that plasma uric acid concentrations associate inversely with some diseases (144), whereas they are independently and significantly associated with other diseases (37).
MAO
MAOs are flavoenzymes (located at the outer mitochondrial membrane) that catalyze the oxidative deamination of both endogenous and exogenous amines, including neurotransmitters and several drugs. They exist as two isoforms, A and B, differing with respect to their tissue distribution, substrate preference, and inhibitor specificity (111). MAO-A reacts preferentially with tyramine, serotonin, and norepinephrine, while dopamine and phenylethylamine are preferential substrates for MAO-B. The imine products are coupled to the reduction of a covalently bound FAD, which in turn is reoxidized by oxygen leading to H2O2. In mitochondria, MAOs thus generate a significant percentage of total H2O2 in addition to that formed by the electron transport chain (5, 64). On the other hand, the imine product can also spontaneously hydrolyze, generating the corresponding aldehyde and ammonia (33, 111). Of note, all the three products of MAO catalysis are potentially toxic, especially at the level of mitochondria (64). In this regard, H2O2 and aldehydes can particularly synergize (62) leading to mitochondrial dysfunction. Moreover, they are directly related to endothelial dysfunction, heart function, and muscular dystrophy (Fig. 5). In addition, ammonia can stimulate further ROS formation by dihydrolipoyl dehydrogenase, the E3 component of pyruvate and oxoglutarate dehydrogenase (67).
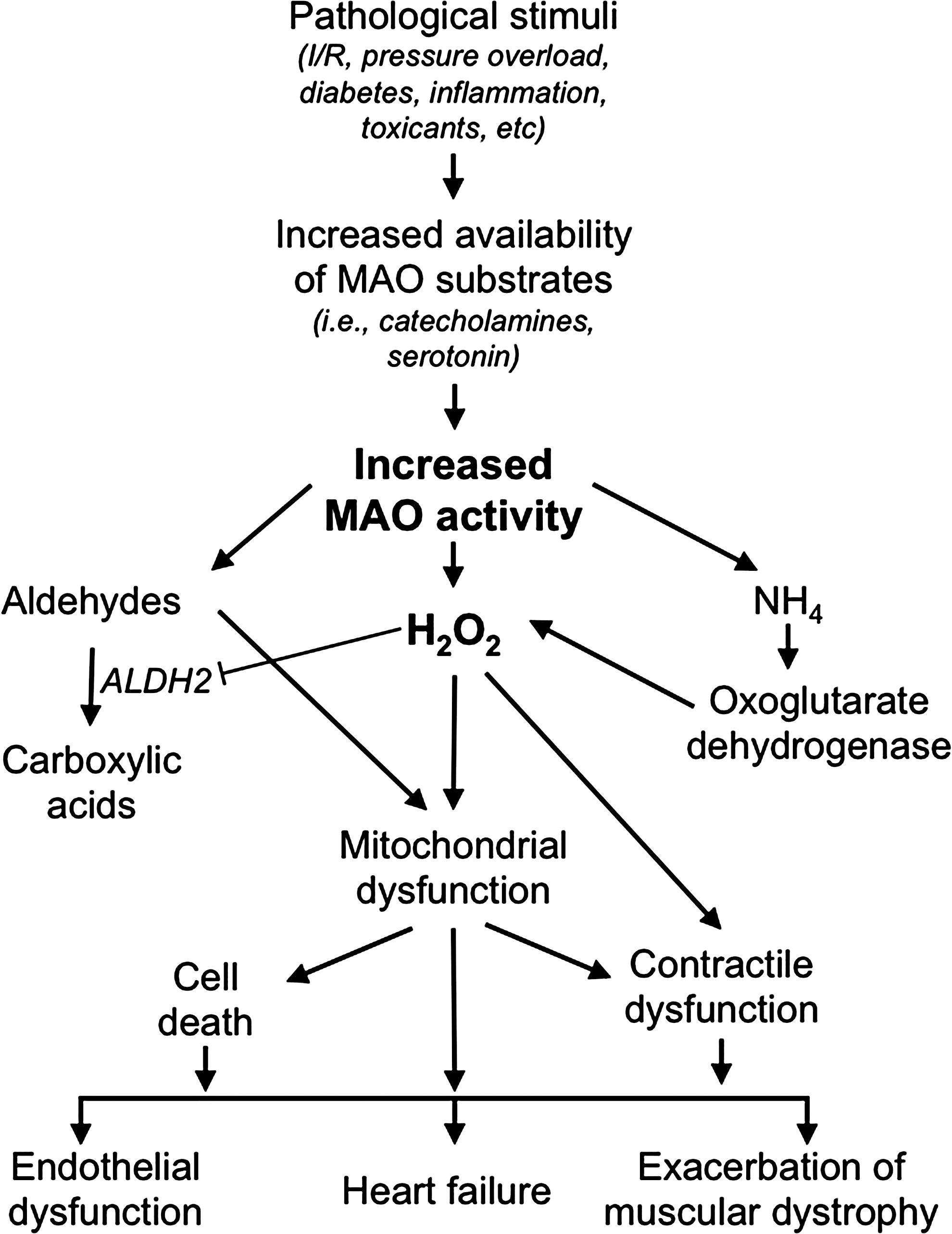
Patients and mice lacking MAO-A activity are characterized by borderline mental retardation and aggressive behavior (17, 19, 20), whereas polymorphisms in the MAO-A gene have been associated with bipolar disorder (40, 83, 114). On the other hand, variations in MAO-B activity in patients have been associated with psychotic disorders, depression, impulsivity, behavioral disinhibition, and attention-deficit/hyperactivity disorder (1, 82, 85, 112). MAO inhibitors have been used for the treatment of affective disorders and their mood-enhancing effect of MAO inhibition is likely related to an increased availability of serotonin, norepinephrine, and dopamine since their decrease is associated with depression (154). Deficit in both MAO-A and -B activity causes severe developmental and intellectual deficits, autistic-like behavior, and stereotypical movements (25, 97, 127, 128, 146).
