
Abstract
Introduction
O
Possible explanations for this paradox may reside in the lack of specificity of antioxidants towards a certain cellular compartment or tissue, and/or the possibility of generating reductive stress, by increasing levels of reducing agents and therefore disturbing redox homeostasis in the opposite direction. Exogenous antioxidants are also likely to interfere with both disease-triggering and physiological ROS levels. The latter regulate extracellular matrix, control vasomotor activity, are involved in the innate immune response, and promote cell differentiation, proliferation, and migration (4, 10, 161, 163).
Another somewhat indirect type of antioxidant therapeutic strategy that could have fewer side effects relies on the activation of endogenous antioxidant responses. In this context, pharmacological activation of the transcription factor NRF2 is promising therapeutic option currently studied clinically. The conceptual difference between these two antioxidant approaches is broad unspecific scavenging versus a localized response at physiological (sub)cellular sites. Only the latter has promise in leaving physiological ROS formation and signaling intact.
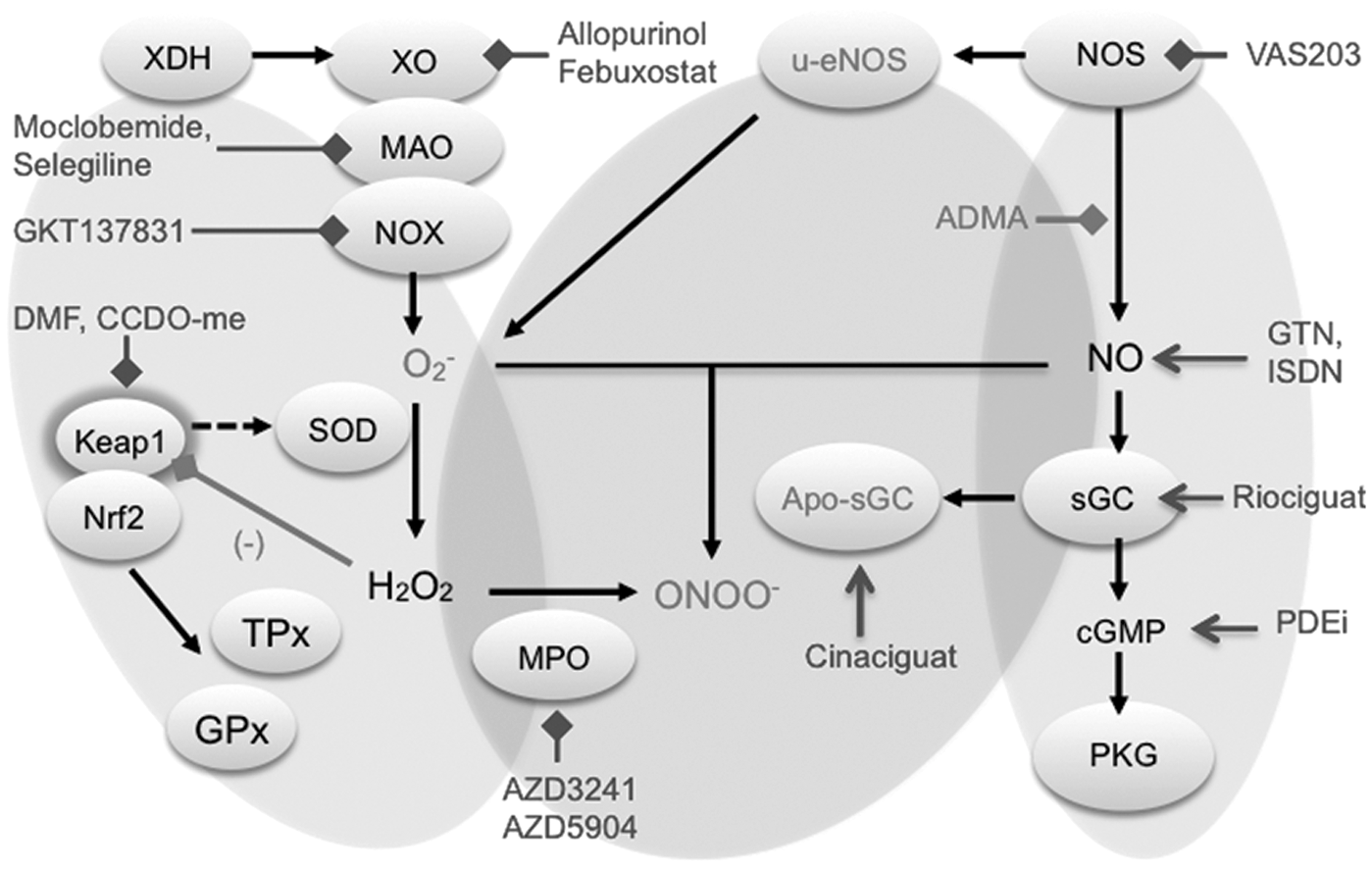
Of far broader relevance is a third approach that involves the specific inhibition of the disease-relevant sources of ROS. In this case, the key question is which enzyme to target. Besides NADPH oxidases (NOXs) (10), xanthine oxidase (XO) (96), uncoupled nitric oxide synthase (uc-NOS) (155), and monoamine oxidases (MAOs) (39), other sources such as cytochrome P450 oxidases (44), lipoxygenases (170), and the mitochondrial electron transport chain (134) are all able to generate ROS. Among these, NOXs stand out as their primary function is to produce ROS. All other enzymes do not form ROS as their primary function, but only as a collateral or side product. Examples include uc-NOS, uncoupled mitochondria, and XO. Additional approaches include the inhibition of ROS-toxifying peroxidases, such as myeloperoxidase (MPO), or the functional repair of oxidatively damaged proteins, such as the redox-sensitive soluble guanylate cyclase (sGC), a principle that has already entered the clinic.