The emphasis on MAO substrates (i.e., neurotransmitters) has curtailed the attention on the relevance of MAO products. Increased MAO-B activity has been correlated with Parkinson's disease (13, 66, 117). MAO expression increases in aging (88, 118) and an increased expression is associated with endothelial dysfunction (134), postoperative atrial fibrillation, muscular dystrophy (91), and prostate cancer (152). A common denominator among all these pathologies is altered ROS. In agreement with this, beneficial effects of MAO inhibition have been demonstrated in these conditions as well as in myocardial ischemia/reperfusion injury (14, 30), heart failure (62, 65, 140), and neurodegenerative disorders (16).
ROS Toxifiers
ROS toxifiers include different peroxidases such us eosinophil peroxidase, lactoperoxidase, and thyroid peroxidase. These enzymes share several similarities with their ortholog, MPO. However, here we mainly focus on MPO due to its clinical relevance and promising preclinical data.
MPO
MPO, a heme peroxidase present in circulating neutrophils, monocytes, and some tissue macrophages, plays an important role in killing invading microbes (71). MPO generates a number of reactive chlorinating and brominating oxidants, including nonradical species (two-electron oxidants) and radical species (50). In fact, MPO acts as a toxifier since in the presence of halides (Cl−, Br−), it transforms the relatively weak two-electron oxidant H2O2 into the more reactive hypohalous acids, (hypochlorous acid, [HOCl]; and hypobromous acid, [HOBr]) (150), as well as chloramines (104). MPO is also a major contributor to protein nitration since inflamed tissues of MPO-deficient mice contain significantly less 3-nitrotyrosine than those in wild-type mice (41). In addition to their role in the innate immune response, MPO-derived oxidants have the potential to cause host tissue injury by promoting post-translational protein modification (107, 150) and lipid oxidation (119). MPO was demonstrated to promote CVD and pharmacological inhibition or genetic deletion partially prevented these adverse effects (79, 141). However, it should be noted that MPO also fulfills an important role in host defense against pathogens and genetic deletion increased the severity of infections in animal models, although no clear increase in susceptibility to infections was observed in humans with MPO polymorphisms (34). Therefore, although MPO is detrimental in the context of CVDs, it also plays a major role in defense against pathogens, thus partial inhibition may be better than a complete blockage of the enzyme (see related review by Dao et al. in this Forum on the New ROS Pharmacology).
MPO is predominately located in inflamed tissue where it is found within or nearby infiltrated neutrophils and certain macrophages. Upon activation, phagocytes release MPO. In the case of circulating neutrophils, released MPO can bind to the endothelium, translocate, and be deposited in the subendothelial space (101). As a consequence of its localization and production of highly reactive oxidants, MPO is thought to contribute to a wide range of chronic inflammatory diseases as well as cardiovascular and neuroinflammatory diseases.
In addition to inhibiting MPO activity directly, an alternative therapeutic strategy is to displace MPO from the vascular endothelium and subendothelial space, that is, the sites where MPO released from circulating phagocytes is thought to bind to and reside. MPO binds to endothelial cells via heparin sulfate glycosaminoglycan, and heparin prevents and reverses such binding (9). The removal of MPO by heparin may help explain its anti-inflammatory actions. In fact, infusion of heparin increases the plasma concentration of MPO and increases flow-mediated dilatation (115), a major indicator of endothelial nitric oxide bioavailability.
Targeting MPO is in the early stages of clinical development, for example, as treatment for neurodegenerative, cardiovascular, and pulmonary diseases. Therefore, the next few years will be crucial in providing a definitive answer on whether inhibition of this toxifier enzyme is a valid strategy for preventing or alleviating various inflammatory diseases.
Proteins Damaged by ROS
In addition to preventing ROS-induced damage, its functional repair is much more than an option and has already entered clinical practice. A key example of this is impaired NO-cyclic guanosine monophosphate (cGMP) signaling.
uc-eNOS, NO scavenging, and apo-sGC
NO is an important cellular signaling molecule, which is involved in many physiological processes (58). NO production and ROS activate PKC, which contributes to cellular proliferation, neoplasia, and cancer (75, 113). Besides, NO interacts with different receptors (i.e., NMDAR and G-protein-coupled receptors) promoting the release of zinc ions from metallothioneins mediated by the nNOS/NO pathway (116). Accumulation of zinc is related to mood disorders, schizophrenia, and both neurological and neurodegenerative diseases (108).
However, almost all physiological effects of NO are mediated through its receptor enzyme, sGC, a heterodimeric heme protein comprising of a larger α subunit and a smaller heme-binding β subunit. Upon binding of NO to sGC heme, the conversion of guanosine-5′-triphosphate to the intracellular signaling molecule, cGMP, is activated. cGMP in turn regulates cGMP-dependent protein kinases and ion channels and is degraded by phosphodiesterases (29). The resulting effects include (acutely) inhibition of blood vessel contraction, improved perfusion, antithrombosis, neurotransmission, and memory formation, as well as (chronically) antiproliferation, antiremodeling, and anti-inflammation effects (123).
Oxidative stress can lead to the deregulation of NO-cGMP signaling (90) either by oxidizing and uncoupling NOS, by chemical scavenging of NO, or by oxidation and loss of heme in sGC. NOS3/eNOS, NOS1/nNOS (93), and to a lesser extent NOS2/iNOS (153) can be oxidatively damaged. This involves a highly redox-sensitive cofactor, tetrahydrobiopterin (H4B). In uc-NOS, oxygen activation is uncoupled from arginine-to-NO metabolism and NOSs become themselves ROS-forming enzymes; another example of ROS-induced ROS formation.