We here review the current status and outlook of the most advanced areas in the field of translational redox medicine by focusing on drugs in four categories: • Activators of endogenous antioxidant defense systems (indirect antioxidants) • Inhibitors of ROS formation • Inhibitors of ROS toxification • Compounds that allow functional repair of ROS-induced damage
Activators of Antioxidant Defense Systems
The main, if not only, representative members of this group of drugs are nuclear factor (erythroid-derived 2)-like 2 (NRF2) activators. NRF2 is a basic region-leucine zipper (bZIP) transcription factor (Fig. 1A) that forms heterodimers with other bZIP partners, of which the small musculoaponeurotic fibrosarcoma proteins are the best characterized. Together, they recognize an enhancer sequence termed Antioxidant Response Element (ARE) that is present in the regulatory regions of over 250 genes (ARE genes), including antioxidant genes such as HMOX1 (coding heme oxygenase-1) (58). These genes encode enzymes involved in antioxidant reactions, including those driven by glutathione and thioredoxin, generation of nicotinamide adenine dinucleotide phosphate (NADPH), biotransformation, proteostasis, and even DNA repair (58, 90, 135).
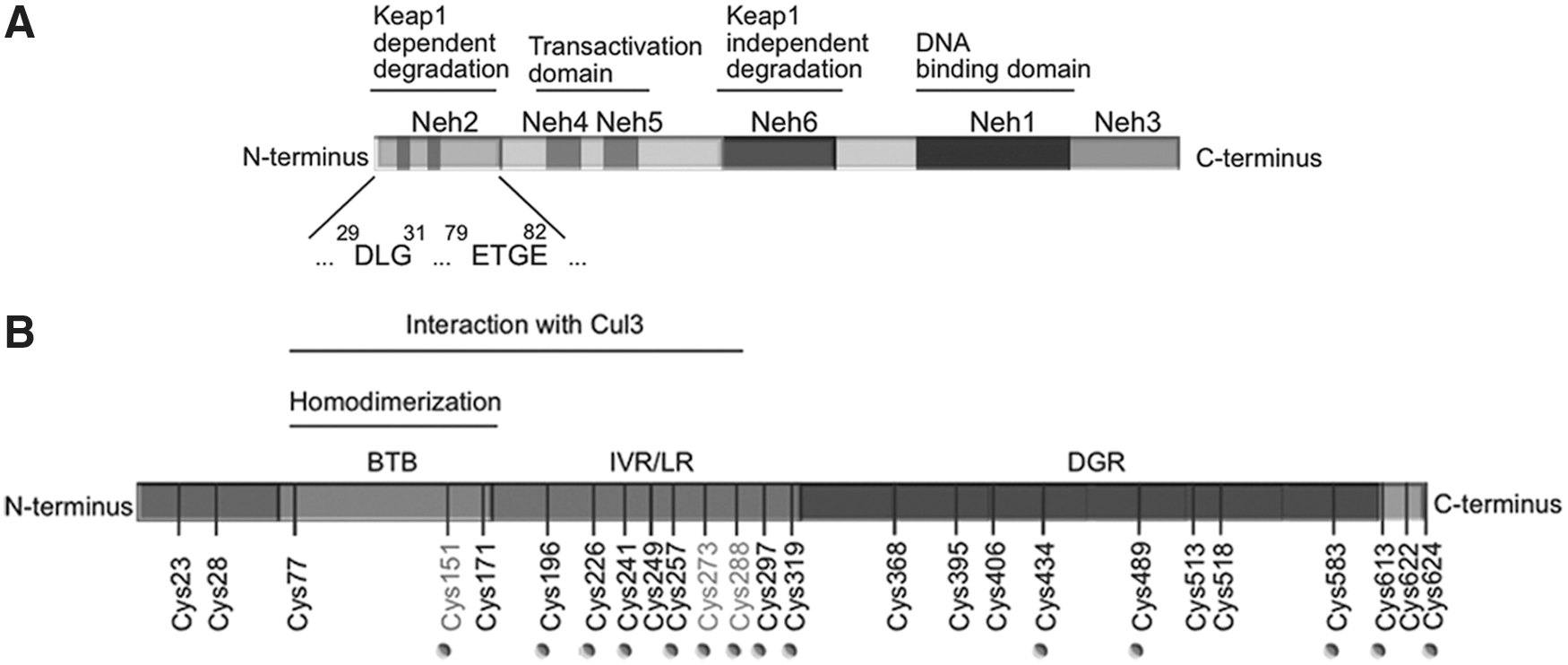
The main mechanism of regulation of NRF2 transcriptional activity is through control of protein stabilization by the E3 ligase adapter Kelch-like ECH-associated protein 1 (KEAP1). This is a homodimeric zinc (Zn) finger protein that bridges NRF2 with the E3 ligase complex formed by Cullin3 and Rbx proteins (Cul3/Rbx). Under homeostatic conditions, the N-terminal domain of the KEAP1 homodimer binds one molecule of NRF2 at two amino acid sequences of low (aspartate leucine glycine) and high glutamate, threonine, glycine, and glutamate (ETGE) affinity, thus presenting NRF2 to ubiquitination by Cul3/Rbx (152). However, in the presence of ectopic or endogenous electrophiles, KEAP1 is inactivated.
Mechanistically, electrophiles modify sulfhydryl groups of specific redox-sensitive cysteines of KEAP1, including C151, C273, and C288 (Fig. 1B). These modifications of KEAP1 lead to changes in NRF2 recognition, alterations in dimer conformation, or interaction with Cul3/Rbx. As a result, NRF2 escapes KEAP1-dependent degradation, accumulates in the nucleus, and activates ARE genes.
KEAP1 is one of the best-suited proteins to act as an electrophilic/redox sensor as it contains a large number of cysteine residues (27 in the human protein) and can function as an electrophile trap. However, other proteins such as phosphatase and tensin homolog (PTEN), which is mutated in a large number of human tumors, are also redox sensitive (55, 79, 84) and affect NRF2 activity. The catalytic C124 residue of PTEN can be modified through adduct formation with strong electrophiles such as synthetic triterpenoids (2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid-imidazolide; CDDO-Im) (114) and tert-butylhydroquinone (121). This modification results in loss of the PTEN lipid phosphatase activity and yields a more sustained activation of signaling events downstream of phosphoinositide 3-kinase, leading to NRF2 activation by a KEAP1-independent mechanism (117, 118). Thus, electrophilic targeting of NRF2 may involve not only KEAP1 but also other redox-sensitive enzymes. Moreover, KEAP1 interacts with other proteins that also contain the high-affinity binding motif, ETGE (57), such as inhibitor of nuclear factor kappa-B kinase subunit beta and Bcl-2 (78, 109). Hence, some results obtained from KEAP1 mutant or -deficient cells may not be necessarily related to the control of NRF2.
Several groups of electrophilic compounds induce NRF2 in cell culture and less frequently in animals or humans (120) [Fig. 2; for a detailed list of KEAP1 ligands, see refs. (37, 59, 91)]. Many of these compounds are used as nutraceuticals, and for some of them, there is evidence of clinical efficacy. The most successful drug of this type is the ester derivative of fumaric acid, dimethyl fumarate (DMF) (87). DMF crosses the gastrointestinal barrier where it is converted into monomethyl fumarate. The first clinical use of DMF was for the topical treatment of psoriasis in 1994 (5). More recently, an oral formulation of DMF, known as BG12, was commercialized for the treatment of relapsing–remitting multiple sclerosis (14, 76). Other autoimmune diseases such as lupus erythematosus, asthma, and arthritis are under investigation with other formulations of fumarate esters (128, 153).
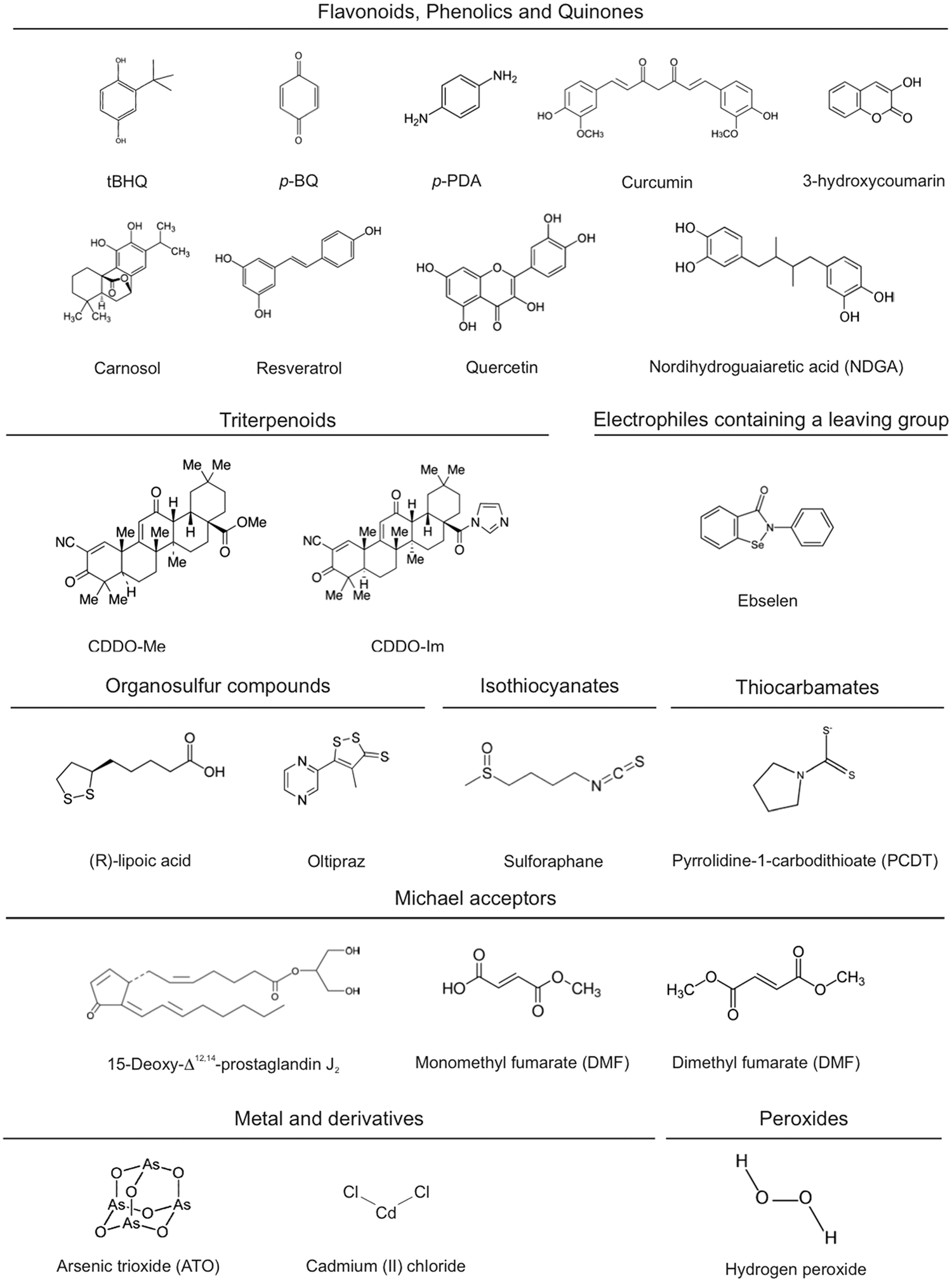
Other lines of research have focused on targeting NRF2 in degenerative diseases where low-grade chronic inflammation is present. One very potent synthetic triterpenoid, CDDO-methyl ester, bardoxolone methyl, has been studied in great detail for treatment of diabetic nephropathy (157). The initial excitement about this compound was set back by a small yet significant increase in the risk of heart failure. Importantly though, this effect appears not to be related to NRF2 targeting, but rather to alteration of endothelin signaling, leading to reduction in urine volume and sodium excretion in some patients with advanced chronic kidney disease (26). Bardoxolone methyl is now being studied in new indications for pulmonary arterial hypertension, melanoma, and Friedreich's ataxia.