Impaired NO-sGC-cGMP signaling can thus be caused by reduced NO bioavailability and/or decreased responsiveness to NO and has been implicated in the pathogenesis of many cardiovascular, pulmonary, endothelial, renal, and neurological diseases (90, 148). Clinical evidence for a role of oxidative H4B depletion is based on recoupling and improvement of endothelial function in chronic smokers by BH4, but not by tetrahydroneopterin (H4N), which shares the antioxidant properties of H4B, but is not a cofactor for NOS3/eNOS (54, 55). Likewise, supplementation with the BH4 analog, folic acid, improves endothelial function in human subjects (6, 47). In fact, in experimental hypertension as well as atherosclerosis treatment with the H4B precursor, sepiapterin restores endothelial function (80, 125). In addition, many studies have reported a positive effect of
In addition to NOS, ROS can also affect the bioavailability of NO by direct chemical scavenging and the redox state of sGC resulting in the oxidation of its heme iron to Fe3+ and/or ultimately in loss of the sGC heme (36, 131). The resulting apo-sGC is completely unresponsive to NO and rapidly degrades (92). Both pathomechanisms can be functionally reversed. So-called sGC stimulators sensitize sGC for lower NO concentrations to yield the same cGMP stimulatory effects as physiological NO levels would cause; sGC activators bind to the oxidized/heme-free form of sGC and reactivate the enzyme to the same Vmax levels as NO-stimulated heme-containing sGC.
sGC stimulators have entered the clinic. The PATENT-1 and PATENT-2 clinical trials in pulmonary arterial hypertension patients showed an increased walking distance (45). Moreover, the CHEST-1 trial in chronic thromboembolic pulmonary hypertension, for which otherwise pulmonary endarterectomy has been the only other curative option, showed improved exercise capacity, mean pulmonary artery pressure, cardiac output, and decreased clinically relevant pulmonary vascular resistance. A second sGC stimulator, vericiguat, is now in clinical development for different forms of heart failure (105). In addition, preclinical data suggest that sGC stimulators may be of benefit in chronic kidney disease (130) and hypertension (26, 129, 142, 143).
The development of sGC activators lags behind that of sGC stimulators due to initial pharmacokinetic setbacks (44), but preclinical data suggest benefit in cardiac hypertrophy (26) and type 2 diabetic nephropathy (102). Importantly, these effects seem to occur at doses that do not affect mean arterial pressure and heart rate and may thus involve preferential microvascular dilation.
Conclusions
For several decades redox imbalances have been suggested to have relevance in neurodegenerative, cardiovascular, metabolic, and neoplastic diseases. Therapeutically, most attempts to translate ROS scavenging by antioxidants into the clinic have yielded mostly disappointing results. However, pharmacological modulation of protein targets to either decrease ROS overproduction or toxification, as well as functional reversal of ROS-induced damage, has lead to several therapeutic breakthroughs (Table 3).
CVD, cardiovascular disease; COPD, chronic obstructive pulmonary disease; MAO, monoamine oxidase; MPO, myeloperoxidase; NOS, nitric oxide synthase; NOX, nicotinamide adenine dinucleotide phosphate oxidase; PAH, pulmonary arterial hypertension; sGC, soluble guanylate cyclase; XO, xanthine oxidase.
Outlook
With the introduction of sGC stimulators for pulmonary hypertension, repurposing of XOi and monoamine oxidase inhibitors for ROS-related cardiovascular indications and the successful development of several new principles such as NOXi, MPOi, and NOSi into phase III translational ROS research are at the verge of major breakthroughs. Several of these candidate compounds are currently in clinical development and are likely to dramatically reshape the perception of the field of ROS and oxidative stress.
Footnotes
Acknowledgments
A.I.C., A.D., F.D.L., V.J., T.S., K.H.K., M.G.L., A.C., P.G., and H.H.H.W.S. were supported by the European Cooperation in Science and Technology (COST Action BM1203/EU-ROS). N.K. is supported by an EFSD/Sanofi Award. H.H.H.W.S. is the recipient of an ERC Advanced Grant and a Marie Curie IRG. R.S. is supported by a Senior Principal Research Fellowship from the National Health and Medical Research Council of Australia.