A third NRF2 inducer that has reached the level of clinical studies is the isothiocyanate sulforaphane (SFN) isolated from broccoli sprout extracts. Contrary to DMF and bardoxolone methyl, a drawback of this compound is the absence of a pure formulation that could be used clinically and the lack of commercial value. Nevertheless, SFN provided proof of concept that NRF2 targeting has a therapeutic potential (40, 129, 132). Furthermore, NRF 2 agonists in clinical development are summarized in Table 1.
DMF, dimethyl fumarate; NRF2, nuclear factor (erythroid-derived 2)-like 2.
Inhibitors of ROS-Forming Enzymes
NADPH oxidase inhibitors
NOXs are transmembrane proteins comprising seven members (NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2). Each NOX isoform has specific tissue expression and regulation (10, 85). The catalytic core of all NOXs contains four conserved C-terminal NADPH-binding subregions and two flavin adenine dinucleotide (FAD)-binding subregions, as well as four conserved histidine residues, which coordinate two nonidentical iron heme prosthetic groups located between transmembrane domains 3 and 5. They are commonly referred to as the inner and outer heme, depending on their proximity to the cytosol and extracellular space, respectively. NOX activity was first described in neutrophils (7) where it forms superoxide anion radical (O2 −) as part of the phagocytic oxidative burst of the innate immune response (65). All NOX enzymes catalyze the reduction of extracytosolic oxygen (i.e., in phagosomes, endosomes, or the extracellular space) with cytosolic NADPH serving as an electron donor. The activity of most NOX isoforms is tightly regulated: NOX1, NOX2, and NOX3 require the presence of cytosolic proteins, while NOX4 generates ROS in a constitutive manner; NOX5 and DUOX isoforms require increased cellular Ca2+ concentrations and binding to N-terminal EF-hand domains for full activity. NOX5 is also a notable exception with respect to preclinical target validation; it is present in several mammals, including humans, but not in mice and rats. Increasing evidence shows that inhibition of NOX activity can be beneficial in multiple models of human diseases [for details, see Casas et al. in the same Forum (24)]. In addition, NOX2-derived ROS can have anti-inflammatory effects under certain conditions such as rheumatoid arthritis and multiple sclerosis (67). This provides a rationale for the development of NOX activators. Many advantages and pitfalls of currently available NOX inhibitors have been recently comprehensively reviewed in (4) and (69). In this study, we focus on three chemical compound families, with one compound currently in clinical development (Table 2).
NO, nitric oxide; NOX, reduced nicotinamide adenine dinucleotide phosphate oxidase.
GKT136901 and GKT137831 were developed by GenKyoTex to explore structure–activity relationship along pyrazolopyridine dione compounds (82). These compounds were selected based on oral bioavailability and beneficial pharmacokinetic parameters (4, 70). They block NOX1, NOX4, NOX5, and DUOX (142) activity in the micromolar range. In terms of off-targets effects, GKT136901 also scavenges peroxynitrite (125), but no interference was identified with other redox-sensitive enzymes, G-protein-coupled receptors, kinases, ion channels, or other enzyme activity (4). However, GKT136901 interacts with Amplex Red fluorescence and dose dependently decreases the signal, thereby complicating the interpretation of in vitro results (4). Preclinical results show that GKT137831 reduces glomerular injury and structural changes, as well as macrophage infiltration and proinflammatory transcription factor expression, in models of diabetic nephropathy (54, 70). GKT137831 has entered a clinical study phase II clinical trial, testing its efficacy in diabetic type 2 patients with diabetic nephropathy (50a) (study completed 2015).
Vasopharm developed the triazolo pyrimidine, VAS2870, following a screening approach for NOX2 inhibitors (139, 148). Its derivative, VAS3947, was later generated to slightly improve VAS solubility while keeping a similar NOX inhibitory profile.
Both compounds are able to inhibit different NOX isoforms, such as NOX2 (4), NOX 4, and NOX5 (3, 81), in the micromolar range. Intrathecal injection of VAS2890 significantly reduced cerebral infarct volume and ROS production in a mouse stroke model, suggesting a crucial contribution of one or more NOX enzymes in stroke (Table 2).
However, VAS2870 presents a number of limitations: (i) it blocks NOX2-derived ROS in neutrophils; (ii) its mode of action is independent of NOX2 (50); (iii) it is cytotoxic at low concentrations (171); and (iv) it exerts thioalkylation of cysteine residues in vitro, with so far unknown functional relevance (4, 144). In terms of drug development, a proof of principle of VAS compounds in humans is currently unfeasible due to their low solubility and unknown oral pharmacokinetic profile.
Recently, GSK2795039, a novel NOX2 inhibitor, abolished NOX2-induced ROS production in a model of paw inflammation and is protective in an animal model of acute pancreatitis (62). A pharmacokinetic/pharmacodynamic evaluation indicates that GSK2795039 is suitable for in vivo use. Further assessment of this compound will provide insights regarding its possible utility for validation of NOX2 as a pharmacological target.
XO inhibitors
Xanthine oxidoreductase (XOR), a 300-kDa homodimer, can exist as an NAD-dependent dehydrogenase (XD) or as an O2-dependent oxidase (XO), depending on the oxidation state of its cysteine thiols (95). XD can be converted into the ROS-generating XO either by formation of intramolecular disulfide bonds (reversible) or by proteolytic cleavage of a loop region connecting the FAD-binding domain and the molybdenum-binding domain (irreversible) (107). While XD depends on NAD+ (31, 140), XO uses O2 as electron acceptor and generates O2 − and H2O2 as products (110). As a consequence, XO conversion from XD could be a direct consequence of increased oxidative stress and results in further production of ROS by XO.
XO may contribute to the pathogenesis of various diseases, such as coronary artery disease, type 2 diabetes, and idiopathic dilated cardiomyopathy (21, 22). The XO inhibitor, allopurinol, an analog of hypoxanthine, and its active metabolite, oxypurinol, have been in clinical use for more than 40 years for the treatment of hyperuricemia and gout (41). A recent meta-analysis of 38 clinical trials with allopurinol or oxypurinol in patients with chronic heart failure and coronary artery disease has concluded that XO inhibition improves endothelial function and circulating markers of oxidative stress in patients with, or at risk of, cardiovascular disease (61). Because heterogeneity in those studies made it impossible to come to a conclusion on the effect of XO inhibitors on cardiac outcome, larger prospective multicenter trials are needed (61). Most recently, a study involving 253 high-risk heart failure patients with elevated uric acid levels failed to show improvement with allopurinol in clinical and functional parameters (53).
In 2009, the XOR inhibitor, febuxostat (TEI-6720, TMX-67), was approved by the Food and Drug Administration and marketed for gout (9) as more selective and potent than allopurinol and oxypurinol (110). In contrast to allopurinol, febuxostat has no structural similarity to a purine. Therefore, it has no effects on the activities of other enzymes involved in purine and pyrimidine metabolism, such as guanine deaminase, hypoxanthine-guanine phosphoribosyltransferase, purine nucleoside phosphorylase, orotate phosphoribosyltransferase, and orotidine-5V-monophosphate decarboxylase, compared with allopurinol (166). Contrary to allopurinol and oxypurinol, febuxostat, a potent inhibitor of both XO and XD (146), forms stable long-lasting complexes with the oxidized XOR (111). Its therapeutic application may be useful in cases of allopurinol incompatibility (8). From an experimental point of view, febuxostat may be a superior tool over allopurinol, which may have intrinsic radical scavenging properties that could make it difficult to distinguish between its antioxidant effects and XO inhibition. For example, it was proposed that the protective effects of allopurinol after hypoxia cannot be entirely explained by XO inhibition alone (104).
Another compound used in preclinical studies is BOF-4272 [sodium-8-(3-methoxy-4-phenylsulfinyl-phenyl) pyrazolo[1,5-a]-1,3,5-triazine-4-olate monohydrate] (112), which specifically inhibits XO-based O2 − generation (94, 100, 123, 145). However, it could not be tested clinically because of unfavorable pharmacokinetics due to both hepatic metabolism and poor intestinal absorption (108).
Other newly introduced XO inhibitors, such as naphtoflavons, 1,3,5-triazine-based purine analogs, and topiroxostat (FYX-051, 4-[5-pyridin-4-yl-1H-[1, 2, 4] triazol-3-yl]pyridine-2-carbonitrile), are currently being tested in preclinical studies (86, 93, 108, 131). A selection of substances in clinical development is shown in Table 3.
PD, Parkinson's Disease; MAO, monoamine oxidase; BOF-4272 sodium,7-[4-(benzenesulfinyl)-3-methoxyphenyl]-1,3,9-triaza-5-azanidabicyclo[4.3.0]nona-3,6,8-trien-2-one; XO, xanthine oxidase; XOR, xanthine oxidoreductase.
MAO inhibitors
The attention on MAO as a drug target has been driven by the serendipitous discovery of the antidepressant effect of the antitubercular agent, iproniazid, which was found to act as an MAO inhibitor (35). This observation paved the way to the clinical use of MAO inhibition in depressive disorders (130). Recently, MAO has become also a drug target for ROS-related pathologies. Due to its localization on the outer mitochondrial membrane, H2O2 and other MAO products [aldehydes and ammonia; for details, see Casas et al. in the same Forum (24)] can accumulate in the mitochondria to a significant extent and affect mitochondrial function (73). This can further lead to amplification of oxidative stress and cell damage so that the inhibition of MAO is beneficial in a number of disease models [for details, see Casas et al. in the same Forum (24); (11, 17, 18, 36, 73, 74, 98, 136, 156, 165)]. With the possible exception of NOX4 [for details, see Hirschhäuser et al. in the same Forum (63)]), MAO is the only known mitochondrial ROS source that can be inhibited pharmacologically without interfering with energy metabolism.
MAO exists in two isoforms, A and B, which generate H2O2 as a by-product during the oxidative deamination of biogenic monoamines. A wide range of MAO inhibitors are in clinical use, targeting one or both isoforms. Clorgyline is the prototypic MAO-A-specific inhibitor, while deprenyl inhibits MAO-B, and pargyline is nonselective. Recently, other more selective MAO inhibitors have been developed for the treatment of depressive disorders (130). Of those, phenelzine, isocarboxazid, and tranylcypromine are nonselective and irreversible MAO inhibitors, while moclobemide, toloxatone, and pirlindole are MAO-A selective and reversible. Selective and irreversible MAO-B inhibitors, such as selegiline and rasagiline, are widely prescribed for the treatment of affective and neurodegenerative disorders (Table 3), for example, mild symptoms of Parkinson's disease (PD) and associated motor fluctuations (30). Recently, specific and reversible MAO-B inhibitor, safinamide, has been launched in Germany for the treatment of mid- to late-stage PD in combination with levodopa or other PD therapies (15, 16). The therapeutic potential of MAO-B inhibitors is currently being evaluated also for the treatment of Alzheimer's disease. GABA formation from reactive astrocytes is mediated by MAO-B and affects synaptic plasticity, learning, and memory (71). Since astrocytic GABA and MAO-B are upregulated also in postmortem brains of individuals affected by Alzheimer's disease, MAO-B inhibition has been proposed as a potentially effective therapeutic strategy for treating memory impairment in this disease. Indeed, ladostigil, a dual acetylcholine butyrylcholine esterase and brain-selective MAO-A and -B inhibitor, was shown to antagonize scopolamine-induced impairment in spatial memory (66). More recently, a new small-molecule MAO-B inhibitor, EVT 302, is currently in phase IIb clinical trial for the treatment of Alzheimer's disease.
MAO inhibition can also be the result of an off-target effect. For example, the PPAR-gamma agonist, pioglitazone, used for the treatment of type 2 diabetes, specifically inhibits MAO-B in a reversible manner (12), a property that is not shared by other members of the glitazone family, such as troglitazone and rosiglitazone. Importantly, this off-target effect may contribute to the beneficial effects of pioglitazone in diabetic cardiomyopathy.
To date, MAO inhibitors have been used in patients to preserve or increase monoamine levels. It remains to be investigated clinically whether MAO inhibitors modulate oxidative stress-based pathologies and whether their use can be extended to other indications. The most relevant hurdle in the clinical development of MAO inhibitors is represented by a hypertensive reaction occurring when selective MAO-A inhibition is combined with intake of tyramine-rich food, such as aged cheese and alcoholic beverages (43). Tyramine is mostly oxidized by intestinal MAO-A; MAO-A inhibition causes an increase in circulating tyramine, which is taken up by postganglionic sympathetic neurons and induces noradrenaline release. However, MAO-B and reversible MAO-A inhibitors are devoid of this potential risk (167). Other minor contraindications and concerns related to MAO inhibitors are listed in (162).
NOS inhibitors
Nitric oxide (NO) is another ROS, although mostly with beneficial effects. However, under certain conditions, overproduction may cause cell death, for example, in neurotrauma and stroke. Most NOS inhibitors are based on displacing the substrate, arginine, off its binding site. However, none of these has been approved as a drug for any indication. The most dramatic failure was NG-mono-methyl-
The most advanced and currently most successful therapeutic approach is to target another and more unique binding site in NOS, the redox-sensitive cofactor, tetrahydrobiopterin (13, 48). Vasopharm's VAS203 has been successfully developed up to phase II for traumatic brain injury (141) (Table 4).
GW273629, (3-[[2-[(1-iminoethyl)amino]ethyl]sulfonyl]-l-alanine); L-NIL-TA, L-N6-(1-iminoethyl)lysine-5-tetrazole-amide; L-NMMA, 1-(4-aminopentyl)-2-methylguanidine; SC-51, L-N6-(1-iminoethyl)lysine 5-tetrazole amide.
Inhibitors of ROS Toxification
These inhibitors target enzymes that do not produce ROS but metabolize ROS to other more toxic species. The most prominent example is myeloperoxidase (MPO).
MPO inhibitors
MPO is a heme protein that can use H2O2 to oxidize Cl− to the highly reactive hypochlorous acid (HOCl), a potent oxidizing agent, but can also generate free radicals through its catalytic peroxidase cycle (77). Besides the major halide Cl−, MPO can also utilize bromide (Br−) to form brominating species, including hypobromous acid (HOBr) (60).
MPO is abundant in neutrophils and certain macrophages where it plays a role in the innate immune response. MPO-derived oxidants also have the potential to cause host tissue injury via initiation of post-translational protein modifications (i.e., chlorination) of proteins (115, 164) and lipid peroxidation (124). As a result, MPO-mediated oxidative damage is thought to contribute to a wide range of chronic inflammatory diseases, including cardiovascular and neuroinflammatory diseases (33, 106). The extracellular Br− concentration is much lower than that of Cl− (149). Thus, the physiological relevance of brominating oxidants such as HOBr, although they elicit antimicrobial effects in vitro (80, 159), has yet to be determined.
However, complete deficiency of MPO can be detrimental. For example, mice deficient in MPO and the low-density lipoprotein receptor (Ldlr), that is, Mpo−/−Ldlr−/−mice, develop larger atherosclerotic lesions compared with Ldlr−/−mice (20), and engraftment of bone marrow from Mpo−/−mice into Ldlr−/−mice increases rather than decreases the size of atherosclerotic lesions (20). Moreover, mice lacking MPO are more susceptible to experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis (19), and are protected from some features of PD (28). As a result of the implied overall benefit of phagocyte MPO, pharmacological strategies to attenuate MPO-mediated inadvertent oxidant damage aim at partial rather than complete inhibition of the enzyme.
Until recently, no specific MPO inhibitors were described that could be considered drug candidates. Although a number of commercially available compounds, including hydroxamic acids, hydrazines, and hydrazides, were used previously to inhibit the catalytic activity of MPO (92), they are not specific and also inhibit other heme peroxidases.
More recently, AstraZeneca found that 2-thioxanthines are potent and selective suicide inhibitors of MPO. Upon oxidation by MPO compound I, the thioxanthine radical forms an adduct with the heme prosthetic group of the enzyme, resulting in inactivation of MPO (150). These new compounds inhibit MPO activity in plasma, decrease protein chlorination in a mouse model of peritonitis, and elicit a range of beneficial effects in various disease models, without interfering with the killing of bacteria by neutrophils or other peroxidases, for example, thyroid peroxidase or lactoperoxidase activity (150). A number of thioxanthines have yielded positive results in preclinical and clinical studies. For example, the thioxanthine, AZD5904, stopped progression of emphysema and small airway remodeling and partially protected against pulmonary hypertension in a guinea pig model of chronic obstructive pulmonary disease (COPD) induced by exposure to cigarette smoke (29). In addition to AZD5904 entering phase I clinical trials for COPD and multiple sclerosis, AstraZeneca has completed a phase IIA clinical trial with another thioxanthine, AZD3241, in patients with PD (116) (Table 5).
MPO, myeloperoxidase.
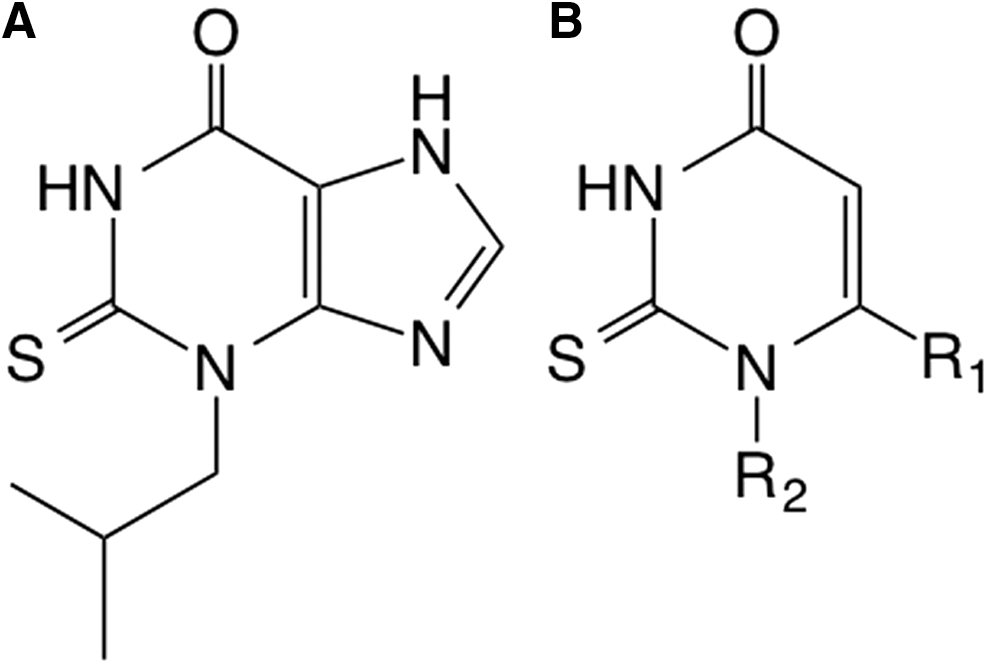
Another small-molecule inhibitor of MPO is INV-315 with a submicromolar IC50 [0.9 μM; (88)]. INV-315 decreases plaque burden and improves endothelial function in apolipoprotein E-deficient mice fed a high-fat diet for 16 weeks, a commonly used mouse model of atherosclerosis (88). However, no direct evidence for MPO inhibition or improved endothelial function was provided in that study. Pfizer, Inc., has also implemented a discovery program targeting MPO in inflammation and has filed patents for 2-thiopyrimidones (Fig. 3), which have a structure that is similar to 2-thioxanthines, suggesting that they may also act as suicide inhibitors by forming adducts with the heme moiety of MPO (23).
Functional Repair of ROS-Induced Protein Damage
This category of ROS-related drugs does not modulate ROS formation, but corrects some of its functional consequences. In the present review, we focus on NO-cGMP signaling, which appears to be one of the major mechanisms of deregulation initiated by oxidative stress (97). ROS can interfere with NO-cGMP signaling in three manners: • by uncoupling NOS, • by chemically scavenging NO, or • by oxidatively damaging the NO receptor, sGC.
This essentially leads to four therapeutic options: • to recouple uc-NOS [e.g., in peripheral arterial disease (Clinical Trials Registry ACTRN12609000882224)] • to replenish scavenged NO via NO donor compounds • to sensitize sGC for lower NO levels [e.g., in pulmonary arterial hypertension (52) and chronic thromboembolic pulmonary hypertension (51)] • to (re-)activate oxidatively damaged and heme-free sGC (apo-sGC) (e.g., in calcific aortic valve stenosis [NCT02481258] and neuropathic pain [NCT00799656])
NO donors
For over 100 years, NO-releasing drugs have been in clinical use. However, they have several serious side effects that will most likely lead to their eventual replacement. First, many NO donors are subject to tolerance, leading to loss of efficacy and requiring treatment interruptions. Moreover, they can lead to systemic hypotension and reflex tachycardia. Another concern is the fact that under oxidative stress, additional NO from NO donors leads to a spillover and sGC activation, but may also be metabolized to peroxynitrite. Thus, with NO donors, NO-cGMP signaling is partially and acutely recovered, but this happens at the expense of a chronic buildup of unwanted post-translational modifications such as protein tyrosine nitration (133). In addition, NO-drug hybrid molecules comprising an established drug and an NO-releasing moiety have been developed with the aim to preserve the pharmacological activity of the lead structure and add possibly beneficial effects of NO. Of the many compounds tested and developed, a series of NO-NSAIDs (nonsteroidal anti-inflammatory drugs), nitrosylated adrenoreceptor antagonist moxisylate (S-NO-moxisylate), and latanoprostene bunod (VESNEO®), an NO-donating prostaglandin F2-alpha analog, are currently most advanced. The latter is currently in phase III clinical development for the reduction of intraocular pressure in patients with glaucoma and ocular hypertension. Results from the phase 2b study confirmed that the drug is safe (158). Whether these combinations will not have similar limitations as other NO donors remains to be seen.
HNO donors
Besides classical NO donors and NO-drug hybrid molecules, recent preclinical studies and a phase IIa study reveal the therapeutic potential of an NO-related species, nitroxyl (HNO), which is developed as an HNO-donating drug (CXL-1020) by Cardioxyl, for acute decompensated heart failure therapy (122). However, serious inflammatory irritation at the injection site led to development of a second-generation HNO donor, CXL-1427, which is currently in clinical phase II testing. In contrast to NO donors, HNO donors such as Angeli's Salt/HNO appear to not induce tolerance, at least preclinically (6, 68, 103). Interestingly, HNO seems resistant towards scavenging by superoxide and retains efficacy after repeated infusions (45, 103, 113, 122, 151). However, further proof-of-concept studies need to be performed with safe HNO donors.
Recoupling uc-NOS
Oxidative damage of NOS is seen predominately not only for NOS3/eNOS but also for NOS1/nNOS (102). For this, two reversible processes are important, the oxidation of the redox-sensitive NOS cofactor, tetrahydrobiopetrin (BH4) (119), and the accumulation of an endogenous antagonist at the arginine substrate binding site, asymmetric-dimethyl-
To replenish the BH4 binding site, BH4 substitution is an option (143). However, BH4 therapy under oxidative stress may also carry the risk of leading to BH2 accumulation, a BH4 antagonist at the NOS BH4 binding site (13). The so-called salvage pathway recycles oxidized BH2 back to BH4 via dihydrofolate reductase (25). Moreover, angiotensin II type 1 receptor blockers and statins may, among other actions, increase the expression of the BH4-forming GTP cyclohydrolase 1 and therefore normalize low BH4 levels (160). High doses of
sGC stimulators and activators
Although stimulation and activation of sGC may sound similar, both innovative drug classes display entirely different modes of action and target different redox and disease states of the NO receptor, sGC. sGC stimulators (sGCs) such as riociguat (BAY 63-2521), vericiguat (BAY 1021189), BAY 41-8543, and BAY 60-4552, and YC-1 (49) bind to an allosteric binding site of Fe(II)heme-containing sGC and allosterically sensitize the enzyme for diminished biophase levels of endogenous NO. In a disease condition where biophase levels of NO are diminished, for example, by oxidative stress, higher or physiological increases in cGMP tissue levels can be achieved. Clinical indications may be similar to NO donors, but without the risk of tolerance and protein nitration as an accumulating by-product.
In contrast, sGC activators (sGCa), such as cinaciguat (BAY 58-2667), ataciguat (HMR 1766), and S3448 (126), activate only Fe(III)heme-oxidized or heme-free (apo-)sGC. They do this by either replacing the weakly bound oxidized heme in (apo-)sGC or by directly occupying the orphaned heme pocket in apo-sGC (138).
Otherwise, apo-sGC would be ubiquitinated at the empty heme binding site and degraded (64, 99). Therefore, sGC activators also stabilize apo-sGC. The ratio of oxidized or apo-sGC to Fe(II)-sGC is increased under oxidative stress conditions (42). In a condition where just NO levels are diminished, but Fe(II)sGC is intact, sGCa would be ineffective. Most recently two other sGCa, GlaxoSmithKline's GSK218123A and Boehringher-Ingelheims's Bl 703704, have been tested preclinically in different animal models of hypertension (32) and kidney diseases (137). The possible benefit of this new compound class and precise mechanism of action, as well as safety, need to be further validated.
Conclusion
In recent years, considerable data have accrued, indicating that disturbances in redox homeostasis are a common mechanism in different cardiovascular, neurological, and metabolic diseases. However, oxidative stress was hitherto not pharmacologically targetable, and the only strategy tested so far, using antioxidants, has been ineffective or even harmful. A possible reason for this is the lack of specificity for disease triggering versus physiological ROS that have a signaling, rather than pathological, role. Furthermore, ROS scavenging by antioxidants takes place in all (sub)cellular locations, not just those relevant for the disease. Innovative drugs need to target disease-relevant ROS-producing enzymes, ROS toxifying enzymes, or proteins damaged by ROS. For all of these, small molecules have become available that are able to perturb specific targets and allow for therapeutic proof-of-concept studies.
These include not only new compounds but also some well-characterized drugs, such as allopurinol and MAO isoform-selective inhibitors, which have been clinically used for decades, although not with the purpose to inhibit ROS formation. In addition, sGC stimulators (in the clinic), NOX inhibitor (entering phase III), NOS inhibitors (phase II-III), sGC activators (phase I-II), and superoxide dismutase mimetics such as GC4410 (phase I) [for details, see Schmidt et al. in the same Forum (127)] are rapidly gaining relevance. Other possible clinical candidates are, for example, mitochondria targeted antioxidants such as mitoquinone and mito TEMPO [for details, see Schmidt et al. in the same Forum (127)].
However, in several cases (e.g., NOX inhibitors), there is an unmet need for isoform-selective drugs. Finally, promising results have been obtained with activators of the transcription factor NRF2, even though in this case the mechanisms are more complex. In particular, one NRF2-activating compound, BG12, is effective and approved for the treatment of multiple sclerosis and, following its success, other NRF2 activators are currently being tested in proof-of-principle studies for various inflammatory diseases (Fig. 4).
Outlook
New, more specific pharmacological agents and future drugs are likely to transform the field of oxidative stress, with its many potential medical implications. Indirectly acting compounds (e.g., sGCs) have already provided proof of concept. The final breakthrough will be achieved when inhibitors of ROS-forming enzymes will enter evidence-based medicine.
Footnotes
Acknowledgments
Several authors of this review were supported by the European Cooperation in Science and Technology (COST Action BM1203/EU-ROS). H.H.H.W.S. is the recipient of an ERC Advanced Grant and Marie-Curie IRG and co-leads a EUROSTARS program. R.S. is supported by a Senior Principal Research Fellowship from the National Health and Medical Research Council of Australia N.K. is supported by an EFSD/Sanofi Award.
Author Disclosure Statement
Vincent Jaquet holds shares in Genkyotex SA and Harald H.H.W. Schmidt in Vasopharm GmbH. For the remaining authors, no competing financial interests exist.